
Submitted:
19 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
Lung adenocarcinoma is the most prevalent form of lung cancer, and drug resistance is a significant obstacle in its treatment. This study aimed to investigate the overexpression of long non-coding RNAs (lncRNAs) as a mechanism that promotes intrinsic resistance in tumor cells from the onset of treatment. Drug-tolerant persister cells (DTP) are a subset of cancer cells that survive and proliferate after exposure to therapeutic drugs, making them an essential object of study for cancer treatment. The molecular mechanisms underlying DTP cell survival are not fully understood; however, long non-coding RNAs (lncRNAs) have been proposed to play a crucial role. DTP cells from lung adenocarcinoma cell lines were obtained after a single exposure to tyrosine kinase inhibitor TKIs (erlotinib or osimertinib). After establishing DTPs, RNA sequencing was performed to investigate the differential expression of the lncRNAs. Some lncRNAs and one mRNA were found to be overexpressed in DTP cells. The clinical relevance of lncRNAs was evaluated in a cohort of patients with lung adenocarcinoma from The Cancer Genome Atlas (TCGA). RT-qPCR validated the overexpression of lncRNAs and mRNA in residual DTP cells and LUAD biopsies. Knockdown of these lncRNAs increases the sensitivity of DTP cells to therapeutic drugs. This study provides an opportunity to investigate the involvement of lncRNAs in the genetic and epigenetic mechanisms underlying intrinsic resistance. The identified lncRNAs and CD74 mRNA may serve as potential prognostic markers or therapeutic targets to improve the overall survival of patients with lung cancer.
Keywords:
lung adenocarcinoma
; drug-tolerant persister (DTP) cells
; intrinsic resistance
; tyrosine kinase inhibitors (TKIs)
; erlotinib
; osimertinib
; senescent cells
; CD74
1. Introduction
According to GLOBOCAN, lung cancer is the leading cause of death in developed and developing countries [1]. Lung cancer is classified into small cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC) [2]. NSCLC is the most frequent type, comprising three major histological types: large-cell carcinoma, squamous cell carcinoma, and adenocarci-noma (the most common histologic type [3]. Identifying genetic alterations has led to the development of several targeted therapies for treating early and advanced lung cancer, resulting in higher overall survival of patients [4].
Based on the clinical guidelines outlined by the National Comprehensive Cancer Network (NCCN) for the treatment of lung cancer, patients with tumors harboring activating mutations in the epidermal growth factor receptor (EGFR) gene require therapy with tyrosine kinase inhibitors (TKIs) [5]. Among the EGFR inhibitors, osimertinib has become the principal option in the TKI regimen [6]. However, approximately 16%–30% of patients treated with TKIs do not show clinical benefits, mainly because of the development of drug resistance, which limits the effectiveness of these therapies [7].
Tumors comprise distinct populations of cells that show genetic and phenotypic heterogeneity, and populations with lower susceptibility to chemotherapy and targeted therapies have been reported. The biological causes of resistance, particularly those associated with acquired resistance, have been extensively investigated. Upon exposure to therapeutic compounds, these drug-tolerant persister (DTP) cells are able to survive and proliferate by exhibiting alterations, such as entering a state of dormancy, showing decreased proliferation and changes in oxidative and lipid metabolism, as well as chromatin remod-eling [8]. Specific changes have been reported, such as increased drug metabolism, alterations in drug efflux systems, and cell cycle arrest [9]. In addition, DTP cells can generate clones with characteristics such as a mesenchymal–epithelial tran-sition (MET) phenotype, increased migration, and promotion of metastatic activity [10].
Intrinsic resistance, an additional phenomenon, has recently been considered as a significant factor in tumor recurrence [11]. In this phenomenon, populations of tumor cells display alterations that allow them to survive treatment before drug exposure or immediately after the initial cycles of treatment, eventually giving rise to tumors with reduced sensitivity to therapeutic drugs [12]. However, the molecular mechanisms associated with intrinsic resistance remain poorly understood [13]. Varia-bility among transcriptomic profiles of tumor cell populations has been proposed as a leading cause of intrinsic resistance [14]. In vitro studies in lung cancer, osteosarcoma, and melanoma cell lines have reported that a fraction of tumor cells survive after initial exposure to therapeutic drugs, and that residual cells are detected in a latent state [15,16,17]. Intrinsic resistance appears to depend on nongenetic mechanisms, as suggested in several types of cancer [18]. Further studies are needed to better understand the molecular mechanisms underlying the emergence of DTP cells in EGFR-mutated lung ade-nocarcinoma and their role in treatment resistance. A possible mechanism for intrinsic resistance is the immediate repro-gramming of the transcriptome mediated by the deregulation of long non-coding RNAs (lncRNAs), which allows tumor cells to modulate gene expression to resist the cytotoxic effects of various antitumor agents [19].
The present study aimed to identify early transcriptomic alterations associated with the DTP state in EGFR-mutated lung adenocarcinoma cell lines after a single exposure to TKIs. Our results demonstrated that cell lines with EGFR mutation show different degrees of susceptibility to TKIs; and mainly induce arrest in the G0/G1 phase of the cell cycle. After exposure to erlotinib and osimertinib, residual β-galactosidase-positive cells were detected, suggesting a senescence-like phenotype in the DTP cells. RNA sequencing and differential gene expression analyses showed that the DTP cells from the cell lines exposed to TKIs displayed specific overexpression of lncRNAs, suggesting that these lncRNAs may act as intrinsic resistance factors that may be deregulated after the onset of treatment. Based on TCGA data, the detected lncRNAs were significantly associated with worse OS in lung adenocarcinoma patients. In addition, the expression levels of some lncRNAs were vali-dated in lung adenocarcinoma biopsies. Concerning CD74, osimertinib–DTP cells overexpressed this gene, and was detected in biopsy material obtained from patients with lung adenocarcinoma. The expression of the identified lncRNAs and CD74 could be employed as novel predictive biomarkers or therapeutic targets to increase the effectiveness of targeted therapy to improve the clinical outcomes of patients with lung adenocarcinoma.
2. Materials and Methods
2.1. Cell Culture
The EGFR-mutant lung adenocarcinoma cell lines employed in this study included HCC827 (harboring the E746-A750 activating mutation) and HCC4006 (harboring the L747-E749 + A750P activating mutation), both of which exhibit EGFR exon 19 deletions and are sensitive to erlotinib. Additionally, the H1975 cell line characterized by the presence of the EGFR mutation (L858R/T790M) and resistance to erlotinib, but sensitivity to osimertinib was incorporated. All cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cells were cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA. Cat. No. R6504) supplemented with 10% fetal calf serum (Hyclone, Issaquah, WA, USA, Cat. No. SH30070.03) and 1% antibiotics (complete medium). The cell cultures were maintained at 37 °C in a humidified incubator with 5% CO2.
2.2. Targeted Next-Generation Sequencing to Confirm Mutations in Lung Adenocarcinoma Cell Lines
To corroborate the mutational profiles of the cell lines obtained from ATCC and employed throughout the experiments, we conducted targeted sequencing using the Illumina TruSight Tumor 15 Kit (Illumina, San Diego, CA, USA, Cat. No. OP-101-1001). DNA extraction was performed using the Pure Link Genomic DNA Mini Kit (Thermo Fisher Scientific). DNA concentration was assessed using a Qubit (Invitrogen, Carlsbad, CA, USA). DNA libraries were prepared according to the manufacturer's guidelines and sequenced using the MiSeq Illumina device. This panel was designed to identify clinically relevant mutations in 15 cancer driver genes: AKT1, BRAF, EGFR, ERBB2, FOXL2, GNA11, GNAQ, KIT, KRAS, MET, NRAS, PDGFRA, PIK3CA, RET, and TP53.
2.3. Therapeutic Compounds
Erlotinib-HCl (Cat. No. S1023) and osimertinib (Cat. No. S7297) were purchased from Selleckchem (TX, USA), dissolved in dimethyl sulfoxide DMSO (Sigma-Aldrich, MO, USA), and diluted in complete media. Cell cultures were exposed to TKI at concentrations ranging from 25 to 200 nM. The range of concentrations of these compounds includes those reported in serum-treated patients [20,21]. Control cultures maintained in complete medium and medium containing DMSO were used as references.
2.4. Dose–Response Curves to TKIs in EGFR-Mutant Lung Adenocarcinoma Cell Lines
Lung adenocarcinoma cell lines were cultured in T-25 flasks until they reached 80%–90% confluency. The cells were then detached by treatment with trypsin, and the viability of the harvested cells always exceeded 95%. Cells were seeded in 96-well plates based on their growth rates: HCC827 and HCC4006 cells at 1 × 105 cells/mL and H1975 cells at 1.5 × 104 cells/mL, respectively. Cells were incubated for 24 h to allow cell adhesion to plastic and to reach 60%–70% confluence. Then, cells were exposed to serial dilutions of TKIs and incubated for an additional 48 h.
Microscopic observations were performed during drug exposure to assess changes on cell density or associated with cell death. These changes were documented using micrographs captured using an EVOS FL digital inverted microscope (Life Technologies, CA, USA). To quantify the effect of treatment in lung adenocarcinoma cell lines, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed following the manufacturer's instruc-tions (Trevigen, MD, USA, Cat. No. 4890-25K). At the end of the drug exposure period, 10 μL MTT was added to each well. The plates were incubated for 4 h in the dark at 37 °C. Then, formazan crystals were dissolved in 150 μL of DMSO, and the absorbance was measured at 560 nm using a Multiskan Ascent plate reader (Thermo Fisher Scientific, MA, USA). Cell viability was calculated relative to diluent-treated cells or media-treated cells as a control and was representative of 100% maximum viability. Three independent experiments were performed in triplicate.
2.5. Cell Cycle Analysis
We considered relevant to examine the effect of TKIs on cell cycle phases. After TKI exposure, the supernatant and adherent cells were meticulously collected, combined, and washed with phosphate-buffered saline (PBS). Subsequently, the cells were fixed in cold 70% (v/v) ethanol and stored at -20 °C for a minimum of 24 h. After washing, the cells were permeabilized with PBS containing 0.1% (v/v) Triton X-100 (Sigma-Aldrich, Cat. No. X100-5ML) and treated with RNase A (30 μg/mL) (Thermo Fisher Scientific, MA, USA, Cat. No. EN0531) to avoid nonspecific RNA staining. We performed DNA staining with 0.25 μg of propidium iodide (PI) (Thermo Scientific, IL, USA, Cat. No. 62248) for 30 min to acquire 25,000 to 30,000 events using a FACS Canto II flow cytometer. After establishing a Forward Scatter Area (FSC-A) vs. Forward Scatter Height (FSC-H) dot plot to discern individual cells (singlets), events were analyzed using an FSC-A vs. SSC-A dot plot. This pop-ulation was analyzed using a PI-A vs. SSC-A dot plot. Finally, histograms were constructed to quantify the percentage of cells in each cell cycle phase using the FlowJo v.10 software (FlowJo, Ashland, OR, USA). Two independent experiments were performed in triplicate to ensure the robustness and reproducibility of each assay. After treatment with TKIs, residual re-sistant cells were mainly arrested in the G0/G1 cell cycle phase, which may suggest early induction of DTP cells.
2.6. Senescent Cells Detected by Beta-Galactosidase Staining
To explore whether the lung adenocarcinoma cell lines acquired a senescent phenotype in response to TKIs, we exposed the cell lines to osimertinib for a 48 h incubation period using a four-chamber slide. Diluent-treated and complete me-dia-treated cells were used as controls. After this time, the cell cultures were washed, and residual cells were cultured in complete media for additional five days. After, cells were washed with PBS, and the senescent beta-galactosidase staining protocol outlined in the beta-galactosidase staining kit from Cell Signaling Technology (MA, USA, Cat. No. 9860) was used according to the manufacturer's instructions. After treating the cells with the fixative solution for 10 min, they were washed and incubated with a beta-galactosidase staining solution (pH 6.0) for 4–5 h in a CO2-free incubator at 37 °C. Fi-nally, cells were washed and maintained in 70% glycerol. Micrographs of senescent cells were acquired using an EVOS FL digital inverted microscope (Life Technologies).
2.7. Quantification of Viable, Apoptotic, and Necrotic Cells
Microscopic observations in some cell cultures showed features of cell death, such as cell shrinkage, membrane blebbing, apoptotic bodies, and cell debris, regardless of the presence or absence of EGFR mutations.
To corroborate these observations and quantify the percentages of viable, apoptotic, and necrotic cells, we used the FITC Annexin-V Kit from BD Pharmingen (Becton Dickinson, NJ, USA, Cat. No. 556570). Briefly, the cells were cultured in T-25 flasks under the same experimental conditions as previously described. After drug exposure to 100 nM (low concentration) or 200 nM (high concentration) of erlotinib or osimertinib, floating dead cells were collected, and adherent cells were harvested by trypsinization. The two cell fractions were mixed, washed with Ca2+-Mg2+-free PBS, and rinsed in the kit binding buffer.
As cell death is time-dependent, this phenomenon was analyzed at different times. This paper shows only the results obtained at the end of culture treatment. Cells (3 × 105) were stained with annexin V/propidium iodide (PI) following the manufac-turer's instructions. After staining, 15,000 events were immediately acquired using a FACS Canto II flow cytometer (BD). An FSC-A vs. FSC-H dot plot was constructed to detect single cells in the gate. An FSC-A vs. SSC-A plot was then constructed, and the distribution of the collected events was analyzed using an Annexin-V vs. propidium iodide dot plot. Finally, the proportions of viable, apoptotic, and necrotic cells were quantified using FlowJo v.10 software. Three independent experi-ments were performed in duplicate.
2.8. Transcriptome Sequencing
Transcriptome sequencing was performed in DTP cells induced from lung adenocarcinoma cell lines to identify transcrip-tional alterations related to intrinsic resistance to TKIs. After exposure to a single drug, we removed floating dead cells after several washes with PBS and microscopically verified that only adherent cells remained in the culture. Adherent cells were harvested by trypsinization, ensuring a cell viability of over 95%. Following the manufacturer's instructions, we extracted total RNA from treated and untreated cells (control) using an RNAeasy Kit (Qiagen, Germany). High-quality RNA samples (RIN > 8) were obtained using the TruSeq RNA Sample Prep Kit v2 (Illumina, USA). We sequenced the libraries on the NextSeq 500 platform (Illumina, USA) at a depth of approximately 40 million reads, generating 75-bp paired-end reads. We sequenced from at least two independent experiments in duplicate for each experimental condition.
2.9. Bioinformatics Analysis of DTP cell RNA-Seq Data
Raw reads were subjected to quality control using FastQC (v.0.11.9) [22]. Trimmomatic (v.0.38) was used to filter reads with a Phred score > 26 [23], and the Cutadapt program (v.2.7) was used to remove adapter sequences as waste products [24]. The resulting clean reads were aligned with the GRCh38.95 reference genome using STAR (v.2.7.3a) [25], and gene abundance was estimated using RSEM (v.1.3.1) [26]. Poorly expressed genes were excluded and only those with a mean count per million >1 in all samples were considered. Differential gene expression (DGE) between DTP cells and untreated (diluent or media control) cells was assessed using the edgeR package (v.3.26.0) [27]. Significant gene expression was considered when they showed log2 fold ≥ 1 and an adjusted p-value of <0.05. Overexpressed genes were annotated using BioMart [28], and overexpressed lncRNAs were identified. In adenocarcinoma cell lines, the shared lncRNAs from erlotinib–treated, osimer-tinib–DTPs cells were identified using Venn diagrams.
2.10. Survival Analysis
The clinical relevance of the expression of the DTP-associated lncRNAs in the overall survival (OS) of lung adenocarcinoma patients was analyzed in the LUAD cohort of the TCGA, by using GEPIA2 software [29]. Kaplan–Meier (KM) analyses and log-rank tests were performed. lncRNAs with a log-rank p-value of <0.05 were considered statistically significant. The expression of the clinically relevant DTP-associated lncRNA and CD74 was further analyzed in the DTP cells.
2.11. RT-qPCR Validation
To validate the results obtained from bioinformatics analysis, total RNA from DTP cells and the corresponding control was obtained using the MagMAX™ mirVana™ Kit (Thermo Fisher Scientific, MA, USA, Cat. No. A27828) according to the manufacturer's instructions. The RNA concentration and purity were analyzed using a Nano Drop spectrophotometer (Thermo Fisher Scientific). The purified RNA was reverse-transcribed into cDNA using SuperScript™ VI-LO™ (Thermo Fisher Scientific, Cat. No. 11754050) according to the manufacturer’s instructions. The expression levels of selected lncRNAs were quantified by real-time PCR using TaqMan® gene expression assays (Thermo Fisher Scientific) (Supplementary Table S1) in the StepOnePlus™ system (Thermo Fisher Scientific). GAPDH was used as a housekeeping gene, and the relative expression of lncRNAs and mRNAs, normalized to the expression of GAPDH, was calculated using the 2-ΔΔCt method. Two independent experiments were performed.
2.12. lncRNA Knockdown
We studied the effects of the knockdown of lncRNA AGAP2-AS1 and LINC01133, on the sensitivity of HCC827 and H1975 cell lines to TKIs. Briefly, cells were seeded in 48-well plates (1 × 105 cells/well) and cultured overnight to allow attachment. After washing with RPMI without FBS, cells were maintained in serum-free media for 4 h. Knockdown was performed with a TriFECTa RNAi Kit in OptiMEM medium (Thermo Fisher, Waltham, MA, USA, Cat. No. 31985) according to the provider's instructions. The cells were incubated with 3 μL of Lipofectamine 3000 (Invitrogen, USA, Cat. No. L3000-015), and AGAP2-AS1, LINC01133, and DsiRNAs were added at a final concentration of 10 nM for 24 h, at which the maximum knockdown was previously detected. For each knockdown assay, we included a control mock (lipofectamine alone), negative control (DsiRNA non-targeting human genes), and DsiRNA for an unrelated gene (HPRT1). The DsiRNA sequences used for knockdown are provided in Supplementary Table S2. RT-qPCR was used to determine the knockdown efficiency. The knockdown assessment was performed after the exposure of transfected cells to TKIs or CDDP for 48 h, and the change in cell viability was analyzed by the MTT assay as described above.
2.13. CD74 Localization in DTP Cells
Transcriptomic analysis highlighted the overexpression of the CD74 protein-coding gene in osimertinib-DTPs cells of the HCC4006 and H1975 cell lines. The localization of CD74 in DTPs cells was determined using indirect immunofluorescence staining. Lung adenocarcinoma cell lines were cultured on four-chamber slides and treated as described above. After treat-ment, the cells were fixed with ethanol, washed, and treated for 30 min with blocking solution to avoid nonspecific binding. The slides were incubated with anti-CD74 antibody (Cell Signaling Tech, MA, USA, Cat. No. 77274) at 1:100 dilution overnight in a humidified atmosphere at 4 °C. After washing, slides were incubated with the Alexa Fluor 488-conjugated secondary antibody (1:500) (Invitrogen, IL, USA, Cat. No. A2731) at 37 °C for 90 min. Raji cells were used as a positive control for CD74 staining (data not shown). Finally, the cells were incubated with DAPI (1:150) (Thermo Scientific, USA; Cat. No. 62248) for nuclear staining for 15 min. The slides were mounted with Vectashield (Vector Laboratories, CA, USA, Cat. No. H-1000). Micrographs were acquired using an EVOS FL microscope (Thermo Fisher Scientific, MA, USA). Two independent experiments were performed.
2.14. Tissue Samples of Lung Adenocarcinoma
Specimens were obtained from untreated patients diagnosed with advanced-stage primary lung adenocarcinoma (stage IIIb or IV). Tissue blocks of patients with lung adenocarcinoma were collected from the repository of residual biological material from the archives of the Pathology Department at the Instituto Nacional de Enfermedades Respiratorias Ismael Cosio Vil-legas. The biopsies selected for this study included those with a high proportion of tumor cells (70-90%). Two independent pathologists verify the diagnosis of lung adenocarcinoma, according to the 2021 WHO Classification of Lung Tumors [30].
2.15. Validation of lncRNA in Lung Adenocarcinoma Biopsies
Four paraffin-embedded tissue biopsies from untreated-LUAD patients were used for RNA extraction. Serial sections of 10 µm were processed using the PureLinkTM FFPE RNA isolation kit (Invitrogen, CA, USA), following the manufacturer’s instructions. RNA concentration and purity were analyzed using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Purified RNA was reverse transcribed to cDNA using SuperScript™ VI-LO™ (Thermo Fisher Scientific, Cat. No. 11754050). Expression levels of the selected lncRNAs were quantified by real-time PCR using TaqMan® gene expression assays (Thermo Fisher Scientific) (Supplementary Table S1), as indicated in section 2.11.
2.16. Immunohistochemical Staining of CD74
For immunostaining, slices were obtained from the cellected biopsies of 15 patients with EGFR-mutated lung adenocarci-nomas. Tissue slices were deparaffinized at 60 °C in an oven for 20 min, followed by rehydration. Subsequently, heat-induced epitope retrieval was carried out using 0.01 M citrate buffer (pH 6.0) in an NxGen decloaking chamber (BioCare Medical, Pacheco, CA, USA) at 110 °C and 6 psi for 20 min. All the slides were treated with 3% (v/v) H2O2 in methanol for 25 min to block endogenous peroxidase activity. After washing with PBS, the slices were incubated with 2% serum-containing PBS for 30 min to block nonspecific binding. Immunostaining was performed using the anti-CD74 human monoclonal antibody (Cell Signaling, MA, USA, Cat. No. 77274) at 1:100 dilution. The slides were incubated overnight at 4 °C in a humidified chamber, and the next day, the slides were washed twice with 0.1% v/v Tween in PBS and twice with PBS. The tissue sections were then incubated with a biotin-labeled anti-rabbit antibody (Thermo Fisher, CA, USA, Cat. No. 65-6140) at a 1:500 dilution in a humidified chamber at 32 °C for 2 h, washed, and incubated with StreptABComplex/HRP (Vector Laboratories, CA, USA, Cat. No. PK6100) was used. Color was developed using H2O2 as the substrate and diaminobenzidine as the chromogen. Slides were mounted with Vectashield (Vector Laboratories, CA, USA, Cat. No. H1000) and microscopy images were captured using a DFC425 C color camera (Leica Microsystem Inc., Wetzlar, Germany) coupled with a Leica DMLB light microscope and Leica Application Systems v. 3.6.0 software (Leica Microsystem Inc.).
2.17. Statistical Analysis
The results obtained under the experimental conditions are presented as mean ± standard deviation (SD). For comparison, the experimental and control groups were analyzed using Student's t-test. For multiple comparisons, one-way analysis of vari-ance (ANOVA) was performed using Prism 10 (GraphPad Software, La Jolla, CA, USA). Statistical significance was set at p < 0.05.
2.18. Data Availability Statement
The accession numbers of RNA-seq raw reads, raw count arrays and target sequencing raw reads, as well as data analyses that were performed in R language using open source packages are available at the following link: https://github.com/maperezm/Intrinsic_CDDP_Resistant_Scripts
3. Results
3.1. Effects of TKIs on EGFR-Mutated Lung Adenocarcinoma Cell Lines
Microscopic observation in the cultures of HCC827 and HCC4006 cell lines, the concentrations of erlotinib tested caused a cytostatic effect (Figure 1A,D) observed by a gradual reduction in cell density after 48 h. The highest concentration reduced cell density by approximately 40% compared to the cell density of the control culture (data not shown). Low (100 nM) and high (200 nM) concentrations of erlotinib induced cell cycle arrest, increasing the proportion of cells in the G0/G1 phase with a concomitant reduction in the S and G2 phases. In addition, highest concentration of erlotinib caused a minimal proportion (10-20%) of cell death, detected as a subG0 peak (Figure 1B and C, 1E and F). Our results suggest that S-phase and G2 cells are more susceptible to TKI-induced cell death (Figure 1F). Several reports indicate that the H1975 cell line resists erlotinib due to the T750M mutation it harbors. We exposed this cell line to erlotinib as a control of resistance and confirmed that, under our experimental conditions, these cells were not susceptible to this drug, since the wide range of concentrations tested did not affect cell viability nor the proportion of cells in the different phases of the cell cycle (Figure 1G,H). Therefore, erlotinib in the HCC827and HCC4006 cell lines caused mainly a cytostatic effect (Supplementary Figure S1).
Clinical studies have recently suggested that patients with lung cancer, regardless of the type of EGFR mutation they harbor, should undergo treatment with osimertinib [31]. Based on this proposal, we studied the effects of osimertinib in the cell lines employed. Compared to the effect induced by erlotinib, osimertinib induced a cytotoxic effect in the HCC827 and HCC4006 cell lines. (Figure 2A,D). The highest osimertinib concentration (200nM) induced around 50% of cytotoxicity in both cell lines (Figure 2A–F). Surprisingly, in the H1975 cell line, which has been reported to be highly susceptible to osimertinib, this drug only altered the cell cycle and arrested cells in the G0/G1 phase without inducing cell death (Figure 2H,I).
Collectively, in the HCC827 and HCC4006 cell lines erlotinib caused alterations in the cell cycle, mainly blocking the transition of the G0/G1 phase to the S phase, while osimertinib induced a cytotoxic effect. The H1975 cell line, that harbors an erlotinib-resistance mutation was not affected by this drug, but osimertinib only induced arrest in the G0/G1 phase. Notably, in both treatments, residual cells were detected.
3.2. Osimertinib Induces β-Galactosidase Expression in Residual DTP Cells
We found that cell cycle arrest is an important characteristic of the residual cells after exposure to TKIs. It has been reported that DTP cells acquire a non-proliferative senescent-like phenotype to survive exposure to therapeutic drugs [32]. Since osimertinib is the treatment of choice for lung cancer harboring EGFR mutations [31], we investigated the expression of β-galactosidase, which is characteristic of a senescent-like phenotype, in the residual cells after osimertinib exposure. After exposing the EGFR-mutated cell lines to osimertinib, the residual cells were culture in fresh medina and labeled for the expression of beta-galactosidase. More than 70% of the residual cells were senescent-associated β-galactosidase (SA-β-gal)-positive (Figure 2J and Supplementary Figure S2), suggesting that these cells may acquire a senescent-like phe-notype, associated with the DTP state.
3.3. NGS-Based Mutation Analysis in Lung Adenocarcinoma Cell Lines
To exclude the possibility that driver mutations, distinct from EGFR mutations, could be associated with the biological response detected in the cell lines studied, we analyzed the cell lines employed for the mutation profiles of 15 solid can-cer-associated genes. The results confirmed the presence of the corresponding TP53 and EGFR-activating mutations in the HCC827, HCC4006, and H1975 cell lines, as previously reported by the ATCC. Since no additional mutations of clinical relevance were detected, we can assume that the biological behavior detected in these cell lines does not depend on any additional genetic alterations.
3.4. Transcriptomic Sequencing of Drug-Tolerant Persister Cells
To analyze the transcriptional changes associated with resistance to TKIs exposure, we performed total RNA sequencing of the residual and control cells. In total, 40 million rea–ds per sample were generated, and read quality was analyzed using FastQC. High-quality reads were aligned, mapped, and estimated for abundance to ensure reproducibility between the bio-logical replicates. Principal component analysis of the normalized counts revealed that treatment was the main factor con-tributing to dataset variability. We used edgeR to identify differentially expressed genes (DEGs). Significant changes in gene expression in erlotinib–and osimertinib–DTP cells were based on |log2 fold change| ≥ 1 and an adjusted p-value <0.05. The volcano plots for each cell line exposed to TKIs are shown in Figure 3A,B.
We used Venn diagrams to identify the overexpressed genes shared among the residual cells from all cell lines (Figure 3C,D). In total, 71 and 87 genes were found to be overexpressed in erlotinib– and osimertinib–DTP cells, respectively. Inter-estingly, we found that some of the overexpressed genes in the residual cells corresponded to lncRNAs. In total, eight and 13 overexpressed lncRNAs were found in erlotinib– and osimertinib–DTP cells, respectively (Supplementary Tables S3 and S4). The overexpression of these RNAs may account for an important mechanism of early transcriptomic reprogramming that promotes intrinsic resistance. Functional annotation of these commonly overexpressed RNAs was performed using the Gene Ontology (GO) biological process database (Figure 3E,F).
3.5. Survival Analysis
To confirm the clinical relevance of the overexpressed lncRNAs, Kaplan–Meier (KM) curves were generated from the LUAD project of the TCGA. High expression of the lncRNAs AGAP2-AS1, NKILA, CERS6-AS1, and LINCO1133 was associated with worse patient survival (Figure 4).
3.6. Gene Expression Validation of Clinically Relevant DTP-Associated Genes
The expression levels of clinically relevant DTP-associated lncRNAs identified by RNA-seq were validated using RT–qPCR to confirm their expression in DTP cells. RT-qPCR results showed overexpression of AGAP2-AS1 and NKILA in erlotinib–DTP cells, consistent with our RNA-seq results. Overexpression of AGAP2-AS1 was higher in HCC827 cells than in HCC4006 cells, which may account for the different sensitivity to erlotinib observed between these cell lines (Figure 5A). However, this proposal requires a more robust study, both in vitro and in vivo.
In osimertinib–DTP cells, the expression levels of CERS6-AS1 and LINC01133 lncRNAs were consistent with the tran-scriptome sequencing results. Interestingly, the HCC827 cell line that showed the highest osimertinib sensitivity among the cell lines displayed the lowest overexpression of lncRNA LINC01133. Additionally, we found that the expression of the protein-coding gene CD74 was notably increased in the osimertinib-DTP cells from the HCC4006 and H1975 cell lines (Figure 5B). These data are concordant with previous findings by Kashima et al., who used a single-cell assay for Trans-posase-Accessible Chromatin with sequencing (scATAC-seq analysis) and found that DTP cells from the H1975 cell line overexpress CD74 after exposure to this TKI [33].
3.7. Knockdown of lncRNAs Associated with the DTP State
To verify the involvement of the lncRNAs identified in the intrinsic resistance of lung adenocarcinoma cell lines, we em-ployed dsiRNA-mediated knockdown to target these lncRNAs. Specifically, AGAP-AS1, associated with erlotinib–DTP cells, was knocked down in the HCC827 cell line; LINC01133, linked to osimertinib–DTP cells was knocked down in the H1975 cell line (Figure 5C). Following transfection, the cells were exposed to the respective TKIs, and cell viability was assessed using the MTT assay, as previously described. Cell viability assays showed that knockdown of the targeted lncRNAs sig-nificantly diminished cell viability in response to a single exposure to TKIs, in comparison to the control conditions (Figure 5D).
These results substantiate the proposition that overexpression of these specific lncRNAs is linked to intrinsic resistance to TKIs. Furthermore, the increased sensitivity of the cells after knockdown of the clinically relevant DTP-associated lncRNAs suggests that these lncRNAs could be used as viable targets to overcome resistance to these therapeutic approaches.
3.8. Tissue Expression of Clinically Relevant lncRNAs Associated with the DTP State
To further explore the relevance of the lncRNAs associated with intrinsic resistance, we analyzed their expression in lung adenocarcinoma biopsies using RT-qPCR. We assessed the expression levels of relevant lncRNAs associated to TKI-DTP cells in four formalin-fixed paraffin-embedded (FFPE) biopsies from nontreated lung adenocarcinoma patients. The ex-pression levels of the osimertinib-DTP-associated lncRNAs in tissue samples were barely detected, compared to those measured for erlotinib-DTP-associated lncRNAs, suggesting a lower proportion of osimertinib-DTP cells in non-treated patients. This observation is in line with clinical trends, since erlotinib resistance is more prevalent than osimertinib re-sistance in lung adenocarcinoma [34]. Since we barely detected osimertinib-DTP-associated lncRNAs in biopsies and our RNA-seq analysis in cell lines showed that the protein-coding gene CD74 is highly expressed in the osimertinib-DTP cells, we analyzed the expression of this protein-coding gene. The expression of CD74 was elevated in tumor biopsies similar to what we found in cell lines. This observation suggests that CD74 participates in an additional mechanism of the cellular response of osimertinib-induced DTP cells, which may involve an intricate interplay between lncRNAs and protein-coding genes (Figure 6).
3.9. Cellular Localization of CD74 in Cell Lines
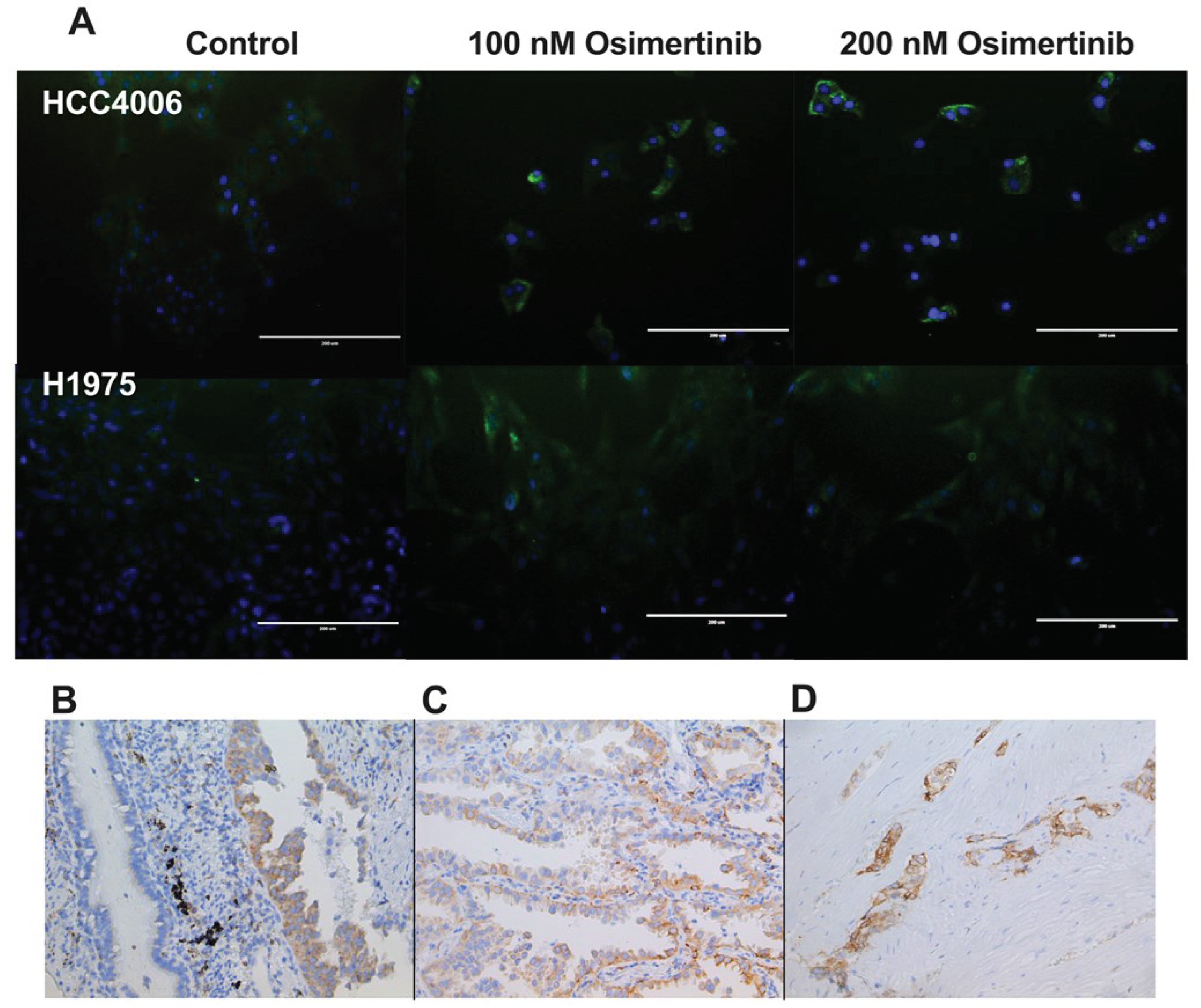
To evaluate the cellular localization of CD74 in the studied cell lines, we performed immunofluorescence staining against CD74. We found that CD74 is mainly present in the cytoplasm. CD74 staining increased in the osimertinib-DTPs, in ac-cordance with the results of RT-qPCR. Interestingly, heterogeneous staining of CD74 was observed in the same cell line, suggesting different expression levels among the tumor cell populations, which could be related to different sensitivities to TKIs in the same cell line (Figure 7A).
3.10. CD74 Staining in Lung Adenocarcinoma Biopsies
To assess CD74 expression within a tumor, we conducted immunohistochemical staining of CD74 in lung adenocarcinoma biopsies obtained from nontreated patients. Notably, IHC staining revealed discernible overexpression of CD74 in tumor cells compared to normal ciliated epithelial cells (Figure 7B). This observation suggests that overexpression of CD74 is associated with the development of lung adenocarcinoma.
Intriguingly, within the tumor cell population, we observed cells with distinct CD74 staining intensity in the tumor mass (Figure 7C,D). This result suggests the existence of subpopulations of tumor cells characterized by varying levels of CD74 expression within tumors, similar to that observed by IF. Such heterogeneity in CD74 expression could be related to the presence of a subset of tumor cells with elevated CD74 levels, which could promote resistance to initial exposure to osi-mertinib.
In summary, our results suggest that within the tumor, there are cells that are susceptible to TKIs, and tumor cells that overexpress molecules, such as CD74 and the DTP-associated lncRNAs, that allow them to resist the initial exposure to these therapeutic drugs. Eventually, these tumor cells could give rise to new tumor clones with that exhibit resistance to TKIs and conventional treatments, consequently promoting tumor relapse.
4. Discussion
Advances in the development of EGFR inhibitors (TKIs) with increased efficacy have encouraged the use of these drugs as first-line therapies for treating NSCLC harboring activating EGFR mutations. However, treatment failure frequently occurs due to the acquisition of molecular events that lead to resistance in tumor cells. Despite the application of new-generation TKIs, resistance to treatment is developed over time, leading to tumor relapse. Numerous studies have explored the mecha-nisms of acquired resistance, usually analyzing this phenomenon after multiple rounds of exposure to therapeutic drugs [35,36,37]. Intrinsic resistance, denoting the inherent or natural resistance of cancer cells to specific treatments or drugs, has recently become a subject of exploration [38]. Initial investigations into this drug resistance mechanism have been conducted in diverse lung cancer cell lines, with additional insights gleaned from information available in public databases [39]. In this context, the exploration of long non-coding RNAs (lncRNAs) has emerged as a focal point of interest. However, few studies have focused on the role of lncRNAs in intrinsic resistance to TKIs in lung cancer [38]. The present study sought to identify the transcriptional changes associated with DTPs cells in lung adenocarcinoma cell lines induced after a single exposure to TKIs.
The assessed lung adenocarcinoma cell lines showed distinct sensitivity to a single exposure to therapeutic drugs. Notably, in EGFR-mutated lung adenocarcinoma cell lines, TKIs mainly inhibit cell proliferation and induce arrest in the G0/G1 phase of the cell cycle. Reduction in cell proliferation and cell cycle arrest have been suggested as mechanisms of drug resistance in tumor cells. Consistent with our results, it has been reported that DTP cells may be arrested with a G1 DNA content in a senescent-like phenotype, after drug exposure [40]. To explore whether the residual cells remaining in the culture after a single drug exposure acquired this phenotype, we assessed the expression of the enzyme β-galactosidase, which is charac-teristic of a senescent-like phenotype [32]. We found that most residual cells produced this enzyme, suggesting the induction of the DTP state. β-Galactosidase-positive DTP cells were consistently observed under experimental conditions, maintaining viability and β-galactosidase staining for several days after drug removal.
To identify the transcriptional changes that led to the development of the DTP state and the phenomenon of intrinsic re-sistance in the cell lines, we analyzed and compared the transcriptomes of the residual and control cells using RNA se-quencing. DEG analysis revealed a significant proportion of overexpressed genes shared by DTP cells in all cell lines tested. Interestingly, lncRNAs were found among the DEGs of DTP cells, and RT-qPCR validated their expression. Overexpression of these lncRNAs may be associated with unique transcriptomic profiles that modulate immediate transcriptome repro-gramming in DTP cells, which promotes the resistance of the cell lines. For instance, the HCC827 cell line showed higher resistance to erlotinib than HCC4006, since erlotinib mainly induced a cytostatic effect in the HCC827 cell line and a slight proportion of cell death. Consistently, erlotinib-DTP cells from the HCC827 cell line displayed higher overexpression of the lncRNA AGAP2-AS1 than those obtained from HCC4006 cells. In contrast, the HCC827 cell line showed the highest sensi-tivity to osimertinib among the cell lines studied, and the osimertinib-DTPs from this cell line displayed the lowest over-expression of lncRNA LINC01133. These results suggest that overexpression of these lncRNAs may be related to the intrinsic resistance to TKIs observed in these cell lines. In this regard, the knockdown of AGAP2-AS1 in the HCC827 cell line and LINC01133 in the H1975 cell line promoted the sensitivity of these cell lines to their respective TKIs. Interestingly, higher expression of AGAP2-AS1 and LINC01133 was associated with a significantly lower OS in lung adenocarcinoma patients from TCGA, which suggests the clinical relevance of these lncRNAs.
AGAP2-AS1 inhibits apoptosis and promotes cell proliferation mediated by the miR-1993a-3p/LOXL4 axis [41,42]. It has also been implicated in epithelial–mesenchymal transition (EMT) through the miR-468-3p/SRSF1 axis [43]. It has been proposed as a potential diagnostic and prognostic biomarker for NSCLC and renal cell carcinoma [44,45]. Dysregulation of LINC01133 in NSCLC has been consistently observed in NSCLC cell lines and samples from both the blood and tissues of lung cancer patients. Zang et al. highlighted a significant upregulation of LINC01133 expression in NSCLC tissues compared to healthy tissues. This upregulation was also evident in the NSCLC cell lines A549, PC-9, and H1975 as opposed to the human bronchial epithelial cell line HBEC [46]. These findings strongly indicated that LINC01133 functions as an oncogenic molecule in NSCLC. Additional studies by Zhang, et al. [47] provided insights into the clinical implications of elevated LINC01133 expression in NSCLC. The higher expression levels of LINC01133 were positively correlated with several ag-gressive tumor characteristics, including larger tumor size, lymph node metastasis, and increased cell proliferation, migra-tion, and invasion. Elevated LINC01133 expression was associated with a higher tumor grade. Interestingly, an inverse correlation was observed in the G1/G0 phase, underscoring its potential role in cell cycle regulation. Importantly, a study linked higher LINC01133 expression to diminished overall survival and progression-free survival in patients with NSCLC [47]. These comprehensive associations emphasize the pivotal role of LINC01133 in NSCLC pathogenesis and its potential use as a prognostic marker.
We next explored the expression of these DTP-associated lncRNAs in tumor tissue. Using RT-qPCR, we evaluated the expression of these lncRNAs in tumor biopsies of patients with untreated primary lung adenocarcinoma collected at our facility. We found that erlotinib-DTP-associated lncRNAs MEG3 and AGAP2-AS1 could be detected in tumor biopsies from non-treated patients, suggesting that these lncRNAs may be overexpressed throughout tumor development and that patients displaying overexpression of these lncRNAs would eventually develop resistance to erlotinib treatment. Interestingly, the osimertinib-DTP-associated lncRNAs LINC01133 and CERS6-AS1 could hardly be detected in tumor biopsies, compared to erlotinib-DTP-associated lncRNAs AGAP2-AS1, MEG3 and NKILA, which is consistent with clinical reports indicating that resistance to osimertinib is less frequent than to erlotinib, supporting the use of osimertinib as a better therapeutic option. It is possible that the tumor cells that express these osimertinib-DTP-associated lncRNAs are present in a low proportion prior to treatment and that the expression of these lncRNAs increases after the first rounds of treatment.
Since we did not find detectable levels of the osimertinib-DTP-associated lncRNAs in tumor biopsies, we examined the expression of a protein-coding gene that could potentially contribute to intrinsic resistance to osimertinib. Our RNA-seq and differential expression analyses revealed CD74 is an overexpressed protein-coding gene shared among osimertinib-DTP cells. CD74 is a protein-coding gene that encodes an invariant chain associated with HLA class II. This molecule is synthe-sized in the endoplasmic reticulum and transported to the membranes of antigen-presenting cells (APC). CD74 is a receptor for macrophage migration inhibitory factor (MIF) secreted by phagocytic cells [48]. In addition, reports indicate that CD74 is expressed mainly by the malignant cells of NSCLC and other cancers. Five in-frame fusion proteins have been reported [49]. In the current study, RT-qPCR analyses confirmed CD74 expression in osimertinib-DTP cells from the cell lines and lung adenocarcinoma biopsies. It is noteworthy that in the HCC827 cell line that exhibited a high sensitivity to osimertinib, the expression of CD74 was the lowest compared to those of the cell lines HCC4006 and H1975, which displayed higher re-sistance to osimertinib.
We analyzed the protein expression of CD74 in cell lines via immunofluorescence. We found that CD74 mainly shows cytoplasmic/membrane staining, and that, similar to what was found at the mRNA level, the expression of CD74 was higher in the osimertinib-DTP cells than in the control. Additionally, in tumor biopsies, we found that tumor cells displayed a high staining of CD74, compared to normal ciliated epithelial cells, which suggests that this molecule is related to tumor devel-opment. Interestingly, a heterogeneous staining pattern among tumor cells was observed in the tumor mass, implying that some tumor cells expressed higher levels of CD74. As indicated by data from the LUAD project of TCGA, elevated CD74 expression correlated with improved OS in patients with lung cancer. Elevated expression of CD74 may be linked to tumors characterized by increased immune infiltration, a factor associated with a favorable impact on overall survival. Further clinical studies are imperative to scrutinize the biological implications of CD74 expression and its fusion proteins in cold lung tumors compared to those exhibiting high immune cell infiltration. Based on this study, tumor cells that express higher levels of CD74 would survive after the initial rounds of osimertinib treatment, and they would then proliferate to promote tumor relapse and resistance to this therapeutic option, making CD74 a possible therapeutic target to overcome osimertinib resistance. Whether the role of CD74 in intrinsic resistance against osimertinib is related to the multiple fusion proteins that CD74 produces in lung cancer remains to be investigated.
5. Conclusions
Long non-coding RNAs (lncRNAs) have been implicated in resistance to drug treatments and they might be participating since the onset of treatment. After a single exposure to TKIs in lung cancer cell lines, transcriptomic analysis detected overexpression of some LncRNAs and of CD74 in DTP cells. The clinical relevance of the lnc RNAs and CD74 were validated in a cohort of patients from the ATCG, in DTP cells using RT-PCR and by knockdown some of them. In addition, they were detected in a small number of biopsies from adenocarcinoma patients. Further basic and clinical practice studies are required to highlight the role that they are playing in intrinsic resistance of lung carcinoma. However, this research provides oppor-tunities to study the involvement of lncRNAs in the intricate genetic and epigenetic mechanisms of intrinsic resistance. Their implications as prognostic markers and potential predictive markers of therapeutic targets that will help to improve the outcomes of lung cancer patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Representative strategy of cytometry analysis for the quantification of viable, apoptotic, and necrotic cells. Effect of erlotinib in EGFR-mutated lung adenocarcinoma cell lines. Figure S2: SA-ß-gal-positive cells evidencing DTP cells showing a senescent-like phenotype. Table S1: TaqMan® gene expression assays. Table S2: DsiRNA sequences employed for knockdown. Table S3: LncRNAs overexpressed in erlotinib-DTP cells. Table S4: LncRNAs overexpressed in Osimertinib-DTP cells. Table S5: LncRNAs overexpressed in CDDP-DTP cells.
Author Contributions
Conceptualization, MAP-M, JSL-G and DA-C (corresponding author). Data curation: MAP-M, AM-T, and RC-D. Formal Analysis: MAP-M, JSL-G and DA-C. Funding acquisition: DA-C, SA-R. Investigation: MAP-M, MS-N, MM-F, MG-V, JJB-L, RC-D Methodology: MAP-M, MS-N, MM-F, MG-V, JJB-L, RC-D. Project administration: DA-C, JSL-G. Resources: SA-R Software: AM-T, SEM-T. Supervision: JSL-G, DA-C Validation: MAP-M, MG-V, SEM-T. Visualization: JSL-G Writing – original draft: MAP-M, JSL-G and DA-C.Writing – review & editing: MAP-Mario, JSL-G, JJB-L, and DA-C.
Funding
Part of this research was funded by the National Council of Humanities, Science, and Technology (CONHACyT) of Mexico (CF-2023-I-179).
Institutional Review Board Statement
This study was approved by the Scientific, Bioethics, and Biosafety Committee of the Instituto Nacional de Enfermedades Respiratorias "Ismael Cosío Villegas," and a waiver request was obtained for the use of tissue blocks from patients with lung adenocarcinoma obtained from the repository of residual biological material from the archives of the Pathology Department of the institution.
Data Availability Statement
The accession numbers of RNA-seq raw reads, raw count arrays and target sequencing raw reads, as well as data analyses that were performed in R language using open source packages are available at the following link: https://github.com/maperezm/Intrinsic_CDDP_Resistant_Scripts
Acknowledgments
MP-M, a Ph.D. student from Posgrado en Ciencias en Biomedicina y Biotecnologia Molecular, Escuela Nacional de Ciencias Biologicas, Mexico, received a fellowship from CONACyT (740805). JJB-L student of Posgrado en Ciencias Biologicas, Universidad Nacional Autonoma de Mexico, received a fellowship from CONACyT (1085486) We would like to thank Erika Monterrubio-Flores and Guadalupe Hiriart Valencia for their excellent technical assistance with histochemical work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung, H. , Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 71, 209–249. [CrossRef]
- Tfayli, A. , and Mohty, R. (2021). EGFR tyrosine kinase inhibitors in non-small cell lung cancer: treatment paradigm, current evidence, and challenges. Tumori 107, 376–384. [CrossRef]
- Jamal-Hanjani, M. , Wilson, G. A., McGranahan, N., Birkbak, N. J., Watkins, T. B. K., Veeriah, S., et al. (2017). Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121. [CrossRef]
- Waarts, M. R. , Stonestrom, A. J., Park, Y. C., and Levine, R. L. (2022). Targeting mutations in cancer. J. Clin. Invest. 132. [CrossRef]
- Nie, N. , Li, J., Zhang, J., Dai, J., Liu, Z., Ding, Z., et al. (2022). First-line osimertinib in patients with EGFR-mutated non-small cell lung cancer: effectiveness, resistance mechanisms, and prognosis of different subsequent treatments. Clin. Med. Insights Oncol. 16, 117955492211347. [CrossRef]
- Pirker, R. (2016). Third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr. Opin. Oncol. 28, 115–121. [CrossRef]
- Wang, J. , Wang, B., Chu, H., and Yao, Y. (2016). Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations. Onco. Targets. Ther. 9, 3711–3726. [CrossRef]
- Sharma, S. V. , Lee, D. Y., Li, B., Quinlan, M. P., Takahashi, F., Maheswaran, S., et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. [CrossRef]
- Sullivan, I. , and Planchard, D. (2016). Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front. Med. 3, 76. [CrossRef]
- Hammerlindl, H. , and Schaider, H. (2018). Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance. J. Cell Commun. Signal. 12, 133–141. [CrossRef]
- De Conti, G. , Dias, M. H., and Bernards, R. (2021). Fighting drug resistance through the targeting of drug-tolerant persister cells. Cancers (Basel). 13, 1–15. [CrossRef]
- Chhouri, H. , Alexandre, D., and Grumolato, L. (2023). Mechanisms of acquired resistance and tolerance to EGFR targeted therapy in non-small cell lung cancer. Cancers (Basel). 15. [CrossRef]
- Wang, X., Zhang, H., and Chen, X. (2019). Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2. 141–160. [CrossRef]
- Gao, X. , Liu, Q., Chen, X., Chen, S., Yang, J., Liu, Q., et al. (2021). Screening of tumor grade-related mRNAs and lncRNAs for esophagus squamous cell carcinoma. J. Clin. Lab. Anal. 35, e23797. [CrossRef]
- Yano, S., Yamada, T., Takeuchi, S., Tachibana, K., Minami, Y., Yatabe, Y., et al. (2011). Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a japanese cohort. J. Thorac. Oncol. 6, 2011–2017. [CrossRef]
- Hsiao, S.-H. , Liu, H. E., Lee, H.-L., Lin, C.-L., Chen, W.-Y., Wu, Z.-H., et al. (2013). Distinct clinical outcomes of non-small cell lung cancer patients with epidermal growth factor receptor (EGFR) mutations treated with EGFR tyrosine kinase inhibitors: Non-responders versus responders. PLoS One 8, e83266. [CrossRef]
- Swayden, M. , Chhouri, H., Anouar, Y., and Grumolato, L. (2020). Tolerant/persister cancer cells and the path to resistance to targeted therapy. Cells 9, 2601. [CrossRef]
- Recasens, A. , and Munoz, L. ( 2019). Targeting cancer cell dormancy. Trends Pharmacol. Sci. 40, 128–141. [CrossRef]
- Ding, D. , Zhang, J., Luo, Z., Wu, H., Lin, Z., Liang, W., et al. (2022). Analysis of the lncRNA–miRNA–mRNA network reveals a potential regulatory mechanism of EGFR-TKI resistance in NSCLC. Front. Genet. 13, 851391. [CrossRef]
- Vishwanathan, K., So, K., Thomas, K., Bramley, A., English, S., and Collier, J. (2019). Absolute bioavailability of osimertinib in healthy adults. Clin. Pharmacol. Drug Dev. 8, 198–207. [CrossRef]
- Le Louedec, F. , Puisset, F., Chatelut, E., and Tod, M. (2023). Considering the oral bioavailability of protein kinase inhibitors: essential in assessing the extent of drug–drug interaction and improving clinical practice. Clin. Pharmacokinet. 62, 55–66. [CrossRef]
- Anders, S. (2010). Babraham Bioinformatics - FastQC a quality control tool for high throughput sequence data. Soil 5. https://www.bioinformatics.babraham.ac.uk/projects/. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bolger, A. M. , Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [CrossRef]
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10. [CrossRef]
- Dobin, A. , Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [CrossRef]
- Li, B. , and Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [CrossRef]
- Robinson, M. D. , McCarthy, D. J., and Smyth, G. K. (2009). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [CrossRef]
- Durinck, S. , Moreau, Y., Kasprzyk, A., Davis, S., De Moor, B., Brazma, A., et al. (2005). BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [CrossRef]
- Tang, Z. , Li, C., Kang, B., Gao, G., Li, C., and Zhang, Z. (2017). GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102. [CrossRef]
- Nicholson, A. G. , Tsao, M. S., Beasley, M. B., Borczuk, A. C., Brambilla, E., Cooper, W. A., et al. (2022). The 2021 WHO classification of lung tumors: impact of advances since 2015. J. Thorac. Oncol. 17, 362–387. [CrossRef]
- Aguiar, P. N. , Haaland, B., Park, W., San Tan, P., del Giglio, A., and de Lima Lopes, G. (2018). Cost-effectiveness of osimertinib in the first-line treatment of patients with EGFR-mutated advanced non–small cell lung cancer. JAMA Oncol. 4, 1080. [CrossRef]
- Leonce, C. , Saintigny, P., and Ortiz-Cuaran, S. (2022). Cell-intrinsic mechanisms of drug tolerance to systemic therapies in cancer. Mol. Cancer Res. 20, 11–29. [CrossRef]
- Kashima, Y. , Shibahara, D., Suzuki, A., Muto, K., Kobayashi, I. S., Plotnick, D., et al. (2021). Single-cell analyses reveal diverse mechanisms of resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Res. 81, 4835–4848. [CrossRef]
- Liu, L. , Lizaso, A., Mao, X., Yang, N., and Zhang, Y. (2020). Rechallenge with erlotinib in osimertinib-resistant lung adenocarcinoma mediated by driver gene loss: a case report. Transl. Lung Cancer Res. 9, 144–147. [CrossRef]
- Meador, C. B. , and Hata, A. N. (2020). Acquired resistance to targeted therapies in NSCLC: updates and evolving insights. Pharmacol. Ther. 210, 107522. [CrossRef]
- Ashrafi, A. , Akter, Z., Modareszadeh, P., Modareszadeh, P., Berisha, E., Alemi, P. S., et al. (2022). Current landscape of therapeutic resistance in lung cancer and promising strategies to overcome resistance. Cancers (Basel). 14, 4562. [CrossRef]
- Al Bakir, M. , Huebner, A., Martínez-Ruiz, C., Grigoriadis, K., Watkins, T. B. K., Pich, O., et al. (2023). The evolution of non-small cell lung cancer metastases in TRACERx. Nature 616, 534–542. [CrossRef]
- Mondal, P. , and Meeran, S. M. (2023). Emerging role of non-coding RNAs in resistance to platinum-based anti-cancer agents in lung cancer. Front. Pharmacol. 14, 1–18. [CrossRef]
- Gupta, S. , Verma, V., and Dwarakanath, B. S. (2022). Emerging concepts in cancer therapy: mechanisms of resistance. Cancer Rep. 5, e1715. [CrossRef]
- Presta, M. (2021). β-Galactosylceramidase in cancer: friend or foe? Trends in Cancer 7, 974–977. [CrossRef]
- Luo, W. , Li, X., Song, Z., Zhu, X., and Zhao, S. (2019). Long non-coding RNA AGAP2-AS1 exerts oncogenic properties in glioblastoma by epigenetically silencing TFPI2 through EZH2 and LSD1. Aging (Albany. NY). 11, 3811–3823. [CrossRef]
- Ren, P. , Niu, X., Zhao, R., Liu, J., Ren, W., Dai, H., et al. (2022). Long non-coding RNA AGAP2-AS1 promotes cell proliferation and invasion through regulating miR-193a-3p/LOXL4 axis in laryngeal squamous cell carcinoma. Cell Cycle 21, 697–707. [CrossRef]
- Li, H. , Guo, S., Zhang, M., Li, L., Wang, F., and Song, B. (2020). Long non-coding RNA AGAP2-AS1 accelerates cell proliferation, migration, invasion and the EMT process in colorectal cancer via regulating the miR-4,668-3p/SRSF1 axis. J. Gene Med. 22, e3250. [CrossRef]
- Fan, K.-J. , Liu, Y., Yang, B., Tian, X.-D., Li, C.-R., and Wang, B. (2017). Prognostic and diagnostic significance of long non-coding RNA AGAP2-AS1 levels in patients with non-small cell lung cancer. Eur. Rev. Med. Pharmacol. Sci. 21, 2392–2396. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28617550.
- Gao, L. , Zhao, A., and Wang, X. (2020). Upregulation of lncRNA AGAP2-AS1 is an independent predictor of poor survival in patients with clear cell renal carcinoma. Oncol. Lett. 19, 3993–4001. [CrossRef]
- Zang, C. , Nie, F. Q., Wang, Q., Sun, M., Li, W., He, J., et al. (2016). Long non-coding RNA LINC01133 represses KLF2, P21 and E-cadherin transcription through binding with EZH2, LSD1 in non small cell lung cancer. Oncotarget 7, 11696–11707. [CrossRef]
- Zhang, J., Zhu, N., and Chen, X. (2015). A novel long noncoding RNA LINC01133 is upregulated in lung squamous cell cancer and predicts survival. Tumor Biol. 36, 7465–7471. [CrossRef]
- McClelland, M. , Zhao, L., Carskadon, S., and Arenberg, D. (2009). Expression of CD74, the receptor for macrophage migration inhibitory factor, in non-small cell lung cancer. Am. J. Pathol. 174, 638–646. [CrossRef]
- Vargas, J. , and Pantouris, G. ( 2023). Analysis of CD74 occurrence in oncogenic fusion proteins. Int. J. Mol. Sci. 24, 15981. [CrossRef]
Figure 1.
Morphology changes and proportion of cells in the cell cycle phases in the EGFR-mutated lung adenocarcinoma cell lines after a single erlotinib exposure. Cellular density changes induced by erlotinib in the cell lines (A, D, and G). Magnification 20x. Histograms showing the profile of cell cycle distribution and graphs of the percentage of cells in the cell cycle phases induced by erlotinib in the HCC827 (B and C), HCC4006 (E and F), and H1975 (H and I) cell lines, respectively. Data are shown as mean ± SD. ****p < 0.0001.
Figure 1.
Morphology changes and proportion of cells in the cell cycle phases in the EGFR-mutated lung adenocarcinoma cell lines after a single erlotinib exposure. Cellular density changes induced by erlotinib in the cell lines (A, D, and G). Magnification 20x. Histograms showing the profile of cell cycle distribution and graphs of the percentage of cells in the cell cycle phases induced by erlotinib in the HCC827 (B and C), HCC4006 (E and F), and H1975 (H and I) cell lines, respectively. Data are shown as mean ± SD. ****p < 0.0001.
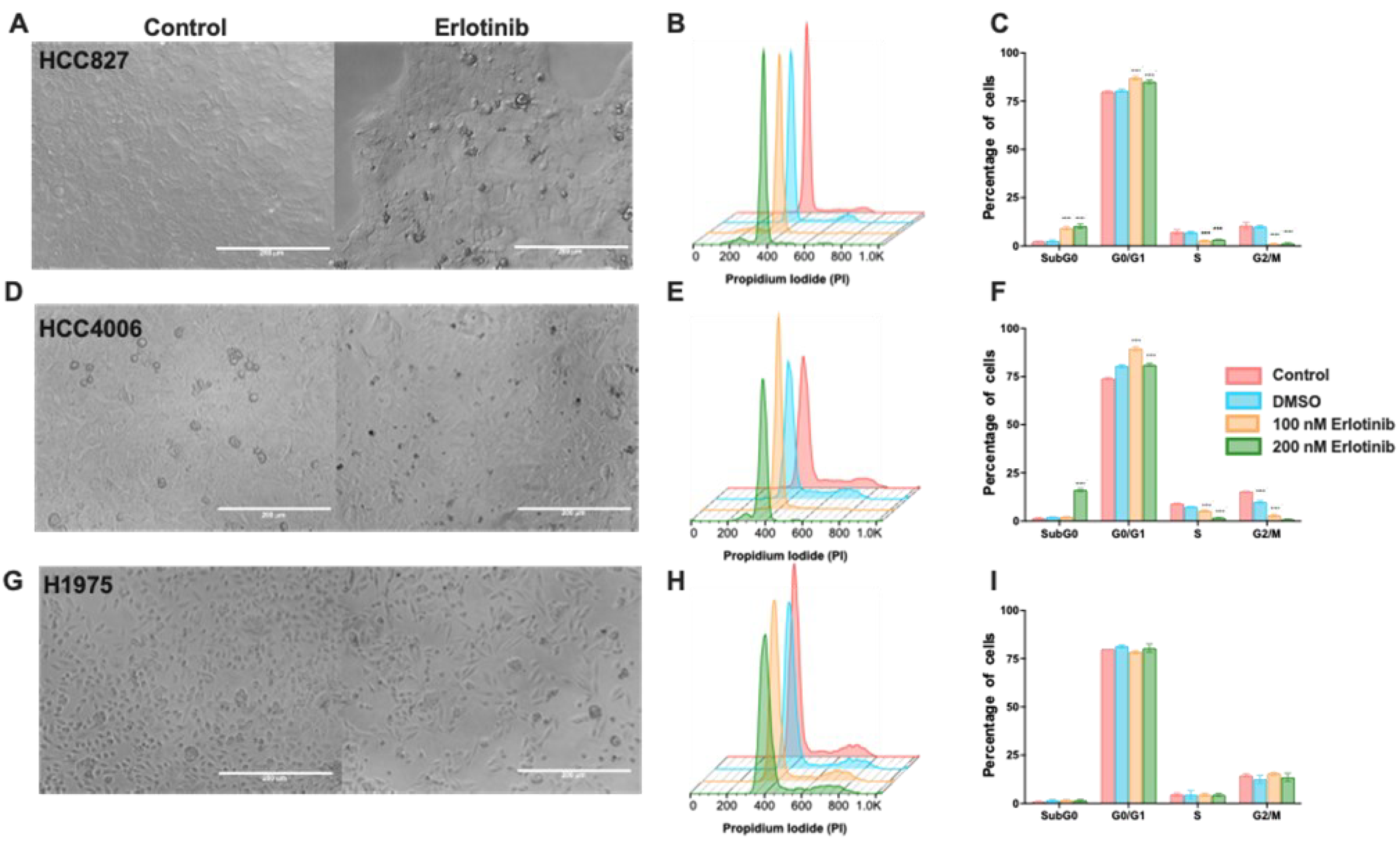
Figure 2.
Effect of osimertinib exposure in the EGFR-mutated lung adenocarcinoma cell lines. Morphologic changes induced by a single Osimertinib exposure in cell lines studied (A, D, and G). Magnification 20x. Histograms show the profile of cell distribution and graphs of the percentage of cells in the cell cycle phases, and the subG0 peak induced by osimertinib in the HCC827 (B and C), HCC4006 (E and F), and H1975 (H and I) cell lines, respectively. Data are shown as mean ± SD. **** p < 0.0001. (J). After a single 48 h exposure to osimertinib, residual cells from the H1975 cell line were cultured in fresh complete media for five days and the expression of be-ta-galactosidase was detected. Micrographs show SA-ß-gal-positive cells evidencing DTP cells showing a senescent-like phenotype. Magnification 40x.
Figure 2.
Effect of osimertinib exposure in the EGFR-mutated lung adenocarcinoma cell lines. Morphologic changes induced by a single Osimertinib exposure in cell lines studied (A, D, and G). Magnification 20x. Histograms show the profile of cell distribution and graphs of the percentage of cells in the cell cycle phases, and the subG0 peak induced by osimertinib in the HCC827 (B and C), HCC4006 (E and F), and H1975 (H and I) cell lines, respectively. Data are shown as mean ± SD. **** p < 0.0001. (J). After a single 48 h exposure to osimertinib, residual cells from the H1975 cell line were cultured in fresh complete media for five days and the expression of be-ta-galactosidase was detected. Micrographs show SA-ß-gal-positive cells evidencing DTP cells showing a senescent-like phenotype. Magnification 40x.
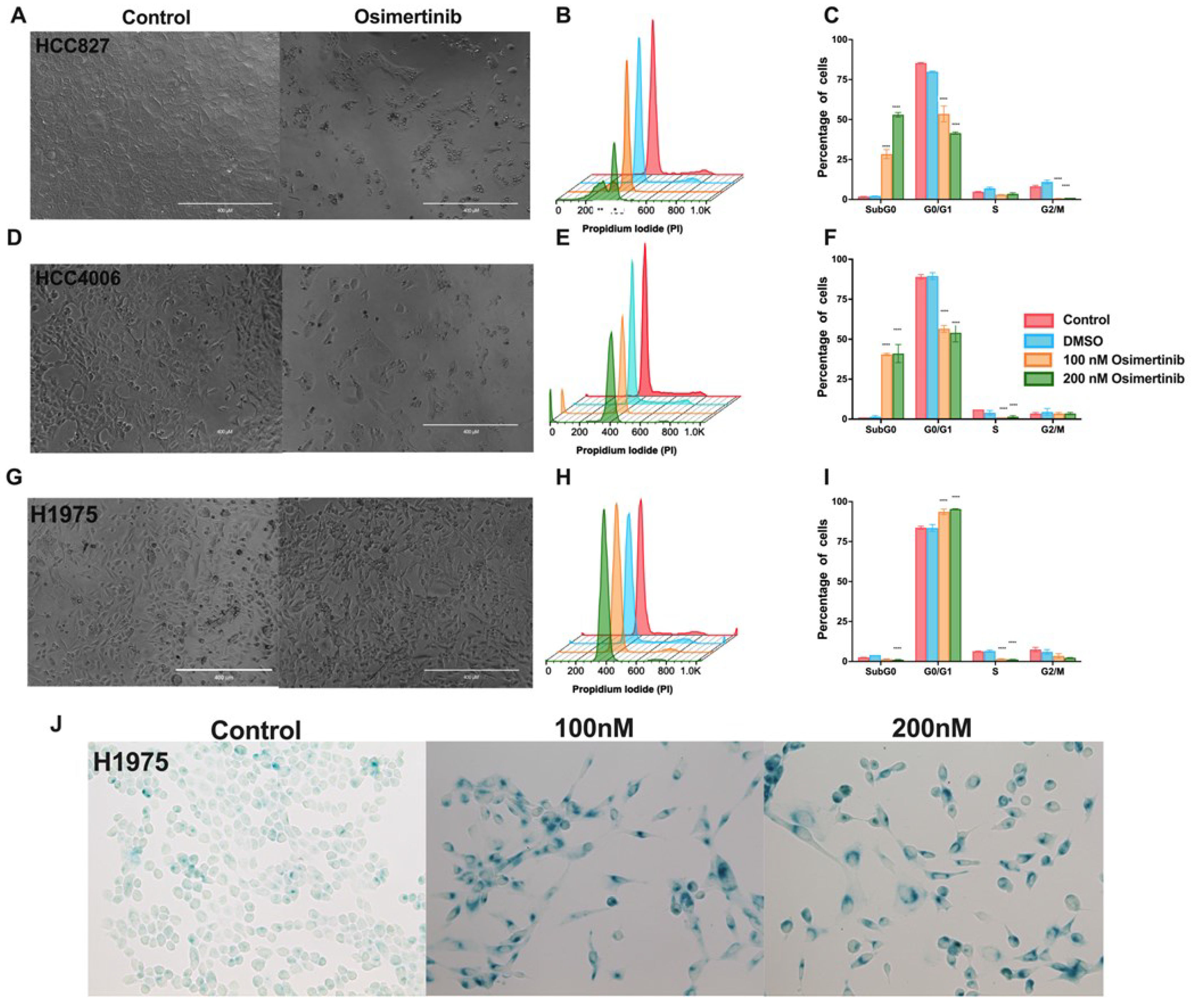
Figure 3.
Differentially expressed genes (DEG) in DTP cells. Volcano plots showing DEG in DTP cells relative to untreated cells (A and B). Gray shading shows overexpressed transcripts. Venn diagram of overexpressed genes shared among DTP cells of the lung adeno-carcinoma cell lines respect to drug exposure (C and D). Functional annotation of the overexpressed RNAs was performed using the Gene Ontology (GO) biological process database (E and F).
Figure 3.
Differentially expressed genes (DEG) in DTP cells. Volcano plots showing DEG in DTP cells relative to untreated cells (A and B). Gray shading shows overexpressed transcripts. Venn diagram of overexpressed genes shared among DTP cells of the lung adeno-carcinoma cell lines respect to drug exposure (C and D). Functional annotation of the overexpressed RNAs was performed using the Gene Ontology (GO) biological process database (E and F).
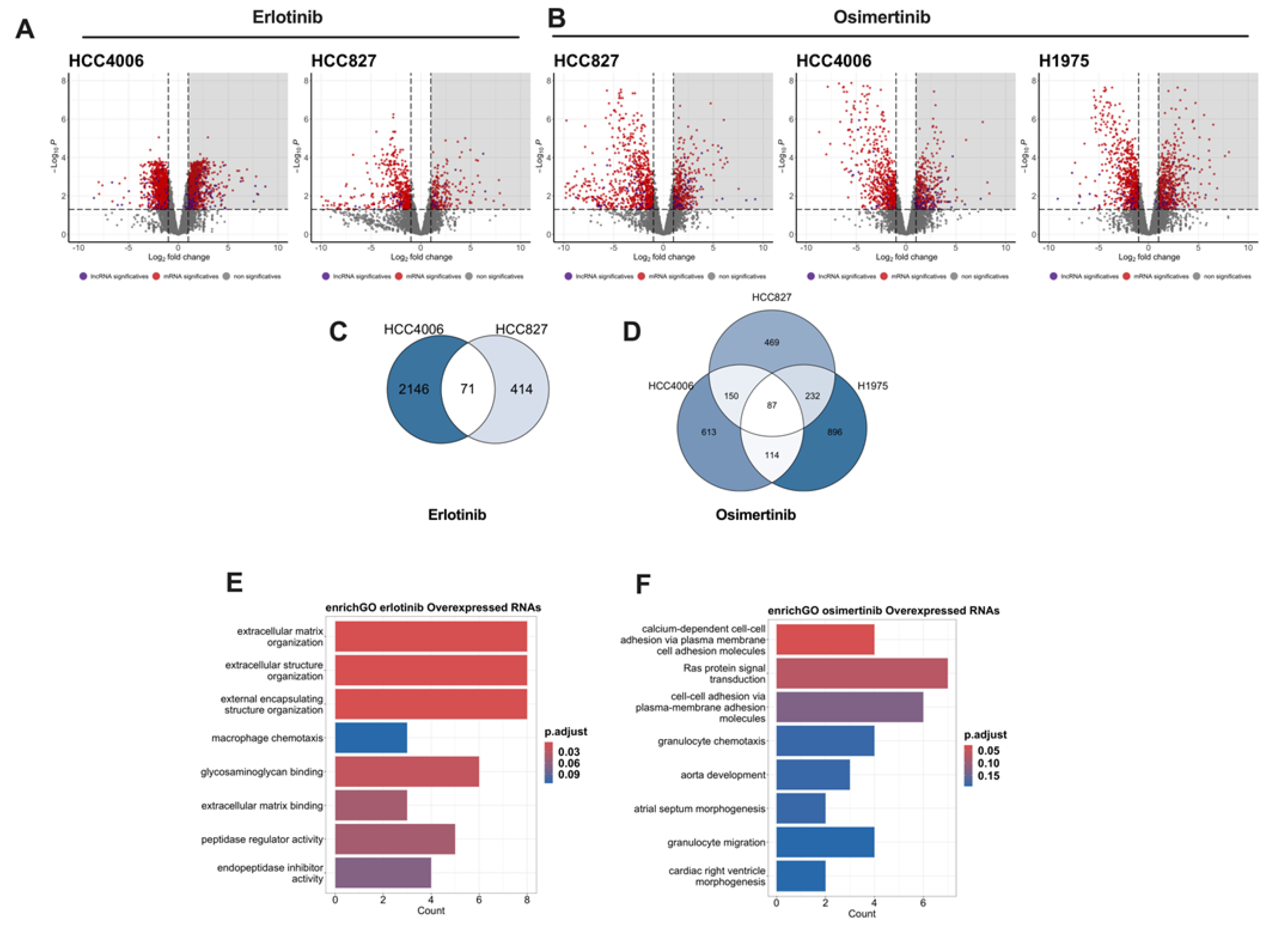
Figure 4.
Clinical relevance of lncRNAs associated with intrinsic resistance. K–M survival analysis according to the LUAD patient cohort from The Cancer Genome Atlas (A). High lncRNA expression is shown in red; low lncRNA expression is shown in blue. Log-rank p-values are indicated.
Figure 4.
Clinical relevance of lncRNAs associated with intrinsic resistance. K–M survival analysis according to the LUAD patient cohort from The Cancer Genome Atlas (A). High lncRNA expression is shown in red; low lncRNA expression is shown in blue. Log-rank p-values are indicated.
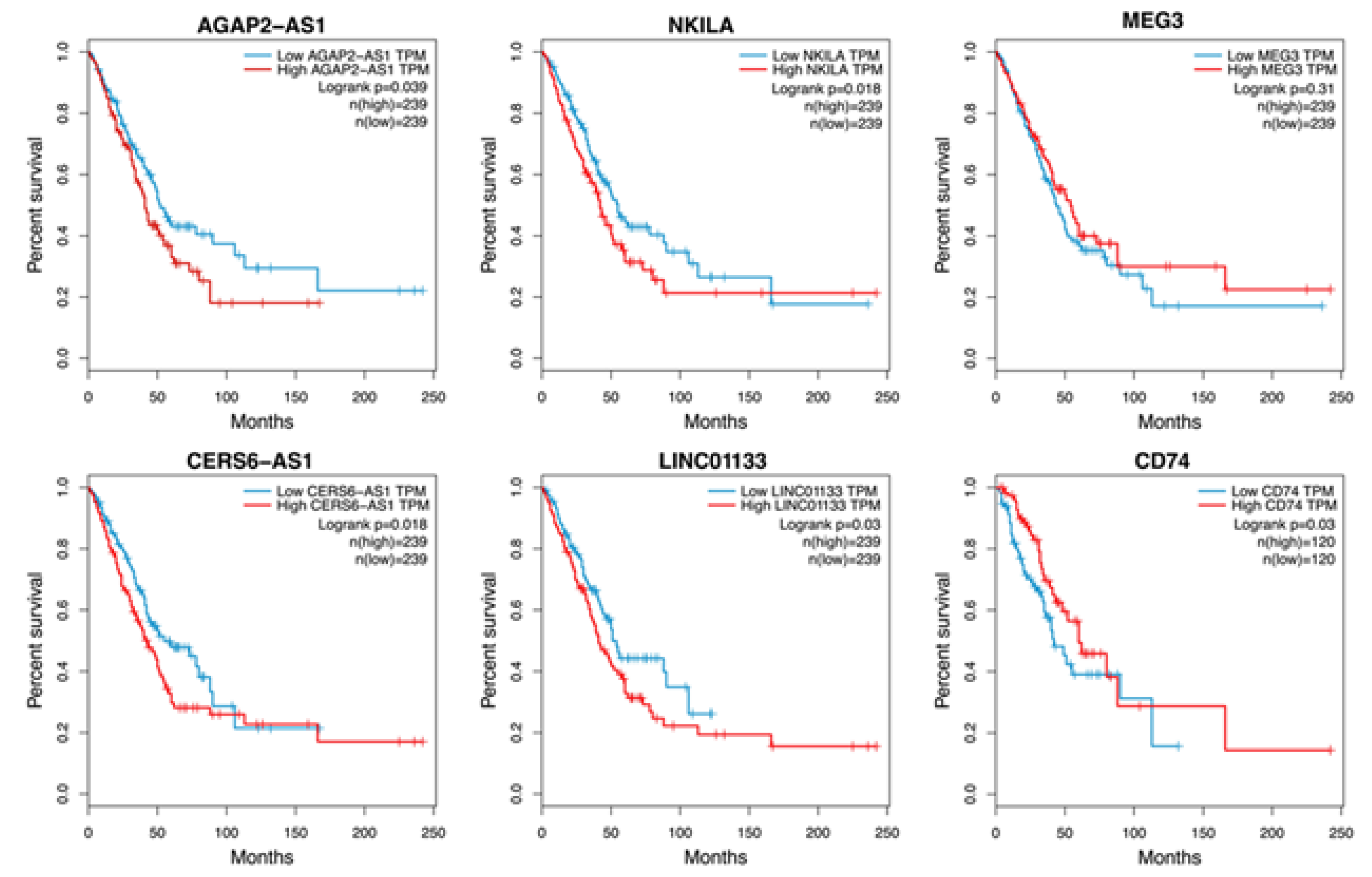
Figure 5.
Gene expression of clinically relevant lncRNAs detected in DTP cells. LncRNAs detected in erlotinib-and osimertinib-DTP cells (A and B). Fold changes relative to the control are represented. DsiRNA-mediated knockdown in the cell lines reduces the expression of the lncRNAs AGAP-AS1 and LINC01133. DsiRNA against HPRT was used as control (C). Knockdown of the DTP-associated lncRNA reduces cell viability after a single exposure to TKIs (D). The data are shown as mean ± SD of three independent experiments.****p-value <0.05.
Figure 5.
Gene expression of clinically relevant lncRNAs detected in DTP cells. LncRNAs detected in erlotinib-and osimertinib-DTP cells (A and B). Fold changes relative to the control are represented. DsiRNA-mediated knockdown in the cell lines reduces the expression of the lncRNAs AGAP-AS1 and LINC01133. DsiRNA against HPRT was used as control (C). Knockdown of the DTP-associated lncRNA reduces cell viability after a single exposure to TKIs (D). The data are shown as mean ± SD of three independent experiments.****p-value <0.05.
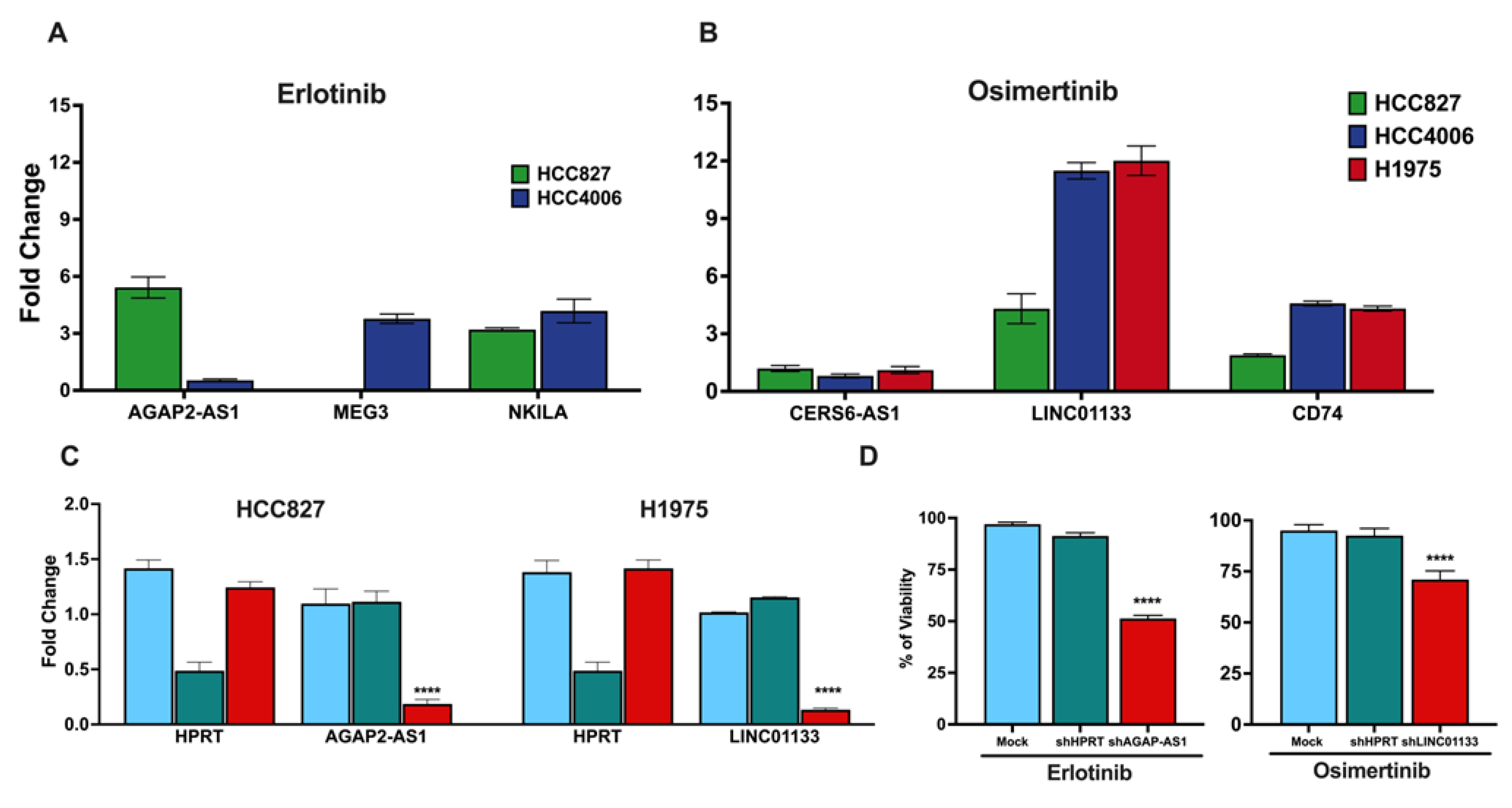
Figure 6.
Gene expression of clinically relevant genes in tumor biopsies. Expression of DTP-associated genes in four FFPE lung adenocarcinoma biopsies, detected by RT-qPCR. Gene expression was normalized respect to GAPDH and compared to patient 1.
Figure 6.
Gene expression of clinically relevant genes in tumor biopsies. Expression of DTP-associated genes in four FFPE lung adenocarcinoma biopsies, detected by RT-qPCR. Gene expression was normalized respect to GAPDH and compared to patient 1.
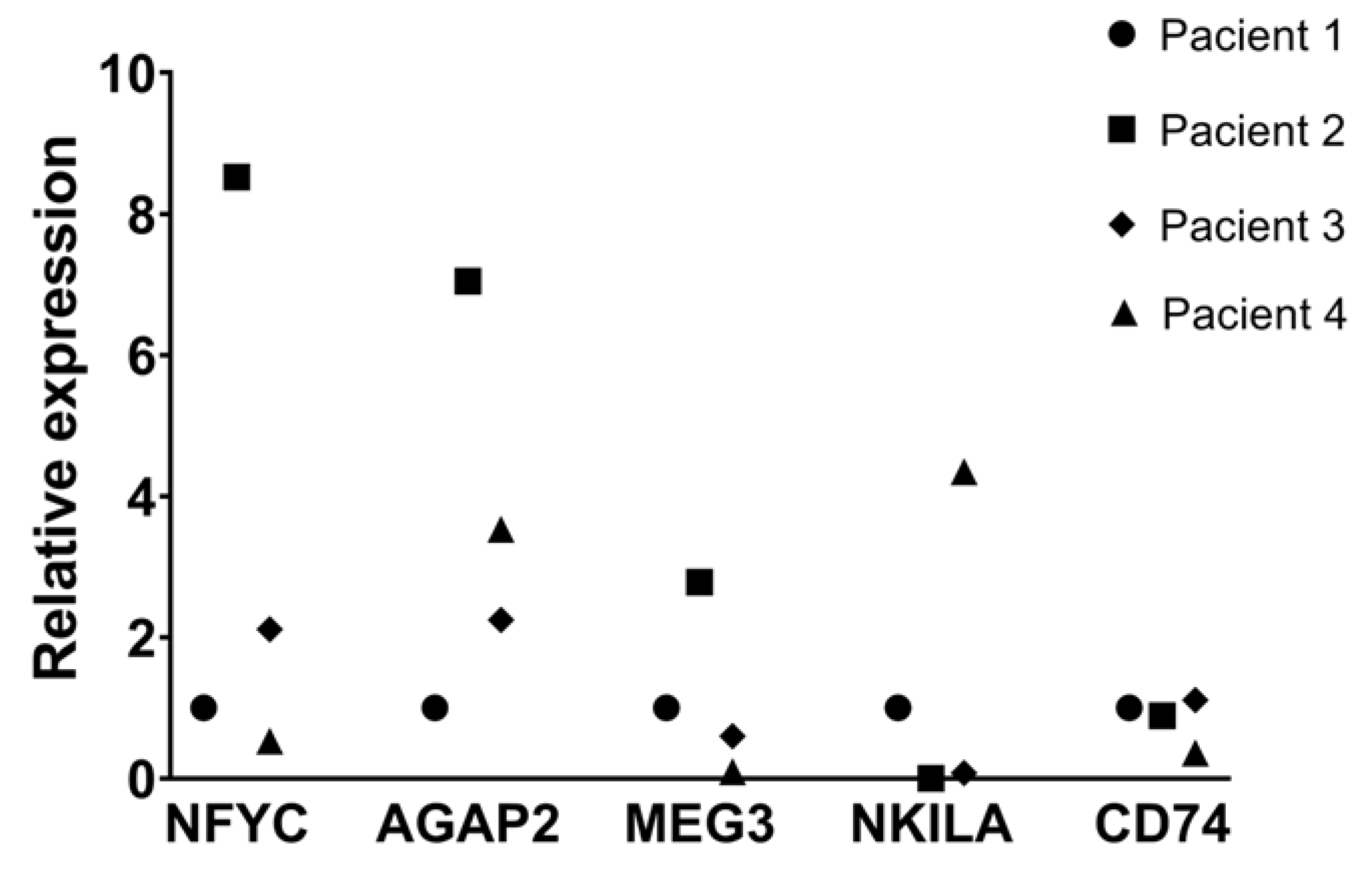
Figure 7.
CD74 expression in lung adenocarcinoma. A) Immunofluorescence against CD74 in osimertinib–DTP cells. CD74 mainly shows cytoplasmic staining, which increases in osimertinib–treated DTP cells. Magnification 20x. Lung adenocarcinoma biopsy. B) Comparison of CD74 staining between normal ciliated-epithelial cells and malignant cells. C). Tumor cells with varied level of CD74 expression. D) High CD74 expression observed in tumor cells with invasive phenotype. Magnification 20x.
Figure 7.
CD74 expression in lung adenocarcinoma. A) Immunofluorescence against CD74 in osimertinib–DTP cells. CD74 mainly shows cytoplasmic staining, which increases in osimertinib–treated DTP cells. Magnification 20x. Lung adenocarcinoma biopsy. B) Comparison of CD74 staining between normal ciliated-epithelial cells and malignant cells. C). Tumor cells with varied level of CD74 expression. D) High CD74 expression observed in tumor cells with invasive phenotype. Magnification 20x.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



