
Submitted:
30 March 2024
Posted:
01 April 2024
You are already at the latest version
Abstract
As a widely used plastic, the aging and degradation of polyethylene (PE) are inevitable problems, whether the goal is to prolong the life of PE products or address the issue of white pollution. Molecular simulation is a vital scientific tool in elucidating the mechanisms and processes of chemical reactions. To obtain the distribution and evolution process of PE's thermal oxidation products, this work employs the self-consistent charge density functional tight binding (SCC-DFTB) method to perform molecular simulations of the thermal oxidation of PE with different crystallinity and branched structures. We discovered that crystallinity does not affect the thermal oxidation mechanism of PE, but higher crystallinity makes PE more susceptible to cross-linking and carbon chain growth, reducing the degree of PE carbon chain breakage. The branched structure of PE results in differences in pore size between the carbon chains, with larger pores leading to a concentrated distribution of O2 and free radicals subsequently formed. The breakdown of PE is slowed down when free radicals are localized in low-density regions of the carbon chain. The specifics and mechanism of PE's thermal oxidation are clearly revealed in this paper, which is essential for understanding the process in-depth and for the development of anti-aging PE products.
Keywords:
polyethylene thermal degradation
; crystallinity
; branched chain
; molecular dynamics simulations
; density functional theory calculation
1. Introduction
As a form of thermoplastic resin that has exceptional corrosion resistance to most acids and alkalis, PE is odorless, chemically stable, non-toxic, and possesses great insulating qualities [1]. Consequently, it finds extensive application in high-frequency insulating materials, containers, wires encapsulation, and packaging materials [2].
Nevertheless, although exhibiting stable chemical characteristics, PE still suffer problems including thermal oxidation [3,4,5] and photo-oxidation [6,7,8]. The PE chains break due to these oxidation processes, and the free radicals will accelerate the reactions between carbon chains [9,10]. While broken carbon chains can recover [11,12], the resulting amorphous and irregular chain structure causes PE to lose its initial stability and other benefits. A thorough grasp of the regularity of oxidation will promote the development of superior PE products and help extend its service life. Thermal oxidation is a frequent phenomenon among the aging reactions that denaturize PE [13] since it simply needs O2 while photo-oxidation reactions need both light and O2. Accordingly, it is crucial to clarify the laws and affecting factors of thermal oxidation.
PE's thermal oxidation process has been explored via experiment [14,15,16,17] and simulations [18,19,20,21]. A valuable method for anticipating the results of thermal oxidation is molecular dynamics simulations. Lijuan Liao et al. employed reactive force field (Reaxff) molecular dynamics to simulate the thermal oxidation of amorphous PE under anaerobic and low oxygen concentration circumstances [18]. The findings demonstrated that hydroxy, aldehyde, and carboxyl groups are typical organic products when O2 is present. The activation energy of PE thermal oxidation varies from 50 to 450 kJ/mol as the reaction progresses, indicating the complexity of thermal oxidation. However, the effects of crystallinity and branched chain structure on oxidation products were not considered in their work.
Additionally, molecular dynamics simulation is an effective way to investigate the formation mechanism of different products. Lihua Chen et al. [19] investigated the formation pathways and energy barrier of olefins, hydroxyl, aldehydes, and ketones. They revealed that the carboxyl group is dehydrated to generate the ketone carbonyl group, and the dehydration process also produces an O-containing free radical. Due to the O-containing free radical, the carbon chain becomes unstable, leading to chain scission that produces aldehydes and hydroxyl. However, Mingxiao Zhu et al [20] got different results. claiming that when PE is thermally oxidized, the carbon chain breaks first—instead of the H atoms dissociating as Lihua had predicted. Subsequently, O2 and the carbon chain breakpoint combine, followed by a dehydration reaction that forms aldehydes. The thermal oxidation mechanism of PE needs further study from a microscopic perspective to resolve this controversy.
The work mentioned above primarily focuses on the thermal oxidation of unbranched PE, although branched chain structures are ubiquitous, resulting in different densities in PE. Jin Woo Park et al. [22] conducted an experimental investigation of the thermal degradation behavior of high-density PE (HDPE), low-density PE (LDPE), and linear low-density PE (LLDPE). They discovered that HDPE has the highest stability, whereas LDPE is the most susceptible. They determined that the HDPE, LDPE, and LLDPE degradation activation energies were 333.2–343.2, 187.5–199.1, and 219.2–230.1 kJ/mol, respectively. As the degree of branching grows, so does the reaction order.
Although the above experimental work studied the thermal oxidation activation energy of PE with different densities, the experiments were examined from a macro perspective, and the mechanism from a micro perspective remains to be explored. Additionally, HDPE is distinct from LDPE and LLDPE since it has both crystalline and amorphous parts, whereas the other two forms of PE mainly have amorphous sections. Variations in crystallinity cause carbon chains to be arranged differently, which might have an impact on the movement of carbon chains migration [23] and diffusion of O2 molecules [24]. Consequently, the thermal oxidation of PE becomes more complicated due to the crystallinity and branched chain structure.
As mentioned above, it is likely that crystallinity and branched chain structure have an important impact on the thermal oxidation behavior of PE, but there are still few simulation studies in this area. To close the gaps and provide clarity on the intricate thermal oxidation of PE with various structures, we established 4 models: high-denstiy crystalline PE (HDPE-C), high-density amorphous unbranched PE (HDPE-A), LDPE with a random and short chain structure, and LLDPE with a regular branched chain structure and did molecular dynamics simulation. Our research demonstrates that higher crystallinity makes PE more susceptible to cross-linking and carbon chain growth, reducing the degree of PE carbon chain breakage. Although crystallinity and branched structure do not affect the thermal oxidation by affecting the thermodynamic energy barrier of the reaction of PE with O2, short and random branched chains contribute to a local pore structure in the carbon chain, leading to the localized distribution of O2 and subsequently formed free radicals, and delaying the chain scission of PE.
This study contributes to an improved comprehension of PE's thermal oxidation process by demonstrating the effects of crystallinity and branching degree. To increase the service life of PE products, it is crucial to design and enhance the molecular structure of PE at the manufacturing level.
2. Methods
We utilized the Materials Studio software for model construction, employing molecular dynamics to simulate the thermal oxidation process of PE. Furthermore, the density functional theory (DFT) method was employed to scrutinize the thermodynamic energy landscape of PE reacting with O2.
Figure 1.
model construction for (a)HDPE-C, (b)HDPE-A, (c)LDPE, (d)LLDPE.
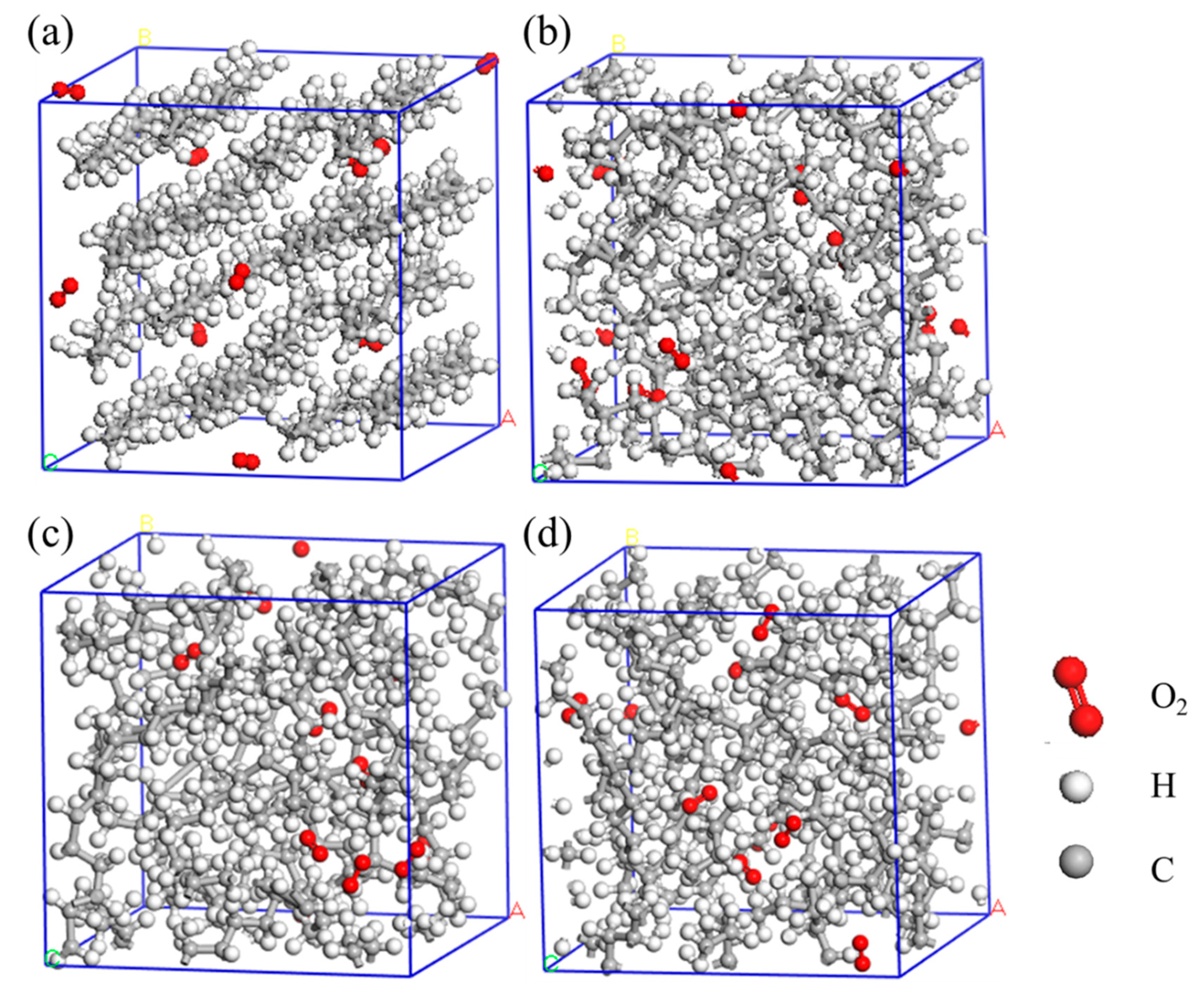
Model construction: In this work, 4 PE models, i.e., HDPE-C, HDPE-A, LDPE, and LLDPE, were constructed. The comparison of HDPE-C and HDPE-A can reflect the effect of crystallinity on the thermo-oxidative behavior of the PE, whereas the comparison of HDPE-A, LDPE, and LLDPE can show the effect of the branched chains. 3 models of HDPE-A, LDPE, and LLDPE were constructed utilizing Amorphous cell module in Materials Studio. It was configured to have a consistently ordered chain structure for the HDPE-C, which has a density of 0.97 g/cc and is set to 16 carbon chains to prevent interaction between periodic lattice. When modeling LDPE and LLDPE, the structural details of PE found in Ref [25] were implemented. LDPE has a branch density of 8CH3/200C and a random amount within 6 branched C atoms, and LLDPE has a branch density of 4CH3/200C, with 6 C atoms in each branch. Both LDPE and LLDPE have densities of 0.92 g/cc. There are no branches in HDPE-A, and the density is 0.94 g/cc. Each of the models has 10 O2 molecules and 224 CH2 units. Using the pcff force field, structural optimization and thermodynamic relaxation were conducted in the Forcite module. The structural optimization was carried out employing the conjugate gradient method. The energy convergence standard was 10-4 kcal mol-1, and the force convergence standard was 0.005 kcal mol-1 Å-1. After structural optimization, thermodynamic relaxation was performed. To keep the set density constant, only NVT ensemble relaxation was used. The relaxation time was 400 ps, with the temperature 323 K. Berendsen thermostat was adopted, and the attenuation constant was set to 0.1 ps.
Molecular dynamics calculation: The free source program CP2K was applied to complete the calculation and the SCC-DFTB approach was chosen. To achieve the integral value that is difficult to determine, SCC-DFTB fitted the system energy expression defined by electron density applying the present functional and a second-order expansion was conducted on the exchange correlation term. Accordingly, the energy of a system can be calculated as[26]:
where is the total energy of the system, the first sum is over occupied Kohn-Sham eigenstates . represents the repulsive two-particle interaction. The second sum is the final second-order energy term. and are two point charges. is an intergral evaluation related to the normalized radial dependence of the density fluctuation on atom α and β ,which can be approximated as a funciton of the Hubbard parameter and distance between the two atoms:
As a result, SCC-DFTB could essentially duplicate DFT accuracy, while for substantial systems the computation speed was increased. It is reasonable to believe that SCC-DFTB is also applicable in PE and O2 systems, as it has been successfully applied thus far in systems such as metal oxide and water systems [27], light element covalent compound systems [28], organic small molecule systems [29], and C-containing polymers [30]. A canonical sampling through velocity rescaling (CSVR) heat bath was employed, with the time constant set to 80 fs. The NVT ensemble was implemented throughout with a simulation temperature of 2400 K to speed up the thermal oxidation. The simulation duration was 200 ps with a time step of 0.25 fs. Numerous earlier studies have demonstrated that raising the temperature in a polymer degradation or oxidation scenario can accelerate the reaction without changing the process's mechanism [31,32,33,34,35]. Simultaneously, we considered spin multiplicity in the computation by setting the RELAX_MULTIPLICITY parameter to 0.01. Long-range van der Waals interactions were also included in the SCC-DFTB technique. The Python application was executed with Anaconda software to process the atomic coordinate data obtained from the molecular simulation. When assessing the bonding status between two atoms, the criteria involved the van der Waals radius, with a bond presumed to have occurred if the distance between two atoms was less than 0.6 times the sum of their van der Waals radii. Determining whether bonds are formed between atoms is to count the amount of various fragments in the system. In statistics, the following expression is used to calculate the amunt of fragments[31]:
is the total amount of n fragments at any moment, is the specific amount of species i.
First-principles computations: VASP software was chosen to execute subsequent computations of the Gibbs free energy change. In the reference literature, the Perdew-Burke-Ernzerhof (PBE) functional was employed as an established technique [19]. In this work the plane wave cutoff energy was set as 450 eV. Since the fragment contains radicals, we considered spin polarization. The energy convergence criteria for structural optimization was 10-5 eV, and the force convergence criterion was 0.02 eV Å-1. The Gibbs free energy change was computed at 298 K, while the Gibbs free energy of O2 was derived from studies published in the literature [36], as the plane wave approach of estimating the electron configuration of O2 has unavoidable errors.
3. Result and Discussion
This article first simulates the thermal oxidative degradation of PE, giving the main products and their distribution, and then discusses the reaction mechanism. After analyzing the distribution of free radicals and carbon chain density, the effects of crystallinity and branched chain structure on the thermal oxidation behavior of PE are discussed.
3.1. Distribution of Oxidation Products of Different PEs
This section compares the thermal oxidation products of the 4 PEs from the perspective of carbon numbers and types of small molecules, and demonstrates the degradation process of PE by showing the change in the amount of C–C bonds.
Figure 2 illustrates how C-containing molecular fragments changed over time. The distribution of the number of C atoms in their C-containing fragments at the end of 200 ps is more clearly shown in Figure S1. The results of HDPE-C and HDPE-A demonstrate that crystalline PE is less prone to fragmentation due to bond breakdown. Comparatively, more fragments are formed in HDPE-A, and HDPE-A produces more C2 products, which may be either C2H4 or C2H6. After around 150 ps, the total quantity of fragments in HDPE-C decreases, suggesting that the system is experiencing carbon chain growth reactions. This is due to the restricted mobility of the carbon chain in the HDPE-C system, causing the alkyl radicals to only migrate along the carbon chain and the relatively sluggish movement of the free radicals[9].
When HDPE-A, LDPE, and LLDPE are compared, it becomes clear that HDPE-A and LLDPE are more susceptible to oxidation and structural degradation. In both systems more C2 products are formed. Compared to HDPE-A and LLDPE, LDPE has not undergone as much deterioration. Although this phenomenon is different from the conclusions of existing experimental studies [22], it is because in the experiment, the density of HDPE, LLDPE and LDPE decreased successively while the permeability of O2 to PE increased, causing more O2 to diffuse into LDPE [37]. To study the influence of branched chain structure, we did not place more O2 in LDPE based on the principle of controlling variables.
Figure 3 shows that compared with the results of HDPE-C, HDPE-A produces more small molecules such as CO and C2H4, indicating that PE with lower crystallinity is more susceptible to thermal oxidation. Comparing the results of HDPE-A, LDPE and LLDPE, it is found that HDPE-A yields the most, LLDPE the second, and LDPE the lowest number of small molecules. It shows that under the same O2 concentration, unbranched and long-branched structures are more susceptible to thermal oxidation.
It is possible to directly uncover the mechanism of PE thermal oxidation through investigating small molecules that emerge or accumulate throughout the oxidation process. Figure 3 illustrates how H2O2 is produced in all systems by rapidly consuming O2. Subsequently, the H2O2 is consumed, suggesting that H2O2 is an essential intermediary product that may be related to the oxidation mechanism. Although H2O2 was not included in previous modeling work [18,19,21], it is present in photo-oxidation [38]. Our research demonstrates that H2O2 is an intermediate product that can be tough to identify experimentally because it exists for a short period. H2O is produced in substantial quantities when H2O2 is consumed, which makes sense since the final products of hydrocarbon oxidation are CO2 and H2O.
Moreover, C2H4, C2H6, CH4, and CO were generated but H2 and CO2 were not. This is because the initial quantity of O2 in this study is set small relative to the amount of C atoms to emphasize the differences in the interactions between various branched structures and O2. We analyzed the system of C20H42+32O2 to demonstrate the precision of the employed methodology, displaying the results in Figure S2. When there is enough O2 present, CO2 will be produced. Inadequate simulation time might be an explanation of the absence of H2 generation, as even in the experiment, little H2 gets generated [21].
It's reasonable that the degree of PE degradation may be inferred from the alteration in the quantity of C–C bonds [39]. Figure 4 illustrates that the C–C bonds in HDPE-A did not decrease much before 25 ps, but decreased sharply after 100 ps, while the amount of C–C bonds in HDPE-C increased slowly after 100 ps. Compared with the results in Figure 2, it shows that in HDPE-C cross-linking of carbon chains occurs, indicating that a decrease in crystallinity leads to more complete carbon chain cleavage of PE, while a higher crystallinity facilitates carbon chain cross-linking. Compared with LDPE and LLDPE, the C–C bonds in HDPE-A are reduced the most, and the C–C bonds in LDPE are broken the least. This result is consistent with Figure 2 and Figure 3, indicating that the unbranched structure is more likely to be thermally oxidized, and the short and random branched chain structure is more stable.
3.2. Reaction Mechanism of Thermal Oxidation of PE
To reveal the reasons why crystallinity and branched chain structure lead to differences in thermal oxidation products of various PEs, this section first gives the thermal oxidation mechanism of PE through simulation at low temperature, pointing out that H2O2 is an important intermediate product. The details and thermodynamic energy barriers of the reactions of different PEs with O2 were then compared.
Given that PE's thermal oxidation process slows down at lower temperature and that temperature merely changes the reaction rate rather than the reaction path, further research is needed to completely understand the PE thermal oxidation mechanism. As a result, we calculated the 140 ps oxidation process for the HDPE-C system at a lower temperature. Figure 5 depicts small molecules' development throughout the system. As low as 1200 K, O2 consumption slows down with only H2O2 formed but not consumed. When the temperature is raised to 1600 K, H2O is formed by the consumption of H2O2. With a temperature of 2000 K, H2O is produced more quickly and C2H4 starts to appear in the system. The oxidation products' carbon number distribution also reveals that within 140 ps the carbon chain breaks around 2000 K, and the generation of the C2, C4, and C10 products begins at 2000 K (Figure S3). It is evident that O2 initially reacts with H atom on the carbon chain, producing an intermediate H2O2 when thermal oxidation of PE takes place. The carbon chain breaks when H2O2 is reduced to H2O.
To acquire a better understanding of the oxidation mechanism of PE, it is necessary to analyze the details of H2O2 formation and consumption. Table 1 summarizes the main reactions involving all O2 and species that include O in HDPE-C, exhibiting the H2O2 generation and consumption paths. The reactions involving H2O2 in HDPE-A, LDPE, and LLDPE systems are listed in Tables S1, S3, and S5, respectively. In essence, the chemical pathways resemble those of HDPE-C. This article will refer to the reaction that generates H2O as a simple reaction and the reaction that combines an O atom with a carbon chain as a complicated reaction for the purpose of simplicity of description.
In HDPE-C, 5 O2 molecules immediately interact with the H atoms on the two neighboring C atoms on the carbon chain to produce H2O2. Through diffusion, the remaining O2 first interact with a H atom to generate ·OOH, which subsequently combines with H atoms in other locations to form H2O2. H2O2 then breaks down into two ·OH or a reactive O atom as an intermediate product.
Because H2O2 displays a smaller O-O bond energy [40] and requires breaking more bonds to yield reactive O atoms, more ·OH are produced. Afterwards, these decomposition products react with PE. H2O emerges when an abundance of ·OH react with H atoms on the carbon chain, as Table 2 illustrates. This process results in the formation of alkyl radicals or unsaturated C atoms. Figure S4 highlights the way certain ·OH and reactive O atoms interact with pre-existing alkyl radicals leading to alcohols, which then transform into enols or CO. In HDPE-C, the transformation of enols into acids or ketones was never detected due to restricted calculation time. The details of the reactions involving O2 in HDPE-A, LDPE and LLDPE are shown in Figures S5-S10 and Tables S1-S6.
The mechanism of PE oxidation can be summarized as follows: O2 combine with H atoms on the carbon chain to form H2O2, and the H2O2 decomposes to ·OH to continue dehydrogenating the carbon chain. Alkyl radicals are formed when the carbon chain is dehydrogenated, and these radicals eventually transform into more stable structures. During the reaction process, alkyl radicals may undergo hydrogen transfer reactions between carbon chains, or they may combine with O2, reactive O atoms, and ·OH to form more complex structures. The process of carbon chain evolution is complex and may be affected by numerous factors such as the distribution of O-containing species, the free volume within the carbon chain, and the density distribution of the carbon chain.
After clarifying the details of the reaction of various PEs with O2, it is necessary to conduct a more in-depth analysis of the reactions listed in Table 1, Table S1, S3, S5. H2O2 is a key intermediate product in the thermal oxidation process of PE, and its formation kinetics has an important impact on the thermal oxidation. As can be seen from the above tables, there are many PE fragments with different structures in each system that react with O2 to form H2O2. To avoid selective calculation of the reaction between a certain structure and O2, we performed DFT calculations on all reactions that form H2O2 in Table 1, Table S3, and S5, and obtained the Gibbs free energy changes of these reactions to examine the influence of branched chains on the energy landscape of the H2O2 formation reaction. Note that since neither HDPE-C nor HDPE-A contains branched chains and the types of H2O2 formation reactions in these two systems are similar, only reactions in the HDPE-C system were selected to represent the unbranched structure. Since there are PE fragments of different structures in the same system, there are various energy paths in Figure 6. Although they are different, these paths are processes that have actually occurred, and all paths need to be considered.
Results in Figure 6 indicate that the process that produces ·OOH from O2 is the rate-determining step, even when certain reactions result in a reduction in the total energy landscape. The three structures have maximum energy barriers of 1.42 eV, 1.61 eV, and 1.41 eV for the rate-determining step, respectively. The other processes' energy barriers fall within the range of maximum values. It is evident that the branched chain structure has little impact on the H2O2 production process and that the energy barrier range of PE with various structures to produce H2O2 is analogous.
3.3. Effects of Branched Chains on PE Oxidation
This section starts from the analysis of the free volume between PE carbon chains and explores the distribution and diffusion behavior of O2 in 4 PEs. We also calculated the distribution of free radicals and the carbon chain density distribution in all systems.
To characterize the continuous but erratic free volume between carbon chains, a sphere sufficiently large was searched to fit between the chains without contacting with any H or C atoms. In the calculation a frame of data is selected every 12.5 fs, and the initial 250 fs time range is used to evaluate the initial maximum pore size of all systems. The results are presented in Figure 7. Among all systems, HDPE-C has the smallest maximum pore size of any system, approximately 4.8~5 Å. The maximum diameter of pores in HDPE-A is around 5.3~5.5 Å, indicating that higher crystallinity is not conducive to the formation of large free volume. LDPE has the greatest maximal pore size, ranging from 5.75 to 5.9 Å. This might be because LDPE has random and short branches, allowing larger free volume to emerge due to intermolecular repulsion between various chains, while the maximum diameter in LLDPE is approximately 5.2~5.4 Å.
The entropy effect of O2 within the free volume varies depending on the size of the pores. Figure 8 displays the distribution diagram of the distance between all O2 molecules in each system at the beginning of the simulation. A system with a uniform distribution of O2 will have a single primary peak that is positioned in the middle of the picture, similar to a normal distribution curve. Uneven O2 distribution is indicated by the presence of multiple peaks. The distribution of O2 is uneven in HDPE-A but uniform in HDPE-C, indicating that a disordered structure leads to disordered O2 distribution. Among all disordered systems, the O2 distribution in LDPE and HDPE-A with larger free volume is uneven, showing that the larger free volume caused by random and short branched structures will also lead to irregular O2 distribution.
Even though the carbon chain structure will result in variations in the homogeneity of O2 distribution in each framework, O2 will diffuse before reacting with PE. This influences the location of the PE-O2 reaction, and the carbon chain's structure additionally regulates the diffusion process. The distance between each O2 molecule in each system from the location at the beginning time to the position at the reaction time was evaluated, which was referred to as the diffusion displacement to characterize the diffusion route length of O2 prior to interacting with PE. Figure 9 illustrates the distribution of O2 diffusion displacement throughout all systems. The average diffusion displacement of O2 in HDPE-C and HDPE-A systems is 8.61 Å and 9.05 Å, respectively. This is because HDPE-C has regular channel structures, and as O2 molecules prefer to migrate in the direction of the channels, there is relatively little diffusion displacement [41]. The average O2 diffusion displacement in LDPE and LLDPE systems is 8.72 Å and 9.85 Å, respectively. This difference is thought to be caused by greater free volume in LDPE, which contributes to an uneven distribution of carbon atom density (Figure S11). Regions with higher carbon atom density are not conducive to the diffusion of O2.
Table 2 and PE directly affects the distribution of free radicals on the PE carbon chain. Since free radicals play a promoter role in the subsequent carbon chain breaking reaction, understanding the distribution of free radicals is of great significance for analyzing the breakage of PE carbon chains.
This work attempts to give the spatial distribution of free radicals and unsaturated carbon atoms at 50 ps. The rationale behind adopting this moment is depicted in Figure 4. The initial stage of C-C bond breaking and the PE-O2 reaction terminate at 50 ps. Defects in the carbon chain are the primary triggers of subsequent cleavage, which predominantly originate from free radicals and unsaturated carbon atoms. Note that each system was split into 4x4x4 small parts to determine the density distribution of free radicals and unsaturated carbon atoms, as shown in Figure 10.
The atomic density of each region was then calculated and expressed by the size and color of the point located in the center of the region. In regions where the density is zero, these points are not shown. We connected neighboring locations with dotted lines to better illustrate the relative positions between each point. Figure 10 illustrates that the free radicals distribution in HDPE-C and HDPE-A is relatively uniform, indicating that crystallinity does not affect the free radicals distribution. Among disordered systems, LDPE exhibits uneven free radicals distribution. Most of the points in LDPE are in the area with Y coordinate less than 10, and only 6 light blue points are in the area with Y coordinate greater than 10. However, there is no spatial location preference for the distribution of free radicals and unsaturated carbon atoms in HDPE-A and LLDPE.
The density distribution of the C and H atoms constituting the PE chain needs to be supplied to analyze how the density distribution of free radicals and unsaturated carbon atoms affects the cracking behavior of PE chains. The spatial distribution of C and H atom densities at 50 ps is displayed in Figure 11. As in Figure 10, the atomic density of a sub-region is represented by the color and size of each point. Figure 11 shows that there is a location in LDPE with an exceptionally high atomic density of 271.3 nm-3. The atomic densities of the other locations range from 100 nm-3 or less, with around 5 places reaching values of 200 nm-3. The carbon chain atom density distribution for other systems is essentially homogeneous and overlaps with the free radicals and unsaturated C atom distribution.
According to the description above, the impact of branched chain structure on the cracking behavior of PE is: random and short branched chains lead to the formation of large pores between PE chains, causing O2 distribute around the pores and react with carbon chains locally, resulting in the free radical density distribution staggered with the carbon chain atomic density distribution, and delaying atomic migration and carbon chain cleavage.
4. Conclusions
The SCC-DFTB method is utilized in this work to simulate the thermal oxidation of crystallized and amorphous PE with various branched chain structures. The thermal oxidation products of these PEs are compared. After assessing the thermodynamic energy barriers of the reaction between various PEs and O2 and analyzing the distribution of free radicals and carbon chain denstiy, the following conclusions were achieved that Higher crystallinity limits the diffusion of O2, and ordered carbon chains are more prone to cross-linking reactions instead of chain scission during thermal oxidation. The branched chain structure does not affect the thermodynamic energy barrier for the reaction between O2 and PE to produce H2O2. Random and short branched chains lead to the formation of pores between PE carbon chains and the free radicals distribution staggered with the dense carbon chain areas, delaying atomic migration and carbon chain breakage.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Shumao Zeng: Conceptualization, Methodology, Software, Validation, Investigation, Data curation, Writing-original draft, Visualization. Diannan Lu: Writing-review & editing, Supervision. Rui Yang: Writing-review & editing, Supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by National Key Research and Development Programs of China (no. U1862204).
Data Availability
Data will be made available on request.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Al-Salem, S. M.; Chandrasekaran, S. R.; Dutta, A.; Sharma, B. K. , Study of the fuel properties of extracted oils obtained from low and linear low density polyethylene pyrolysis. Fuel 2021, 304, 121396. [Google Scholar] [CrossRef]
- Geyer, R., Chapter 2 - Production, use, and fate of synthetic polymers. In Plastic Waste and Recycling, Letcher, T. M., Ed. Academic Press: 2020; pp 13-32.
- Suresh, B.; Maruthamuthu, S.; Khare, A.; Palanisamy, N.; Muralidharan, V. S.; Ragunathan, R.; Kannan, M.; Pandiyaraj, K. N. , Influence of thermal oxidation on surface and thermo-mechanical properties of polyethylene. Journal of Polymer Research 2011, 18(6), 2175–2184. [Google Scholar] [CrossRef]
- Yang, R.; Liu, Y.; Yu, J.; Wang, K. , Thermal oxidation products and kinetics of polyethylene composites. Polymer Degradation and Stability 2006, 91(8), 1651–1657. [Google Scholar] [CrossRef]
- Jelinski, L. W.; Dumais, J. J.; Luongo, J. P.; Cholli, A. L. , Thermal oxidation and its analysis at low levels in polyethylene. Macromolecules 1984, 17(9), 1650–1655. [Google Scholar] [CrossRef]
- Cruz-Pinto, J. J. C.; Carvalho, M. E. S.; Ferreira, J. F. A. , The kinetics and mechanism of polyethylene photo-oxidation. Die Angewandte Makromolekulare Chemie 1994, 216(1), 113–133. [Google Scholar] [CrossRef]
- Chew, C. H.; Gan, L. M.; Scott, G. , Mechanism of the photo-oxidation of polyethylene. European Polymer Journal 1977, 13(5), 361–364. [Google Scholar] [CrossRef]
- Suresh, B.; Maruthamuthu, S.; Kannan, M.; Chandramohan, A. , Mechanical and surface properties of low-density polyethylene film modified by photo-oxidation. Polymer Journal 2011, 43(4), 398–406. [Google Scholar] [CrossRef]
- Bracco, P.; Costa, L.; Luda, M. P.; Billingham, N. , A review of experimental studies of the role of free-radicals in polyethylene oxidation. Polymer Degradation and Stability 2018, 155, 67–83. [Google Scholar] [CrossRef]
- O’Neill, P.; Birkinshaw, C.; Leahy, J. J.; Barklie, R. , The role of long lived free radicals in the ageing of irradiated ultra high molecular weight polyethylene. Polymer Degradation and Stability 1999, 63(1), 31–39. [Google Scholar] [CrossRef]
- Chen, T. B. Y.; Yuen, A. C. Y.; Lin, B.; Liu, L.; Lo, A. L. P.; Chan, Q. N.; Zhang, J.; Cheung, S. C. P.; Yeoh, G. H. , Characterisation of pyrolysis kinetics and detailed gas species formations of engineering polymers via reactive molecular dynamics (ReaxFF). Journal of Analytical and Applied Pyrolysis 2021, 153, 104931. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Liu, J.; Wang, Z.; Kong, B.; Gong, X.; Yang, X.; Lin, W.; Guo, L. , Study of high density polyethylene (HDPE) pyrolysis with reactive molecular dynamics. Polymer Degradation and Stability 2014, 104, 62–70. [Google Scholar] [CrossRef]
- Oluwoye, I.; Altarawneh, M.; Gore, J.; Dlugogorski, B. Z. , Oxidation of crystalline polyethylene. Combustion and Flame 2015, 162(10), 3681–3690. [Google Scholar] [CrossRef]
- Hawkins, W. L.; Chan, M. G.; Link, G. L. Factors influencing the thermal oxidation of polyethylene. Polymer Engineering & Science 1971, 11(5), 377–380. [Google Scholar]
- Iring, M.; Földes, E.; Barabás, K.; Kelen, T.; Tüdős, F.; Ódor, L. , Thermal oxidation of Linear Low Density Polyethylene. Polymer Degradation and Stability 1986, 14(4), 319–332. [Google Scholar] [CrossRef]
- Gugumus, F. , Re-examination of the thermal oxidation reactions of polymers3. Various reactions in polyethylene and polypropylene. Polymer Degradation and Stability 2002, 77(1), 147–155. [Google Scholar] [CrossRef]
- Bravo, A.; Hotchkiss, J. H. , Identification of volatile compounds resulting from the thermal oxidation of polyethylene. Journal of Applied Polymer Science 1993, 47(10), 1741–1748. [Google Scholar] [CrossRef]
- Liao, L.; Meng, C.; Huang, C. , Thermal decomposition behaviour of polyethylene in oxygen-free and low oxygen content circumstances by reactive molecular dynamic simulation. Molecular Simulation 2018, 44(12), 954–964. [Google Scholar] [CrossRef]
- Chen, L.; Tran, H. D.; Ramprasad, R. , Atomistic mechanisms for chemical defects formation in polyethylene. The Journal of Chemical Physics 2018, 149(23), 234902. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M. X.; Li, J. C.; Song, H. G.; Chen, J. M.; Zhang, H. Y. , Determination of trap energy in polyethylene with different aging status by molecular dynamics and density function theory. IEEE Transactions on Dielectrics and Electrical Insulation 2019, 26(6), 1823–1830. [Google Scholar] [CrossRef]
- Kong, J.; Zhou, K.; Ren, X.; Chen, Y.; Li, Y.; Meng, P. , Insight into gaseous product distribution of cross-linked polyethylene pyrolysis using ReaxFF MD simulation and TG-MS. Journal of Analytical and Applied Pyrolysis 2023, 169, 105847. [Google Scholar] [CrossRef]
- Woo Park, J.; Cheon Oh, S.; Pyeong Lee, H.; Taik Kim, H.; Ok Yoo, K. , A kinetic analysis of thermal degradation of polymers using a dynamic method. Polymer Degradation and Stability 2000, 67(3), 535–540. [Google Scholar] [CrossRef]
- Tsuge, K.; Enjoji, H.; Terada, H.; Ozawa, Y.; Wada, Y. , Mechanical Dispersion and Molecular Motion in Crystals of Polyethylene and Other Polymers. Japanese Journal of Applied Physics 1962, 1(5), 270. [Google Scholar] [CrossRef]
- Hori, Y.; Fukunaga, Z.; Shimada, S.; Kashiwabara, H. E.s.r. studies on oxidation processes in irradiated polyethylene: 2. Diffusion of oxygen into crystalline regions. Polymer 1979, 20(2), 181–186. [Google Scholar] [CrossRef]
- Yao, Z.; Seong, H. J.; Jang, Y.-S. , Environmental toxicity and decomposition of polyethylene. Ecotoxicology and Environmental Safety 2022, 242, 113933. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. , Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Physical Review B 1998, 58(11), 7260–7268. [Google Scholar] [CrossRef]
- Prasetyo, N.; Hofer, T. S. , Adsorption and dissociation of water molecules at the α-Al2O3(0001) surface: A 2-dimensional hybrid self-consistent charge density functional based tight-binding/molecular mechanics molecular dynamics (2D SCC-DFTB/MM MD) simulation study. Computational Materials Science 2019, 164, 195–204. [Google Scholar] [CrossRef]
- Ploysongsri, N.; Vchirawongkwin, V.; Ruangpornvisuti, V. , Hydrogen boride nanotubes and their C, N, O decoration and doping derivatives as materials for hydrogen-containing gases storage and sensing: A SCC–DFTB study. Vacuum 2021, 187, 110140. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Liu, R.; Guo, W. , Thermal decomposition mechanisms and stability of NTO crystal involving molecular vacancy and surface effects: DFTB-MD and DFT studies. Journal of Materials Science 2023, 58(8), 3641–3656. [Google Scholar] [CrossRef]
- Fan, W.-J.; Kang, Z.; Zhu, W.-Q.; Ding, Y.-N.; Xu, H.-Y.; Tan, D.-Z.; Chen, Y.-G. , Conjugated microporous polymers as novel adsorbent materials for VOCs capture: A computational study. Computational Materials Science 2019, 170, 109207. [Google Scholar] [CrossRef]
- Zhang, F.; Cao, Y.; Liu, X.; Xu, H.; Lu, D.; Yang, R., How Small Molecules Affect the Thermo-Oxidative Aging Mechanism of Polypropylene: A Reactive Molecular Dynamics Study. In Polymers, 2021; Vol. 13.
- Lu, X.; Wang, X.; Li, Q.; Huang, X.; Han, S.; Wang, G. , A ReaxFF-based molecular dynamics study of the pyrolysis mechanism of polyimide. Polymer Degradation and Stability 2015, 114, 72–80. [Google Scholar] [CrossRef]
- Bhoi, S.; Banerjee, T.; Mohanty, K. , Insights on the combustion and pyrolysis behavior of three different ranks of coals using reactive molecular dynamics simulation. RSC Advances 2016, 6(4), 2559–2570. [Google Scholar] [CrossRef]
- Zhao, T.; Li, T.; Xin, Z.; Zou, L.; Zhang, L., A ReaxFF-Based Molecular Dynamics Simulation of the Pyrolysis Mechanism for Polycarbonate. Energy & Fuels 2018, 32, (2), 2156-2162.
- Wang, Q.-D.; Wang, J.-B.; Li, J.-Q.; Tan, N.-X.; Li, X.-Y. , Reactive molecular dynamics simulation and chemical kinetic modeling of pyrolysis and combustion of n-dodecane. Combustion and Flame 2011, 158(2), 217–226. [Google Scholar] [CrossRef]
- Fang, Y.-H.; Liu, Z.-P. , Mechanism and Tafel Lines of Electro-Oxidation of Water to Oxygen on RuO2(110). Journal of the American Chemical Society 2010, 132(51), 18214–18222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Easteal, A. J.; Chen, X. D. , Ethylene and oxygen permeability through polyethylene packaging films. Packaging Technology and Science 1998, 11(4), 169–178. [Google Scholar] [CrossRef]
- Singh, A. , Irradiation of polyethylene: Some aspects of crosslinking and oxidative degradation. Radiation Physics and Chemistry 1999, 56(4), 375–380. [Google Scholar] [CrossRef]
- He, X.-c.; Chen, D.-z. , ReaxFF MD study on the early stage co-pyrolysis of mixed PE/PP/PS plastic waste. Journal of Fuel Chemistry and Technology 2022, 50(3), 346–356. [Google Scholar] [CrossRef]
- Qu, Y.; Bian, X.; Zhou, Z.; Gao, H. , Existence of hydroperoxy and hydrogen peroxide radical complex (HO2·H2O2). Chemical Physics Letters 2002, 366(3), 260–266. [Google Scholar] [CrossRef]
- Meng, X.; Jin, G.; Yang, R. , A quantum chemical and molecular dynamics simulation study on photo-oxidative aging of polyethylene: Mechanism and differences between crystalline and amorphous phases. Polymer Degradation and Stability 2023, 217, 110536. [Google Scholar] [CrossRef]
Figure 2.
Carbon fragments distribution of (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE.
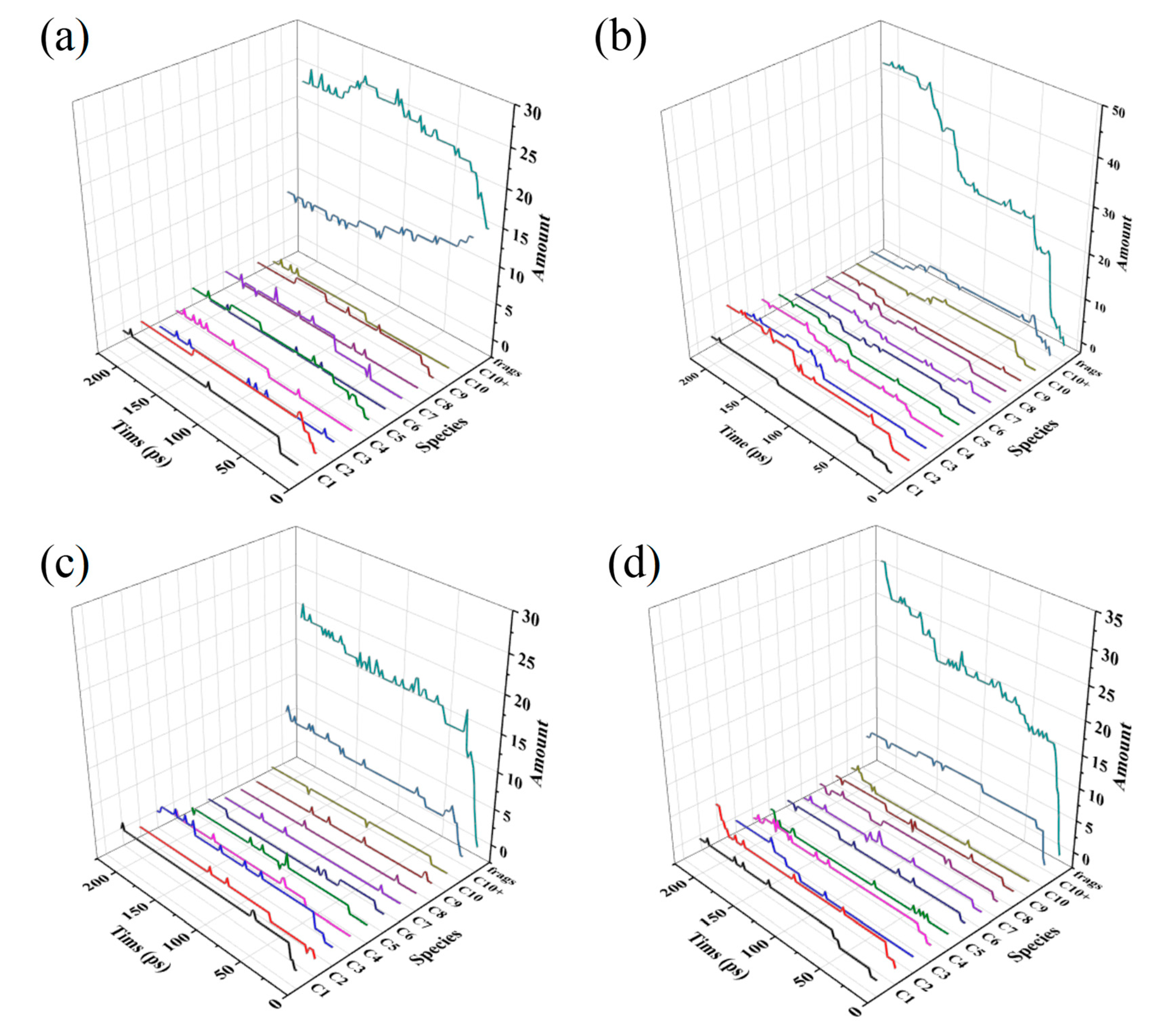
Figure 3.
Changes of the amount of small molecules in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE.
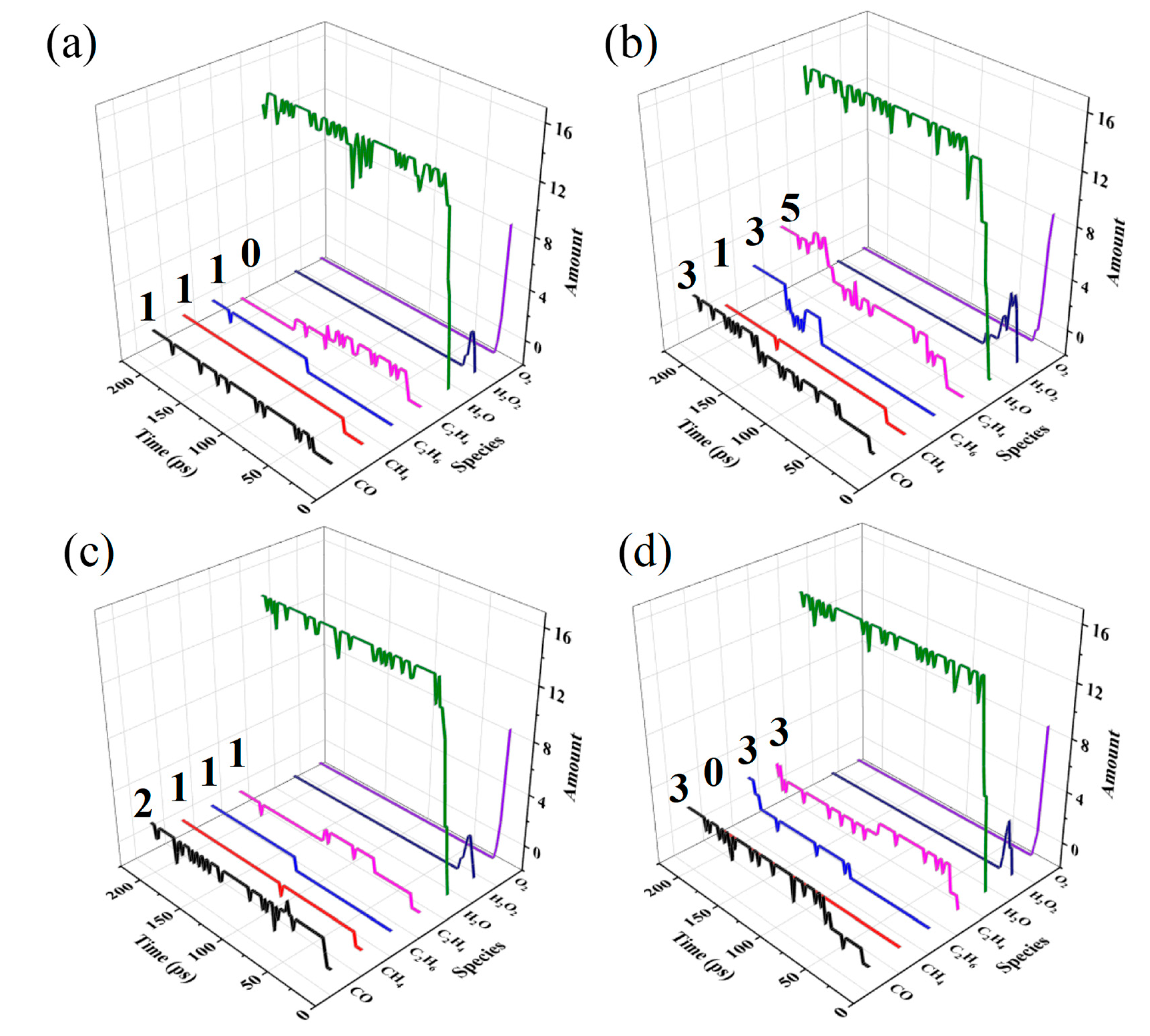
Figure 4.
Changes of C-C bond amount.
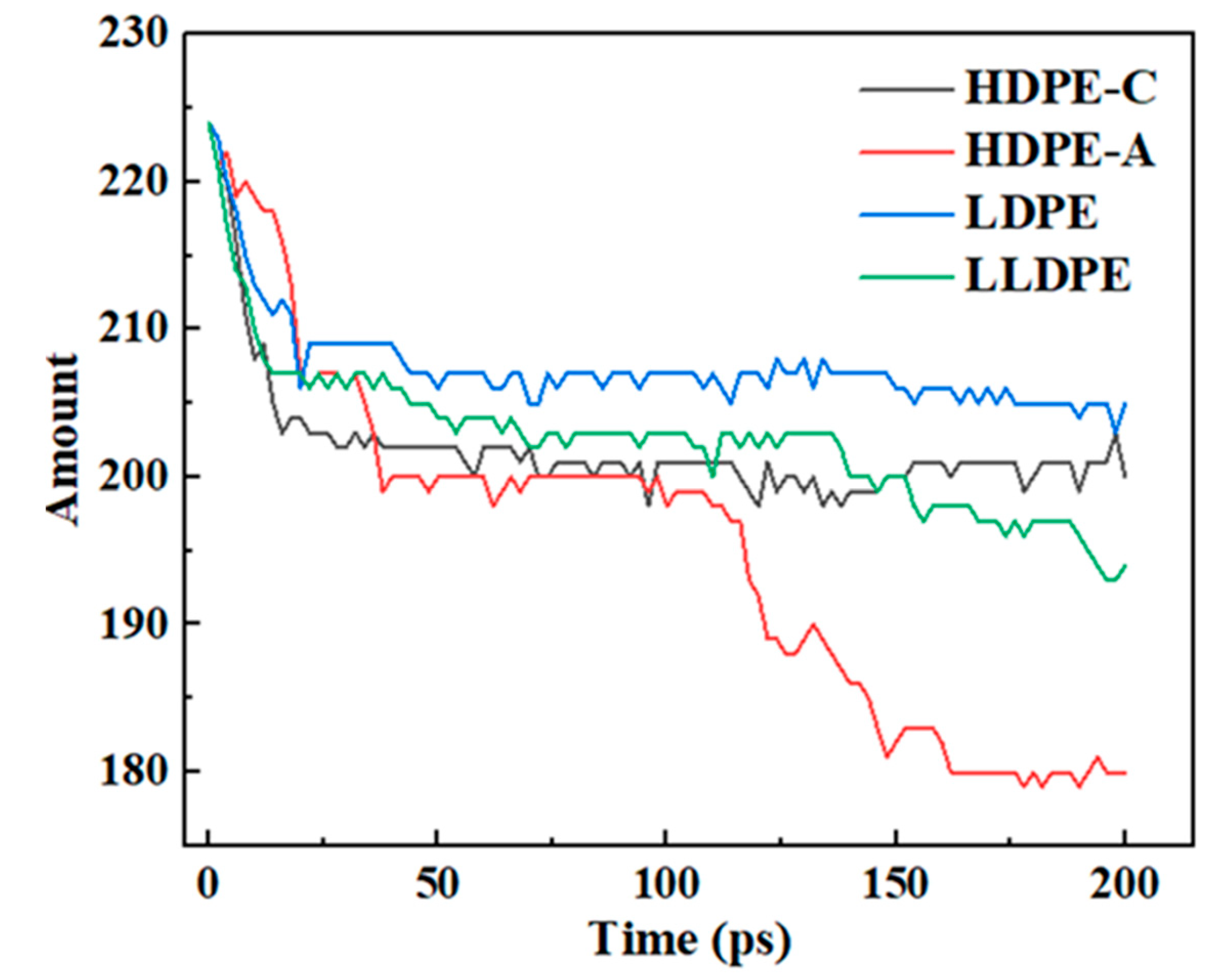
Figure 5.
Carbon fragments distribution of thermal oxidation of HDPE-C system at (a) 1200 K, (b) 1600 K, (c) 2000 K.
Figure 5.
Carbon fragments distribution of thermal oxidation of HDPE-C system at (a) 1200 K, (b) 1600 K, (c) 2000 K.
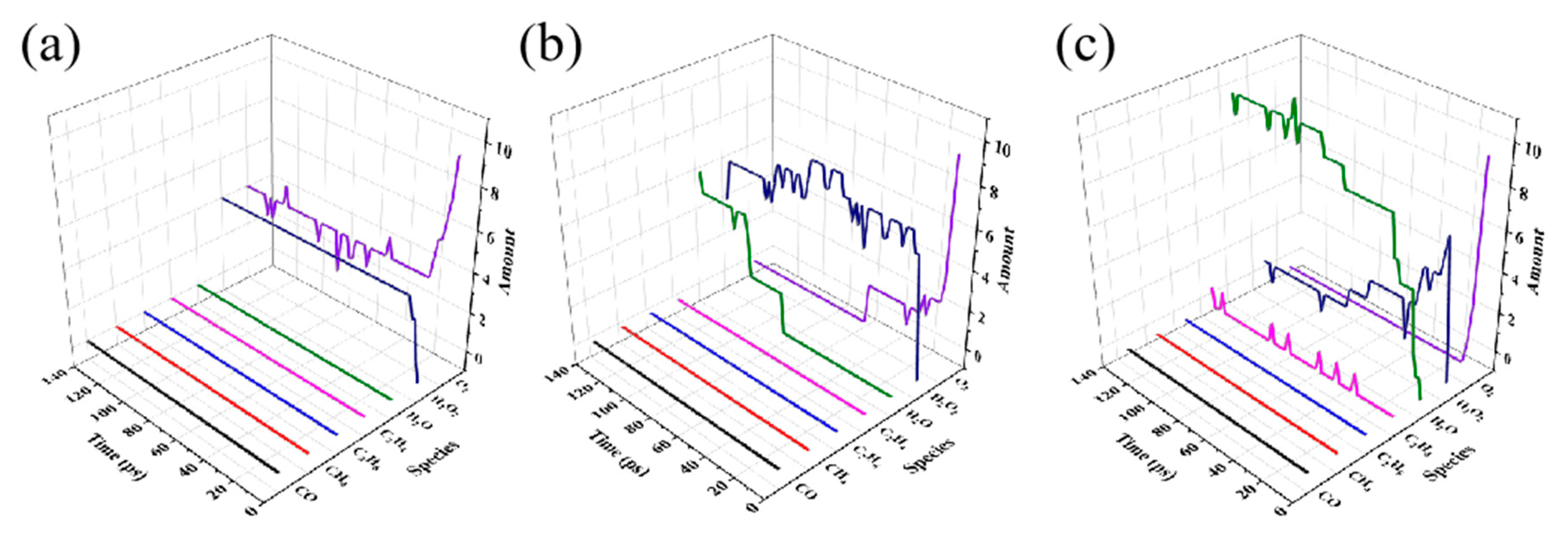
Figure 6.
Gibbs free energy change for H2O2 generation reaction in (a) HDPE-C, (b) LDPE, (c) LLDPE.
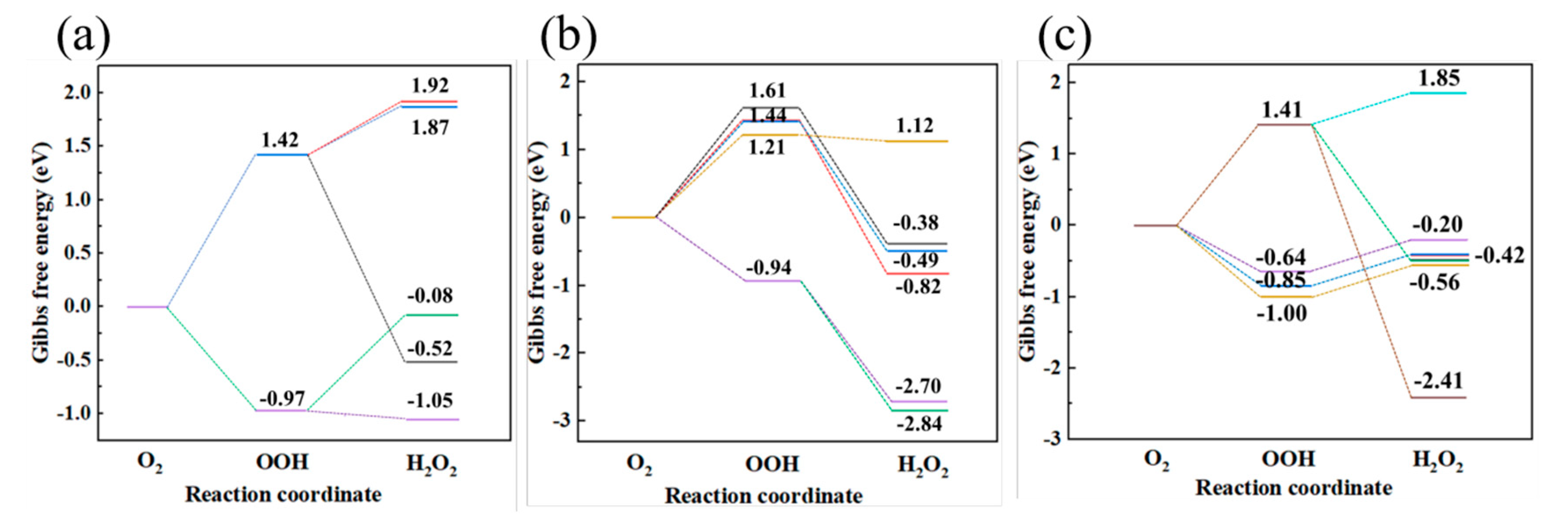
Figure 7.
Maximum spherical pore size distribution in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE.
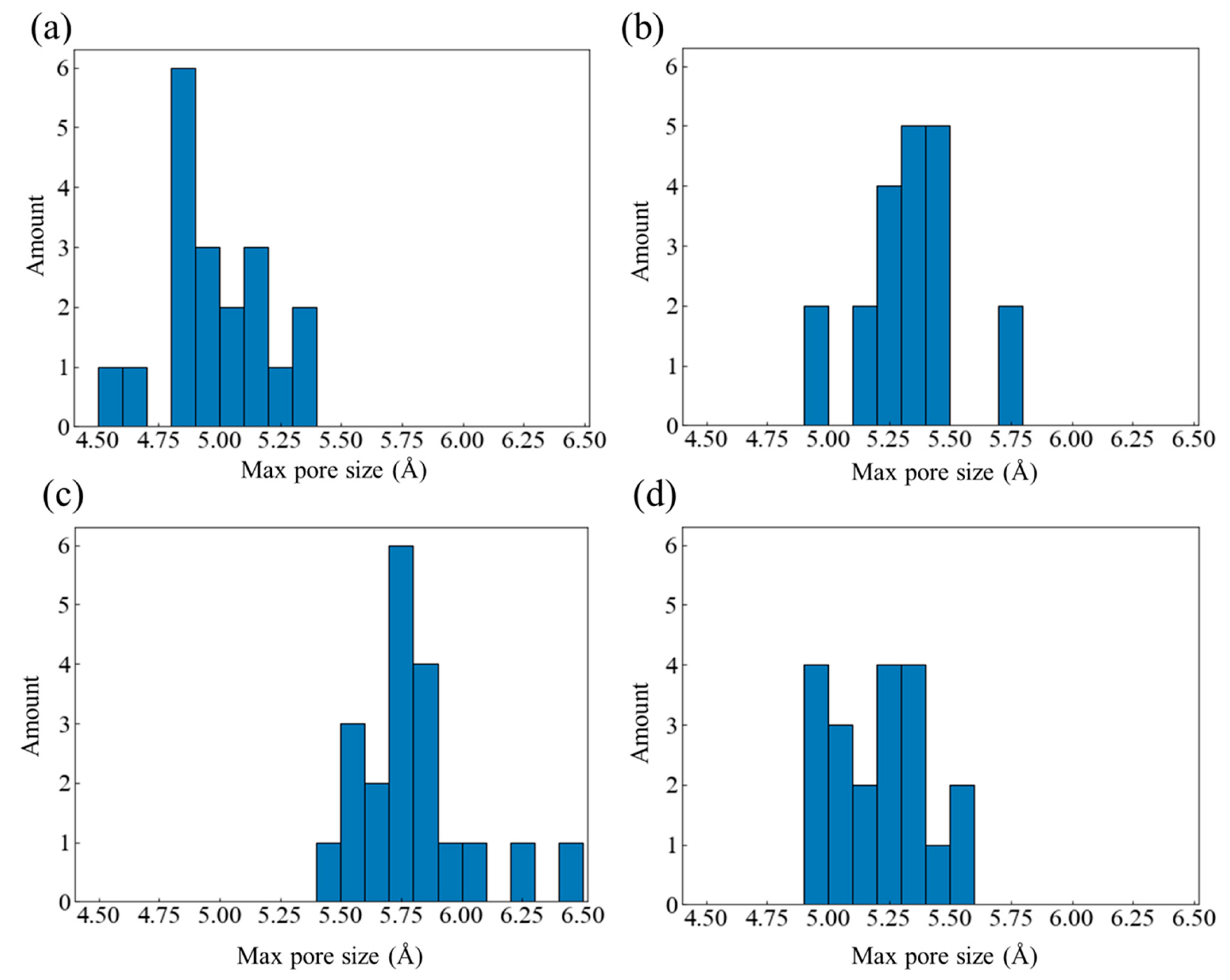
Figure 8.
(a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE oxygen spacing distribution diagram at the initial moment.
Figure 8.
(a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE oxygen spacing distribution diagram at the initial moment.
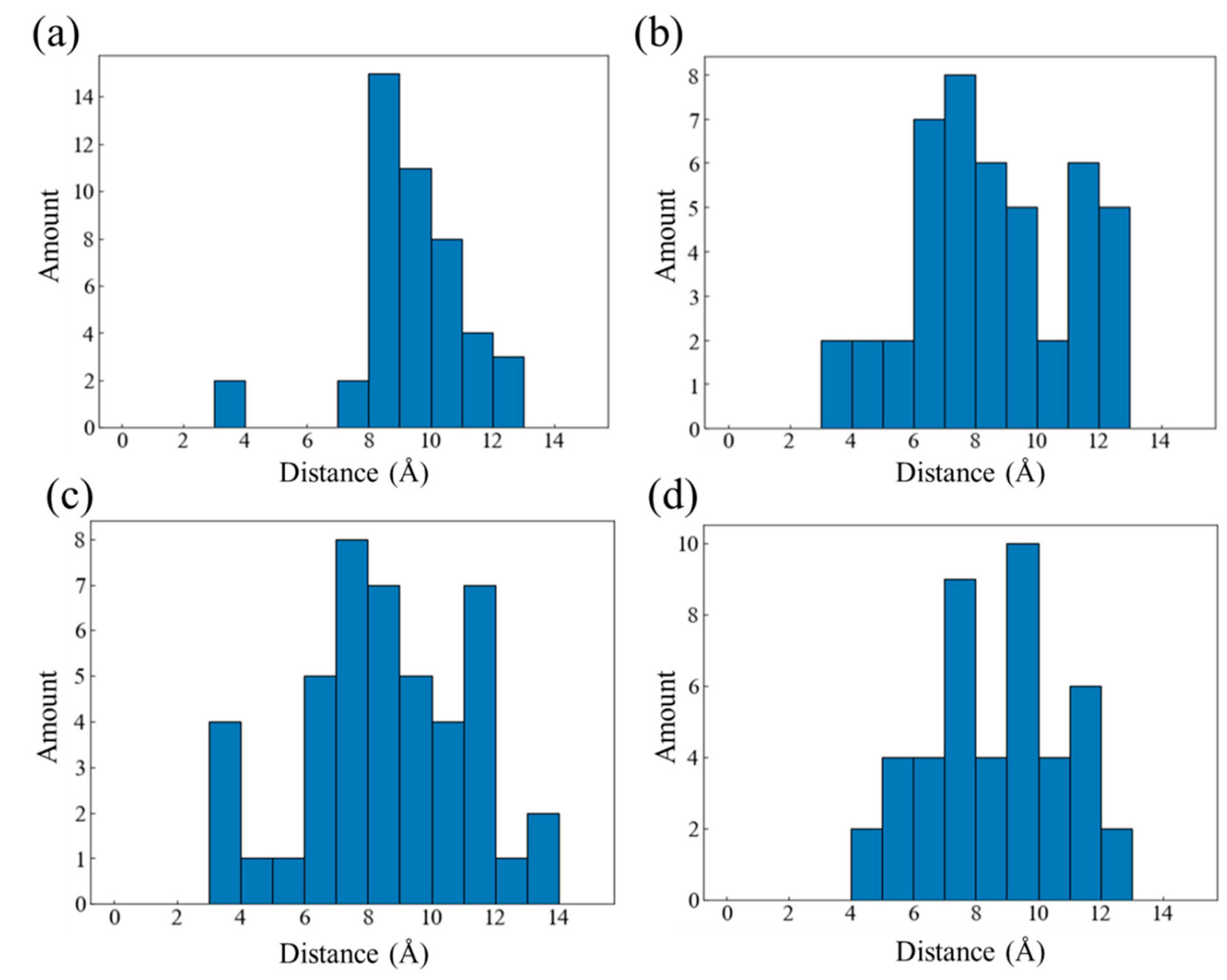
Figure 9.
oxygen diffusion displacement distribution in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE.
Figure 9.
oxygen diffusion displacement distribution in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE.
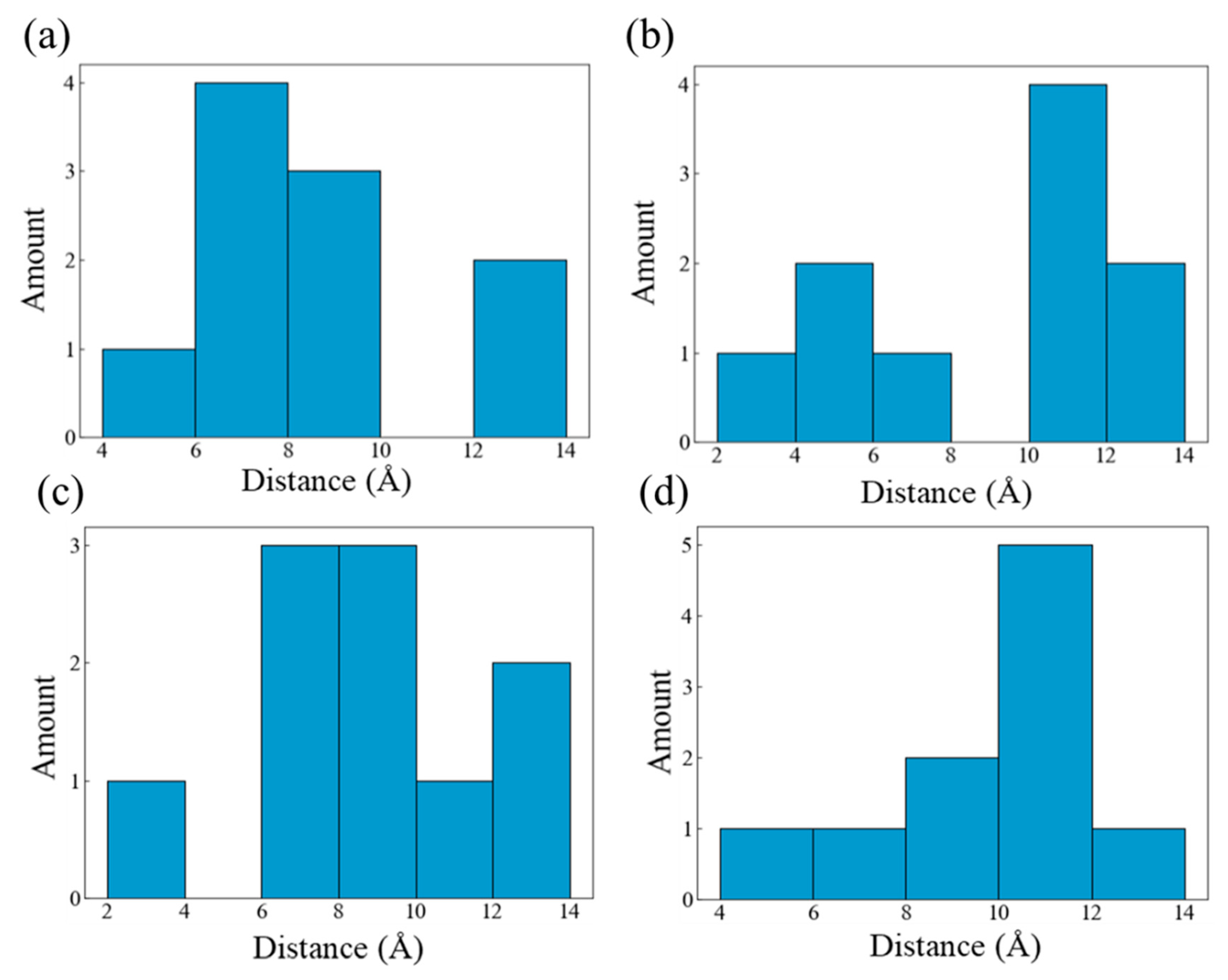
Figure 10.
Distribution of free radicals and unsaturated carbon atoms in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE system at t = 50 ps (unit: nm-3).
Figure 10.
Distribution of free radicals and unsaturated carbon atoms in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE system at t = 50 ps (unit: nm-3).
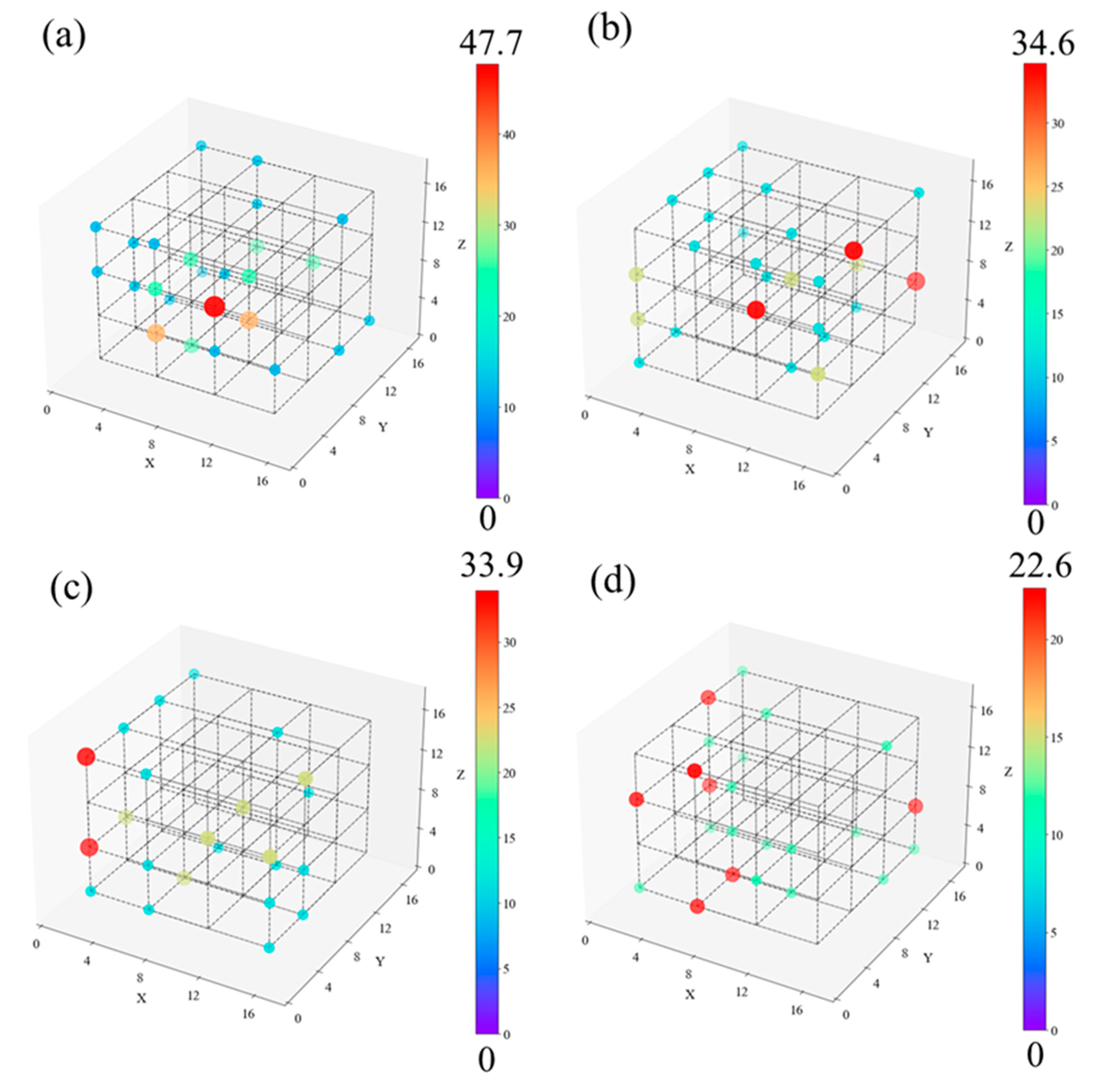
Figure 11.
Distribution of atoms constituting the carbon chain in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE system at t = 50 ps (unit: nm-3).
Figure 11.
Distribution of atoms constituting the carbon chain in (a) HDPE-C, (b) HDPE-A, (c) LDPE, (d) LLDPE system at t = 50 ps (unit: nm-3).
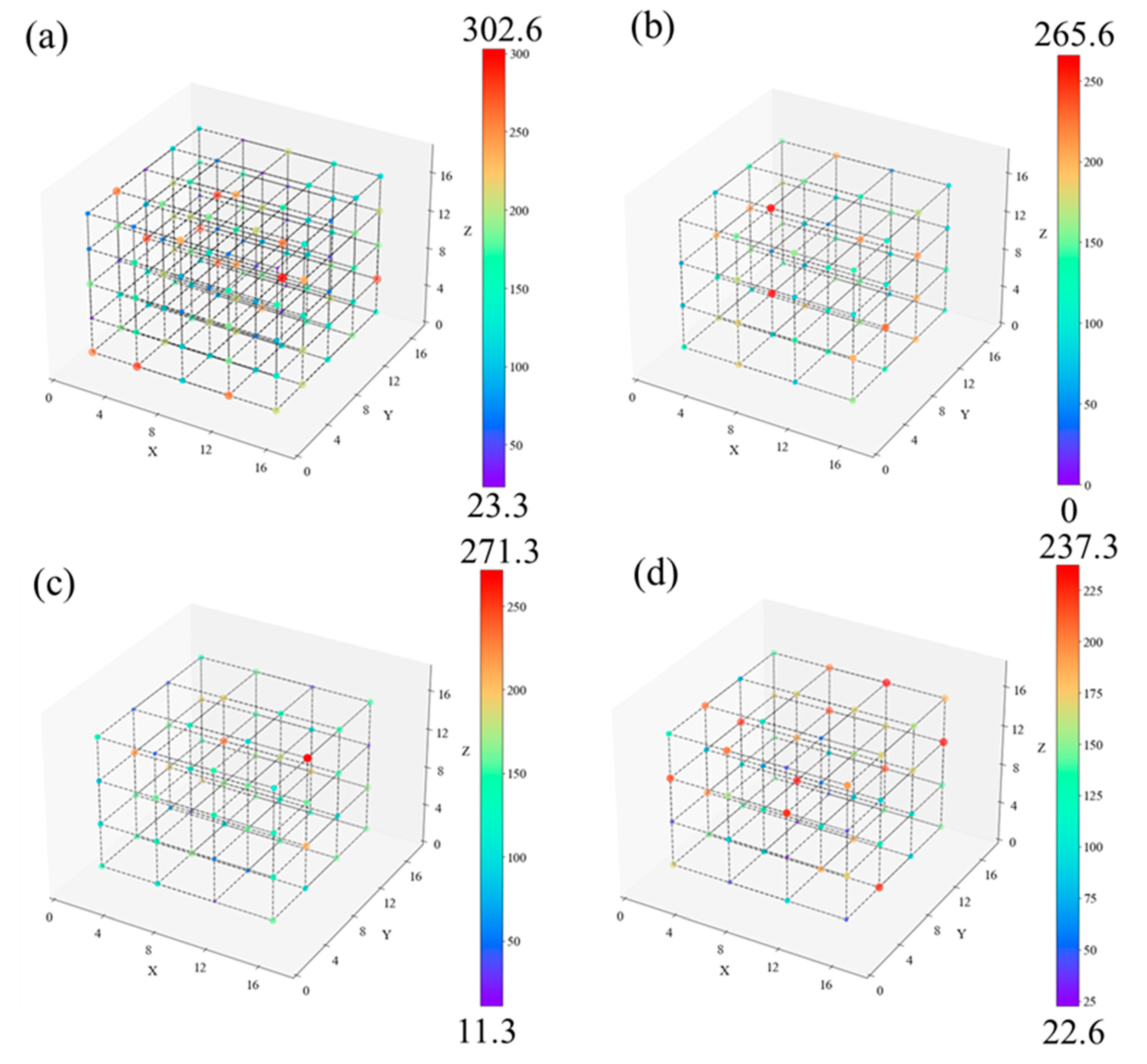

Table 1.
Reactions involving O2 and the formation and consumption of H2O2 in HDPE-C.
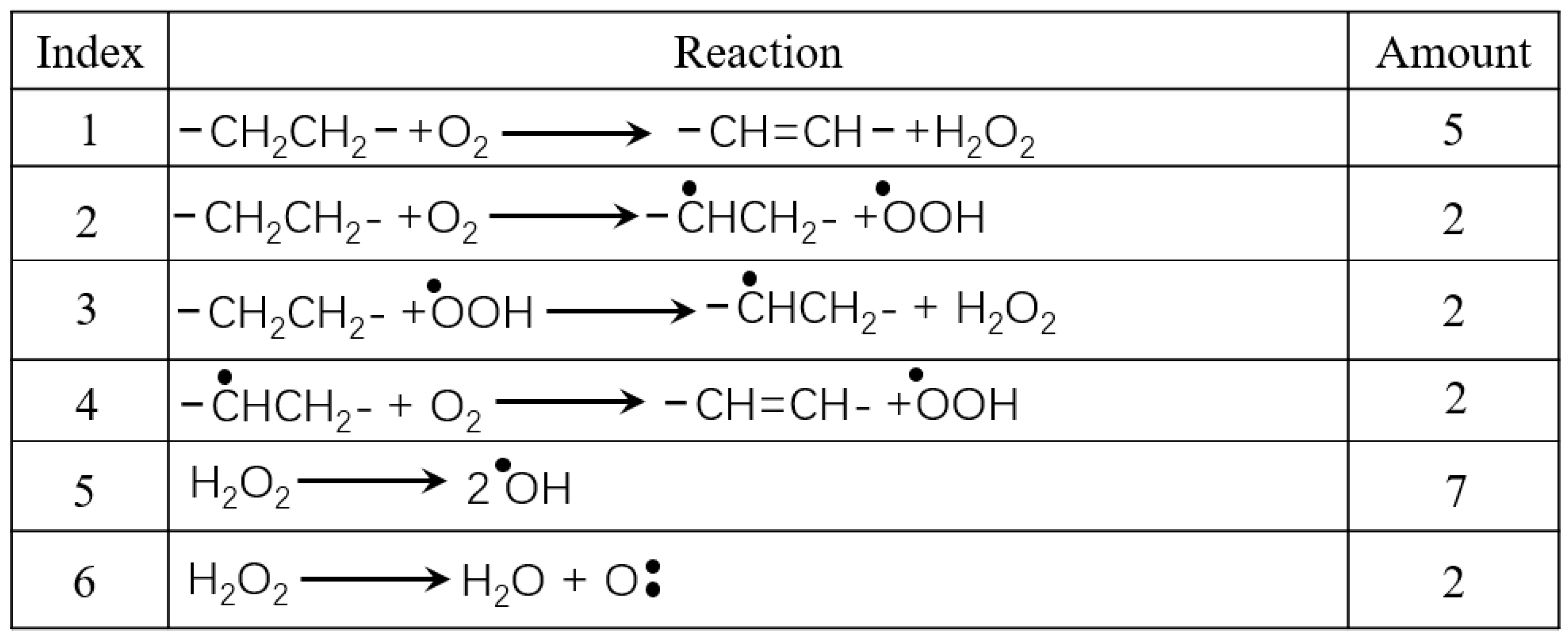 |
Table 2.
Simple reactions involving ·OH radicals in HDPE-C.
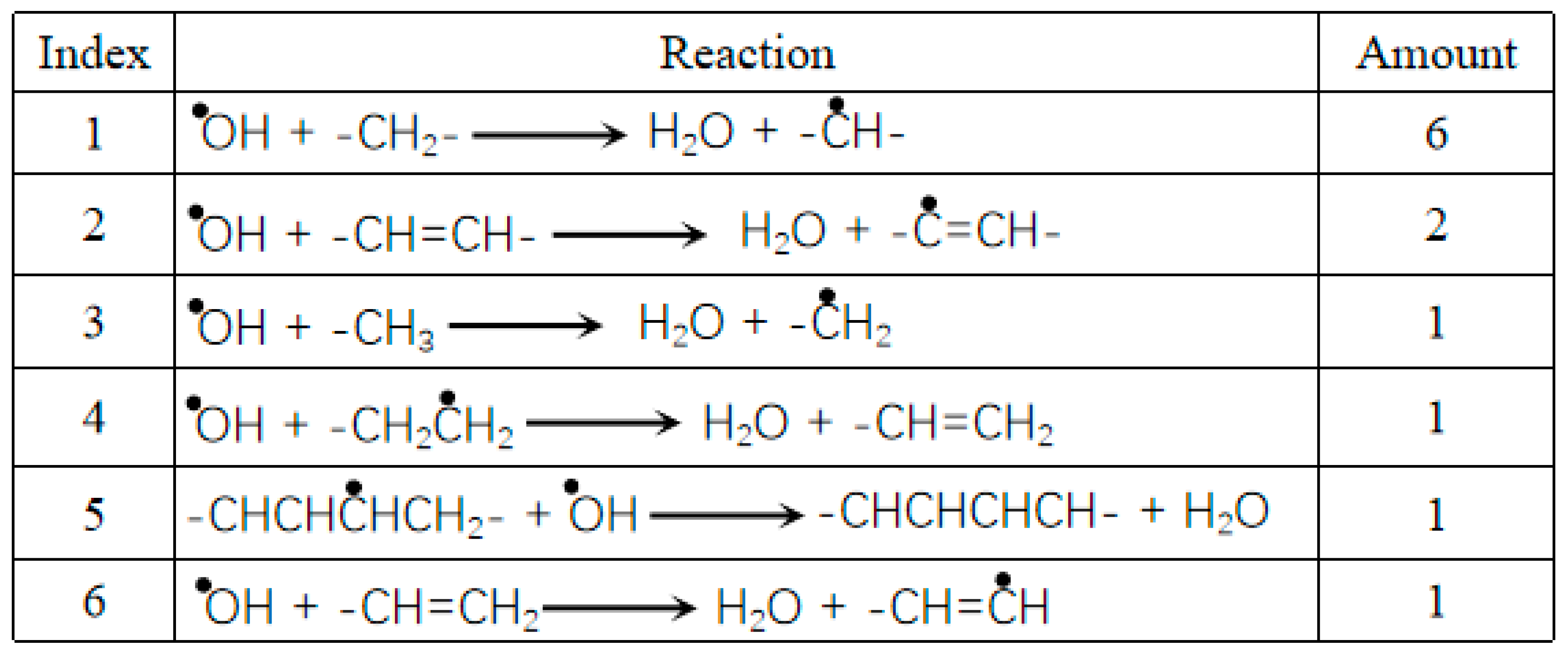 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



