
Submitted:
27 March 2024
Posted:
28 March 2024
You are already at the latest version
Abstract
In genetic conflicts, driver and killer elements achieve biased survival, replication, or transmission over sensitive and targeted elements through a wide range of molecular mechanisms, including mimicry. Driving mechanisms manifest at all organismal levels, from the biased propagation of individual genes, as demonstrated by transposable elements, to the biased transmission of genomes, as illustrated by viruses, to the biased transmission of cell lineages, as in cancer. Targeted genomes are vulnerable to molecular mimicry through the conserved motifs they use for their own signaling and regulation. Mimicking these motifs enables a selfish element to control core target processes, and can occur at the sequence, structure, or functional level. Molecular mimicry was first appreciated as an important phenomenon more than twenty years ago. Modern genomics technologies, databases, and machine learning approaches offer tremendous potential to study the distribution of molecular mimicry across genetic conflicts in nature. Here, we explore the theoretical expectations for molecular mimicry between conflicting genomes, the trends in molecular mimicry mechanisms across known genetic conflicts, and outline how new examples can be gleaned from population genomic datasets. We discuss how mimics involving short sequence-based motifs or gene duplications can evolve convergently from new mutations. Whereas, processes that involve divergent domains or fully-folded structures occur among genomes by horizontal gene transfer. These trends are largely based on a small number of organisms and should be reevaluated in a general, phylogenetically independent framework. Currently, publicly available databases can be mined for genotypes driving non-mendelian inheritance patterns, epistatic interactions, and convergent protein structures. A subset of these conflicting elements may be molecular mimics. We propose approaches for detecting genetic conflict and molecular mimicry from these datasets.
Keywords:
genetic conflict
; selfish element
; molecular mimicry
; horizontal gene transfer
; convergent evolution
1. Introduction to Genetic Conflict and Molecular Mimicry
Genetic conflicts between genes, genomes, and individuals drove the evolution of many emergent biological phenomena, from mobile element-repressing heterochromatin, to endosymbiont-derived organelles, to protein mimicry [1,2,3,4]. Extant selfish genetic elements include intraspecific selfish chromosomes, plasmids, organelles, cell lineages (cancer), and transposable elements. Interspecific genetic conflicts occur between viruses, symbiotic bacteria, and symbiotic fungi and their hosts (Figure 1A). In all of these examples, conflicts are waged at the molecular level to promote the survival, abundance, and transmission of a selfish driving element to the detriment of sensitive elements. These are also termed killer and target elements, respectively. Although essential proteins involved in replication and transmission can theoretically be targeted through novel mechanisms, mimicry of existing molecular mechanisms provides a fast-track for driving elements to bias cellular functions [2,3]. For example, diverse bacterial pathogens and viruses gain access to host cells by co-opting small receptor-binding motifs and full domains to induce cellular uptake or niche formation.
Many forms of molecular mimicry exist for mediating interspecific conflicts between pathogens and hosts, as well as intraspecific conflicts between elements in a single genome. Mimicry may be based on shared short motifs or complex structures. The genes encoding molecular mimics can arise in divergent organisms de novo by mutation and convergence on target functions and/or structures. Or they can arise through horizontal transmission from another lineage [2,5]. We define molecular mimics to be DNA, RNA, or proteins that have converged on a function, structure or sequence, to co-opt a process imparted by a target element, resulting in the redirection of the products, rewards, or implications of that process to the genetic element encoding the mimic. Molecular mimics are distinguished as “perfect” or “imperfect” based on how closely they reproduce the full mechanism of the element or phenotype being imitated. Studying mechanistic trends among these elements provides insight into how likely molecular mimics are to evolve, which functional strategies they employ, and how we might identify them in genetic datasets.
Molecular mimic detection has historically been challenging because validation involves carefully controlled experiments. Genetic conflict distorts allele frequencies from mendelian expectations, permitting signatures of conflict to be detected from genome-wide population sequencing data [7,8]. However, only a subset of these elements will use molecular mimicry. Driving and sensitive haplotypes can be large and in strong linkage due to structural variation, such as chromosomal inversions, that prevents recombination. This linkage makes ascertaining the causative distortion loci in these multigenic haplotypes difficult. Elucidating the functional molecular basis of the distortion, which is required for molecular mimic identification, is exceedingly challenging because few organisms are genetically and developmentally tractable. Thus, knowledge of molecular mimics in genetic conflicts is limited and biased towards genetically tractable systems.
Until genome-wide, culture-independent screens are leveraged to find mimics broadly, the occurrence and distribution of molecular mimicry across life is informed through careful experiments in a subset of model systems. Model systems for genetic conflict, ranging from selfish Drosophila chromosomes to SARS-CoV-2 viruses, have been studied well enough at the mechanistic level to inform on the possible forms of molecular mimicry. These investigations have revealed diverse manifestations of mimicry, prompting its description since the early 2000s [5,9,10,11]. By the 2010s, the power of whole genome sequencing and accumulating genomic data in public databases made in silico screens for molecular mimicry possible [12,13,14,15]. The power of these approaches strengthens every year, as more genomes across the tree of life are sequenced and as new tools, such as AlphaFold [16], are developed for computationally predicting and comparing molecular function [17]. Full knowledge about the distribution of molecular mimicry in nature awaits genome mining studies across the tree of life. Soon the necessary genomic data and protein-interaction tools will be available to address this question organism-agnostically.
In the sections below, we explore 1) what types of molecular mimics are expected to occur based on evolutionary theories, 2) empirical examples of mimicry in nature, and 3) approaches for identifying genetic conflicts involving molecular mimics from genome-wide sequencing data (full list of literature in Table S1). Through this theoretical and empirical survey, we conclude that convergently evolved mimics are generally limited to short motifs in interspecific conflicts. Full-length sequence and structure molecular mimics arise in intraspecific conflicts via gene duplication events and in intraspecific conflicts via horizontal gene transfer (HGT). Functional mimics tend to involve entirely novel genes and pathways to reconstruct the mimicked molecular function. The few well-characterized examples of functional mimicry occur in intraspecific conflicts. Through leveraging the bioinformatic approaches for mimicry detection we present in the final section, we hope that more molecular mimics will be identified and these hypotheses about their distributions can be empirically tested in the future.
2. Distribution of Molecular Mimicry across Life: Expectations from Molecular Evolutionary Theory
Genetic conflicts can be mediated by molecular mimicry at functional, structural, and genetic (i.e., sequence) levels [5] (Figure 1B–E). Mimicry of cellular and molecular functions at its highest level does not require similar protein structures or genetic sequences. If a conflicting element has evolved an entirely new pathway to accomplish the same functional goals, this is termed functional mimicry [18]. Often, molecular mimics evolve similar protein structures as their target due to constraints on binding and interaction with other factors in the functional molecular network. Proteins engaging in structural mimicry share similar 3-dimensional conformations, despite not sharing extensive sequence similarity at the amino acid level. They arise convergently, from divergent elements that fold into conformations similar to the structure and activity of mimicked elements [7]. Compared to coevolving convergent functions and structures, sequence-based molecular mimicry provides a quick path to coopting target functions. Sequence-level mimicry can occur via single domains, multi-domain repeats, or full-length homology that translates to identical structures upon expression of those elements [5,18,19,20].
The expected distribution of molecular mimics across life depends on phylogenetic constraints, the complexity of the mimicked element, and the relationship between conflicting elements (i.e., intraspecific vs interspecific conflict). The gene content of every genome is structured by its phylogenetic history, with potential exceptions due to HGT. The infinite sites model of molecular evolution [21] predicts that each mutation will occur only once across evolutionary history because mutation rates are generally low enough to make a mutation at any given position exceedingly unlikely [21,22]. Although there are key examples of recurrent mutations at specific residues due to strong convergent selection [23,24,25]. Thus, stretches of high sequence identity between divergent genomes are more likely to be due to the reshuffling of genetic variation among genomes by recombination and HGT than by random mutations. Overall, these factors inform our expectations that the rate of convergent sequence-based mimic evolution to be inversely proportional to gene length in interspecific conflicts. These trends do not apply to intraspecific conflicts, as mimics can be created by sequence duplication events in one genome (discussed in Section 3). The frequency of structural and functional mimics may also be related to the length of the underlying genetic elements in interspecific conflicts, as the evolution of long elements may require more time and coevolutionary pressures than short elements to acquire the necessary substitutions.
Driver elements experience evolutionary pressures to mimic target functions, structures, or sequences, which stem from the conserved, endogenous motifs used by the target genome itself [26]. Endogenous molecular networks rely on conserved binding sites and signaling motifs to make efficient use of gene products across diverse molecular pathways. This conservation of interaction parameters enabled the evolution of diverse tissue types, developmental programs, and environmentally-driven phenotypic plasticity across multicellular eukaryotes [27,28,29]. However, it also selected for selfish elements that happened across conserved motifs, structures, or functions and could use them for their own advantage. Viruses and host-associated bacteria contain a high abundance of eukaryotic-like domains due to both convergence and HGT [18,20,30]. The HGT domains often function to mimic homologous elements in the target genome. For example, the eukaryotic-like sphingosine-phosphate lyase from Legionella pneumophila intracellular bacteria was horizontally transferred from one of its protozoan host ancestors and functions to inhibit macrophage autophagy [18].
In the next section, we detail examples of molecular mimicry in genetic conflicts at sequence, structural, and functional levels to assess whether the current empirical data support their expected distributions. These examples are organized by mechanistic impact on their target’s cell biology. Overall, we observe distinct molecular mimic distributions between interspecific and intraspecific conflicts that are consistent with theoretical expectations (summarized in Figure 1F).
3. Empirical examples of molecular mimicry in nature
3.1. Sequence-Based Mimics
The evolution of sequence-based molecular mimicry is largely structured by selection for convergent short motifs, full sequence duplications, and elements that have undergone HGT. Convergent mimicry of short motifs is a common trend among viruses, pathogenic bacteria, and cancerous cell lineage proteins [19,31,32]. Whereas, convergent full-length sequence mimics are more common in intraspecific conflicts [33,34,35]. The number of substitutions required to reach high sequence identity bound these distributions. Full-length homology between a mimic and a divergent target’s nucleic or amino acid sequence suggests that the driving element arose via HGT, and subsequently coevolved for interspecific conflict [5].
3.1.1. Recurrently Evolved SLiM Ligand Mimics Hijack Eukaryotic Cell Biology
Eukaryotic proteomes leverage short linear motifs (SLiMs) that are three to 15 residues long for protein-protein interactions including signaling, post-translational modifications (PTMs), and localization. These motifs are highly conserved, but are surrounded by lower-conservation sequences that modify the specificity of SLiM-protein interactions. SLiMs often occur in intrinsically disordered regions of proteins, allowing the binding surfaces to be accessed [26,31]. These short elements, and their DNA and RNA precursors, enable genomes to leverage existing molecular networks to evolve new functions and phenotypic plasticity [39]. Indeed, the human proteome is estimated to encode more than 100,000 SLiMs [26]. These endogenous mimics potentially drive mimicry in genetic conflict indirectly through increased selection pressures for selfish elements to convergently evolve highly-utilized target/host motifs.
A common theme among bacterial pathogens seeking to modify eukaryotic cells is mimicry of the ligand RGD (arginine-glycine-aspartate) SLiM domain that binds to integrin, an essential host cell-adhesion receptor [32]. Integrin signaling induces actin polymerization to strengthen cellular adhesion to the bound substrate (Figure 2A). This is critical to normal tissue differentiation and is co-opted in many cancers for metastasis and tumor formation [40]. By recognizing the small RGD domain in various ligands, host integrin is able to bind many factors, including laminin, fibronectin, and tenascin glycoproteins. Selfish elements mimicking the integrin ligands include the gastrointestinal pathogen Helicobacter pylori, which manipulates host integrin signaling through a RGD motif in its CagL protein [41] (Figure 2B). CagL is an external component of the H. pylori type IV secretion system (T4SS) that induces CagA effector protein injection into host cells upon integrin binding to induce cellular differentiation [32,42]. The mammalian cell entry protein (Mce) Mce3C from Mycobacterium tuberculosis contains a RGD motif that binds β2 integrin to induce local actin rearrangements that permit bacterial invasion [37] (Figure 2C). In addition to the RGD motif, many of these proteins contain other motifs mimicking other extracellular matrix (ECM) interactions that modulate the outcome of integrin binding [41]. For example, the SARS-CoV-2 spike protein S1 contains an RGD motif that binds to host integrin and ACE2 receptors to induce cell spreading, adhesion, and proliferation [43,44].
Histone tail SLiM mimicry is a common trend among viruses and cancer cell lineages. These short, degenerate, fast-evolving sequences facilitate histone-protein binding to modulate chromatin compaction and transcriptional regulation [19,45]. Numerous viral proteins mimic histone motifs to hijack host gene expression, thereby repressing antiviral gene expression and upregulating their own replication [45]. The histone H3 lysine 4 (H3K4)-like motif of the Influenza non-structural protein 1 (NS1) binds to the human PAF1 transcription elongation complex (hPAF1C) to suppress transcriptional elongation [46]. The NS1 histone mimic is imperfect, as its methylation can not be reversed by histone H3K4 demethylase [6]. SARS-CoV-2 encodes at least two proteins that mimic host histone SLiMs, the ORF8 and envelope (E) proteins. ORF8 mimics host histone H3 ARKS (alanine, arginine, lysine, serine) motifs to disrupt epigenetic regulation, promoting chromatin compaction and increasing virus copy number [38] (Figure 3A,B). Mimicry of healthy histone tail binding is also leveraged by intraspecific conflicts, including cancer. Basal-like breast cancer cells overexpress the transcription activator Twist, which contains a histone H4 motif. Inappropriate twist expression mimics the epithelial-mesenchymal transition (EMT) pathway, which is required for altered cell-to-cell contacts in tumors and metastatic cells [47]. Mimicking an EMT transition is strongly selected in cancer because migration to new somatic niches enables cancer cells to increase their frequency in the population.
3.1.2. De novo Full-Length Sequence Mimics Arise from Gene Duplications
Intraspecific conflicts provide examples of full-length sequence mimicry that arose via gene duplication. Many of these duplication-based drivers operate via imperfect molecular mimicry of the original, functional element to obstruct its function. Gene duplications can result in inhibitory interactions between the original gene and its duplicate through dominant negative interactions [34] or RNA-interference (RNAi) from the negative-sense strand. If these duplicates are expressed in the appropriate tissues at the right times, these interactions can promote the transmission of the driving element and create endogenous genomic conflict de novo.
In Drosophila, many meiotic driver, responder, and suppressor elements arose from gene duplication events on autosomes or sex chromosomes [33] and some of these duplicated genes function as molecular mimics. Meiotic drive is an intraspecific conflict strategy by which a selfish chromosome biases its transmission into the gametes that will contribute genetic material to the next generation, either through disrupting segregation or poisoning target chromosomes. Drosophila melanogaster’s autosomal Segregation Distorter system is one of the best-studied examples of male meiotic drive that employs a driver-responder system. In normal gametogenesis, sperm chromatin must be unwound from large nuclear histones and repackaged with small protamines to fit into the small confines of the sperm head nucleus. Nuclear import of protamines and other cofactors requires high amounts of nuclear RAs-related Nuclear protein (Ran) bound to GTP (RanGTP), which is maintained by the RanGTPase Activating Protein (RanGAP). The selfish SD haplotype encodes a driver locus derived from a partial duplication of RanGAP (Sd-RanGAP), lacking the nuclear export signal and SUMO domain. This truncation traps Sd-RanGAP in the nucleus where it mimics RanGAP function in the wrong compartment: Sd-RanGAP catalyzes the hydrolysis of nuclear RanGTP, disrupting RanGTP/GDP gradients, and blocking transport through nuclear pore channels (NPCs) (Figure 3C,D). Excess RanGDP blocks nuclear envelope and heterochromatin formation, which explains how this single gene can disrupt both protamine import and chromatin repackaging during spermatogenesis, respectively [33,48]. This protein localizes to both spermatids during cellularization, but it is ineffective on the SD haplotype, which also encodes an insensitive responder locus. Although the mechanism is still being elucidated, it is clear the the tandem repeat length of the satellite responder locus underlies the severity of the target haplotype’s response to drive [49].
The D. melanogaster genome contains examples of gene duplication-derived mimics in intraspecific conflicts that are older than the SD system, and have reached fixation across populations. These include the Stellate (Ste) system, which is derepressed in hybrid matings between males lacking the Y-linked repressor element and females encoding the X-linked driving element. In normal spermatogenesis, the X-linked testis-specific regulatory beta subunit of casein kinase II (CkIIβ) controls casein kinase II (CkII) phosphorylation of Wnt pathway components. Extant D. melanogaster X chromosomes carry up to 400 partial copies of the N-terminus of CkIIβ (termed the Ste locus) that bind to the alpha subunit to produce crystalline structures in male premeiotic germ cells that prevent their development. These inhibitory mimic proteins are blocked by the expression of a Y-linked repressor locus, termed suppressor of stellate (Su(Ste)). This repressor is a duplication of Ste that contains a transposon insertion positioned downstream. This insertion event created a promoter that induces the transcription of the opposite strand of Su(Ste), leading to double-stranded RNA (dsRNAs) accumulation. These dsRNAs are cleaved into repeat-associated small RNAs (rasi-RNAs), which function in the piwi-interacting RNA pathway to inhibit Ste expression [50].
3.1.3. Horizontally Transmitted Full-Gene Mimics Are Repurposed for Conflict across Life
Despite the low likelihood of identical genes evolving multiple times by chance, many elements are well-distributed across genomes and taxa, indicating that HGT is a powerful force in shuffling genetic variation that can lead to molecular mimicry. Sequence-based molecular mimics of eukaryotic repeats and domains are found in many bacterial and viral genomes, including ankyrin repeats, leucine rich repeats, tetratricopeptide repeats, and pentatricopeptide repeats, SRC Homology 3 (SH3) domains, and RING domains [18,30]. Similarly, toxin-antitoxin (TA) systems likely evolved among mobile genetic elements, and have subsequently transferred to bacterial, archaeal, and eukaryotic chromosomes [51].
Receptor proteins enable cellular life to sense and respond to stimuli, but leave cells vulnerable to viral entry and manipulation. The Vibrio cholerae genome encodes the cytoplasmic receptor-transcription factor Vibrio quorum modulator A (VqmAVC) that binds the quorum sensing autoinducer ligand 3,5-dimethylpyrazin-2-ol (DPO). DPO binding to VqmAvc, enables V. cholerae to sense and respond to population size changes. At high V. cholerae densities, excess DPO binding suppresses the expression of virulence effectors and biofilm factors. The lysogenic temperate vibriophage of V. cholerae VP882 also encodes a copy of VqmA, termed VqmAphage, that it acquired through a HGT event. Phage-encoded VqmAphage binds DPO, induces the viral lytic program at peak bacterial densities [52]. Thus, through acquisition of a bacterial quorum sensing receptor by HGT, vibriophage are able to mimic host density-dependent growth and transmission responses.
TA systems are excellent examples of highly effective, compact, and versatile elements for controlling cellular outcomes that are present at high frequencies in mobile elements such as transposons and plasmids. These two-element systems consist of a protein-encoded toxin and an antitoxin that is an RNA or protein that directly or indirectly neutralizes the toxin [53]. TA systems induce plasmid addition and virally-induced abortions in bacteria and post-segregational killing of cells that lack the antidote in bacteria and eukaryotes [51]. While TA systems are common players in genomic conflicts, molecular mimics are more common in suppression mechanisms than in the TA systems themselves. For example, the bacterium Pectobacterium atrosepticum hosts bacteriophage in the family Myoviridae and encodes a TA system to induce altruistic suicide of virus-infected clonal lineages. However, the bacteriophage acquired the RNA-based antidote gene from the TA system by HGT, and uses it to mimic the host’s antitoxin activity to repress suicide, permitting its replication and lysis from host cells [53,54]. Similarly, in Schizosaccharomyces pombe fungi, a TA system is employed by selfish chromosomes to poison spore haplotypes lacking the antidote gene. Through non-allelic gene conversion, a non-selfish chromosome acquired the protein-based antidote, endowing it with toxin resistance through antidote mimicry [55].
3.2. Structural Mimics
Similar protein structures can be encoded by highly divergent amino acid sequences, enabling convergent evolutionary routes to shared molecular functions. Viruses and pathogenic bacteria have explored this structural space in their evolution, producing a plethora of novel strategies for mimicking host molecular interactions [8]. However, eukaryotic hosts may be more restricted in the structures they can evolve, as protein mimics can be antigenic and induce autoimmunity or coevolutionary responses [5,56]. Thus, intraspecific selfish element structural mimicry may select for proteins that reappropriate functions without triggering cellular or humoral immunity.
Structural mimicry also enables genetic elements in conflict to diverge from their own endogenous interaction networks, while converging on the function of their target. This strategy simultaneously enables mimicked function while preventing the disruption of existing essential networks in the conflicting genome [57]. Examples of structural mimics have been known for decades, despite the difficulty in detecting them prior to accurate ab initio protein structure prediction tools, through protein crystal structures and binding data. Recent advances in protein structure and interaction predictions are enabling the discovery of many more structural mimics, both across genomes and taxa [17,58,59].
3.2.1. Repurposed Ancient Domains for Molecular Mimicry
Ancient, conserved molecular machinery essential to housekeeping functions provides a slow-moving target for genetic conflict to co-opt function. For example, both bacteria and eukaryotes use PTMs, such as phosphate, to modulate the activity of their enzymes and signaling proteins. Through employing their own PTMs on host proteins, selfish elements can induce large downstream impacts on host signaling pathways. In contrast to the small motifs that trigger PTMs on bacterial and viral effector proteins by eukaryotic enzymes, larger repeats and domains are required for exogenous elements to conjugate PTMs onto eukaryotic factors [32].
Protein tyrosine phosphatase (PTP) domains catalyze the removal of phosphate from activated residues, are present in all domains of life, and have been leveraged in genetic conflicts. For example, PTP domains are encoded on Salmonella’s SptP and Yersinia’s YopH bacterial effector proteins. These effectors are injected into the host cytoplasm through protein secretion systems, where they dephosphorylate host proteins to restore cellular homeostasis and prevent phagocytosis, respectively [60,61]. Some viruses encode type III dual function PTP domains, which can dephosphorylate activated serine and threonine residues, in addition to tyrosine. Overall the structures of these enzymes strongly resemble their eukaryotic targets [60], which likely reflects both deep conservation of PTP function as well as convergence on the host structural conformation.
3.2.2. Convergent Structural Mimics among Membrane-Bound and Secreted Effector Proteins
Ligand structural mimicry enables genetic elements in conflict to bind target receptors in order to block immune activation, invoke cellular signaling, and gain access to targeted cells. As discussed above, eukaryotic extracellular matrix (ECM) proteins bind to integrin and other cell surface receptors to induce normal development and cellular maintenance. Structural mimicry of ECM binding surfaces is a common strategy leveraged by intraspecific selfish elements to promote internalization, and provides an alternative mechanism to sequence-based RGD-motif mimicry. For example, the Yersinia pseudotuberculosis bacterial Invasin protein is a membrane-embedded trimeric autotransporter adhesin that mimics the structure of eukaryotic integrin αv to bind to integrin 𝛽1 for cell surface attachment. Similarly, West Nile Virus glycoprotein E mimics the fibronectin FN10 domain for integrin αV𝛽3 binding [15]. Through searching proteomic databases for potential binding partners of the coronavirus spike protein receptor binding motifs, a new epidermal growth factor (EGF) receptor binding motif was discovered that mimics host receptors and may determine cellular tropism [62].
Cell surface glycan modifications are commonly mimicked among cancer cell lineages, viruses, and bacterial toxins. In non-selfish in vivo cell biology, glycan modifications play significant roles in cellular adhesion and signaling cascades that impact differentiation, metabolism, immunity, and other processes [63]. Loss of cell surface glycans enables cancer cells to evade immune detection. Glycan co-option bestows different metastatic lineages with the ability to colonize new tissues and form tumors [64]. Bacterial pathogens such as Campylobacter jejuni, Neisseria species, H. pylori, and some E. coli strains possess cell walls composed of lipooligosaccharides (LOS), lipopolysaccharides (LPS), or capsular polysaccharides (CPS) decorated with carbohydrate modifications that mimic eukaryotic lectin-binding glycans. For example, an altered glycan linkage in Neisseria meningitidis serogroup B capsules enables recruitment of host factor H, which is required to deactivate the immune complement pathway induced by bacterial infection. In Neisseria gonorrhoeae, LOS with broken sialic acid linkages bind to the lactosamine residues of eukaryotic asialoglycoprotein receptors, triggering clathrin-mediated cell entry. The O-antigen of H. pylori’s LPS membrane is a mimic of Lewis blood group antigens, which enables colonization and invasiveness [65]. In viruses, glycan mimicry often takes the form of a glycan shield that enables immune evasion. For example, HIV envelope proteins and Influenza A hemagglutinin glycoproteins add host-derived N-linked glycans near critical residues to sterically block neutralizing antibody binding [66] (Figure 4A). Enveloped viruses are able to mimic apoptosis cues by collecting phosphatidylserine from organelle membranes and displaying it to receptors such as T cell immunoglobulin and mucin receptor 1 (TIM1) to trigger uptake by phagocytic cells [67].
Vesicle traffic is vital for cellular homeostasis and is commonly manipulated in interspecific conflicts. Through the regulation of membrane budding, cytoskeletal transport, and fusion with cellular organelles, vesicle traffic plays vital roles in many cellular processes. SNARE (soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor) proteins mediate and regulate vesicle fusion with target membranes. Emerging evidence indicates that diverse pathogens, including Chlamydia and Legionella, use SNARE protein mimicry to prevent fusion with autophagosomes, expand the replicative niche, and control membrane traffic (Figure 4B). SNARE proteins consist of transmembrane domains connected with linker sequences to coiled-coil motifs that enable membrane fusion. Based on the sequence and structural diversity among Legionella LegC SNARE mimics, and their ability to complement the loss of endogenous SNAREs, the fusion mechanism appears to be highly permissible to structural variation [68]. LegC fusion with liposome vesicles containing the endosomal arginine (R)-SNARE vesicle-associated membrane protein 4 (VAMP4), providing extra membrane and cytosolic resources for bacterial niche expansion. However, LegC is an imperfect SNARE mimic. Once LegC fuses with the VAMP4 protein, it cannot be disassembled by the disassembly machinery and rendered inactive.
Many different actin and microtubule-associated proteins are mimicked by pathogenic bacteria and viruses for movement within a eukaryotic cell [69]. Actin nucleation proteins, such as the Wiskott-Aldrich syndrome (WASP) family of proteins, are essential for building and recycling the cytoskeleton and vesicles. Microtubule-associated proteins permit long-range directed transport of cellular cargos in normal cell biology. Intraspecific conflicts have evolved mimics for both of these processes. The membrane-associated ActA protein from Listeria monocytogenes is a structural mimic of WASP actin nucleation proteins, which enables the bacteria to polymerize actin, propelling themselves from cell to cell. Following phosphorylation by host casein kinase II (CK2), ActA interacts with the actin-related protein (ARP) 2/3 complex through a region that displays structural similarity to the C region of the VCA domain of WASP proteins to induce polymerization. While these phosphorylation sites are shared among divergent WASP proteins, the rest of the protein is unique to L. monocytogenes, indicating that this mimic evolved by convergence [70]. The microtubule cytoskeleton also offers opportunities for interspecific conflicts to co-opt cellular transport: vaccinia virus encodes a linker protein mimic that enables it to commandeer host Kinesin heavy chain motor proteins for transport to the plasma membrane from the cytoplasm for cell exit (Figure 4C). Similarly, HIV’s capsid protein contains a region that is a structural mimic of the microtubule-associated End-binding1 (Eb1) protein, enabling it to bind to Cytoplasmic linker protein-170 (CLIP-170) for dynein-mediated minus-ended microtubule transport to the nucleus [69].
3.3. Functional Mimics
The molecular armaments of genetic elements in conflict do not need to share sequence or structural similarity with the factors they are mimicking to hijack cellular functions. This functional mimicry is likely the only available strategy for some element types. For example, a sequence-based or structural mimic may not be possible for endogenous proteins that leverage intrinsically disordered regions for dynamic interactions. Importantly, mimic categories are not mutually exclusive. The bacterial CagA protein from H. pylori mimics a tyrosine phosphorylation SLiM motif at the sequence level for proximal molecular interactions, but mimics a Grb2-associated binder (Gab) family adapter protein at the functional level for signaling consequences [36]. Thus, more examples of functional mimicry will likely be reported as the phenotypic consequences of genetic conflict are fully characterized in different systems.
3.3.1. Convergent Charge Distributions for DNA Mimicry
More than a dozen independently evolved bacterial and viral proteins have been reported to mimic host DNA to subvert protein-DNA binding. DNA-binding proteins are required by cellular genomes to regulate genome replication, gene expression, and epigenetic modifications. These binding proteins have evolved to recognize DNA phosphate backbones generally or a sequence of nucleotide residues specifically [71]. Proteins that functionally mimic DNA binding do so by evolving helical strands of negatively charged phosphate residues, with one or two rows of either aspartic acid or glutamic acid residues. The majority of viral DNA mimics are for interspecific conflict: they inhibit host proteins from targeting invading genomes. For example, bacteriophage T7 uses its Overcome classical restriction (Ocr) protein to block the host type I restriction modification (R/M) system from cleavage. Ocr proteins dimerize to form a curved structure that mimics the angle of bent B-form DNA and its negatively charged surface to bind and block the R/M enzyme [72].
3.3.2. Horizontally Transferred Extracellular Matrix Functional Mimics
Selection for extracellular ligand mimics is strong in interspecific conflicts, resulting in the evolution of functional ligand mimics, in addition to the sequence and structural ligand mimics discussed previously. The adherens junctions is an emergent structural complex created through protein-protein binding between adjacent cells. Binding proteins include the transmembrane glycoprotein E-cadherin and intracellular components including p120-catenin, β-catenin and α-catenin. In normal adherens junctions, repeating E-cadherin subunits bind themselves across the junction (Figure 2A). These cellular adhesion structures not only provide scaffolding and structure for cells, they communicate environmental signals and mediate cooperative cellular behaviors, such as epithelial barrier formation and maintenance [73,74]. The food-borne pathogen L. monocytogenes uses its internalin protein InlA as a functional mimic of E-cadherin to gain entry to the host cell. LnlA binding to E-cadherin triggers junction recycling through clathrin or caveolin-mediated endocytosis, engulfing the bacteria in the process [75] (Figure 4D). The InlA protein evolved as a novel structure consisting of fifteen leucine-rich repeats (LRRs) surrounding an inter-repeat region that binds E-cadherin, which arose by ancient horizontal gene transfer, followed by divergence and high rates of recombination among Listeria strains [76].
3.3.3. Bacteria Convergently Evolved a Functional Mimic System for Modifying Ubiquitination
Some host-associated bacteria have evolved a structurally and mechanistically novel set of proteins to leverage eukaryotic ubiquitination pathways for their own use, illustrating deep convergent functional mimicry.
Post-translational protein ubiquitination for signaling and protein-turnover is a eukaryotic innovation that is not employed by bacteria [77]. Despite this, two novel families of bacterial effector ligases, the HECT-like and NEL families of E3 Lys48-specific ligases from Salmonella’s SopA and Shigella’s IpaH9.8 proteins, can polyubiquitinate host inflammatory proteins. Novelty often makes these ubiquitin linkages irreversible because host proteins cannot cleave them. Ubiquitin cleavage is also leveraged by bacterial functional mimics to block immune signaling. For example, Legionella uses its effector transglutaminase MvcA to catalyze the non-canonical deubiquitination of the E2 ubiquitin-conjugating enzyme UBE2N, blocking its activity, preventing it from ubiquitinating bacterial proteins [78]. Given that these bacterial-evolved strategies of modifying ubiquitin signaling are entirely novel and interact with elements of eukaryotic ubiquitination pathways that are still poorly understood, such as through Lys6 polyubiquitin linkages, study of pathogenic mechanisms will reveal endogenous mechanisms [61].
4. Detecting molecular mimicry in genetic conflict computationally
Detecting evidence of genetic conflict and molecular mimicry computationally requires robust coevolutionary theory, ample empirical data, and well-developed interaction models (Figure 5, Supplemental Figures S1 and S2). While molecular mimics have been sought through sequence-based data mining for at least 30 years [11,61], advancements in protein structural prediction models recently enabled screens for mimics at the structure level [8]. In silico approaches for screening for genetic conflict and molecular mimicry are important because they expand our knowledge of the distribution and frequency of elements among taxa and the likelihood for mimicry to evolve. Indeed, expanded knowledge of protein domain distributions among bacteria corrected the assumption that bacteria lack protein tyrosine phosphatase domains; now it is well understood that they arose in bacteria, and not by horizontal gene transfer [61].
4.1. Detecting Sequence-Level Mimics
Molecular mimic identification at the sequence level is a powerful approach for identifying selfish genetic elements that may encode the ability to co-opt the cell biology of their target [31,32]. Indeed, these approaches have identified tapeworm candidate mimics of stickleback proteins [79], as well as malaria, filarial nematode, and Legionella mimic candidates of human proteins [12,80]. Sequence-level mimics of target elements can be detected at the gene, domain, or motif level, either among protein, RNA, or DNA sequences, depending on the mimic type, degree of relatedness, and functional sequence requirement. Therefore, motif, domain, and BLAST searches of candidate sequence-level mimics should be performed as a first-pass search in conflict-driven molecular mimic screens. However, there is a lower limit on the length of sequence mimicry that can be detected bioinformatically. For example, PTM-triggering sequences are likely too small and heterogeneous outside of the modified residue to detect bioinformatically [26].
Databases for short motifs, repeats, and domains exist, which can be leveraged to look for shared sequences between conflicting elements and their targets. The wealth of genomic resources made available by next-generation genome sequencing and decades of functional assays enabled researchers to inventory the molecular interaction modules that manifest organismal phenotypes in the Eukaryotic Linear Motif (ELM) resource [31]. Using the ELM prediction tool, selfish mimics of host protein SLiMs can be predicted in conflicting genomes [81]. Mimic proteins that travel through nuclear pore complexes require nuclear localization signals (NLSs) [82], which exhibit a variety of forms. NLSs can be predicted from amino acid sequences by hidden markov models or machine learning approaches [83]. In contrast to NLSs, nuclear export signals (NESs) only share leucine-rich regions, and may be better predicted structurally.
Genomic surveys for large structural changes, such as duplicated genes, tandem repeats, mobile element insertions, and novel chromosomes or plasmids, can be employed to detect endogenous selfish elements that may use molecular mimicry to achieve a transmission advantage. For example, gene duplications are one of the hallmarks of genetic conflict in meiotic drive systems [33], and can be easily detected by deviations in reference re-sequencing depth [34]. Toxin-antitoxin genes often evolve on plasmids and spread via transposable elements and viruses to chromosomes, making their sequence-based detection relatively simple through alignment to toxin and mobile element databases and ab initio repeat finders [84]. As antidote-mimicking resistance loci spread by horizontal gene transfer and gene conversion, genomes should also be screened for partial TA elements. Accessory chromosomal sequences, such as B chromosomes, that may encode mimic elements can be identified by their variable presence among tissues and their divergence relative to the endogenous chromosomes [4].
4.2. Detecting Structure-Level Mimics
The advent of accurate protein structural prediction models such as AlphaFold [85,86] enabled whole proteome screens for structural mimic candidates, ab initio. Protein structural predictions and structural alignment algorithms have been in development for decades, and have been instrumental in identifying similar protein structures between pathogens and hosts [15]. Structural mimic candidates are recognizable through 1) alignment between mimic and target protein structures [87], 2) interactions between the mimic and its interacting partners [16], and 3) competitive interactions between the mimic and targets for the interacting partners (e.g., [17,59]). Through combining these well-developed conceptual frameworks with recent innovations in structural prediction, more structural mimics can be identified.
Regional structural mimic prediction was useful for understanding the evolution of host tissue infection affinities, i.e., tropisms, in the rapidly evolving viral SARS-CoV2 pandemic. In addition to the integrin-binding fibronectin-like RGD motif, the coronavirus spike glycoprotein also contains a receptor binding motif (RBM) that modulates viral cellular tropism. Structural prediction and alignment allowed SARS-CoV2 researchers to identify host protein structures that are similar to viral spike protein RBMs, resulting in highly specific epidermal growth factor (EGF)-like hits that bind similarly to the factors they mimic in docking simulations [58]. Many other regional structures could be mined for in viral and bacterial genomic data through receptor-informed reverse searches for mimicked ligand binding surfaces, and other specific interaction predictions.
While structural protein predictions are now quite reliable for many secondary and tertiary structures, intrinsically disordered domains [88] and genome-wide protein-protein docking interactions are still difficult to predict [16]. Disordered proteins may mediate imperfect mimicry mechanisms through their conformational flexibility and SLiMs enrichment [26]. So, this subset of the proteome should be further mined for motif-based mimics, such as histone mimics [45]. Signatures of molecular mimicry have been detected in aggregate across conflicting genomes through interaction predictions [8], suggesting that at least some mimicked interactions can be detected ab initio.
4.3. Detecting Function-Level Mimics
Functional mimicry is far more difficult to detect from individual genomic datasets than sequence or structural mimics because predictions about the nature of mimicry need to be identified experimentally before they can be linked to their genetic determinants. Fortunately, genetic conflict leaves marks on populations that can be leveraged to detect unknown elements and mechanisms. The short and long-term consequences of selfish element proliferation and transmission can be seen through phenomena such as repetitive element expansions, skews in allele frequencies, changes in chromosome number, and alterations in sex ratio [3]. Correlated coevolutionary patterns, such as accelerated evolution or constraint, between genes encoded in interacting genomes can also be leveraged to detect signals of genetic conflict and mimicry.
Through leveraging genome-wide signals of biased genetic transmission, recent studies have detected evidence of genetic conflicts that were unresolvable with smaller datasets. Through a scan across thousands of publically available eukaryotic genomes, transposable element expansions were shown to drive intron gain in diverse eukaryotes, from basal protists to copepods [89]. By leveraging bulk gamete genome sequencing and comparing the parental allele frequencies to those of somatic tissue, researchers have been able to detect skews in parental allele frequency transmission in Arabidopsis hybrid plants that are due to unknown segregation distortion mechanisms [90]. Using a similar approach, Zea mays ssp. mexicana was recently shown to encode a male-specific segregation distorter that functions through RNAi-mediated targeting of an essential pollen lipase [91]. While these results do not resolve mechanistic details about the nature of the genetic elements in conflict or mimicry, they do provide candidate loci for further experimental investigation.
Advances in multi-omics approaches enable the identification of specific interacting factors between conflicting genomes in medium to high-throughput. If conflicting genomes are known a priori, then patterns of correlated coevolution can be investigated through phylogenetic branch length tests. Tests such as this have been used to detect molecular mimicry in sexual conflicts between abalone egg coat proteins and their sperm lysin binding partners, which trigger vitelline envelope breakdown and fertilization [92]. Targeted assays for post-translational modification mimics, such as lectin microarrays to assay glycan binding, can identify structural mimics of non-protein factors [93]. If epitopes and binding surfaces are known, targeted structural searches and in silico docking assays can be run on the conflicting genome to identify candidate binding partners among the expressed ligands, receptors, and other proteins.
5. Conclusion
Broad knowledge about the distribution and occurrence of molecular mimics across known intraspecific and interspecific genetic conflicts informs on the likelihood of these strategies evolving in newly discovered interactions. Recent advances in database mining, evolutionary inference, and protein structure prediction offer new strategies for identifying molecular mimics in genetic conflict computationally. Here, we linked empirical data from these limited, yet growing, datasets with theoretical predictions about the likelihood of convergent evolution to show: In interspecific conflicts deploying molecular mimicry, convergence drives the evolution of short genetic elements for sequence-based mimicry. Longer interspecific sequence mimics are generally obtained through HGT. Structural and functional mimics exhibit more variable trends that are likely attributed to the duration and strength of selection pressures on the coevolved function (e.g., short ligand-binding sequence mimics versus long ligand structural mimics). Importantly, functional and structural convergence in interspecific conflicts may be ancient. For example, deeply rooted bacterial protein tyrosine phosphatases and ubiquitin ligases coevolved to mimic eukaryotic processes [60,61,78]. These trends do not hold for intraspecific conflicts that leverage molecular mimicry because the close relationship between driver and target enables sequence duplication events to drive the rapid evolution of any length mimic.
While an impressive number of molecular mimics are known to play a role in genetic conflicts, our knowledge about their taxonomic distribution is still limited by the availability of genomic data and tractable systems for functional testing. With machine-learning approaches improving our ability to predict patterns from observed data, our capacity to detect mimicry at the DNA, RNA, protein, and post-translational levels is likely to improve rapidly. Once we have a better sense of the global distribution of molecular mimics, these phenomena can be explored for emergent properties suggestive of common selective regimes and adaptive potential. For example, mimicry rings, in which divergent lineages converge on a similar mimicry pattern due to similar selective pressures [5], may exist for molecules and shape taxon-specific pressures on host sequence and structural variation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
SLR: study conception and design, literature analysis, figure illustration, and writing. GP: literature analysis and writing CC: literature analysis and writing.
Acknowledgments
This work was supported by UC Santa Cruz, NIH R00GM135583 to SLR, a UCSC Cota-Robles Fellowship and R35GM128932 to (RBC) provided support for CC, NIH T32HG012344 to GP.
References
- Werren, J.H. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc. Natl. Acad. Sci. 2011, 108, 10863–10870. [Google Scholar] [CrossRef]
- Massey, S.E.; Mishra, B. Origin of biomolecular games: deception and molecular evolution. J. R. Soc. Interface 2018, 15, 20180429. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.N.; Malik, H.S. Genetic conflicts: the usual suspects and beyond. J. Exp. Biol. 2017, 220, 6–17. [Google Scholar] [CrossRef]
- Ågren, J.A.; Clark, A.G. Selfish genetic elements. PLOS Genet. 2018, 14, e1007700. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Malik, H.S. The evolutionary conundrum of pathogen mimicry. Nat. Rev. Microbiol. 2009, 7, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Liu, Y.; Tempel, W.; Eram, M.S.; Bian, C.; Liu, K.; Senisterra, G.; Crombet, L.; Vedadi, M.; Min, J. Structural basis for histone mimicry and hijacking of host proteins by influenza virus protein NS1. Nat. Commun. 2014, 5, 3952. [Google Scholar] [CrossRef]
- Aravind, L.; Iyer, L.M.; Burroughs, A.M. Discovering Biological Conflict Systems Through Genome Analysis: Evolutionary Principles and Biochemical Novelty. Annu. Rev. Biomed. Data Sci. 2022, 5, 367–391. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Burgas, M.T. Bacteria use structural imperfect mimicry to hijack the host interactome. PLOS Comput. Biol. 2020, 16, e1008395. [Google Scholar] [CrossRef]
- Holen, H.; Johnstone, R.A. The Evolution of Mimicry under Constraints. Am. Nat. 2004, 164, 598–613. [Google Scholar] [CrossRef]
- Alcami, A. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Galán, J.E. Structural mimicry in bacterial virulence. Nature 2001, 412, 701–705. [Google Scholar] [CrossRef]
- Burstein, D.; Zusman, T.; Degtyar, E.; Viner, R.; Segal, G.; Pupko, T. Genome-Scale Identification of Legionella pneumophila Effectors Using a Machine Learning Approach. PLOS Pathog. 2009, 5, e1000508. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, C.; Miao, Y.; Gao, J.; Xu, D. Effector prediction in host-pathogen interaction based on a Markov model of a ubiquitous EPIYA motif. BMC Genom. 2010, 11, 1–15. [Google Scholar] [CrossRef]
- Doxey, A.C.; McConkey, B.J. Prediction of molecular mimicry candidates in human pathogenic bacteria. Virulence 2013, 4, 453–466. [Google Scholar] [CrossRef]
- Drayman, N.; Glick, Y.; Ben-Nun-Shaul, O.; Zer, H.; Zlotnick, A.; Gerber, D.; Schueler-Furman, O.; Oppenheim, A. Pathogens Use Structural Mimicry of Native Host Ligands as a Mechanism for Host Receptor Engagement. Cell Host Microbe 2013, 14, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Muthye, V.; Wasmuth, J.D. Proteome-wide comparison of tertiary protein structures reveals molecular mimicry in Plasmodium-human interactions. Front. Parasitol. 2023, 2, 1162697. [Google Scholar] [CrossRef]
- Mondino, S.; Schmidt, S.; Buchrieser, C. Molecular Mimicry: a Paradigm of Host-Microbe Coevolution Illustrated by Legionella. mBio 2020, 11. [Google Scholar] [CrossRef]
- Davey, N.E.; Travé, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar] [CrossRef]
- Frank, A.C. Molecular host mimicry and manipulation in bacterial symbionts. FEMS Microbiol. Lett. 2019, 366. [Google Scholar] [CrossRef]
- Ma, J.; Ratan, A.; Raney, B.J.; Suh, B.B.; Miller, W.; Haussler, D. The infinite sites model of genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 14254–14261. [Google Scholar] [CrossRef] [PubMed]
- Harpak, A.; Bhaskar, A.; Pritchard, J.K. Mutation Rate Variation is a Primary Determinant of the Distribution of Allele Frequencies in Humans. PLOS Genet. 2016, 12, e1006489. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.L. The genetic causes of convergent evolution. Nat. Rev. Genet. 2013, 14, 751–764. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Giulieri, S.G.; Guérillot, R.; Duchene, S.; Hachani, A.; Daniel, D.; Seemann, T.; Davis, J.S.; Tong, S.Y.; Young, B.C.; Wilson, D.J.; et al. Niche-specific genome degradation and convergent evolution shaping Staphylococcus aureus adaptation during severe infections. eLife 2022, 11. [Google Scholar] [CrossRef]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A Million Peptide Motifs for the Molecular Biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef]
- Kajala, K.; Gouran, M.; Shaar-Moshe, L.; Mason, G.A.; Rodriguez-Medina, J.; Kawa, D.; Pauluzzi, G.; Reynoso, M.; Canto-Pastor, A.; Manzano, C.; et al. Innovation, conservation, and repurposing of gene function in root cell type development. Cell 2021, 184, 3333–3348.e19. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.R.; Jolma, A.; Yin, Y.; Morgunova, E.; Kivioja, T.; Akhtar, J.; Hens, K.; Toivonen, J.; Deplancke, B.; Furlong, E.E.M.; et al. Conservation of transcription factor binding specificities across 600 million years of bilateria evolution. eLife 2015, 4, e04837. [Google Scholar] [CrossRef]
- Li, S.; Dohlman, H.G. Evolutionary conservation of sequence motifs at sites of protein modification. J. Biol. Chem. 2023, 299, 104617. [Google Scholar] [CrossRef]
- E Martyn, J.; Gomez-Valero, L.; Buchrieser, C. The evolution and role of eukaryotic-like domains in environmental intracellular bacteria: the battle with a eukaryotic cell. FEMS Microbiol. Rev. 2022, 46. [Google Scholar] [CrossRef]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Mészáros, B.; Sámano-Sánchez, H.; Zeke, A.; Dobson, L.; Lazar, T.; Örd, M.; Nagpal, A.; et al. The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res. 2021, 50, D497–D508. [Google Scholar] [CrossRef] [PubMed]
- Sámano-Sánchez, H.; Gibson, T.J. Mimicry of Short Linear Motifs by Bacterial Pathogens: A Drugging Opportunity. Trends Biochem. Sci. 2020, 45, 526–544. [Google Scholar] [CrossRef] [PubMed]
- Courret, C.; Chang, C.-H.; Wei, K.H.-C.; Montchamp-Moreau, C.; Larracuente, A.M. Meiotic drive mechanisms: lessons fromDrosophila. Proc. R. Soc. B: Biol. Sci. 2019, 286, 20191430. [Google Scholar] [CrossRef]
- Levasseur, A.; Pontarotti, P. The role of duplications in the evolution of genomes highlights the need for evolutionary-based approaches in comparative genomics. Biol. Direct 2011, 6, 11. [Google Scholar] [CrossRef]
- A Vogan, A.; Ament-Velásquez, S.L.; Granger-Farbos, A.; Svedberg, J.; Bastiaans, E.; Debets, A.J.; Coustou, V.; Yvanne, H.; Clavé, C.; Saupe, S.J.; et al. Combinations of Spok genes create multiple meiotic drivers in Podospora. eLife 2019, 8. [Google Scholar] [CrossRef]
- Botham, C.M.; Wandler, A.M.; Guillemin, K. A Transgenic Drosophila Model Demonstrates That the Helicobacter pylori CagA Protein Functions as a Eukaryotic Gab Adaptor. PLOS Pathog. 2008, 4, e1000064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Li, B.; Wang, J.; Liu, C.H. Mycobacterium tuberculosisMce3C promotes mycobacteria entry into macrophages through activation of β2 integrin-mediated signalling pathway. Cell. Microbiol. 2017, 20, e12800. [Google Scholar] [CrossRef]
- Kee, J.; Thudium, S.; Renner, D.M.; Glastad, K.; Palozola, K.; Zhang, Z.; Li, Y.; Lan, Y.; Cesare, J.; Poleshko, A.; et al. SARS-CoV-2 disrupts host epigenetic regulation via histone mimicry. Nature 2022, 610, 381–388. [Google Scholar] [CrossRef]
- Davey, N.E.; Cyert, M.S.; Moses, A.M. Short linear motifs – ex nihilo evolution of protein regulation. Cell Commun. Signal. 2015, 13, 1–15. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Conradi, J.; Tegtmeyer, N.; Woźna, M.; Wissbrock, M.; Michalek, C.; Gagell, C.; Cover, T.L.; Frank, R.; Sewald, N.; Backert, S. An RGD Helper Sequence in CagL of Helicobacter pylori Assists in Interactions with Integrins and Injection of CagA. Front. Cell. Infect. Microbiol. 2012, 2, 70. [Google Scholar] [CrossRef]
- Hayashi, T.; Morohashi, H.; Hatakeyama, M. Bacterial EPIYA effectors - Where do they come from? What are they? Where are they going? Cell. Microbiol. 2012, 15, 377–385. [Google Scholar] [CrossRef]
- Norris, E.G.; Pan, X.S.; Hocking, D.C. Receptor-binding domain of SARS-CoV-2 is a functional αv-integrin agonist. J. Biol. Chem. 2023, 299, 102922. [Google Scholar] [CrossRef] [PubMed]
- Simons, P.; Rinaldi, D.A.; Bondu, V.; Kell, A.M.; Bradfute, S.; Lidke, D.S.; Buranda, T. Integrin activation is an essential component of SARS-CoV-2 infection. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, U.; Ho, J.S.; Prinjha, R.K.; Tarakhovsky, A. The "Histone Mimicry" by Pathogens. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 81–90. [Google Scholar] [CrossRef]
- Marazzi, I.; Ho, J.S.Y.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Gingell, L.F.; McLean, J.R. A Protamine Knockdown Mimics the Function ofSdinDrosophila melanogaster. G3 Genes|Genomes|Genetics 2020, 10, 2111–2115. [Google Scholar] [CrossRef]
- Herbette, M.; Wei, X.; Chang, C.-H.; Larracuente, A.M.; Loppin, B.; Dubruille, R. Distinct spermiogenic phenotypes underlie sperm elimination in the Segregation Distorter meiotic drive system. PLOS Genet. 2021, 17, e1009662. [Google Scholar] [CrossRef]
- Malone, C.D.; Lehmann, R.; Teixeira, F.K. The cellular basis of hybrid dysgenesis and Stellate regulation in Drosophila. Curr. Opin. Genet. Dev. 2015, 34, 88–94. [Google Scholar] [CrossRef]
- Burga, A.; Ben-David, E.; Kruglyak, L. Toxin-Antidote Elements Across the Tree of Life. Annu. Rev. Genet. 2020, 54, 387–415. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell 2018, 176, 268–280.e13. [Google Scholar] [CrossRef]
- Jurėnas, D.; Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Biology and evolution of bacterial toxin–antitoxin systems. Nat. Rev. Microbiol. 2022, 20, 335–350. [Google Scholar] [CrossRef]
- Blower, T.R.; Evans, T.J.; Przybilski, R.; Fineran, P.C.; Salmond, G.P.C. Viral Evasion of a Bacterial Suicide System by RNA–Based Molecular Mimicry Enables Infectious Altruism. PLOS Genet. 2012, 8, e1003023. [Google Scholar] [CrossRef] [PubMed]
- Núñez, M.A.B.; Lange, J.J.; Zanders, S.E. A suppressor of a wtf poison-antidote meiotic driver acts via mimicry of the driver’s antidote. PLOS Genet. 2018, 14, e1007836. [Google Scholar] [CrossRef]
- English, J.; Patrick, S.; Stewart, L.D. The potential role of molecular mimicry by the anaerobic microbiota in the aetiology of autoimmune disease. Anaerobe 2023, 80, 102721. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Burgas, M.T. Bacteria use structural imperfect mimicry to hijack the host interactome. PLOS Comput. Biol. 2020, 16, e1008395. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, C.A.; Jamasb, A.R.; Alsulami, A.F.; Copoiu, L.; van Tonder, A.J.; Hala, S.; Bannerman, B.P.; Thomas, S.E.; Vedithi, S.C.; Torres, P.H.; et al. Predicted structural mimicry of spike receptor-binding motifs from highly pathogenic human coronaviruses. Comput. Struct. Biotechnol. J. 2021, 19, 3938–3953. [Google Scholar] [CrossRef] [PubMed]
- Lasso, G.; Honig, B.; Shapira, S.D. A Sweep of Earth’s Virome Reveals Host-Guided Viral Protein Structural Mimicry and Points to Determinants of Human Disease. Cell Syst. 2020, 12, 82–91.e3. [Google Scholar] [CrossRef]
- Böhmer, F.; Szedlacsek, S.; Tabernero, L.; Östman, A.; Hertog, J.D. Protein tyrosine phosphatase structure–function relationships in regulation and pathogenesis. FEBS J. 2012, 280, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Salomon, D.; Orth, K. What Pathogens Have Taught Us About Posttranslational Modifications. Cell Host Microbe 2013, 14, 269–279. [Google Scholar] [CrossRef]
- Beaudoin, C.A.; Jamasb, A.R.; Alsulami, A.F.; Copoiu, L.; van Tonder, A.J.; Hala, S.; Bannerman, B.P.; Thomas, S.E.; Vedithi, S.C.; Torres, P.H.; et al. Predicted structural mimicry of spike receptor-binding motifs from highly pathogenic human coronaviruses. Comput. Struct. Biotechnol. J. 2021, 19, 3938–3953. [Google Scholar] [CrossRef] [PubMed]
- Custódio, C.A.; Mano, J.F. Cell Surface Engineering to Control Cellular Interactions. Chemnanomat 2016, 2, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Kannagi, R.; Toole, B.; Stanley, P. Glycosylation Changes in Cancer. In Essent. Glycobiol., 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., Schnaar, R.L., Seeberger, P.H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; Available online: http://www.ncbi.nlm.nih.gov/books/NBK453023/ (accessed on 31 October 2023).
- de Jong, H.; Wösten, M.M.S.M.; Wennekes, T. Sweet impersonators: Molecular mimicry of host glycans by bacteria. Glycobiology 2021, 32, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.L.; Clark, T.; Raman, R.; Sasisekharan, R. Glycans in Virus-Host Interactions: A Structural Perspective. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef]
- Shi, X.; Halder, P.; Yavuz, H.; Jahn, R.; Shuman, H.A. Direct targeting of membrane fusion by SNARE mimicry: Convergent evolution ofLegionellaeffectors. Proc. Natl. Acad. Sci. 2016, 113, 8807–8812. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Gammon, D.B. Manipulation of Host Microtubule Networks by Viral Microtubule-Associated Proteins. Viruses 2022, 14, 979. [Google Scholar] [CrossRef] [PubMed]
- Lamason, R.L.; Welch, M.D. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr. Opin. Microbiol. 2016, 35, 48–57. [Google Scholar] [CrossRef]
- Yesudhas, D.; Batool, M.; Anwar, M.A.; Panneerselvam, S.; Choi, S. Proteins Recognizing DNA: Structural Uniqueness and Versatility of DNA-Binding Domains in Stem Cell Transcription Factors. Genes 2017, 8, 192. [Google Scholar] [CrossRef]
- Wang, H.-C.; Ho, C.-H.; Hsu, K.-C.; Yang, J.-M.; Wang, A.H.-J. DNA Mimic Proteins: Functions, Structures, and Bioinformatic Analysis. Biochemistry 2014, 53, 2865–2874. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. et Biophys. Acta (BBA) - Biomembr. 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Shibata-Seki, T.; Nagaoka, M.; Goto, M.; Kobatake, E.; Akaike, T. Direct visualization of the extracellular binding structure of E-cadherins in liquid. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Bonazzi, M.; Lecuit, M.; Cossart, P. Listeria monocytogenesinternalin and E-cadherin: from structure to pathogenesis. Cell. Microbiol. 2009, 11, 693–702. [Google Scholar] [CrossRef]
- Ragon, M.; Wirth, T.; Hollandt, F.; Lavenir, R.; Lecuit, M.; Le Monnier, A.; Brisse, S. A New Perspective on Listeria monocytogenes Evolution. PLOS Pathog. 2008, 4, e1000146. [Google Scholar] [CrossRef] [PubMed]
- Grau-Bové, X.; Sebé-Pedrós, A.; Ruiz-Trillo, I. The Eukaryotic Ancestor Had a Complex Ubiquitin Signaling System of Archaeal Origin. Mol. Biol. Evol. 2014, 32, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.G.; Pruneda, J.N. Bacteria make surgical strikes on host ubiquitin signaling. PLOS Pathog. 2021, 17, e1009341. [Google Scholar] [CrossRef]
- Hebert, F.O.; Phelps, L.; Samonte, I.; Panchal, M.; Grambauer, S.; Barber, I.; Kalbe, M.; Landry, C.R.; Aubin-Horth, N. Identification of candidate mimicry proteins involved in parasite-driven phenotypic changes. Parasites Vectors 2015, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Ludin, P.; Nilsson, D.; Mäser, P. Genome-Wide Identification of Molecular Mimicry Candidates in Parasites. PLOS ONE 2011, 6, e17546. [Google Scholar] [CrossRef]
- Hraber, P.; O’maille, P.E.; Silberfarb, A.; Davis-Anderson, K.; Generous, N.; McMahon, B.H.; Fair, J.M. Resources to Discover and Use Short Linear Motifs in Viral Proteins. Trends Biotechnol. 2019, 38, 113–127. [Google Scholar] [CrossRef]
- Lu, J.; Wu, T.; Zhang, B.; Liu, S.; Song, W.; Qiao, J.; Ruan, H. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 2021, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bernhofer, M.; Goldberg, T.; Wolf, S.; Ahmed, M.; Zaugg, J.; Boden, M.; Rost, B. NLSdb—major update for database of nuclear localization signals and nuclear export signals. Nucleic Acids Res. 2017, 46, D503–D508. [Google Scholar] [CrossRef] [PubMed]
- Goubert, C.; Craig, R.J.; Bilat, A.F.; Peona, V.; Vogan, A.A.; Protasio, A.V. A beginner’s guide to manual curation of transposable elements. Mob. DNA 2022, 13, 1–19. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2023, 42, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Choy, W.-Y.; Karttunen, M. AlphaFold2: A Role for Disordered Protein/Region Prediction? Int. J. Mol. Sci. 2022, 23, 4591. [Google Scholar] [CrossRef]
- Gozashti, L.; Roy, S.W.; Thornlow, B.; Kramer, A.; Ares, M.; Corbett-Detig, R. Transposable elements drive intron gain in diverse eukaryotes. Proc. Natl. Acad. Sci. 2022, 119. [Google Scholar] [CrossRef]
- Corbett-Detig, R.; Medina, P.; Frérot, H.; Blassiau, C.; Castric, V. Bulk pollen sequencing reveals rapid evolution of segregation distortion in the male germline of Arabidopsis hybrids. Evol. Lett. 2019, 3, 93–103. [Google Scholar] [CrossRef]
- Berube, B.; Ernst, E.; Cahn, J.; Roche, B.; de Santis Alves, C.; Lynn, J.; Scheben, A.; Siepel, A.; Ross-Ibarra, J.; Kermicle, J.; et al. Teosinte Pollen Drive guides maize domestication and evolution by RNAi. BioRxiv 2023. [Google Scholar] [CrossRef]
- Aagaard, J.E.; Springer, S.A.; Soelberg, S.D.; Swanson, W.J. Duplicate Abalone Egg Coat Proteins Bind Sperm Lysin Similarly, but Evolve Oppositely, Consistent with Molecular Mimicry at Fertilization. PLOS Genet. 2013, 9, e1003287. [Google Scholar] [CrossRef]
- Chen, S.; Qin, R.; Mahal, L.K. Sweet systems: technologies for glycomic analysis and their integration into systems biology. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 301–320. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Models of molecular conflict evolution and selection for molecular mimics. A) Ecological molecular mimic categories: Molecular mimics are distinguished by the nature of the interaction, i.e., whether they conflict or synergize with organismal fitness, and the genetic relationship with the selfish element. B-E) Idealized illustrations of the three types of molecular mimics: B,C) sequence-based, D) structure-based, and E) functional mimics. F) Molecular evolution mimic categories: Molecular mimics are distinguished by the source of their encoding genetic material (endogenous convergence or HGT from an exogenous source) and how closely they match the original element they evolved to mimic in proximal and downstream phenotypes.
Figure 1.
Models of molecular conflict evolution and selection for molecular mimics. A) Ecological molecular mimic categories: Molecular mimics are distinguished by the nature of the interaction, i.e., whether they conflict or synergize with organismal fitness, and the genetic relationship with the selfish element. B-E) Idealized illustrations of the three types of molecular mimics: B,C) sequence-based, D) structure-based, and E) functional mimics. F) Molecular evolution mimic categories: Molecular mimics are distinguished by the source of their encoding genetic material (endogenous convergence or HGT from an exogenous source) and how closely they match the original element they evolved to mimic in proximal and downstream phenotypes.
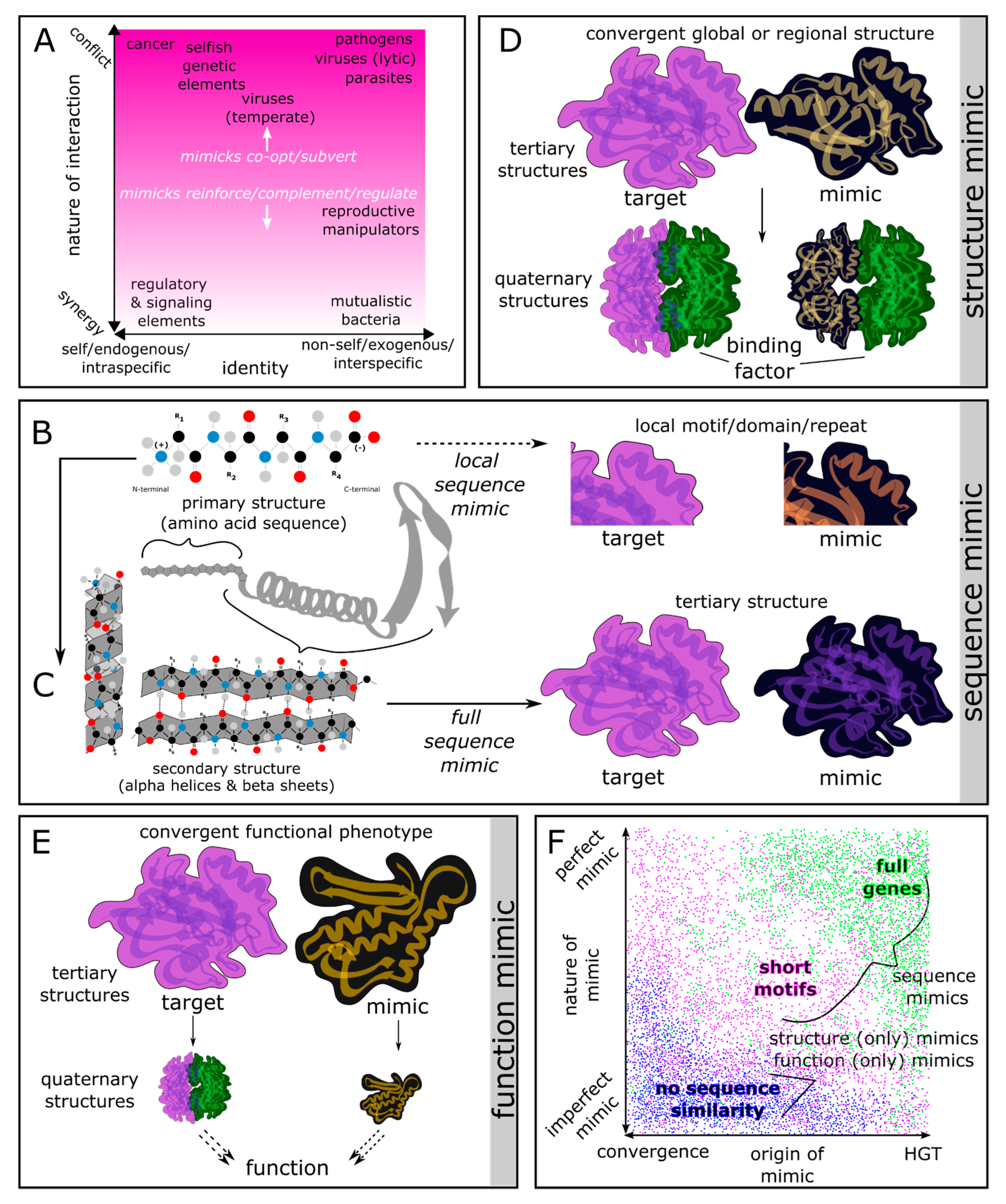
Figure 2.
Molecular mimicry of SLiM motifs mediate intraspecific and interspecific genetic conflicts at the cell surface. A) Fibronectin-integrin binding in the extracellular matrix (ECM) mediates cellular adhesion, migration, and differentiation in normal cells, and can be co-opted by metastatic cancerous cell lineages for metastasis. B) The CagL protein in the H. pylori T4SS mimics integrin-binding RGD motifs. Once injected into the host cytoplasm, the CagA protein’s EPIYA motifs trigger phosphorylation, leading to functional mimicry of the Ras signaling pathway [36]. C) The M. tuberculosis Mce3C membrane protein mimics RGD-integrin binding, inducing actin polymerization and bacterial engulfment [37].
Figure 2.
Molecular mimicry of SLiM motifs mediate intraspecific and interspecific genetic conflicts at the cell surface. A) Fibronectin-integrin binding in the extracellular matrix (ECM) mediates cellular adhesion, migration, and differentiation in normal cells, and can be co-opted by metastatic cancerous cell lineages for metastasis. B) The CagL protein in the H. pylori T4SS mimics integrin-binding RGD motifs. Once injected into the host cytoplasm, the CagA protein’s EPIYA motifs trigger phosphorylation, leading to functional mimicry of the Ras signaling pathway [36]. C) The M. tuberculosis Mce3C membrane protein mimics RGD-integrin binding, inducing actin polymerization and bacterial engulfment [37].
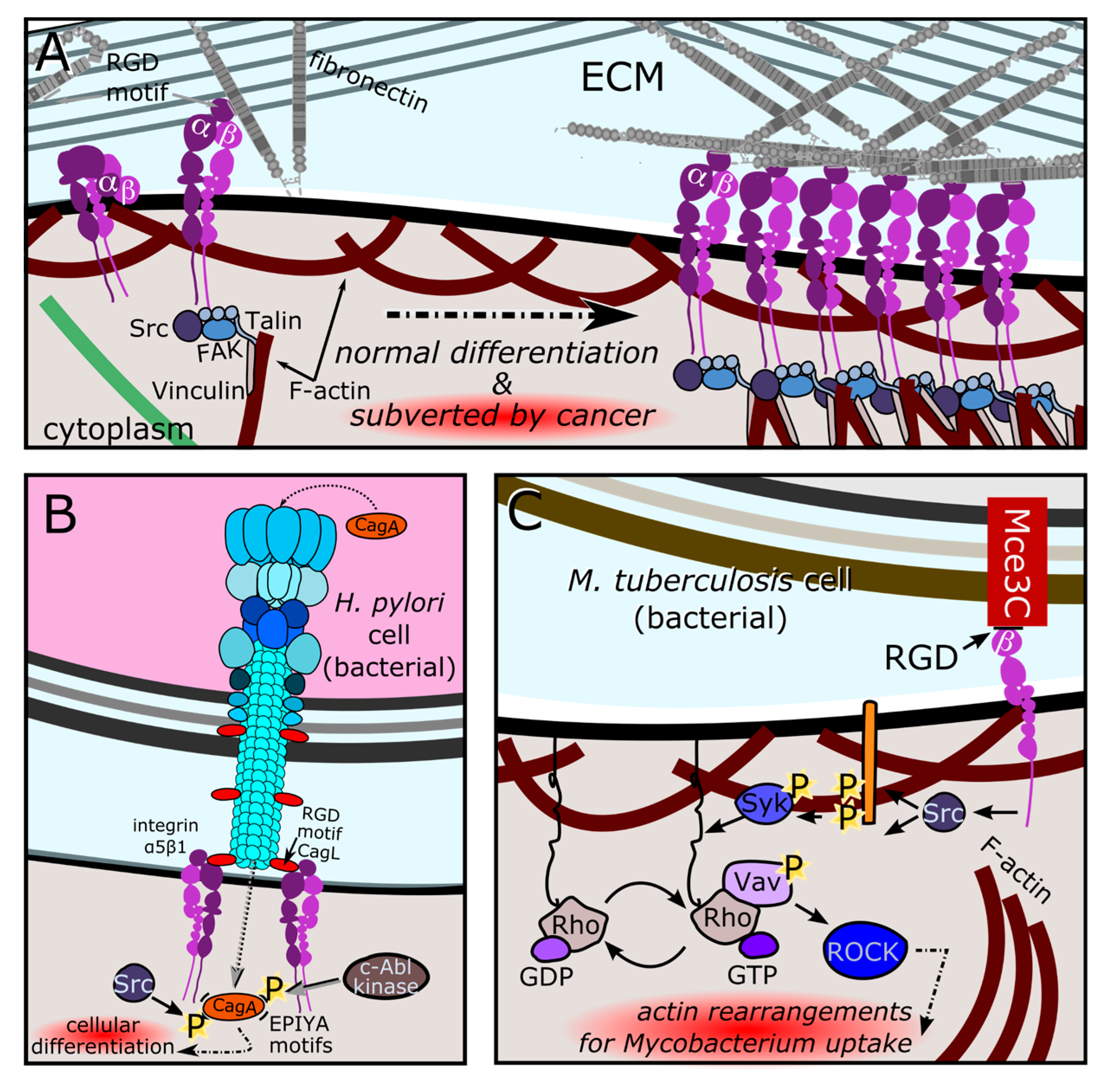
Figure 3.
Molecular mimicry of SLiM motifs and large domains mediate genetic conflicts in the somatic and germline nucleus. A) Eukaryotic cell infected with SARS-CoV2 virions, which express B) ORF8 proteins containing ARKS motifs. This motif mimics histone tails and alters histone acetylation rates by co-opting the KAT2A lysine acetyltransferase for viral acetylation [38] (note wider rate arrow for viral acetylation compared to host acetylation). C) Male germline autosomal segregation distortion in Drosophila depends on two loci: the Sd driver (red chromosome) and the sensitive Rsp locus (cyan chromosome). Sd-RanGAP is produced by Sd chromosomes, and diffuses into non-Sd-containing sperm through cytoplasmic bridges prior to cellularization. D) In the presence of the Rsp(sensitive) sequence, truncated Sd-RanGAP localizes to and is trapped in the nucleus, mimicking GTP hydrolysis, but in the wrong compartment. Disrupted RanGTP gradients block protamine import and stop spermiogenesis in sensitive haplotypes (blue).
Figure 3.
Molecular mimicry of SLiM motifs and large domains mediate genetic conflicts in the somatic and germline nucleus. A) Eukaryotic cell infected with SARS-CoV2 virions, which express B) ORF8 proteins containing ARKS motifs. This motif mimics histone tails and alters histone acetylation rates by co-opting the KAT2A lysine acetyltransferase for viral acetylation [38] (note wider rate arrow for viral acetylation compared to host acetylation). C) Male germline autosomal segregation distortion in Drosophila depends on two loci: the Sd driver (red chromosome) and the sensitive Rsp locus (cyan chromosome). Sd-RanGAP is produced by Sd chromosomes, and diffuses into non-Sd-containing sperm through cytoplasmic bridges prior to cellularization. D) In the presence of the Rsp(sensitive) sequence, truncated Sd-RanGAP localizes to and is trapped in the nucleus, mimicking GTP hydrolysis, but in the wrong compartment. Disrupted RanGTP gradients block protamine import and stop spermiogenesis in sensitive haplotypes (blue).
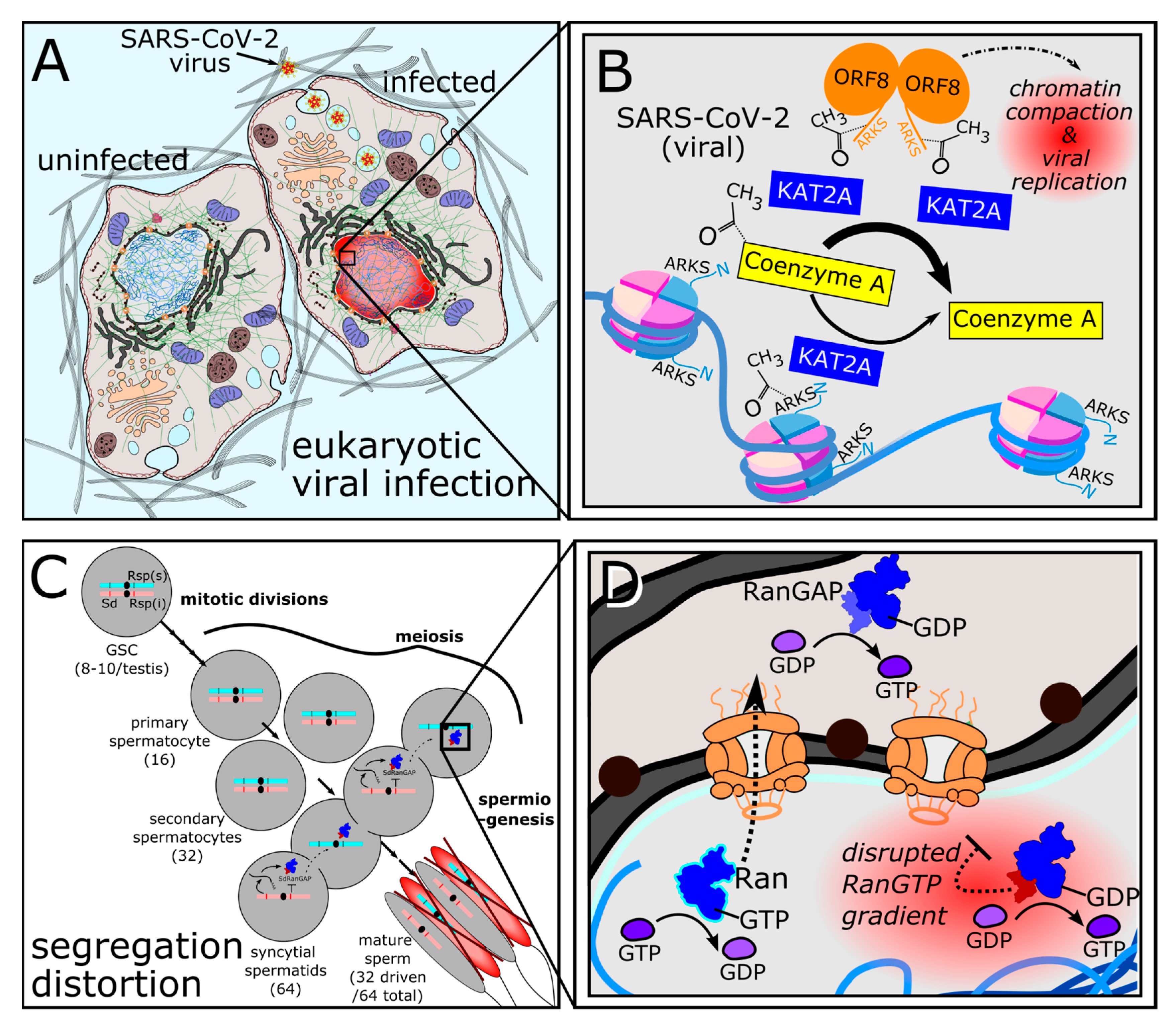
Figure 4.
Interspecific conflicts leverage numerous examples of structural and functional molecular mimicry for entry to and egress from host cells. A) HIV lipid envelopes are decorated with proteins that undergo N-linked glycosylation as they mature, enabling them to mimic self signals and evade immune detection. B) Intracellular L. pneumophila encodes SNARE protein mimics that control host vesicle fusion. C) Mature vaccinia virus encodes motor protein adapter mimic proteins that co-opt KHC-mediated plus-end microtubule transport for egress. D) To promote intracellular uptake, L. monocytogenes functionally mimics adherens junction membrane tension to restructure the actin cytoskeleton.
Figure 4.
Interspecific conflicts leverage numerous examples of structural and functional molecular mimicry for entry to and egress from host cells. A) HIV lipid envelopes are decorated with proteins that undergo N-linked glycosylation as they mature, enabling them to mimic self signals and evade immune detection. B) Intracellular L. pneumophila encodes SNARE protein mimics that control host vesicle fusion. C) Mature vaccinia virus encodes motor protein adapter mimic proteins that co-opt KHC-mediated plus-end microtubule transport for egress. D) To promote intracellular uptake, L. monocytogenes functionally mimics adherens junction membrane tension to restructure the actin cytoskeleton.
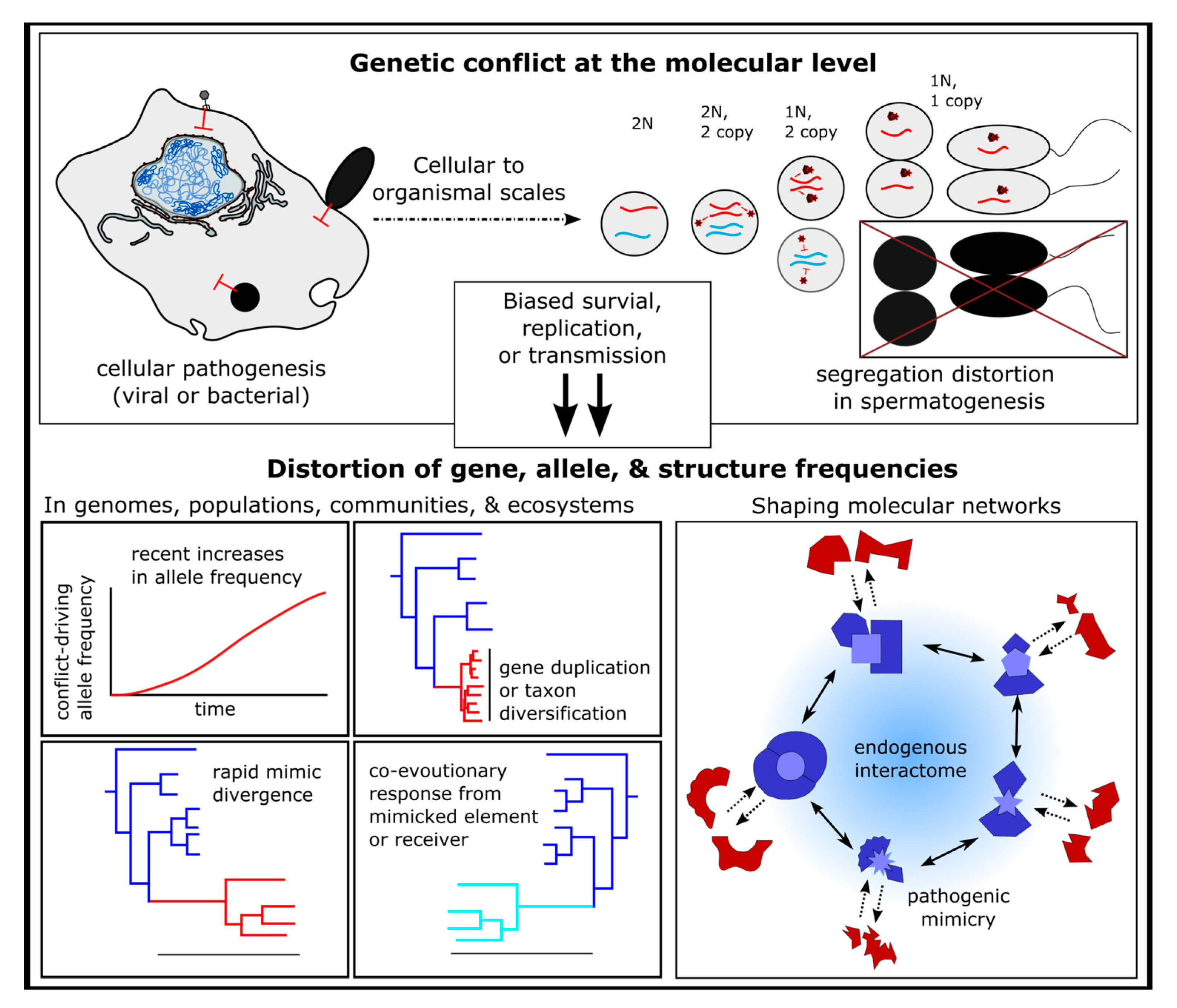
Figure 5.
Computational approaches for detecting molecular mimicry. Genetic conflicts at the cellular and organismal levels can be detected through population genomic data and evolutionary inference because of the influence biased survival, replication, and transmission have on allele frequencies. Protein structure and interactions predictions from genomic data can be leveraged to detect structural arms races. See Supplemental Figures S1 and S2 for summaries of molecular mimic evolutionary sources and screening guidelines, respectively.
Figure 5.
Computational approaches for detecting molecular mimicry. Genetic conflicts at the cellular and organismal levels can be detected through population genomic data and evolutionary inference because of the influence biased survival, replication, and transmission have on allele frequencies. Protein structure and interactions predictions from genomic data can be leveraged to detect structural arms races. See Supplemental Figures S1 and S2 for summaries of molecular mimic evolutionary sources and screening guidelines, respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



