
Submitted:
30 March 2024
Posted:
01 April 2024
You are already at the latest version
Abstract
Thyroid hormones are essential for regulating the metabolic rate, growth, development, and maintenance of many physiological processes in vertebrates. They are synthesized by the thyroid gland, which is an ancient organ that evolved early in vertebrate evolution. This study aims to characterize the evolutionary path of polymorphic variants associated with the hypothalamus-pituitary-thyroid (HPT) axis by comparing ancient DNA (aDNA) sequences of Late Pleistocene-Holocene hominin populations with those of modern humans. We evaluated the genetic sequences of several populations, including Neanderthals, Denisovans, Palaeolithic hunter-gatherers from European Russia and Sunghir, and Neolithic hunter-gatherers/farmers from Anatolia focusing on genes involved in the development of the thyroid gland and thyroid hormone (TH) biosynthesis, secretion, and transport. Results showed interesting variants in the DIO2 (rs225014), TPO (rs4927611), and TG (rs2069556 and rs1133076) genes. All SNPs seemed to confirm that Neanderthals, and in part Denisovas, were physiologically hypothyroidic. Probably they had lower T3 levels due to defective production or peripheral conversion. From the moment that the most favourable alleles in terms of T3 production appear during the Paleolithic, we are inclined to assume that their selection was linked to environmental pressure.Our study supports insight into the evolutionary history of the endocrine system and can help in providing understanding into the evolution of physiological systems and their adaptation to changing environments. Understanding the evolution of thyroid hormones can also shed light on the mechanisms underlying thyroid disorders in humans.
Keywords:
thyroid
; Neanderthals
; Denisovas
; Anatomiccaly modern human
; hypothyroidism
Introduction
Thyroid hormones (TH) play an essential role in growth and differentiation, control of metabolism, and other physiological functions in virtually every human tissue. Thyroxine (T4), the major prohormone secreted by the thyroid gland, is largely converted into the active hormone 3,3′,5-triiodothyronine (T3) in peripheral tissues by specific enzymes called deiodinases [Deiodinase 1 (DIO1) and Deiodinase 2 (DIO2)] [1]. Circulating TH levels are regulated via a negative feedback mechanism by the hypothalamus–pituitary–thyroid (HPT) axis. Thyrotropin-releasing hormone (TRH), produced by hypothalamus, stimulates anterior pituitary to release thyroid-stimulating hormone (TSH) that, binding TSH receptor (TSHR) expressed on the thyroid follicular cells, stimulates the TH synthesis. In turn, T4 and T3 negatively regulate TSH production [2]. Hypothyroidism and hyperthyroidism are thyroid dysfunctions caused by an increase or decrease of TSH levels with or without decrease or increase of T4 levels, respectively. Hypothyroidism may be due to a number of causes including autoimmune disease, thyroid surgery, iodine deficiency, pituitary disorders and congenital diseases [3]. Congenital hypothyroidism (CH) is a thyroid hormone deficiency present at birth for permanent or transient causes [4]. Permanent CH can be primary, secondary (for TRH resistance or defects in TSH production) or peripheral [for defects in thyroid hormone transport genes, as solute carrier family 16 member 2 (SLC16A2) and solute carrier family 16 member 10 (SLC16A10), or resistance to TH]. Primary CH is related to conditions of dysgenesis or dyshormonogenesis, caused by alterations in genes involved in the growth and development of thyroid and in the TH biosynthesis and secretion, respectively [4]. Genes associated with dysgenesis are thyroid transcription factor-1 (TTF-1), thyroid transcription factor-2 (TTF-2), paired box 8 (PAX-8) and TSHR, while dyshormonogenesis is associated with gene defects in thyroperoxidase (TPO), thyroglobulin (TG), dual oxidase 2 (DUOX2), sodium-iodide symporter (NIS), solute carrier family 5 member 5 (SLC5A5), and pendrin (SLC26A4) [5,6]. In addition to known mutations, the role of single nucleotide polymorphisms (SNPs) has been also investigated in CH. Different studies reported as polymorphic variants in TPO, TSHR and TG genes can represent a genetic risk factor for dishormonogentic CH [7,8,9,10].
SNPs may be the basis of the evolutionary process, since they are heritable and modified by natural selection. During human evolution, interaction with factors such as climatic conditions, nutrients availability, and lifestyle habits have allowed the selection of favourable features [11]. Natural selection has led to a gradual modification of genetic composition of population; individuals who were better adapted passed on their genes, including those conferring these benefits with an increasing frequency [12]. In the process of adaptation, hormones and the endocrine system certainly have played a crucial role. Thanks to genome analysis of ancestral humans [12,13,14,15,16,17] it is possible to better understand human evolution and deepen the functionality of TH in our ancestors. In a previous work, we showed an evolutionary perspective for the rs225014 (p.Thr92Ala) variant of the DIO2 gene from Neanderthals to Anatomically Modern Humans (AMH) [18]. In fact, Neanderthals and Denisovans, with a diet characterised by low carbohydrate intake, displayed the alanine amino acid at position 92, associated with a reduced production of T3, while Modern Humans, with a high carbohydrate diet, exhibited threonine that is related to an increased production of T3 [18]. These findings have shown that the DIO2 rs225014 variant has been positively selected under particular living conditions and food habits. Considering this evidence, we investigated other variants in genes involved in the development of thyroid gland and TH biosynthesis, secretion and transport, comparing ancestral genomes of different human populations with contemporary humans. By studying a larger panel of genes, we will have the possibility to reconstruct with increasing accuracy the changes that TH and endocrine system have undergone in the course of human evolution.
Methods
For the present study, we analysed genomic and/or the protein sequence data for the DIO1, DIO2, DIO3, TPO, TG, DUOX2, PAX8, SLC5A5, SLC16A2, SLC16A10 and TSHR genes. These data were extracted from publically available genome/exome sequence databases including H. neanderthalensis, H. denisovans and representatives of Anatomically Modern Humans (AMH) of Palaeolithic and Neolithic. For Neanderthals (https://projects.ensembl.org/neandertal/) and Denisovans (https://www.eva.mpg.de/genetics/genome-projects/denisova/) we referred to public databases. The Neandertal Genome Project has sequenced six samples: three specimens from the Vindija Cave in Croatia; one individual from Mezmaiskaya in the Altai Mountains (Russia), a fossil found in El Sidron cave in Asturias (Spain) and a little fraction from Neander valley (Germany). The Denisova DNA sequence derived from a phalanx bone excavated from Denisova Cave in the Altai Mountains in southern Siberia. For Palaeolithic, we employed the sequence data of 1 subject from Kostenki 14 in European Russia, dating to 38,700-36,200 years ago, one of the oldest fossils of Anatomically Modern Humans from Europe (PRJEB7618) [19], and 5 individuals from the archaeological site of Sunghir (Russia), dated to about 34.000 BP (PRJEB22592) [20]. The Neolithic population was represented by 9 central Anatolian Neolithic individuals (PRJEB14675) (available sequences from 5 subjects only) [21]. Locations of archaeological sites are depicted in Figure 1. The raw sequence data were downloaded either from the Sequence Read Archive (SRA) [22] of NCBI (www.ncbi.nlm.nih.gov) or from the Department of Evolutionary Genetics (www.eva.mpg.de) and then processed using the SRA toolkit for further analysis. A blast similarity-based search with the ncbi-blast-2.9.0+ program was carried out to extract gene sequences of ancient humans [23].
Allele/genotype frequencies for contemporary humans were obtained from the Ensemble genome browser (www.ensembl.org). Frequencies in different extant populations were derived from the 1000 Genomes Project (www.internationalgenome.org), the largest public catalogue of human variation and genotype data, and from The Genome Aggregation Database - gnomAD (https://gnomad.broadinstitute.org), a resource that aggregates both exome and genome sequencing data from a wide variety of large-scale sequencing projects.
Principal component analysis (PCA) was used to obtain a dimensionality reduction, increasing the interpretability of data while preserving as much of information as possible, and improving the visualisation of multidimensional data. In this system, each dimension corresponds to a SNP and the genomes of all the individuals are included in the study. The analysis was performed using the web tool ClustVis (https://biit.cs.ut.ee/clustvis/#mathematics) [24].
Results
We analysed several populations belonging to different periods and regions (Figure 1). Sequences from Neanderthal and Denisova databases, Palaeolithic hunter-gatherers from European Russia (n=1) and Sunghir (n=5) and Neolithic hunter-gatherers/farmers from Anatolia (n=5) were analysed for DIO1, DIO2, DIO3, TPO, TG, DUOX2, PAX8, SLC5A5, SLC16A2, SLC16A10 and TSHR genes. For DIO1, DIO3, DUOX2, PAX8, SLC5A5, SLC16A2 and SLC16A10 we did not find any differences in SNPs described to be associated with congenital hypothyroidism, thyroid defects and/or inhibited enzyme activity. On the contrary, for DIO2, TPO, TG, and TSHR gene sequences displayed relevant variants. Results from alignments are described below.
Type II Iodothyronine Deiodinase (DIO2)
We have already reported (18) that Neanderthals and Denisovans displayed only the G allele at the rs225014 SNP (c.274A>G), which encodes for an alanine on the amino acid level. We observed evidence of the AA and heterozygous AG genotypes in the population from Sunghir (Table 1), in which the presence of the homozygous GG genotype persisted. Thus, threonine as the prevalent amino acid seems to have been established since Palaeolithic (Table 1).
Thyroperoxidase (TPO)
TPO gene sequence was analysed, and three polymorphisms were found: rs2175977 c.1193G>C (p.Ser398Thr); rs4927611 c.796G>T (p.Ala257Ser) and rs732609 c.2173A>C (p.Thr725Pro). For rs2175977, Neanderthal displayed the CC genotype corresponding to threonine at position 398 (Table 1). The region was not covered in the Denisova database. Both hunter-gathered populations from the Palaeolithic had GG, CC or GC genotypes. Similar findings were observed for the populations from Neolithic (Table 1). For rs4927611, Neanderthals were homozygous for the rare allele T (corresponding to serine), whereas Denisova were homozygous for the allele G (corresponding to alanine). In the analysed populations, the alanine appeared for the first time in the group of hunter-gatherers from Sunghir during the Paleolithic (Table 1) and confirmed its presence during Neolithic (Table 1). The last SNP, rs732609, was present in the Neanderthal population as CC genotype (proline). On the contrary, Denisova displayed the AA genotype (threonine). During the Palaeolithic, in the hunter-gatherer population from Sunghir we observed a coexistence of AA, CC and AC genotypes. Again, the three genotypes were present in Anatolia during the Neolithic in hunter-gatherer/farmers population.
Tyreoglobulin (TG)
We also found three interesting SNPs for TG: rs2069556 c.3935A>G (p.Asp1312Gly); rs180223 c.2200T>G (p.Ser734Ala) and rs1133076 c.1988G>A (p.Arg986Gln). For rs2069556, Neanderthal displayed only the GG genotype and Denisova the AA. The heterozygous asset was evident starting from Palaeolithic in the population from Sunghir. In the Neolithic populations we found both the AA and GG genotypes corresponding to aspartate and glycine amino acids, respectively (Table 1). For rs180223, Neanderthal were homozygous for the allele G (alanine); Palaeolithic individuals and Neolithic population from Anatolia had both alanine and serine at residue 734 (without the evidence of heterozygous subjects) (Table 1). For the last SNP, rs1133076, Neanderthal showed the GG genotype, corresponding to arginine, while Denisovas were homozygous for the allele A, corresponding to glycine. For the other analysed populations, the sequence of this region was available for very few individuals. So, two out of ten subjects from Sunghir (Palaeolithic period) were genotyped, one showing the GG genotype and the other the AA genotype, 1 Palaeolithic from Russia homozygous for the allele G, and 3 out of 7 subjects belonging to the Neolithic, with hunter-gatherer/farmer habits, with both GG genotype (n=2) and AA genotypes (n=1) (Table 1).
Thyroid-Stimulating Hormone Receptor (TSHR)
For TSHR we found SNP rs1991517 c.2181G>C (p.Glu727Asp). Both Neanderthals and Denisovans were homozygous for the allele C, as well as most of the analysed subjects belonging to different periods (Table 1). Being, the C allele (corresponding to aspartic acid) the most represented, according with 1000 Human Genome, we found of interest that 2 out of 10 Palaeolithic hunters-gatherers from Sunghir displayed the heterozygous G/C genotype and 1 out of 9 Neolithic hunter-gatherers/farmers from Anatolia was homozygous for the rare allele G (glutamic acid) (Table 1).
PCA Plot
Based on the SNPs included in the study, we visualised and compared the genomes of the individuals with available sequences by plotting them in an 8-dimensional space, where each dimension corresponds to a SNP. In this multidimensional space, the closer two individuals appear, the more similar their SNP profiles are. To improve graphical visualisation, we reduced the space by principal component analysis (PCA) (24). The SNP data clearly separated H. neanderthalensis from Anatomically Modern Humans and from H. denisovans (Figure 2A and 2B), the latest showing a pattern similar to AMH. In addition, Neolithic individuals displayed a specific profile and localised in a subgroup, together with some Palaeolithic individuals (SI and SIV in Table 1). Analysis of the PCA axes also showed that the most important factors explaining the first dimension of the SNP projection separating H. neanderthalensis from AMH were rs225014 in DIO2 gene and rs2069556 in TG gene. The second principal component was mainly explained by rs4927611 in TPO and rs1133076 in TG genes, respectively (Figure 2A and 2B).
Discussion
The aim of this study was to characterise the evolutionary path of polymorphic variants associated with HPT axis by comparing available ancient DNA (aDNA) sequences of Late Pleistocene-Holocene hominin populations, who lived in different geographical areas and had different habits (such as Neanderthals, Denisovans), with those of AMH from Upper Paleolithic until Contemporary Humans. In this way, we have analysed a long period spanning from Paleolithic to modern times contributing to shed some light on the evolutionary history of the endocrine system. The most interesting variants that arise from our analysis were found in DIO2, TPO and TG genes.
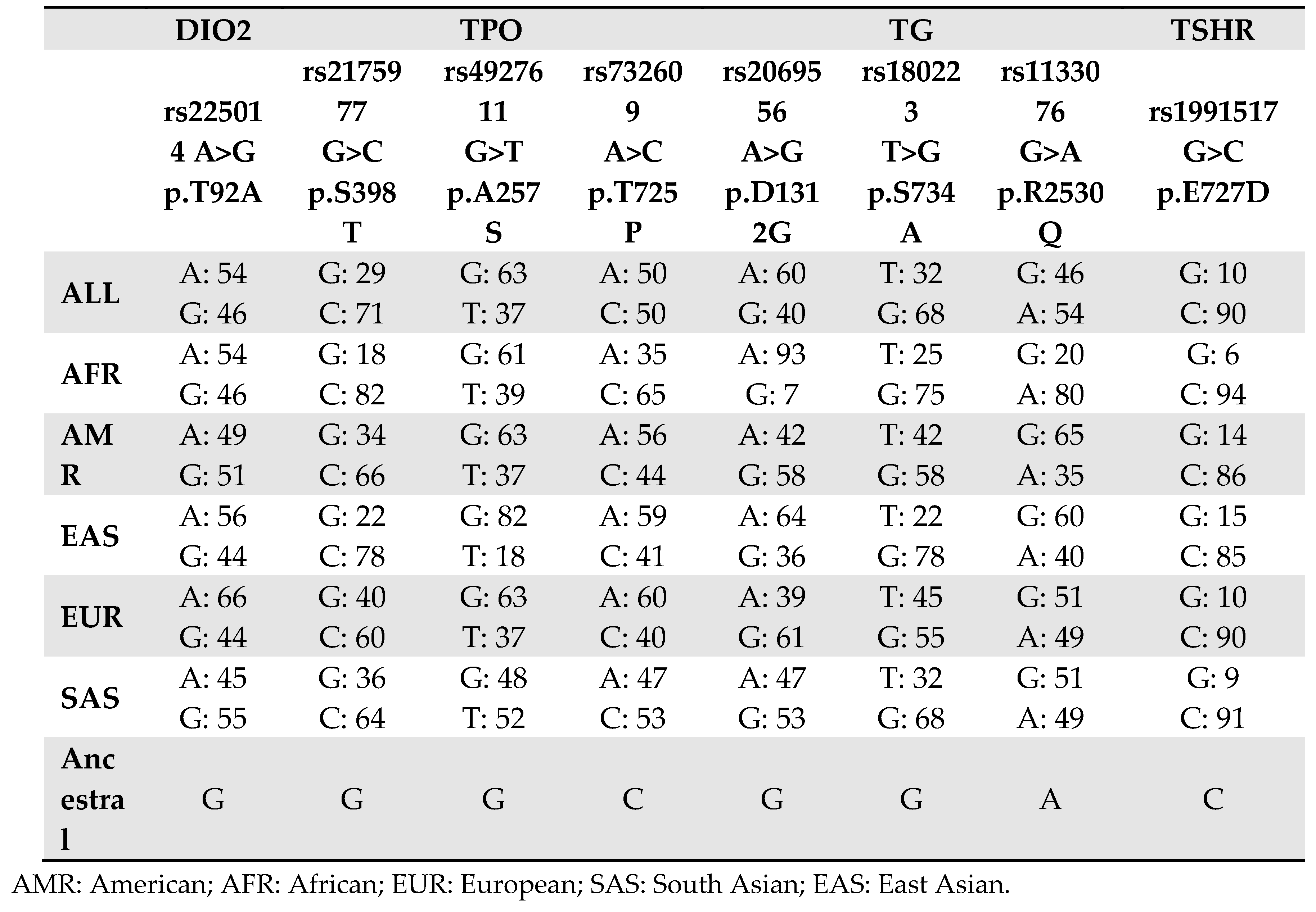
Regarding rs225014 SNP in the DIO2 gene, we have already described (18) results obtained for Neanderthal DNA isolated from three individuals found in the Vindija Cave (Croatia). In the present article, we expand the analysis to three more individuals from different area, and one individual from the Altai Mountains (Russia), a fossil found in El Sidron cave (Spain) and a fragment from Neander valley (Germany), avoiding like that the bias that fossils found at the same site might originate from genetically related individuals. Again, we found that Neanderthals (and Denisovas) had the ancestral allele (G) corresponding to Alanine, the amino acid responsible for a decreased activity of desiodase 2 enzyme (18). The A allele, associated with a more performant enzyme with increased conversion of T4 in T3, has been present since the Paleolithic and appears to be a characteristic trait of HS on all continents (Table 2).
Regarding the rs2069556 (p.Asp1312Gly) SNP in the TG gene, it is interesting to note that minor allele (G) was present in Neanderthals and is now the more frequent in Eurasia, while is rare in Africa. A similar situation is also observed for the rs1133076 (p.Arg2530Gln) SNP, suggesting that these polymorphisms could be of Neanderthal origin in the European-Asiatic populations, where they asserted themselves over the other allele still predominant in Africa. The role of these SNPs remains however unclear. Both of them have been reported to be associated with iodotyrosyl coupling defect, also known as CH due to dyshormonogenesis (https://www.ncbi.nlm.nih.gov/clinvar/RCV000280505/ and https://www.ncbi.nlm.nih.gov/clinvar/RCV000329782/), as “benign” variants. Their high frequencies in the modern populations seem to rule out the hypothesis of a coupling defect, resulting in ineffective formation of T4 and T3 again with a reduction in total T3 and the development of hypothyroidism [25]. However, their role still needs to be elucidated.
Similarly to what observed for TG, the rs4927611 (p.Ala257Ser) SNP in the TPO gene has been reported to be associated with dyshormonogenesis in congenital hypothyroidism (CH) [7,26,27,28,29]. In this case, structural modelling showed that the SNP is lying in the entrance of the active site of TPO enzyme. TPO is a 933 amino acid, type I transmembrane glycoprotein found as a dimer on the apical surface of thyroid follicular cells, which participates in the iodination of tyrosine residues in thyroglobulin and phenoxy-ester formation between pairs of iodinated tyrosines to generate TH. For rs4927611, the non-ancestral T allele (associated with serine), found in Neanderthals, generally represents, today, the minor allele, in all world populations, including Africa (Table 2). In TPO, we found also of interest the variant rs732609 corresponding to a p.Thr725Pro substitution. In this case Neanderthal (but not Denisova) displayed the ancestral allele C which represents the minor allele in modern populations except Africa. Several studies report an association of the C allele with autoimmune hypothyroidism, correlated anti-TPO levels and disease severity [7,28]. Moreover, Begum and colleagues [29], using an in silico approach, evaluated the p.Thr725Pro binding energy and the interactions between the crucial residues His239, Arg396, Glu399, and His494 of TPO protein and heme, demonstrating that the p.Thr725Pro affected the interactions more severely than the other SNPs concluding for a damaging effect on the TPO protein activity. Evidence of an inactivated effect of p.Thr725Pro on the enzyme was provided also at functional level [28].
It is interesting to note that these SNPs allow clearly separating H. neanderthalensis from Anatomically Modern Humans. On the other hand, H. denisovans shows a pattern quite similar to AMH, mainly to Paleolithic individuals. Denisovans in Altai are a western extension of a much larger population originating in central and southwestern Asia [30,31]. Thus, they could have adapted to a climate more similar to that typical of the regions where AMH evolved, with a diet already partially changed [32,33]. Further analyses are required to confirm this hypothesis, when more Denisova genomes become available.
Based on our genome comparisons, again Neanderthals, and in part Denisovas, appear to be physiologically hypothyroidic. Polymorphisms found in genes crucial for TH production, confirm that they may probably have lower T3 levels due to defective production or peripheral conversion. From the moment that the most favourable alleles in terms of T3 production appear during the Paleolithic, we are inclined to assume that their selection was linked to environmental pressure. Neanderthals diet was mainly based on dark meat with low intake of fruit and vegetables [34,35] and limited (but probably sufficient) intake of iodine. During the Paleolithic, the diet was more diversified and included plants (i.e. tubers, seeds, nuts, wild-grown barley, legumes, and flowers), shellfish and other smaller fish, a variety of insects and their products (i.e. honey, and honeycombs) while only a 3% of the whole diet was constituted by meat [36]. Finally, the transition to Neolithic food producers, associated with a high carbohydrate diet, may have concluded the positive selection of alleles associated with higher T3 production and their attestation in modern populations.
Funding
No funds were received for this research.
Conflict of Interest
The authors have no conflict of interest to declare.
References
- Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122(9):3035-43. [CrossRef]
- Ortiga-Carvalho TM, Chiamolera MI, Pazos-Moura CC, Wondisford FE. Hypothalamus-Pituitary-Thyroid Axis. Compr Physiol. 2016;6(3):1387-428. [CrossRef]
- Chaker L, Razvi S, Bensenor IM, Azizi F, Pearce EN, Peeters RP. Hypothyroidism. Nat Rev Dis Primers. 2022;8(1):30. Erratum in: Nat Rev Dis Primers. 2022;8(1):39. [CrossRef]
- Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. 2010;5:17. [CrossRef]
- Park SM, Chatterjee VK. Genetics of congenital hypothyroidism. J Med Genet. 2005;42(5):379-89. [CrossRef]
- Grasberger H, Refetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr. 2011;23(4):421-8. [CrossRef]
- Su Y, Wang J, Zhou J, Chen Y, Zhao H, Zeng Y, Lin F, Zhang H, Zhu W, Chen H. Association of thyroperoxidase gene polymorphisms with dyshormonogenesis in congenital hypothyroidism. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2015;32(6):861-5. Chinese. [CrossRef]
- Kollati Y, Akella RRD, Naushad SM, Thalla M, Reddy GB, Dirisala VR. The rs1991517 polymorphism is a genetic risk factor for congenital hypothyroidism. 3 Biotech. 2020;10(6):285. [CrossRef]
- Kollati Y, Akella RRD, Naushad SM, Borkar D, Thalla M, Nagalingam S, Lingappa L, Patel RK, Reddy GB, Dirisala VR. Newborn screening and single nucleotide variation profiling of TSHR, TPO, TG and DUOX2 candidate genes for congenital hypothyroidism. Mol Biol Rep. 2020;47(10):7467-7475. [CrossRef]
- Kollati Y, Akella RRD, Naushad SM, Patel RK, Reddy GB, Dirisala VR. Molecular insights into the role of genetic determinants of congenital hypothyroidism. Genomics Inform. 2021;19(3):e29. [CrossRef]
- Cavalli-Sforza LL. Human evolution and its relevance for genetic epidemiology. Annu Rev Genomics Hum Genet. 2007;8:1-15. PMID: 17408354. [CrossRef]
- Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat Rev Genet. 2003;4(2):99-111. [CrossRef]
- Poinar HN (1999) DNA from fossils: the past and the future. Acta Paediatr Suppl 88:133–140. https ://doi.org/10.1111/j.1651-2227.1999.tb144 23.x.
- Scholz M, Bachmann L, Nicholson GJ, Bachmann J, Giddings I, Rüschoff-Thale B, Czarnetzki A, Pusch CM. Genomic differentiation of Neanderthals and anatomically modern man allows a fossil-DNA-based classification of morphologically indistinguishable hominid bones. Am J Hum Genet 2020;66:1927–1932. https ://doi.org/10.1086/302949.
- Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J, Chen F, Platt D, Pääbo S, Pritchard JK, Rubin EM. Sequencing and analysis of Neanderthal genomic DNA. Science 2006;314:1113–1118. https ://doi.org/10.1126/scien ce.11314 12.
- Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, Du L, Egholm M, Rothberg JM, Paunovic M, Pääbo S. Analysis of one million base pairs of Neanderthal DNA. Nature2006; 444:330–336. https ://doi.org/10.1038/natur e0533 6.
- Wakefield MJ. Genomics from Neanderthals to high throughput sequencing. Genome Biol 2006;7:326. https ://doi.org/10.1186/gb-2006-7-8-326.
- Ricci C, Kakularam KR, Marzocchi C, Capecchi G, Riolo G, Boschin F, Kuhn H, Castagna MG, Cantara S. Thr92Ala polymorphism in the type 2 deiodinase gene: an evolutionary perspective. J Endocrinol Invest. 2020;43(12):1749-1757. [CrossRef]
- Seguin-Orlando A, Korneliussen TS, Sikora M, Malaspinas AS, Manica A, Moltke I, Albrechtsen A, Ko A, Margaryan A, Moiseyev V, Goebel T, Westaway M, Lambert D, Khartanovich V, Wall JD, Nigst PR, Foley RA, Lahr MM, Nielsen R, Orlando L, Willerslev E. Paleogenomics. Genomic structure in Europeans dating back at least 36,200 years. Science. 2014;346(6213):1113-8. [CrossRef]
- Sikora M, Seguin-Orlando A, Sousa VC, Albrechtsen A, Korneliussen T, Ko A, Rasmussen S, Dupanloup I, Nigst PR, Bosch MD, Renaud G, Allentoft ME, Margaryan A, Vasilyev SV, Veselovskaya EV, Borutskaya SB, Deviese T, Comeskey D, Higham T, Manica A, Foley R, Meltzer DJ, Nielsen R, Excoffier L, Mirazon Lahr M, Orlando L, Willerslev E. Ancient genomes show social and reproductive behavior of early Upper Paleolithic foragers. Science. 2017;358(6363):659-662. [CrossRef]
- Kılınç GM, Omrak A, Özer F, Günther T, Büyükkarakaya AM, Bıçakçı E, Baird D, Dönertaş HM, Ghalichi A, Yaka R, Koptekin D, Açan SC, Parvizi P, Krzewińska M, Daskalaki EA, Yüncü E, Dağtaş ND, Fairbairn A, Pearson J, Mustafaoğlu G, Erdal YS, Çakan YG, Togan İ, Somel M, Storå J, Jakobsson M, Götherström A. The Demographic Development of the First Farmers in Anatolia. Curr Biol. 2016;26(19):2659-2666. [CrossRef]
- Leinonen R, Sugawara H, Shumway M; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011;39:D19-21. [CrossRef]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990;21:403–410. [CrossRef]
- Tauno Metsalu, Jaak Vilo, ClustVis: a web tool for visualising clustering of multivariate data using Principal Component Analysis and heatmap, Nucleic Acids Research, Volume 43, Issue W1, 1 July 2015, Pages W566–W570. [CrossRef]
- Hannoush ZC, Weiss RE. Defects of Thyroid Hormone Synthesis and Action. Endocrinol Metab Clin North Am. 2017;46(2):375-388. [CrossRef]
- Wajdi Safi, Faten Hadj Kacem, Hana Charfi, Mouna Mnif Feki, Mohamed Abid and Noura Bougacha-Elleuch. Genetic investigation of thyroid dyshormonogenesis in a Tunisian consanguineous family. Endocrine Abstracts (2021) 73 AEP707 | DOI: 10.1530/endoabs.73.AEP707.
- Hashemipour M, Soheilipour F, Karimizare S, Khanahmad H, Karimipour M, Aminzadeh S, Kokabee L, Amini M, Hovsepian S, Hadian R. Thyroid peroxidase gene mutation in patients with congenital hypothyroidism in isfahan, iran. Int J Endocrinol. 2012;2012:717283. [CrossRef]
- Guria S, Bankura B, Balmiki N, Pattanayak AK, Das TK, Sinha A, Chakrabarti S, Chowdhury S, Das M. Functional analysis of thyroid peroxidase gene mutations detected in patients with thyroid dyshormonogenesis. Int J Endocrinol. 2014;390121. [CrossRef]
- Begum MN, Islam MT, Hossain SR, Bhuyan GS, Halim MA, Shahriar I, Sarker SK, Haque S, Konika TK, Islam MS, Rahat A, Qadri SK, Sultana R, Begum S, Sultana S, Saha N, Hasan M, Hasanat MA, Banu H, Shekhar HU, Chowdhury EK, Sajib AA, Islam ABMMK, Qadri SS, Qadri F, Akhteruzzaman S, Mannoor K. Mutation Spectrum in TPO Gene of Bangladeshi Patients with Thyroid Dyshormonogenesis and Analysis of the Effects of Different Mutations on the Structural Features and Functions of TPO Protein through In Silico Approach. Biomed Res Int. 2019;2019:9218903. [CrossRef]
- Kuhlwilm M, Gronau I, Hubisz MJ, de Filippo C, Prado-Martinez J, Kircher M, Fu Q, Burbano HA, Lalueza-Fox C, de la Rasilla M, Rosas A, Rudan P, Brajkovic D, Kucan Ž, Gušic I, Marques-Bonet T, Andrés AM, Viola B, Pääbo S, Meyer M, Siepel A, Castellano S. Ancient gene flow from early modern humans into Eastern Neanderthals. Nature. 2016;530(7591):429-33. [CrossRef]
- Slon V, Mafessoni F, Vernot B, de Filippo C, Grote S, Viola B, Hajdinjak M, Peyrégne S, Nagel S, Brown S, Douka K, Higham T, Kozlikin MB, Shunkov MV, Derevianko AP, Kelso J, Meyer M, Prüfer K, Pääbo S. The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature. 2018;561(7721):113-116. [CrossRef]
- Inchley CE, Larbey CD, Shwan NA, Pagani L, Saag L, Antão T, Jacobs G, Hudjashov G, Metspalu E, Mitt M, Eichstaedt CA, Malyarchuk B, Derenko M, Wee J, Abdullah S, Ricaut FX, Mormina M, Mägi R, Villems R, Metspalu M, Jones MK, Armour JA, Kivisild T. Selective sweep on human amylase genes postdates the split with Neanderthals. Sci Rep. 2016;6:37198. [CrossRef]
- Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, Werner J, Villanea FA, Mountain JL, Misra R, Carter NP, Lee C, Stone AC. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007t;39(10):1256-60. [CrossRef]
- Eaton SB. The ancestral human diet: what was it and should it be a paradigm for contemporary nutrition? Proc Nutr Soc. 2006;65(1):1-6. [CrossRef]
- Jaouen K, Richards MP, Le Cabec A, Welker F, Rendu W, Hublin JJ, Soressi M, Talamo S. Exceptionally high δ15N values in collagen single amino acids confirm Neandertals as high-trophic level carnivores. Proc Natl Acad Sci U S A. 2019;116(11):4928-4933. [CrossRef]
- Singh A, Singh D. The Paleolithic Diet. Cureus. 2023;15(1):e34214. [CrossRef]
Figure 1.
Geographical distribution of archaeological sites.
Figure 2.
PCA plot considering the distribution of both alleles (A) and genotypes (B).
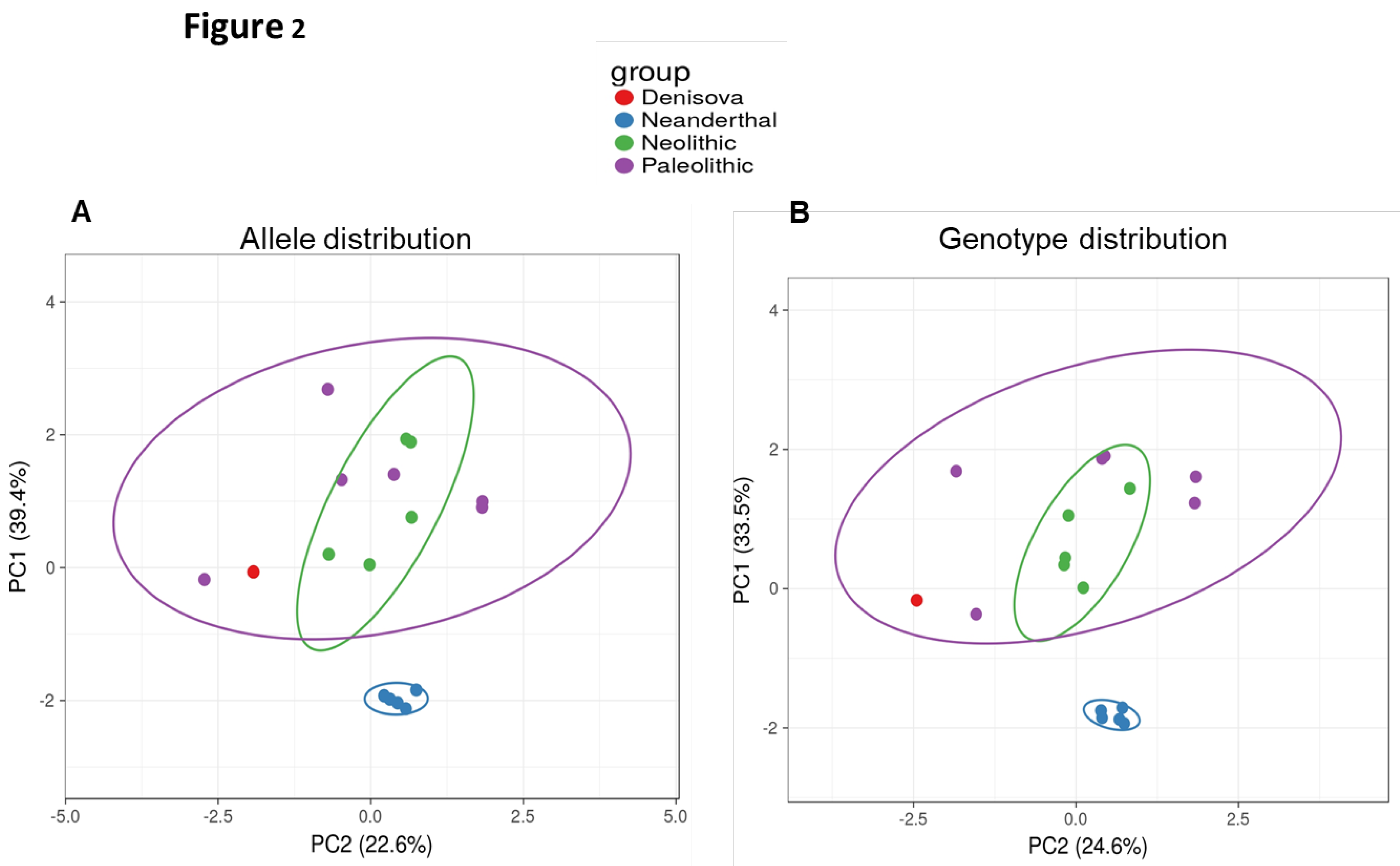

Table 1.
Results from sequence alignment for DIO2, TPO, TG and TSHR genes.
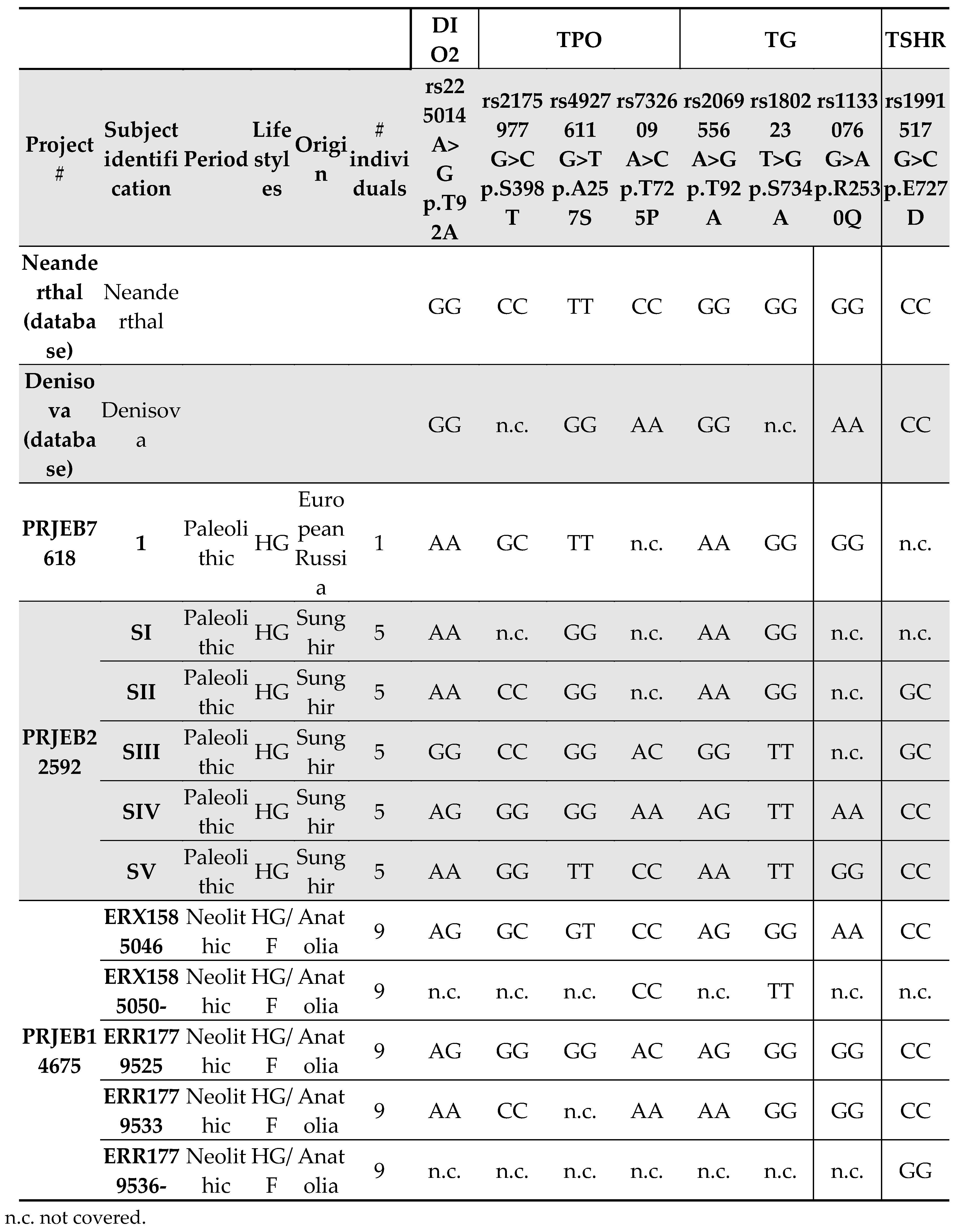
Table 2.
Allele distribution for each SNPs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



