
Submitted:
22 March 2024
Posted:
25 March 2024
You are already at the latest version
Abstract
Hepatitis C virus (HCV) is a major medical health burden and leading cause of chronic liver disease and cancer worldwide. More than 58 million people are chronically infected with HCV, with 1.5 million new infections occurring each year. An effective HCV vaccine is a major public health and medical need as recognized by the World Health Organization. However, due to the high variability of the virus and its ability to escape the immune response, HCV rapidly accumulates mutations making a vaccine a formidable challenge. An effective vaccine must elicit broadly neutralizing antibodies (bnAbs) in a consistent fashion. After decades of studies from basic research through clinical development, the antigen of choice is considered to be the E1E2 envelope glycoprotein due to conserved, broadly neutralizing antigenic domains located in the constituent subunits of E1, E2, and the E1E2 heterodimeric complex itself. The challenge has been elicitation of robust humoral and cellular responses leading to broad virus neutralization due to the relatively low immunogenicity of this antigen. In view of this challenge, structure-based vaccine design approaches to stabilize key antigenic domains have been hampered due to the lack of E1E2 atomic-level resolution structures to guide them. Another challenge has been development of a delivery platform in which a multivalent form of the antigen can be presented in order to elicit a more robust anti-HCV immune response. Recent nanoparticle vaccines are gaining prominence in the field due to their ability to facilitate a controlled multivalent presentation and trafficking to lymph nodes, where they can interact with both the cellular and humoral components of the immune system. This review focuses on recent advances in understanding the E1E2 heterodimeric structure to facilitate a rational design approach and the potential for development of a multivalent nanoparticle-based HCV E1E2 vaccine. Both aspects are considered important in the development of an effective HCV vaccine that can effectively address viral diversity and escape.
Keywords:
HCV E1E2
; structure
; nanoparticles
; vaccine
1. Introduction
An estimated 58 million people are infected with hepatitis C virus (HCV) worldwide with 1.5 million new infections occurring each year [6]. It is a major medical and public health burden in which approximately 75% of infections become chronic, which lead can to liver cirrhosis or hepatocellular carcinoma, and a leading cause of liver-related deaths. The number of deaths associated with HCV infection in the United States has been increasing, and it is the primary indication for liver transplantation in the Western world [7,8,9]. The development of direct-acting antivirals (DAA) have led to have led to cure rates over 90% with HCV treatment, but does not prevent reinfection. Moreover, DAA treatment is inaccessible to those infected in developing and in developed countries due to high costs and/or infrastructure limitations [10,11,12]. Additionally, diagnosis of HCV infection often occurs at a late stage after infection due to the asymptomatic “silent nature” of the virus, and successful DAA treatment may not alter the risk for cancer. Reinfection also remains a problem even after successful treatment in subjects with continued at risk behavior such as injection drug use. For these reasons, the most viable method for controlling HCV infections worldwide is through development of an effective prophylactic vaccine[13].
Structural characterization of the E2 glycoprotein has provided substantial information on the major antigenic sites that are the targets of bnAb-binding, particularly as it pertains to binding to primary CD81 receptor binding domain [14,15,16,17,18,19,20,21,22]. More recently, the cryo-EM structural characterization of the E1E2 heterodimer, either as a membrane-extracted E1E2 heterodimer [23] or as a soluble, secreted E1E2 heterodimer ectodomain [1], was a major advance in the field to more fully understand the structural antigenic features of this complex molecule. This knowledge will greatly facilitate a structure-based design approach in order to optimize immune responses to the major bnAb antigenic determinants. However, development of a vaccine will be challenging for multiple reasons [24,25,26,27]. These include the genetic diversity of HCV of at least seven HCV genotypes that differ up to 30% in nucleotide sequence, which can be further subdivided into over 90 subtypes, flexibility of the conformational regions, glycan shielding of neutralizing epitopes, the presence of immunodominant non-neutralizing “decoy” epitopes, and the tendency for membrane solubilized E1E2 antigen preparations to form aggregates [25,28,29,30,31,32,33]. Moreover, direct cell-to-cell transmission of the virus, systemic circulation of virions associate with lipoproteins, and downregulation of major histocompatibility complex (MHC) expression are other mechanisms for the virus to escape protective immunity [30,34,35,36]. Although immune correlates of protection have yet to be defined for HCV, there is broad agreement that both B and T cell immunity contribute to the control of acute HCV infection [26,37,38]. Thus, the expectation is that an ideal vaccine will elicit high bnAb titers directed against multiple conserved E1E2 epitopes to ensure broad neutralization [39,40,41,42,43], in conjunction with cytotoxic and tissue-resident memory T cells, in order to achieve immunity and protection against a high diversity of HCV isolates.
For this review, we will focus on structure-guided approaches to enhance a B cell immune response to E1E2 antigenic determinants that is a primary component of the host defense against HCV infection. It is worth highlighting that such approaches is more complex than sterilizing immunity as observed for hepatitis A, B and E vaccines [28]. During an acute infection, spontaneous viral clearance occurs in about 25 percent of individuals that is typically correlated with a robust neutralizing antibody response early in infection [44,45,46,47]. The rate of clearance of a reinfection is improved with a shorter course of infection and an increased likelihood of viral clearance compared with primary infection suggesting that pre-existing immunity is important [48,49,50,51,52]. In support of this observation, bnAbs passively administered to humanized mice or chimpanzees protect against HCV infection [42,53,54] Also, passive immunization with anti-HCV antibodies before HCV challenge prevented infection in animal models [54,55,56,57,58]. However, passive immunization of chimpanzees with antibodies from an HCV-infected patient that neutralized infectivity of several HCV genotypes in cell culture only suppressed infection with homologous virus challenge and failed to protect against heterologous virus strains [55]. Moreover, in a human clinical study using membrane-extracted E1E2 formulated with MF59, an oil-in-water adjuvant, broadly neutralizing antibodies were observed but only in a small fraction of patients in which most patients had low titers and limited breadth of neutralization [59,60]. Therefore, approaches to enhance immunogenicity such as the development of adjuvant systems that enhance both cellular and humoral immunity and design of nanoparticle platforms that permit multivalent presentation of E1E2 and use of novel adjuvants will be essential to achieve immunity against the broad diversity of HCV isolates.
2. HCV E1E2 Diversity
As previously noted in the literature [24,35], the HCV E1E2 glycoproteins possess high sequence diversity, which is a major reason that HCV is a challenging vaccine target. This diversity is highlighted in Figure 1, which shows a phylogenetic tree generated with representative E1E2 sequences for 69 HCV genotypes and subtypes. As can be seen in the figure, between subtypes of the same genotype, there is often 10% or more in sequence divergence, while greater divergence is observed between genotypes (20-30%). E1E2 sequence variability is not uniform across the glycoprotein residues and domains [24,35]; the sequences are punctuated by highly variable regions, including hypervariable region (HVRs) 1 and 2 and the inter-genotypic variable region (IgVR) in E2. However, other E1E2 regions and sites are highly conserved [24,35], including cysteine residues that form known or putative disulfide bonds, and the majority of the N-glycosylation sites (4 sites in E1 and 11 sites in E2 in the genotype 1a H77 strain). Many conserved sites have been confirmed to be important for E1E2 folding and/or function through systematic mutagenesis studies of E2 and E1E2 [61,62,63]. As shown in Figure 1, currently available experimentally determined E1E2 and E2 glycoprotein structures, which are discussed in greater detail in the next section, only account for a few genotypes and subtypes, while they do represent three out of four globally prevalent subtypes noted by others (1a, 1b, 2a, and 3a) [64]. Of note, a recently described but currently unreleased cryo-EM structure contains a modified genotype 3a (S52 isolate) E1E2 [65].
A key feature of HCV diversity is the virus’s capability to form quasispecies in infected individuals and to actively escape the immune response [66]. This immune escape was highlighted in a clinical trial of a neutralizing monoclonal antibody, HCV1, that targets the highly conserved AS412 site (residues 412-423) in E2 [67,68]. Following liver transplantation and monoclonal antibody therapy, which led to dramatic viral reduction, viral rebound was observed in all treated patients along with rare mutations directly within the epitope, at residues 415 or 417 [68,69]. These mutations disrupted HCV1 antibody binding through mutation of a key epitope side chain or shift of a glycosylation site within the epitope [69]; the latter escape mechanism was also observed for another antibody targeting that E2 site [70]. Other E1E2 polymorphisms associated with resistance and escape have been noted previously [35], with certain polymorphisms outside known epitope sites leading to broad viral resistance or sensitivity [71,72], possibly in some cases due to effects on E1E2 dynamics and putative “open” and “closed” conformational states [73].
Recent observations have highlighted that HCV phenotypic diversity, based on analysis of viral neutralization sensitivity and resistance, often does not directly map onto overall E1E2 sequence identities and known genotypes and subtypes [74,75,76], likely due in part to the importance of polymorphisms noted above. These studies have separately identified representative reference panels of HCV strains spanning levels of sensitivity and resistance that can prospectively be used to perform standardized assessments of antibodies and immune sera, analogous to a commonly used global panel of HIV strains [77], however a single coordinated panel of HCV strains, versus multiple panels, would be advantageous for the HCV research community.
3. HCV E2 and E1E2 Structure
Antibody responses against the HCV E1E2 glycoprotein have been mapped to various antigenic domains and regions on the E1 and E2 subunits, which include targets of broadly-neutralizing, strain-specific, and non-neutralizing antibodies [78]. Antigenic targets on the E2 subunit have been named using different nomenclatures, including antigenic domains A-E, antigenic regions 1-5, or epitopes I-III (Figure 2) [42,79]. The most characterized broadly neutralizing antibodies inhibit entry by restricting host CD81 receptor binding and map to a region known as the neutralizing face of E2, which includes overlapping antigenic domains B, D, and E and antigenic region 3 (Figure 2). A contiguous helical region within the stem of E1 also has been defined as a neutralizing antibody target [80,81,82,83]. Antigenic regions 4 and 5, defined by neutralizing antibodies AR4A and AR5A, also confer broad recognition across HCV genotypes but, in contrast to other sites, depend on an intact E1E2 heterodimer for antibody recognition [84].
To date, numerous structures of truncated or modified forms of the E2 subunit ectodomain in complex with antibodies have been determined, including those belonging to genotypes 1a, 1b, 2a, and 6b [14,15,16,85,86,87,88]. The first of these structures, which utilized a truncated E2 protein spanning residues 412-645, defined the overall architecture of the globular E2 core, which was found to contain a central β-sandwich flanked by front and back layers made of loops and short stretches of secondary structure elements [14]. While the core structure of E2 has been found to be largely conserved across genotypes despite sequence diversity, some regions of E2 have been observed to adopt conformational differences, even within the same genotype. These include β-hairpin and extended conformations of the AS412 (domain E) epitope and conformational flexibility of the E2 front layer [18,80,81,87]. Moreover, the CD81 binding site of E2 (residues 418-422 and 520-539) undergoes substantial conformational changes when bound to the large extracellular loop of CD81 [89]. These findings coupled with previous studies showing substantial flexibility and functional interplay between the E2 HVR1, the front layer, and the CD81 binding site, underscore inherent structural plasticity of some regions of E2 that are thought to underlie differences in susceptibility to antibody neutralization [73,90,91].
In order to structurally characterize the HCV E1E2 envelope complex in a more native-like form, cryo-EM has recently become the technique of choice (Figure 3). Notably, this technique yielded the first structure of a full-length membrane-extracted form of the E1E2 heterodimer of genotype 1a bound by neutralizing antibodies [23]. This structure resolved the overall architecture of the E1E2 heterodimer, including regions of E1 and the C-terminus of E2, and defined for the first time the AR4 site of vulnerability. Subsequently, our group determined a cryo-EM structure of an engineered soluble form of the E1E2 heterodimer ectodomain (of genotype 1b) that we developed for vaccine efforts by replacing the transmembrane domains of E1 and E2 with a soluble self-assembling coiled-coil scaffold (Figure 3) [1]. Both heterodimer structures were found to share a similar E1E2 fold and architecture despite being of different genotypes possessing significant differences in amino acid sequences (22.8% between E1 subunits and 19.6% between E2 subunits), and one being completely liberated from the membrane [1]. Comparative analysis of the structures revealed several common features of the E1E2 heterodimer. The E1-E2 interface was made up entirely of non-covalent, predominantly hydrophobic interactions. A substantial contribution of two conserved E1 N-linked glycans, N196 and N305, to the interface accounted for roughly a third of the E1 interface with E2 [1]. Both E1E2 structures also allowed for a portion of the C-terminal domain of E2 to be resolved (residues 646-704), termed the base or bridging domain, which was found to pack against the back layer of the E2 core and against E1, and accounted for roughly 70% of the E2 interface with E1. This domain also contained the epitope of E1E2-specific neutralizing antibody AR4A, which was found to be one of the most highly conserved epitopes defined to date [1].
Several domains of E1 were also resolved in the cryo-EM structures [1,23]. The E1 N-terminal domain (NTD), spanning residues 192-205, was found to consist of two anti-parallel β-strands that packed against variable region 2, post-variable region 3, and the bridging/base domain of E2. The E1 NTD also contained the N-linked glycan at position N196, which, along with N305, was previously found to be critical for the E1-E2 interface and heterodimer integrity [63,82,92]. The E1 core domain, residues 206-255, consisted of a cluster of β-strands and contained two N-linked glycans, N209 and N250. The C-terminal loop (CTL) domain of E1, spanning residues 295-312, contained the N305 glycosylation site that, as noted above, packed against the bridging/base domain of E2 and was critical for heterodimer integrity. The CTL also contained the epitope for E1-specific neutralizing antibodies IGH520/IGH505 and IGH526 [23,80,81,82,83]. Missing from either of the structures, however, was the region linking the E1 core and the C-terminal loop, residues 256-294, which has been predicted to contain the putative fusogenic machinery [93,94,95].
Recently, a cryo-EM structure of a full-length membrane-extracted dimer of E1E2 heterodimers has also been reported, although not yet released, indicating possible higher order oligomers may represent the native state of the E1E2 glycoprotein on the HCV virion [65]. Interestingly, flexible regions of E2 and E1 that were not resolved in previous structures, including parts of HVR1, AS412, and membrane embedded portions of the E2 and E1 stem regions, were reportedly resolved in this structure. Structural definition of these regions will likely provide further insight into underlying structural features of E1E2 that play a role in membrane fusion and possibly those that define phenotypic differences in susceptibility to antibody neutralization [65].
4. Structure-Based Vaccine Design
To generate an effective vaccine for HCV and overcome the challenges of HCV diversity and immune evasion [35], recent efforts have increasingly explored the use of reverse vaccinology and structure-based vaccine design [96,97] to generate optimized vaccine antigens that will elicit broadly neutralizing antibodies that target key conserved sites on E1E2 [98]. These follow successful structure-based designs of antigens for other variable or dynamic viruses, including prefusion RSV F [99] and influenza hemagglutinin stem [100] antigens, which have both been in clinical trials, and recently approved for use in the case of prefusion RSV F [101,102]. Structure-based HCV antigen designs have included stabilized and scaffolded conserved epitopes [103,104,105], optimized E2 antigens with truncated or removed variable regions [106,107] or a targeted proline substitution to stabilize a key epitope [108], as well as display of E2 self-assembling protein nanoparticles [106]; these and other HCV antigen designs and strategies are described in a recent review [98]. Some E2 and E1E2 design strategies are shown in Figure 4.
Several recent advances and findings provide possible avenues to pursue in future HCV antigen design efforts. Importantly, the current availability of experimentally determined E1E2 structures, as discussed in the previous section, enables structure-based design of E1E2, rather than E2 alone or individual epitopes as in previous work, to optimize its stability, antigenicity, or other features. Additionally, possible “open” and “closed” states of E2 and E1E2 [73], or HVR1 entropy [90], which have been associated with viral neutralization sensitivity, can be utilized to tune E1E2 antigenicity, particularly if sufficient structural and dynamic details underlying those states can be defined. Of relevance, a recently described cryo-EM structure with E1E2 in dimeric form seems to provide details of a preferred conformation of HVR1 which was corroborated by AlphaFold2 structural modeling [65].
Other (non-structural) rational antigen design approaches represent promising strategies to address HCV diversity and escape. Frumento et al. identified E1E2 ectodomains associated with spontaneous viral clearance and improved neutralizing antibody breadth [109] that may useful in a vaccine, versus the H77 glycoprotein sequences which are commonly used in E2 and E1E2 antigens. Another strategy is the use of consensus ectodomains, which was utilized in HCV E2 [110] and HIV envelope glycoprotein [111,112] antigen designs. Finally, an additional means to address HCV diversity could be the use of multiple representative designed or natural E1E2 antigens in a vaccine that are representative of prevalent genotypes; given the previous success of self-assembling mosaic nanoparticles to display diverse coronavirus spike receptor binding domain antigens [113], an analogous strategy could be explored for displaying diverse representative HCV antigens.
5. Multivalent Delivery Platforms and Considerations
Subunit vaccines are often poorly immunogenic, a phenomenon routinely attributed to their size as particulate antigens have been known for quite some time to be highly immunogenic [114,115,116,117,118]. There is recent data in a study by Aung et al. [119] that puts a finer point on this concept. In that study, the authors showed that subunit vaccines were trafficked primarily to the subcapsular sinus or extracellular regions of lymph nodes and subsequently degraded by metalloproteases. Such degradation eliminates conformation-dependent epitopes on the associated antigens and hampers the immune response. Nanoparticle-sized antigens were localized instead to follicular dendritic cells (FDCs) where they remained intact and preserved such conformation-dependent epitopes and thus elicited a more robust immune response. In light of these recent observations and the historical data, it seems clear that increasing the size of a subunit vaccine is beneficial and thus a number of strategies have been developed toward that end. One strategy is to employ an adjuvant system that, when formulated with the antigen, produces a nano- or microparticulate vaccine. Common adjuvant systems in use to make particulate subunit vaccines are aluminum salts (Alum), polymers like poly(D,L-lactic-co-glycolic) acid (PLG), oil and water emulsions like MF59, liposomes and other vesicles, and micelle forming adjuvants. Micelles can be particularly advantageous for membrane-anchored antigens as the formulation process creates rosettes of antigens covering the exterior of the micelle. These kinds of formulations have been used in vaccines for influenza (Flublok) and SARS-CoV-2 (NVX-CoV2373) [120,121,122,123,124,125,126]. A second strategy to develop particulate vaccines is to construct virus-like particles (VLPs). VLPs are non-infectious but more closely mimic the native virion than subunit vaccines and other particulate platforms by using the viral structural proteins to self-assemble in a similar manner to the virus. Energix (hepatitis B virus) and Gardasil (human papilloma virus) are two prominent VLP-based vaccines. A third strategy for increasing the size of a subunit vaccine is the use of protein-based nanoparticles. These nanoparticle platforms are typically naturally occurring or engineered protein shells that allow multivalent display of subunit vaccines on the exterior [127,128,129,130,131,132,133]. These assemblies can be formed in cis, where the subunit vaccine and nanoparticle protomer is expressed as a single open reading frame and assembly yields a 100% occupied nanoparticle or in trans where the nanoparticle shell and subunit vaccine are produced separately and coupled post hoc, as in the case of the SpyCatcher-SpyTag system [134]. Each of these platforms is being explored for a potential HCV vaccine candidate [106,135,136]. The first studies, by Yan et al. [136] and by He et al. [106], used the E2 ectodomain and a modified E2 core ectodomain respectively as proof-of-principle antigens to be appended to nanoparticles. Given the importance of the E1E2 complex, and in particular the AR4/AR5 antigenic region in viral clearance [41,137], a nanoparticle presenting native E1E2 should be a high priority for HCV vaccine development. One nanoparticle study has been conducted with E1 and E2 [135], but this used a permuted E2-E1 version of the antigen which does not retain the native AR4/AR5 antigenic domain.
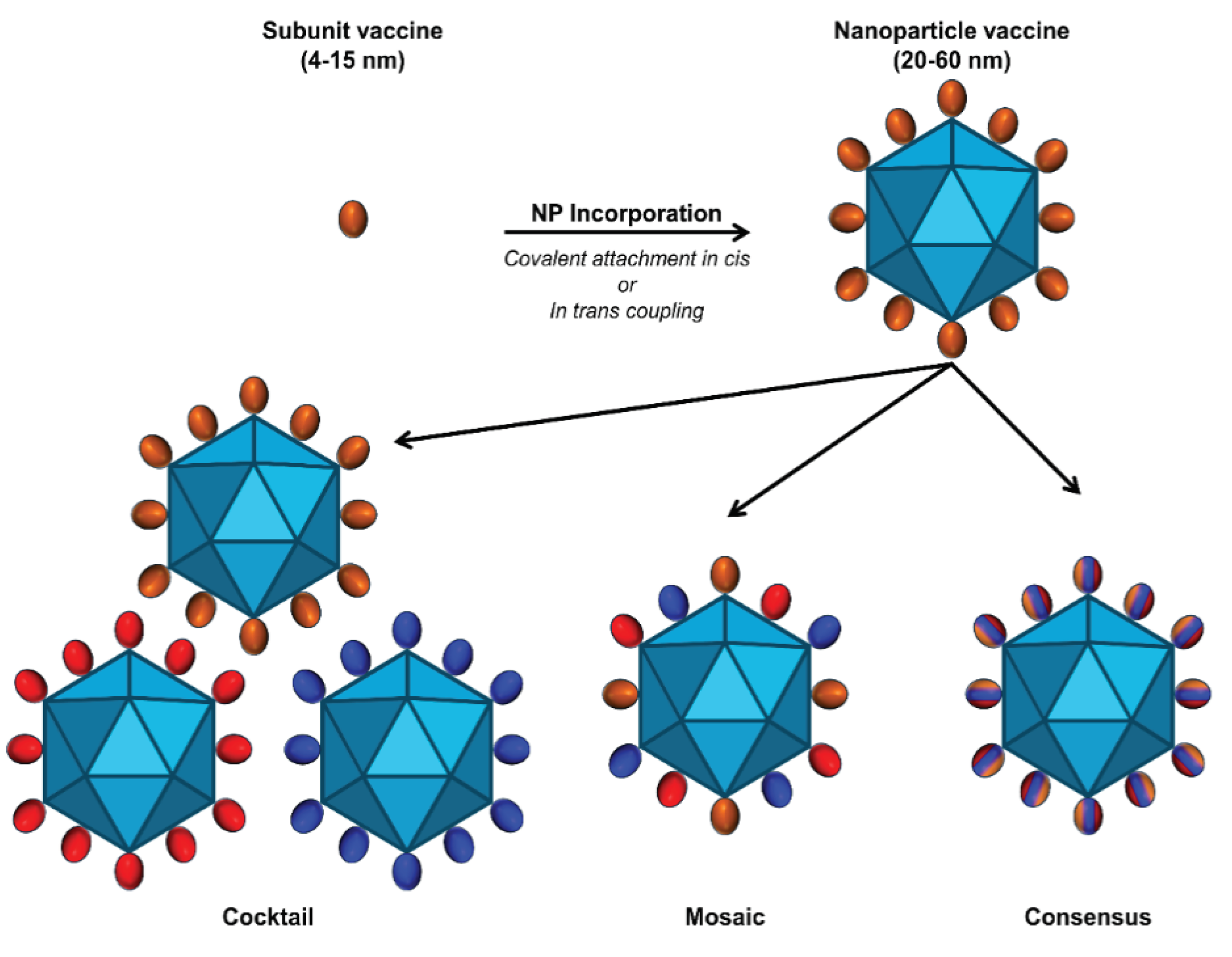
While the above platforms address the question of size in the context of a vaccine against HCV, the question that still remains is how to accommodate HCV diversity in the context of a particulate adjuvant, a VLP, or a nanoparticle. As described in the previous section, this problem can be overcome by the use of consensus sequences or mosaic vaccines composed of multiple antigens encompassing different genotypes or phenotypes contained within a single vaccine (Figure 5). These approaches have been used for vaccine trials against HIV and SARS-CoV-2 [113,138,139,140,141,142,143,144,145,146] and, importantly, are compatible with the adjuvant and nanoparticle platforms, but it is unclear if such approaches are compatible with VLPs. An additional approach is to use cocktails of different genotypic or phenotypic representatives mixed together after preparation and validation. This is the most straightforward approach, but requires a brute-force regimen of multiple separate preparations and validations for each different member of the cocktail. Which one of the above approaches is most likely to yield an effective HCV vaccine is currently an open question that will need to be evaluated experimentally.
6. Conclusions
Since its discovery in 1989, HCV has been a particularly vexing pathogen for vaccine development, in large part due to its high sequence diversity. However, significant advances have provided avenues to potentially overcome this sequence diversity. First, despite significant sequence variability among the different genotypes and subtypes, highly-conserved regions have been defined and characterized as antigenic regions [42,84,147,148,149,150,151,152,153,154,155] that give rise to bnAbs and can serve as potential targets for rational vaccine design. Second, after significant struggles, structural studies were successful, first for modified truncated versions of the E2 ectodomain [14,15], and subsequently for a more complete E2 ectodomain [16] in complex with neutralizing and non-neutralizing antibodies. More recently, the structure of membrane-extracted E1E2 in complex with multiple bnAbs was determined by cryo-EM [23], providing the first look at the antigenic domain AR4, which is bound by the bnAb AR4A and correlates with viral clearance [41,137]. In addition, our group developed a soluble, secreted form of the E1E2 complex, with the idea that an E1E2 antigen liberated from the membrane would prove more amenable to vaccine design efforts. The structure of the secreted E1E2 complex [1] shows that it preserves the native architecture of the E1E2 ectodomain outside the context of the membrane. This catalogue of structures (plus other structures likely to be determined) can be used for structure-based vaccine design for an E1E2-based HCV vaccine. Third, the development of nanoparticle platforms, both protein- and adjuvant-based, allows a multivalent presentation of E1E2 as a means to enhance its immunogenicity. Moreover, advances in these nanoparticle platforms such as plug-and-display technology [134] allows the potential development of mosaic E1E2 vaccines. It is not known what means of incorporating sequence diversity into an E1E2 vaccine will be successful, so making available as many options as possible is critical for successful vaccine development. Additional improvements such as incorporating the PADRE peptide [156,157,158,159], which activates CD4+ T cells, into nanoparticles could potentially boost the cellular immune response to an E1E2-based vaccine, thereby further enhancing its potency. Like pieces of a puzzle, these and other breakthroughs from research on HCV and other pathogens have come together to put the field of HCV vaccine development in a position to overcome the hurdle of HCV sequence diversity. This progress has the potential to deliver an HCV vaccine that elicits the breadth of neutralization required to achieve containment and eventual eradication.
Funding
This work was supported by NIH R01s AI132213 (B.G.P., E.A.T., and T.R.F.) and AI168048 (to B.G.P., G.A.O., E.A.T., and T.R.F.), R21 AI154100 (to B.G.P., E.A.T., and T.R.F.), and the University of Maryland Strategic Partnership: MPowering the State (T.R.F.).
Acknowledgments
None to declare.
Conflicts of Interest Statement
The authors have no conflicts of interest to declare.
References
- Metcalf, M.C.; Janus, B.M.; Yin, R.; Wang, R.; Guest, J.D.; Pozharski, E.; Law, M.; Mariuzza, R.A.; Toth, E.A.; Pierce, B.G.; et al. Structure of engineered hepatitis C virus E1E2 ectodomain in complex with neutralizing antibodies. Nat. Commun. 2023, 14, 3980. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Yusim, K.; Boykin, L.; Richardson, R. The Los Alamos hepatitis C sequence database. Bioinformatics 2005, 21, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.Y.; Wang, R.; Lau, P.; Garagusi, K.; Elkholy, K.; Toth, E.A.; Urbanowicz, R.A.; Guest, J.D.; Agnihotri, P.; et al. Structure-Based Design of Hepatitis C Virus E2 Glycoprotein Improves Serum Binding and Cross-Neutralization. Journal of virology 2020, 94, epublish. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- WHO, Hepatitis C Key Facts (WHO, 2022).
- Rosen, H.R. Chronic Hepatitis C Infection. New England Journal of Medicine 2011, 364, 2429–2438. [Google Scholar] [CrossRef]
- Ly, K.N.; Hughes, E.M.; Jiles, R.B.; Holmberg, S.D. Rising Mortality Associated With Hepatitis C Virus in the United States, 2003-2013. Clin Infect Dis 2016, 62, 1287–1288. [Google Scholar] [CrossRef] [PubMed]
- Spach, D.H. HCV Epidemiology in the United States. 2024.
- Chung, R.T.; Baumert, T.F. Curing chronic hepatitis C--the arc of a medical triumph. New England Journal of Medicine 2014, 370, 1576–1578. [Google Scholar] [CrossRef]
- Cox, A.L.; Thomas, D.L. Hepatitis C virus vaccines among people who inject drugs. Clin. Infect. Dis. 2013, 57 Suppl. 2, S46–S50. [Google Scholar] [CrossRef]
- Callaway, E. Hepatitis C drugs not reaching poor. Nature 2014, 508, 295–296. [Google Scholar] [CrossRef]
- Cox, A.L. MEDICINE. Global control of hepatitis C virus. Science 2015, 349, 790–791. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef]
- Flyak, A.I.; Ruiz, S.; Colbert, M.D.; Luong, T.; Crowe, J.E., Jr.; Bailey, J.R.; Bjorkman, P.J. HCV Broadly Neutralizing Antibodies Use a CDRH3 Disulfide Motif to Recognize an E2 Glycoprotein Site that Can Be Targeted for Vaccine Design. Cell Host Microbe 2018, 24, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Tzarum, N.; Giang, E.; Kong, L.; He, L.; Prentoe, J.; Augestad, E.; Hua, Y.; Castillo, S.; Lauer, G.M.; Bukh, J.; et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci. Adv. 2019, 5, eaav1882. [Google Scholar] [CrossRef]
- Kong, L.; Lee, D.E.; Kadam, R.U.; Liu, T.; Giang, E.; Nieusma, T.; Garces, F.; Tzarum, N.; Woods, V.L., Jr.; Ward, A.B.; et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc. Natl. Acad. Sci. U S A 2016, 113, 12768–12773. [Google Scholar] [CrossRef]
- Tzarum, N.; Wilson, I.A.; Law, M. The Neutralizing Face of Hepatitis C Virus E2 Envelope Glycoprotein. Frontiers in immunology 2018, 9, 1315. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pierce, B.G.; Wang, Q.; Keck, Z.Y.; Fuerst, T.R.; Foung, S.K.; Mariuzza, R.A. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J. Biol. Chem. 2015, 290, 10117–10125. [Google Scholar] [CrossRef]
- Yechezkel, I.; Law, M.; Tzarum, N. From Structural Studies to HCV Vaccine Design. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Aleman, F.; Tzarum, N.; Kong, L.; Nagy, K.; Zhu, J.; Wilson, I.A.; Law, M. Immunogenetic and structural analysis of a class of HCV broadly neutralizing antibodies and their precursors. Proc. Natl. Acad. Sci. U S A 2018, 115, 7569–7574. [Google Scholar] [CrossRef]
- Torrents de la Pena, A.; Sliepen, K.; Eshun-Wilson, L.; Newby, M.L.; Allen, J.D.; Zon, I.; Koekkoek, S.; Chumbe, A.; Crispin, M.; Schinkel, J.; et al. Structure of the hepatitis C virus E1E2 glycoprotein complex. Science 2022, 378, 263–269. [Google Scholar] [CrossRef]
- Fuerst, T.R.; Pierce, B.G.; Keck, Z.Y.; Foung, S.K.H. Designing a B Cell-Based Vaccine against a Highly Variable Hepatitis C Virus. Frontiers in microbiology 2017, 8, 2692. [Google Scholar] [CrossRef]
- Bailey, J.R.; Barnes, E.; Cox, A.L. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology 2019, 156, 418–430. [Google Scholar] [CrossRef]
- Duncan, J.D.; Urbanowicz, R.A.; Tarr, A.W.; Ball, J.K. Hepatitis C Virus Vaccine: Challenges and Prospects. Vaccines 2020, 8. [Google Scholar] [CrossRef]
- Law, M. Antibody Responses in Hepatitis C Infection. Cold Spring Harb. Perspect. Med. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.M. Designing an HCV vaccine: a unique convergence of prevention and therapy? Curr. Opin. Virol. 2017, 23, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Lavie, M.; Hanoulle, X.; Dubuisson, J. Glycan Shielding and Modulation of Hepatitis C Virus Neutralizing Antibodies. Frontiers in immunology 2018, 9, 910. [Google Scholar] [CrossRef]
- Cashman, S.B.; Marsden, B.D.; Dustin, L.B. The Humoral Immune Response to HCV: Understanding is Key to Vaccine Development. Frontiers in immunology 2014, 5, 550. [Google Scholar] [CrossRef] [PubMed]
- Prentoe, J.; Bukh, J. Hypervariable Region 1 in Envelope Protein 2 of Hepatitis C Virus: A Linchpin in Neutralizing Antibody Evasion and Viral Entry. Frontiers in immunology 2018, 9, 2146. [Google Scholar] [CrossRef]
- Bhattarai, N.; McLinden, J.H.; Xiang, J.; Kaufman, T.M.; Stapleton, J.T. Conserved Motifs within Hepatitis C Virus Envelope (E2) RNA and Protein Independently Inhibit T Cell Activation. PLoS Pathog. 2015, 11, e1005183. [Google Scholar] [CrossRef]
- Vijayamahantesh, V.; Patra, T.; Meyer, K.; Alameh, M.G.; Reagan, E.K.; Weissman, D.; Ray, R. Modified E2 Glycoprotein of Hepatitis C Virus Enhances Proinflammatory Cytokines and Protective Immune Response. J. Virol. 2022, 96, e0052322. [Google Scholar] [CrossRef]
- Dunlop, J.; Owsianka, A.; Cowton, V.; Patel, A. Current and future prophylactic vaccines for hepatitis C virus. Vaccine: Development and Therapy 2015, 2015, 31–44. [Google Scholar] [CrossRef]
- Pierce, B.G.; Keck, Z.Y.; Foung, S.K. Viral evasion and challenges of hepatitis C virus vaccine development. Curr. Opin. Virol. 2016, 20, 55–63. [Google Scholar] [CrossRef]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Fauvelle, C.; Colpitts, C.C.; Keck, Z.Y.; Pierce, B.G.; Foung, S.K.; Baumert, T.F. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert. Rev. Vaccines 2016, 15, 1535–1544. [Google Scholar] [CrossRef]
- Walker, C.M.; Grakoui, A. Hepatitis C virus: why do we need a vaccine to prevent a curable persistent infection? Current opinion in immunology 2015, 35, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, T.H.; Pedersen, J.; Prentoe, J.C.; Giang, E.; Keck, Z.Y.; Mikkelsen, L.S.; Law, M.; Foung, S.K.; Bukh, J. Breadth of neutralization and synergy of clinically relevant human monoclonal antibodies against HCV genotypes 1a, 1b, 2a, 2b, 2c, and 3a. Hepatology 2014, 60, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- de Jong, Y.P.; Dorner, M.; Mommersteeg, M.C.; Xiao, J.W.; Balazs, A.B.; Robbins, J.B.; Winer, B.Y.; Gerges, S.; Vega, K.; Labitt, R.N.; et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med. 2014, 6, 254ra129. [Google Scholar] [CrossRef] [PubMed]
- Kinchen, V.J.; Massaccesi, G.; Flyak, A.I.; Mankowski, M.C.; Colbert, M.D.; Osburn, W.O.; Ray, S.C.; Cox, A.L.; Crowe, J.E., Jr.; Bailey, J.R. Plasma deconvolution identifies broadly neutralizing antibodies associated with hepatitis C virus clearance. J. Clin. Invest. 2019, 129, 4786–4796. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef]
- Mankowski, M.C.; Kinchen, V.J.; Wasilewski, L.N.; Flyak, A.I.; Ray, S.C.; Crowe, J.E., Jr.; Bailey, J.R. Synergistic anti-HCV broadly neutralizing human monoclonal antibodies with independent mechanisms. Proc. Natl. Acad. Sci. U S A 2018, 115, E82–E91. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Laskey, S.; Wasilewski, L.N.; Munshaw, S.; Fanning, L.J.; Kenny-Walsh, E.; Ray, S.C. Constraints on viral evolution during chronic hepatitis C virus infection arising from a common-source exposure. J. Virol. 2012, 86, 12582–12590. [Google Scholar] [CrossRef]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J.A. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U S A 2004, 101, 10149–10154. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Snider, A.E.; Wells, B.L.; Latanich, R.; Bailey, J.R.; Thomas, D.L.; Cox, A.L.; Ray, S.C. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 2014, 59, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U S A 2007, 104, 6025–6030. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Walker, C.M. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 2005, 436, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.H.; Cox, A.; Hoover, D.R.; Wang, X.H.; Mao, Q.; Ray, S.; Strathdee, S.A.; Vlahov, D.; Thomas, D.L. Protection against persistence of hepatitis C. Lancet 2002, 359, 1478–1483. [Google Scholar] [CrossRef]
- Osburn, W.O.; Fisher, B.E.; Dowd, K.A.; Urban, G.; Liu, L.; Ray, S.C.; Thomas, D.L.; Cox, A.L. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010, 138, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Hahn, J.A.; Evans, J.; Shiboski, S.; Lum, P.; Delwart, E.; Tobler, L.; Andrews, W.; Avanesyan, L.; Cooper, S.; et al. Acute hepatitis C virus infection in young adult injection drug users: a prospective study of incident infection, resolution, and reinfection. J. Infect. Dis. 2009, 200, 1216–1226. [Google Scholar] [CrossRef]
- Grebely, J.; Prins, M.; Hellard, M.; Cox, A.L.; Osburn, W.O.; Lauer, G.; Page, K.; Lloyd, A.R.; Dore, G.J.; International Collaboration of Incident, H.I.V.; et al. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: towards a vaccine. Lancet Infect. Dis. 2012, 12, 408–414. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Wang, Y.; Lau, P.; Lund, G.; Rangarajan, S.; Fauvelle, C.; Liao, G.C.; Holtsberg, F.W.; Warfield, K.L.; Aman, M.J.; et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute hepatitis C virus infection in mice. Hepatology 2016, 64, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Morin, T.J.; Broering, T.J.; Leav, B.A.; Blair, B.M.; Rowley, K.J.; Boucher, E.N.; Wang, Y.; Cheslock, P.S.; Knauber, M.; Olsen, D.B.; et al. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog. 2012, 8, e1002895. [Google Scholar] [CrossRef]
- Bukh, J.; Engle, R.E.; Faulk, K.; Wang, R.Y.; Farci, P.; Alter, H.J.; Purcell, R.H. Immunoglobulin with High-Titer In Vitro Cross-Neutralizing Hepatitis C Virus Antibodies Passively Protects Chimpanzees from Homologous, but Not Heterologous, Challenge. J. Virol. 2015, 89, 9128–9132. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar] [CrossRef]
- Farci, P.; Shimoda, A.; Wong, D.; Cabezon, T.; De Gioannis, D.; Strazzera, A.; Shimizu, Y.; Shapiro, M.; Alter, H.J.; Purcell, R.H. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. U S A 1996, 93, 15394–15399. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef]
- Law, J.L.; Chen, C.; Wong, J.; Hockman, D.; Santer, D.M.; Frey, S.E.; Belshe, R.B.; Wakita, T.; Bukh, J.; Jones, C.T.; et al. A hepatitis C virus (HCV) vaccine comprising envelope glycoproteins gpE1/gpE2 derived from a single isolate elicits broad cross-genotype neutralizing antibodies in humans. PLoS One 2013, 8, e59776. [Google Scholar] [CrossRef]
- Stamataki, Z.; Coates, S.; Abrignani, S.; Houghton, M.; McKeating, J.A. Immunization of human volunteers with hepatitis C virus envelope glycoproteins elicits antibodies that cross-neutralize heterologous virus strains. The Journal of infectious diseases 2011, 204, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.Y.; Lau, P.; Fauvelle, C.; Gowthaman, R.; Baumert, T.F.; Fuerst, T.R.; Mariuzza, R.A.; Foung, S.K.H. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: Implications for vaccine design. Proc. Natl. Acad. Sci. U S A 2016, 113, E6946–E6954. [Google Scholar] [CrossRef]
- Gopal, R.; Jackson, K.; Tzarum, N.; Kong, L.; Ettenger, A.; Guest, J.; Pfaff, J.M.; Barnes, T.; Honda, A.; Giang, E.; et al. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog. 2017, 13, e1006735. [Google Scholar] [CrossRef]
- Pfaff-Kilgore, J.M.; Davidson, E.; Kadash-Edmondson, K.; Hernandez, M.; Rosenberg, E.; Chambers, R.; Castelli, M.; Clementi, N.; Mancini, N.; Bailey, J.R.; et al. Sites of vulnerability in HCV E1E2 identified by comprehensive functional screening. Cell Rep. 2022, 39, 110859. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef] [PubMed]
- Augestad, E.H.; Olesen, C.H.; Grønberg, C.; Soerensen, A.; Velázquez-Moctezuma, R.; Fanalista, M.; Bukh, J.; Wang, K.; Gourdon, P.; Prentoe, J. Structure of the hepatitis C virus E1/E2 envelope proteins in a homodimeric complex. bioRxiv 2023. [Google Scholar] [CrossRef]
- von Hahn, T.; Yoon, J.C.; Alter, H.; Rice, C.M.; Rehermann, B.; Balfe, P.; McKeating, J.A. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 2007, 132, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Broering, T.J.; Garrity, K.A.; Boatright, N.K.; Sloan, S.E.; Sandor, F.; Thomas, W.D., Jr.; Szabo, G.; Finberg, R.W.; Ambrosino, D.M.; Babcock, G.J. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J. Virol. 2009, 83, 12473–12482. [Google Scholar] [CrossRef]
- Chung, R.T.; Gordon, F.D.; Curry, M.P.; Schiano, T.D.; Emre, S.; Corey, K.; Markmann, J.F.; Hertl, M.; Pomposelli, J.J.; Pomfret, E.A.; et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant. Surgeons 2013, 13, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Iyer, S.; Smith, H.L.; Wang, Y.; Rowley, K.; Ambrosino, D.M.; Zamore, P.D.; Pierce, B.G.; Molrine, D.C.; Weng, Z. High-throughput sequencing analysis of post-liver transplantation HCV E2 glycoprotein evolution in the presence and absence of neutralizing monoclonal antibody. PLoS One 2014, 9, e100325. [Google Scholar] [CrossRef]
- Pantua, H.; Diao, J.; Ultsch, M.; Hazen, M.; Mathieu, M.; McCutcheon, K.; Takeda, K.; Date, S.; Cheung, T.K.; Phung, Q.; et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 2013, 425, 1899–1914. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Wasilewski, L.N.; Snider, A.E.; El-Diwany, R.; Osburn, W.O.; Keck, Z.; Foung, S.K.; Ray, S.C. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J. Clin. Invest. 2015, 125, 437–447. [Google Scholar] [CrossRef]
- El-Diwany, R.; Cohen, V.J.; Mankowski, M.C.; Wasilewski, L.N.; Brady, J.K.; Snider, A.E.; Osburn, W.O.; Murrell, B.; Ray, S.C.; Bailey, J.R. Extra-epitopic hepatitis C virus polymorphisms confer resistance to broadly neutralizing antibodies by modulating binding to scavenger receptor B1. PLoS Pathog. 2017, 13, e1006235. [Google Scholar] [CrossRef]
- Augestad, E.H.; Castelli, M.; Clementi, N.; Stroh, L.J.; Krey, T.; Burioni, R.; Mancini, N.; Bukh, J.; Prentoe, J. Global and local envelope protein dynamics of hepatitis C virus determine broad antibody sensitivity. Sci. Adv. 2020, 6, eabb5938. [Google Scholar] [CrossRef]
- Salas, J.H.; Urbanowicz, R.A.; Guest, J.D.; Frumento, N.; Figueroa, A.; Clark, K.E.; Keck, Z.; Cowton, V.M.; Cole, S.J.; Patel, A.H.; et al. An Antigenically Diverse, Representative Panel of Envelope Glycoproteins for Hepatitis C Virus Vaccine Development. Gastroenterology 2022, 162, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Bankwitz, D.; Bahai, A.; Labuhn, M.; Doepke, M.; Ginkel, C.; Khera, T.; Todt, D.; Stroh, L.J.; Dold, L.; Klein, F.; et al. Hepatitis C reference viruses highlight potent antibody responses and diverse viral functional interactions with neutralising antibodies. Gut 2020. [Google Scholar] [CrossRef]
- Chumbe, A.; Grobben, M.; Capella-Pujol, J.; Koekkoek, S.M.; Zon, I.; Slamanig, S.; Merat, S.J.; Beaumont, T.; Sliepen, K.; Schinkel, J.; et al. A panel of hepatitis C virus glycoproteins for the characterization of antibody responses using antibodies with diverse recognition and neutralization patterns. Virus Res. 2024, 341, 199308. [Google Scholar] [CrossRef]
- deCamp, A.; Hraber, P.; Bailer, R.T.; Seaman, M.S.; Ochsenbauer, C.; Kappes, J.; Gottardo, R.; Edlefsen, P.; Self, S.; Tang, H.; et al. Global panel of HIV-1 Env reference strains for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2014, 88, 2489–2507. [Google Scholar] [CrossRef] [PubMed]
- Sevvana, M.; Keck, Z.; Foung, S.K.; Kuhn, R.J. Structural perspectives on HCV humoral immune evasion mechanisms. Curr. Opin. Virol. 2021, 49, 92–101. [Google Scholar] [CrossRef]
- Keck, M.L.; Wrensch, F.; Pierce, B.G.; Baumert, T.F.; Foung, S.K.H. Mapping Determinants of Virus Neutralization and Viral Escape for Rational Design of a Hepatitis C Virus Vaccine. Frontiers in immunology 2018, 9, 1194. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Robbins, J.B.; Deller, M.C.; Stanfield, R.L.; Wilson, I.A.; Law, M. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J. Virol. 2012, 86, 13085–13088. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Giang, E.; Robbins, J.B.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A.; Law, M. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. U S A 2012, 109, 9499–9504. [Google Scholar] [CrossRef]
- Meunier, J.C.; Russell, R.S.; Goossens, V.; Priem, S.; Walter, H.; Depla, E.; Union, A.; Faulk, K.N.; Bukh, J.; Emerson, S.U.; et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J. Virol. 2008, 82, 966–973. [Google Scholar] [CrossRef]
- Wahid, A.; Helle, F.; Descamps, V.; Duverlie, G.; Penin, F.; Dubuisson, J. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J. Virol. 2013, 87, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Giang, E.; Dorner, M.; Prentoe, J.C.; Dreux, M.; Evans, M.J.; Bukh, J.; Rice, C.M.; Ploss, A.; Burton, D.R.; Law, M. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. U S A 2012, 109, 6205–6210. [Google Scholar] [CrossRef] [PubMed]
- Flyak, A.I.; Ruiz, S.E.; Salas, J.; Rho, S.; Bailey, J.R.; Bjorkman, P.J. An ultralong CDRH2 in HCV neutralizing antibody demonstrates structural plasticity of antibodies against E2 glycoprotein. eLife 2020, 9. [Google Scholar] [CrossRef]
- Kumar, A.; Rohe, T.C.; Elrod, E.J.; Khan, A.G.; Dearborn, A.D.; Kissinger, R.; Grakoui, A.; Marcotrigiano, J. Regions of hepatitis C virus E2 required for membrane association. Nat. Commun. 2023, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Tzarum, N.; Giang, E.; Kadam, R.U.; Chen, F.; Nagy, K.; Augestad, E.H.; Velazquez-Moctezuma, R.; Keck, Z.Y.; Hua, Y.; Stanfield, R.L.; et al. An alternate conformation of HCV E2 neutralizing face as an additional vaccine target. Sci. Adv. 2020, 6, eabb5642. [Google Scholar] [CrossRef] [PubMed]
- Tzarum, N.; Giang, E.; Kong, L.; He, L.; Prentoe, J.; Augestad, E.; Hua, Y.; Castillo, S.; Lauer, G.M.; Bukh, J.; et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human V(H)1-69 antibodies. Sci. Adv. 2019, 5, eaav1882. [Google Scholar] [CrossRef]
- Kumar, A.; Hossain, R.A.; Yost, S.A.; Bu, W.; Wang, Y.; Dearborn, A.D.; Grakoui, A.; Cohen, J.I.; Marcotrigiano, J. Structural insights into hepatitis C virus receptor binding and entry. Nature 2021, 598, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, L.; Kalemera, M.D.; Lewis, C.B.; Palor, M.; Walker, L.; Daviter, T.; Lees, W.D.; Moss, D.S.; Kremyda-Vlachou, M.; Kozlakidis, Z.; et al. An entropic safety catch controls hepatitis C virus entry and antibody resistance. eLife 2022, 11. [Google Scholar] [CrossRef]
- Stejskal, L.; Lees, W.D.; Moss, D.S.; Palor, M.; Bingham, R.J.; Shepherd, A.J.; Grove, J. Flexibility and intrinsic disorder are conserved features of hepatitis C virus E2 glycoprotein. PLoS Comput. Biol. 2020, 16, e1007710. [Google Scholar] [CrossRef]
- Helle, F.; Vieyres, G.; Elkrief, L.; Popescu, C.I.; Wychowski, C.; Descamps, V.; Castelain, S.; Roingeard, P.; Duverlie, G.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 2010, 84, 11905–11915. [Google Scholar] [CrossRef]
- Drummer, H.E.; Boo, I.; Poumbourios, P. Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J. Gen. Virol. 2007, 88 Pt 4, 1144–1148. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J. Exp. Med. 2016, 213, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Byrne, P.O.; McLellan, J.S. Principles and practical applications of structure-based vaccine design. Curr. Opin. Immunol. 2022, 77, 102209. [Google Scholar] [CrossRef] [PubMed]
- Guest, J.D.; Pierce, B.G. Structure-Based and Rational Design of a Hepatitis C Virus Vaccine. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.J.; Kanekiyo, M.; Kong, W.P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G.; et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015, 21, 1065–1070. [Google Scholar] [CrossRef]
- Widge, A.T.; Hofstetter, A.R.; Houser, K.V.; Awan, S.F.; Chen, G.L.; Burgos Florez, M.C.; Berkowitz, N.M.; Mendoza, F.; Hendel, C.S.; Holman, L.A.; et al. An influenza hemagglutinin stem nanoparticle vaccine induces cross-group 1 neutralizing antibodies in healthy adults. Sci. Transl. Med. 2023, 15, eade4790. [Google Scholar] [CrossRef]
- Ruckwardt, T.J. The road to approved vaccines for respiratory syncytial virus. NPJ Vaccines 2023, 8, 138. [Google Scholar] [CrossRef]
- Sandomenico, A.; Leonardi, A.; Berisio, R.; Sanguigno, L.; Foca, G.; Foca, A.; Ruggiero, A.; Doti, N.; Muscariello, L.; Barone, D.; et al. Generation and Characterization of Monoclonal Antibodies against a Cyclic Variant of Hepatitis C Virus E2 Epitope 412-422. J. Virol. 2016, 90, 3745–3759. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Cheng, Y.; Kong, L.; Azadnia, P.; Giang, E.; Kim, J.; Wood, M.R.; Wilson, I.A.; Law, M.; Zhu, J. Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci. Rep. 2015, 5, 12501. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Boucher, E.N.; Piepenbrink, K.H.; Ejemel, M.; Rapp, C.A.; Thomas, W.D.; Sundberg, E.J.; Weng, Z.; Wang, Y. Structure-Based Design of Hepatitis C Virus Vaccines That Elicit Neutralizing Antibody Responses to a Conserved Epitope. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Tzarum, N.; Lin, X.; Shapero, B.; Sou, C.; Mann, C.J.; Stano, A.; Zhang, L.; Nagy, K.; Giang, E.; et al. Proof of concept for rational design of hepatitis C virus E2 core nanoparticle vaccines. Sci. Adv. 2020, 6, eaaz6225. [Google Scholar] [CrossRef]
- Vietheer, P.T.; Boo, I.; Gu, J.; McCaffrey, K.; Edwards, S.; Owczarek, C.; Hardy, M.P.; Fabri, L.; Center, R.J.; Poumbourios, P.; et al. The core domain of hepatitis C virus glycoprotein E2 generates potent cross-neutralizing antibodies in guinea pigs. Hepatology 2017, 65, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Keck, Z.Y.; Wang, R.; Lau, P.; Garagusi, K.; Elkholy, K.; Toth, E.A.; Urbanowicz, R.A.; Guest, J.D.; Agnihotri, P. Structure-Based Design of Hepatitis C Virus E2 Glycoprotein Improves Serum Binding and Cross-Neutralization. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Frumento, N.; Figueroa, A.; Wang, T.; Zahid, M.N.; Wang, S.; Massaccesi, G.; Stavrakis, G.; Crowe, J.E.; Flyak, A.I.; Ji, H. Repeated exposure to heterologous hepatitis C viruses associates with enhanced neutralizing antibody breadth and potency. J. Clin. Invest. 2022, 132. [Google Scholar] [CrossRef]
- Tarr, A.W.; Backx, M.; Hamed, M.R.; Urbanowicz, R.A.; McClure, C.P.; Brown, R.J.P.; Ball, J.K. Immunization with a synthetic consensus hepatitis C virus E2 glycoprotein ectodomain elicits virus-neutralizing antibodies. Antiviral research 2018, 160, 25–37. [Google Scholar] [CrossRef]
- Liao, H.X.; Tsao, C.Y.; Alam, S.M.; Muldoon, M.; Vandergrift, N.; Ma, B.J.; Lu, X.; Sutherland, L.L.; Scearce, R.M.; Bowman, C.; et al. Antigenicity and immunogenicity of transmitted/founder, consensus, and chronic envelope glycoproteins of human immunodeficiency virus type 1. J. Virol. 2013, 87, 4185–4201. [Google Scholar] [CrossRef]
- Sliepen, K.; Han, B.W.; Bontjer, I.; Mooij, P.; Garces, F.; Behrens, A.J.; Rantalainen, K.; Kumar, S.; Sarkar, A.; Brouwer, P.J.M.; et al. Structure and immunogenicity of a stabilized HIV-1 envelope trimer based on a group-M consensus sequence. Nat. Commun. 2019, 10, 2355. [Google Scholar] [CrossRef]
- Cohen, A.A.; Gnanapragasam, P.N.P.; Lee, Y.E.; Hoffman, P.R.; Ou, S.; Kakutani, L.M.; Keeffe, J.R.; Wu, H.J.; Howarth, M.; West, A.P.; et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science 2021, 371, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Al-Halifa, S.; Gauthier, L.; Arpin, D.; Bourgault, S.; Archambault, D. Nanoparticle-Based Vaccines Against Respiratory Viruses. Front. Immunol. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Wu, Y.; Kim, Y.B.; Oh, Y.K. Advances in vaccine delivery systems against viral infectious diseases. Drug Deliv. Transl. Res. 2021, 11, 1401–1419. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, N.; Streatfield, S.J.; Yusibov, V. Virus-like particles as a highly efficient vaccine platform: diversity of targets and production systems and advances in clinical development. Vaccine 2012, 31, 58–83. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kroll, A.V.; Holay, M.; Fang, R.H.; Zhang, L. Biomimetic Nanotechnology toward Personalized Vaccines. Adv. Mater. 2020, 32, e1901255. [Google Scholar] [CrossRef] [PubMed]
- De Temmerman, M.L.; Rejman, J.; Demeester, J.; Irvine, D.J.; Gander, B.; De Smedt, S.C. Particulate vaccines: on the quest for optimal delivery and immune response. Drug Discov. Today 2011, 16, 569–582. [Google Scholar] [CrossRef]
- Aung, A.; Cui, A.; Maiorino, L.; Amini, A.P.; Gregory, J.R.; Bukenya, M.; Zhang, Y.; Lee, H.; Cottrell, C.A.; Morgan, D.M.; et al. Low protease activity in B cell follicles promotes retention of intact antigens after immunization. Science 2023, 379, eabn8934. [Google Scholar] [CrossRef] [PubMed]
- Agger, E.M.; Rosenkrands, I.; Hansen, J.; Brahimi, K.; Vandahl, B.S.; Aagaard, C.; Werninghaus, K.; Kirschning, C.; Lang, R.; Christensen, D.; et al. Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): a versatile adjuvant for vaccines with different immunological requirements. PLoS One 2008, 3, e3116. [Google Scholar] [CrossRef]
- Coeshott, C.M.; Smithson, S.L.; Verderber, E.; Samaniego, A.; Blonder, J.M.; Rosenthal, G.J.; Westerink, M.A. Pluronic F127-based systemic vaccine delivery systems. Vaccine 2004, 22, 2396–2405. [Google Scholar] [CrossRef]
- Cox, M.M.; Patriarca, P.A.; Treanor, J. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir. Viruses 2008, 2, 211–219. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Laupeze, B.; Di Pasquale, A.; Hergli, N.; Collignon, C.; Garcon, N. Adjuvant system AS01: helping to overcome the challenges of modern vaccines. Expert. review of vaccines 2017, 16, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Holtz, K.M.; Robinson, P.S.; Matthews, E.E.; Hashimoto, Y.; McPherson, C.E.; Khramtsov, N.; Reifler, M.J.; Meghrous, J.; Rhodes, D.G.; Cox, M.M.; et al. Modifications of cysteine residues in the transmembrane and cytoplasmic domains of a recombinant hemagglutinin protein prevent cross-linked multimer formation and potency loss. BMC Biotechnol. 2014, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Schwendener, R.A. Liposomes as vaccine delivery systems: a review of the recent advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.H.; Patel, N.; Haupt, R.; Zhou, H.; Weston, S.; Hammond, H.; Logue, J.; Portnoff, A.D.; Norton, J.; Guebre-Xabier, M.; et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 2021, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Bruun, T.U.J.; Andersson, A.C.; Draper, S.J.; Howarth, M. Engineering a Rugged Nanoscaffold To Enhance Plug-and-Display Vaccination. ACS Nano 2018, 12, 8855–8866. [Google Scholar] [CrossRef] [PubMed]
- Domingo, G.J.; Orru, S.; Perham, R.N. Multiple display of peptides and proteins on a macromolecular scaffold derived from a multienzyme complex. J. Mol. Biol. 2001, 305, 259–267. [Google Scholar] [CrossRef]
- Hsia, Y.; Bale, J.B.; Gonen, S.; Shi, D.; Sheffler, W.; Fong, K.K.; Nattermann, U.; Xu, C.; Huang, P.S.; Ravichandran, R.; et al. Design of a hypersTable 60-subunit protein dodecahedron. [corrected]. Nature 2016, 535, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68. [Google Scholar] [CrossRef]
- Wang, W.; Liu, Z.; Zhou, X.; Guo, Z.; Zhang, J.; Zhu, P.; Yao, S.; Zhu, M. Ferritin nanoparticle-based SpyTag/SpyCatcher-enabled click vaccine for tumor immunotherapy. Nanomedicine 2019, 16, 69–78. [Google Scholar] [CrossRef]
- Wei, Y.; Kumar, P.; Wahome, N.; Mantis, N.J.; Middaugh, C.R. Biomedical Applications of Lumazine Synthase. J. Pharm. Sci. 2018, 107, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-Display: decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef] [PubMed]
- Sliepen, K.; Radic, L.; Capella-Pujol, J.; Watanabe, Y.; Zon, I.; Chumbe, A.; Lee, W.H.; de Gast, M.; Koopsen, J.; Koekkoek, S.; et al. Induction of cross-neutralizing antibodies by a permuted hepatitis C virus glycoprotein nanoparticle vaccine candidate. Nat. Commun. 2022, 13, 7271. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, X.; Lou, P.; Hu, Z.; Qu, P.; Li, D.; Li, Q.; Xu, Y.; Niu, J.; He, Y.; et al. A Nanoparticle-Based Hepatitis C Virus Vaccine With Enhanced Potency. J. Infect. Dis. 2020, 221, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Frumento, N.; Figueroa, A.; Wang, T.; Zahid, M.N.; Wang, S.; Massaccesi, G.; Stavrakis, G.; Crowe, J.E., Jr.; Flyak, A.I.; Ji, H.; et al. Repeated exposure to heterologous hepatitis C viruses associates with enhanced neutralizing antibody breadth and potency. J. Clin. Invest. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.A.; van Doremalen, N.; Greaney, A.J.; Andersen, H.; Sharma, A.; Starr, T.N.; Keeffe, J.R.; Fan, C.; Schulz, J.E.; Gnanapragasam, P.N.P.; et al. Mosaic RBD nanoparticles protect against challenge by diverse sarbecoviruses in animal models. Science 2022, 377, eabq0839. [Google Scholar] [CrossRef]
- Cohen, A.A.; Yang, Z.; Gnanapragasam, P.N.P.; Ou, S.; Dam, K.A.; Wang, H.; Bjorkman, P.J. Construction, characterization, and immunization of nanoparticles that display a diverse array of influenza HA trimers. PLoS One 2021, 16, e0247963. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.B.; Kim, H.; Jeong, J.H.; Jang, U.S.; Jang, Y.; Roh, S.; Jeon, H.; Kim, E.J.; Han, S.Y.; Maeng, J.Y.; et al. Mosaic RBD nanoparticles induce intergenus cross-reactive antibodies and protect against SARS-CoV-2 challenge. Proc. Natl. Acad. Sci. U S A 2023, 120, e2208425120. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, S.; Zhang, Y.; Yuan, L.; Zheng, Q.; Wei, M.; Shi, Y.; Wang, Z.; Ma, J.; Wang, K.; et al. Lineage-mosaic and mutation-patched spike proteins for broad-spectrum COVID-19 vaccine. Cell Host Microbe 2022, 30, 1732–1744. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, S.; Liu, J.; Chen, R.; Zhang, Y.; Lin, Y.; Xi, Z.; Deng, J.; Pu, Z.; Liang, C.; et al. A Mosaic Nanoparticle Vaccine Elicits Potent Mucosal Immune Response with Significant Cross-Protection Activity against Multiple SARS-CoV-2 Sublineages. Adv Sci (Weinh) 2023, 10, e2301034. [Google Scholar] [CrossRef]
- Baden, L.R.; Walsh, S.R.; Seaman, M.S.; Cohen, Y.Z.; Johnson, J.A.; Licona, J.H.; Filter, R.D.; Kleinjan, J.A.; Gothing, J.A.; Jennings, J.; et al. First-in-Human Randomized, Controlled Trial of Mosaic HIV-1 Immunogens Delivered via a Modified Vaccinia Ankara Vector. J. Infect. Dis. 2018, 218, 633–644. [Google Scholar] [CrossRef]
- Barouch, D.H.; Tomaka, F.L.; Wegmann, F.; Stieh, D.J.; Alter, G.; Robb, M.L.; Michael, N.L.; Peter, L.; Nkolola, J.P.; Borducchi, E.N.; et al. Evaluation of a mosaic HIV-1 vaccine in a multicentre, randomised, double-blind, placebo-controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13-19). Lancet 2018, 392, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Maciel, M., Jr.; Amara, R.R.; Bar, K.J.; Crotty, S.; Deeks, S.G.; Duplessis, C.; Gaiha, G.; McElrath, M.J.; McMichael, A.; Palin, A.; et al. Exploring synergies between B- and T-cell vaccine approaches to optimize immune responses against HIV-workshop report. NPJ Vaccines 2024, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Murji, A.A.; Qin, J.S.; Hermanus, T.; Morris, L.; Georgiev, I.S. Elicitation of Neutralizing Antibody Responses to HIV-1 Immunization with Nanoparticle Vaccine Platforms. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Drakenberg, K.; Beyene, A.; Rosa, D.; Abrignani, S.; Houghton, M.; Widell, A.; Grillner, L.; Persson, M.A. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen. Virol. 2000, 81 Pt 10, 2451–2459. [Google Scholar] [CrossRef]
- Bugli, F.; Mancini, N.; Kang, C.Y.; Di Campli, C.; Grieco, A.; Manzin, A.; Gabrielli, A.; Gasbarrini, A.; Fadda, G.; Varaldo, P.E.; et al. Mapping B-cell epitopes of hepatitis C virus E2 glycoprotein using human monoclonal antibodies from phage display libraries. J. Virol. 2001, 75, 9986–9990. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E. Challenges to the development of vaccines to hepatitis C virus that elicit neutralizing antibodies. Frontiers in microbiology 2014, 5, 329. [Google Scholar] [CrossRef]
- Habersetzer, F.; Fournillier, A.; Dubuisson, J.; Rosa, D.; Abrignani, S.; Wychowski, C.; Nakano, I.; Trepo, C.; Desgranges, C.; Inchauspe, G. Characterization of human monoclonal antibodies specific to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties. Virology 1998, 249, 32–41. [Google Scholar] [CrossRef]
- Hadlock, K.G.; Lanford, R.E.; Perkins, S.; Rowe, J.; Yang, Q.; Levy, S.; Pileri, P.; Abrignani, S.; Foung, S.K. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 2000, 74, 10407–10416. [Google Scholar] [CrossRef]
- Keck, Z.; Wang, W.; Wang, Y.; Lau, P.; Carlsen, T.H.; Prentoe, J.; Xia, J.; Patel, A.H.; Bukh, J.; Foung, S.K. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J. Virol. 2013, 87, 37–51. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Li, T.K.; Xia, J.; Bartosch, B.; Cosset, F.L.; Dubuisson, J.; Foung, S.K. Analysis of a highly flexible conformational immunogenic domain a in hepatitis C virus E2. J. Virol. 2005, 79, 13199–13208. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.Y.; Li, T.K.; Xia, J.; Gal-Tanamy, M.; Olson, O.; Li, S.H.; Patel, A.H.; Ball, J.K.; Lemon, S.M.; Foung, S.K. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 2008, 82, 6061–6066. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.Y.; Xia, J.; Wang, Y.; Wang, W.; Krey, T.; Prentoe, J.; Carlsen, T.; Li, A.Y.; Patel, A.H.; Lemon, S.M.; et al. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 2012, 8, e1002653. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Sidney, J.; Southwood, S.; Ruppert, J.; Oseroff, C.; Maewal, A.; Snoke, K.; Serra, H.M.; Kubo, R.T.; Sette, A.; et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity 1994, 1, 751–761. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Chaudhary, A.; Lin, X.; Sou, C.; Alkutkar, T.; Kumar, S.; Ngo, T.; Kosviner, E.; Ozorowski, G.; Stanfield, R.L.; et al. Single-component multilayered self-assembling nanoparticles presenting rationally designed glycoprotein trimers as Ebola virus vaccines. Nat. Commun. 2021, 12, 2633. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Lin, X.; Wang, Y.; Abraham, C.; Sou, C.; Ngo, T.; Zhang, Y.; Wilson, I.A.; Zhu, J. Single-component, self-assembling, protein nanoparticles presenting the receptor binding domain and stabilized spike as SARS-CoV-2 vaccine candidates. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Paynter, J.; Antanasijevic, A.; Allen, J.D.; Eldad, M.; Lee, Y.Z.; Copps, J.; Newby, M.L.; He, L.; Chavez, D.; et al. Single-component multilayered self-assembling protein nanoparticles presenting glycan-trimmed uncleaved prefusion optimized envelope trimmers as HIV-1 vaccine candidates. Nat. Commun. 2023, 14, 1985. [Google Scholar] [CrossRef]
Figure 1.
HCV sequence diversity and representative available structures. HCV E1E2 genotype and subtype reference sequences were downloaded from the LANL HCV database [2] and are shown as an unrooted phylogenetic tree, with branches and labels colored by genotype. Subtypes with currently available experimentally determined E2 or E1E2 glycoprotein structures are labeled, and representative structures (Protein Data Bank codes, HCV strain, and glycoprotein contained) are shown. Multiple sequence alignment and phylogenetic clustering was performed with the MAFFT web server [4], and the unrooted tree was generated with iTOL [5].
Figure 1.
HCV sequence diversity and representative available structures. HCV E1E2 genotype and subtype reference sequences were downloaded from the LANL HCV database [2] and are shown as an unrooted phylogenetic tree, with branches and labels colored by genotype. Subtypes with currently available experimentally determined E2 or E1E2 glycoprotein structures are labeled, and representative structures (Protein Data Bank codes, HCV strain, and glycoprotein contained) are shown. Multiple sequence alignment and phylogenetic clustering was performed with the MAFFT web server [4], and the unrooted tree was generated with iTOL [5].
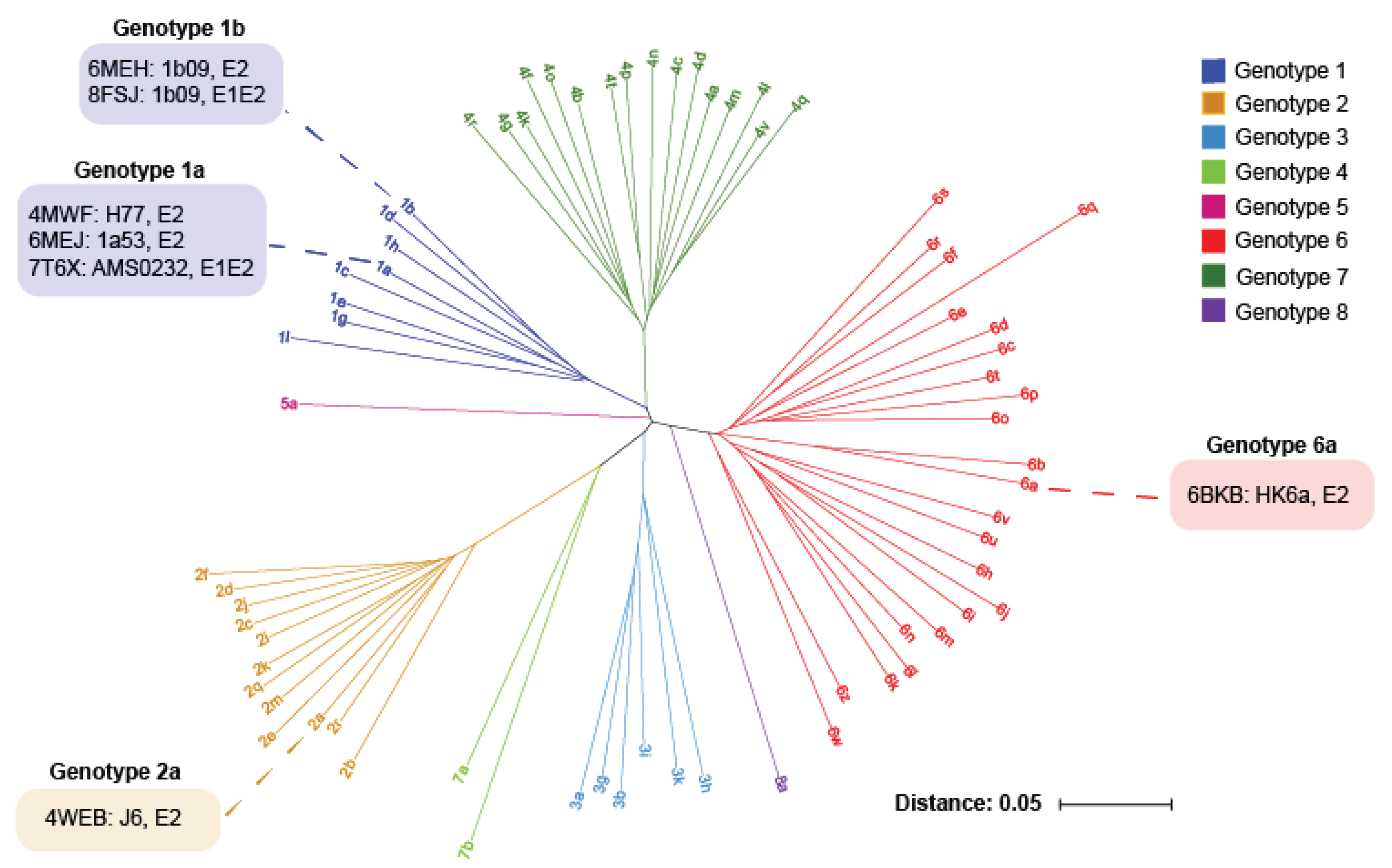
Figure 2.
Structure and antigenic regions/domains of the E1E2 heterodimer. Shown is the structure of engineered E1E2 heterodimer ectodomain1, shown in cartoon and semi-transparent surface representation. The E1 and E2 subunits are colored blue and orange, respectively, with mapped antigenic surfaces colored by region/domain targeted: neutralizing face (purple), AR-4 (pink), AR-5 (dark blue), AD-C (teal), and AD-A (light green). The CD81 binding loop and the bridging/base domains are colored yellow and wheat, respectively. N-linked glycans are depicted as gray sticks. AD, antigenic domain; AR, antigenic region.
Figure 2.
Structure and antigenic regions/domains of the E1E2 heterodimer. Shown is the structure of engineered E1E2 heterodimer ectodomain1, shown in cartoon and semi-transparent surface representation. The E1 and E2 subunits are colored blue and orange, respectively, with mapped antigenic surfaces colored by region/domain targeted: neutralizing face (purple), AR-4 (pink), AR-5 (dark blue), AD-C (teal), and AD-A (light green). The CD81 binding loop and the bridging/base domains are colored yellow and wheat, respectively. N-linked glycans are depicted as gray sticks. AD, antigenic domain; AR, antigenic region.
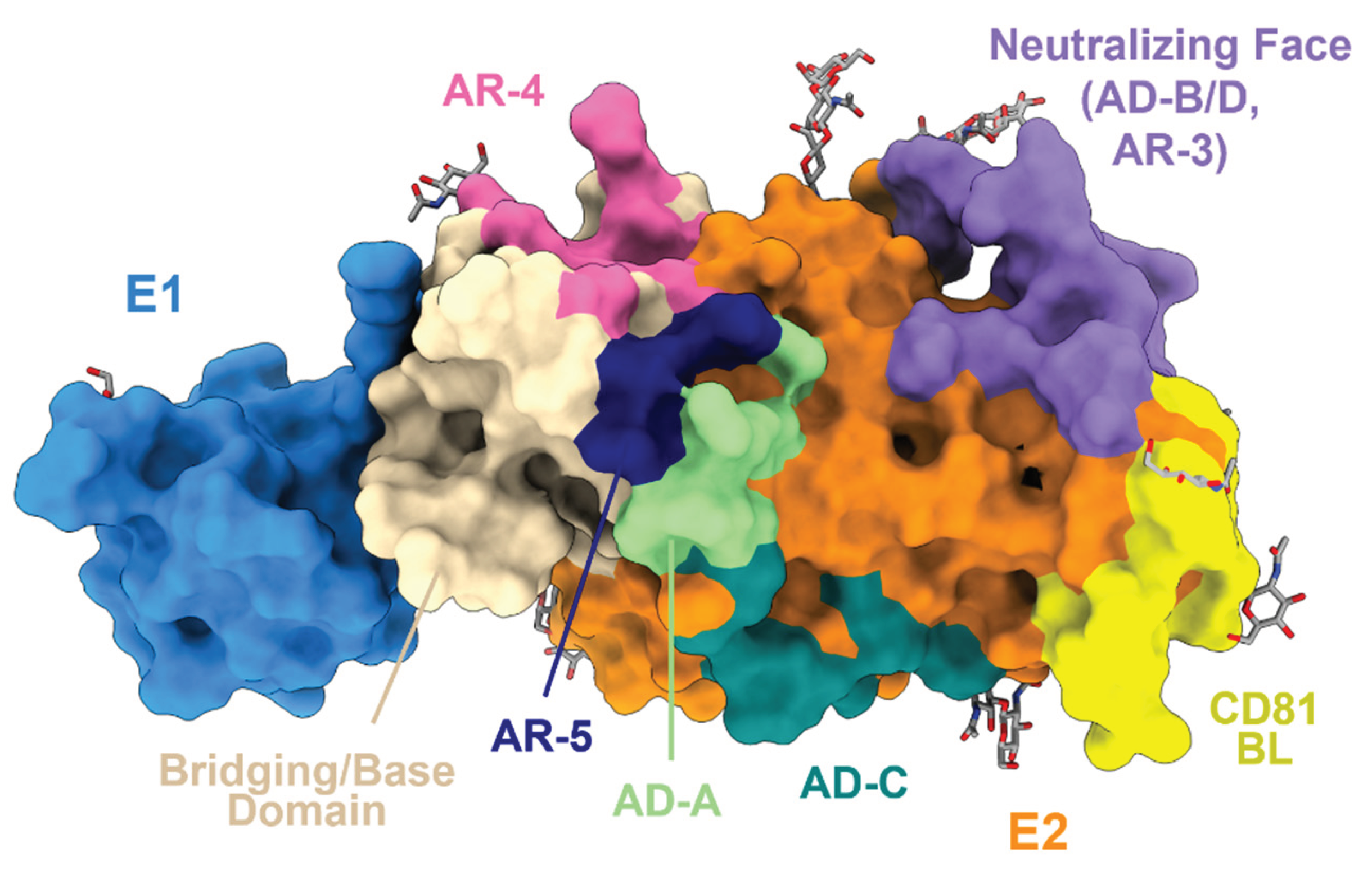
Figure 3.
Sub-regions of E1 and E2. Structure of E1E2 heterodimer shown in cartoon representation and colored by sub-region (based on PDB ID 8FSJ). E2 sub-regions depicted include: Front layer (residues 420-458), variable region 2 (VR2, residues 459-483), β-sandwich core (residues 484-517 and 535-568), CD81 binding loop (CD81 BL, residues 518-534), variable region 3 (VR3, residues 569-579), post-variable 3 region (pVR3, residues 580-595), back layer (BL, residues 596-645), and the bridging (or base) domain (BD, residues 646-701). E1 sub-regions depicted include: N-terminal domain (residues 192-205), E1 core (residues 206-255 and 295-299), pFP-containing region (residues 256-298), and C-terminal loop (residues 300-314).
Figure 3.
Sub-regions of E1 and E2. Structure of E1E2 heterodimer shown in cartoon representation and colored by sub-region (based on PDB ID 8FSJ). E2 sub-regions depicted include: Front layer (residues 420-458), variable region 2 (VR2, residues 459-483), β-sandwich core (residues 484-517 and 535-568), CD81 binding loop (CD81 BL, residues 518-534), variable region 3 (VR3, residues 569-579), post-variable 3 region (pVR3, residues 580-595), back layer (BL, residues 596-645), and the bridging (or base) domain (BD, residues 646-701). E1 sub-regions depicted include: N-terminal domain (residues 192-205), E1 core (residues 206-255 and 295-299), pFP-containing region (residues 256-298), and C-terminal loop (residues 300-314).
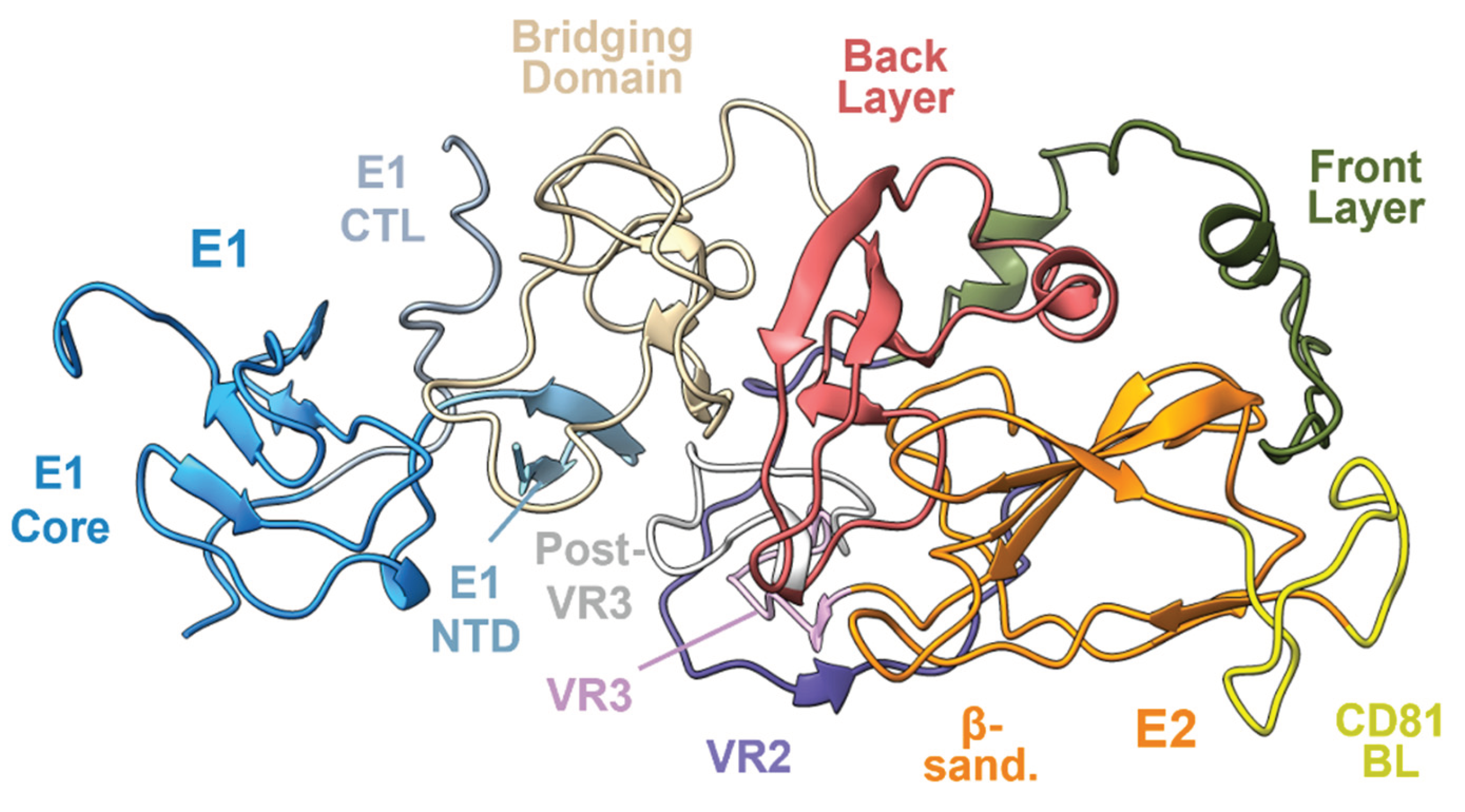
Figure 4.
E2 and E1E2 structure-based and rational design strategies. Selected reported E2 and E1E2 design approaches are shown on an E1E2 structure (PDB code 8FSJ), with E1, E2 and neutralizing face (antigenic domain B/D, antigenic region 3) colored as in Figure 2. E2 position 445, the site of a previously reported neutralizing face proline design[3], is colored green. E1E2 scaffolding and nanoparticle fusion are shown at the E1 and E2 C-termini, while loop removal is shown at the E2 HVR2 loop.
Figure 4.
E2 and E1E2 structure-based and rational design strategies. Selected reported E2 and E1E2 design approaches are shown on an E1E2 structure (PDB code 8FSJ), with E1, E2 and neutralizing face (antigenic domain B/D, antigenic region 3) colored as in Figure 2. E2 position 445, the site of a previously reported neutralizing face proline design[3], is colored green. E1E2 scaffolding and nanoparticle fusion are shown at the E1 and E2 C-termini, while loop removal is shown at the E2 HVR2 loop.
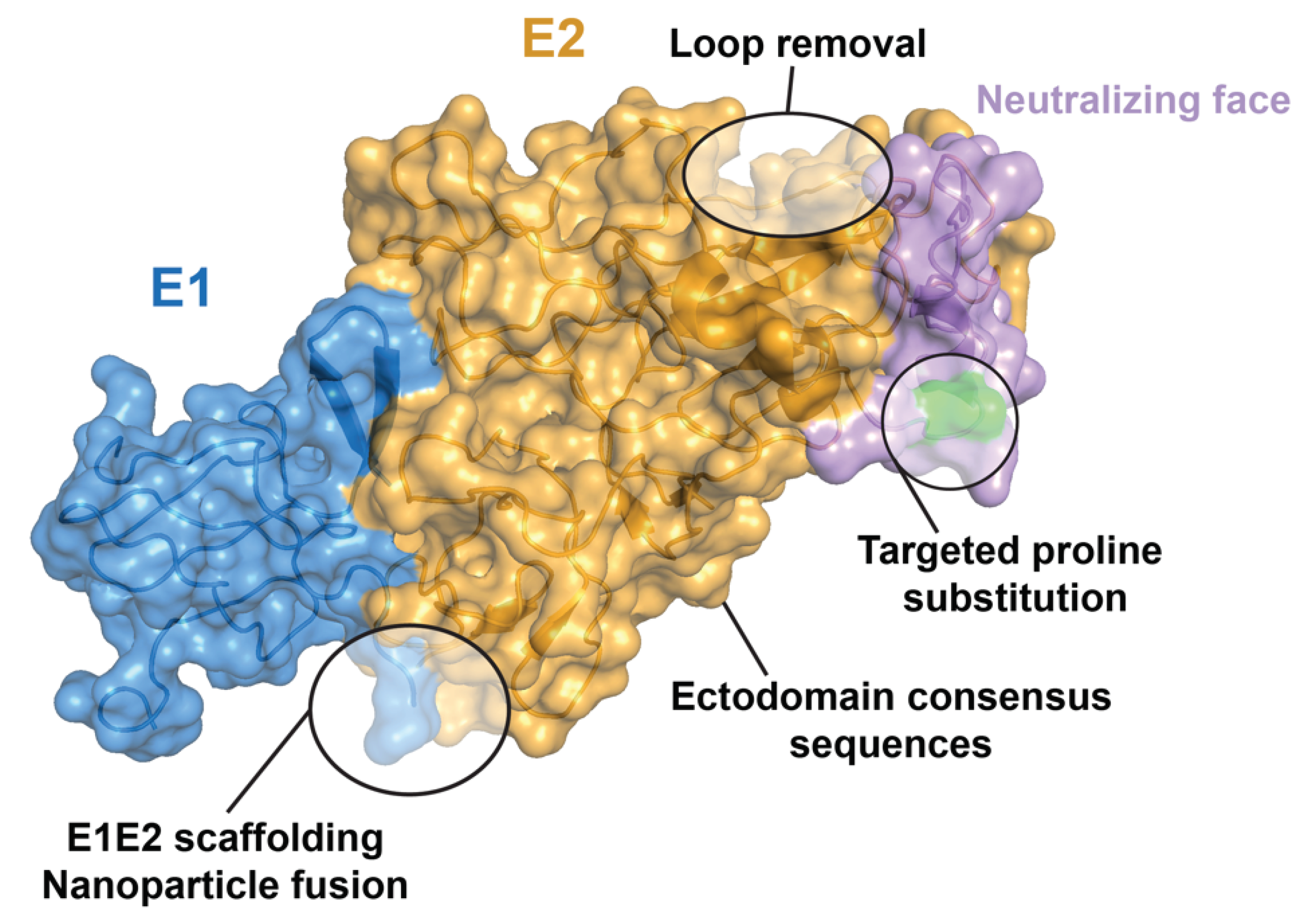
Figure 5.
Multivalent presentation of E1E2 antigens. Coupling to a nanoparticle increases the size and valency of E1E2 antigen presentation. Flexible attachment methods allow for the presentation of genetically diverse sequences as consensus (striped), mosaic (multiple colors), or cocktails of multiple representative genome sequences.
Figure 5.
Multivalent presentation of E1E2 antigens. Coupling to a nanoparticle increases the size and valency of E1E2 antigen presentation. Flexible attachment methods allow for the presentation of genetically diverse sequences as consensus (striped), mosaic (multiple colors), or cocktails of multiple representative genome sequences.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



