
Submitted:
25 March 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
The “circular bioeconomy” offers numerous opportunities for the health, cosmetic, and nutrition sectors through the potential reuse of Castanea sativa by-products. This abundant and sustainable resource provides valuable bioactive secondary metabolites with antioxidants and anti-inflammatory properties. The transformation of these by-products into high-value products for human health can contribute to sustainable economic growth and reduce the environmental impact of traditional waste disposal practices, thus creating added value from previously underutilized resources. In the present study, we investigated the antioxidant capacity, phytochemical composition and in vitro antioxidant and anti-inflammatory activity of C. sativa burrs (CSB) aqueous extract. The spectrophotometric study yielded optimal total phenolic content (TPC) values, along with significant antioxidant and anti-radical properties. The utilization of UPLC-MS-MS techniques for phytochemical investigation resulted in the identification of a total of 56 metabolites, providing confirmation of the presence of phenolic compounds in CSB. In addition, CSB significantly downregulated pro-inflammatory mediators in LPS-stimulated RAW 264.7 macrophage cells without significant cell toxicity.
Keywords:
Castanea sativa
; waste repurposing
; polyphenols
; UPLC-MS-MS
; inflammation
; RAW 264.7
; molecular modeling
; docking simulations
1. Introduction
C. sativa (Mill.) is a key fruit crop in Southern Europe, bearing notable economic significance. Worldwide, its production is predominantly concentrated in the two macro-regions of Asia and Europe [1,2]. Italy stands as the leading chestnut producer in Europe, with five regions specializing in cultivation [3]. The C. sativa specimens investigated in this study originate from chestnut groves in Monte Amiata (Tuscany) and bear the " Protected Geographical Indication (PGI) Castagna del Monte Amiata" certification. Harvesting typically occurs from September to November, with most of the fruit earmarked for industrial processing, while the remainder is designated for fresh consumption. At this stage, a considerable quantity of protective burrs surrounding the chestnuts is discarded as waste or burned to prevent pest infestation, which could harm future crops.
With an increasing focus on sustainable and circular economies, there is a rising interest in recovering bioactive molecules from waste and by-products within the agrifood and forestry supply chain. Currently, plant metabolites contribute significantly to the pharmaceutical industry's revenue. Given the anticipated expansion of the global biomedical market to reach $232,280 million by 2028, the economic significance of employing plant materials for bioactive compounds becomes considerable [4,5]. Furthermore, with the deepening understanding of the immune system, it becomes increasingly apparent that the state of inflammation plays a pivotal role in determining health outcomes and disease progression, reaching beyond conditions conventionally linked to the immune system. Inflammatory states are involved in the occurrence and prognosis of cancer but also influences gut dysbiosis and various other facets [6,7,8], showcasing its extensive impact on health beyond conventional expectations. Consequently, there is a growing imperative to explore natural sources for developing new anti-inflammatory agents. Chestnut by-products present an excellent opportunity to meet this demand while addressing the issue of waste reduction. Scientific research has revealed that chestnut by-products contain functional substances with antioxidant and anti-inflammatory properties [9,10,11], making them valuable and readily available raw materials for the pharmaceutical, nutraceutical, and cosmetic sectors. Among the compounds of interest, polyphenols have gained growing commercial value in the nutrition, cosmetic, and pharmaceutical industries. The global polyphenol market has experienced rapid growth in recent years, with a projected compound annual growth rate (CAGR) of 7.4% from 2023 to 2030 [12]. Several reports have demonstrated the potential advantages of using crop waste as a natural source of polyphenols [13,14,15,16], replacing expensive chemically synthesized antioxidants, anti-inflammatory, and artificial dye compounds.
The purpose of this study was to emphasize the potential for recovering added-value products from PGI C. sativa Monte Amiata spiny burrs, which can act as an innovative, cost-effective, and readily available raw material for applications in the health sector. An aqueous extract obtained from the spiny burrs of C. sativa, utilizing a total green ultrasound-assisted extraction method, was employed to assess the in vitro anti-inflammatory properties of chestnut burrs. In silico studies were performed to obtain the 3D structures of the entire RAW 264.7 anti-inflammatory target complement, and docking simulations provided findings about the potential target/compound interactions. This method has been proven effective in extracting essential secondary metabolites from these valuable by-products, making it a viable natural source for use in nutraceutical or cosmeceutical applications.
2. Materials and Methods
2.1. Materials
RPMI medium, Dulbecco’s Modified Eagle’s Medium (DMEM), trypsin solution, and all the solvents used for cell culture were purchased from Merck (Germany). Mouse immortalized fibroblasts (NIH3T3) and RAW 264.7 cells were from American Type Culture Collection (Manassas, VA, USA). Ames test kit was supplied from Xenometrix (Allschwil, Switzerland).
2.2. Preparation of C. sativa Burrs (CSB) Extract
The spiny burrs from C. sativa Mill. certified as PGI Castagna del Monte Amiata (Italian IGP Reg. Cee n. 2081/92), Tuscany, an important area of chestnut production in Italy. After being thoroughly cleaned, the material was dried at Room Temperature (RT) until it reached a constant weight. It was then ground into a fine powder, and the pulverized burrs were stored in dark sealed plastic bags at -80°C in a dark environment until extraction. Extraction was performed in an ultrasonic bath applying 20 kHz frequency ultrasounds to pulverized chestnut burrs, for three hours at RT. The obtained aqueous extract, designated as CSB (C. sativa burrs), was freeze-dried and stored at -32°C for further studies.
2.3. Total Phenolic Content (TPC)
Total TPC was quantified by the Folin-Ciocalteu (FC) method [17] with some modifications. A calibration curve was generated using gallic acid (GA) solutions in the concentration range of 20-120 μg/mL. CSB samples were prepared by diluting the stock solution (1 mg/mL) in milli-Q water. Standard and sample tubes were then mixed with 1 mL of 1N FC reagent in milli-Q water. After 3 minutes, 1 mL of saturated Na2CO3 and 7 mL of milli-Q water were added. All tubes were incubated for 90 minutes at room temperature, shielded from light, before measuring absorbance at 725 nm. Simultaneously, a solution containing all reagents with the extract solvent alone were prepared as blank. TPC was expressed as milligrams of GA equivalent (GAE) per gram of dry extract.
2.4. Total Flavonoid Content (TFC)
The determination of the TFC was carried out according to the aluminum chloride (AlCl3) colorimetric method [18]. A calibration curve was generated using quercetin (Q) solutions in the concentration range of 20-200 μg/mL. CSB samples were prepared by diluting the stock solution (1 mg/mL) in milli-Q water. 500 μL of standard/sample were mixed to 100 μL of 10% AlCl3 in 1M potassium acetate and 3.3 mL of ethanol. Each solution was prepared in triplicate. After 30 min of incubation, the absorbance was measured at 430 nm using an EnVision system (PerkinElmer). The results were expressed as mg of Q equivalent (QE) per gram of extract.
2.5. Determination of Reducing Power
The total reducing power (TRP) of CSB extracts was assessed using the potassium ferricyanide reducing power assay, following a modified version of the method described by [19]. A calibration curve was generated using ascorbic acid (AA) solutions in the concentration range of 20-140 μg/mL. CSB samples were prepared by diluting the stock solution (1 mg/mL) in milli-Q water. A blank was created with water.
The samples, standards, and blank were treated with 1 mL of 0.2 M phosphate buffer (K2HPO4:KH2PO4) at pH 6.6 and 1 mL of 1% potassium ferricyanide (K3[Fe(CN)6]), followed by incubation at 50°C for 20 minutes. Subsequently, 1 mL of 10% (w/v) trichloroacetic acid was added to each solution, allowing an additional incubation at room temperature for 10 minutes. After this step, 2.5 mL of milli-Q water and 0.5 mL of 0.1% (w/v) ferric chloride (FeCl3) solution were added to 2.5 mL of the mixture before measuring the absorbance at 700 nm. The antioxidant power was quantified as mg AA equivalents (AAE) per gram of dry extract.
2.6. ABTS∙+ Free-Radical Scavenging Activity
Trolox equivalent antioxidant capacity (TEAC) assay is based on the conversion of oxidized ABTS∙+ radicals to ABTS by molecules able to neutralize the radical [20]. The assay was performed using the OxiSelect™ TEAC Assay Kit (Cell Biolabs Inc., San Diego, CA, USA) according to the manufacturer's instructions. Briefly, 25 µL of different concentrations of sample were added to 150 µL of freshly prepared ABTS•+ reagent diluted 1:50 in the appropriate diluent in a 96-well plate. After 5 min incubation on an orbital shaker, the absorbance was measured at 405 nm. Results were expressed as IC50 (µg/mL) (i.e., Inhibitory Concentration causing a 50% decrease of the absorbance).
2.7. DPPH∙+ Free-Radical Scavenging Activity
DPPH∙+ free-radical scavenging activity was estimated by dosing the free DPPH (2,2-diphenyl-1-picrylhydrazyl) radical. In this form, the DPPH• radical is in a stable, intensely red-colored form, able to absorb at 515 nm, which decolors when reduced by an antioxidant molecule. 100µM DPPH was added to each sample and the solutions were incubated 30’ in the dark, 37°C. The reaction was monitored at 517 nm to determine the percentage of discoloration. Τrolox (T) was used to set the standard curve. The capability to scavenge the DPPH radical was reported as IC50 (µg/mL) (i.e., Inhibitory Concentration causing a 50% decrease of the absorbance).
2.8. UPLC-MS-MS
To investigate the non-volatile profile of CSB extract, an Ultimate 3000 UPLC system (Thermo Fisher Scientific, Waltham, MA, USA) controlled with Thermo Xcalibur software (Thermo Fisher Scientific, Waltham, MA, USA) was used. The dry CSS extract was dissolved in the ethanol-water (70:30 v/v) mixture and injected into the UPLC-Q-Exactive Plus system. The samples were separated using a column Acquity UPLC BEH C18 (2.1 mm × 15 cm, 1.7 μm, Waters, Waltham, MA, USA). The mobile phases consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The gradient started with 2% of B, which was maintained constant for 1 min. Then, the organic phase was increased up to 100% in 50 min. The phase B was maintained at 100% for other 2 min and then returned to the initial condition. The flow rate was maintained at 0.2 mL/min and the injection volume of the sample was 10 μL. Additionally, the column temperature was kept at 35 °C. A Q-Exactive Plus™ quadrupole Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) was used to perform mass spectrometry analyses in the negative and positive ion modes, with a scan mass range set at m/z 200–2000. HR-MS spectra were recorded in the positive and negative ion modes using the following parameters: spray voltage 3.5 kV (positive) and 3.0 kV (negative), sheath gas 20 (arbitrary units), auxiliary gas 5.0 (arbitrary units), capillary temperature 320 °C and resolution 35,000. MS/MS spectra were obtained by a Higher Energy Collision Dissociation (HCD) of 30 (arbitrary units). The accuracy error threshold was fixed at 5 ppm. The final annotated metabolome dataset was generated by Compound Discoverer 3.3 (Thermo Fisher Scientific, Waltham, MA, USA). The Compound Discoverer 3.0 software is fully integrated with the ChemSpider database and the mzCloud for automated and expedited data processing. The retention time tolerance (RT) was set to 0.2 min, with mass tolerance equal to 5 ppm, and other parameters were selected as the default values for peak extraction and peak alignment.
2.9. In vitro Cytotoxicity and Anti-Inflammatory Activity
The evaluation of in vitro acute toxicity does not depend on the final use for which the product is intended, and the document ISO 10995-5:2009 (Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity) [ISO 10993-5; Biological Evaluation of Medical Devices. Part 5: Tests for In vitro Cytotoxicity. ISO: Geneva, Switzerland, 2009.] recommends some cell lines from American Type Collection as models to screen the cytotoxicity of novel materials or compounds. Among them, NIH3T3 mouse fibroblasts were chosen to test CSB extract in vitro cytotoxicity.
2.9.1. Cell Cultures
NIH3T3 and RAW 264.7 macrophage cells were obtained from ATCC (ATCC, Manassas, VA, United States) and cultured in DMEM containing 10% v/v FBS, 100 mg/mL penicillin and 100 mg/mL streptomycin. Cultures were maintained at 37◦C in a humidified atmosphere of 5% CO2. Comparative analysis was performed with cell populations at the same generation.
2.9.2. NIH3T3 Cytotoxicity
Once at confluence, NIH3T3 were washed with 0.1 M PBS, separated with trypsin-EDTA solution, centrifuged at 1200 RCF for 5 min., re-suspended in complete medium at a density of 1.5 × 104 cells/mL, seeded in each well of 24-well plates and incubated at 37 °C in an atmosphere of 5% CO2. After 24 h of culture, the test compounds, properly diluted in completed medium were added to each well. The following concentrations of CSB extract were tested: 0.025, 0.050, 0.10, 10,0 mg/mL for 24, 48, and 72 h. The experiments were repeated three times, and all samples were set up in six replicates. Complete medium was used as negative control. After 24, 48 and 72 h of incubation, cell viability was evaluated by neutral red uptake (NRU) assay using the procedure already reported [21].
2.9.3. Cell Viability
RAW 264.7 cells were seeded at 1×104 cells/well, in 96-well plates and cultured until sub-confluence (80–85% confluence). Cells were treated with different concentrations (6, 12, 25, 50, and 100 µg/mL) of CSB prepared in DMSO (Sigma-Aldrich) and diluted in medium, and the final DMSO concentration was kept below 0.1% v/v throughout the experiment. The control was performed by treating cells with DMSO 0.1% v/v, corresponding to the higher concentration of the compound. After 24 h of treatment, cells were washed with sterile PBS and MTT was added to a final concentration of 1 mg/mL. After a 2 h incubation, cells were lysed with 150 µL DMSO. The absorbance was measured at 550 nm using an EnVision system (PerkinElmer) and percentage of cell viability was calculated relative to control. The percentage of viable cells was determined relative to the vehicle control.
2.9.4. Cell Stimulation
Cells were treated with CSB prior to stimulation with Lipopolysaccharide (LPS) (obtained from Escherichia coli O111:B4, Sigma-Aldrich). Dexamethasone (DEX) (Sigma-Aldrich), commonly used to treat inflammation, was used as a positive control at a concentration of 5 µg/mL.
2.9.5. Quantification of Intracellular ROS Formation
The generation of reactive oxygen species (ROS) within RAW 264.7 cells was determined in 96-well plates with 2′,7′-dichlorodihydrofluorescein diacetate (DCFH2-DA), which is intracellularly deacetylated and oxidized to highly fluorescent 2′,7′-dichlorofluorescein (DCF) [22]. After pre-treatment with different concentrations of CSB, cells were stimulated with LPS (200 ng/mL) for 5 h. DCFH2-DA (10 µM) dissolved in HBSS was applied to the cells and incubated for 10 min at 37 °C. The plate was scanned using an EnVision system (PerkinElmer) with excitation λ of 485 nm and emission λ of 535 nm. Afterwards, the number of cells in each well was determined by Crystal Violet assay [23]. Briefly, after removing the medium the cells were washed and stained with 0.1% crystal violet at RT for 20 min under stirring. Next, cells were washed and incubated with 200µL pure ethanol for another 20 min at RT under stirring. OD was measured at 570 nm. Results were normalized to the relative cell count for each well and expressed as the relative ROS production % (RFI) with respect to the LPS group.
2.9.6. Determination of NO Production
The production of nitric oxide (NO) in the supernatant of RAW 264.7 cells was determined in 6-well plates (1×106 cells/well) cultured until sub-confluence (80-85%). After treatment with CSB at different concentrations (25, 50 and 100 μg/mL) for 4 h, the cells were stimulated with LPS (200 ng/mL) for 24 h. Following stimulation, 100 µL of conditioned medium from each well was transferred to a new 96-well plate and mixed with an equal volume of Griess reagent composed of 1% sulfanilamide and 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in 5% phosphoric acid. After incubation at RT for 10 min, the absorbance was measured at 540 nm using an EnVision system (PerkinElmer). The nitrite concentration was assessed by a sodium nitrite standard curve.
2.9.7. Protein Extraction
Whole-cell lysates were obtained using RIPA buffer, added with phosphate and protease inhibitors, and then disrupted by sonication for 15 min in an ice bath. Protein concentration was assessed by BCA protein assay. Nuclear fractionations were obtained with NE-PER™ Cytoplasmic and Nuclear Protein Extraction Kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol.
2.9.8. Western Blotting
20 μg of protein were resolved by 8% SDS–PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked in PBS 10% w/v nonfat dry milk at RT with gentle shaking for 2h. The membrane was incubated with anti-iNOS (rabbit polyclonal IgG, 1:10.000 Sigma-Aldrich), anti-NF-κB p65 (clone 1G10.2, 1:500) mouse monoclonal antibody (Sigma-Aldrich) and anti-GAPDH HRP-conjugated (1:50.000) primary antibodies, ON at 4◦C. The blots were washed three times and incubated with anti-rabbit HRP-conjugated secondary antibody (Sigma-Aldrich) 1:80,000 and anti-mouse HRP-conjugated secondary antibody (Sigma-Aldrich) 1:50,000 for 1 h, RT. After washing three times, immunoreactive bands were detected using ECL (LuminataCrescendo, Merck Millipore, Burlington, MA, USA) and images acquired by LAS4000 (GE Healthcare, Chicago, IL, USA). The optical densities of immunoreactive bands were analyzed by ImageQuantTL software (GE Healthcare, Chicago, IL, USA, V 7.0) using GAPDH as a loading normalizing factor.
2.9.9. Immunofluorescence Study
RAW 264.7 cells were grown on glass coverslips for 24 h. Then the cells were pre-treated with CSB at 100 μg/mL for 4 h and were stimulated with LPS for 1 h. The cells were fixed with 4% paraformaldehyde dissolved in PBS for 15 min. After washing three times with PBS, cells were permeabilized using 0.5% TritonX-100 in PBS for 5 min. Then cells were incubated with 5% NGS in PBS solution for 20 min, and with 1:200 of anti-NF-κB p65 (clone 1G10.2) mouse monoclonal antibody (Sigma-Aldrich) at 4 ◦C overnight. Cells were washed three times with PBS for 10 min each time, and incubated with a 1:100 dilution of Alexa 594-conjugated goat anti-Mouse Ig (Life Technologies, USA) for 1 h, in the dark, at RT. After washing three times with PBS and one time with distilled water, cells were mounted with fluoroshield mounting medium containing DAPI. Images were captured by fluorescence microscopy (Zeiss AxioLabA1, Oberkochen, Germany).
2.10. Mutagenicity Assay: Ames Test
The TA100 and TA98 strains of Salmonella typhimurium were utilized for mutagenicity assay in absence and presence of metabolic activation, i.e. with and without S9 liver fraction. The tester strains used were selected because they are sensitive and detect a large proportion of known bacterial mutagens and are most commonly used routinely within the pharmaceutical industry [24]. The following specific positive controls were used, respectively, with and without S9 fraction: 2-Nitrofluorene (2-NF) 2 µg/mL + 4-Nitroquinoline N-oxide (4-NQO) 0.1 µg/mL, and 2-aminoanthracene (2-AA) 5 µg/mL. The final concentration of S9 in the culture was 4.5%.
Approximately 107 bacteria were exposed to 6 concentrations (0,025, 0,050, 0,10, 0,50, 1,0 and 10,0 mg/mL) of the CSB extract, as well as to positive and negative controls, for 90 minutes in medium containing sufficient histidine to support approximately two cell divisions. After 90 minutes, the exposure cultures were diluted in pH indicator medium lacking histidine, and aliquoted into 48 wells of a 384-well plate. Within two days, cells which had undergone the reversion to His grew into colonies. Metabolism by the bacterial colonies reduced the pH of the medium, changing the colour of that well. This colour change can be detected visually. The number of wells containing revertant colonies were counted for each dose and compared to a zero dose control. Each dose was tested in six replicates.
The material was regarded mutagenic if the number of histidine revertant colonies was twice or more than the spontaneous revertant colonies.
2.11. Statistical Analysis
Experiments were performed in triplicate. Statistical analyses were performed with GraphPad Prism 9.0 software (GraphPad Software, San Diego, CA, USA). Data are presented as mean±SD and were compared using the unpaired t-test or the one-way ANOVA with an appropriate post hoc test. A P value of 0.05 or less was considered significant.
2.12. In Silico Studies
2.12.1. Structural Optimization and Resources
The anti-inflammatory target complement was retrieved from DrugBank [25,26] using the "target section" with the keyword "inflammatory". To select the RAW 264.7 anti-inflammatory target complement, we analyzed the RAW 264.7 transcriptome using the Harmonizome 3.0 database [27], and extracted all targets present in the anti-inflammatory target complement list of DrugBank. Their 3D structures and FASTA sequences were retrieved from the RCSB Protein Data Bank [26] and UniProt database [28], respectively. The 3D structures were obtained by performing a multiple sequence alignment with BLASTp v.2.15.0 and choosing PDB as the search database; all parameters were used as default [29]. All targets considered in this study are reported in Table S1.
To avoid errors during the docking simulations, the potential missing side chains and steric clashes in the 3D structures reported in PDB files were added/resolved with molecular/homology modelling using MODELLER v.9.3 implemented in PyMOD3.0 (PyMOL2.5 plugin) [30]. The three-dimensional structures were then analyzed and validated using PROCHECK v.3.5.4 [31]. Prior to conducting the docking simulations, high-energy intramolecular interactions were minimized using GROMACS 2019.3 [32] with the charmm36 force field. CHARMM-GUI v.3.8 [33] was used to assign all parameters to the biological targets and ligands.
In detail, prior to conducting further simulations, the starting conformation sequence was aligned against its primary structure, allowing for the addition of potential missing side chains to the structure. Furthermore, loop modeling implemented in MODELLER v.9.3 was employed to optimize the best starting orientation of each loop within the structure. Lastly, each structure was analyzed using the PROCHECK tool, where a Ramachandran plot (which analyzes the backbone of ϕ and ψ angles and Chi1–Chi2 plots for side chains) confirmed the validity of the starting conformation. Then, we minimized the energy of each structure by performing energy minimization using GROMACS 2019.3 with the charmm36 force field. This step was taken to prevent the possibility of the structures to sterically hinder potential clashes and/or to optimize the energy values. The resulting structures were then immersed in a cubic box filled with TIP3P water molecules, and the system was neutralized with the addition of counter ions. Simulations were run by applying periodic boundary conditions. Energy minimization was performed with 5000 steps using the steepest descent as the algorithm, which converged to a minimum energy with forces less than 10 kJ/mol/nm.
To enhance the reliability of our simulations, we conducted docking simulations based on in vitro evidence. Therefore, we selectively choose targets for which their experimental 3D structures were in complex with an active compound. In cases where multiple 3D structures of the same target were combined with different ligands in different binding regions, such as allosteric pockets, we created a box capable of enclosing such binding regions. Consequently, a box was created for each target, and we set the grid box at the center of mass of the ligand in the experimental 3D structure of the target, using AutoDock/VinaXB v.1.1.2. and MGLTOOLS v.1.5.7 [34] scripts. To provide a more consistent result for our docking simulation, we changed the default exhaustiveness from 8 to 32 and only selected binding poses with a root mean square deviation (RMSD) 2 Å lower than that of the best-docked pose. All parameters were used as default.
Three-dimensional structures of compounds were retrieved and downloaded in sdf format from the PubChem database [35] (detailed information provided in Table S2). Then, a virtual screening was carried out using the extracted compounds on the targets. OpenBabel v.3.1.0 [36] was used to convert protein and ligand files and to assign gasteiger partial charges, as proposed in previous works [37,38]. The interaction network was explored with PLIP tool [39]. As suggested in a previous work [27,40] an alignment of sequences was performed to identify potential key residues of targets using ClustalW v.2.1 [41].
3. Results
3.1. Chemical Composition and Antioxidant Capacity of C. sativa Extract
The extract of C. sativa burrs (CSB) under investigation, produced by ultrasound-assisted extraction using water as a solvent, was first characterized for its TPC and TFC using appropriate colorimetric dosages to evaluate the extraction process's efficiency. The antioxidant capacity of the extract was then evaluated by estimating the reducing power using the potassium ferricyanide method and the radical scavenging activity using the ABTS•+ and DPPH assays. Table 1 shows the calculated TPC (mg GAE/g dry extract), TFC (mg QE/g dry extract), reducing power (mg AAE/g dry extract) and radical scavenging activity (IC50 µg/mL) of CSB.
CSB exhibited an exceptionally elevated TPC of 243.98±17.77 mg GAE/g, with a calculated TFC of 27.54±0.60 mg QE/g (Table 1), as visually depicted in Figure 1a. Accordingly, CSB showcased notable reducing power, determined by the potassium ferricyanide method (272.12±4.64 mg AAE/g of dry extract, Table 1), correlating with an elevated radical scavenging activity. Figure 1b illustrates a more pronounced impact on the ABTS radical compared to DPPH, notably revealing that CSB demonstrated an IC50 (µg/mL) (i.e., Inhibitory Concentration causing a 50% decrease of the absorbance) significantly lower (p<0.0001) than the standard compound Trolox.
CSB was subsequently subjected to UPLC-MS-MS analysis, which provided confirmation of the presence of phenolic compounds, representing the 71.43% of the total CSB (Table 2). The presence of amino acids (22.23%), terpenes (1.69%), and hormones (3.64%) was also observed.
A total of 56 metabolites were identified using Compound Discoverer 3.3 software integrated with the ChemSpider database and the mzCloud for data processing and compared with literature data. The results are listed in Table 3, along with their retention time, molecular formulae, observed and theoretical m/z, and error (ppm). For all matched compounds, the error was lower than 5 ppm (Table 3). The most abundant compound was the phenolic Ellagic Acid (51.7%), followed by the amino acid Betaine (22.0%) and the flavonol 5,7-dihydroxy-3,8-dimethoxy-2-phenyl-4h-chromen-4-one (15.8%).
3.2. NIH3T3 Viability and Proliferation
Non-confluent adherent mouse fibroblasts (NIH3T3) were incubated with increasing concentrations of CSB extract ranging from 0.025 to 10 mg/mL. Cells were analyzed after 24, 48, and 72 h of incubation with the extract, and the results in terms of cell viability (%) as a function of both samples’concentration and incubation time are reported in Figure 2.
The data reported in Figure 2 show that CSB extract was unable to influence the viability and proliferation of mouse fibroblasts at any of the tested concentrations and incubation times. ISO standard states that a material can be considered non-cytotoxic if it allows cell viability greater than 70% after exposure for 24 h. In this regard, CSB extract result highly cytocompatible both in terms of viability and cell proliferation.
3.3. CSB Inhibits LPS-Induced ROS Generation
The cytotoxicity of CSB was assessed by the MTT assay. Results are reported in Fig. 3A as a percentage of cell viability compared to control, obtained by treating the cells with DMSO, as a vehicle, at the concentration corresponding to the highest dose used; the final concentration never exceeded 0.1% (v/v) in both treated and untreated cells and did not adversely affect the parameters analyzed. Results showed that no concentration of CSB used had an adverse effect on cell viability of RAW 264.7 cells (Figure 3a). We used 25, 50 and 100 µg/mL as the optimal concentrations in the subsequent experiments. A microplate-based DCFH-DA assay showed that LPS-induced RAW 264.7 cells exhibited the highest fluorescence intensity DCFH-DA staining, which indicates ROS production; however, CSB significantly inhibited the cellular ROS generation at any tested concentration (Figure 3b).
3.4. CSB Reduced LPS-Induced Inflammation in RAW 264.7 Cells
Macrophages are the main cells responsible for the initiation of the inflammatory cascade. We treated RAW 264.7 murine macrophages with different concentrations of CSB and measured the amount of the main pro-inflammatory mediators. As expected, the NO production was strongly inhibited by DEX (5 μg/mL). LPS-induced NO production was significantly decreased by the presence of CSB extract as well. The strongest effect was observed at 100 µg/mL, with significant reductions in NO levels also obtained at concentrations of 25 and 50 µg/mL (Figure 4). DMSO vehicle control demonstrated no effect on suppressing LPS-induced NO production in RAW264.7 cells.
Protein expression of inducible NO synthase (iNOS), the precursor enzyme of NO, was determined by Western Blotting. Figure 5 shows a dose-dependent reduction of expression levels when compared to the LPS-treated group.
The Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is implicated in the regulation of inflammatory mediators in LPS-stimulated macrophages [42]. Therefore, this study investigated whether CSB has the ability to block the activation of NF-κB pathway. The analysis of NF-κB localization by immunostaining and fluorescence microscopy showed that pre-treatment with CSB extract significantly reduced the nuclear expression of NF-κB p65 in RAW 264.7 cells (Figure 6a). The analysis of NF-κB localization by immunostaining and fluorescence microscopy showed that in normal RAW 264.7 cells, the p65 protein was localized outside the nucleus, and LPS stimulation induced the translocation of p65 from the outside to inside the nucleus. However, CSB extract at concentration 100 µg/mL retained NF-κB in the cytoplasm of cells (Figure 6b).
3.5. Mutagenicity Assay: Ames Test
In Salmonella mutagenicity assay, six different concentrations of the CSB extract were tested by Ames test on TA98, and TA100 strains with and without S9 metabolic activation. The results for the mutagenic effect of the samples, reported in Figure 7a,b, demonstrated that CSB extract at all the concentrations tested was not genotoxic towards both TA98 and TA100 with and without S9 fraction. In fact, also at the highest concentration (10 mg/mL), the number of revertants was lower and statistically different in comparison to positive control (p<0.01). The background level, as well as positive control values, were in all cases within the normal limit found in our laboratory.
2.2. In Silico Results
2.2.1. Target/Compound Virtual Screening
To identify potential targets involved in interactions with our compounds, we conducted a ligand-based virtual screening against the entire RAW 264.7 cell anti-inflammatory target complement identified through the "target section" in the DrugBank database. Our investigation provided 41 targets indirectly/directly involved in the cell anti-inflammatory condition, and the primary structure of each target was downloaded from the UniProt database. To verify the availability of 3D structures for these targets, a multiple sequence alignment (MSA) was performed using BLASTp with the “Protein Data Bank” database. From the MSA results, we obtained and downloaded a total of 32 protein 3D structures (Table S1).
Each structure underwent optimization through molecular modeling, to resolve potential structural gaps and steric clashes. Subsequently, a virtual screening was performed among all targets and compounds extracted from C. sativa. To standardize our analyses, and bolster the reliability of our in silico results, we employed two different strategies to select the best three complexes—(i) binding free energy (docking score) and (ii) evolution approach considering the inter-action network consensus binding residue, as suggested in a previous work [43]. We selected the first three complexes with the highest binding free energy: the crystal structure of Jnk1 (PDB code: 3ELJ), the crystal structure of MAPK11 (PDB code: 8YGW), and the structure of human ERK1 (PDB code: 4QTB). Remarkably, all targets were in the same complex with ellagic acid, showing a binding free energy from −9.5 Kcal/mol to −8 kcal/mol. Interaction network analyses showed that the ellagic acid formed a wide polar and hydrophobic interaction network within the target binding pocket (Figure 8). Furthermore, the MSA results revealed the ability of our compounds to trigger strong polar interactions with target key residues: Lys-53 (ATP binding site), Glu-71 (inhibitor binding site) (residue number based on the crystal structure of MAPK11 with UniProtKB entry- Q15759-) (Figure S1). Taken together, our findings suggest the potential of our compound to inhibit the targets by forming strong interactions with critical residues.
4. Discussion
The demonstrated benefits of utilizing plant-derived metabolites, especially those obtained from waste biomasses, as a source of potential therapeutics are now widely acknowledged. C. sativa main fruit or by-products’ extracts are a valuable source of bioactive secondary metabolites with outstanding antioxidant, anti-inflammatory, and anti-microbial properties [1,44,45,46,47,48,49]. These characteristics have led to the proposition of the application of C. sativa in the health and cosmetic industry, as demonstrated by several studies [1,9,10,11,50,51].
The aim of this study was to extract bioactive compounds from the spiny burrs of Monte Amiata PGI C. sativa using a total-green ultrasound-assisted extraction method with water as the solvent. Traditionally, polyphenols and other antioxidant or potential therapeutic compounds are extracted using mixtures of methanol, ethanol, or acetone and water [52]. However, ultrasound-water extractions have shown promising efficiency in extracting phenolic compounds [53,54]. The effectiveness of the procedure was evaluated by measuring the TPC, TFC, and antioxidant capacity of the extract. Our findings indicated that CSB demonstrated a notable abundance of phenolic compounds and displayed an overall optimal antioxidant capacity, as assessed through various assays including TRP, ABTS•+, and DPPH. Notably, the radical scavenging activity of the ABTS•+ radical exhibited by CSB surpassed that of the standard Trolox, indicating a particularly robust antioxidant potential. These results suggest that the extraction method effectively retained a significant quantity of phenolic compounds in the CSB extract, contributing to its remarkable antioxidant properties. The superiority of CSB's radical scavenging activity over the standard further emphasized its potential as a valuable source of natural antioxidants. The comprehensive assessment using multiple assays provides a robust understanding of the antioxidant capacity of the extract, however reporting the phenolic content and antioxidant capacity of extracts in literature is not standardized, making the comparative analysis challenging; this highlights the need for standardized normalization factors [52]. Since information is scarce in the literature regarding the aqueous extraction of C. sativa burrs [1], and no in-depth investigations on the phytochemical composition of PGI C. sativa burrs from Monte Amiata have been conducted, we analyzed CSB by UPLC-MS-MS which confirmed the presence of high abundance of phenolic compounds, such as ellagic acid and other phenol glucoside derivatives, flavonoids and flavonol derivatives and their glycosides, as well as triterpenoids and plant hormones. Moreover, the analysis led to the identification of a wide range of secondary metabolites with numerous biological activities such as antioxidant, antiviral, and anti-inflammatory activities, among others, suggesting a possible reuse of these by-products as a natural source of bioactive phytochemicals [55]. In vitro assessment of the cytotoxicity of CSB towards NIH3T3 mouse fibroblasts and Salmonella mutagenicity assay demonstrated our extract is not cytotoxic and not mutagenic. Therefore, it was tested on LPS-challenged RAW 264.7 macrophage cells. Oxidative stress and inflammation are strictly related. In the inflammatory process, macrophages play pivotal roles such as antigen presentation, phagocytosis, and immunomodulation by generating a variety of cytokines and growth factors [42]. It is well known that iNOS plays an important role in inflammation. NO, downstream signaling factor of iNOS, and several other pro-inflammatory cytokines and chemokines are involved in the regulation of immune and inflammatory responses causing symptoms such as pain, fever, and edema [56]. NO, which is regulated by iNOS, is a potent reactive factor in inflammatory responses found in stimulated macrophages and in the sites of inflammation [57]. For this reason, therapeutic interventions that targeted macrophages and their products could open new avenues for anti-inflammatory treatments.
Numerous experimental models have been developed to aid in the development of new anti-inflammatory drugs. However, many instances in the literature emphasize the use of the in vitro model employing LPS-stimulated RAW 264.7 cells to explore the anti-inflammatory properties of natural extracts and anti-inflammatory medications. This model has proven to be a suitable and reliable in vitro approach for studying inflammation [58]. Results first showed that CSB did not affect the cell viability of macrophages at any tested concentration, demonstrating no cytotoxic effects. Moreover, pretreatment with CSB significantly reduced the production of ROS and NO in the supernatants of RAW 264.7 cells and LPS-induced increased levels of iNOS protein in a dose-dependent manner. NF-κB plays an important role in the control of the gene encoding pro-inflammatory cytokines as well as inducible enzymes, including iNOS [42]. Increased activation of the NF-κB signaling pathway triggers the production of downstream inflammation-related factors. Since we observed that CSB modulated NF-κB downstream pro-inflammatory markers, we further elucidated if the extract could interfere with the activation of NF-κB signaling by examining its effect on inhibiting LPS-induced translocation of NF-κB into the nucleus. Our results showed that CSB treatment caused a significant decrease of NF-κB p65 expression in the nucleus indicating that signal transduction pathways mediated by NF-κB may be effectively blocked by CSB.
The most abundant compound found in CSB was ellagic acid, which has gained attention for its potential therapeutic effects in treating human diseases. Its properties include antioxidant, anti-inflammatory, antimutagenic, and antiproliferative. Alone, or combined with other antioxidants, has shown positive therapeutic effects [59].
Our in silico investigations, directed at pinpointing potential targets in RAW 264.7 cells implicated in anti-inflammatory conditions, supported existing literature. We retrieved the complete anti-inflammatory target complex from the "target section" within the DrugBank database. Subsequently, we conducted molecular modeling to acquire and refine the 3D structures of the pre-selected targets. To define the potential compound(s) derived from chestnut burrs possessing anti-inflammatory proprieties and its biological target(s), a virtual screening of bioactive compounds was performed. Each target was subjected to computational virtual screening. As a result of the docking simulation, ellagic acid emerged as the compound with the highest binding free energy score among all compounds on the best three targets, triggering strong polar interactions with target critical residues [60,61]. Furthermore, ellagic acid shared both a similar binding region and pose with known experimental inhibitors of these kinases.
Other phenolic compounds identified in CSB were gallic acid and derivatives, especially n-propyl gallate and ethyl gallate. Gallic acid has been acknowledged for its therapeutic properties, such as its ability to act as an antioxidant and anti-inflammatory agent [62]. According to Mard et al., gallic acid pretreatment of a gastric mucosal lesion decreased inflammatory responses via inhibiting iNOS [63]. Moreover, another phenolic compound found in CSB, Protocatechuic aldehyde (PCA), was previously reported to suppress inflammatory effects. More specifically, it was found that PCA reduced the production of NO and the expression level of the iNOS gene induced by LPS in RAW 264.7 cells [1]. Overall, numerous studies have reported the ability of phenolic compounds to repress inflammation-related genes in various types of cells [64,65,66,67,68].
Flavonoids was the main subclass of phenolic compounds in CSB. It was reported that flavonoids can exert their anti-inflammatory action largely by modulating the expression of proinflammatory molecules, including iNOS, and proinflammatory cytokines [64,65,69,70]. Studies have suggested that flavonoid dietary intake is inversely associated with age-related diseases such as cardiovascular disease, neurodegeneration, and type 2 diabetes [69,70]. Moreover, Lim et al., reported studies demonstrating that uptake of flavonoids, including kaempferol, also lowered the elevated level of inflammatory cytokines and NF-κB activation in aged animal model [65]. Worth mentioning is the presence of Betaine in CSB, also known as trimethylglycine, as it is a modified amino acid that is considered an important human nutrient [71]. This dietary supplement obtained from various foods has demonstrated anti-inflammatory potential [72,73]. Go et al., conducted a study on Sprague-Dawley (SD) rats to evaluate the in vivo anti-inflammatory effect of betaine on NF-κB. The results showed that betaine suppressed NF-κB and related gene expression of iNOS and attenuated oxidative stress-induced NF-κB in YPEN-1 cells, suggesting its potential as a preventive agent against the activation of NF-κB induced during inflammation and aging [73].
Although some compounds were already reported in the burrs of C. sativa [9,10,14,55], to the best of our knowledge, this is the first report on the detailed composition of C. sativa burrs from Monte Amiata. The high antioxidant potential of chestnut burr extracts is due to their phenolic contents, commonly extracted using hydroalcoholic solvents [52]. Nonetheless, we found high content of phenolic compounds in C. sativa spiny burrs extracted with an innovative water-ultrasound method, which has the advantages of being non-toxic, environmentally friendly, and safe. It is worth mentioning that other extracts from C. sativa by-products have demonstrated anti-inflammatory potential. An extract tested on BV-2 microglia cells showed cytoprotective activity and reduction of the transcriptional levels of cytokines, and NF-kB expression following LPS stimulation, imputable to the presence of flavonoids such as astragalin, isorhamnetin glucoside, and myricitrin, [14]. Moreover, GA and PCA were identified as the primary phenolic components in a chestnut shell extract that effectively protected against inflammation, dehydration, and photoaging caused by external agents such as UV radiation. This was demonstrated by evaluating the expression of proteins involved in water balance and collagen stability in human keratinocytes [1].
The discovery that an aqueous extract from the spiny burrs of C. sativa, rich in antioxidant compounds can significantly suppress the main inflammatory players in macrophagic cells opens many possibilities for treating various inflammatory illnesses.
5. Conclusions
Our research provides valuable information regarding the phytochemical composition, antioxidant capacity and therapeutic properties of C. sativa burrs with PGI status from Monte Amiata. The bioactive compounds extracted using an innovative water-ultrasound method demonstrated antioxidant activities and anti-inflammatory properties. This promising outcome suggests that these by-products could be employed in commercial products as a natural source of beneficial compounds. The high phenolic content, extracted with a totally green method, is noteworthy due to its non-toxic, environmentally friendly, and safe nature. This perspective on waste valorization offers a solution to the economic and environmental challenges associated with the disposal of chestnut by-products, potentially generating income. Our findings strongly encourage the recycling and valorization of C. sativa by-products, which should no longer be considered as mere "waste".
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Sequence alignment results between human MAPK11, ERK1, and JNK1. Identical and positively conserved amino acid residues are marked with a star and colons, respectively. The red boxes highlight all the amino acids involved in hydrogen bonds and/or salt bridges with ellagic acid.; Table S1: List of targets considered in this study; Table S2: List of compounds considered in this study.
Author Contributions
Conceptualization, L.F., A.S., and M.G; methodology, L.F, M.G., S.L. and L.S.; validation, L.F., M.G. and A.S.; investigation, L.F., T.O., P.M., S.L., L.S. and M.G.; formal analysis, A.T.; resources, A.S.; data curation, L.F.; writing—original draft preparation, L.F.; writing—review and editing, L.F., A.S., M.G., S.L.; visualization, L.F., M.G. and A.T.; supervision, A.S., and O.S.; project administration, A.S.;. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors thank: Associazione per la Valorizzazione della Castagna del Monte Amiata IGP; NEXT Technologies Tecnotessile, Prato, Italy; PRIN: PROGETTI DI RICERCA DI RILEVANTE INTERESSE NAZIONALE – Bando 2022 Prot. 2022LW54KC; PRIN: PROGETTI DI RICERCA DI RILEVANTE INTERESSE NAZIONALE – Bando 2022 PNRR Prot. P2022RYR5W; CHEBAPACK Programma di Sviluppo Rurale 2014-2020 Regione Toscana; PSR 2014-2020 Regione Toscana OPENRICCIO; F-Cur funds to M.G.; UE – FSE REACT-EU, PON Ricerca e Innovazione 2014-2020; Progetto ERICA ARTES 4.0, E87G23000100001 Agenzia Coesione Territoriale; Progetto PNRR THE Spoke 6.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Squillaci, G.; Apone, F.; Sena, L.M.; Carola, A.; Tito, A.; Bimonte, M.; Lucia, A. De; Colucci, G.; Cara, F. La; Morana, A. Chestnut ( Castanea Sativa Mill.) Industrial Wastes as a Valued Bioresource for the Production of Active Ingredients. Process Biochemistry 2018, 64, 228–236. [Google Scholar] [CrossRef]
- Pinto, D.; Braga, N.; Silva, A.M.; Costa, P.; Delerue-Matos, C.; Rodrigues, F. Chestnut. In Valorization of Fruit Processing By-products; Elsevier, 2020; pp. 127–144. [Google Scholar]
- Battisti, A.; Benvegnù, I.; Colombari, F.; Haack, R.A. Invasion by the Chestnut Gall Wasp in Italy Causes Significant Yield Loss in Castanea Sativa Nut Production. Agric For Entomol 2014, 16, 75–79. [Google Scholar] [CrossRef]
- Savchenko, T.; Degtyaryov, E.; Radzyukevich, Y.; Buryak, V. Therapeutic Potential of Plant Oxylipins. International Journal of Molecular Sciences 2022, Vol. 23, Page 14627 2022, 23, 14627. [Google Scholar] [CrossRef] [PubMed]
- Https://Www.Marketgrowthreports.Com/Global-Biomedical-Materials-Market-21051012.
- Li, L.; Peng, P.; Ding, N.; Jia, W.; Huang, C.; Tang, Y. Oxidative Stress, Inflammation, Gut Dysbiosis: What Can Polyphenols Do in Inflammatory Bowel Disease? Antioxidants 2023, 12, 967. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y. Regulation of the Gut Microbiome by Inflammasomes. Free Radic Biol Med 2017, 105, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Lillo, S.; Saleh, M. Inflammasomes in Cancer Progression and Anti-Tumor Immunity. Front Cell Dev Biol 2022, 10. [Google Scholar] [CrossRef]
- Pinto, D.; Braga, N.; Rodrigues, F.; Oliveira, M. Castanea Sativa Bur: An Undervalued By-Product but a Promising Cosmetic Ingredient. Cosmetics 2017, 4, 50. [Google Scholar] [CrossRef]
- Pinto, D.; Rodrigues, F.; Braga, N.; Santos, J.; Pimentel, F.B.; Palmeira-de-Oliveira, A.; Oliveira, M.B.P.P. The Castanea Sativa Bur as a New Potential Ingredient for Nutraceutical and Cosmetic Outcomes: Preliminary Studies. Food Funct 2017, 8, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Costa, P.C.; Delerue-Matos, C.; Rodrigues, F. Assessment of a Formulation Containing a Castanea Sativa Shells Extract on Skin Face Parameters: In Vivo Evaluation. Processes 2022, 10, 2230. [Google Scholar] [CrossRef]
- Nema, N.; Rajan, N.; Babu, M.; Sabu, S.; Khamborkar, S.; Sarojam, S.; Sajan, L.; Peter, A.; Chacko, B.; Jacob. Viju Plant Polyphenols as Nutraceuticals and Their Antioxidant Potentials. In Polyphenols; Wiley, 2023; pp. 21–44. [Google Scholar]
- Squillaci, G.; Apone, F.; Sena, L.M.; Carola, A.; Tito, A.; Bimonte, M.; Lucia, A. De; Colucci, G.; Cara, F. La; Morana, A. Chestnut ( Castanea Sativa Mill.) Industrial Wastes as a Valued Bioresource for the Production of Active Ingredients. Process Biochemistry 2018, 64, 228–236. [Google Scholar] [CrossRef]
- Chiocchio, I.; Prata, C.; Mandrone, M.; Ricciardiello, F.; Marrazzo, P.; Tomasi, P.; Angeloni, C.; Fiorentini, D.; Malaguti, M.; Poli, F.; et al. Leaves and Spiny Burs of Castanea Sativa from an Experimental Chestnut Grove: Metabolomic Analysis and Anti-Neuroinflammatory Activity. Metabolites 2020, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Fernando, A.L.; Duarte, M.P.; Vatsanidou, A.; Alexopoulou, E. Environmental Aspects of Fiber Crops Cultivation and Use. Ind Crops Prod 2015, 68, 105–115. [Google Scholar] [CrossRef]
- Durán-Zuazo, V.H.; Rodríguez, B.C.; García-Tejero, I.F.; Ruiz, B.G. Suitability and Opportunities for Cannabis Sativa L. as an Alternative Crop for Mediterranean Environments. In Current Applications, Approaches, and Potential Perspectives for Hemp; Elsevier, 2023; pp. 3–47. [Google Scholar]
- Song, F.L.; Gan, R.Y.; Zhang, Y.; Xiao, Q.; Kuang, L.; Li, H. Bin Total Phenolic Contents and Antioxidant Capacities of Selected Chinese Medicinal Plants. Int J Mol Sci 2010, 11, 2362–2372. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Yang, M.-H.; Wen, H.-M.; Chern, J.-C. Estimation of Total Flavonoid Content in Propolis by Two Complementary Colometric Methods. J Food Drug Anal 2020, 10. [Google Scholar] [CrossRef]
- Jayaprakasha, G.K.; Singh, R.P.; Sakariah, K.K. Antioxidant Activity of Grape Seed (Vitis Vinifera) Extracts on Peroxidation Models in Vitro. Food Chem 2001, 73, 285–290. [Google Scholar] [CrossRef]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP Decolorization Assay of Antioxidant Capacity Reaction Pathways. Int J Mol Sci 2020, 21, 1131. [Google Scholar] [CrossRef] [PubMed]
- Lamponi, S. Preliminary In Vitro Cytotoxicity, Mutagenicity and Antitumoral Activity Evaluation of Graphene Flake and Aqueous Graphene Paste. Life 2022, 12, 242. [Google Scholar] [CrossRef]
- Ng, N.; Ooi, L. A Simple Microplate Assay for Reactive Oxygen Species Generation and Rapid Cellular Protein Normalization. Bio Protoc 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb Protoc 2016, 2016, pdbprot087379. [Google Scholar] [CrossRef]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/Microsome Mutagenicity Assay. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A Better Web Interface. Nucleic Acids Res 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Janson, G.; Paiardini, A. PyMod 3: A Complete Suite for Structural Bioinformatics in PyMOL. Bioinformatics 2021, 37, 1471–1472. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput Phys Commun 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J Comput Chem 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, New Empirical Halogen Bond Scoring Function, into AutoDock Vina. J Cheminform 2016, 8, 27. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J Cheminform 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Fusi, F.; Trezza, A.; Spiga, O.; Sgaragli, G.; Bova, S. Ca v 1.2 Channel Current Block by the PKA Inhibitor H-89 in Rat Tail Artery Myocytes via a PKA-Independent Mechanism: Electrophysiological, Functional, and Molecular Docking Studies. Biochem Pharmacol 2017, 140, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Fusi, F.; Durante, M.; Spiga, O.; Trezza, A.; Frosini, M.; Floriddia, E.; Teodori, E.; Dei, S.; Saponara, S. In Vitro and in Silico Analysis of the Vascular Effects of Asymmetrical N,N-Bis(Alkanol)Amine Aryl Esters, Novel Multidrug Resistance-Reverting Agents. Naunyn Schmiedebergs Arch Pharmacol 2016, 389, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein–Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Carullo, G.; Ahmed, A.; Trezza, A.; Spiga, O.; Brizzi, A.; Saponara, S.; Fusi, F.; Aiello, F. A Multitarget Semi-Synthetic Derivative of the Flavonoid Morin with Improved in Vitro Vasorelaxant Activity: Role of CaV1.2 and KCa1.1 Channels. Biochem Pharmacol 2021, 185, 114429. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol Syst Biol 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional Profiling of the LPS Induced NF-ΚB Response in Macrophages. BMC Immunol 2007, 8, 1–17. [Google Scholar] [CrossRef]
- Trezza, A.; Geminiani, M.; Cutrera, G.; Dreassi, E.; Frusciante, L.; Lamponi, S.; Spiga, O.; Santucci, A. A Drug Discovery Approach to a Reveal Novel Antioxidant Natural Source: The Case of Chestnut Burr Biomass. Int J Mol Sci 2024, 25, 2517. [Google Scholar] [CrossRef]
- Barreira, J.C.M.; Ferreira, I.C.F.R.; Oliveira, M.B.P.P.; Pereira, J.A. Antioxidant Potential of Chestnut (Castanea Sativa L.) and Almond (Prunus Dulcis L.) By-Products. Food Science and Technology International 2010, 16, 209–216. [Google Scholar] [CrossRef]
- Fernández-Agulló, A.; Freire, M.S.; Antorrena, G.; Pereira, J.A.; González-Álvarez, J. Effect of the Extraction Technique and Operational Conditions on the Recovery of Bioactive Compounds from Chestnut ( Castanea Sativa ) Bur and Shell. Sep Sci Technol 2014, 49, 267–277. [Google Scholar] [CrossRef]
- Silva, V.; Falco, V.; Dias, M.I.; Barros, L.; Silva, A.; Capita, R.; Alonso-Calleja, C.; Amaral, J.S.; Igrejas, G.; C. F. R. Ferreira, I.; et al. Evaluation of the Phenolic Profile of Castanea Sativa Mill. By-Products and Their Antioxidant and Antimicrobial Activity against Multiresistant Bacteria. Antioxidants 2020, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.; Santos, J.; Pimentel, F.B.; Braga, N.; Palmeira-de-Oliveira, A.; Oliveira, M.B.P.P. Promising New Applications of Castanea Sativa Shell: Nutritional Composition, Antioxidant Activity, Amino Acids and Vitamin E Profile. Food Funct 2015, 6, 2854–2860. [Google Scholar] [CrossRef] [PubMed]
- Vella, F.M.; Laratta, B.; La Cara, F.; Morana, A. Recovery of Bioactive Molecules from Chestnut ( Castanea Sativa Mill.) by-Products through Extraction by Different Solvents. Nat Prod Res 2018, 32, 1022–1032. [Google Scholar] [CrossRef]
- Braga, N.; Rodrigues, F.; P.P. Oliveira, M.B. Castanea Sativa by-Products: A Review on Added Value and Sustainable Application. Nat Prod Res 2015, 29, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Flórez-Fernández, N.; Torres, M.D.; Gómez, S.; Couso, S.; Domínguez, H. Potential of Chestnut Wastes for Cosmetics and Pharmaceutical Applications. Waste Biomass Valorization 2020, 11, 4721–4730. [Google Scholar] [CrossRef]
- Cadau, S.; Gault, M.; Berthelemy, N.; Hsu, C.-Y.; Danoux, L.; Pelletier, N.; Goudounèche, D.; Pons, C.; Leprince, C.; André-Frei, V.; et al. An Inflamed and Infected Reconstructed Human Epidermis to Study Atopic Dermatitis and Skin Care Ingredients. Int J Mol Sci 2022, 23, 12880. [Google Scholar] [CrossRef] [PubMed]
- Bioactive Compounds in Underutilized Fruits and Nuts; Murthy, H.N., Bapat, V.A., Eds.; Springer International Publishing: Cham, 2020; ISBN 978-3-030-30181-1. [Google Scholar]
- Wen, C.; Zhang, J.; Zhang, H.; Dzah, C.S.; Zandile, M.; Duan, Y.; Ma, H.; Luo, X. Advances in Ultrasound Assisted Extraction of Bioactive Compounds from Cash Crops – A Review. Ultrason Sonochem 2018, 48, 538–549. [Google Scholar] [CrossRef]
- Chemat, F.; Rombaut, N.; Sicaire, A.-G.; Meullemiestre, A.; Fabiano-Tixier, A.-S.; Abert-Vian, M. Ultrasound Assisted Extraction of Food and Natural Products. Mechanisms, Techniques, Combinations, Protocols and Applications. A Review. Ultrason Sonochem 2017, 34, 540–560. [Google Scholar] [CrossRef]
- Cerulli, A.; Napolitano, A.; Hošek, J.; Masullo, M.; Pizza, C.; Piacente, S. Antioxidant and In Vitro Preliminary Anti-Inflammatory Activity of Castanea Sativa (Italian Cultivar “Marrone Di Roccadaspide” PGI) Burs, Leaves, and Chestnuts Extracts and Their Metabolite Profiles by LC-ESI/LTQOrbitrap/MS/MS. Antioxidants 2021, 10, 278. [Google Scholar] [CrossRef]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and Biochemical Demonstration of the Role of Cyclooxygenase 2 in Inflammation and Pain. Proc Natl Acad Sci U S A 1994, 91, 12013–12017. [Google Scholar] [CrossRef]
- Zamora, R.; Vodovotz, Y.; Billiar, T.R. Inducible Nitric Oxide Synthase and Inflammatory Diseases. Molecular Medicine 2000 6:5 2000, 6, 347–373. [Google Scholar] [CrossRef]
- Facchin, B.M.; dos Reis, G.O.; Vieira, G.N.; Mohr, E.T.B.; da Rosa, J.S.; Kretzer, I.F.; Demarchi, I.G.; Dalmarco, E.M. Inflammatory Biomarkers on an LPS-Induced RAW 264.7 Cell Model: A Systematic Review and Meta-Analysis. Inflammation Research 2022, 71, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Quispe, C.; Castillo, C.M.S.; Caroca, R.; Lazo-Vélez, M.A.; Antonyak, H.; Polishchuk, A.; Lysiuk, R.; Oliinyk, P.; De Masi, L.; et al. Ellagic Acid: A Review on Its Natural Sources, Chemical Stability, and Therapeutic Potential. Oxid Med Cell Longev 2022, 2022, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-H.; Shiao, H.-Y.; Tu, C.-H.; Liu, P.-M.; Hsu, J.T.-A.; Amancha, P.K.; Wu, J.-S.; Coumar, M.S.; Chen, C.-H.; Wang, S.-Y.; et al. Protein Kinase Inhibitor Design by Targeting the Asp-Phe-Gly (DFG) Motif: The Role of the DFG Motif in the Design of Epidermal Growth Factor Receptor Inhibitors. J Med Chem 2013, 56, 3889–3903. [Google Scholar] [CrossRef] [PubMed]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural Features of the Protein Kinase Domain and Targeted Binding by Small-Molecule Inhibitors. Journal of Biological Chemistry 2022, 298, 102247. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhang, Y.; Tang, C.; Hou, Y.; Ai, X.; Chen, X.; Zhang, Y.; Wang, X.; Meng, X. Gallic Acid: Pharmacological Activities and Molecular Mechanisms Involved in Inflammation-Related Diseases. Biomedicine & Pharmacotherapy 2021, 133, 110985. [Google Scholar] [CrossRef]
- Mard, S.A.; Mojadami, S.; Farbood, Y.; Kazem, M.; Naseri, G.; Ali, S.; Phd, M. The Anti-Inflammatory and Anti-Apoptotic Effects of Gallic Acid against Mucosal Inflammation- and Erosions-Induced by Gastric Ischemia-Reperfusion in Rats. Veterinary Research Forum 2015, 6, 305. [Google Scholar] [PubMed]
- Chagas, M. do S.S.; Behrens, M.D.; Moragas-Tellis, C.J.; Penedo, G.X.M.; Silva, A.R.; Gonçalves-de-Albuquerque, C.F. Flavonols and Flavones as Potential Anti-Inflammatory, Antioxidant, and Antibacterial Compounds. Oxid Med Cell Longev 2022, 2022, 1–21. [Google Scholar] [CrossRef]
- Lim, H.; Heo, M.Y.; Kim, H.P. Flavonoids: Broad Spectrum Agents on Chronic Inflammation. Biomol Ther (Seoul) 2019, 27, 241–253. [Google Scholar] [CrossRef]
- Ahmed, M.; -Ji, M.; * -Qin; -Gu, Z.; -Liu, Y.; -Sikandar; -Iqbal, M.F.; -Javeed, A. PHYTOCHEMICAL SCREENING, TOTAL PHENOLIC AND FLAVONOIDS CONTENTS AND ANTIOXIDANT ACTIVITIES OF CITRULLUS COLOCYNTHIS L. AND CANNABIS SATIVA L. [CrossRef]
- Mkpenie, V.N.; Essien, E.E.; Udoh, I.I. Effect of Extraction Conditions on Total Polyphenol Contents, Antioxidant and Antimicrobial Activities of Cannabis Sativa L. Electronic Journal of Environmental, Agricultural and Food Chemistry 2012, 11, 300–307. [Google Scholar]
- Liu, H.-M.; Cheng, M.-Y.; Xun, M.-H.; Zhao, Z.-W.; Zhang, Y.; Tang, W.; Cheng, J.; Ni, J.; Wang, W. Possible Mechanisms of Oxidative Stress-Induced Skin Cellular Senescence, Inflammation, and Cancer and the Therapeutic Potential of Plant Polyphenols. Int J Mol Sci 2023, 24, 3755. [Google Scholar] [CrossRef] [PubMed]
- Vernarelli, J.A.; Lambert, J.D. Flavonoid Intake Is Inversely Associated with Obesity and C-Reactive Protein, a Marker for Inflammation, in US Adults. Nutrition & Diabetes 2017 7:5 2017, 7, e276–e276. [Google Scholar] [CrossRef]
- Lefèvre-Arbogast, S.; Gaudout, D.; Bensalem, J.; Letenneur, L.; Dartigues, J.F.; Hejblum, B.P.; Féart, C.; Delcourt, C.; Samieri, C. Pattern of Polyphenol Intake and the Long-Term Risk of Dementia in Older Persons. Neurology 2018, 90, e1979–e1988. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; He, F.; Wu, C.; Li, P.; Li, N.; Deng, J.; Zhu, G.; Ren, W.; Peng, Y. Betaine in Inflammation: Mechanistic Aspects and Applications. Front Immunol 2018, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.K.; Paal, M.C.; Donohue, T.M.; Ganesan, M.; Osna, N.A.; Kharbanda, K.K. Beneficial Effects of Betaine: A Comprehensive Review. Biology 2021, Vol. 10, Page 456 2021, 10, 456. [Google Scholar] [CrossRef]
- Go, E.K.; Jung, K.J.; Kim, J.Y.; Yu, B.P.; Chung, H.Y. Betaine Suppresses Proinflammatory Signaling During Aging: The Involvement of Nuclear Factor- B via Nuclear Factor-Inducing Kinase/I B Kinase and Mitogen-Activated Protein Kinases. J Gerontol A Biol Sci Med Sci 2005, 60, 1252–1264. [Google Scholar] [CrossRef]
Figure 1.
(a) Phenolic composition of CSB measured as TPC (mg GAE/g dry extract) and TFC (mg QE/g dry extract); (b) % Radical Scavenging Activity of CSB on ABTS and DPPH radicals with relative IC50 (µg/mL). All experiments were performed in triplicate. Data are presented as mean ± SD. Unpaired t-test was used to assess statistically significant differences, ****p<0.0001, ***p=0.0003, df=2.
Figure 1.
(a) Phenolic composition of CSB measured as TPC (mg GAE/g dry extract) and TFC (mg QE/g dry extract); (b) % Radical Scavenging Activity of CSB on ABTS and DPPH radicals with relative IC50 (µg/mL). All experiments were performed in triplicate. Data are presented as mean ± SD. Unpaired t-test was used to assess statistically significant differences, ****p<0.0001, ***p=0.0003, df=2.
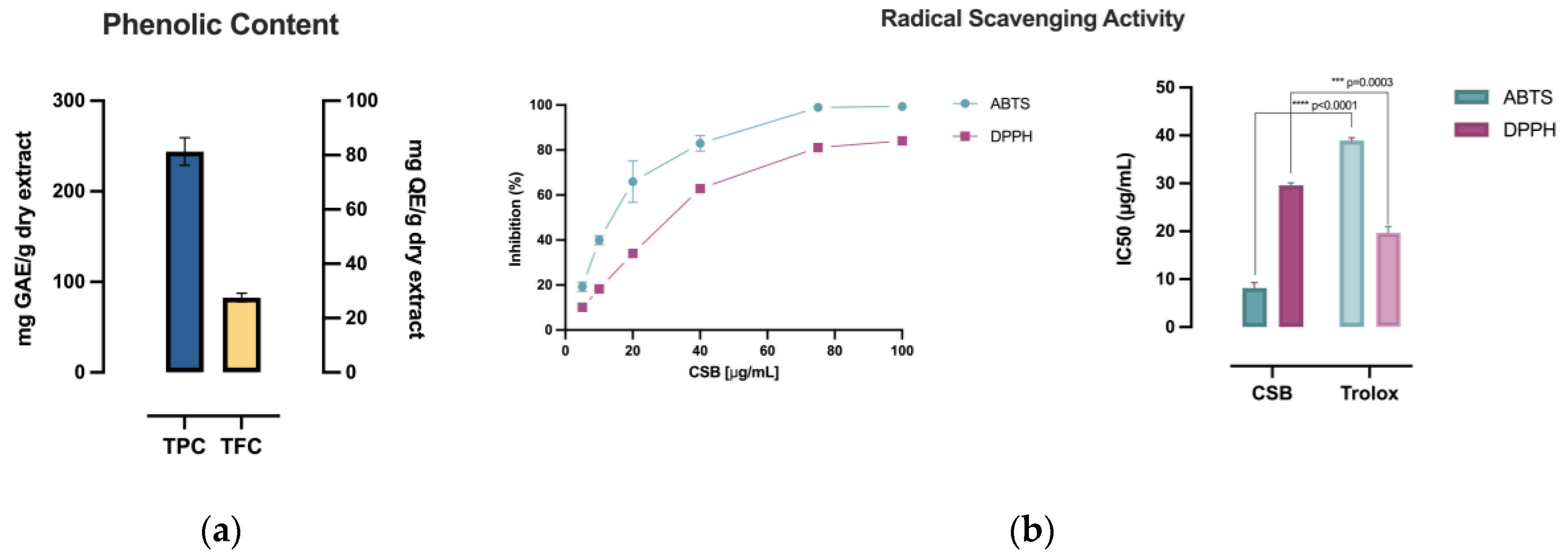
Figure 2.
Percentage of viable NIH3T3 cells as a function of incubation time (24, 48, and 72 h) and CSB extract concentration, as determined by the neutral red uptake. Data are mean ± SD of three experiments run in six replicates. No value is statistically different versus negative control (complete medium), p < 0.05.
Figure 2.
Percentage of viable NIH3T3 cells as a function of incubation time (24, 48, and 72 h) and CSB extract concentration, as determined by the neutral red uptake. Data are mean ± SD of three experiments run in six replicates. No value is statistically different versus negative control (complete medium), p < 0.05.
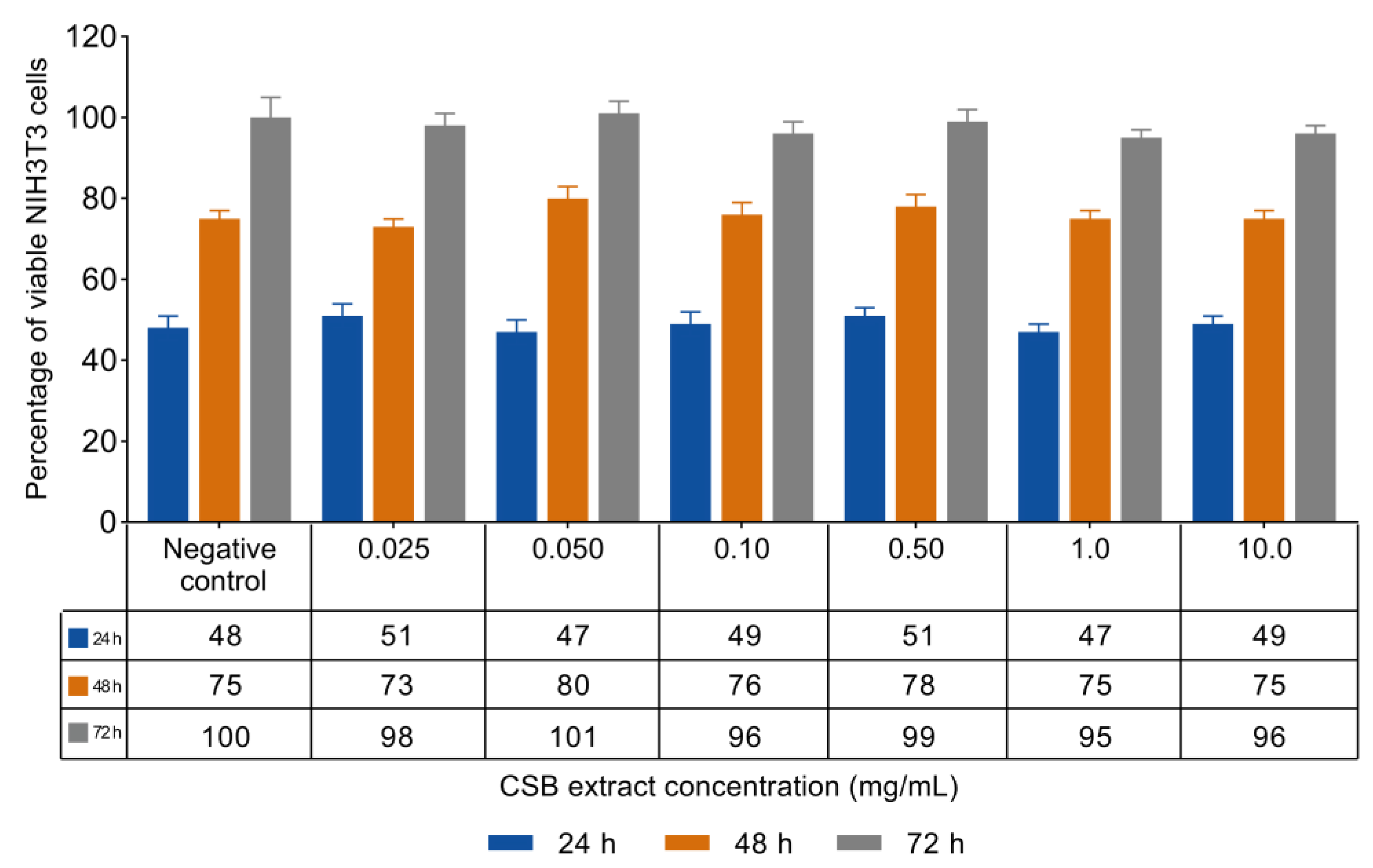
Figure 3.
(a) Viability of RAW 264.7 cells following 24 h treatment with CSB; (b) Intracellular ROS levels were quantified after pre-treatment with different concentrations of CSB followed by LPS stimulation (200 ng/mL) for 5 h Data are presented as bar graphs for ROS level measured from relative fluorescence intensity normalized to cell count with Crystal Violet assay. Statistically significant differences are denoted by ****p<0.0001 (vs. LPS) or ####p<0.0001 (vs. DEX as positive control). All experiments were performed in triplicate. Data are expressed as a percentage of control and presented as mean ± SD. P-values were calculated, by one-way ANOVA with Tukey’s post-hoc test.
Figure 3.
(a) Viability of RAW 264.7 cells following 24 h treatment with CSB; (b) Intracellular ROS levels were quantified after pre-treatment with different concentrations of CSB followed by LPS stimulation (200 ng/mL) for 5 h Data are presented as bar graphs for ROS level measured from relative fluorescence intensity normalized to cell count with Crystal Violet assay. Statistically significant differences are denoted by ****p<0.0001 (vs. LPS) or ####p<0.0001 (vs. DEX as positive control). All experiments were performed in triplicate. Data are expressed as a percentage of control and presented as mean ± SD. P-values were calculated, by one-way ANOVA with Tukey’s post-hoc test.
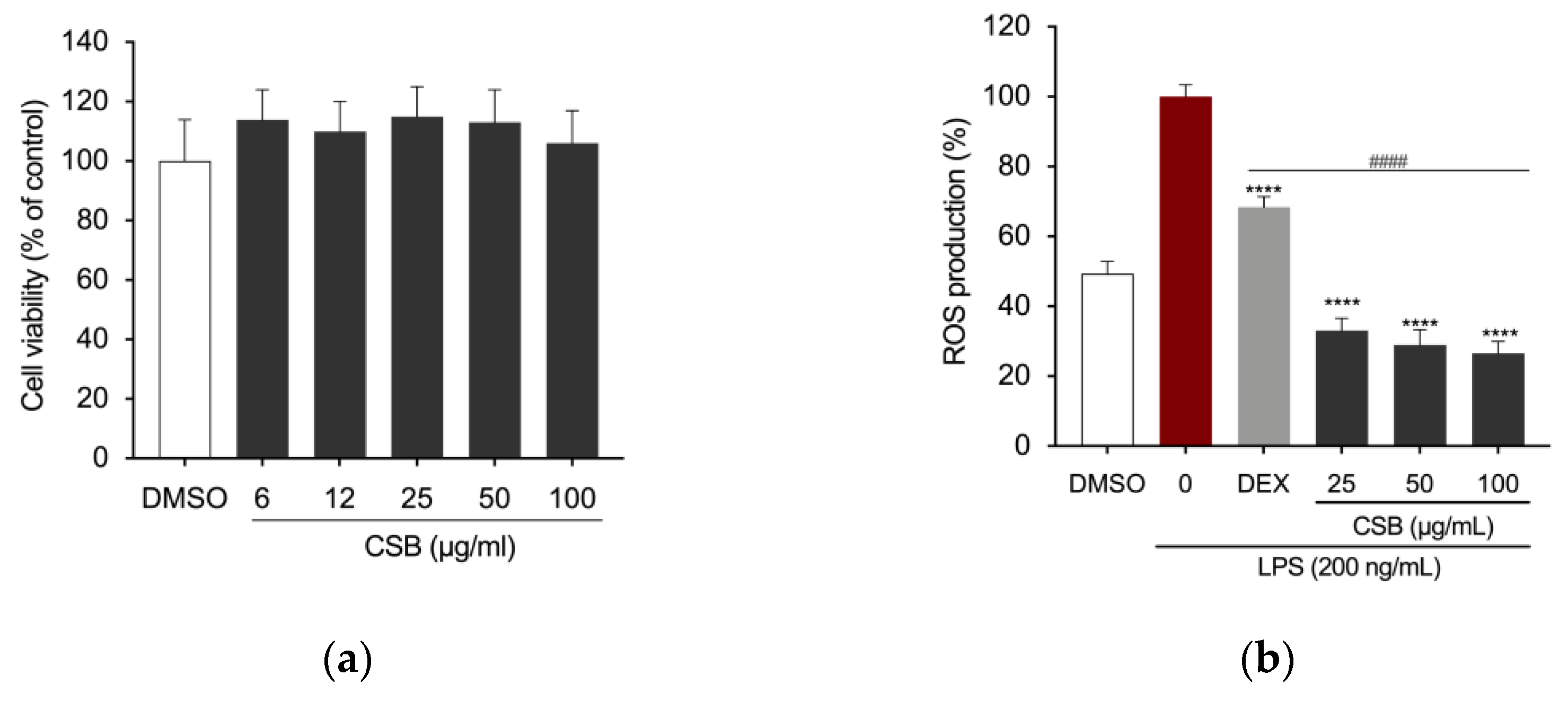
Figure 4.
Effects of CSB on LPS-induced NO production in stimulated RAW264.7 cells. After pre-treating the cells with DEX or CSB for 4 h, the cells were stimulated with 200 ng/mL LPS for 24h. The culture supernatants were analyzed for NO production. Data show mean ± SD values of three independent experiments. ****p=0.0001 (vs. LPS); #p=0.0282 (vs. DEX as a positive control). P-values were calculated by one-way ANOVA with Tukey’s post-hoc test.
Figure 4.
Effects of CSB on LPS-induced NO production in stimulated RAW264.7 cells. After pre-treating the cells with DEX or CSB for 4 h, the cells were stimulated with 200 ng/mL LPS for 24h. The culture supernatants were analyzed for NO production. Data show mean ± SD values of three independent experiments. ****p=0.0001 (vs. LPS); #p=0.0282 (vs. DEX as a positive control). P-values were calculated by one-way ANOVA with Tukey’s post-hoc test.
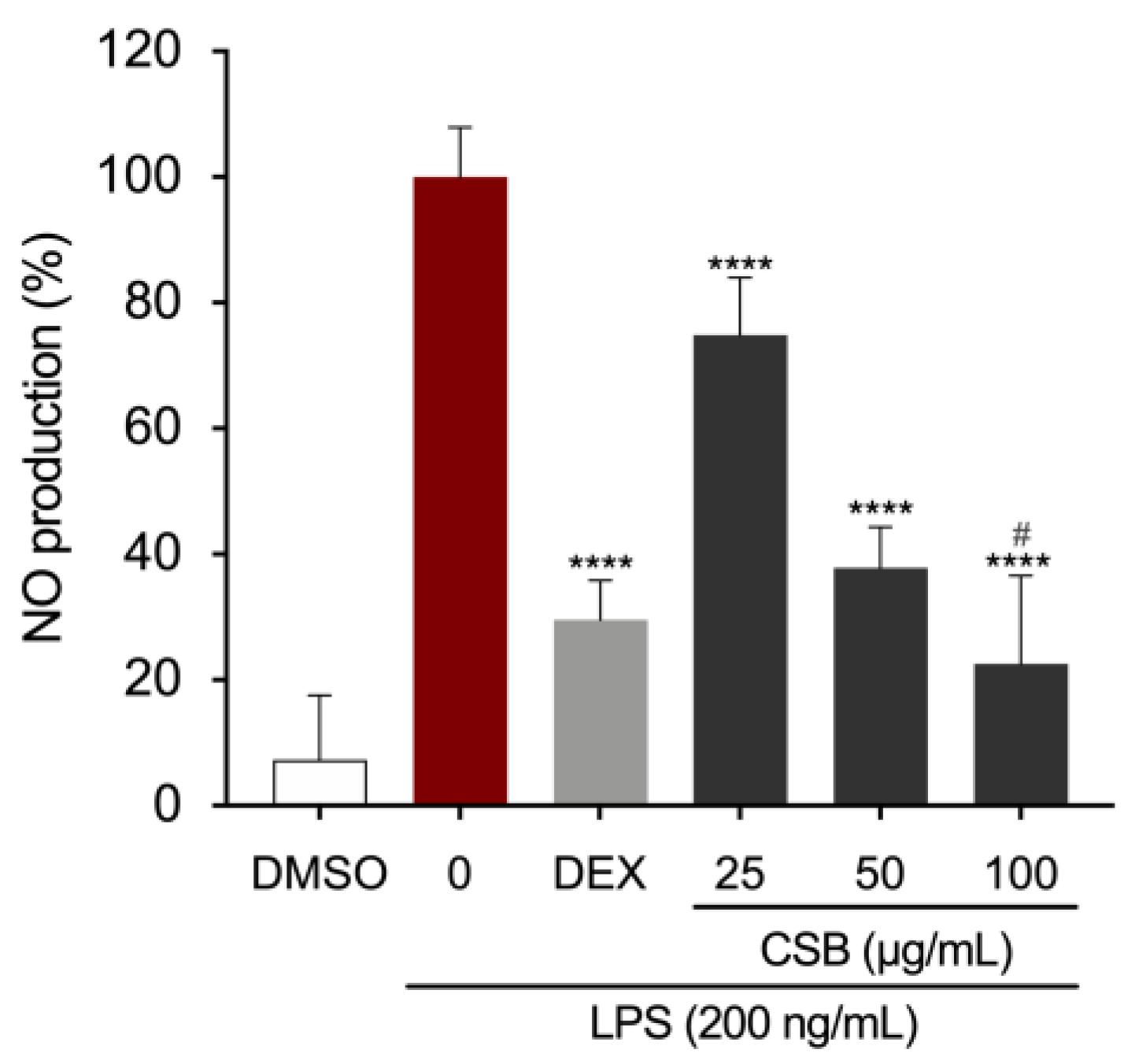
Figure 5.
Effects of CSB on iNOS protein expression levels in stimulated RAW264.7 cells. After pre-treating the cells with DEX or CSB for 4 h, the cells were stimulated with 200 ng/mL LPS for 24h. The iNOS expression levels were determined by Western blotting. Data show mean ± SD values of three independent experiments. ****p<0.0001 (vs. LPS). P-values were calculated by one-way ANOVA with Tukey’s post-hoc test.
Figure 5.
Effects of CSB on iNOS protein expression levels in stimulated RAW264.7 cells. After pre-treating the cells with DEX or CSB for 4 h, the cells were stimulated with 200 ng/mL LPS for 24h. The iNOS expression levels were determined by Western blotting. Data show mean ± SD values of three independent experiments. ****p<0.0001 (vs. LPS). P-values were calculated by one-way ANOVA with Tukey’s post-hoc test.
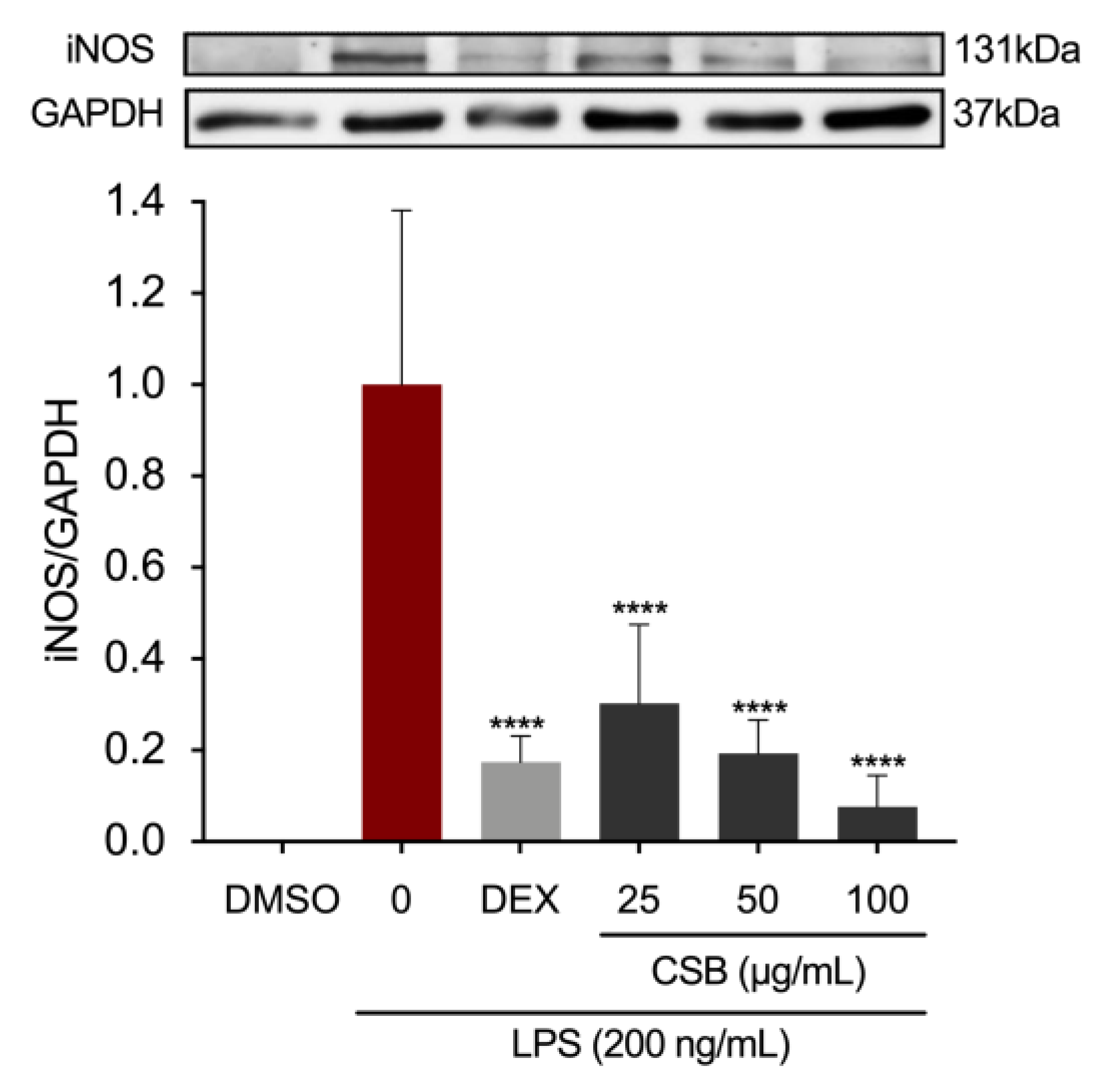
Figure 6.
Effects of CSB on suppressing the up-stream signaling for NF-κB activation in LPS- induced RAW264.7 cells. Cells were pre-treated with DEX or 100 μg/mL of CSB for 4 h, and then incubated with LPS (200 ng/mL) for 1 h. (a) NF-κB in LPS-stimulated RAW264.7 cells by western blotting. Quantification of relative band intensities from three independent experimental results determined by densitometry. Data are presented as mean±SD of three independent experiments. *p=0.0218, ***p=0.0002 (vs. LPS). P-values were calculated by one-way ANOVA with Tukey’s post-hoc test; (b) Localization of NF-κB visualized by a fluorescent microscope after staining for NF-κB (red). The nuclei of cells were stained with DAPI (blue). Micrographs were captured with 40X magnification.
Figure 6.
Effects of CSB on suppressing the up-stream signaling for NF-κB activation in LPS- induced RAW264.7 cells. Cells were pre-treated with DEX or 100 μg/mL of CSB for 4 h, and then incubated with LPS (200 ng/mL) for 1 h. (a) NF-κB in LPS-stimulated RAW264.7 cells by western blotting. Quantification of relative band intensities from three independent experimental results determined by densitometry. Data are presented as mean±SD of three independent experiments. *p=0.0218, ***p=0.0002 (vs. LPS). P-values were calculated by one-way ANOVA with Tukey’s post-hoc test; (b) Localization of NF-κB visualized by a fluorescent microscope after staining for NF-κB (red). The nuclei of cells were stained with DAPI (blue). Micrographs were captured with 40X magnification.
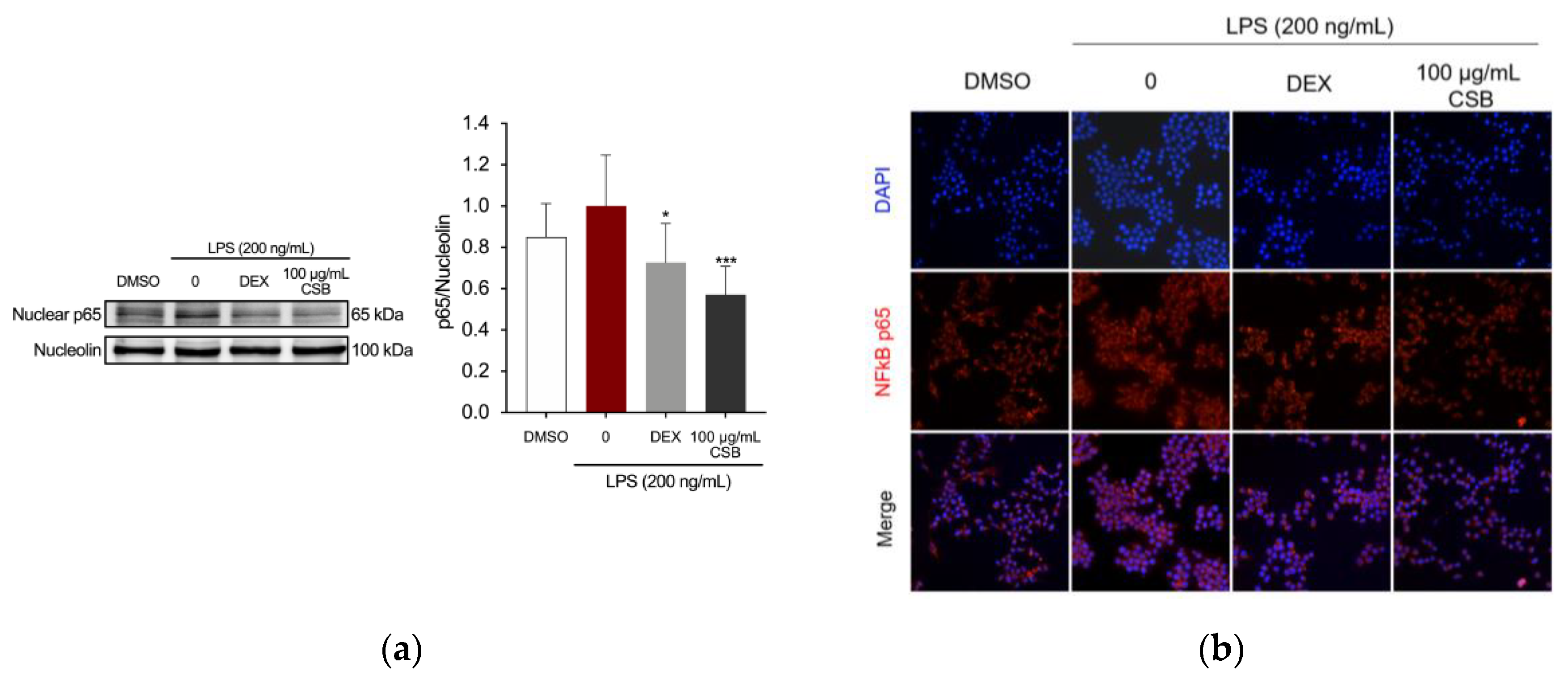
Figure 7.
Number of revertants in TA98 (a) and TA100 (b) S. typhimurium strain exposed to different concentrations of CSB with S9 fraction and without S9 fraction; The results are reported as the mean of revertants ± SD, p≤0.01.
Figure 7.
Number of revertants in TA98 (a) and TA100 (b) S. typhimurium strain exposed to different concentrations of CSB with S9 fraction and without S9 fraction; The results are reported as the mean of revertants ± SD, p≤0.01.
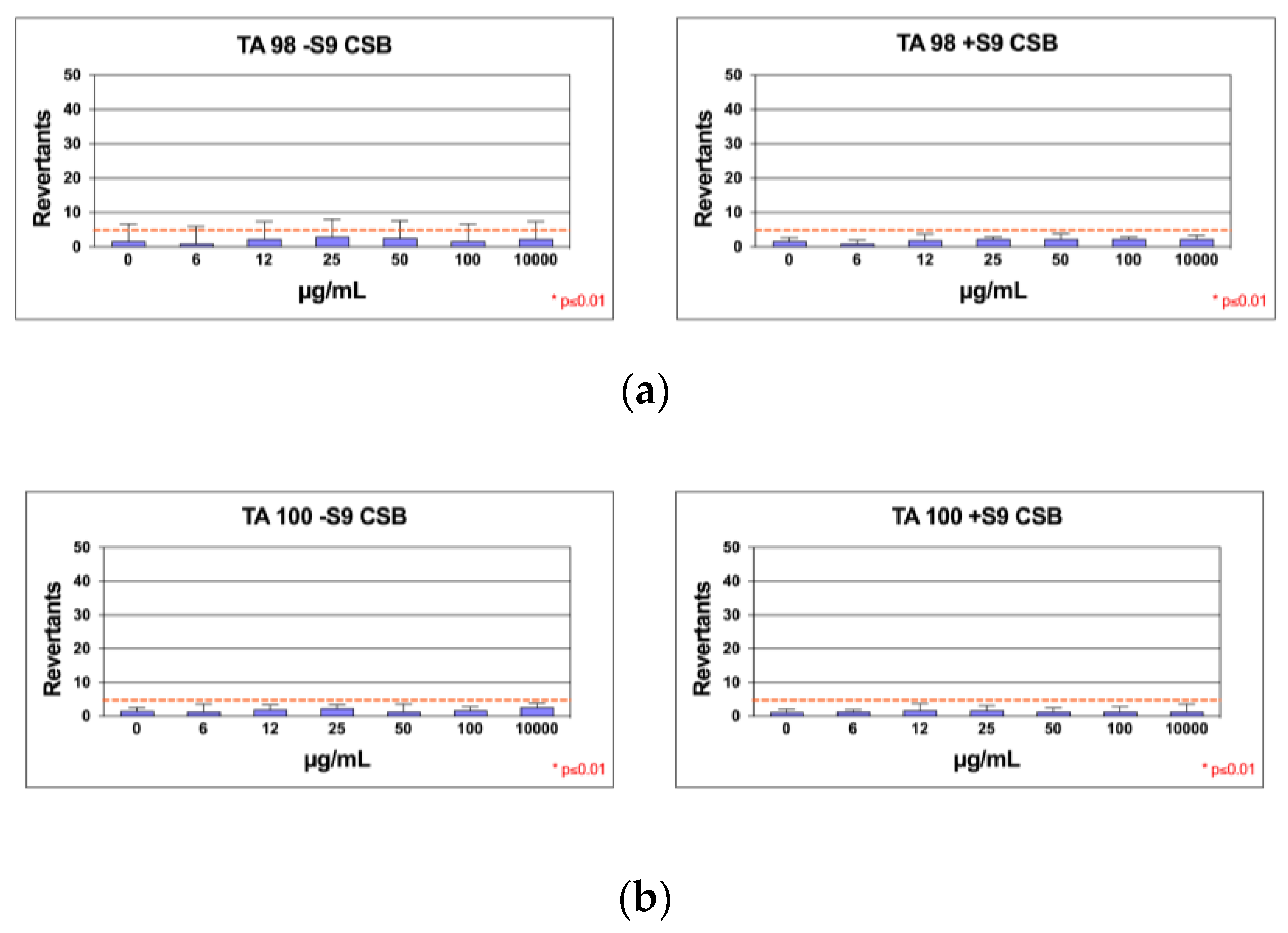
Figure 8.
Overview of target/ellagic acid complexes. 3D structures of the crystal structure of Jnk1 (PDB code: 3ELJ), MAPK11 (PDB code: 8YGW), and ERK1 (PDB code: 4QTB), are reported in the same complex with ellagic acid (shown in sticks) in green, blue, and magenta, respectively. The binding residues involved in hydrophobic interactions, hydrogen bonds, and salt bridges are represented as grey, pink, and depth green sticks, respectively. The hydrogen and salt brides bonds are indicated as yellow and purple dotted lines. The table displayed on the bottom right side shows the binding free energy (kCal/mol) of ellagic acid in complex with the targets.
Figure 8.
Overview of target/ellagic acid complexes. 3D structures of the crystal structure of Jnk1 (PDB code: 3ELJ), MAPK11 (PDB code: 8YGW), and ERK1 (PDB code: 4QTB), are reported in the same complex with ellagic acid (shown in sticks) in green, blue, and magenta, respectively. The binding residues involved in hydrophobic interactions, hydrogen bonds, and salt bridges are represented as grey, pink, and depth green sticks, respectively. The hydrogen and salt brides bonds are indicated as yellow and purple dotted lines. The table displayed on the bottom right side shows the binding free energy (kCal/mol) of ellagic acid in complex with the targets.
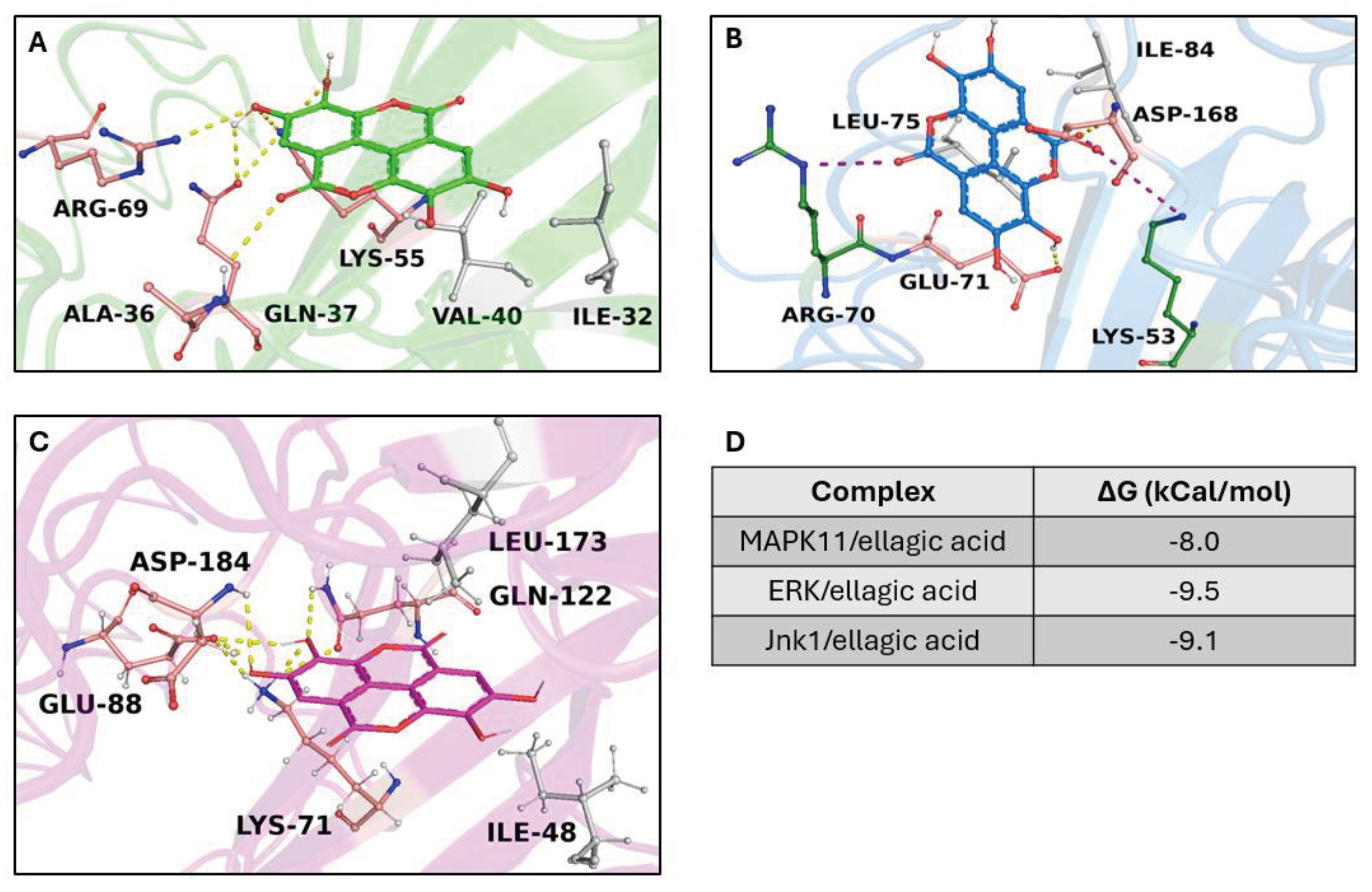

Table 1.
TPC, TFC and Antioxidant Capacity of CSB.
| Antioxidant Capacity | |||||
|---|---|---|---|---|---|
| TPC (mg GAE/g) |
TFC (mg QE/g) |
TRP (mg AAE/g) |
ABTS•+ (IC50 µg/mL) |
DPPH (IC50 µg/mL) |
|
| CSB | 243.98±17.77 | 27.54±0.60 | 272.12±4.64 | 8.16±1.11 | 29.57±0.57 |
Note: Data are expressed as mean±SD (n = 3).
Table 2.
Main classes of metabolites found in CSB US-water extract.
| Chemical class | % of CSB |
|---|---|
| Phenolic compounds | 71.43 |
| Polyphenols | 54.01 |
| Flavonoids | 16.95 |
| Phenolic aldehydes | 0.47 |
| Amino acids | 22.0 |
| Plant hormones | 3.64 |
| Terpenes | 1.69 |
| Others | tr.* |
* tr. = traces (Area<0.1%).
Table 3.
Matched metabolites in C. sativa burrs US-water extract. .
| No. | Name | Retention time (min) | Formula | Calculated MW |
Theoretical m/z |
Reference ion |
Mass error (ppm) | Area (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Ellagic acid | 15.388 | C14H6O8 | 302.00617 | 300.9988 | [M-H]-1 | -0.33 | 51.7 |
| 2 | Betaine | 1.863 | C5H11NO2 | 117.07923 | 118.0865 | [M+H]+1 | 2.17 | 22.0 |
| 3 | 5,7-dihydroxy-3.8-dimethoxy-2-phenyl-4h-chromen-4-one | 29.172 | C17H14O6 | 314.07985 | 315.0871 | [M+H]+1 | 2.59 | 15.8 |
| 4 | Mollioside | 25.285 | C26H40O10 | 512.26256 | 513.2698 | [M+H]+1 | 0.81 | 1.7 |
| 5 | (±)-(2e)-abscisic acid | 11.733 | C15H20O4 | 264.13625 | 265.1435 | [M+H]+1 | 0.34 | 1.6 |
| 6 | 3,8-di-o-methylellagic acid | 21.233 | C16H10O8 | 330.03838 | 331.0457 | [M+H]+1 | 2.46 | 1.4 |
| 7 | 12-hydroxyjasmonic acid | 18.885 | C12H18O4 | 226.12108 | 227.1284 | [M+H]+1 | 2.53 | 0.9 |
| 8 | Epi-jasmonic acid | 15.442 | C12H18O3 | 210.12618 | 211.1335 | [M+H]+1 | 2.77 | 0.7 |
| 9 | Protocatechuic aldehyde | 6.164 | C7H6O3 | 138.03177 | 139.0391 | [M+H]+1 | 0.56 | 0.4 |
| 10 | Gibberellin A2 o-beta-d-glucoside | 23.076 | C25H36O11 | 512.22551 | 513.2328 | [M+H]+1 | -0.49 | 0.3 |
| 11 | Sinapaldehyde | 21.782 | C11H12O4 | 208.07372 | 209.081 | [M+H]+1 | 0.76 | 0.3 |
| 12 | 12-hydroxyjasmonic acid 12-o-beta-d-glucoside | 19.078 | C19H30O8 | 386.1931 | 387.2004 | [M+H]+1 | -2.5 | 0.3 |
| 13 | 5,7-dihydroxy-3’,4’,5’-trimethoxyflavanone | 18.774 | C18H18O7 | 346.10628 | 347.1136 | [M+H]+1 | 2.97 | 0.2 |
| 14 | (+)-Gibberellic acid | 16.236 | C19H22O6 | 346.14242 | 347.1497 | [M+H]+1 | 2.25 | 0.2 |
| 15 | N-propyl galiate | 10.343 | C10H12O5 | 212.06876 | 235.058 | [M+Na]+1 | 1.35 | 0.2 |
| 16 | Syringaldehyde | 12.269 | C9H10O4 | 182.05821 | 183.0655 | [M+H]+1 | 1.68 | 0.2 |
| 17 | Retusin (flavonol) | 18.648 | C19H18O7 | 358.10628 | 359.1136 | [M+H]+1 | 2.87 | 0.2 |
| 18 | Acaciin | 11.453 | C28H32O14 | 592.17833 | 593.1856 | [M+H]+1 | -1.48 | 0.2 |
| 19 | Kaempferol | 17.111 | C15H10O6 | 286.04805 | 287.0553 | [M+H]+1 | 1.1 | 0.2 |
| 20 | Scopoletin | 16.019 | C10H8O4 | 192.04282 | 193.0501 | [M+H]+1 | 2.91 | 0.2 |
| 21 | Isorhamnetin 3-rhamnosyl-(1->2)-gentiobiosyl- (1->6) -glucoside | 28.24 | C40H52O26 | 948.27548 | 949.2828 | [M+H]+1 | 0.84 | 0.1 |
| 22 | 5,7-methoxyflavanone | 12.653 | C17H16O4 | 284.10549 | 285.1128 | [M+H]+1 | 2.21 | 0.1 |
| 23 | 4’.5.7-trimethoxyflavone | 20.486 | C18H16O5 | 312.10053 | 313.1079 | [M+H]+1 | 2.44 | 0.1 |
| 24 | Ethyl gallate | 13.29 | C9H10O5 | 198.05319 | 199.0605 | [M+H]+1 | 1.87 | 0.1 |
| 25 | 2-(2,6-dimethoxyphenyl)-5,6-dimethoxy-4h-chromen-4-one (zapotin) | 16.541 | C19H18O6 | 342.11131 | 343.1186 | [M+H]+1 | 2.85 | 0.1 |
| 26 | Helichrysoside | 20.13 | C30H26O14 | 610.13403 | 633.1237 | [M+Na]+1 | 2.91 | 0.1 |
| 27 | 1,4-dihydro-4-oxo-3-(2-pyrrolidinyl)-2-quinolinecarboxylic acid | 17.834 | C14H14N2O3 | 258.10066 | 259.1079 | [M+H]+1 | 0.86 | 0.1 |
| 28 | Afrormosin | 16.195 | C17H14O5 | 298.08489 | 299.0922 | [M+H]+1 | 2.56 | 0.1 |
| 29 | Gibberellin A17 | 6.043 | C20H26O7 | 378.16813 | 377.1609 | [M-H]-1 | 0.75 | 0.1 |
| 30 | Quercetin | 17.258 | C15H10O7 | 302.04329 | 303.0506 | [M+H]+1 | 2.11 | 0.1 |
| 31 | 1,3-bis-(5-carboxypentyl)-urea | 11.099 | C13H24N2O5 | 288.16887 | 289.1762 | [M+H]+1 | 1.21 | 0.1 |
| 32 | 5-carboxyvanillic acid | 13.026 | C9H8O6 | 212.0318 | 211.0245 | [M-H]-1 | -1.37 | tr. |
| 33 | 3-hydroxyflavone | 18.639 | C15H10O3 | 238.06363 | 239.0709 | [M+H]+1 | 2.68 | tr. |
| 34 | Coniferaldehyde | 19.593 | C10H10O3 | 178.0632 | 179.0705 | [M+H]+1 | 1.18 | tr. |
| 35 | Gibberellin A1/A34 | 39.565 | C19H24O6 | 348.15815 | 349.1654 | [M+H]+1 | 2.47 | tr. |
| 36 | Isorhamnetin 3-o-alpha-l-[6’’’’-p-coumaroyl-beta-d-glucopyranosyl-(1->2)-rhamnopyranoside] | 20.825 | C37H38O18 | 770.20801 | 793.1978 | [M+Na]+1 | 2.85 | tr. |
| 37 | Gibberellin A53 | 38.444 | C20H28O5 | 348.19405 | 349.2013 | [M+H]+1 | 1.07 | tr. |
| 38 | 2’,5-digalloylhamamelofuranose | 12.064 | C20H20O14 | 484.08397 | 485.0913 | [M+H]+1 | -2.76 | tr. |
| 39 | (+)-Catechin 7-o-beta-d-xyloside | 18.011 | C20H22O10 | 422.12248 | 445.1117 | [M+Na]+1 | 2.81 | tr. |
| 40 | Digallic acid | 2.807 | C14H10O9 | 322.03294 | 321.0257 | [M-H]-1 | 1.43 | tr. |
| 41 | (E)-ferulic acid | 29.676 | C10H10O4 | 194.05788 | 195.0652 | [M+H]+1 | -0.16 | tr. |
| 42 | 1,3-dibutyl-1,3-dimethylurea | 13.625 | C11H24N2O | 200.18829 | 199.181 | [M-H]-1 | -2.86 | tr. |
| 43 | Kaempferol-3-o-(6’’’-trans-p-coumaroyl-2’’-glucosyl)rhamnoside | 21.66 | C36H36O17 | 740.19518 | 741.2025 | [M+H]+1 | -0.1 | tr. |
| 44 | Tomentosin | 15.583 | C15H20O3 | 248.14121 | 249.1487 | [M+H]+1 | -0.15 | tr. |
| 45 | Catechin gallate. (-)- | 18.618 | C22H18O10 | 442.08926 | 443.0965 | [M+H]+1 | -1.68 | tr. |
| 46 | Coniferyl aldehyde | 24.95 | C10H10O3 | 178.0632 | 179.0705 | [M+H]+1 | 1.18 | tr. |
| 47 | Quercetin-3-o-(6’’’-trans-p-coumaroyl-2’’-glucosyl)rhamnoside | 18.932 | C36H36O18 | 756.19158 | 779.1812 | [M+Na]+1 | 1.87 | tr. |
| 48 | Gibberellin A24 | 37.98 | C20H26O5 | 346.17818 | 347.1855 | [M+H]+1 | 0.45 | tr. |
| 49 | Glucogallin | 3.747 | C13H16O10 | 332.07488 | 331.0676 | [M-H]-1 | 1.61 | tr. |
| 50 | 5’-desgalloylstachyurin | 11.878 | C34H24O22 | 784.0757 | 783.0684 | [M-H]-1 | -0.29 | tr. |
| 51 | Gibberellin A12 | 44.209 | C20H28O4 | 332.19787 | 333.2052 | [M+H]+1 | -2.67 | tr. |
| 52 | (+)-Gallocatechin | 2.929 | C15H14O7 | 306.07341 | 305.0661 | [M-H]-1 | -1.78 | tr. |
| 53 | Isorhamnetin | 23.872 | C16H12O7 | 316.05908 | 315.0518 | [M-H]-1 | 2.47 | tr. |
| 54 | Myricetin-3-o-glucoside | 8.465 | C21H20O13 | 480.09172 | 479.0844 | [M-H]-1 | 2.77 | tr. |
| 55 | 1,6-bis-o-galloyl-beta-d-glucose | 8.974 | C20H20O14 | 484.0862 | 483.0789 | [M-H]-1 | 1.84 | tr. |
| 56 | Castalagin/vescalagin | 15.21 | C41H26O26 | 934.06952 | 933.0623 | [M-H]-1 | -1.83 | tr. |
| Tot | 100 |
tr. = traces (Area<0.1%)
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



