
Submitted:
28 March 2024
Posted:
29 March 2024
You are already at the latest version
Abstract
Dipeptidyl peptidase 9 (DPP9) is a multifunctional intracellular protease with roles in tumour growth, inflammation and mitochondrial function. We developed a hepatocyte-specific DPP9 knockout mouse (DPP9-KO) to explore DPP9 in a mouse model of hepatocellular carcinoma (HCC). DPP9-KO mice were generated by crossing Albumin-Cre mice (Wt/Wt Cre+/+) with DPP9 floxed mice (Fl/Fl Cre-/-). Mice were treated with Diethylnitrosamine and Thioacetamide then an atherogenic High Fat Diet until 28 weeks of age. DPP9-KO mice had reduced liver and subcuta-neous adipose tissue mass and lower fasting plasma glucose, fewer small macroscopic liver nod-ules compared to DPP9-WT mice. However, there were no differences in the total number of mac-roscopic liver nodules, tumour burden, inflammation score and steatosis score. Consistent with the known ability of DPP9 to suppress NLRP1 activation, activated caspase-1 protein was ele-vated in DPP9-KO mouse liver. Additionally, Nfkbib, Cxcl10 and Ccl5 mRNA and protein levels of autophagy marker beclin1 and tumour suppressor p53 were increased. In conclusion, DPP9 de-pletion in hepatocytes may reduce liver cancer initiation, via mechanisms that may include in-creased autophagy and innate tumour suppression in this experimental model. Finally, the data supports DPP9 having a role in glucose regulation by the liver.
Keywords:
dipeptidyl peptidase 9
; serine protease
; hepatocellular carcinoma
; inflammasome
; autophagy
; histopathology
1. Introduction
Liver cancer accounts for 8.2% of cancer-associated mortality [1] and is the second leading cause of cancer related death in male patients [2]. In Australia, liver cancer is associated with a death-to-incidence ratio of 0.98 [3]. Over 90% of liver cancer is hepatocellular carcinoma (HCC), which has a median survival rate of 6.1 to 10.3 months [4,5]. The few therapeutic medicines for liver cancer patients include the multi-kinase inhibitors sorafenib, lenvatinib and brivanib which can upregulate tumour cell apoptosis and restrain angiogenesis [6,7], and immune checkpoint and angiogenesis inhibitors [8,9,10]. Drug-associated adverse events are common [8,9,11]. Therefore, there is a need to investigate novel medicines. Metabolic liver disease is the most rapidly rising risk factor for HCC, alongside rising global obesity and metabolic syndrome [12]. Therefore, our HCC mouse model incorporates a high-fat, high-sucrose, high-cholesterol diet.
A malignant tumour in the liver has distinct cytological and architectural features. Trabecular HCC hepatic cord width is above three nuclei while normal hepatic cord width is under two nuclei [13]. In humans, the histopathological differences between HCC and normal liver, and the variety of HCC nodules, have been established. There are two types of unusual liver lesions: dysplastic foci (< 1 mm) and dysplastic nodules (> 1 mm). The former can be divided into large cell change and small cell change, while the latter can be classified as low-grade dysplastic nodules and high-grade dysplastic nodules. Small cell dysplastic foci are regarded as premalignant [14]. Initial stages of HCC are marked by stromal invasion. In mouse and human tumours, there are many similarities among lesion patterns and histopathological details. Therefore, it is effective to classify liver lesion similarly in animal models as is done in human pathology [14,15,16,17].
The Dipeptidyl Peptidase 4 (DPP4) protease family is involved in cancers, inflammation and collagen turnover [18,19,20,21]. The DPP4 family is composed of six proteins, four of them (FAP, DPP4, DPP8 and DPP9) have enzymatic activities. Of these enzymes, FAP and DPP4 are on the cell surface and released as soluble proteins [22,23,24]. DPP8 and DPP9 are intracellular [25,26,27,28,29,30,31]. The enzymatic activities of DPP8 and DPP9 are very similar to DPP4 and the expression of these three proteases is ubiquitous in immune, endothelial, and epithelial cells. The differentiation of the complex roles of DPP4 with the more recently discovered DPP8 and DPP9 is essential [10,26,28,32].
Gene expression profiling of HCC has shown that DPP9 is upregulated in HCC and tumour-bearing liver tissue compared with normal liver tissue [33]. Moreover, protein expression of DPP9 is upregulated in chronically injured liver [34]. Overexpression of DPP9 is independently associated with lower 5-year survival in non-small lung cancer (NSCLC) [35]. Conversely, DPP9 inhibition has been associated with slower tumour growth, enhanced intratumoral immune response and increased macrophage pyroptosis [36,37].
DPP9 is a unique intracellular serine protease with diverse roles that encompass tumour growth, monocyte/macrophage death, inflammation, DNA repair, mitochondrial function and metabolism [38,39,40,41,42,43]. We previously found that pan-DPP inhibition in mice can reduce the volume and number of primary liver tumours [44]. However, the roles and functions of DPP9 in HCC and in pan-DPP inhibition are not understood. DPP9 suppresses inflammasome activation in epithelial cells [45,46]. This study specifically depleted DPP9 from a crucial and abundant liver epithelial cell, the hepatocyte, to examine whether that action upregulates inflammation markers and decreases tumour burden in a HCC model. This study also asked whether DPP9 is a dominant target of pan-DPP inhibition in mouse HCC.
With the development of DPP9 selective inhibitory compounds as potential therapeutics [47], the extent to which mammals can tolerate DPP9 depletion becomes a more important question. We discovered that loss of DPP9 is neonate lethal in mice [48,49] and that DPP9 Loss-of-Function SNPs is not tolerated in humans[49,50]. The present study showed that loss of DPP9 from hepatocytes is benign.
2. Materials and Methods
Mouse handling and maintenance
All mice were maintained in the Centenary Institute animal facility under specific, pathogen free conditions, co-housed with ad libitum food and water, filtered air and exposed to a 12 h light-dark cycle. C57BL/6 mice were purchased from either Animal Resource Centre (Perth, WA, Australia) or Australian Bio Resources (Moss Vale, NSW, Australia). All experiments were approved and monitored by animal ethics committees of The University of Sydney and Sydney Local Health District (animal welfare approvals 2013/030 and 2017/030) and conducted in accordance with applicable laws and regulations.
Generation of hepatocyte-specific DPP9 knockout primary HCC mouse model
The Dpp9fl/fl allele was provided by the European Mouse Mutant Archive (C57BL/6N-Dpp9tm1a(EUCOMM)Hmgu/Cnrm; EMMA ID EM:04611). The FRT-flanked LacZ reporter/neomycin resistance cassette was removed by FLPe recombinase transgenic mice [51]. Consequently, Dpp9 exons 5-7 were flanked by loxP sites and ready for deletion after breeding with mice expressing a Cre-recombinase such as Alb-Cre.
Female B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J (Alb-Cre) mice (wt/wt Cre+/+) (JAX stock #003547) [31], were crossed with male DPP9fl/fl (Cre -/-) to generate hepatocyte-specific DPP9 knockout mice (fl/fl Cre+/-, DPP9-KO). The littermates were used as controls (fl/fl Cre -/-, DPP9-WT). DPP9 depletion was validated in hepatocyte purification and in the liver at mRNA level.
In order to generate primary HCC, DEN was injected intraperitoneally at 25 mg/kg body weight at 12 days of age into male DPP9-KO (n = 17) and DPP9-WT mice (n = 13). At weaning (approximately 3 weeks old), mice were fed a High Fat Diet (HFD). The HFD was 45% kcal fat, 20% kcal protein, 35% kcal carbohydrate (Table 1). At 8 weeks of age, mice were given the hepatotoxin thioacetamide (TAA) (Alfa Aesar, Shanghai, China; catalogue number A12926) at 200 mg/L in drinking water, twice a week. Mice were euthansed and organs harvested at 28 weeks of age (Figure 1).
High fat diet (HFD)
The HFD was prepared in a fume hood and based on rodent diet no. D12451 [52]. The HFD ingredients are presented in Table 1. In a clean container, 182.88 g casein, 156.96 g sucrose, 150.48 g starch, 35.72 g AIN mineral mix, 39.92 g bran, 2.4 g methionine, 16.12 g gelatine, 3.24 g choline bitartrate, 10.36 g AIN vitamins, 4.2 g cholesterol were mixed thoroughly. Following that, 175.16 g of room temperature lard, 20.8 mL safflower oil and 2 drops of strawberry flavouring were added and mixed together to form a dough like texture. The mixture was stored under nitrogen at 4 for up to two weeks.
Glucose Tolerance Test (GTT)
The intraperitoneal GTT was performed as previously described [53]. All mice were fasted for 6 hours and then administered α-D-glucose at 5 g/kg (Gibco TM, Auckland, NZ) via intraperitoneal injection, respectively. Glucose levels were measured in blood from the tail vein before (0 min) and at 15, 30, 45, 60, 90 and 120 min after the glucose bolus. All glucose concentrations were measured using an Accu-Chek Glucometer (Roche, Diagnostics GmbH, Mannheim, Germany).
Caspase-1 assay
The activity of caspase-1 in liver lysates was determined using the Caspase-1 Assay Kit (Fluorometric) (Abcam, ab39412) according to the manufacturer’s protocols. Fluorescence values were measured with a fluorescence microplate reader at excitation (400 nm) and emission (505 nm). The fold change in caspase-1 activity was determined by comparing the readings of induced samples with the results of the non-induced control.
Hepatocytes perfusion
Buffers used are: A: 50 mL HBSS (pH 7.4), B: 49.55 mL HBSS with 0.05 mL 0.5 mM EDTA, C: 50 mL HBSS with 0.25% (v/v) 5 mM CaCl2 and 0.05% (w/v) Collagenase, D: 90 mL isotonic Percoll dissolved in 10 mL 10 X PBS, E: 250 mL RPMI with 5 mL FBS and 2.5 mL P/S, and F: 6 mL isotonic Percoll dissolved in 10 mL 1X PBS. Water bath was prewarmed to 40 and the pump tube was prewashed by HBSS to minimise bubbles. Mice were killed by CO2 asphyxiation and dissected. A blunt needle was inserted into the IVC and clamped down inside the vessel. The pump rate was set at 50 and circulated buffers A-B-A-C. The liver was removed and placed into buffer E, chopped and kept on ice. The liver mixture was passed through a 70 M cell strainer and centrifuged at 50 x g for 3 minutes at 4 . The supernatant was collected as non-hepatocyte suspension and the pellet was resuspended into buffer E as hepatocyte suspension [54].
Following, the hepatocyte suspension was centrifuged at 50 x g for 3 minutes at 4 . The pellet was resuspended into buffer D and centrifuged at 50 x g for 10 minutes at 4 . The pellet was resuspended into buffer E to perform cell counting. After that, the cell suspension was centrifuged at 50 x g for 3 minutes at 4 and washed with 1X PBS.
107 cells were resuspended into 1 mL lysis buffer and stored at -20 for future enzyme assay or Western blot. 5 x 106 cells were stored as snap frozen cell pellet in RNAase-free tubes at -80 for future qPCR use.
Western blot
Snap frozen mouse liver pieces were lysed in lysis buffer containing 50 mM Tris-HCl pH 7.6, 1 mM EDTA, 10% glycerol, 1% Triton-X100 and complete mini inhibitor cocktail. Protein concentrations were determined using the Micro BCATM Protein Assay Kit (ThermoFisher Scientific, Rockford, 23235) following manufactures instructions.
Following separation of proteins by SDS-PAGE, samples were transferred to PVDF membranes. Following the transfer, PVDF membranes were stained with Ponceau S to visualise proteins and subsequently de-stained in successive PBST washes. PVDF membranes were blocked in 5% (w/v) skim milk in PBST for an hour at room temperature. After blocking, PVDF membranes were incubated with primary antibodies overnight at 4 on a roller (Table 2). The following day, PVDF membranes were washed in PBST for 5 minutes 3 times, then incubated with secondary antibodies (Table 2) conjugated to horse-radish peroxidase (HRP) in 5% skim milk in PBST for 1-2 hours at room temperature. Proteins were then visualised using Immobilon® Forte Western HRP in Chemi Doc MP imaging system.
qPCR
Total liver RNA was isolated using PureLink RNA Mini kit (ThermoFisher, 12183018A), and reverse transcribed to cDNA using Superscript VILO cDNA synthesis kit (Invitrogen, 11756050) following manufacturer’s instructions. The expression of genes was measured by qPCR using custom TaqMan array cards (format 384-well microfluidic card, Applied Biosystems, Foster City, CA), which were pre-spotted with custom designed, dried-down TaqManTM probes including housekeeping control Hprt1, as listed (Table 3). Real time qPCR used the QuantStudioTM 12K Flex Real-Time PCR System (Applied Biosystems) and Expression Suite v1.0 (Applied Biosystems), utilizing the comparative Cτ (ΔΔCτ) method for data analyses. Gene expressions were as a percentage to the housekeeping control.
Histology
Tissue samples were collected and fixed in 10% neutral buffered formalin overnight, then stored in 70% ethanol. Samples were then processed by the Histopathology Core Facility, Charles Perkins Centre, The University of Sydney.
5 M sections were cut and incubated at 65 °C for 1 hour prior to Haematoxylin and Eosin (H&E), immunohistochemistry and immunofluorescence staining (Table 2 as described (Gall et al. 2013). Bright-field imaging was performed at 20x magnification on a Leica DM6000B microscope and analysed on the Mosaic software (Leica, Wetzlar, Germany) to calculate % area of total tissue stained.
Haematoxylin and Eosin (H&E) staining
Paraffin-embedded liver tissue was sectioned at 5 µm thickness. Briefly, slides were de-paraffinised with histolene and rehydrated. Sections were then stained with Harris Haematoxylin for 1.5 min, washed in ddH20 and 0.3% acetic alcohol. Slides were then incubated in Scott’s bluing solution for 3 mins, washed again in ddH20 and stained with Eosin Y for 1 min. Slides were dehydrated with ethanol, cleared with histolene and mounted with Eukitt, as previously described [44,55]. Histology was assessed by a certified pathologist.
Immunohistochemistry (IHC)
IHC was performed as described previously [44,56]. Briefly, paraffin-embedded liver tissue was sectioned at 5 M. Slides were then deparaffinised with histolene, rehydrated and then antigen retrieved for 20 minutes using a pressure cooker and Sodium Citrate Buffer (pH 6.0). Sections were incubated with 0.3% H2O2 for 10 mins to inhibit endogenous peroxidases and rinsed with PBS. Sections were then incubated for 1 hour at room temperature with blocking solution (10% BSA, 10% in normal serum in PBST) and then incubated in primary antibody with 1% BSA in PBST at 4 °C overnight. After washing thoroughly in PBST, sections were incubated for 1.5 hours with secondary antibody conjugated to HRP, washed again thoroughly in PBS then incubated with 3,3- diaminobenzidine (DAB) dissolved in triple distilled water (TDW). Bright-field imaging was performed using a Leica DM6000B microscope.
Image analysis
Bright-field imaging was performed at 20x magnification on a DM6000B microscope and analysed using Mosaic software (Leica, Wetzlar, Germany) to calculate % area of total tissue stained. Entire tissue sections were scanned and individual tiles analysed using Leica application suite v4.8.0 (Leica, Wetzlar, Germany). Tiles with damage or artefacts were excluded from the final analysis. Measuring the immunostained area used a section exposed only to isotype-control immunoglobulin and the secondary antibody as negative control. The thresholds for total tissue were set with all non-tissue areas of the section including blood vessels excluded, at the following thresholds; H:3-5, S:48-255, I:0-243. The image tiles were then automatically analysed by the software and % tissue stain was determined using the following formula, as described [56]: % tissue stained = total stain area 𝑥 100 / total tissue area. Analysis of Sirius red staining was similar, as described previously [56].
To derive steatosis and inflammation scores, multiple photomicrograph tiles of each H&E stained slide, were scored, blinded, by two experienced researchers, using scoring criteria described elsewhere [52]. Lesions were categorised as either HCC, high grade dysplasia or low grade dysplasia [52].
Statistics and Data analysis
A two-way analysis of variance (ANOVA) with Tukey’s multiple comparison test, Kruskal-Wallis comparison test, or one or two-tailed Mann Whitney U test was used to compare data between groups. Data was plotted on GraphPad Prism (GraphPad v. 9.9, San Diego, CA, USA). Significance was assigned to p values; * = 0.05, ** = 0.01, *** = 0.001, **** = 0.0001.
3. Results
3.1. Hepatocyte-Specific DPP9 Knockout Primary HCC Mouse Model Validation
To validate the hepatocyte-specific DPP9 knockout mice, RT-qPCR was performed. As shown in Figure 2, DPP9 mRNA expression in the whole liver was greatly reduced, but not in spleen and kidney. qPCR analyses of RNA extracted from the perfused hepatocytes further validated that the reduced DPP9 mRNA expression occured in the hepatocytes. The results showed that DPP9 was successfully and specifically depleted in the hepatocytes in DPP9-KO mice.
In our mouse model of primary HCC, the body weight of mice was recorded every week until organs were collected at 28 weeks of age. Compared to DPP9-WT mice, DPP9-KO mice appeared to gain less weight (p = 0.053) (Figure 3B). DPP9-KO mice had significantly less liver and subcutaneous white adipose tissue (WAT) to body weight ratios at harvest (Figure 3A).
After mice were treated to generate HCC, depletion of DPP9 was retested. At the mRNA level, DPP9 was successfully depleted in mice livers, without compensation from other DPP4 family genes (Figure 4A). At the protein level, DPP9 depletion in mice livers was also shown upon western blotting (Figure 4B,C). Moreover, IHC showed dense brown stain in the nuclei for DPP9 in DPP9-WT mice, which was absent from DPP9-KO mice, both inside and separate from lesions (Figure 4D–G).
m. DPP9-WT n = 13; DPP9-KO n = 17. Individual data presented with mean. Mann-Whitney statistical test; ****p < 0.0001.
3.2. DPP9 and Glucose Metabolism
The metabolic effects of DPP9 deficiency in the liver at 22 weeks of age were investigated by GTT. The results showed that there was no difference between genotypes in glucose clearance and glucose AUC (Figure 5A, B). However, DPP9-KO mice had significantly lower Fasting Plasma Glucose (FPG) levels compared to DPP9-WT mice (Figure 5C). Interestingly, FPG and body weight presented a positive correlation in DPP9-KO mice (rho = 0.627, p < 0.01), while glucose AUC and body weight presented a moderately positive correlation in DPP9-KO mice (rho = 0.45, p < 0.05), indicating a possible function of DPP9 in glucose metabolism that requires further investigation (Figure 5D, E).
3.3. DPP9 and Cancer Burden
Mice livers from the primary HCC model were harvested at 28 weeks of age. Macroscopic nodules were observed on the surfaces of the mouse livers (Figure 6A). Hepatocyte-specific depletion of DPP9 in the primary HCC model caused fewer macroscopic nodules in DPP9-KO mice but without statistical difference compared with DPP9-WT mice (Figure 6B). These nodules were then classified by size, either as ≤ 3 mm and > 3 mm, whereby significantly fewer small macroscopic nodules (≤ 3 mm) were found in the DPP9-KO livers (Figure 6C). There was no significant difference in large macroscopic nodule numbers between the two genotypes (Figure 6D). These livers were sectioned, stained with H&E and histology assessed by a trained pathologist (Figure 7). There were no significant differences observed in the number of HCC, high grade dysplasia and low grade dysplasia, as well as inflammation and steatosis scores, between DPP9-KO and DPP9-WT mice (Figure 8). As a fibrosis measurement, Sirius red staining of the HCC bearing livers showed no difference in crosslinked collagen between DPP9-KO and DPP9-WT mice (Figure 9).
3.4. DPP9 and Inflammasomes Regulation
Caspase-1 is the hallmark of canonical NLRP1 inflammasome pathway which responds to the inhibition of the DPP9 [57,58,59]. Pro-inflammatory cytokines such as IL1β and Il18 can be cleaved by activated caspase-1 and leads to pyroptosis in cells [60,61,62]. There was a significant increase in the activated caspase-1 (p20) in DPP9-KO mice, revealing potential roles of DPP9 in tumour-bearing livers via the inflammasome activation pathway (Figure 10A,B). NLRP3 is an inflammasome that is highly involved in HCC but not regulated by DPP9. NLRP3 expression is dynamic during HCC development, with a low expression in normal liver tissue, higher expression during liver injury and downregulated after HCC developed [63]. Exported data suggested that the protein expression of NLRP3 remained the same between DPP9-KO and DPP9-WT mice in the HCC livers (Figure 10C, D). The mRNA levels of Nlrp1b, Caspase-1, Il18 and Il1β between DPP9-KO and DPP9-WT mice showed no difference (Figure 11A–D). Correlation analysis revealed that the mRNA expression of Caspase-1 is moderately positive associated with the mRNA expression of NLRP1b (rho = 0.5810, p < 0.05) and IL18 (rho = 0.5980, p < 0.05) in DPP9-KO mice livers (Figure 11E,F).
The enzyme levels of IL-1β and IL-18 were not significantly different between DPP9-WT and DPP9-KO livers, suggesting other drivers of inflammation (Figure 12A,C). Further, IL-1β and IL-18 did not correlate with activated caspase-1 (Spearman’s rho = -0.22, p = 0.399 for IL-1β in DPP9-KO; Spearman’s rho = -0.22, p = 0.470 for IL-1β in DPP9-WT; Spearman’s rho = -0.06, p = 0.809 for IL-18 in DPP9-KO; Spearman’s rho = 0.09, p = 0.765 for IL-18 in DPP9-WT) (Figure 12B,D).
3.5. Intrahepatic Gene Expression Other than Inflammasome Related Genes
Intrahepatic expression of genes associated with HCC, fibrosis, immune response, macrophages and TLR pathway was also assessed. There was no significant difference in the mRNA expression of HCC associated genes: Afp, Gpc3, Birc5, Braf, Trp5 and Ccnd1; immune response associated genes: Tnf, Itgam, Nfkbib, Ccl5, Ccl2, Cx3cr1, Il-6, Cxcl10, Ccr2 and Itgax; inflammasome associated genes: Nlrp3, Caspase-3 and Gasdermin D; macrophage associated genes: CD163, CD47 and CD68; TLR genes: Tlr7, Tlr8 and Tlr9; and extracellular matrix associated genes: Col1a2 and Col3a1. Conversely, there was significantly greater expression of the immune associated genes: Nfkbia and Cxcr3 and macrophage associated gene CD64 in the livers of DPP9-KO mice compare to DPP9-WT mice (Figure 13).
Tumour infiltrating CD8+ cells in the HCC bearing livers showed no difference between DPP9-WT and DPP9-KO mice (Figure 14).
3.6. Autophagy and DNA Repair
Beclin1 is a marker for autophagy [64,65]. Beclin1 protein showed significant upregulation in DPP9-KO mice compared to DPP9-WT mice (Figure 15A, B). The tumour suppressor protein p53 can arrest cell cycle progression in cellular stress (Soussi 2000). Here, protein levels of p53 were elevated in the livers of DPP9-KO mice compared to their DPP9-WT counterparts (Figure 15C, D).
4. Discussion
This study successfully established non-lethal hepatocyte-specific knockout of the DPP9 protease in an epithelial cell type using Cre recombinase driven by the albumin promoter. This approach permits studies of DPP9 knockdown following our previous discovery that universal knockout of DPP9 is neonate lethal [48]. Applying our multi-insult DEN/TAA/HFD primary liver cancer model [66] to this novel mouse strain revealed that DPP9-KO mice had improved energy metabolism, evidenced by smaller subcutaneous adipose tissues and livers and better glucose control. Our observation of fewer small liver lesions suggests depressed levels of tumour initiation. The DPP9-KO mice exhibited more intrahepatic cleaved caspase-1 and a few inflammation markers but not increases in cellular inflammation or downstream pyroptosis markers, so NLRP1 inflammasome activation was not a clear mechanism in this model. However, p53 and Beclin-1 levels were increased compared with littermate control livers. These data are consistent with known roles of DPP9 in adipogenesis and in NLRP1 inflammasome activation as well as with previously unappreciated roles in glucose metabolism, tumour suppression and autophagy (Figure 16).
Validation of DPP9-KO Model
Cre enzyme expression in the Alb-Cre transgenic mouse strain that we used is controlled by the albumin promoter and so is restricted to hepatocytes [31,67]. DPP9 mRNA and protein expression was greatly depressed in our DPP9-KO mice in whole liver and in isolated hepatocytes, thus validating the model.
DPP9 and Liver Inflammation
Our data is consistent with the well characterised role of DPP9 in suppressing NLRP1 activation [68]. NLRP1 inflammasome activation in response to intracellular stress signals results in self-cleavage activation of caspase-1. Activated caspase-1 (p20) can convert the precursor of inflammatory cytokines IL-1β and IL-18 into mature and active cytokines and release them by inducing cell apoptosis [69,70,71]. DPP9 is ubiquitous and is prominent in both epithelial and leucocytic cells [72,73]. Showing that caspase-1 cleavage was greater in DPP9-KO mice, and thus in liver epithelial cells, concords with a recent finding that DPP9 suppresses NLRP1 in corneal epithelial cells [45]. So, this action of DPP9 is common to both epithelia and the monocyte/macrophage lineage. Our observation of increased intrahepatic caspase-1 cleavage in mice treated with an inhibitor of DPP9 confers additional support for this conclusion [44]. In contrast to that previous study, in which inflammation score and expression of some inflammatory markers was greater with DPP9 inhibition, lifelong DPP9 depletion from hepatocytes did not alter inflammation score, but did increase several inflammation markers, NFkbia, NFkbib, CCL5 and CXCL10 (IP10). Our data supports the already expanded view of an anti-inflammatory role for DPP9: that is, establishment of the pro-inflammatory effects of DPP9 down-regulation derived from hepatocytes and other epithelial cell types, in addition to the monocyte/macrophage lineage [45,62,68,74].
The transcription factor NF-κB can mediate the expression of cytokines and inflammatory mediators [75,76]. Both Nfkbia and Nfkbib are inhibitors of NF-κB via IκB [77,78]. When NF-κB is activated, it enters the cell nucleus and binds to the promoters of chemokines for promoting their transcription [79,80]. In this study, all measured cytokines exhibited no change in expression between DPP9-KO and DPP9-WT mice, however, two important chemokines were upregulated. CCL5 and CXCL10 induce migration, mainly of monocytes and lymphocytes, into injured tissues [81,82,83]. However, in contrast to systemic administration of a DPP9 inhibitor to hepatocyte-specific depletion in this liver cancer model [44], we did not observe significant differences in CD8+ cell numbers or locations in the livers.
DPP9 and Adipose Tissue
Knowledge of a relationship between DPP9 and adipose tissue is limited. Global depletion of DPP9 enzyme activity influences metabolic gene expression levels in newborn mouse liver, including increased AMPK [84], which could increase fat utilization [85]. DPP9 downregulation in vitro prevents preadipocyte differentiation in 3T3-L1 cells and downregulates expression of the nuclear receptor PPARγ [86], a regulator of adipocyte differentiation and lipid metabolism [87]. Therefore, the lower liver:body weight and subcutaneous white adipose tissue (WAT):body weight ratios in DPP9-KO mice suggest that DPP9 has an important role in the normal development and accumulation of adipose tissue and possibly in intrahepatic lipid deposition. Despite no difference in steatosis scores, the DPP9-KO livers were smaller and therefore stored less total lipid. Inhibition of the DPP9 related protease DPP4 lowers plasma glucose, and protects against diet-induced liver and adipose tissue inflammation through decreasing the expression of fatty acid synthase [88]. However, there is no rationale for suggesting that the intracellular molecule DPP9 could act similarly to extracellular DPP4.
DPP9 and Glucose Metabolism
DPP9 is not an established glucose metabolism regulator, so the improved FPG in DPP9-KO mice was unexpected. Our finding is possibly a consequence of reduced WAT, or a consequence of the same, unknown mechanism that caused reduced WAT. The FPG effect was significant but small and was insufficient to significantly affect glucose tolerance. Nevertheless, DPP9 has been associated with metabolic processes in the only study of gut and liver gene expression in DPP9 deficient mice [40]. That study observed associations of DPP9 deficiency with long chain fatty acid uptake, lipoprotein metabolism, mitochondrial function, adipokine transport and gluconeogenesis in vivo, and, in vitro, showed a link with AMPK and that insulin and palmitate cause altered DPP9 levels [40]. Furthermore, proteomics has shown that DPP8/DPP9 inhibition regulates glycolysis in the THP-1 cell line [39]. Thus, a variety of data implicate DPP9 in energy storage and metabolism. Our new data supports that role of DPP9 in hepatocytes in vivo.
The impact of DPP9 on FPG could be mediated by an unidentified binding activity, but would more probably be mediated by DPP9 enzyme activity on a substrate. The only potential substrate that has been identified is nucleobindin-2/nesfatin-1 [89], which influences appetite and is regulated by PPARγ [90]. Other metabolism/appetite – associated DPP4 substrates, such as NPY, PYY, GlP-1 and GLP-1 can be cut by DPP9 [91,92,93] but are extracellular, so their exposure to DPP9 is very unlikely.
Another possible mechanism for energy regulation by DPP9 may be through mitochondrial protein degradation. Recently, Finger et al. (2020) identified a novel pathway in which cytosolic DPP8 and DPP9 have a quality control function to prevent abnormal cytosolic accumulation of mitochondrial precursors [38]. Notably, DPP8/9 activity ensures stable processing of intracellular AK2 [38]. Attenuation of DPP9, but not DPP8, leads to excess cellular levels of AK2 [38]. AK2 is associated with oxidative phosphorylation [94] and metabolic signaling [95]. Therefore, these studies suggest a possible role for DPP9 in energy regulation by processing mitochondrial proteins.
Taken together, our data and the literature suggest an under-recognized role for DPP9 in energy regulation.
DPP9 and Beclin-1
The upregulation of intrahepatic Beclin1 protein levels in DPP9-KO mice is a novel observation suggesting that DPP9 has a role in autophagy. This finding concords with the very recent discovery that DPP9 downregulation in the MCF-7 breast cancer cell line increases autophagy [96]. Beclin1 regulates autophagy by forming complexes through interactions with other proteins and participating in various stages of autophagy [97,98,99,100]. Beclin1 can inhibit autophagy by binding to Bcl-2 family proteins to regulate cell growth and apoptosis [101,102]. In addition, Beclin-1 up-regulates USP10 and USP13, which stabilize p53 by de-ubiquitination [103]. p53 is an important tumor suppressor that plays crucial roles in cell cycle regulation, DNA damage repair, apoptosis, and metabolic regulation [104]. Upon cellular stress, p53 can halt cell cycle progression, promote cellular senescence or promote apoptosis (reviewed in: [105]). Mutations in p53 are present in almost 50% of all cancers, making it an attractive target in cancer therapeutics and diagnostics [105]. DPP9 downregulation has been found to increase p53 protein levels in human non-small cell lung cancer cells [106], analogous with our observation that DPP9 depletion promoted p53 protein expression in mouse hepatocytes. Concordantly, tumoral overexpression of DPP9 is linked to poor survival in humans [35,50]. These findings suggest that inhibition of DPP9 may stabilize or enhance mammalian p53 expression, suggesting a possible avenue for tumour therapeutics.
Therefore, our data suggests that DPP9 might both regulate hepatocyte autophagy and influence tumor suppressor p53 levels via beclin-1.
DPP9 and Liver Tumor Growth
Tumourigenesis was assessed by histopathology as well as quantifying macroscopic liver nodules. DPP9-KO mice showed no difference in total macroscopic nodule numbers or tumor burden, but had fewer small liver nodules of < 3 mm diameter compared with their DPP9-WT littermates. This HCC mouse model uses DEN, HFD and TAA, which cause continuous chronic hepatic inflammation, chronic hepatocyte cell death and metabolic dysregulation. The high fat, high sucrose, high cholesterol diet (HFD) causes expansion of adipose tissue and lipid deposition in hepatocytes whereas TAA causes some weight loss and lipid depletion from hepatocytes, but, as again seen here, the TAA in this model does not reverse all the HFD induced lipid accumulation [44,66]. Effects upon inflammation markers, beclin-1, lipid storage and blood glucose were seen in this model when DPP9 was depleted from hepatocytes, and all four parameters could have influenced tumorigenesis and nodule development. The reduced number of small nodules may reflect a reduction in liver cancer initiation in the mice with downregulated hepatocyte DPP9, while progression into larger liver tumors was unaltered. The latter was unexpected and suggests that derepressing NLRP1 was not the dominant outcome of DPP9 depletion in hepatocytes. These findings are also consistent with an overall interpretation that inhibition of DPP9 stabilizes or enhances mammalian p53 expression.
5. Conclusions
We established a hepatocyte-specific DPP9 knockout mouse model to investigate functions of DPP9 in this crucial epithelial cell type. We observed effects on WAT and liver mass, blood glucose, hepatocellular carcinoma development, inflammation markers, p53 and beclin-1. These results suggest that DPP9 exerts effects in hepatocytes in multiple aspects in vivo, rather than just suppressing activation of the NLRP1 inflammasome. This study initiates a deeper and broader understanding of the functions and mechanisms of DPP9 in epithelial cells, in the liver, and in liver cancer.
Author Contributions
Conceptualization, M.D.G, H.E.Z, and Y.Y.; methodology, J.C.H, L.T, H.E.Z; validation, J.C.H, L.T, M.S.W.X, B.B.B and T.R; formal analysis, J.C.H, L.T, M.S.W.X, M.Z, B.B.B and T.R; investigation, J.C.H, L.T, M.S.W.X, M.Z, B.B.B and T.R; resources, M.D.G and G.W.M; data curation, J.C.H and L.T; writing—original draft preparation, J.C.H and L.T; writing—review and editing, J.C.H, L.T, M.S.W.X, B.B.B, G.W.M, T.R, H.E.Z and M.D.G.; visualization, J.C.H, L.T and B.B.B.; supervision, M.D.G and H.E.Z; project administration, M.D.G and H.E.Z.; funding acquisition, M.D.G and T.R. All authors have read and agreed to the published version of the manuscript.
Funding
J.C.H. and M.Z. hold International Postgraduate Scholarships from The University of Sydney. This work was supported by the Deutsche Forschungsgemeinschaft (DFG), under Germany’s Excellence Strategy (BIOSS-EXC-294), the Collaborative Research Centre 850 (project B7 to TR), and GRK2606 (Project ID 423813989; to T.R.). The work was further supported by the German Cancer Consortium DKTK (FR01-371 to T.R.), by a Gastroenterological Society of Australia Project Grant (H.E.Z), a Perpetual Trustee IMPACT Philanthropy grant (H.E.Z), a Centenary Institute grant (M.D.G.), a University of Sydney Kickstart grant (H.E.Z) and Rebecca L. Cooper Medical Research Foundation Equipment Grant RLC_10303 (M.D.G.).
Institutional Review Board Statement
All experiments were approved and monitored by animal ethics committees of The University of Sydney and Sydney Local Health District (animal welfare approvals 2013/030 and 2017/030) and conducted in accordance with applicable laws and regulations.
Data Availability Statement
The data presented in this study is available upon request from the corresponding author.
Acknowledgments
The authors would like to thank Dr. Diarmid Foulis from NSW Health Pathology at the Royal Prince Alfred Hospital for assistance with histopathological analyses as well as the Centenary Institute BioResources unit for animal care. The authors would also like to thank Nicole Klemm and Susanne Dollwet-Mack of the Institute of Molecular Medicine and Cell Research, Freiburg, for excellent technical assistance.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018, 68, 394-424. [CrossRef]
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: the trends projection analysis. Chemical Biology Letters 2023, 10, 451-451.
- Wallace, M.C.; Preen, D.B.; Short, M.W.; Adams, L.A.; Jeffrey, G.P. Hepatocellular carcinoma in Australia 1982-2014: Increasing incidence and improving survival. Liver Int 2019, 39, 522-530. [CrossRef]
- Brar, G.; Greten, T.F.; Graubard, B.I.; McNeel, T.S.; Petrick, J.L.; McGlynn, K.A.; Altekruse, S.F. Hepatocellular carcinoma survival by etiology: a SEER-Medicare database analysis. Hepatology communications 2020, 4, 1541-1551. [CrossRef]
- Chrysavgis, L.; Giannakodimos, I.; Diamantopoulou, P.; Cholongitas, E. Non-alcoholic fatty liver disease and hepatocellular carcinoma: Clinical challenges of an intriguing link. World Journal of Gastroenterology 2022, 28, 310. [CrossRef]
- Llovet, J.M.; Decaens, T.; Raoul, J.-L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.-K.; Assenat, E.; Lim, H.-Y.; Boige, V. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. Journal of Clinical Oncology 2013, 31, 3509-3516. [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Cheng, A.-L.; Piscaglia, F.; Ueshima, K.; Aikata, H.; Vogel, A. Analysis of survival and objective response (OR) in patients with hepatocellular carcinoma in a phase III study of lenvatinib (REFLECT). 2019. [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. The Lancet 2017, 389, 2492-2502. [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New England Journal of Medicine 2020, 382, 1894-1905. [CrossRef]
- Dunaevsky, Y.E.; Tereshchenkova, V.F.; Oppert, B.; Belozersky, M.A.; Filippova, I.Y.; Elpidina, E.N. Human proline specific peptidases: A comprehensive analysis. Biochimica et Biophysica Acta (BBA) - General Subjects 2020, 1864, 129636. [CrossRef]
- Man, K.-F.; Ma, S. Mechanisms of resistance to tyrosine kinase inhibitors in liver cancer stem cells and potential therapeutic approaches. Essays in biochemistry 2022, 66, 371-386. [CrossRef]
- Zhang, C.h.; Cheng, Y.; Zhang, S.; Fan, J.; Gao, Q. Changing epidemiology of hepatocellular carcinoma in Asia. Liver International 2022, 42, 2029-2041. [CrossRef]
- Brunt, E.M. Histopathologic features of hepatocellular carcinoma. Clinical liver disease 2013, 1, 194-199. [CrossRef]
- WHO Classification of Tumours Editorial Board. Digestive System Tumours in WHO Classification of Tumours, 5th Edition; IARC Publications: Lyon, France, 2019; Chapter 8, pp. 235-237, ISBN 978-92-832-4499-8.
- Dhanasekaran, R.; Bandoh, S.; Roberts, L. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances [version 1; peer review: 4 approved]. F1000Research 2016, 5. [CrossRef]
- Libbrecht, L.; Desmet, V.; Roskams, T. Preneoplastic lesions in human hepatocarcinogenesis. Liver International 2005, 25, 16-27. [CrossRef]
- Thoolen, B.; Ten Kate, F.J.W.; van Diest, P.J.; Malarkey, D.E.; Elmore, S.A.; Maronpot, R.R. Comparative histomorphological review of rat and human hepatocellular proliferative lesions. Journal of toxicologic pathology 2012, 25, 189-199. [CrossRef]
- Röhrborn, D.; Eckel, J.; Sell, H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Letters 2014, 588, 3870-3877. [CrossRef]
- Sakamoto, Y.; Mafune, K.; Mori, M.; Shiraishi, T.; Imamura, H.; Mori, M.; Takayama, T.; Makuuchi, M. Overexpression of MMP-9 correlates with growth of small hepatocellular carcinoma. Int J Oncol 2000, 17, 237-243. [CrossRef]
- Zhang; Chen, X.; Zhou, J.; Zhang, L.; Zhao, Q.; Chen, G.; Xu, J.; Qian, F.; Chen, Z. Zhang Q, Chen X, Zhou J, Zhang L, Zhao Q, Chen G, Xu J, Qian F, Chen ZNCD147, MMP-2, MMP-9 and MVD-CD34 are significant predictors of recurrence after liver transplantation in hepatocellular carcinoma patients. Cancer Biol Ther 5: 808-814. Cancer biology & therapy 2006, 5, 808-814. [CrossRef]
- Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L.; Puré, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest 2009, 119, 3613-3625. [CrossRef]
- Williams, K.H.; Viera de Ribeiro, A.J.; Prakoso, E.; Veillard, A.S.; Shackel, N.A.; Bu, Y.; Brooks, B.; Cavanagh, E.; Raleigh, J.; McLennan, S.V.; et al. Lower serum fibroblast activation protein shows promise in the exclusion of clinically significant liver fibrosis due to non-alcoholic fatty liver disease in diabetes and obesity. Diabetes Res Clin Pract 2015, 108, 466-472. [CrossRef]
- Zhang; Chen; Wadham, C.; McCaughan, G.W.; Keane, F.M.; Gorrell, M.D. Dipeptidyl peptidase 9 subcellular localization and a role in cell adhesion involving focal adhesion kinase and paxillin. Biochim Biophys Acta 2015, 1853, 470-480. [CrossRef]
- Zhang; Maqsudi, S.; Rainczuk, A.; Duffield, N.; Lawrence, J.; Keane, F.M.; Justa-Schuch, D.; Geiss-Friedlander, R.; Gorrell, M.D.; Stephens, A.N. Identification of novel dipeptidyl peptidase 9 substrates by two-dimensional differential in-gel electrophoresis. Febs j 2015, 282, 3737-3757. [CrossRef]
- Abbott, C.A.; Gorrell, M.D. Dipeptidyl peptidases. 2001.
- Ajami; Abbott, C.A.; McCaughan, G.W.; Gorrell, M.D. Dipeptidyl peptidase 9 has two forms, a broad tissue distribution, cytoplasmic localization and DPIV-like peptidase activity. Biochim Biophys Acta 2004, 1679, 18-28. [CrossRef]
- Qi; Riviere, P.J.; Trojnar, J.; Junien, J.L.; Akinsanya, K.O. Cloning and characterization of dipeptidyl peptidase 10, a new member of an emerging subgroup of serine proteases. Biochem J 2003, 373, 179-189. [CrossRef]
- Abbott, C.A.; Yu, D.M.; Woollatt, E.; Sutherland, G.R.; McCaughan, G.W.; Gorrell, M.D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur J Biochem 2000, 267, 6140-6150. [CrossRef]
- Olsen, C.; Wagtmann, N. Identification and characterization of human DPP9, a novel homologue of dipeptidyl peptidase IV. Gene 2002, 299, 185-193. [CrossRef]
- Zhang; Chen; Keane, F.M.; Gorrell, M.D. Advances in understanding the expression and function of dipeptidyl peptidase 8 and 9. Mol Cancer Res 2013, 11, 1487-1496. [CrossRef]
- Postic, C.; Magnuson, M.A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000, 26, 149-150. [CrossRef]
- Zhang, H.; Chen, Y.; Keane, F.M.; Gorrell, M.D. Advances in understanding the expression and function of dipeptidyl peptidase 8 and 9. Molecular Cancer Research 2013, 11, 1487-1496. [CrossRef]
- Kurokawa; Matoba, R.; Takemasa, I.; Nakamori, S.; Tsujie, M.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; Ueno, N.; et al. Molecular features of non-B, non-C hepatocellular carcinoma: a PCR-array gene expression profiling study. J Hepatol 2003, 39, 1004-1012. [CrossRef]
- Chowdhury, S.; Chen, Y.; Yao, T.W.; Ajami, K.; Wang, X.M.; Popov, Y.; Schuppan, D.; Bertolino, P.; McCaughan, G.W.; Yu, D.M.; et al. Regulation of dipeptidyl peptidase 8 and 9 expression in activated lymphocytes and injured liver. World J Gastroenterol 2013, 19, 2883-2893. [CrossRef]
- Tang; Li, J.; Shen, Q.; Feng, J.; Liu, H.; Wang, W.; Xu, L.; Shi, G.; Ye, X.; Ge, M.; et al. Contribution of upregulated dipeptidyl peptidase 9 (DPP9) in promoting tumoregenicity, metastasis and the prediction of poor prognosis in non-small cell lung cancer (NSCLC). Int J Cancer 2017, 140, 1620-1632. [CrossRef]
- Fitzgerald, A.A.; Wang, S.; Agarwal, V.; Marcisak, E.F.; Zuo, A.; Jablonski, S.A.; Loth, M.; Fertig, E.J.; MacDougall, J.; Zhukovsky, E.; et al. DPP inhibition alters the CXCR3 axis and enhances NK and CD8+ T cell infiltration to improve anti-PD1 efficacy in murine models of pancreatic ductal adenocarcinoma. Journal for ImmunoTherapy of Cancer 2021, 9, e002837. [CrossRef]
- Duncan, B.B.; Highfill, S.L.; Qin, H.; Bouchkouj, N.; Larabee, S.; Zhao, P.; Woznica, I.; Liu, Y.; Li, Y.; Wu, W.; et al. A pan-inhibitor of DASH family enzymes induces immune-mediated regression of murine sarcoma and is a potent adjuvant to dendritic cell vaccination and adoptive T-cell therapy. J Immunother 2013, 36, 400-411. [CrossRef]
- Finger, Y.; Habich, M.; Gerlich, S.; Urbanczyk, S.; van de Logt, E.; Koch, J.; Schu, L.; Lapacz, K.J.; Ali, M.; Petrungaro, C.; et al. Proteasomal degradation induced by DPP9-mediated processing competes with mitochondrial protein import. EMBO Journal 2020, e103889. [CrossRef]
- Suski, M.; Wiśniewska, A.; Kuś, K.; Kiepura, A.; Stachowicz, A.; Stachyra, K.; Czepiel, K.; Madej, J.; Olszanecki, R. Decrease of the pro-inflammatory M1-like response by inhibition of dipeptidyl peptidases 8/9 in THP-1 macrophages – quantitative proteomics of the proteome and secretome. Molecular Immunology 2020, 127, 193-202. [CrossRef]
- Chen, Y.; Gall, M.G.; Zhang, H.; Keane, F.M.; McCaughan, G.W.; Yu, D.M.; Gorrell, M.D. Dipeptidyl peptidase 9 enzymatic activity influences the expression of neonatal metabolic genes. Experimental Cell Research 2016, 342, 72-82. [CrossRef]
- Bachovchin, D.A. NLRP1: a jack of all trades, or a master of one? Molecular Cell 2021, 81, 423-425. [CrossRef]
- Bolgi, O.; Silva-Garcia, M.; Ross, B.; Pilla, E.; Kari, V.; Killisch, M.; Spitzner, M.; Stark, N.; Lenz, C.; Weiss, K.; et al. Dipeptidyl peptidase 9 triggers BRCA2 degradation and promotes DNA-damage repair. EMBO Reports 2022, 23, e54136. [CrossRef]
- Duncan, B.B.; Highfill, S.L.; Qin, H.; Bouchkouj, N.; Larabee, S.; Zhao, P.; Woznica, I.; Liu, Y.; Li, Y.; Wu, W.; et al. A pan-inhibitor of DASH family enzymes induces immune-mediated regression of murine sarcoma and is a potent adjuvant to dendritic cell vaccination and adoptive T-cell therapy. Journal of Immunotherapy 2013, 36, 400-411. [CrossRef]
- Henderson; Polak, N.; Chen, J.; Roediger, B.; Weninger, W.; Kench, J.G.; McCaughan, G.W.; Zhang, H.E.; Gorrell, M.D. Multiple liver insults synergize to accelerate experimental hepatocellular carcinoma. Sci Rep 2018, 8, 10283. [CrossRef]
- Griswold, A.R.; Huang, H.-C.; Bachovchin, D.A. The NLRP1 Inflammasome Induces Pyroptosis in Human Corneal Epithelial Cells. Investigative Ophthalmology & Visual Science 2022, 63, 2-2. [CrossRef]
- He, H.; Wang, W.; Li, L.; Zhang, X.; Shi, H.; Chen, J.; Shi, D.; Xue, M.; Feng, L. Activation of the NLRP1 Inflammasome and Its Role in Transmissible Gastroenteritis Coronavirus Infection. J Virology 2023, e0058923. [CrossRef]
- Benramdane, S.; De Loose, J.; Filippi, N.; Espadinha, M.; Beyens, O.; Rymenant, Y.V.; Dirkx, L.; Bozdag, M.; Feijens, P.-B.; Augustyns, K.; et al. Highly Selective Inhibitors of Dipeptidyl Peptidase 9 (DPP9) Derived from the Clinically Used DPP4-Inhibitor Vildagliptin. Journal of Medicinal Chemistry 2023, in press. [CrossRef]
- Gall, M.G.; Chen, Y.; Ribeiro, A.J.V.d.; Zhang, H.; Bailey, C.G.; Spielman, D.; Yu, D.M.; Gorrell, M.D. Targeted inactivation of Dipeptidyl Peptidase 9 enzyme activity causes mouse neonate lethality. PLOS ONE 2013, 8, e0078378. [CrossRef]
- Harapas, C.R.; Robinson, K.S.; Lay, K.; Wong, J.; Moreno Traspas, R.; Nabavizadeh, N.; Rass-Rothschild, A.; Boisson, B.; Drutman, S.B.; Laohamonthonkul, P.; et al. DPP9 deficiency: An inflammasomopathy that can be rescued by lowering NLRP1/IL-1 signaling. Science Immunology 2022, 7, eabi4611. [CrossRef]
- Huang, J.C.; Emran, A.A.; Endaya, J.M.; McCaughan, G.W.; Gorrell, M.D.; Zhang, H.E. DPP9: Comprehensive In Silico Analyses of Loss of Function Gene Variants and Associated Gene Expression Signatures in Human Hepatocellular Carcinoma. Cancers 2021, 13, 1637. [CrossRef]
- Rodríguez, C.I.; Buchholz, F.; Galloway, J.; Sequerra, R.; Kasper, J.; Ayala, R.; Stewart, A.F.; Dymecki, S.M. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nature Genetics 2000, 25, 139-140. [CrossRef]
- Henderson, J.M.; Xiang, M.S.W.; Huang, J.C.; Wetzel, S.; Jiang, L.; Lai, J.H.; Wu, W.; Kench, J.G.; Bachovchin, W.W.; Roediger, B.; et al. Dipeptidyl Peptidase Inhibition Enhances CD8 T Cell Recruitment and Activates Intrahepatic Inflammasome in a Murine Model of Hepatocellular Carcinoma. Cancers 2021, 13, 5495. [CrossRef]
- Nunemaker, C.S.; Chen, M.; Pei, H.; Kimble, S.D.; Keller, S.R.; Carter, J.D.; Yang, Z.; Smith, K.M.; Wu, R.; Bevard, M.H.; et al. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab 2008, 295, E1065-E1075. [CrossRef]
- Warren, A.; Le Couteur, D.G.; Fraser, R.; Bowen, D.G.; McCaughan, G.W.; Bertolino, P. T lymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 2006, 44, 1182-1190. [CrossRef]
- Gall; Chen, Y.; Vieira de Ribeiro, A.J.; Zhang, H.; Bailey, C.G.; Spielman, D.S.; Yu, D.M.; Gorrell, M.D. Targeted inactivation of dipeptidyl peptidase 9 enzymatic activity causes mouse neonate lethality. PLoS One 2013, 8, e78378. [CrossRef]
- Wang; Holz, L.E.; Chowdhury, S.; Cordoba, S.P.; Evans, K.A.; Gall, M.G.; Vieira de Ribeiro, A.J.; Zheng, Y.Z.; Levy, M.T.; Yu, D.M.; et al. The pro-fibrotic role of dipeptidyl peptidase 4 in carbon tetrachloride-induced experimental liver injury. Immunol Cell Biol 2017, 95, 443-453. [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: host cell death and inflammation. Nature Reviews. Microbiology 2009, 7, 99-109. [CrossRef]
- Zhong, F.L.; Robinson, K.; Teo, D.E.T.; Tan, K.-Y.; Lim, C.; Harapas, C.R.; Yu, C.-H.; Xie, W.H.; Sobota, R.M.; Au, V.B.; et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem 2018, 293, 18864-18878. [CrossRef]
- Johnson, D.C.; Okondo, M.C.; Orth, E.L.; Rao, S.D.; Huang, H.-C.; Ball, D.P.; Bachovchin, D.A. DPP8/9 inhibitors activate the CARD8 inflammasome in resting lymphocytes. Cell Death & Disease 2020, 11, 628. [CrossRef]
- Biswas; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature Immunology 2010, 11, 889-896. [CrossRef]
- Franchi; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozören, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol 2006, 7, 576-582. [CrossRef]
- de Vasconcelos, N.M.; Vliegen, G.; Gonçalves, A.; De Hert, E.; Martín-Pérez, R.; Van Opdenbosch, N.; Jallapally, A.; Geiss-Friedlander, R.; Lambeir, A.-M.; Augustyns, K.; et al. DPP8/DPP9 inhibition elicits canonical Nlrp1b inflammasome hallmarks in murine macrophages. Life Science Alliance 2019, 2, e201900313. [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Laboratory Investigation 2014, 94, 52-62. [CrossRef]
- Dickey, J.S.; Redon, C.E.; Nakamura, A.J.; Baird, B.J.; Sedelnikova, O.A.; Bonner, W.M. H2AX: functional roles and potential applications. Chromosoma 2009, 118, 683-692. [CrossRef]
- Kang; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death & Differentiation 2011, 18, 571-580. [CrossRef]
- Henderson, J.M.; Polak, N.; Chen, J.; Roediger, B.; Weninger, W.; Kench, J.G.; McCaughan, G.W.; Zhang, H.E.; Gorrell, M.D. Multiple liver insults synergize to accelerate experimental hepatocellular carcinoma. Scientific Reports 2018, 8, 10283. [CrossRef]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 1999, 274, 305-315.
- Okondo, M.C.; Johnson, D.C.; Sridharan, R.; Go, E.B.; Chui, A.J.; Wang, M.S.; Poplawski, S.E.; Wu, W.; Liu, Y.; Lai, J.H.; et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nature Chemical Biology 2017, 13, 46-53. [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821-832. [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002, 10, 417-426. [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013-1022. [CrossRef]
- Ajami, K.; Abbott, C.A.; McCaughan, G.W.; Gorrell, M.D. Dipeptidyl peptidase 9 has two forms, a broad tissue distribution, cytoplasmic localization and DPIV-like peptidase activity. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression 2004, 1679, 18-28. [CrossRef]
- Yu, D.M.T.; Ajami, K.; Gall, M.G.; Park, J.; Lee, C.S.; Evans, K.A.; McLaughlin, E.A.; Pitman, M.R.; Abbott, C.A.; McCaughan, G.W.; et al. The in vivo expression of dipeptidyl peptidases 8 and 9. Journal of Histochemistry and Cytochemistry 2009, 57, 1025-1040. [CrossRef]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chemical Biology 2017, 24, 507-514.e504. [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344-362. [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007, 8, 49-62. [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res 2011, 21, 71-85. [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 2009, 1, a000034. [CrossRef]
- Ohmori, Y.; Hamilton, T.A. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J Immunol 1995, 154, 5235-5244. [CrossRef]
- Tian, B.; Nowak, D.E.; Jamaluddin, M.; Wang, S.; Brasier, A.R. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem 2005, 280, 17435-17448. [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: a new classification system and their role in immunity. Immunity 2000, 12, 121-127. [CrossRef]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 2011, 89, 207-215. [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 2014, 32, 659-702. [CrossRef]
- Chen, Y.; Gall, M.G.; Zhang, H.; Keane, F.M.; McCaughan, G.W.; Yu, D.M.T.; Gorrell, M.D. Dipeptidyl peptidase 9 enzymatic activity influences the expression of neonatal metabolic genes. Experimental Cell Research 2016, 342, 72-82. [CrossRef]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int 2018, 42, 384-392. [CrossRef]
- Han, R.; Wang, X.; Bachovchin, W.; Zukowska, Z.; Osborn, J.W. Inhibition of dipeptidyl peptidase 8/9 impairs preadipocyte differentiation. Scientific Reports 2015, 5, 12348. [CrossRef]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism--novel insights from genome-wide studies. FEBS Lett 2010, 584, 3242-3249. [CrossRef]
- Shirakawa, J.; Fujii, H.; Ohnuma, K.; Sato, K.; Ito, Y.; Kaji, M.; Sakamoto, E.; Koganei, M.; Sasaki, H.; Nagashima, Y.; et al. Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 2011, 60, 1246-1257. [CrossRef]
- Zhang, H.; Maqsudi, S.; Rainczuk, A.; Duffield, N.; Lawrence, J.; Keane, F.M.; Justa-Schuch, D.; Geiss-Friedlander, R.; Gorrell, M.D.; Stephens, A.N. Identification of novel dipeptidyl peptidase 9 substrates by two-dimensional differential in-gel electrophoresis. FEBS Journal 2015, 282, 3737 -3757. [CrossRef]
- Yamada, M.; Horiguchi, K.; Umezawa, R.; Hashimoto, K.; Satoh, T.; Ozawa, A.; Shibusawa, N.; Monden, T.; Okada, S.; Shimizu, H.; et al. Troglitazone, a ligand of peroxisome proliferator-activated receptor-{gamma}, stabilizes NUCB2 (Nesfatin) mRNA by activating the ERK1/2 pathway: isolation and characterization of the human NUCB2 gene. Endocrinology 2010, 151, 2494-2503. [CrossRef]
- Lu, C.; Tilan, J.U.; Everhart, L.; Czarnecka, M.; Soldin, S.J.; Mendu, D.R.; Jeha, D.; Hanafy, J.; Lee, C.K.; Sun, J.; et al. Dipeptidyl Peptidases as Survival Factors in Ewing Sarcoma Family of Tumors: IMPLICATIONS FOR TUMOR BIOLOGY AND THERAPY *<sup> </sup>. Journal of Biological Chemistry 2011, 286, 27494-27505. [CrossRef]
- Ajami, K.; Pitman, M.R.; Wilson, C.H.; Park, J.; Menz, R.I.; Starr, A.E.; Cox, J.H.; Abbott, C.A.; Overall, C.M.; Gorrell, M.D. Stromal cell-derived factors 1alpha and 1beta, inflammatory protein-10 and interferon-inducible T cell chemo-attractant are novel substrates of dipeptidyl peptidase 8. FEBS Lett 2008, 582, 819-825. [CrossRef]
- Bjelke, J.R.; Christensen, J.; Nielsen, P.F.; Branner, S.; Kanstrup, A.B.; Wagtmann, N.; Rasmussen, H.B. Dipeptidyl peptidases 8 and 9: specificity and molecular characterization compared with dipeptidyl peptidase IV. Biochem J 2006, 396, 391-399. [CrossRef]
- Burkart, A.; Shi, X.; Chouinard, M.; Corvera, S. Adenylate kinase 2 links mitochondrial energy metabolism to the induction of the unfolded protein response. J Biol Chem 2011, 286, 4081-4089. [CrossRef]
- Dzeja, P.; Terzic, A. Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int J Mol Sci 2009, 10, 1729-1772. [CrossRef]
- Bettecken, A.; Hess, L.; Hoelzen, L.; Reinheckel, T. Dipeptidyl-Aminopeptidases 8 and 9 Regulate Autophagy and Tamoxifen Response in Breast Cancer Cells. Cells 2023, 12, 2031. [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death & Differentiation 2011, 18, 571-580. [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009, 11, 385-396. [CrossRef]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol 2010, 20, 355-362. [CrossRef]
- Ma, B.; Cao, W.; Li, W.; Gao, C.; Qi, Z.; Zhao, Y.; Du, J.; Xue, H.; Peng, J.; Wen, J.; et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Research 2014, 24, 912-924. [CrossRef]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600-606. [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561-568. [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223-234. [CrossRef]
- Boutelle, A.M.; Attardi, L.D. p53 and Tumor Suppression: It Takes a Network. Trends Cell Biol 2021, 31, 298-310. [CrossRef]
- Soussi, T. The p53 Tumor Suppressor Gene: From Molecular Biology to Clinical Investigation. Annals of the New York Academy of Sciences 2000, 910, 121-139. [CrossRef]
- Tang, Z.; Li, J.; Shen, Q.; Feng, J.; Liu, H.; Wang, W.; Xu, L.; Shi, G.; Ye, X.; Ge, M.; et al. Contribution of upregulated dipeptidyl peptidase 9 (DPP9) in promoting tumoregenicity, metastasis and the prediction of poor prognosis in non-small cell lung cancer (NSCLC). Int J Cancer 2017, 140, 1620-1632. [CrossRef]
Figure 1.
Overview of experimental model of primary HCC. Mice were treated with N-nitrosodiethylamine (DEN; d12), thioacetamide (TAA; weeks 8 - 28) and a high fat high sucrose atherogenic diet (HFD; weeks 3 - 28), with the endpoint at 28 weeks of age.
Figure 1.
Overview of experimental model of primary HCC. Mice were treated with N-nitrosodiethylamine (DEN; d12), thioacetamide (TAA; weeks 8 - 28) and a high fat high sucrose atherogenic diet (HFD; weeks 3 - 28), with the endpoint at 28 weeks of age.

Figure 2.
RT-qPCR on DPP9 fl/fl Cre+/- mice (DPP9-KO) and DPP9 wt/wt Cre-/- mice (DPP9-WT). DPP9 gene expression normalized to the mean of housekeeping genes 18S rRNA and Hprt1 of individual mice. Data plotted with mean with standard deviation, from isolated primary hepatocytes, liver, kidney and spleen. (n = 2-3). Mann-Whitney test showed p < 0.05 for hepatocytes and liver.
Figure 2.
RT-qPCR on DPP9 fl/fl Cre+/- mice (DPP9-KO) and DPP9 wt/wt Cre-/- mice (DPP9-WT). DPP9 gene expression normalized to the mean of housekeeping genes 18S rRNA and Hprt1 of individual mice. Data plotted with mean with standard deviation, from isolated primary hepatocytes, liver, kidney and spleen. (n = 2-3). Mann-Whitney test showed p < 0.05 for hepatocytes and liver.
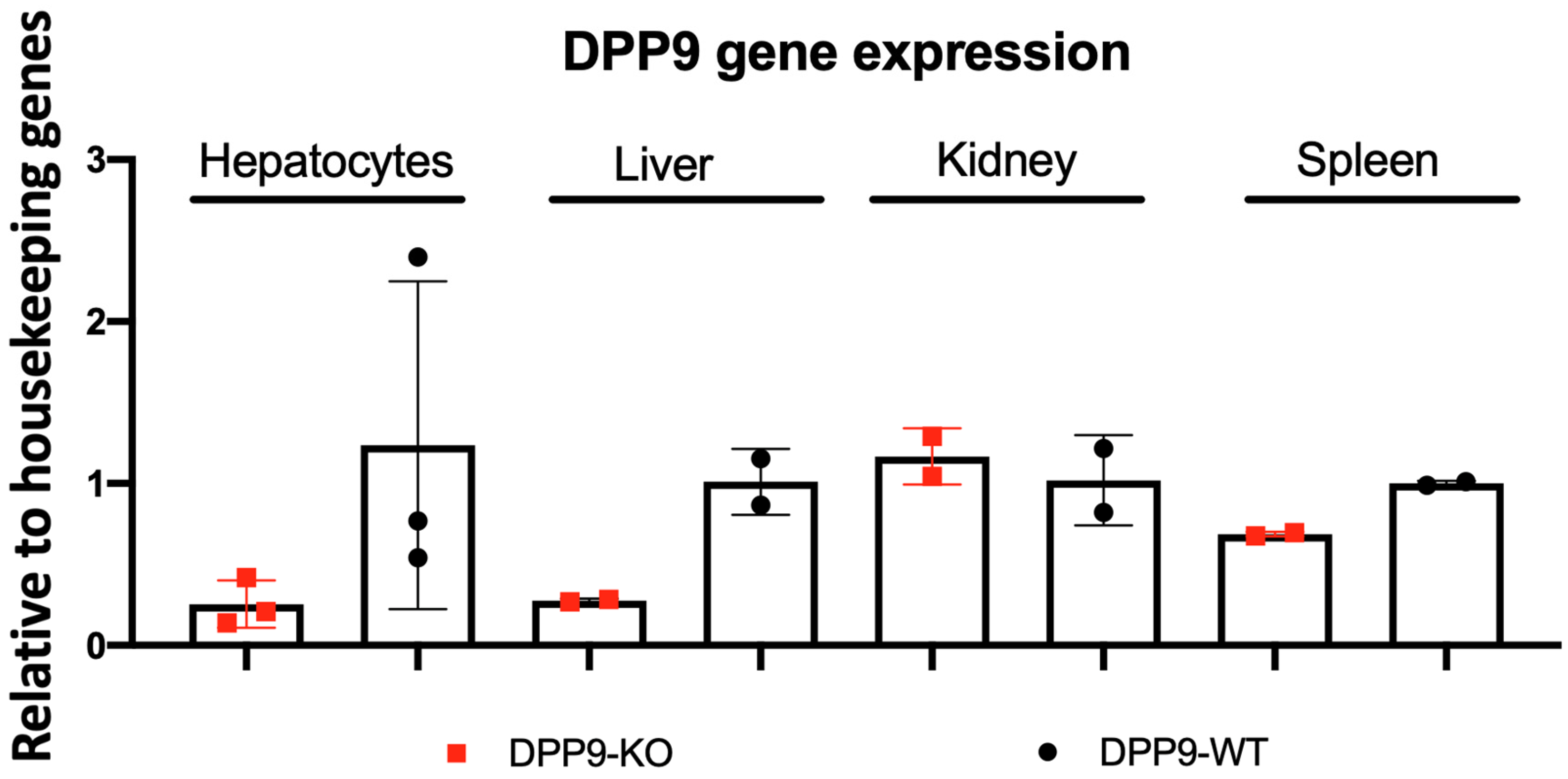
Figure 3.
Body weight and organ weights of DPP9-KO and DPP9-WT mice. Mouse body weight gain (A) and organ weights as ratio to body weight (BW) (B) measured over time and at time of death, respectively. DPP9-WT (n = 13); DPP9-KO (n = 17). Mean ± SD. Mann-Whitney statistical test, *p < 0.5.
Figure 3.
Body weight and organ weights of DPP9-KO and DPP9-WT mice. Mouse body weight gain (A) and organ weights as ratio to body weight (BW) (B) measured over time and at time of death, respectively. DPP9-WT (n = 13); DPP9-KO (n = 17). Mean ± SD. Mann-Whitney statistical test, *p < 0.5.
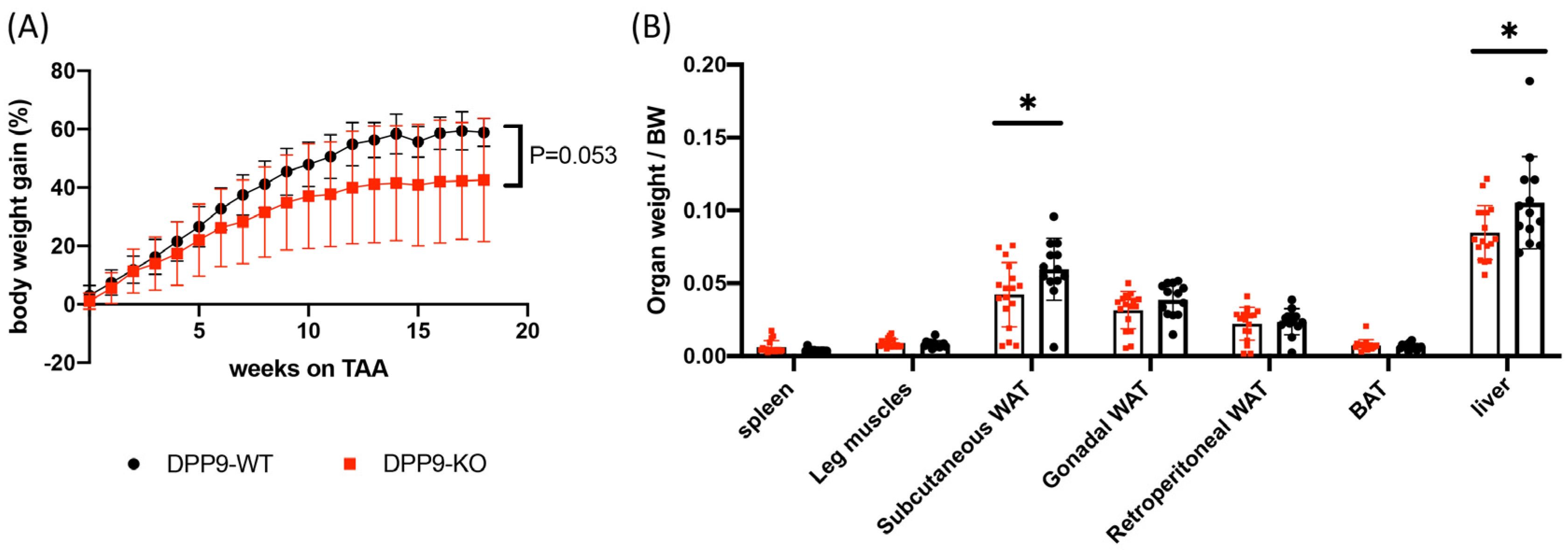
Figure 4.
Validation of DPP9 depletion in the liver. (A) DPP4 family gene expression normalized to housekeeper Hprt1 of individual mice plotted with mean and standard deviation. (B) Representative immunoblot of DPP9 (Antibody catalogue number Origene #TA504019) on protein extracts of liver. (C) Densitometry of immunoblots. (D-G) Representative images of DPP9 (Antibody catalogue number Abcam #42080) immunostaining (brown) of liver region inside lesions in paraffin sections from mice after DEN/TAA/HFD treatment. (D,E) DPP9-WT mouse. (F,G) DPP9-KO mouse. Scale Bars = 200
Figure 4.
Validation of DPP9 depletion in the liver. (A) DPP4 family gene expression normalized to housekeeper Hprt1 of individual mice plotted with mean and standard deviation. (B) Representative immunoblot of DPP9 (Antibody catalogue number Origene #TA504019) on protein extracts of liver. (C) Densitometry of immunoblots. (D-G) Representative images of DPP9 (Antibody catalogue number Abcam #42080) immunostaining (brown) of liver region inside lesions in paraffin sections from mice after DEN/TAA/HFD treatment. (D,E) DPP9-WT mouse. (F,G) DPP9-KO mouse. Scale Bars = 200
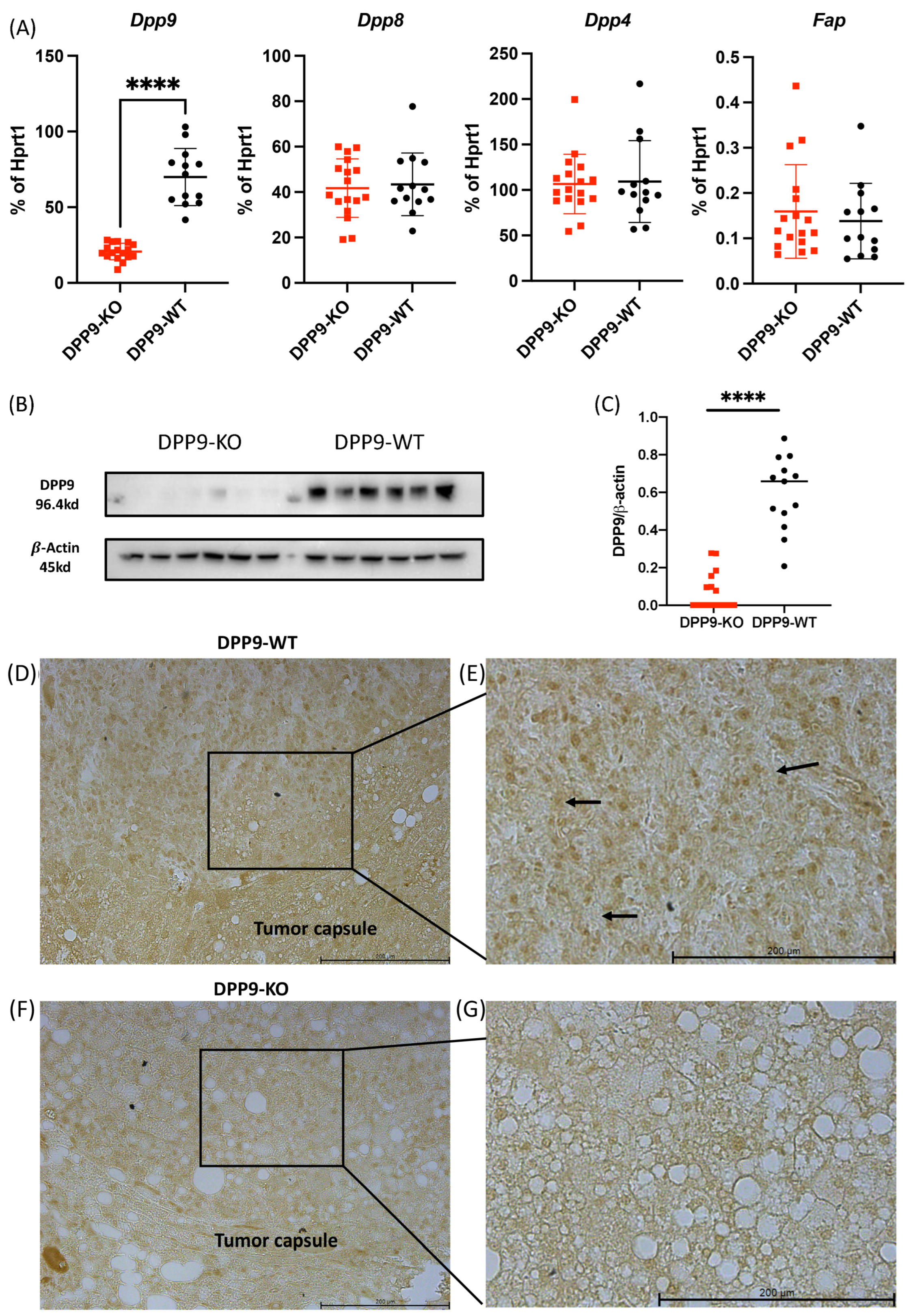
Figure 5.
Glucose measurements of DPP9-KO mice and DPP9-WT mice in the primary HCC model. (A) Blood glucose following intraperitoneal glucose tolerance test (ipGTT). (B) Area under the curve (AUC) for 0-120 minutes of ipGTT. (C) Fasting glucose levels in blood samples. (D) Correlation analysis of Fasting Plasma Glucose (FPG) and body weight (BW). (E) Correlation analysis of ipGTT AUC and body weight. Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Statistical analyses used Two-way ANOVA with Tukey’s multiple comparisons test (A), Mann-Whitney test (B, C), nonparametric Spearman correlation test (D, E), *p < 0.5, **p < 0.01.
Figure 5.
Glucose measurements of DPP9-KO mice and DPP9-WT mice in the primary HCC model. (A) Blood glucose following intraperitoneal glucose tolerance test (ipGTT). (B) Area under the curve (AUC) for 0-120 minutes of ipGTT. (C) Fasting glucose levels in blood samples. (D) Correlation analysis of Fasting Plasma Glucose (FPG) and body weight (BW). (E) Correlation analysis of ipGTT AUC and body weight. Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Statistical analyses used Two-way ANOVA with Tukey’s multiple comparisons test (A), Mann-Whitney test (B, C), nonparametric Spearman correlation test (D, E), *p < 0.5, **p < 0.01.
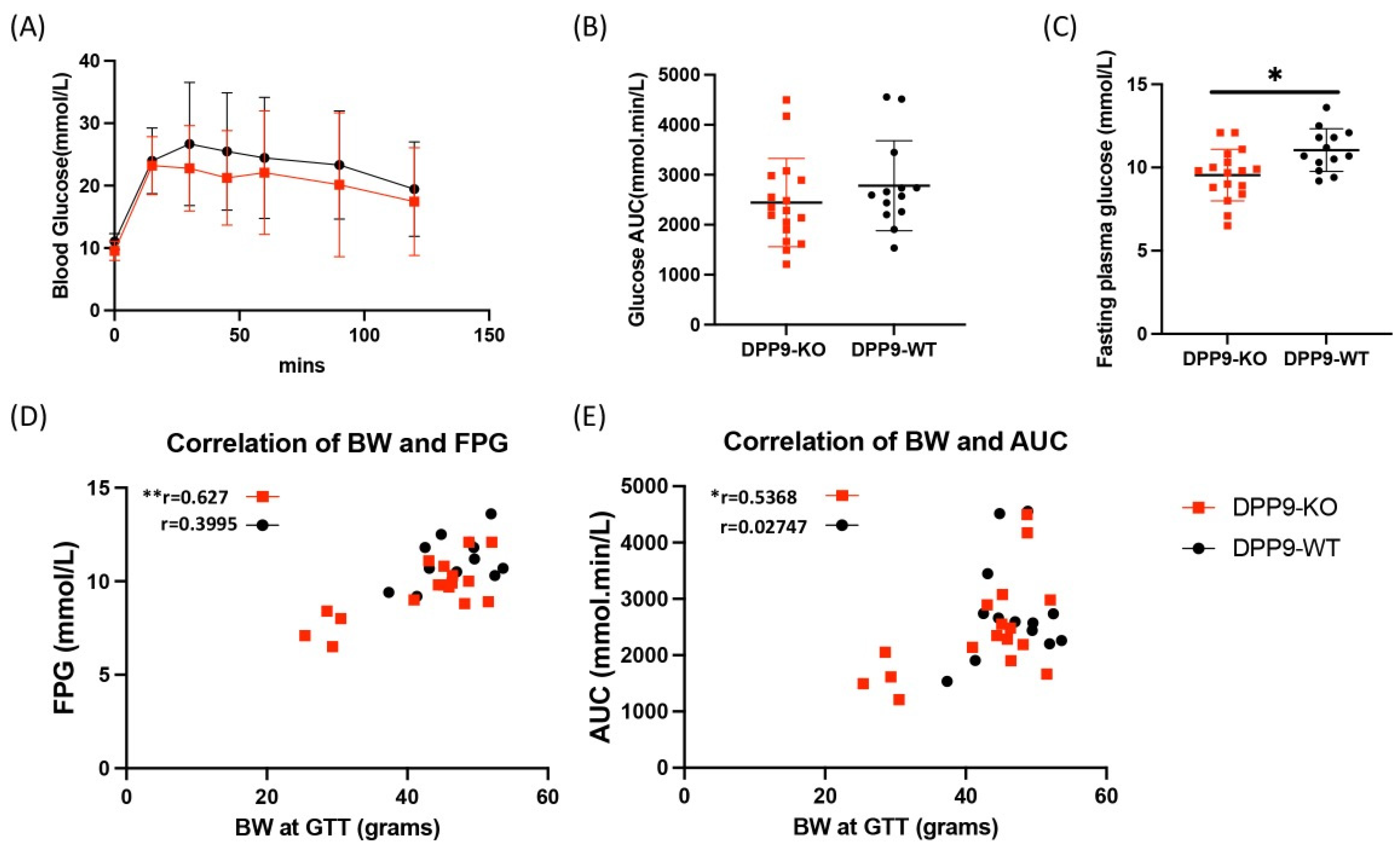
Figure 6.
Primary cancer burden assessment. (A) Representative images of macroscopic nodules (spots) that were observed in primary HCC model mouse livers. (B) Quantification of macroscopic nodules. (C) Quantification of macroscopic nodules (≤ 3 mm). (D) Quantification of macroscopic nodul es (> 3 mm). Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Statistical analyses used Mann-Whitney test, *p < 0.5.
Figure 6.
Primary cancer burden assessment. (A) Representative images of macroscopic nodules (spots) that were observed in primary HCC model mouse livers. (B) Quantification of macroscopic nodules. (C) Quantification of macroscopic nodules (≤ 3 mm). (D) Quantification of macroscopic nodul es (> 3 mm). Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Statistical analyses used Mann-Whitney test, *p < 0.5.
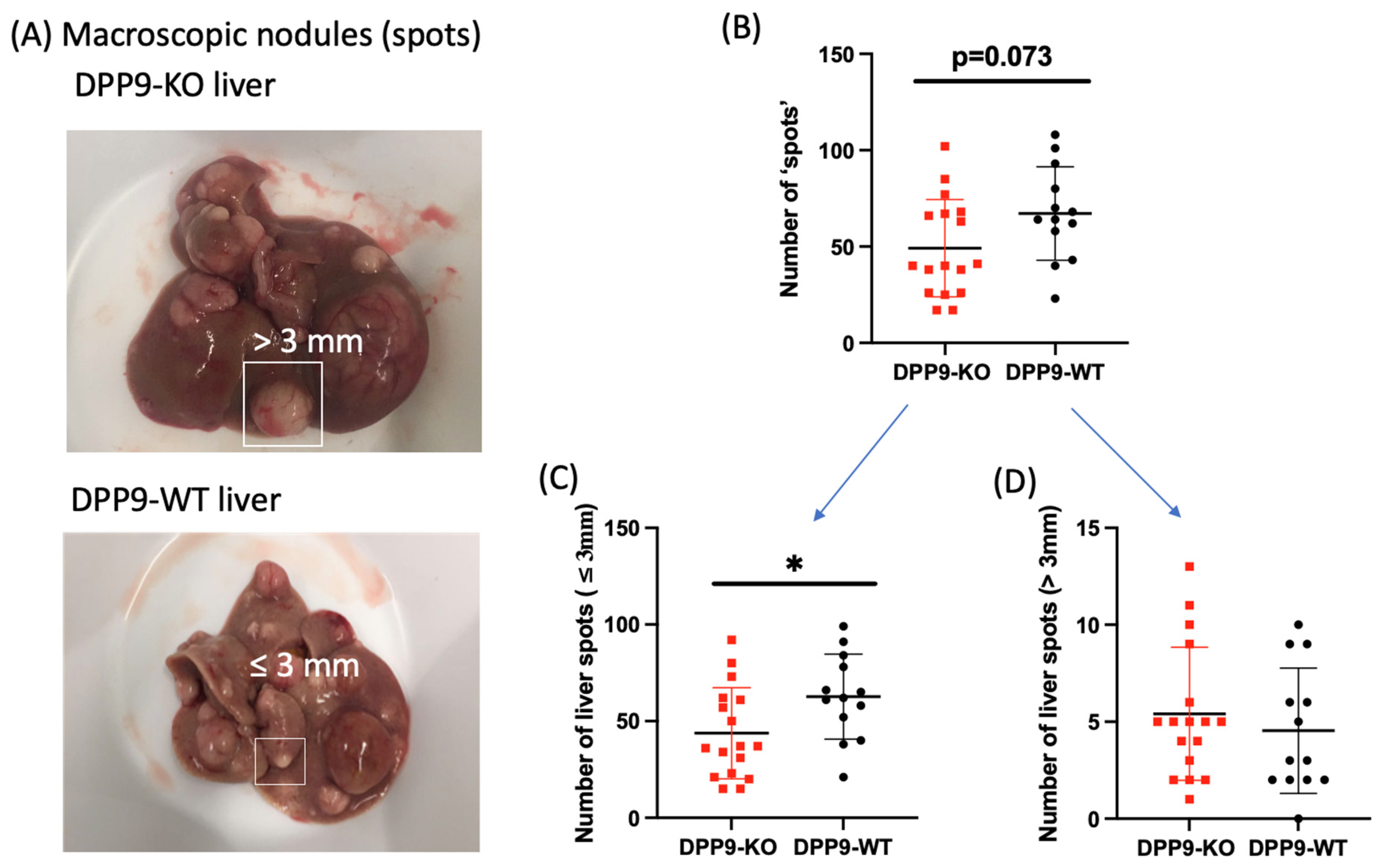
Figure 7.
Liver histology. Boxed area shows an area of either (A) HCC in DPP9-WT mouse liver. (B) HCC in DPP9-KO mouse liver. (C) Dysplasia with large cell change. (D). High grade dysplasia with small cell change (E) Arrowed area shows an area of macrosteatosis (1) or microsteatosis (2). Scale Bars = 200 m.
Figure 7.
Liver histology. Boxed area shows an area of either (A) HCC in DPP9-WT mouse liver. (B) HCC in DPP9-KO mouse liver. (C) Dysplasia with large cell change. (D). High grade dysplasia with small cell change (E) Arrowed area shows an area of macrosteatosis (1) or microsteatosis (2). Scale Bars = 200 m.
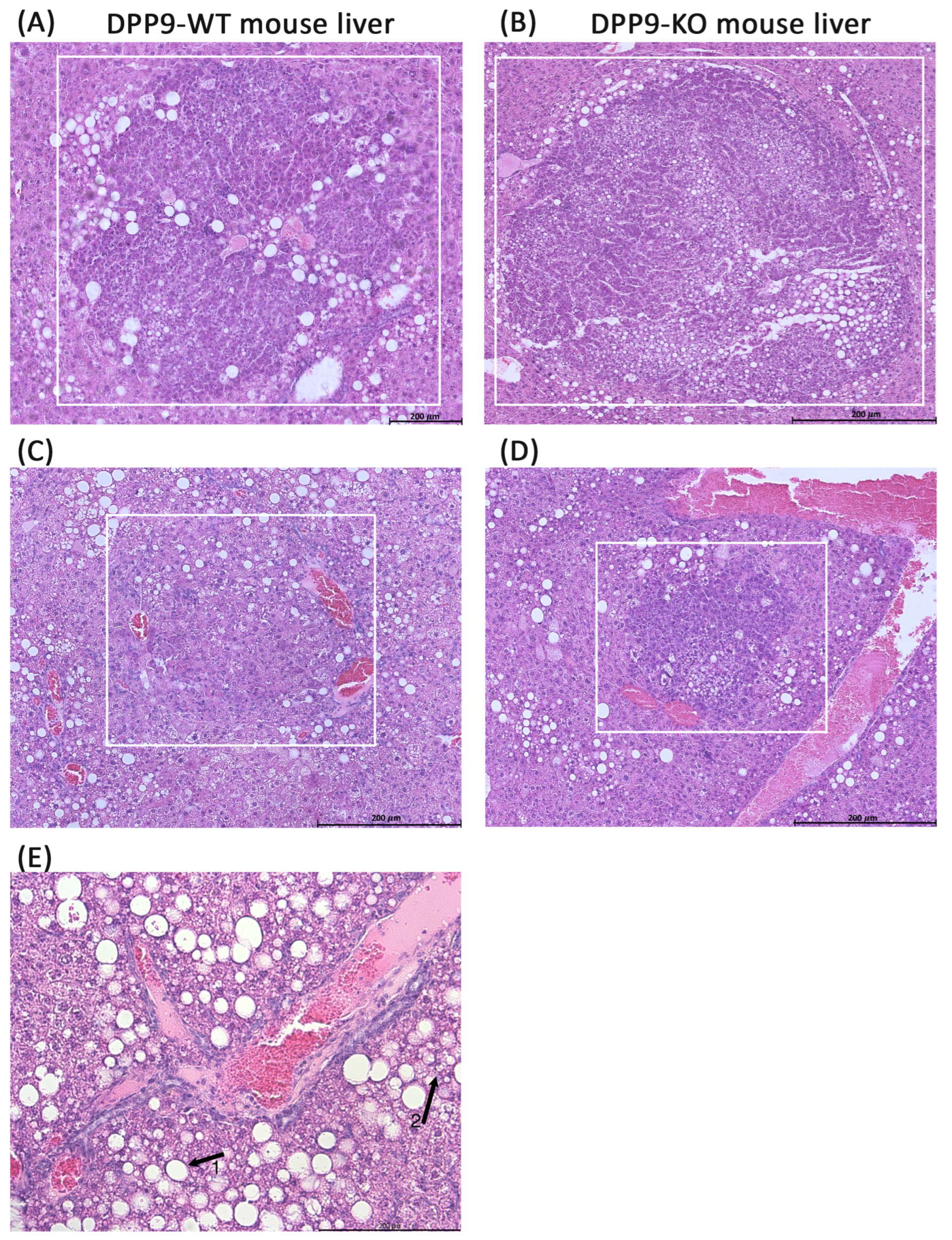
Figure 8.
Cancer burden and histological scores in the primary HCC model. (A-E) The types of lesions observed and enumerated. Mean ± SEM, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test.
Figure 8.
Cancer burden and histological scores in the primary HCC model. (A-E) The types of lesions observed and enumerated. Mean ± SEM, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test.
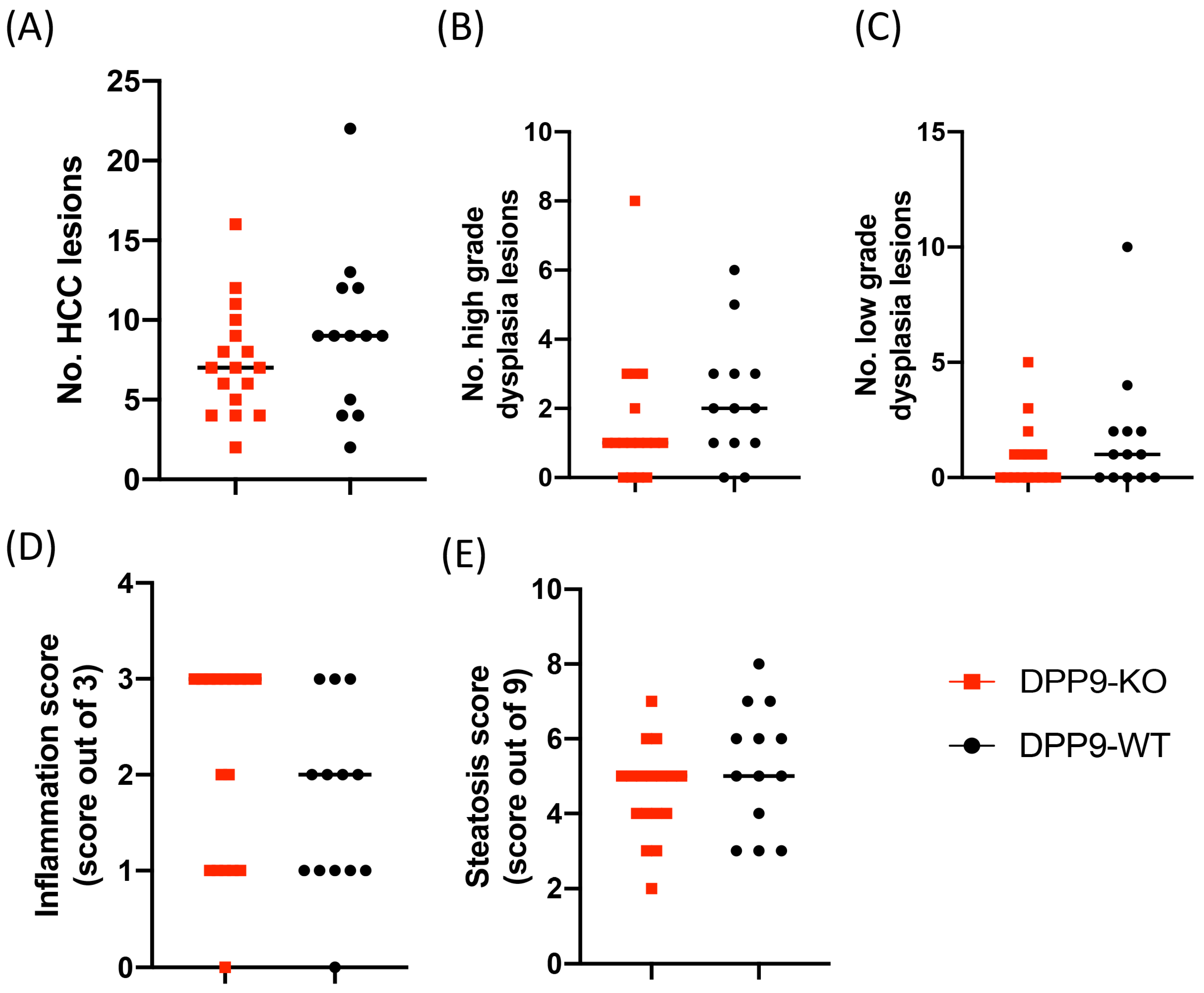
Figure 9.
Crosslinked collagen in the primary HCC model. Quantified by image analysis of Sirius red stain. Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test.
Figure 9.
Crosslinked collagen in the primary HCC model. Quantified by image analysis of Sirius red stain. Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test.
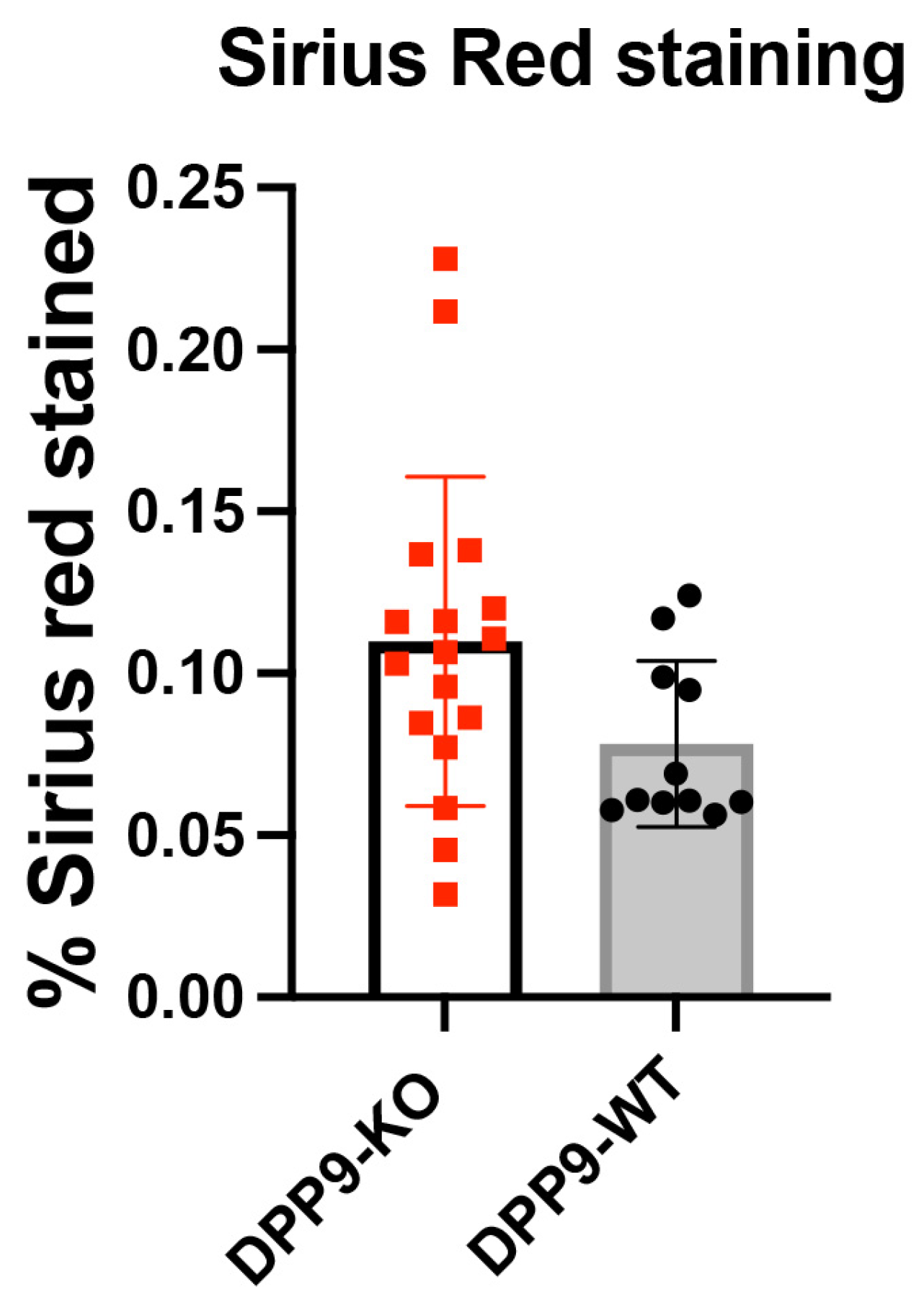
Figure 10.
Inflammasomes assessment. (A) Representative image of caspase-1 immunoblot. (B) Quantification of caspase-1 normalized to loading control -Actin. (C) Representative image of NLRP3 immunoblot. (D) Quantification of NLRP3 expression normalized to loading control -Actin.Mean ± SEM, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test, ***p < 0.001. The centre lane of each gel contained the molecular mass markers.
Figure 10.
Inflammasomes assessment. (A) Representative image of caspase-1 immunoblot. (B) Quantification of caspase-1 normalized to loading control -Actin. (C) Representative image of NLRP3 immunoblot. (D) Quantification of NLRP3 expression normalized to loading control -Actin.Mean ± SEM, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test, ***p < 0.001. The centre lane of each gel contained the molecular mass markers.
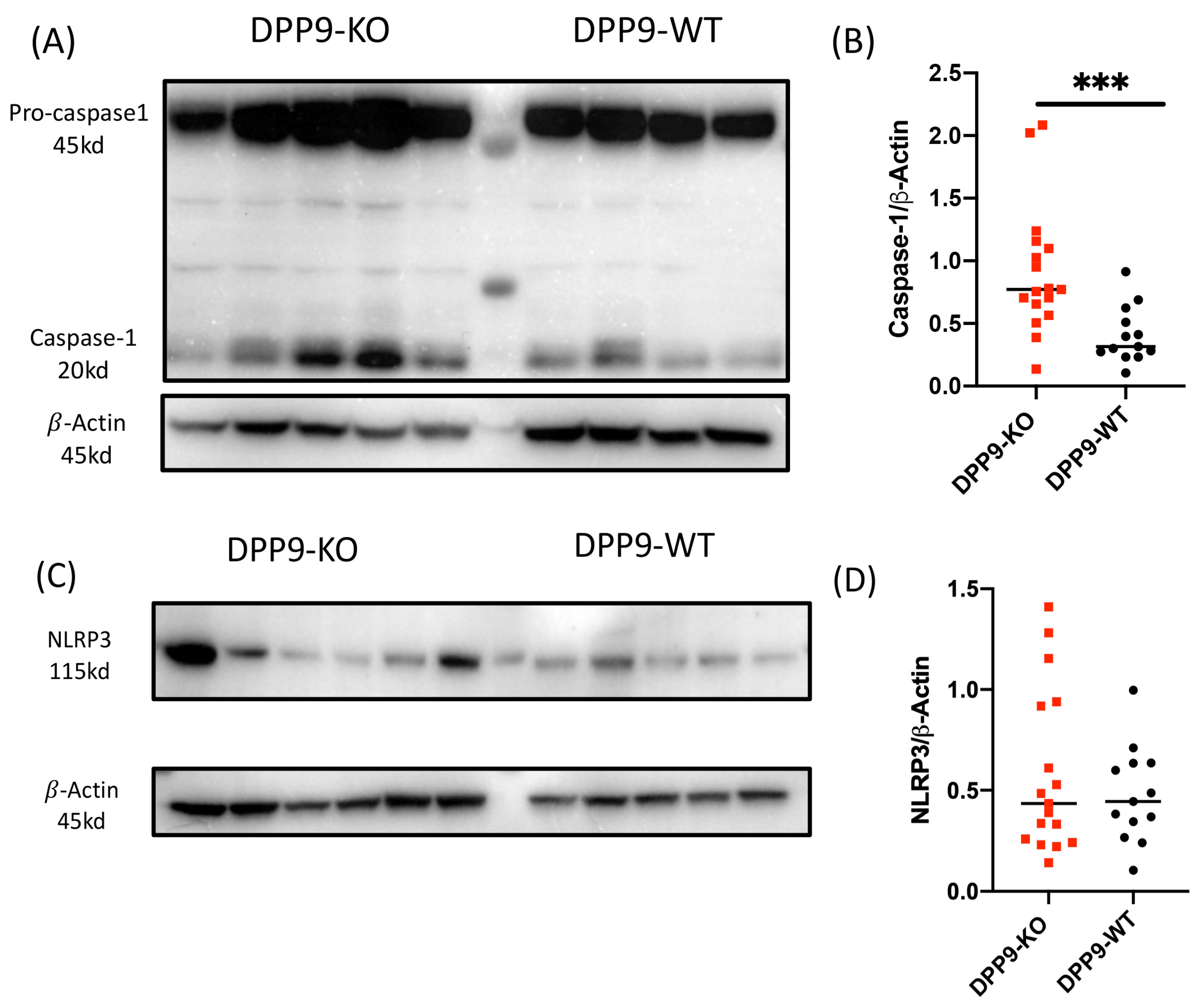
Figure 11.
Quantitative assessment of intrahepatic gene expression. (A-D) Gene expression normalized to housekeeper Hprt1 of individual mice plotted with mean and standard deviation. (E, F) mRNA level of Caspase-1, Nlrp1b and Il18 in primary HCC mouse liver samples and regression analysis. DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test and nonparametric Spearman correlation test, *p < 0.05.
Figure 11.
Quantitative assessment of intrahepatic gene expression. (A-D) Gene expression normalized to housekeeper Hprt1 of individual mice plotted with mean and standard deviation. (E, F) mRNA level of Caspase-1, Nlrp1b and Il18 in primary HCC mouse liver samples and regression analysis. DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test and nonparametric Spearman correlation test, *p < 0.05.
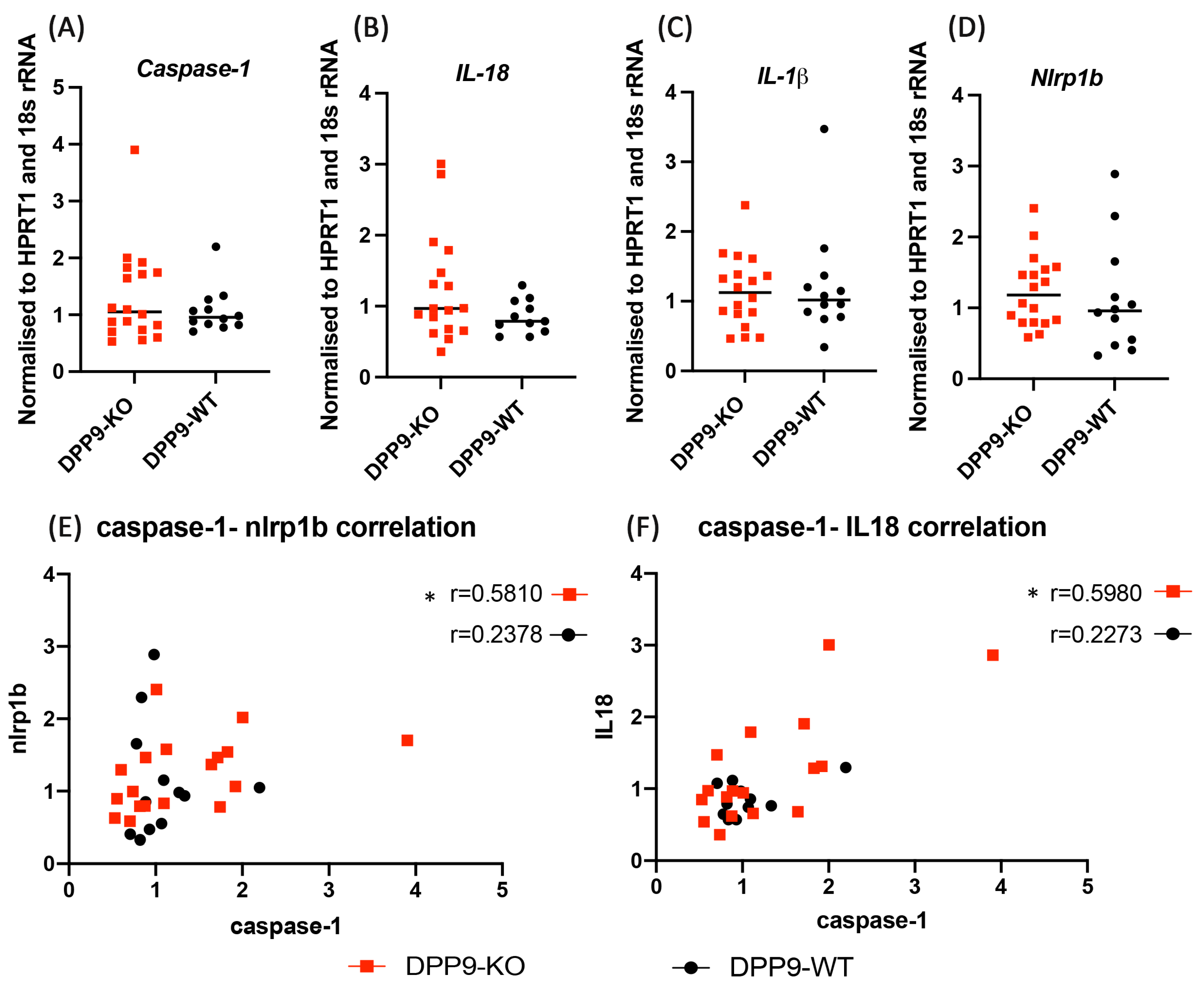
Figure 12.
Intrahepatic IL-1β and IL-18 proteins quantified by ELISA. (A) IL-1β ELISA. (B) Regression analysis of caspase-1 with IL-1β proteins in the primary HCC model. (C) IL-18 ELISA. (D) Regression analysis of caspase-1 with IL-18 proteins in the primary HCC model. Mean ± SD, DPP9-KO n = 17, DPP9-WT n =13. Mann-Whitney test and nonparametric Spearman correlation test.
Figure 12.
Intrahepatic IL-1β and IL-18 proteins quantified by ELISA. (A) IL-1β ELISA. (B) Regression analysis of caspase-1 with IL-1β proteins in the primary HCC model. (C) IL-18 ELISA. (D) Regression analysis of caspase-1 with IL-18 proteins in the primary HCC model. Mean ± SD, DPP9-KO n = 17, DPP9-WT n =13. Mann-Whitney test and nonparametric Spearman correlation test.
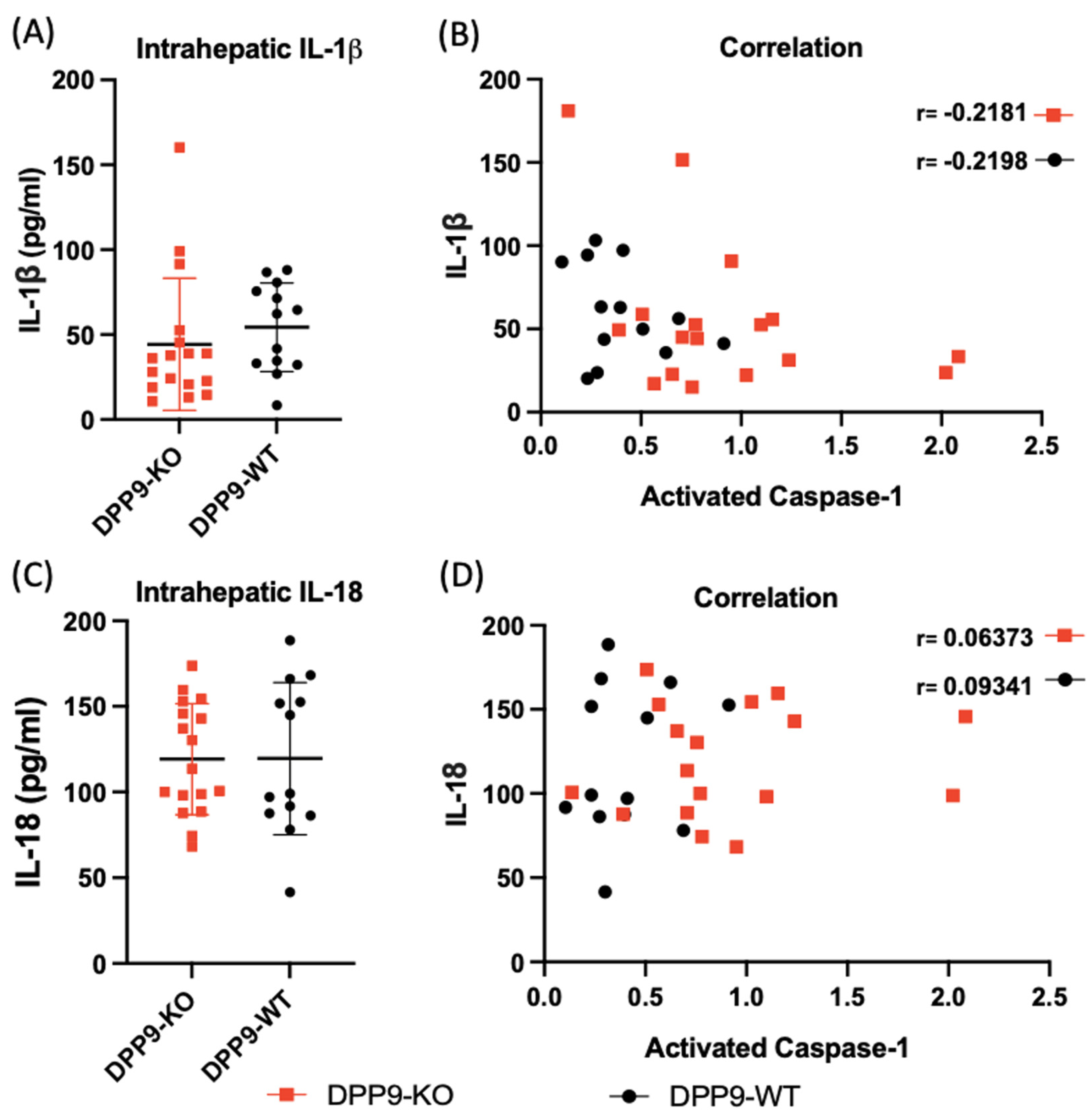
Figure 13.
Quantitative assessment of intrahepatic gene expression. Gene expression normalized to housekeepers Hprt1 of individual mice plotted with mean and standard deviation. DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test, *p < 0.05. **p < 0.01.
Figure 13.
Quantitative assessment of intrahepatic gene expression. Gene expression normalized to housekeepers Hprt1 of individual mice plotted with mean and standard deviation. DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test, *p < 0.05. **p < 0.01.
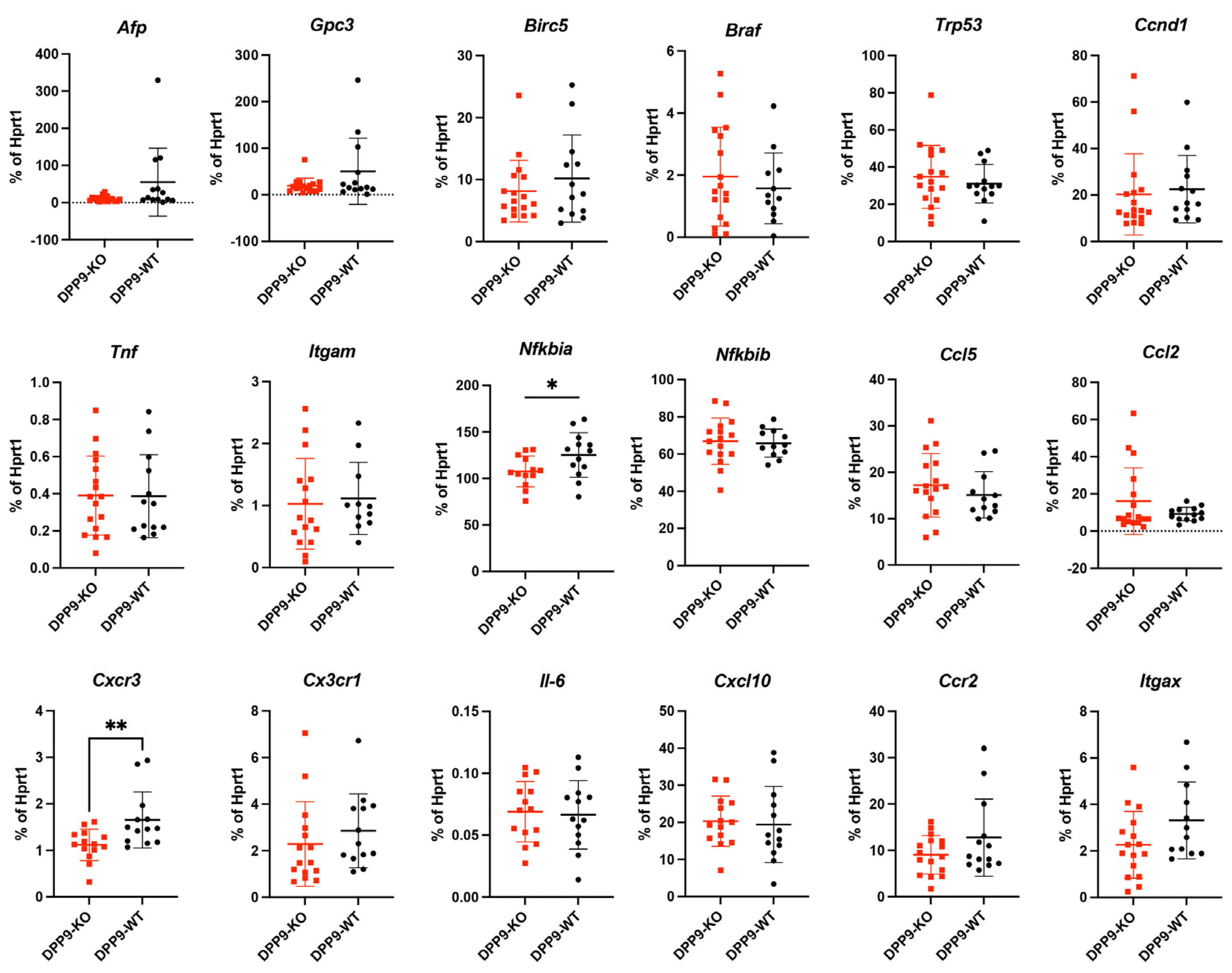
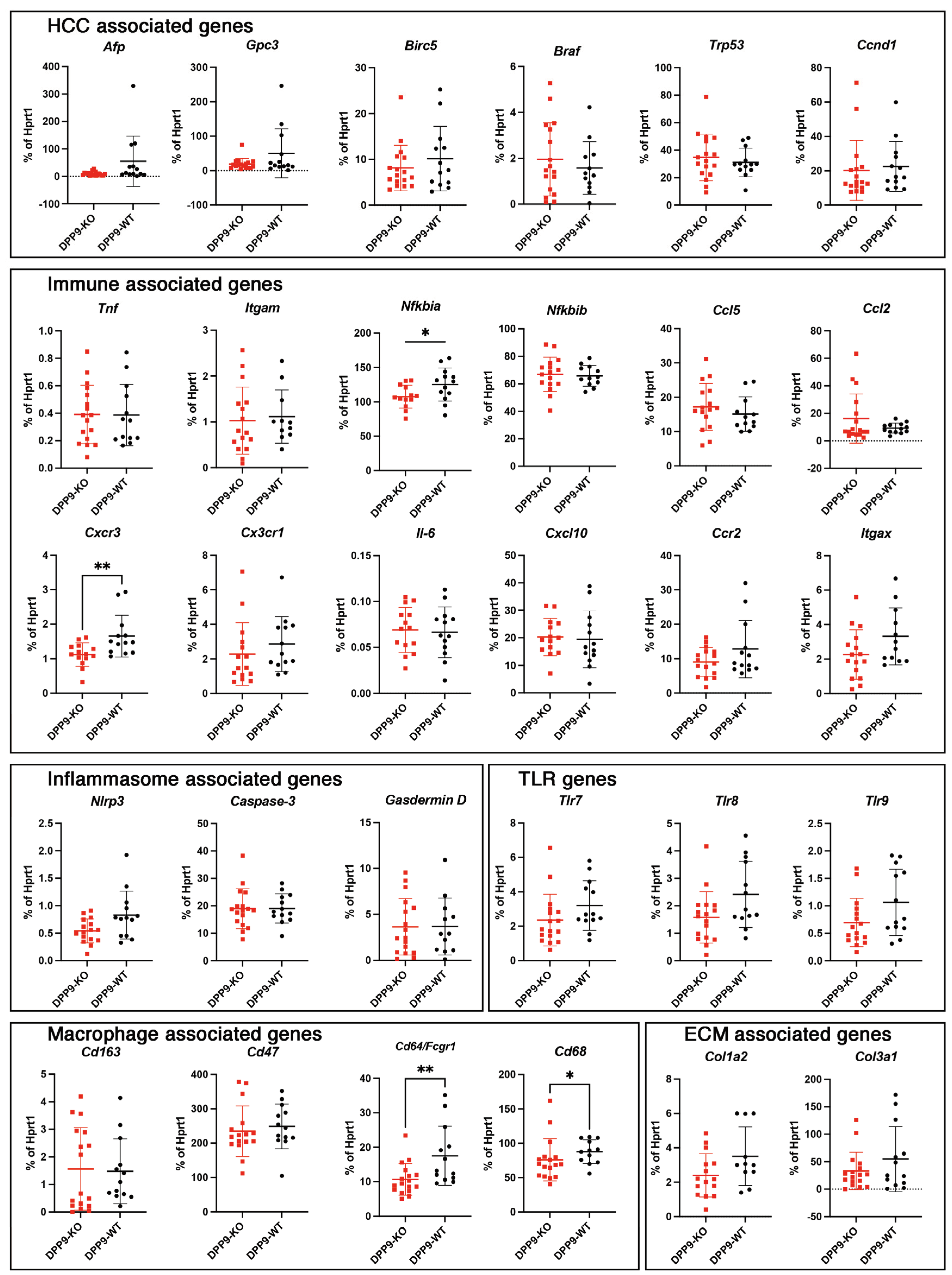
Figure 14.
Quantitation of tumour infiltrating CD8+ T cells. CD8+ T cells were counted on immunostained sections. Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test.
Figure 14.
Quantitation of tumour infiltrating CD8+ T cells. CD8+ T cells were counted on immunostained sections. Mean ± SD, DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney test.
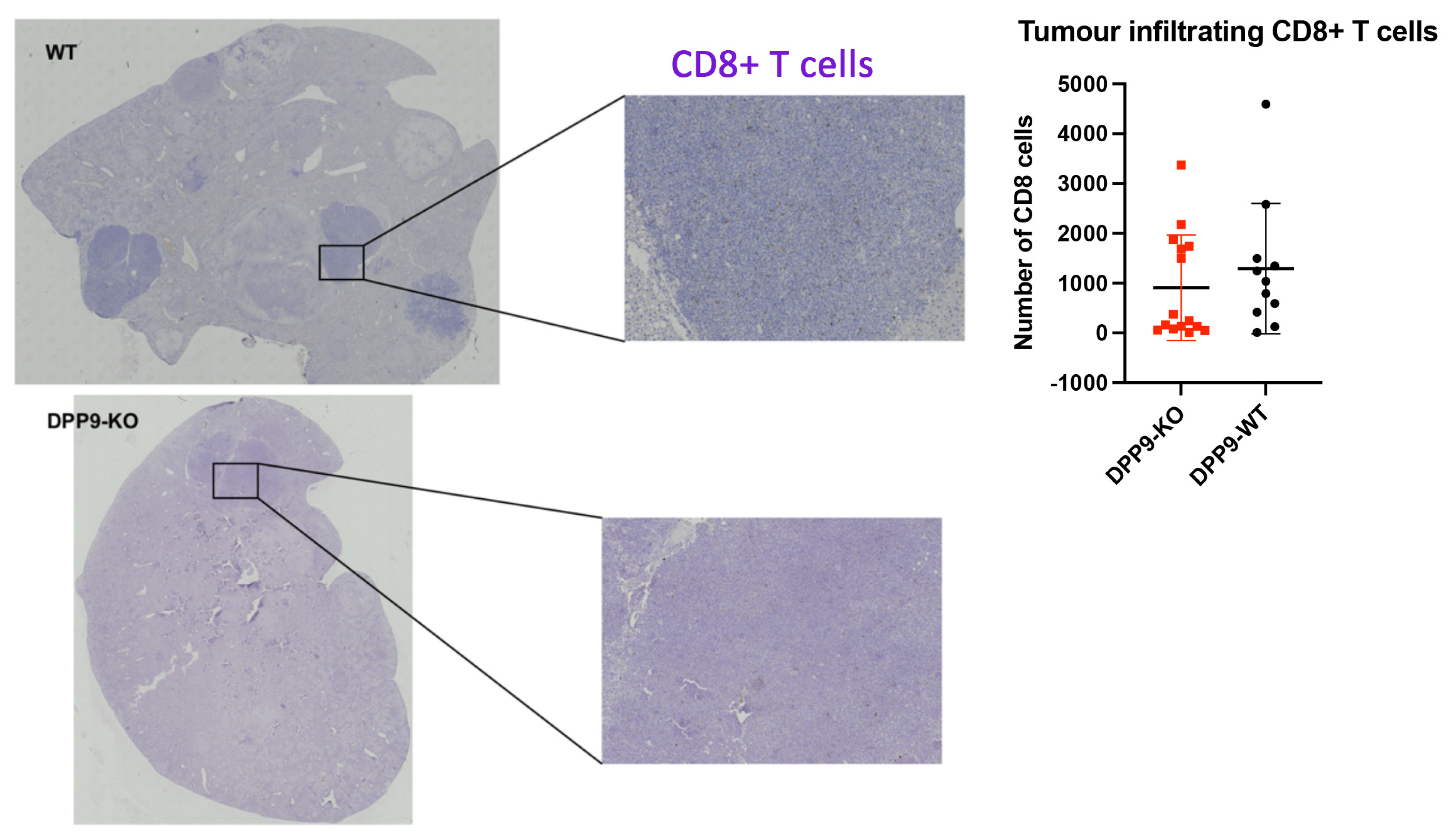
Figure 15.
Measurement of beclin1 and p53. Representative images of Beclin1 (A) and p53 (C) immunoblots. Quantification by densitometry of immunoblots of Beclin1 (B) and p53 (D), normalized to loading control -Actin. Data with mean ± SEM. DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney statistical test, *p < 0.05.
Figure 15.
Measurement of beclin1 and p53. Representative images of Beclin1 (A) and p53 (C) immunoblots. Quantification by densitometry of immunoblots of Beclin1 (B) and p53 (D), normalized to loading control -Actin. Data with mean ± SEM. DPP9-KO n = 17, DPP9-WT n = 13. Mann-Whitney statistical test, *p < 0.05.
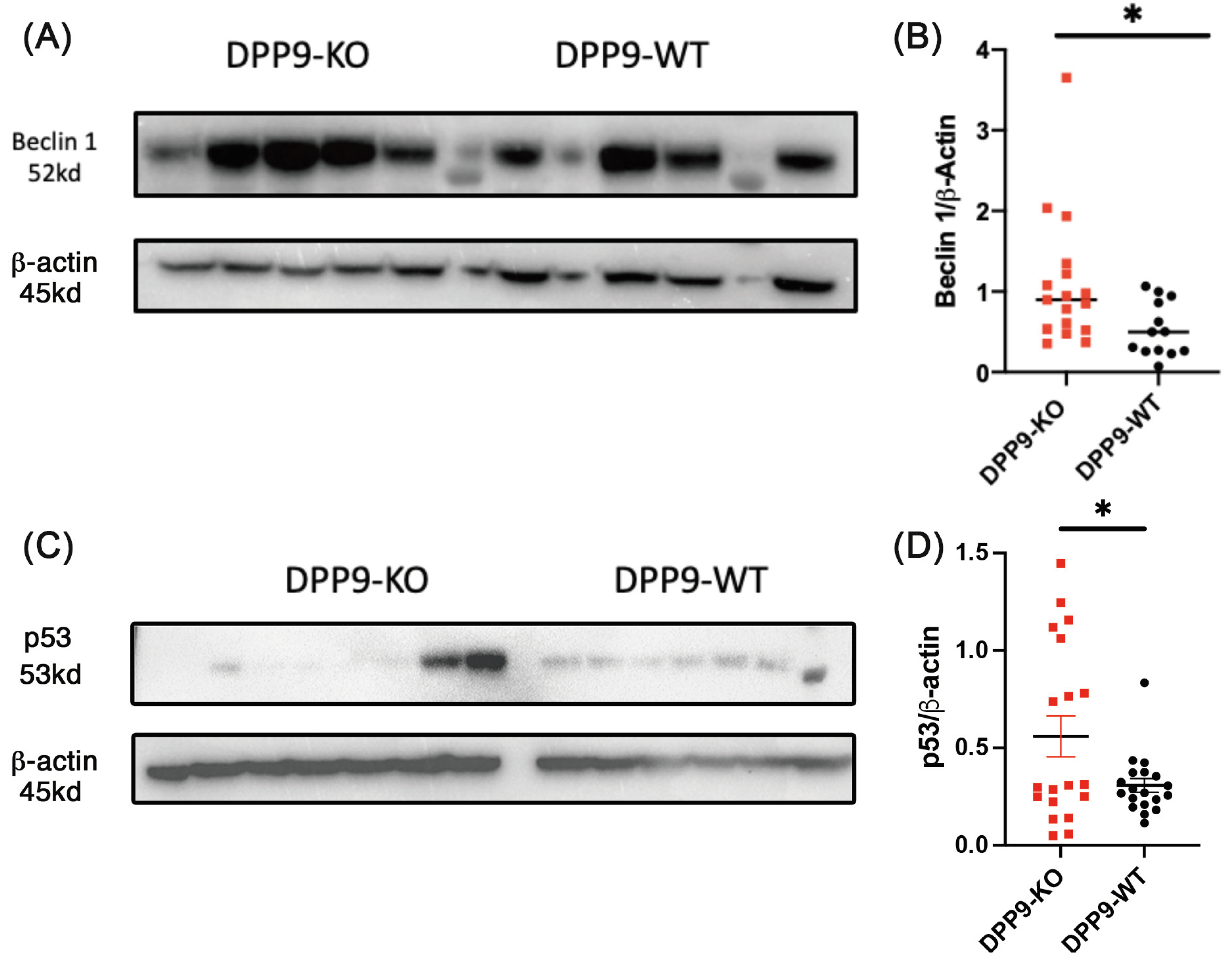
Figure 16.
Summary of outcomes of hepatocyte-specific depletion of DPP9 expression in our experimental model of primary HCC.
Figure 16.
Summary of outcomes of hepatocyte-specific depletion of DPP9 expression in our experimental model of primary HCC.
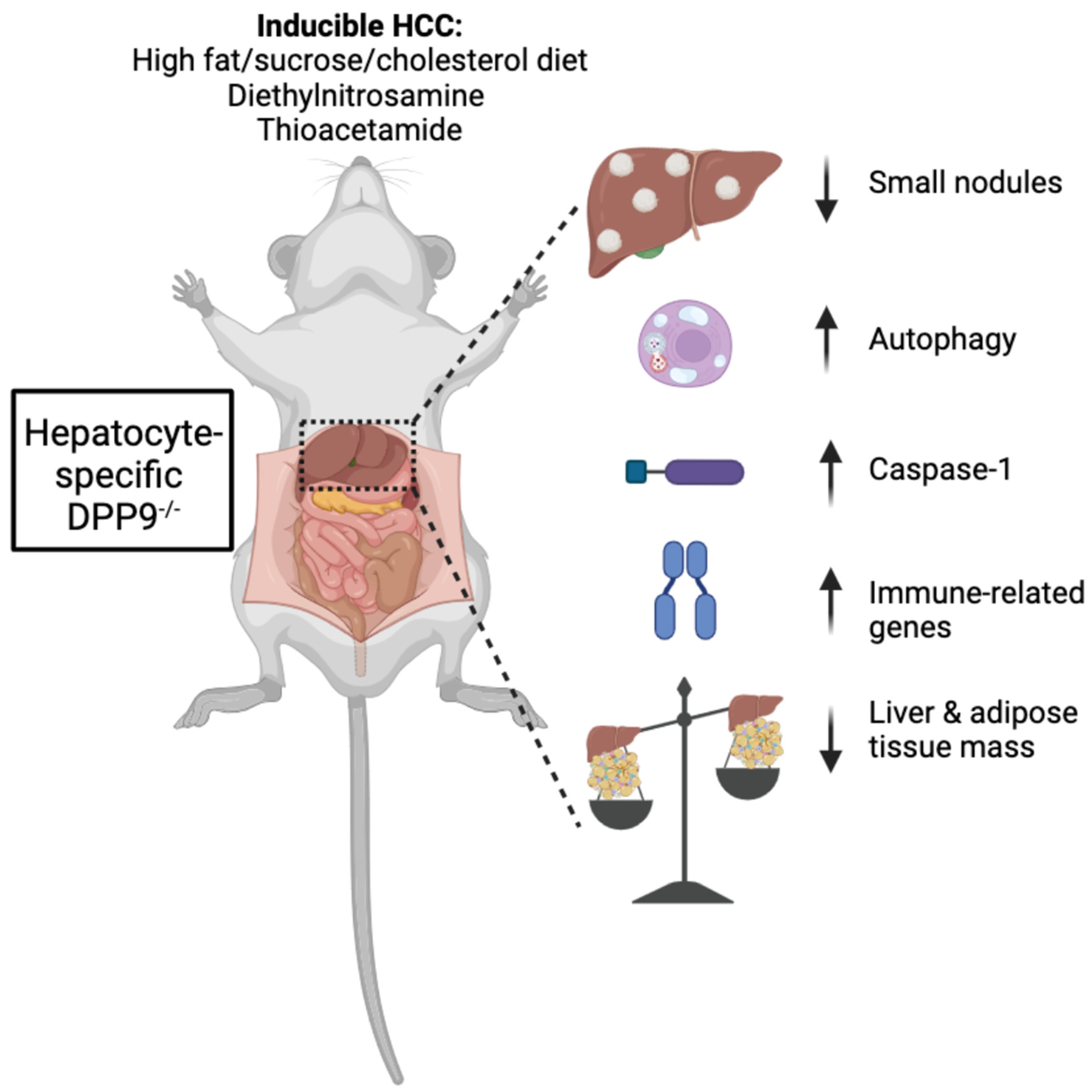

Table 1.
The ingredients of the high fat high sucrose high cholesterol diet.
| Ingredient | Quantity | Catalogue No. | Supplier/source |
|---|---|---|---|
| Lard | 175.16 g | Yorkfoods; Goulburn NSW | |
| Casein | 182.88 g | C7078 | Sigma-Aldrich; St Louis, MO |
| Sucrose | 156.96 g | 904713 | MP biomedical |
| Starch | 150.48 g | 102955 | MP biomedical |
| AIN mineral Mix | 35.72 g | 905455 | MP biomedical |
| Bran | 39.92 g | Coles; Bella Vista, NSW | |
| Methionine | 2.4 g | M9625 | Sigma-Aldrich; St Louis, MO |
| Gelatine | 16.12 g | 041941 | McKenzies; Altona, VIC |
| Choline bitartrate | 3.24 g | C1629 | Sigma-Aldrich; St Louis, MO |
| AIN vitamins | 10.36 g | 960098 | MP Biomedical |
| Cholesterol | 4.2 g | 8503 | Sigma-Aldrich; St louis, MO |
| Safflower Oil | 20.8 mL | oil057 | Melrose; Mt Waverly, VIC |
| Natural Strawberry | 2 drops | 646045 | Queens; Aldery, QLD |
| Essence |
Table 2.
Antibodies. Primary antibodies were diluted in 0.05% (w/v) BSA, 0.1% NaN3 in PBST.
| Antibody | Host species | Supplier | Catalogue No. | Working dilution |
|---|---|---|---|---|
| Dipeptidyl peptidase 9 | Rabbit | Abcam | Ab42080 | 1:100 |
| Dipeptidyl peptidase 9 | Mouse | Origene | TA504019 | 1:1000 |
| Rabbit IgG-HRP | Goat | Dako | P0448 | 1:5000; 1:200 |
| Mouse IgG-HRP | Rabbit | Dako | P0161 | 1:5000; 1:200 |
| Beta actinHRP | Abcam | Ab49900 | 1:50000 | |
| Caspase1 | Mouse | AdipoGen | AG-20B-0048-C100 | 1:1000 |
| Becin1 | Mouse | Genetex | GTX34055 | 1:1000 |
| NLRP3 | Rabbit | Cell Signalling | 1510S | 1:1000 |
| p53 | Rabbit | Cell Signalling | 9282S | 1:1000 |
Table 3.
Taqman probes for quantitative PCR.
| Gene symbol |
Gene name | Primer/probe assay | Gene function | Amplicon length |
|---|---|---|---|---|
| Hprt1 | Hypoxanthine guanine phosphoribosyl transferase1 | Mm00446968_m1 | Housekeeping gene | 65 |
| Afp | Alpha fetoprotein | Mm00431715_m1 | HCC associated | 96 |
| Gpc3 | Glypican 3 | Mm00516722_m1 | HCC associated | 91 |
| Birc5 | Baculoviral IAP repeat-containing 5 | Mm00599749_m1 | HCC associated | 83 |
| Braf | Braf transforming gene | Mm01165837_m1 | HCC associated | 94 |
| Trp53 | Transformation related protein 53 | Mm01731290_g1 | HCC associated | 119 |
| Ccnd1 | Cyclin D1 | Mm00432359_m1 | HCC associated | 58 |
| Il1 | interleukin 1 beta | Mm00434228_m1 | Immune system | 90 |
| Il18 | interleukin 18 | Mm00434226_m1 | Immune system | 141 |
| Nlrp1 | NLR family, pyrin domain containing 1 | Mm01241387_m1 | Immune system | 93 |
| Nlrp3 | NLR family, pyrin domain containing 3 | Mm00840904_m1 | Immune system | 84 |
| TNF | Tumour necrosis factor | Mm00443258_m1 | Immune system | 81 |
| Itgam | Integrin alpha M | Mm00434455_m1 | Immune system | 69 |
| Nfkbia | Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha | Mm00477798_m1 | Immune system | 70 |
| Nfkbib | Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, beta | Mm00456853_m1 | Immune system | 64 |
| Ccl2 | Chemokine (C-C motif) ligand 2 |
Mm99999056_m1 | Immune system | 96 |
| Ccl5 | Chemokine (C-C motif) ligand 5 |
Mm01302427_m1 | Immune system | 103 |
| Cxcr3 | Chemokine (C-C motif) receptor 3 |
Mm99999054_s1 | Immune system | 57 |
| Cx3cr1 | Chemokine (C-X3-X motif) receptor 1 |
Mm02620111_s1 | Immune system | 107 |
| Il6 | Interleukin 6 | Mm00446190_m1 | Immune system | 78 |
| Cxcl10 | Chemokine (C-X-C motif) ligand 10 |
Mm00445235_m1 | Immune system | 59 |
| Ccr2 | Chemokine (C-C motif) receptor 2 |
Mm99999051_gH | Immune system | 60 |
| Itgax | Integrin alpha X | Mm00498701_m1 | Immune system | 93 |
| Tlr7 | Toll-like receptor 7 | Mm00446590_m1 | Immune system | 125 |
| Tlr8 | Toll-like receptor 8 | Mm04209873_m1 | Immune system | 82 |
| Tlr9 | Toll-like receptor 9 | Mm00446193_m1 | Immune system | 60 |
| Nlrp3 | NLR family, pyrin domain containing 3 | Mm00840904_m1 | Inflammasome | 84 |
| Gasdermin D | Gasdermin D | Mm00509958_m1 | Inflammasome | 94 |
| Dpp9 | dipeptidyl peptidase 9 | Mm00841122_m1 | Protease | 61 |
| Dpp8 | dipeptidyl peptidase 8 | Mm00547049_m1 | Protease | 95 |
| Fap | Fibroblast activation protein | Mm00484254_m1 | Protease | 107 |
| Dpp4 | dipeptidyl peptidase 4 | Mm00494538_m1 | Protease | 88 |
| Casp1 | Caspase 1 | Mm00438023_m1 | Apoptosis/pyroptosis | 99 |
| Casp3 | Caspase 3 | Mm01195085_m1 | Apoptosis | 100 |
| Cd163 | CD163 antigen | Mm00474091_m1 | Macrophage associated | 83 |
| Cd47 | CD47 antigen | Mm00495011_m1 | Macrophage associated | 77 |
| CD64/Fcgr1 | Fc receptor, IgG, high affinity I | Mm00438874_m1 | Macrophage associated | 58 |
| Cd68 | CD68 antigen | Mm00839636_g1 | Macrophage associated | 86 |
| Col1a2 | Collagen, type I, alpha 2 | Mm00483888_m1 | ECM | 67 |
| Col3a1 | Collagen, type III, alpha 1 | Mm00802300_m1 | ECM | 88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



