
Submitted:
31 March 2024
Posted:
02 April 2024
You are already at the latest version
Abstract
Inhaled particulate matter (PM) is a key player in the millions of yearly deaths worldwide related to air pollution. The oxidative potential of PM provides an indication of its ability to promote an oxidative environment. When the human lungs are exposed to PM, the reactive oxygen species (ROS) generated in such oxidative environment, if excessive, may lead to cell damage via oxidative stress. Inflammation, endoplasmic reticulum stress, airway remodeling, and many different cell death modes (apoptosis, ferroptosis, pyroptosis, among others) can be triggered by oxidative stress via complex molecular pathways, which also involve the adaptive cellular response. But ROS can also directly interact with macromolecules such as proteins and lipids, as well as nucleic acids, having been reported to induce DNA damage and epigenetic modifications, which jeopardizes homeostasis. These facts have been extensively studied in in vitro models and confirmed in in vivo models.In this review, we delve beyond conventional assessments of airborne particles, discussing their oxidative potential and the PM-induced ROS-mediated cellular damage observed in in vitro models. The close link between oxidative stress and inflammation, and the manifestation of different cell death modes is highlighted by reviewing the latest literature. The effects of ROS on the balance of DNA damage and repair, carcinogenicity and epigenetics interplay are then analyzed. Finally, we expand on the latest reports on the antioxidants’ potential to counteract the deleterious effects of ROS, and disclose future perspectives for the topic.
Keywords:
oxidative stress
; oxidative potential
; air pollution
; particulate matter
; PM
1. Introduction
Air pollution has emerged as the predominant threat to human health among environmental hazards, with particulate matter (PM) playing a central role in driving these adverse effects. The World Health Organization (WHO) estimates that air pollution is accountable for a staggering 7 million premature deaths per year (World Health Organization, 2021). These deaths are linked to a range of conditions including ischemic heart disease, stroke, chronic obstructive pulmonary disease, and lung cancer. Additionally, air pollution has been identified as a risk factor for infectious diseases, with PM being the pollutant that exhibits the strongest correlation with these health outcomes according to WHO. Awareness of the harmful effects of inhaling PM dates back decades, with historical events like the London fog incidents in 1952 underscoring the dangers (LOGAN, 1953). Since the 1990s, research has elucidated the role of reactive oxygen species (ROS) in driving these detrimental effects (Gilmour et al., 1996), by triggering oxidative stress. Oxidative stress is conventionally described as a situation of unbalance in the redox homeostasis towards the accumulation of oxidative species leading to cell damage. In recent times, the concept has been updated to accommodate the “time-course” (besides the concentration) component of the increase in the steady-state ROS levels, distinguishing transient from chronic oxidative stress (Lushchak and Storey, 2021). Also, based on the magnitude of the increase in the steady-state ROS levels, the oxidative stress pioneer Helmut Sies recently hypothesized that a moderate oxidative challenge can actually be beneficial, a status termed oxidative eustress (where the Greek-derived prefix “eu” means “good”). However, when exacerbated and if not timely counteracted, the accumulated ROS can directly interact with the cell’s lipids, proteins, and even DNA, resulting in damage known as oxidative distress (Sies, 2017). Within the context of this review, the expression “oxidative stress” refers to any situation where ROS increases were detected together with signs of cell damage and, eventually, cell death.
Oxidative stress in the lung seems to be exacerbated by not only involuntary exposure to air pollutants but also by natural sources, the utilization of nanoparticle-containing products, and habits such as smoking. While ROS are fundamental for cell homeostasis, regulation and defense, an unbalance between the cellular redox systems towards the accumulation of ROS may trigger a whole variety of pathways leading to molecular changes that can lead to different deleterious health effects (Figure 1). This concept is further detailed in sections 3 to 6.
Over time, two fundamental questions have come to the forefront of research: which specific characteristics of PM induce oxidative stress, and what are the primary health impacts related to oxidative stress? With regard to the former, a substantial body of data points to factors like metal content, polycyclic aromatic hydrocarbons (PAHs), surface properties, and particle size as key elements (Costabile et al., 2023; Oberdörster et al., 2005a) (Table 1). Concerning the latter question, the focus has traditionally been on cardiovascular and respiratory effects (Cho et al., 2018). However, growing evidence is indicating links between inhaling PM and conditions such as neurodegenerative disorders (Calderón-Garcidueñas et al., 2023), heightened susceptibility to Sars-cov-2 infection (Marchetti et al., 2023), prenatal and neonatal complications (Janssen et al., 2017), and even fertility issues (Bongaerts et al., 2023) (Figure 2). In the present review we will focus on three main areas: determining the OP of PM (section 2), the oxidative effects and mechanisms triggered by PM (section 3), and the genotoxic and epigenetic effects related to the oxidative stress triggered by PM (sections 4-6). The review is finalized with a section on antioxidants used to counteract PM-induced oxidative stress-derived adverse effects (section 7) as well as the authors’ perspectives (section 8) on the topic.
2. Oxidative Potential of Particulate Matter
As highlighted earlier, the potential of PM to elicit effects through oxidative reactions has been acknowledged since the 1990s, and there is evidence that the OP of PM has a higher correlation with health effects, than their standard measurement in mass/m3 (Weichenthal et al., 2016). Extensive efforts have been directed towards unraveling the precise mechanisms by which PM induces oxidative stress. The exploration of characteristics that initiate oxidative stress has been a subject of investigation for an extended period, with a focus on elements such as metal ions like Cu+ and Fe2+, along with other chemical components capable of instigating Fenton-like reactions (Quintana et al., 2011; Quintana-Belmares R, 2015; Shi et al., 2003). The overarching objective has been to develop a rapid, and ideally, real-time method for assessing whether PM can contribute to oxidative environments, consequently inducing oxidative stress. Recent years have witnessed some progress in this pursuit. A study comparing the most used acellular assays to detect OP in extracts of recovered PM10 collected in Chamonix, France, showed that the dithiothreitol (DTT) assay has the highest correlation with OP comparatively to the ascorbic acid assay, to electron spin resonance (also known as electron paramagnetic resonance, or EPR), and to the respiratory tract lining fluid assay (Calas et al., 2018). The DTT assay evaluates reactions with ions such as Fe2+, Cu+, Mn3+, and other molecules such as quinones, following Fenton-like reactions, which in the presence of H2O2 produce the highly reactive species superoxide (O2•−) and hydroxyl radicals (·OH) (Jiang et al., 2019). Recently, real-time measuring instruments have been developed, which are capable of collecting PM2.5 and, with a clever approach, mix the particles with a solution of DTT to generate and detect the oxidative species (Eiguren-Fernandez et al., 2017; Puthussery et al., 2018). A limitation of the DTT assay is that some short life oxidative species may not be detected, and in this case, the use of electron spin resonance might be a better option (Jiang et al., 2019). Even though the study by Calas (Calas et al., 2018) may point out that DTT is the best method to do an assessment of OP, the DTT is very sensitive to light, so working with it on the field requires conditions excluding the interaction with light. This is very critical considering that the DTT method measures the extinction coefficient of DTT due to its oxidation.
The transformation of ascorbic acid into dehydroascorbic acid due to oxidation has also been used to assess the OP of PM. Based on this concept, a method for real-time monitoring of OP in PM was recently developed, displaying notable sensitivity to the presence of certain metals such as Cu2+ and Fe2+ (Campbell et al., 2019). A large study comparing the ascorbic acid, DTT, diclhlorofluorescein (DCFH) and EPR methods, demonstrated that ascorbic acid method is the one that better correlates with the sources contributing to the complex mixture of PM (Campbell et al., 2021).
Another widely used method to assess the OP of PM involves the transformation of the non-fluorescent 2′,7′-dichlorodihydrofluorescein (DCFH2) into the fluorescent 2′,7′-dichlorofluorescein (DCF) (Crobeddu et al., 2017). Drawing from this principle, researchers at Peking University devised a system to measure the OP of both gaseous and particulate air pollutants (Huang et al., 2016). The authors were able to identify seasonal variations in the OP, and interestingly, higher levels of OP were observed during winter, correlating with the use of larger amounts of coal and biomass fuel. A limitation of this approach is that the method depends on measurement of fluorescence and that it presents very low correlations when predicting the sources of PM (Campbell et al., 2021).
So far, the evidence indicates that the ascorbic acid and the DTT methods are the more reliable approaches to assess the OP in PM, and it would be a great step forward to create a guideline that could be coupled with the PM2.5 guideline (Goshua et al., 2022) to enhance the protection of the population.
3. Mechanisms of PM-Induced Oxidative Stress-Related Respiratory Toxicity
As previously mentioned, it is widely recognized that ambient PM can cause different health effects, including pulmonary, cardiovascular and neurological diseases as a consequence of triggering oxidative stress and inflammation (Figure 2). In recent years, new mechanisms of PM-induced toxicity have been investigated and described, namely inflammation, DNA damage and genotoxicity, as well as different types of cell death (apoptosis, ferroptosis, pyroptosis). Interestingly, most of those mechanisms seem to share a common denominator: PM-induced oxidative stress.
All the recent in vitro studies reviewed in this section are identified and summarized in Table 2 and Table S1.
In all the studies, increased ROS levels were observed upon exposure to the tested PM (mostly PM2.5, cigarette smoke (CS) extracts (CSE) and organic/inorganic extracts of PM), which supports the role of oxidative stress as a major mediator of PM-induced toxicity. While low to moderate levels of ROS are an integral part of regular homeostasis and under the basal regulation of the antioxidant response element (ARE) enhancer sequence, high concentrations of ROS overload the cellular antioxidant defense mechanisms, such as reduced glutathione (GSH), glutathione peroxidase (GPx), superoxide dismutase (SOD) and catalase (CAT), and trigger a state of redox imbalance towards the oxidation (oxidative stress), which is often harmful for the cell (Ngo and Duennwald, 2022). In fact, these accumulated ROS can induce direct damage to DNA, proteins (protein carbonylation) or lipids (lipid peroxidation), and/or initiate a multifaceted, complex cascade of events such as inflammasome activation, NF-kB induction, Nrf2 regulation, endoplasmic reticulum (ER) stress and dysfunctional autophagy. In turn, these events lead to a multitude of toxic responses among which are exacerbated inflammation, epithelial-mesenchymal transition, airway remodeling, apoptosis, ferroptosis and pyroptosis. These ultimately limit tissue health and tissue functionality.
3.1. Nrf2: the Master Regulator of Antioxidant Response
The participation of the nuclear factor erythroid 2-related factor 2 (Nrf2) in the oxidative stress triggered by PM has been extensively reported in recent years (Badran et al., 2020; Bagam et al., 2021; Housseiny et al., 2020; Ito et al., 2020; Liu et al., 2022b; Saha et al., 2022; Shi et al., 2023; Shrestha et al., 2021; Son et al., 2020; Tian et al., 2021; Wang et al., 2022a, 2022b, 2022c; Xue et al., 2021b). This is not surprising given the key role of Nrf2 in the protection against oxidative stimuli (Saha et al., 2020), acting as a sensor to protect the cells against oxidative damage induced by PM-generated ROS. In fact, activated Nrf2 directly regulates the expression of potent antioxidants, while suppressing the production of pro-oxidant enzymes such as NADPH oxidases (NOX) and xanthine oxidase (XO), in an attempt to restore the redox balance. Simultaneously, Nrf2 can negatively regulate NF-kB, this being one of the main mechanisms explored to control ROS-triggered inflammation (See section 7). Though much less reported, a down-regulation of Nrf2 leading to increased ROS following PM-exposure has also been observed (Wang et al., 2022c).
3.2. Activation of the Inflammatory Response and Its Consequences
Inflammation is, by far, the most common effect reported together with oxidative stress (Table 2), underlying their interdependent character. In fact, there are many pathways by which ROS seem to trigger the production and release of inflammatory mediators. For example, PM-induced ROS has been shown to stimulate the assembly of the NLRP3 inflammasome (Liu et al., 2022b; Mahalanobish et al., 2020; Ren et al., 2022; Shi et al., 2023; Tian et al., 2021; Zhang et al., 2021), a multiprotein complex composed of the nucleotide-binding oligomerization domain-like receptor with a pyrin domain (NLRP), the apoptosis-associated speck-like protein (ASC) and cysteine-aspartic protease 1 (Caspase-1). The latter cleaves pro-interleukins 1 beta and 18 (pro-IL-1b, pro-IL-18) into their mature forms, and Gasdermin D (GSDMD) into an active N-terminal domain (GSDMD-NT) that oligomerizes and forms membrane pores, allowing the release of IL-1b and IL-18 and other damage-associated molecular pattern (DAMP) molecules in a process known as pyroptosis. Being a lytic type of programmed cell death that culminates with the release of inflammatory mediators, pyroptosis is characterized by a highly inflammatory profile that has been associated with PM-induced toxicity in several recent works (Liu et al., 2022b; Ren et al., 2022; Shi et al., 2023; Tian et al., 2021). Occasionally, PM-induced ROS-mediated NRLP3 inflammasome activation that leads to pyroptosis may also involve endoplasmic reticulum (ER) stress (Shi et al., 2023). This is because elevated ROS levels cause the accumulation of unfolded or misfolded proteins in the ER triggering the unfolded protein response (UPR), which aims at degrading the misfolded proteins, halting protein translation and stimulating the production of protein chaperones, such as the binding immunoglobulin protein (BIP/GRP78) (Cui et al., 2022). The UPR process is regulated by ER stress sensor-proteins, namely protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1a (IRE1a) and activating factor 6 (ATF6), which have been found overexpressed following PM-exposure (Guan et al., 2021; Shi et al., 2023; Wang et al., 2022a). In case of continuous PM-induced ROS production and failure of UPR to correct the accumulation of misfolded proteins, ER stress is installed, often leading to apoptosis (Guan et al., 2021; Shi et al., 2023; Wang et al., 2022a). Even though we did not find any report of clear necrosis in our recent literature search, it has been previously reported that high PM2.5 concentrations could lead to autophagy-mediated necrosis (Zhou et al., 2017).
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) is a master regulator of inflammation and immune homeostasis with additional intricated roles in cell survival, proliferation and differentiation (Liu et al., 2017). NF-kB regulates the transcription of inflammatory mediators such as cytokines (IL-6 and tumor necrosis factor alpha, TNF-a), chemokines (IL-8 and monocyte chemoattractant protein 1, MCP-1) and key enzymes (e.g. cyclooxygenase-2, COX-2), which trigger the onset of the inflammatory process (Millar et al., 2022). While increased expression and activation of NF-kB has been widely reported as a consequence of PM-exposure (Chen et al., 2020; Kodali et al., 2022; Meganathan et al., 2022; Ren et al., 2022; Shrestha et al., 2021; Wang et al., 2022a, 2022d) only a few works have proven its derivation from increased ROS, as compounds with antioxidant activity could reverse such effect (Meganathan et al., 2022; Ren et al., 2022; Wang et al., 2022a). Along with its pivotal role in inflammation, NF-kB has shown to promote tissue repair and airway remodeling via epithelial-mesenchymal transition (EMT), an underlying process in many fibrotic lung diseases and cancer (Rout-Pitt et al., 2018). EMT is identified by a decrease in epithelial markers of tight junctions, such E-cadherin, and an increase in mesenchymal markers such as alpha smooth muscle actin (a-SMA) and vimentin. Several recent studies have reported changes in these markers following PM-exposure (Chen et al., 2020; Liu et al., 2022a; Pan et al., 2021; Saha et al., 2022; Wang et al., 2022d; Zhongyin et al., 2022), but only one clearly demonstrated the cascade of events, from ROS generation to NF-kB activation to EMT induction, using antioxidants and an NF-kB inhibitor (Chen et al., 2020). The transforming growth factor beta 1 (TGF-b1) is yet another important player in EMT-mediated airway remodeling induced by PM exposure. In fact, core PM2.5 particles and certain components, such as polyaromatic hydrocarbons (PAH), as well as the PM2.5-generated ROS, are known to activate the TGF-b1/Smad2/3 canonical pathway (Liu et al., 2022a; Xu et al., 2019). This also holds true for exposures to CSE, in which a downregulation of the peroxisome proliferator-activated receptor gamma (PPAR-g) upstream of TGF-b1 has been implicated in the process (Pan et al., 2021). Interestingly, indirect exposure of epithelial cells to medium conditioned by PM2.5-exposed macrophages intensified the inflammatory response and EMT induction comparatively to direct exposure (Wang et al., 2022d).
3.3. Other Relevant Mechanisms Triggered by PM-Induced Oxidative Stress
Less studied, but nonetheless relevant, processes associated with PM-induced ROS generation are autophagy (Bagam et al., 2021; Takizawa et al., 2020; Wang et al., 2022c) and ferroptosis (Wang et al., 2022c). While these are often protective mechanisms, they may be exacerbated upon overwhelming ROS levels that lead to the accumulation of oxidized lipids (Liu et al., 2020a). The autophagic process relies on the formation of autophagosomes and their fusion with lysosomes in a well-orchestrated, complex operation involving a multitude of autophagy-related genes (Atg) and other proteins, where microtubule-associated protein 1 light chain 3 (LC3)-phosphatidyl ethanolamine (PE) conjugate (LC3B-II) plays a central role (Liu et al., 2020b). Therefore, the increased levels of LC3B-II measured in BEAS-2B and A549 cells following exposure to CSE or PM2.5 are suggestive of autophagy induction (Bagam et al., 2021; Takizawa et al., 2020; Wang et al., 2022c). A specific type of autophagy that has been found induced by PM2.5 in bronchial epithelial cells (Wang et al., 2022c) is ferritinophagy, the mechanism by which cells balance the ferritin levels by degrading it to generate iron. When ferritinophagy is deregulated, the subsequent iron overload further feeds the intracellular ROS pool via Fenton reaction and increases the activity of iron-containing enzymes, such as lipoxygenases (LOX) that positively contribute to the pool of oxidized lipids, including lipid peroxide (LPO). Ferritinophagy creates the perfect conditions for the onset of ferroptosis, described as an iron-dependent regulated cell death mode caused by excessive intracellular LPO accumulation. The massive release of oxidized lipid mediators following ferroptosis results in a pro-inflammatory environment, closing the loop to inflammation and airway remodeling. The work by Wang et al. provides a rare demonstration of the crosstalk between these different mechanisms of toxicity, confirming that PM2.5-induced ROS can deregulate autophagy, namely inducing ferritinophagy, leading to ferroptosis and inflammation (Wang et al., 2022c). Besides the direct or indirect damage to cell lipids and DNA (Section 5), PM-induced ROS has also been reported to induce oxidative damage to proteins, resulting in protein carbonylation (Badran et al., 2020; Housseiny et al., 2020; Takizawa et al., 2020).
Because PM can translocate from the small airways into the bloodstream (Oberdörster et al., 2005b), systemic effects of PM have been reported (Thangavel et al., 2022). Noteworthy, ROS-induced immune cell senescence following CS exposure has been reported in vitro (Baskara et al., 2020), and endothelial cells became dysfunctional when exposed to plasma from study participants that had been exposed to high PM2.5 levels and had increased levels of IL-18 in their plasma (Prunicki et al., 2020). Altogether, these findings highlight the importance of cell communication and the need to consider indirect exposures more often in in vitro studies.
This section provides but a summary of the possible paths and outcomes that PM-induced oxidative stress can lead to in vitro, which are summarily schematized in Figure 3.
4. Oxidative Stress and DNA Damage-Repair
In this and the following sections (5 and 6), we will focus on investigating the impact of inhaled particulate matter on DNA damage, carcinogenicity, and epigenetic changes, as illustrated in Figure 4. Our emphasis was exclusively on human in vitro systems, excluding epidemiological studies and in vivo investigations.
When it comes to exposure to PM, the significance of oxidative DNA damage and DNA adducts caused by PAHs cannot be overstated. Various forms of DNA damage, including modifications to the DNA base, the formation of substantial DNA adducts, and the occurrence of single and double strand breaks, have been identified in both epidemiological and toxicological studies involving PM. The repair of oxidative DNA damage is primarily facilitated by the base excision repair (BER) process. Even in the absence of external influences, a quantifiable baseline level of oxidative DNA lesions persists due to the endogenous oxygen metabolism. Hence, the exposure to particles could overwhelm the capacity for repair. The stable DNA adducts induced by PAHs are addressed by nucleotide excision repair (NER), a process that is typically slower and unevenly distributed within the genome, potentially resulting in the accumulation of unrepaired damage. It is also essential to consider other factors such as the possible hindrance of repair processes due to PM-associated metal compounds. Consequently, the degree of DNA repair stands as a crucial determinant in the genotoxic and carcinogenic effects stemming from airborne particle-induced actions. Nonetheless, there remains a scarcity of comprehensive information concerning the influence of PM on DNA repair pathways. It remains unclear which pathways are most significantly affected by exposure to PM or whether DNA damage is adequately addressed (Hartwig, 2002; Quezada-Maldonado et al., 2021).
ROS induce a range of DNA lesions, encompassing base modifications, sugar lesions, tandem and clustered lesions, single- and double-strand breaks, DNA-protein and DNA interstrand crosslinks. Using sensitive techniques, nearly 100 distinct lesions have been detected. Notably, common endogenous base modifications include 8-oxo-7, 8-dihydroguanine (8-oxoGua) and 2, 6-diamino-4-hydroxy-5-formamidopyrimidine, all originating from hydroxyl radical interactions at the C8 position of the guanine ring. The frequency of these lesions hinges on factors encompassing oxidative stress quality, level, and diverse contributors. The modifications in DNA prompted by 8-Oxo-2'-deoxyguanosine (8-oxodG) function as recognition sites for DNA glycosylases, which aid in detecting damaged guanine bases. Interestingly, the formation of FapydG—a prevalent guanine-derived lesion—is accentuated under low oxygen (hypoxia) conditions. Additionally, interactions between hydroxyl radicals and pyrimidines (thymine and cytosine) at the 5 or 6 positions of the ring culminate in various base lesions, including prominent products like thymine glycol (Tg) and cytosine glycol. Evidently, 8-oxodG and Tg stand as reliable markers of oxidative stress across diverse biological systems, ranging from bacteria to human cancer patients (Cadet et al., 2017; Hartwig, 2002; Kryston et al., 2011).
Our current focus revolves around examining research that substantiates oxidative DNA damage induced by PM, establishing a clear association with oxidative stress. This involves assessing biomarkers of oxidative DNA damage, such as 8-Hydroxy-2′-Deoxyguanosine (8-OhdG) or FapydG or expressions of genes involved in BER and NER in human lung, vascular and neuronal cell lines. Common techniques employed to detect such DNA damage include assessing strand breaks through simple or enzyme-modified comet assays, direct quantification of 8-OHdG using liquid chromatography/ mass spectrometry (LC/MS) or enzyme-linked immunosorbent assay (ELISA) and evaluating phosphorylated H2A histone family member X (γH2AX) expression through immunostaining. Significant oxidative DNA damage in BEAS-2B cells exposed to PM10 from different sources in Flanders, Belgium, was attributed to variations in PM characteristics and metal compositions (Van Den Heuvel et al., 2016). A549 cells exposed to traffic-related PM demonstrated oxidative DNA damage (8-OHdG), underscoring PM's impact on genetic integrity (Vattanasit et al., 2014). Exposure to PM2.5 (collected from Beijing University of Technology, China) induced DNA damage and altered DNA repair gene expression in 16HBE cells (Niu et al., 2020b). Overexpressing 8-Oxoguanine glycosylase (OGG1) in BEAS-2B cells counteracted PM2.5-induced disruptions, showcasing its potential protective role (Yang et al., 2015). PM10 exposure led to PAH-DNA adducts and altered NER pathway components in A549 cells, linking PM-induced DNA damage to potential carcinogenesis (Quezada-Maldonado et al., 2022). Reference PM (urban dust SRM1649 and diesel PM SRM2975) samples elicited comparable oxidative DNA damage but differed in persistence and chromosomal instability, emphasizing the role of specific particle types (Cao et al., 2022). Interestingly, the diesel exhaust particles-induced oxidative DNA damage in A549 cells was attenuated by THP-1a macrophage co-culture (Jantzen et al., 2012). Wood smoke PM-induced dose-dependent DNA damage and oxidative stress in A549 cells were reported (Danielsen et al., 2011). Sidestream smoke exposure increased ROS, triggering oxidative DNA damage in BEAS-2B cells; Endonuclease VIII-like 2 (NEIL2) knockdown intensified the damage (Sarker et al., 2014). DNA polymerase β played a crucial role in repairing oxidative DNA damage from CS (Cui et al., 2012). PM2.5 induced DNA damage and senescence in corneal epithelial cells was alleviated by ROS inhibition (Gao et al., 2016). The NAC prevented CS-induced double-strand breaks, while the glutathione synthesis inhibitor, DL-Buthionine-[S,R]-sulfoximin (BSO), exacerbated damage in A549 cells (Albino et al., 2006). In brief, in vitro studies highlight PM-induced oxidative DNA damage, underlining the need for comprehensive understanding to mitigate health risks linked to airborne PM exposure.
5. Oxidative Stress and Carcinogenicity
Carcinogenesis refers to the disruption of normal cellular function caused by genomic instability, leading to uncontrolled cellular growth and the invasion of surrounding tissues, a process known as cellular transformation. This transformation of normal cells into malignant ones is a multi-stage process involving initiation, promotion, and progression (Hanahan and Weinberg, 2011). PM-mediated oxidative stress significantly contributes as a crucial driving force across the spectrum of cancer, spanning initiation, promotion, and progression stages (Santibáñez-Andrade et al., 2023).
Substances that disrupt pathways connected to cancer hallmarks are generally considered carcinogenic. In 2012, the International Agency for Research on Cancer (IARC) identified ten key characteristics commonly shared among human carcinogens, including electrophilic behaviour, genotoxicity, DNA repair alteration, epigenetic changes, oxidative stress induction, chronic inflammation, immunosuppression, receptor-mediated effects modulation, immortalization, and influence on cell proliferation, cell death, or nutrient supply (Smith et al., 2016). Airborne PM exhibits most, if not all, of these characteristics. For instance, diesel engine exhaust was classified as carcinogenic to humans (Group 1) and associated PAHs mixture components range from group 1 (carcinogenic to humans) to group 2B (possibly carcinogenic to humans).
Experimental evidence of PM-induced in vitro cell transformation demonstrated the carcinogenic potentiality of airborne PM. The long non-coding RNA (lncRNA) SOX2 overlapping transcript (SOX2-OT) , microRNA (miR)-345-5p and epidermal growth factor receptor (EGFR) cascade drove HBE cell transformation by PM2.5, while lncRNA Nuclear paraspeckle assembly transcript 1 (NEAT1), miR-582-5p and Hypoxia-inducible factor (HIF-1α) axis induced EMT and cancer stem cell traits in BEAS-2B cells by PM2.5 (Fu et al., 2021; Jiang et al., 2021). The carcinogenicity of CS is underscored by its ability to disrupt gene expression, impede DNA adduct repair, and inhibit apoptosis, culminating in neoplastic transformation within BEAS-2B cells (Du et al., 2012). The intricate landscape of CS-induced carcinogenesis involves aberrant DNA methylation, encompassing global hypomethylation and gene-specific modifications, as key drivers in the malignant transformation of BEAS-2B cells (Huang et al., 2017). In vitro models of CS-induced BEAS-2B cell transformation revealed that RNA-binding motif protein 5 (RBM5) overexpression curbs the growth of these transformed cells by imposing cell cycle arrest and apoptosis (Lv et al., 2016). Additionally, prolonged CS exposure amplified miR-200b levels, instigating BEAS-2B cell migration through targeted E26 transformation-specific sequence-1 (ETS1) modulation (J. Wang et al., 2021). In the realm of HBE cells, a convergence of factors underscores CS-induced malignancy. The lncRNA HOX transcript antisense RNA (HOTAIR) emerged as a pivotal nexus linking inflammation, EMT, and lung carcinogenesis (Liu et al., 2015). In a parallel line, miR-217 orchestrated the regulation of enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) and in turn the tri-methylation of lysine 27 on histone H3 protein (H3K27me3) via the lncRNA metastasis associated in lung adenocarcinoma transcript 1 (MALAT1), further contributing to the intricate network governing CS-induced malignancy (Lu et al., 2015). Furthermore, the orchestrated interplay of miR-218, lncRNA colon cancer-associated transcript-1 (CCAT1), and BMI1 proto-oncogene polycomb ring finger (BMI1) exerted its influence, propelling a distinct cell cycle transition in the context of CS extract-induced malignancy within HBE cells (Lu et al., 2016). These in vitro-based findings collectively illuminate the intricate mechanisms underpinning CS-induced malignancy.
6. Oxidative Stress and Epigenetics Interplay
The interplay between oxidative stress (ROS), and various epigenetic factors is a complex and dynamic process that significantly influences cellular functions, development, and disease progression. Epigenetic mechanisms, involving DNA methylation, histone modifications, and non-coding RNAs (miRNAs, lncRNAs, circRNAs), interact with ROS to shape gene expression, driving phenotypic variation beyond the DNA sequence. ROS can directly oxidize DNA bases, such as cytosine and guanosine, leading to the formation of oxidized DNA lesions. These lesions, including 5-hydroxymethylcytosine (5hmC) derived from 5-methylcytosine (5mC), can interfere with DNA methylation inheritance and affect the binding of DNA methyltransferases (DNMTs). Indirectly, ROS can affect DNA methylation by acting on either activity or expression of DNMTs and ten-eleven translocation methylcytosine dioxygenases (TETs) enzymes. On the other hand, DNA methylation can influence ROS homeostasis through epigenetic activation or silencing of pivotal genes involved in ROS regulation, such as NADPH oxidases. This regulation can occur via hypo- or hypermethylation of their promoter regions. Thus, ROS play a critical role in modulating DNA methylation which leads to the disruptions in gene silencing and activation (Bhat et al., 2018; Hayes and Knaus, 2013; Kietzmann et al., 2017). ROS directly influence histone modifications, affecting chromatin structure and gene expression. ROS-induced oxidative stress can modify histone proteins, including H1, H2B, and H3, through processes like nitration and oxidation. These modifications can alter the chromatin architecture and accessibility, ultimately influencing gene transcription. Additionally, ROS-sensitive histone-modifying enzymes, dependent on metabolites such as acetyl-CoA, ketoglutarate, NAD+, and S-adenosylmethionine, establish a critical link between cellular metabolism and epigenetic changes. This dynamic balance between activating marks (e.g., di/tri-methylation of lysine 4 on histone H3 protein H3K4me2/3) and repressive marks (e.g., di/tri-methylation of lysine 9 on histone H3 protein H3K9me2/3, tri-methylation of lysine 27 on histone H3 protein H3K27me3) can be modulated by ROS, impacting gene expression (Guillaumet-Adkins et al., 2017). The interplay between ROS and non-coding RNAs, particularly miRNAs, adds another layer to the regulatory network. ROS-responsive miRNAs play a pivotal role in fine-tuning cellular ROS levels by targeting genes involved in ROS production and scavenging. Conversely, miRNAs that regulate ROS-related pathways can modulate redox homeostasis. Furthermore, ROS can influence the expression of non-coding RNAs through the activation of ROS-sensitive transcription factors, creating a feedback loop between ROS and miRNA-mediated gene regulation (He and Jiang, 2016; Lin, 2019). In summary, the intricate interplay between ROS and epigenetic factors forms a bidirectional regulatory loop. ROS influence epigenetic modifications, and epigenetic mechanisms modulate ROS-related pathways. This dynamic interplay impacts gene expression, cellular responses, and disease progression.
Evidences suggest PM-induced oxidative stress caused epigenetics modification in the in vitro system. The exposure to both inhaled and non-inhaled PM samples (PM2.5: SRM1650b, PM10: ERM-CZ100 and ERM-CZ120, and PM10-PAH) triggered DNA hypomethylation in normal human bronchial epithelial cells (NHBE) and epidermal keratinocytes (NHEK). Interestingly, NHBE cells exposed to PM10-PAH and NHEK cells exposed to PM10 exhibited a more pronounced hypomethylation of repetitive transposable element Alu and long interspersed nuclear elements 1 (LINE1), showcasing differential susceptibility (Lee et al., 2022b). In a separate context, the treatment of lung cells (16HBE) with PM2.5 (collected from Shijiazhuang, Hebei province, China), revealed specific m6A-modified sites on Nrf2 mRNA, located relative to the three prime untranslated region (3'UTR)'s first nucleotide. Furthermore, the YTH N6-methyladenosine RNA binding protein F1 (YTHDF1) and the insulin like growth factor 2 mRNA binding protein 1 (IGF2BP1) interact with m6A sites on Nrf2 mRNA, thereby facilitating Nrf2 translation, which plays a role in mitigating oxidative stress induced by PM2.5 exposure (Ji et al., 2023). PM2.5 (collected at Chongqing south road, China) instigated a significant redox imbalance within cellular environments, leading to the depletion of the intracellular methyl donor S-adenosylmethionine. This depletion triggered widespread DNA hypomethylation in the human neuroblastoma (SH-SY5Y) cell line. Furthermore, PM2.5 exposure disrupted gene-specific promoter DNA methylation patterns, resulting in the abnormal mRNA expression of candidate genes associated with autism. Notably, the phenomenon of PM2.5-induced DNA hypermethylation within gene promoters related to synapses correlated with diminished mRNA and protein expression levels. The intriguing aspect of this interplay is underscored by interventions involving antioxidative reagents, a methylation-supporting agent, and a DNA methyltransferase inhibitor. These interventions highlight the intricate collaboration between redox and methylation mechanisms, contributing to the emergence of abnormal DNA methylation patterns and altered expression of synaptic proteins (Wei et al., 2016). In a related study, the significance of oxidative stress-mediated abnormal DNA hydroxymethylation in neuronal impairments induced by the same PM2.5 exposure became evident. Using the SH-SY5Y cell line, both PM2.5 and its organic extracts triggered a notable increase in global DNA hydroxymethylation and gene-specific DNA hydroxymethylation, particularly within neuronal genes. This perturbation in DNA hydroxymethylation patterns has detrimental effects on mRNA expression. Notably, the compelling aspect emerged when antioxidants, such as NAC and GSH, were employed. Their use validated the role of oxidative stress-induced hydroxymethylation abnormalities in the context of PM2.5-induced deficiencies in neurite outgrowth and synapse formation (Wei et al., 2017). Exposure of human keratinocytes (HaCaT and HEK100) to diesel PM2.5 (SRM1650b), triggered skin senescence by orchestrating ROS-mediated epigenetic alterations that impact the expression of the senescence-associated gene, p16INK4A. Extensive analysis of epigenetic markers revealed reduced DNMT activity, increased DNA demethylase (TET) activity, decreased EZH2 histone methyltransferase activity, and elevated expression of the epigenetic transcriptional activator MLL1. Consequently, interactions of DNMT1, DNMT3B, and EZH2 with the p16INK4A promoter were reduced, while TET1 and MLL1 binding was enhanced. These changes led to decreased histone H3 lysine 27 trimethylation (H3K27Me3), heightened H3 lysine 4 trimethylation (H3K4Me3), and DNA hypomethylation in the p16INK4A promoter region. Significantly, the ROS-scavenger NAC effectively mitigated cellular senescence by modulating these epigenetic modifications (Ryu et al., 2019). Exposure of A549 cells to PM10 (collected from Marylebone and Bloomsbury in London, UK) led to increased activity of histone acetyltransferases (HATs) and elevated levels of acetylated histone 4 (H4). PM10-induced enhancement of H4 acetylation was linked to oxidative stress, as demonstrated by inhibition using thiol antioxidants (NAC and mannitol). Notably, PM10-mediated acetylation of H4 occurred in the promoter region of the IL-8 gene, highlighting the role of histone acetylation-mediated chromatin remodeling in the lung's response to PM10 exposure (Gilmour et al., 2003). The long noncoding RNA, lnc-IL7R, demonstrated an intriguing connection, exhibiting an inverse relationship with both emphysema and exposure to PM2.5. Upon exposure to PM2.5 (SRM 1650b), normal lung epithelial cells (HSAEpCs and BEAS-2B) exhibited heightened expression of lnc-IL7R along with increased ROS levels. Additionally, a mechanistic insight emerged, revealing epigenetic modulation through EZH2-mediated recruitment of H3K27me3, H3K9me3, and acetylation of lysine 9 on histone H3 protein (H3K9ac). This mechanism highlighted how lnc-IL7R orchestrates the regulation of PM2.5-induced cell senescence genes, particularly p21CIP1/WAF1 (Lee et al., 2022c). Functional genetics studies have provided compelling evidence for the critical role of the lncRNA linc01515 in PM2.5-induced oxidative stress (ROS and MDA level, SOD enzyme activity, etc.) in both HBE and BEAS-2B cells. Intriguingly, the enrichment level of m6A on linc01515 was found to increase after exposure to PM2.5, subsequently leading to the upregulation of Nrf2. The PM2.5 treatment may enhance the expression of linc01515 through augmented m6A modification, thereby influencing Nrf2 regulation and inducing oxidative damage in airway epithelial cells (Wang et al., 2023b). The initiation of oxidative stress mediates the PM2.5-activated inflammatory response in human bronchial epithelial cells (16HBE), facilitated by the novel circular RNA (circRNA) circ_0008553 (Wang et al., 2023a). Several circRNA-miRNA-mRNA axes have been identified that promote apoptosis, inflammation, and oxidative stress in Human pulmonary microvascular endothelial cells (HPMECs), BEAS-2B cells, and 16HBE cells exposed to CSE. These include hsa_circ_0006872/miR-145-5p/NF-kB (Xue et al., 2021a), circ_0006892/miR-24/ H domain and leucine rich repeat protein phosphatase 2 (PHLPP2) (Zhang et al., 2022), LINC00612/miR-31-5p/Notch1 (Luo et al., 2020), Circ-RBMS1/miR-197-3p/ F-box protein 11 (FBXO11) (Qiao et al., 2021), and circANKRD11/miR-145-5p/bromodomain containing 4 (BRD4) (Wang et al., 2021), circ-HACE1/miR-485-3p/ toll like receptor 4 (TLR4) (Zhou et al., 2021). The miRNA let-7a plays a pivotal role in counteracting oxidative stress by directly regulating arginase 2 (ARG2) mRNA expression levels. BEAS-2B cells exposure to PM2.5 led to decreased let-7a miRNA levels, resulting in increased ARG2 expression and heightened oxidative stress, as indicated by elevated ROS, MDA, and SOD activity levels (Song et al., 2016). Moreover, exposure to PM2.5 reduced miR-331 expression via the ROS/PI3K/Akt pathway, subsequently increasing the expression of inhibitor of nuclear factor kappa-B kinase subunit beta (IKK-β) and sustaining NF-kB activation in BEAS-2B cells (Song et al., 2017). The miR-486 counteracted PM2.5-induced apoptosis and oxidative stress by targeting phosphatase and tensin homolog (PTEN) and forkhead box O1 (FOXO1) (Li et al., 2018), while exposure to PM2.5 in A549 cells increased DNA damage response-related miRNAs (miR-222, miR-210, miR-101, miR-34a, and miR-93). Pre-treatment with the natural antioxidant morin effectively reduced PM2.5 toxicity and miRNAs expressions (Veerappan et al., 2019). Taken together, the diverse array of in vitro studies outlined here underscores the intricate interplay between epigenetic alterations and the detrimental effects induced by PM exposure, shedding light on the complex mechanisms through which PM impacts cellular function and health.
7. Compounds with Antioxidant Activity against PM-Induced Oxidative Stress
Antioxidants are compounds that can prevent or delay the oxidation of a substrate. For a long time, antioxidants were thought to act by directly scavenging free radicals, but the short half-life of free radicals kinetically restrict the effectiveness of this mechanism (Hrelia and Angeloni, 2021). Many antioxidants trigger the adaptive response to oxidative stress by activating the Nrf2 signaling pathway, which results in up-regulation of the endogenous antioxidant systems. Some polyphenols can down-regulate PI3K/Akt, activate AMPK and sirtuins, or impair NF-κB signaling (Hunyadi, 2019). Interestingly, all of these pathways are suspected to be involved in some PM-induced oxidative stress-related effects (Table 2). Others act by generating bioactive metabolites (Hunyadi, 2019). Besides their pharmacological and therapeutic relevance, antioxidant compounds are also a useful tool to establish the mechanistic dependence of a specific pathway on ROS production. In this section, molecules whose antioxidant activity against PM-induced oxidative stress was explored in vitro in recent years will be addressed, grouped according to their origin. For some of these antioxidants, the in vitro findings are supported by in vivo data (see Table S1).
i) Antioxidants based on synthetic drugs
N-acetylcysteine (NAC), a pro-drug of the GSH precursor, L-cysteine, is by far the most often referred to antioxidant in recent literature (see Table S1). NAC is used medically to relieve mucus production and to fight acetaminophen overdose, and has been used preclinically to underpin the relationship between PM-induced ROS generation and inflammation (Wang et al., 2022b), autophagy regulation (Bagam et al., 2021a; Zhang et al., 2021), ER stress (Guan et al., 2021), cell senescence (Baskara et al., 2020), M2 polarization (Liu et al., 2022a), EMT promotion (Chen et al., 2020), ROS-mediated epigenetic alterations (Gilmour et al., 2003; Wei et al., 2017), as well as apoptosis and pyroptosis (Ren et al., 2022). Therefore, NAC has been a key tool to study the oxidative stress-related effects induced by PM.
The free radical scavenger edaravone protected HBECs against PM-induced ROS generation, mitochondrial dysfunction and inflammation via downregulation of NF-κB (Zeng et al., 2022). The anti-inflammatory drug methylprednisolone is widely used to treat lung inflammatory diseases, such as COPD. Interestingly, besides reducing the inflammatory response, methylprednisolone has been shown to restore the redox homeostasis and prevent excessive apoptosis in CS-exposed epithelial cells, with involvement of the 5' adenosine monophosphate-activated protein kinase (AMPK) pathway (Yu and Zhang, 2022). Targeting the mitochondria, where it reinstates mitochondria membrane potential, the antioxidant drug SS-31 has shown potential to alleviate CS-induced ROS-mediated toxicity in epithelial cells (Yang et al., 2021). SS-31 attenuated the CS-induced activation of the p38 mitogen-activated protein kinase (MAPK) pathway, which has implications in the regulation of pro-oxidative enzymes and NF-κB activity.
ii) Antioxidants extracted from plants or algae
There are many algae or plant-derived compounds that have been tested as antioxidants in PM-induced oxidative stress. This is, indeed, one of the classes of compounds most explored for this purpose, taking advantage of the intrinsic properties of naturally occurring chemicals. Classical examples are the flavonoid quercetin and the alkaloid piperine, that have reversed the oxidative stress induced by PM in lung cells and macrophages (Lee et al., 2022a; Saha et al., 2022; da Silva Araújo et al., 2020). Also, sulforaphane, a compound found in cruciferous vegetables, and its primary metabolite sulforaphane N-acetylcysteine, have been described as cytoprotective chemicals due to their antioxidant, anti-inflammatory and anti-apoptotic properties. Accordingly, they have been found to protect BEAS-2B cells against CS-induced oxidative stress and inflammation by promoting the Nrf2 pathway (Son et al., 2020). Ephedrine (an alkaloid), (-)-epicatechin and biochanin A (flavonoids), and glycyrrhizin (a saponin) proved to have similar Nfr2 induction activity that helped bronchial and alveolar epithelial cells counteract the PM-induced cytotoxic effects (Shi et al., 2023; Tian et al., 2021; Wang et al., 2022a; Xue et al., 2021b). The green tea derived polyphenols gallocatechin gallate and epigallocatechin gallate have demonstrated their antioxidant activity to counteract PM-induced oxidative potential in vitro, possibly by inhibiting the protein kinase B/ mammalian target of rapamycin (Akt/mTOR) pathway, therefore alleviating oxidative stress and EMT induced by PM (Tanaka et al., 2022; Zhongyin et al., 2022). Treatment with antioxidants based on traditional Chinese and Northeast Asian medicine, such as Tiaobu Feishen formulae, effective-component compatibility of Bufei Yishen formula (ECC-BYF), and Citrus junos peel extracts have also shown to be effective in overcoming PM-induced oxidative stress (Haoran et al., 2020; Lee et al., 2022a; Li et al., 2021). It has been suggested that ECC-BYF is able to downregulate the FOXO3a modulator miR-155, leading to FOXO3a upregulation and, consequently, decreased ROS production (Li et al., 2021). Also acting on miRNAs, the natural antioxidant morin, a flavonoid isolated from the Moracea plants family, has demonstrated activity against PM2.5-induced toxicity, as it decreased the expression of DNA damage-related miRNAs (Veerappan et al., 2019). Likewise, andrographolide, a diterpenoid lactone isolated from Andrographis paniculata, has been shown to mitigate CS-induced inflammation in A549 cells via upregulation of miR-218, which in turn inhibited NF-kB activation (Li et al., 2013).
Both vitamin C (ascorbic acid) and hesperidin are compounds found in citrus fruits which are recognized for their antioxidant activity. Extended PM exposure triggered DNA damage and senescence in human lung fibroblasts, which was countered by vitamin C (Jin et al., 2023), while hesperidin mitigated DNA damage, cell cycle arrest, and senescence in human keratinocytes (Herath et al., 2022). Similar ability to reverse the oxidative DNA damage induced by PM has been reported for Apo-9’-fucoxanthinone (Jang et al., 2016), and the ethanolic extract Cornus officinalis fruit, also known as Japanese cornelian cherry (Fernando et al., 2020). Interestingly, the latter was similarly able to revoke the direct effects of PM2.5-induced ROS on lipids (lipid peroxidation) and proteins (protein carbonylation).
iii) Antioxidants based on endogenous molecules
Fibroblast growth factor 10 (FGF10) is an endogenous paracrine signaling molecule that has a role in the maintenance of lung homeostasis, as well as epithelial regeneration and repair. Exogenous FGF10 has also demonstrated to protect against lung injury, and in vitro works on bronchial epithelial cells exposed to PM have confirmed that FGF10 triggered Nrf2 induction (Liu et al., 2022b), as well as NF-κB downregulation (Wang et al., 2022b), resulting in antioxidant and anti-inflammatory activities. The amino acid proline is yet another endogenous molecule that has recently been studied in the context of PM-induced oxidative stress, due to its inherent broad antioxidant profile. Proline supplementation prevented PM-induced DNA damage in epithelial cells, emphasizing that proline supplementation as an antioxidant approach merits further investigation in future works (Barzgar et al., 2023). The natural compound melatonin protected Cs-exposed human lung cells against ER-stress-induced inflammasome activation and death, but it could only partially reverse the oxidative stress comparatively to NAC (Mahalanobish et al., 2020). Being one of the main endogenous antioxidant molecules, GSH could reverse the PM2.5-induced increase in global DNA hydroxymethylation and Tet1 mRNA expression, while also preventing G2/M cell cycle arrest and cell apoptosis in SH-SY5Y cells (Wei et al., 2017).
Despite its well-known toxicity, H2S has been described as potentially protective against oxidative damage by activating the Nrf2/PPAR-g axis and inhibiting the NCOA4-mediated ferritinophagy, which resulted in decreased lipid peroxidation, therefore blocking ferroptosis in epithelial cells (Wang et al., 2022c). However, in another study, the same molecule failed to overcome the inflammatory potential of PM in the same cell line (Shrestha et al., 2021).
iv) New antioxidant approaches
On an interesting final note, the use of adipose mesenchymal stem cells-derived extracellular vesicles (ADSC-EVs) as potential antioxidants has been explored in recent works (Gao et al., 2021, 2020). The results showed decreased ROS and apoptosis and favored M2 phenotype when PM-exposed rat epithelial cells and macrophages were treated with ADSC-EVs, which supports their antioxidant and anti-inflammatory properties.
Despite the abundant literature on the antioxidant activity of many different compounds, the exact mechanism by which they exert their antioxidant activity is not always studied in detail and mostly assumed to be a general one. Moreover, the antioxidant effects mentioned in this section were studied in in vitro settings and a translation to the human in vivo context is usually not direct due to bioavailability issues, interaction with gut microbiota, or metabolic transformations (Hrelia and Angeloni, 2021). Regardless of the advancements, the quest for safe and effective antioxidants that can be therapeutically used still persists.
8. Perspectives and Conclusions
Significant steps have been made in recent years advancing the understanding of the detrimental consequences associated with inhaling PM. Particularly, the significance of OP has emerged as a robust indicator for predicting the potential toxicity of PM (Campbell et al., 2021). Innovative technologies capable of real-time assessment of the OP of PM carries substantial promise. If generally adopted, this progress would introduce an additional layer of protection for the population, not only enabling prompt assessment of how regulatory adjustments, which should include the evaluation of the OP, could rapidly influence air quality, but also providing real-time information to the population about the OP of the airborne particles.
Regarding the oxidative stress that is triggered in cellular models, it is interesting to notice that most studies still use conventional monocultures, which are known to have many limitations, such as lack of tissue complexity, absence of organ-organ interaction, among others. This is surprising, considering the current availability of complex advanced models of the lung. Besides being physiologically more relevant, these advanced models can be cultivated for longer periods, allowing repeated exposures to lower concentrations of PM, therefore allowing to recreate more realistic scenarios. Also, most exposures were performed in submerged conditions, with only 2 out of 53 studies described in Table 2 daring to perform air-liquid interface or aerosolized exposures (Upadhyay et al., 2022b; Xiong et al., 2021b). To advance the field, more studies are needed on human-based realistic models that test real-case scenarios. Even though many of the mechanisms discussed in this review have been confirmed in vivo (Table S1), the understanding we have now on how PM triggers oxidative stress may have significant adjustments in the future with the advancement of the existing complex human-based models. Considering the intricacies of the PM-induced oxidative stress-related effects highlighted in this work (Figure 3), we advocate for multiplex approaches including real-time and high-throughput analysis of a broader panel of biomarkers covering multiple pathways. The same concept applies to DNA damage-repair, carcinogenicity, and epigenetic effects (Figure 4).
Finally, there is continuous interest in testing antioxidants from sources related to diet, with in vitro tests demonstrating their ability to mitigate the oxidative effects triggered by PM. Many of the studies herein discussed have performed parallel tests in animal models mostly confirming the in vitro results (Table S1). More studies are required to understand the full potential of dietary supplements or increased consumption of antioxidant-rich food as effective and safe prophylactic actions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgements
This work was funded by the European Union’s H2020 project Sinfonia (N.857253) and Horizon Europe Projects LEARN (N.101057510) and iCare (N.101092971). SbDToolBox, with reference NORTE-01-0145-FEDER-000047, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement through the European Regional Development.
References
- Albino AP, Huang X, Jorgensen ED, et al. Induction of DNA Double-Strand Breaks in A549 and Normal Human Pulmonary Epithelial Cells by Cigarette Smoke Is Mediated by Free Radicals. Int J Oncol 2006;28(6):1491–1505.
- Badran G, Verdin A, Grare C, et al. Toxicological Appraisal of the Chemical Fractions of Ambient Fine (PM2.5-0.3) and Quasi-Ultrafine (PM0.3) Particles in Human Bronchial Epithelial BEAS-2B Cells. Environmental Pollution 2020;263. [CrossRef]
- Bagam P, Kaur G, Singh DP, et al. In Vitro Study of the Role of FOXO Transcription Factors in Regulating Cigarette Smoke Extract-Induced Autophagy. Cell Biol Toxicol 2021;37(4):531–553. [CrossRef]
- Barzgar F, Sadeghi-Mohammadi S, Aftabi Y, et al. Oxidative Stress Indices Induced by Industrial and Urban PM2.5-Bound Metals in A549 Cells. Science of the Total Environment 2023;877. [CrossRef]
- Baskara I, Kerbrat S, Dagouassat M, et al. Cigarette Smoking Induces Human CCR6+Th17 Lymphocytes Senescence and VEGF-A Secretion. Sci Rep 2020;10(1). [CrossRef]
- Bhat A V, Hora S, Pal A, et al. Stressing the (Epi)Genome: Dealing with Reactive Oxygen Species in Cancer. Antioxid Redox Signal 2018;29(13):1273–1292. [CrossRef]
- Bongaerts E, Mamia K, Rooda I, et al. Ambient Black Carbon Particles in Human Ovarian Tissue and Follicular Fluid. Environ Int 2023;179:108141. [CrossRef]
- Cadet J, Davies KJA, Medeiros MH, et al. Formation and Repair of Oxidatively Generated Damage in Cellular DNA. Free Radic Biol Med 2017;107:13–34. [CrossRef]
- Calas A, Uzu G, Kelly FJ, et al. Comparison between Five Acellular Oxidative Potential Measurement Assays Performed with Detailed Chemistry on PM≪Sub≫10≪/Sub≫ Samples from the City of Chamonix (France). Atmos Chem Phys 2018;18(11):7863–7875. [CrossRef]
- Calderón-Garcidueñas L, Torres-Jardón R, Greenough GP, et al. Sleep Matters: Neurodegeneration Spectrum Heterogeneity, Combustion and Friction Ultrafine Particles, Industrial Nanoparticle Pollution, and Sleep Disorders—Denial Is Not an Option. Front Neurol 2023;14. [CrossRef]
- Campbell SJ, Utinger B, Lienhard DM, et al. Development of a Physiologically Relevant Online Chemical Assay To Quantify Aerosol Oxidative Potential. Anal Chem 2019;91(20):13088–13095. [CrossRef]
- Campbell SJ, Wolfer K, Utinger B, et al. Atmospheric Conditions and Composition That Influence PM≪Sub≫2.5≪/Sub≫ Oxidative Potential in Beijing, China. Atmos Chem Phys 2021a;21(7):5549–5573. [CrossRef]
- Campbell SJ, Wolfer K, Utinger B, et al. Atmospheric Conditions and Composition That Influence PM≪Sub≫2.5≪/Sub≫ Oxidative Potential in Beijing, China. Atmos Chem Phys 2021b;21(7):5549–5573. [CrossRef]
- Cao X, Padoan S, Binder S, et al. A Comparative Study of Persistent DNA Oxidation and Chromosomal Instability Induced in Vitro by Oxidizers and Reference Airborne Particles. Mutation Research/Genetic Toxicology and Environmental Mutagenesis 2022;874–875:503446. [CrossRef]
- Chen YC, Chuang TY, Liu CW, et al. Particulate Matters Increase Epithelial-Mesenchymal Transition and Lung Fibrosis through the ETS-1/NF-ΚB-Dependent Pathway in Lung Epithelial Cells. Part Fibre Toxicol 2020;17(1). [CrossRef]
- Cho C-C, Hsieh W-Y, Tsai C-H, et al. In Vitro and In Vivo Experimental Studies of PM2.5 on Disease Progression. Int J Environ Res Public Health 2018;15(7):1380. [CrossRef]
- Costabile F, Gualtieri M, Rinaldi M, et al. Exposure to Urban Nanoparticles at Low PM$$_1$$ Concentrations as a Source of Oxidative Stress and Inflammation. Sci Rep 2023;13(1):18616. [CrossRef]
- Coyle JP, Derk RC, Kornberg TG, et al. Carbon Nanotube Filler Enhances Incinerated Thermoplastics-Induced Cytotoxicity and Metabolic Disruption in Vitro. Part Fibre Toxicol 2020;17(1). [CrossRef]
- Crobeddu B, Aragao-Santiago L, Bui L-C, et al. Oxidative Potential of Particulate Matter 2.5 as Predictive Indicator of Cellular Stress. Environmental Pollution 2017;230:125–133. [CrossRef]
- Crobeddu B, Baudrimont I, Deweirdt J, et al. Lung Antioxidant Depletion: A Predictive Indicator of Cellular Stress Induced by Ambient Fine Particles. Environ Sci Technol 2020;54(4):2360–2369. [CrossRef]
- Cui J, Zhao W, Xu X, et al. DNA Polymerase Beta Is Involved in the Protection against the Cytotoxicity and Genotoxicity of Cigarette Smoke. Environ Toxicol Pharmacol 2012;34(2):370–380. [CrossRef]
- Cui X, Zhang Y, Lu Y, et al. ROS and Endoplasmic Reticulum Stress in Pulmonary Disease. Front Pharmacol 2022;13. [CrossRef]
- Danielsen PH, Møller P, Jensen KA, et al. Oxidative Stress, DNA Damage, and Inflammation Induced by Ambient Air and Wood Smoke Particulate Matter in Human A549 and THP-1 Cell Lines. Chem Res Toxicol 2011;24(2):168–184. [CrossRef]
- Du H, Sun J, Chen Z, et al. Cigarette Smoke-Induced Failure of Apoptosis Resulting in Enhanced Neoplastic Transformation in Human Bronchial Epithelial Cells. J Toxicol Environ Health A 2012;75(12):707–720. [CrossRef]
- Eiguren-Fernandez A, Kreisberg N and Hering S. An Online Monitor of the Oxidative Capacity of Aerosols (o-MOCA). Atmos Meas Tech 2017;10(2):633–644. [CrossRef]
- Fernando P, Piao MJ, Zhen AX, et al. Extract of Cornus Officinalis Protects Keratinocytes from Particulate Matter-Induced Oxidative Stress. Int J Med Sci 2020;17(1):63–70. [CrossRef]
- Figliuzzi M, Tironi M, Longaretti L, et al. Copper-Dependent Biological Effects of Particulate Matter Produced by Brake Systems on Lung Alveolar Cells. Arch Toxicol 2020;94(9):2965–2979. [CrossRef]
- Fu Y, Li B, Yun J, et al. LncRNA SOX2-OT CeRNA Network Enhances the Malignancy of Long-Term PM(2.5)-Exposed Human Bronchial Epithelia. Ecotoxicol Environ Saf 2021;217:112242. [CrossRef]
- Gao Y, Huang X, Lin H, et al. Adipose Mesenchymal Stem Cell-Derived Antioxidative Extracellular Vesicles Exhibit Anti-Oxidative Stress and Immunomodulatory Effects under PM2.5 Exposure. Toxicology 2021;447. [CrossRef]
- Gao ZX, Song XL, Li SS, et al. Assessment of DNA Damage and Cell Senescence in Corneal Epithelial Cells Exposed to Airborne Particulate Matter (PM2.5) Collected in Guangzhou, China. Invest Ophthalmol Vis Sci 2016;57(7):3093–3102. [CrossRef]
- Gilmour PS, Brown DM, Lindsay TG, et al. Adverse Health Effects of PM10 Particles: Involvement of Iron in Generation of Hydroxyl Radical. Occup Environ Med 1996;53(12):817–822. [CrossRef]
- Gilmour PS, Rahman I, Donaldson K, et al. Histone Acetylation Regulates Epithelial IL-8 Release Mediated by Oxidative Stress from Environmental Particles. Am J Physiol Lung Cell Mol Physiol 2003;284(3):L533-40. [CrossRef]
- Goshua A, Akdis CA and Nadeau KC. World Health Organization Global Air Quality Guideline Recommendations: Executive Summary. Allergy 2022;77(7):1955–1960. [CrossRef]
- Guan M, Tang S, Chang H, et al. Development of Alveolar-Capillary-Exchange (ACE) Chip and Its Application for Assessment of PM2.5-Induced Toxicity. Ecotoxicol Environ Saf 2021;223. [CrossRef]
- Guerra e Oliveira T, Trancoso IA, Lorençoni MF, et al. Toxicological Effects of Air Settled Particles from the Vitoria Metropolitan Area Mediated by Oxidative Stress, pro-Inflammatory Mediators and NFΚB Pathway. Environ Res 2022;204. [CrossRef]
- Guillaumet-Adkins A, Yañez Y, Peris-Diaz MD, et al. Epigenetics and Oxidative Stress in Aging. Oxid Med Cell Longev 2017;2017:9175806. [CrossRef]
- Haggadone MD, Mancuso P and Peters-Golden M. Oxidative Inactivation of the Proteasome Augments Alveolar Macrophage Secretion of Vesicular SOCS3. Cells 2020;9(7). [CrossRef]
- Hanahan D and Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell 2011;144(5):646–674. [CrossRef]
- Haoran D, Xuefang L, Wanchun Z, et al. Three Tiaobu Feishen Formulae Reduces Cigarette Smoke-Induced in-Flammation in Human Airway Epithelial Cells. J Tradit Chin Med 2020;40(3):386–392.
- Hartwig A. Role of DNA Repair in Particle- and Fiber-Induced Lung Injury. Inhal Toxicol 2002;14(1):91–100. [CrossRef]
- Hayes P and Knaus UG. Balancing Reactive Oxygen Species in the Epigenome: NADPH Oxidases as Target and Perpetrator. Antioxid Redox Signal 2013;18(15):1937–1945. [CrossRef]
- He J and Jiang BH. Interplay between Reactive Oxygen Species and MicroRNAs in Cancer. Curr Pharmacol Rep 2016;2(2):82–90. [CrossRef]
- Herath H, Piao MJ, Kang KA, et al. Hesperidin Exhibits Protective Effects against PM(2.5)-Mediated Mitochondrial Damage, Cell Cycle Arrest, and Cellular Senescence in Human HaCaT Keratinocytes. Molecules 2022;27(15). [CrossRef]
- Van Den Heuvel R, Den Hond E, Govarts E, et al. Identification of PM10 Characteristics Involved in Cellular Responses in Human Bronchial Epithelial Cells (Beas-2B). Environ Res 2016;149:48–56. [CrossRef]
- Housseiny H Al, Singh M, Emile S, et al. Identification of Toxicity Parameters Associated with Combustion Produced Soot Surface Chemistry and Particle Structure by in Vitro Assays. Biomedicines 2020;8(9). [CrossRef]
- Hrelia S and Angeloni C. New Mechanisms of Action of Natural Antioxidants in Health and Disease II. Antioxidants 2021;10(8):1200. [CrossRef]
- Huang H, Ji Y, Zhang J, et al. Aberrant DNA Methylation in Radon and/or Cigarette Smoke-Induced Malignant Transformation in BEAS-2B Human Lung Cell Line. J Toxicol Environ Health A 2017;80(23–24):1321–1330. [CrossRef]
- Huang W, Zhang Y, Zhang Y, et al. Development of an Automated Sampling-Analysis System for Simultaneous Measurement of Reactive Oxygen Species (ROS) in Gas and Particle Phases: GAC-ROS. Atmos Environ 2016;134:18–26. [CrossRef]
- Hunyadi A. The Mechanism(s) of Action of Antioxidants: From Scavenging Reactive Oxygen/Nitrogen Species to Redox Signaling and the Generation of Bioactive Secondary Metabolites. Med Res Rev 2019;39(6):2505–2533. [CrossRef]
- Ito Y, Oshinden K, Kutsuzawa N, et al. Heat-Not-Burn Cigarette Induces Oxidative Stress Response in Primary Rat Alveolar Epithelial Cells. PLoS One 2020;15(11 November). [CrossRef]
- Jang JH, Lee JH, Chand HS, et al. APO-9’-Fucoxanthinone Extracted from Undariopsis Peteseniana Protects Oxidative Stress-Mediated Apoptosis in Cigarette Smoke-Exposed Human Airway Epithelial Cells. Mar Drugs 2016;14(7). [CrossRef]
- Janssen BG, Madlhoum N, Gyselaers W, et al. Cohort Profile: The ENVIRonmental Influence ON Early AGEing (ENVIR ON AGE): A Birth Cohort Study. Int J Epidemiol 2017;dyw269. [CrossRef]
- Jantzen K, Roursgaard M, Desler C, et al. Oxidative Damage to DNA by Diesel Exhaust Particle Exposure in Co-Cultures of Human Lung Epithelial Cells and Macrophages. Mutagenesis 2012;27(6):693–701. [CrossRef]
- Ji D, Hu C, Ning J, et al. N(6)-Methyladenosine Mediates Nrf2 Protein Expression Involved in PM2.5-Induced Pulmonary Fibrosis. Ecotoxicol Environ Saf 2023;254:114755. [CrossRef]
- Jiang, Ahmed, Canchola, et al. Use of Dithiothreitol Assay to Evaluate the Oxidative Potential of Atmospheric Aerosols. Atmosphere (Basel) 2019;10(10):571. [CrossRef]
- Jiang P, Hao S, Xie L, et al. LncRNA NEAT1 Contributes to the Acquisition of a Tumor Like-Phenotype Induced by PM 2.5 in Lung Bronchial Epithelial Cells via HIF-1α Activation. Environ Sci Pollut Res Int 2021;28(32):43382–43393. [CrossRef]
- Jin S, Yoon SJ, Jung NY, et al. Antioxidants Prevent Particulate Matter-Induced Senescence of Lung Fibroblasts. Heliyon 2023;9(3):e14179. [CrossRef]
- Kietzmann T, Petry A, Shvetsova A, et al. The Epigenetic Landscape Related to Reactive Oxygen Species Formation in the Cardiovascular System. Br J Pharmacol 2017;174(12):1533–1554. [CrossRef]
- Kodali V, Afshari A, Meighan T, et al. In Vivo and in Vitro Toxicity of a Stainless-Steel Aerosol Generated during Thermal Spray Coating. Arch Toxicol 2022;96(12):3201–3217. [CrossRef]
- Kryston TB, Georgiev AB, Pissis P, et al. Role of Oxidative Stress and DNA Damage in Human Carcinogenesis. Mutat Res 2011;711(1–2):193–201. [CrossRef]
- Lee DH, Woo JK, Heo W, et al. Citrus Junos Tanaka Peel Extract and Its Bioactive Naringin Reduce Fine Dust-Induced Respiratory Injury Markers in BALB/c Male Mice. Nutrients 2022a;14(5). [CrossRef]
- Lee JY, Lee WK and Kim DS. Particulate Matter-Induced Hypomethylation of Alu and LINE1 in Normal Human Bronchial Epithelial Cells and Epidermal Keratinocytes. Genes Environ 2022b;44(1):8. [CrossRef]
- Lee KY, Ho SC, Sun WL, et al. Lnc-IL7R Alleviates PM(2.5)-Mediated Cellular Senescence and Apoptosis through EZH2 Recruitment in Chronic Obstructive Pulmonary Disease. Cell Biol Toxicol 2022c;38(6):1097–1120. [CrossRef]
- Li H, Zhao Z, Luo XS, et al. Insight into Urban PM2.5 Chemical Composition and Environmentally Persistent Free Radicals Attributed Human Lung Epithelial Cytotoxicity. Ecotoxicol Environ Saf 2022;234. [CrossRef]
- Li J, Wang J, Li Y, et al. Effective-Component Compatibility of Bufei Yishen Formula Protects COPD Rats against PM2.5-Induced Oxidative Stress via MiR-155/FOXO3a Pathway. Ecotoxicol Environ Saf 2021;228. [CrossRef]
- Li J, Zhou Q, Liang Y, et al. MiR-486 Inhibits PM2.5-Induced Apoptosis and Oxidative Stress in Human Lung Alveolar Epithelial A549 Cells. Ann Transl Med 2018;6(11):209. [CrossRef]
- Li YJ, Yu CH, Li JB, et al. Andrographolide Antagonizes Cigarette Smoke Extract-Induced Inflammatory Response and Oxidative Stress in Human Alveolar Epithelial A549 Cells through Induction of MicroRNA-218. Exp Lung Res 2013;39(10):463–471. [CrossRef]
- Lin Y-H. MicroRNA Networks Modulate Oxidative Stress in Cancer. Int J Mol Sci 2019;20(18):4497.
- Liu H, Nie H, Lai W, et al. Different Exposure Modes of PM2.5 Induces Bronchial Asthma and Fibrosis in Male Rats through Macrophage Activation and Immune Imbalance Induced by TIPE2 Methylation. Ecotoxicol Environ Saf 2022a;247. [CrossRef]
- Liu J, Guo Z-N, Yan X-L, et al. Crosstalk Between Autophagy and Ferroptosis and Its Putative Role in Ischemic Stroke. Front Cell Neurosci 2020a;14. [CrossRef]
- Liu J, Kuang F, Kroemer G, et al. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem Biol 2020b;27(4):420–435. [CrossRef]
- Liu L, Shi Q, Wang K, et al. Fibroblast Growth Factor 10 Protects against Particulate Matter-Induced Lung Injury by Inhibiting Oxidative Stress-Mediated Pyroptosis via the PI3K/Akt/Nrf2 Signaling Pathway. Int Immunopharmacol 2022b;113. [CrossRef]
- Liu T, Zhang L, Joo D, et al. NF-ΚB Signaling in Inflammation. Signal Transduct Target Ther 2017;2. [CrossRef]
- Liu Y, Luo F, Xu Y, et al. Epithelial-Mesenchymal Transition and Cancer Stem Cells, Mediated by a Long Non-Coding RNA, HOTAIR, Are Involved in Cell Malignant Transformation Induced by Cigarette Smoke Extract. Toxicol Appl Pharmacol 2015;282(1):9–19. [CrossRef]
- LOGAN W. MORTALITY IN THE LONDON FOG INCIDENT, 1952. The Lancet 1953;261(6755):336–338. [CrossRef]
- Lu L, Luo F, Liu Y, et al. Posttranscriptional Silencing of the LncRNA MALAT1 by MiR-217 Inhibits the Epithelial-Mesenchymal Transition via Enhancer of Zeste Homolog 2 in the Malignant Transformation of HBE Cells Induced by Cigarette Smoke Extract. Toxicol Appl Pharmacol 2015;289(2):276–285. [CrossRef]
- Lu L, Xu H, Luo F, et al. Epigenetic Silencing of MiR-218 by the LncRNA CCAT1, Acting via BMI1, Promotes an Altered Cell Cycle Transition in the Malignant Transformation of HBE Cells Induced by Cigarette Smoke Extract. Toxicol Appl Pharmacol 2016;304:30–41. [CrossRef]
- Luo J, Li L, Hu D, et al. LINC00612/MiR-31-5p/Notch1 Axis Regulates Apoptosis, Inflammation, and Oxidative Stress in Human Pulmonary Microvascular Endothelial Cells Induced by Cigarette Smoke Extract. Int J Chron Obstruct Pulmon Dis 2020;15:2049–2060. [CrossRef]
- Lushchak VI and Storey KB. Oxidative Stress Concept Updated: Definitions, Classifications, and Regulatory Pathways Implicated. EXCLI J 2021;20:956–967. [CrossRef]
- Lv XJ, Du YW, Hao YQ, et al. RNA-Binding Motif Protein 5 Inhibits the Proliferation of Cigarette Smoke-Transformed BEAS-2B Cells through Cell Cycle Arrest and Apoptosis. Oncol Rep 2016;35(4):2315–2327. [CrossRef]
- Mahalanobish S, Dutta S, Saha S, et al. Melatonin Induced Suppression of ER Stress and Mitochondrial Dysfunction Inhibited NLRP3 Inflammasome Activation in COPD Mice. Food and Chemical Toxicology 2020;144. [CrossRef]
- Marchetti S, Gualtieri M, Pozzer A, et al. On Fine Particulate Matter and COVID-19 Spread and Severity: An in Vitro Toxicological Plausible Mechanism. Environ Int 2023;179:108131. [CrossRef]
- Marques dos Santos M, Tan Pei Fei M, Li C, et al. Cell-Line and Culture Model Specific Responses to Organic Contaminants in House Dust: Cell Bioenergetics, Oxidative Stress, and Inflammation Endpoints. Environ Int 2022;167. [CrossRef]
- Martikainen MV, Tossavainen T, Täubel M, et al. Toxicological and Microbiological Characterization of Cow Stable Dust. Toxicology in Vitro 2021;75. [CrossRef]
- Meganathan V, Hamilton CE, Natarajan K, et al. NADPH and Xanthine Oxidases Control Induction of Inflammatory Mediator Expression by Organic Dust in the Lung. The FASEB Journal 2022;36(7). [CrossRef]
- Millar MW, Fazal F and Rahman A. Therapeutic Targeting of NF-ΚB in Acute Lung Injury: A Double-Edged Sword. Cells 2022;11(20):3317. [CrossRef]
- Ngo V and Duennwald ML. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022;11(12):2345. [CrossRef]
- Niu BY, Li WK, Li JS, et al. Effects of Dna Damage and Oxidative Stress in Human Bronchial Epithelial Cells Exposed to Pm2.5 from Beijing, China, in Winter. Int J Environ Res Public Health 2020a;17(13):1–14. [CrossRef]
- Niu BY, Li WK, Li JS, et al. Effects of Dna Damage and Oxidative Stress in Human Bronchial Epithelial Cells Exposed to Pm2.5 from Beijing, China, in Winter. Int J Environ Res Public Health 2020b;17(13):1–14. [CrossRef]
- Oberdörster G, Oberdörster E and Oberdörster J. Nanotoxicology: An Emerging Discipline Evolving from Studies of Ultrafine Particles. Environ Health Perspect 2005a;113(7):823–839. [CrossRef]
- Oberdörster G, Oberdörster E and Oberdörster J. Nanotoxicology: An Emerging Discipline Evolving from Studies of Ultrafine Particles. Environ Health Perspect 2005b;113(7):823–839. [CrossRef]
- Pan K, Lu J and Song Y. Artesunate Ameliorates Cigarette Smoke-Induced Airway Remodelling via PPAR-γ/TGF-Β1/Smad2/3 Signalling Pathway. Respir Res 2021;22(1). [CrossRef]
- Pang Y, Huang W, Luo XS, et al. In-Vitro Human Lung Cell Injuries Induced by Urban PM2.5 during a Severe Air Pollution Episode: Variations Associated with Particle Components. Ecotoxicol Environ Saf 2020;206. [CrossRef]
- Pradhan SH, Gibb M, Kramer AT, et al. Peripheral (Lung-to-Brain) Exposure to Diesel Particulate Matter Induces Oxidative Stress and Increased Markers for Systemic Inflammation. Environ Res 2023;231. [CrossRef]
- Prunicki M, Cauwenberghs N, Ataam JA, et al. Immune Biomarkers Link Air Pollution Exposure to Blood Pressure in Adolescents. Environ Health 2020;19(1). [CrossRef]
- Puthussery J V., Zhang C and Verma V. Development and Field Testing of an Online Instrument for Measuring the Real-Time Oxidative Potential of Ambient Particulate Matter Based on Dithiothreitol Assay. Atmos Meas Tech 2018;11(10):5767–5780. [CrossRef]
- Qiao D, Hu C, Li Q, et al. Circ-RBMS1 Knockdown Alleviates CSE-Induced Apoptosis, Inflammation and Oxidative Stress via Up-Regulating FBXO11 Through MiR-197-3p in 16HBE Cells. Int J Chron Obstruct Pulmon Dis 2021;16:2105–2118. [CrossRef]
- Quezada-Maldonado EM, Chirino YI, Gonsebatt ME, et al. Nucleotide Excision Repair Pathway Activity Is Inhibited by Airborne Particulate Matter (PM(10)) through XPA Deregulation in Lung Epithelial Cells. Int J Mol Sci 2022;23(4). [CrossRef]
- Quezada-Maldonado EM, Sánchez-Pérez Y, Chirino YI, et al. Airborne Particulate Matter Induces Oxidative Damage, DNA Adduct Formation and Alterations in DNA Repair Pathways. Environ Pollut 2021;287:117313. [CrossRef]
- Quintana R, Serrano J, Gómez V, et al. The Oxidative Potential and Biological Effects Induced by PM10 Obtained in Mexico City and at a Receptor Site during the MILAGRO Campaign. Environmental Pollution 2011;159(12):3446–3454. [CrossRef]
- Quintana-Belmares R A-MEG-CCG-VVV-LIS-SMR-PIO-VA. Evaluation of the Oxidative Potential of Urban PM and Its Relation to in Vitro Induced DNA Damage: A Spatial and Temporal Comparison. Revista internacional de contaminación ambiental 2015;31(2):145–154.
- Ren F, Xu J, Zhang J, et al. PM2.5 Induced Lung Injury through Upregulating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis. Immunobiology 2022;227(3). [CrossRef]
- Rout-Pitt N, Farrow N, Parsons D, et al. Epithelial Mesenchymal Transition (EMT): A Universal Process in Lung Diseases with Implications for Cystic Fibrosis Pathophysiology. Respir Res 2018;19(1):136. [CrossRef]
- Ryu YS, Kang KA, Piao MJ, et al. Particulate Matter-Induced Senescence of Skin Keratinocytes Involves Oxidative Stress-Dependent Epigenetic Modifications. Exp Mol Med 2019;51(9):1–14. [CrossRef]
- Saha P, Durugkar S, Jain S, et al. Piperine Attenuates Cigarette Smoke-Induced Oxidative Stress, Lung Inflammation, and Epithelial–Mesenchymal Transition by Modulating the SIRT1/Nrf2 Axis. Int J Mol Sci 2022;23(23). [CrossRef]
- Saha S, Buttari B, Panieri E, et al. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020;25(22):5474. [CrossRef]
- Santibáñez-Andrade M, Quezada-Maldonado EM, Rivera-Pineda A, et al. The Road to Malignant Cell Transformation after Particulate Matter Exposure: From Oxidative Stress to Genotoxicity. Int J Mol Sci 2023;24(2). [CrossRef]
- Sarker AH, Chatterjee A, Williams M, et al. NEIL2 Protects against Oxidative DNA Damage Induced by Sidestream Smoke in Human Cells. PLoS One 2014;9(3):e90261. [CrossRef]
- Shi Q, Qian Y, Wang B, et al. Glycyrrhizin Protects against Particulate Matter-Induced Lung Injury via Regulation of Endoplasmic Reticulum Stress and NLRP3 Inflammasome-Mediated Pyroptosis through Nrf2/HO-1/NQO1 Signaling Pathway. Int Immunopharmacol 2023;120. [CrossRef]
- Shi T, Schins RPF, Knaapen AM, et al. Hydroxyl Radical Generation by Electron Paramagnetic Resonance as a New Method to Monitor Ambient Particulate Matter Composition. Journal of Environmental Monitoring 2003;5(4):550. [CrossRef]
- Shrestha D, Bhat SM, Massey N, et al. Pre-Exposure to Hydrogen Sulfide Modulates the Innate Inflammatory Response to Organic Dust. Cell Tissue Res 2021;384(1):129–148. [CrossRef]
- Sies H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol 2017;11:613–619. [CrossRef]
- da Silva Araújo NP, de Matos NA, Leticia Antunes Mota S, et al. Quercetin Attenuates Acute Lung Injury Caused by Cigarette Smoke Both In Vitro and In Vivo. COPD: Journal of Chronic Obstructive Pulmonary Disease 2020;17(2):205–214. [CrossRef]
- Smith MT, Guyton KZ, Gibbons CF, et al. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ Health Perspect 2016;124(6):713–721. [CrossRef]
- So B, Park J, Jang J, et al. Effect of Aerobic Exercise on Oxidative Stress and Inflammatory Response During Particulate Matter Exposure in Mouse Lungs. Front Physiol 2022;12. [CrossRef]
- Son ES, Park JW, Kim YJ, et al. Effects of Antioxidants on Oxidative Stress and Inflammatory Responses of Human Bronchial Epithelial Cells Exposed to Particulate Matter and Cigarette Smoke Extract. Toxicology in Vitro 2020;67. [CrossRef]
- Song L, Li D, Gu Y, et al. Let-7a Modulates Particulate Matter (≤ 2.5 Μm)-Induced Oxidative Stress and Injury in Human Airway Epithelial Cells by Targeting Arginase 2. J Appl Toxicol 2016;36(10):1302–1310. [CrossRef]
- Song L, Li D, Li X, et al. Exposure to PM2.5 Induces Aberrant Activation of NF-ΚB in Human Airway Epithelial Cells by Downregulating MiR-331 Expression. Environ Toxicol Pharmacol 2017;50:192–199. [CrossRef]
- Takizawa M, Nakano M, Fukami T, et al. Decrease in ADAR1 Expression by Exposure to Cigarette Smoke Enhances Susceptibility to Oxidative Stress. Toxicol Lett 2020;331:22–32. [CrossRef]
- Tanaka KI, Nakaguchi S, Shiota S, et al. Preventive Effect of Epigallocatechin Gallate, the Main Component of Green Tea, on Acute Lung Injury Caused by Air Pollutants. Biomolecules 2022;12(9). [CrossRef]
- Thangavel P, Park D and Lee Y-C. Recent Insights into Particulate Matter (PM2.5)-Mediated Toxicity in Humans: An Overview. Int J Environ Res Public Health 2022;19(12):7511. [CrossRef]
- Tian X, Xue Y, Xie G, et al. (−)-Epicatechin Ameliorates Cigarette Smoke-Induced Lung Inflammation via Inhibiting ROS/NLRP3 Inflammasome Pathway in Rats with COPD. Toxicol Appl Pharmacol 2021;429. [CrossRef]
- Upadhyay S, Chakraborty A, Thimraj TA, et al. Establishment of Repeated In Vitro Exposure System for Evaluating Pulmonary Toxicity of Representative Criteria Air Pollutants Using Advanced Bronchial Mucosa Models. Toxics 2022a;10(6). [CrossRef]
- Upadhyay S, Chakraborty A, Thimraj TA, et al. Establishment of Repeated In Vitro Exposure System for Evaluating Pulmonary Toxicity of Representative Criteria Air Pollutants Using Advanced Bronchial Mucosa Models. Toxics 2022b;10(6). [CrossRef]
- Vattanasit U, Navasumrit P, Khadka MB, et al. Oxidative DNA Damage and Inflammatory Responses in Cultured Human Cells and in Humans Exposed to Traffic-Related Particles. Int J Hyg Environ Health 2014;217(1):23–33. [CrossRef]
- Veerappan I, Sankareswaran SK and Palanisamy R. Morin Protects Human Respiratory Cells from PM(2.5) Induced Genotoxicity by Mitigating ROS and Reverting Altered MiRNA Expression. Int J Environ Res Public Health 2019;16(13). [CrossRef]
- Wang C, Meng X, Meng M, et al. Oxidative Stress Activates the TRPM2-Ca2+-NLRP3 Axis to Promote PM2.5-Induced Lung Injury of Mice. Biomedicine and Pharmacotherapy 2020;130. [CrossRef]
- Wang HL, Chen FQ and Wu LJ. Ephedrine Ameliorates Chronic Obstructive Pulmonary Disease (COPD) through Restraining Endoplasmic Reticulum (ER) Stress in Vitro and in Vivo. Int Immunopharmacol 2022a;103. [CrossRef]
- Wang J, Jia J, Wang D, et al. Zn(2+) Loading as a Critical Contributor to the Circ_0008553-Mediated Oxidative Stress and Inflammation in Response to PM(2.5) Exposures. J Environ Sci (China) 2023a;124:451–461. [CrossRef]
- Wang Q, Shi Q, Liu L, et al. FGF10 Mediates Protective Anti-Oxidative Effects in Particulate Matter-Induced Lung Injury through Nrf2 and NF-ΚB Signaling. Ann Transl Med 2022b;10(22):1203–1203. [CrossRef]
- Wang X, Zhu H, Sun G, et al. Linc01515 Regulates PM(2.5)-Induced Oxidative Stress via Targeting NRF2 in Airway Epithelial Cells. Environ Pollut 2023b;331(Pt 2):121798. [CrossRef]
- Wang Y, Liao S, Pan Z, et al. Hydrogen Sulfide Alleviates Particulate Matter-Induced Emphysema and Airway Inflammation by Suppressing Ferroptosis. Free Radic Biol Med 2022c;186:1–16. [CrossRef]
- Wang Y, Zuo X, Jiang F, et al. A Comparative Study on the Model of PM2.5 Direct or Indirect Interaction with Bronchial Epithelial Cells. Environmental Science and Pollution Research 2022d;29(27):41567–41576. [CrossRef]
- Wang Z, Zuo Y and Gao Z. CircANKRD11 Knockdown Protects HPMECs from Cigarette Smoke Extract-Induced Injury by Regulating MiR-145-5p/BRD4 Axis. Int J Chron Obstruct Pulmon Dis 2021;16:887–899. [CrossRef]
- Wei H, Feng Y, Liang F, et al. Role of Oxidative Stress and DNA Hydroxymethylation in the Neurotoxicity of Fine Particulate Matter. Toxicology 2017;380:94–103. [CrossRef]
- Wei H, Liang F, Meng G, et al. Redox/Methylation Mediated Abnormal DNA Methylation as Regulators of Ambient Fine Particulate Matter-Induced Neurodevelopment Related Impairment in Human Neuronal Cells. Sci Rep 2016;6:33402. [CrossRef]
- Weichenthal S, Lavigne E, Evans G, et al. Ambient PM2.5 and Risk of Emergency Room Visits for Myocardial Infarction: Impact of Regional PM2.5 Oxidative Potential: A Case-Crossover Study. Environmental Health 2016;15(1):46. [CrossRef]
- World Health Organization, (ed). WHO Global Air Quality Guidelines: Particulate Matter (PM2.5 and PM10), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide. 2021.
- Xiong R, Wu Y, Wu Q, et al. Integration of Transcriptome Analysis with Pathophysiological Endpoints to Evaluate Cigarette Smoke Toxicity in an in Vitro Human Airway Tissue Model. Arch Toxicol 2021a;95(5):1739–1761. [CrossRef]
- Xiong R, Wu Y, Wu Q, et al. Integration of Transcriptome Analysis with Pathophysiological Endpoints to Evaluate Cigarette Smoke Toxicity in an in Vitro Human Airway Tissue Model. Arch Toxicol 2021b;95(5):1739–1761. [CrossRef]
- Xu Z, Ding W and Deng X. PM2.5, Fine Particulate Matter: A Novel Player in the Epithelial-Mesenchymal Transition? Front Physiol 2019;10. [CrossRef]
- Xue M, Peng N, Zhu X, et al. Hsa_circ_0006872 Promotes Cigarette Smoke-Induced Apoptosis, Inflammation and Oxidative Stress in HPMECs and BEAS-2B Cells through the MiR-145-5p/NF-ΚB Axis. Biochem Biophys Res Commun 2021a;534:553–560. [CrossRef]
- Xue Z, Gao X, Yu W, et al. Biochanin A Alleviates Oxidative Damage Caused by the Urban Particulate Matter. Food Funct 2021b;12(5):1958–1972. [CrossRef]
- Yang DQ, Zuo QN, Wang T, et al. Mitochondrial-Targeting Antioxidant SS-31 Suppresses Airway Inflammation and Oxidative Stress Induced by Cigarette Smoke. Oxid Med Cell Longev 2021;2021. [CrossRef]
- Yang L, Wang Y, Lin Z, et al. Mitochondrial OGG1 Protects against PM2.5-Induced Oxidative DNA Damage in BEAS-2B Cells. Exp Mol Pathol 2015;99(2):365–373. [CrossRef]
- Yu C and Zhang L. Methylprednisolone Up-Regulates Annexin A1 (ANXA1) to Inhibit the Inflammation, Apoptosis and Oxidative Stress of Cigarette Smoke Extract (CSE)-Induced Bronchial Epithelial Cells, a Chronic Obstructive Pulmonary Disease in Vitro Model, through the Formyl Peptide Receptor 2 (FPR2) Receptors and the Adenosine 5’-Monophosphate (AMP)-Activated Protein Kinase (AMPK) Pathway. Bioengineered 2022;13(2):4028–4038. [CrossRef]
- Zeng Y, Zhu G, Zhu M, et al. Edaravone Attenuated Particulate Matter-Induced Lung Inflammation by Inhibiting ROS-NF- κ B Signaling Pathway. Oxid Med Cell Longev 2022;2022. [CrossRef]
- Zhang C, Gu S and Kang X. CircRNA Circ_0006892 Regulates MiR-24/PHLPP2 Axis to Mitigate Cigarette Smoke Extract-Induced Bronchial Epithelial Cell Injury. Biotechnol Appl Biochem 2022;69(2):735–748. [CrossRef]
- Zhang MY, Jiang YX, Yang YC, et al. Cigarette Smoke Extract Induces Pyroptosis in Human Bronchial Epithelial Cells through the ROS/NLRP3/Caspase-1 Pathway. Life Sci 2021;269. [CrossRef]
- Zhao C, Wang Y, Su Z, et al. Respiratory Exposure to PM2.5 Soluble Extract Disrupts Mucosal Barrier Function and Promotes the Development of Experimental Asthma. Science of the Total Environment 2020;730. [CrossRef]
- Zhongyin Z, Wei W, Juan X, et al. Epigallocatechin Gallate Relieved PM2.5-Induced Lung Fibrosis by Inhibiting Oxidative Damage and Epithelial-Mesenchymal Transition through AKT/MTOR Pathway. Oxid Med Cell Longev 2022;2022. [CrossRef]
- Zhou F, Cao C, Chai H, et al. Circ-HACE1 Aggravates Cigarette Smoke Extract-Induced Injury in Human Bronchial Epithelial Cells via Regulating Toll-Like Receptor 4 by Sponging MiR-485-3p. Int J Chron Obstruct Pulmon Dis 2021;16:1535–1547. [CrossRef]
- Zhou W, Yuan X, Zhang L, et al. Overexpression of HO-1 Assisted PM2.5-Induced Apoptosis Failure and Autophagy-Related Cell Necrosis. Ecotoxicol Environ Saf 2017;145:605–614. [CrossRef]
Figure 1.
Schematization of the main topics addressed in this review, showcasing the adverse outcomes related to the oxidative stress triggered by particulate matter. Consistently accumulated ROS are the step stone for direct DNA damage and epigenetic modifications, and the trigger of many different pathways leading to a variety of deleterious outcomes. Details related to the pathways are described more in depth in the different sections of this review. Created with BioRender.com.
Figure 1.
Schematization of the main topics addressed in this review, showcasing the adverse outcomes related to the oxidative stress triggered by particulate matter. Consistently accumulated ROS are the step stone for direct DNA damage and epigenetic modifications, and the trigger of many different pathways leading to a variety of deleterious outcomes. Details related to the pathways are described more in depth in the different sections of this review. Created with BioRender.com.
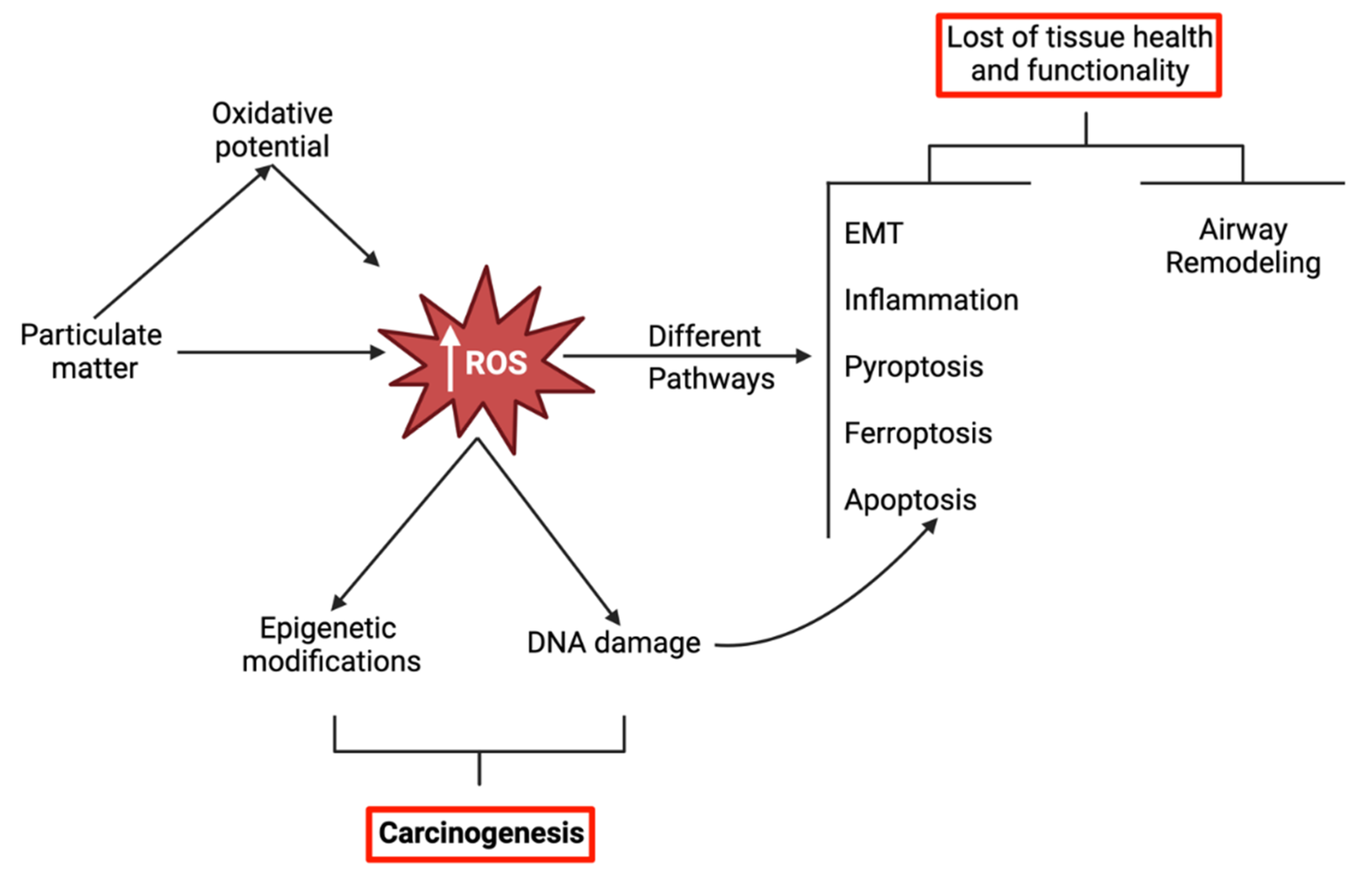
Figure 2.
Anthropogenic origins of airborne pollutants constitute the primary source of PM in the environment, inhalation being the main route of exposure. The inhalation of PM has an immediate effect on the airways and lung, and growing body of evidence indicates that inhaled PM sized under 100 nm can translocate and reach distant organs including the heart, brain, among others. Created with BioRender.com.
Figure 2.
Anthropogenic origins of airborne pollutants constitute the primary source of PM in the environment, inhalation being the main route of exposure. The inhalation of PM has an immediate effect on the airways and lung, and growing body of evidence indicates that inhaled PM sized under 100 nm can translocate and reach distant organs including the heart, brain, among others. Created with BioRender.com.
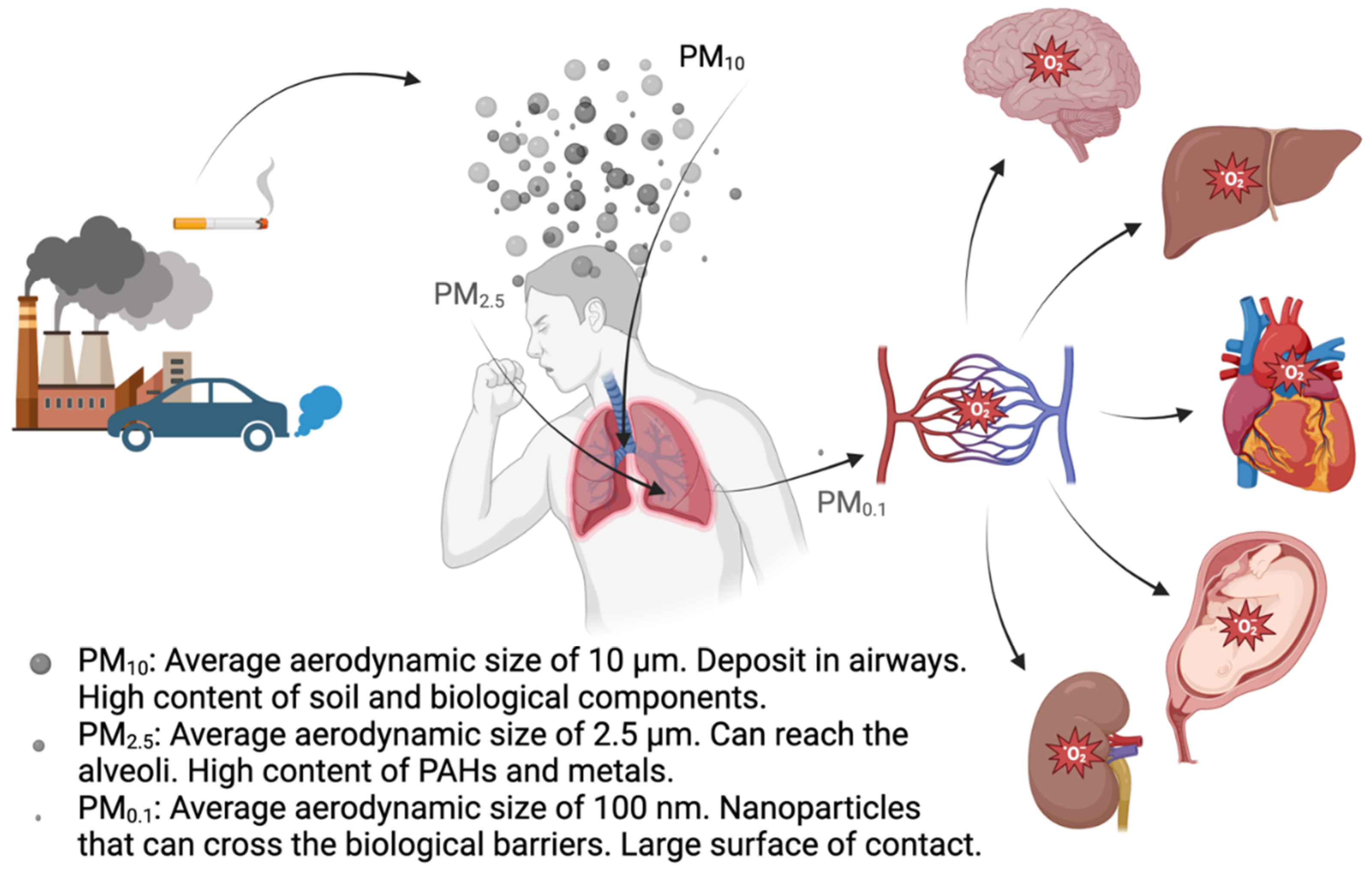
Figure 3.
Summary of the particulate matter-induced oxidative stress-related pathways described in the latest literature. Besides the direct effects on lipids, proteins and DNA, elevated ROS levels are involved in a number of complex pathways that lead to diverse outcomes (e.g. inflammation, tissue remodeling, autophagy), including different modes of cell death (e.g. ferroptosis, pyroptosis, apoptosis). Created with BioRender.com. Abbreviations: a-SMA: alpha smooth muscle actin; AKT: protein kinase B; AMPK: 5' adenosine monophosphate-activated protein kinase; ASC: Apoptosis-associated speck-like protein containing a caspase recruitment domain; ATF: activating transcription factor; ATG: autophagy-related; CD: cluster of differentiation; CHOP: CCAAT/enhancer-binding protein homologous protein; COX: cycloxigenase; eIF: eukaryotic initiation factor; EMT: epithelial-mesenchymal transition; ERK: extracellular signal-regulated kinase; FOXO: forkhead box protein class O; FTH: ferritin heavy chain; GRP78/Bip: 78 kDa glucose-regulated protein/ binding immunoglobulin protein; GSDMD: gasdermin D; HO: heme oxygenase; IL: interleukin; IRE: inositol-requiring enzyme; JNK: c-Jun N-terminal kinases; LC3B: microtubule-associated proteins 1A/1B light chain; M2Pol: M2 polarization; MAPK: mitogen-activated protein kinase; miR: microRNA; MMP: matrix metallopeptidase; mTOR: mammalian target of rapamycin; NCOA: selective cargo receptor nuclear receptor coactivator; NF-kB: nuclear factor kappa beta; NLRP: NOD-like receptor family pyrin domain containing; NO: nitric oxide; NOX: NADPH oxidase; NQO1: NAD(P)H dehydrogenase (quinone); Nrf2: nuclear factor erythroid 2-related factor 2; PERK: protein kinase R-like endoplasmic reticulum kinase; PM: particulate matter; PPAR: peroxisome proliferator-activated receptor; ROS: reactive oxygen species; Smad: mothers against decapentaplegic homolog; SOCS: suppressor of cytokine signaling; SOD: superoxide dismutase; STAT: signal transducer and activator of transcription; TGF: transforming growth factor; TNF: tumor necrosis factor; TRPM: transient receptor potential cation channel, subfamily M; TXNIP: thioredoxin-interacting protein; TXNRD1: thioredoxin reductase; VEGF: vascular endothelial growth factor; XO: xanthine oxidase.
Figure 3.
Summary of the particulate matter-induced oxidative stress-related pathways described in the latest literature. Besides the direct effects on lipids, proteins and DNA, elevated ROS levels are involved in a number of complex pathways that lead to diverse outcomes (e.g. inflammation, tissue remodeling, autophagy), including different modes of cell death (e.g. ferroptosis, pyroptosis, apoptosis). Created with BioRender.com. Abbreviations: a-SMA: alpha smooth muscle actin; AKT: protein kinase B; AMPK: 5' adenosine monophosphate-activated protein kinase; ASC: Apoptosis-associated speck-like protein containing a caspase recruitment domain; ATF: activating transcription factor; ATG: autophagy-related; CD: cluster of differentiation; CHOP: CCAAT/enhancer-binding protein homologous protein; COX: cycloxigenase; eIF: eukaryotic initiation factor; EMT: epithelial-mesenchymal transition; ERK: extracellular signal-regulated kinase; FOXO: forkhead box protein class O; FTH: ferritin heavy chain; GRP78/Bip: 78 kDa glucose-regulated protein/ binding immunoglobulin protein; GSDMD: gasdermin D; HO: heme oxygenase; IL: interleukin; IRE: inositol-requiring enzyme; JNK: c-Jun N-terminal kinases; LC3B: microtubule-associated proteins 1A/1B light chain; M2Pol: M2 polarization; MAPK: mitogen-activated protein kinase; miR: microRNA; MMP: matrix metallopeptidase; mTOR: mammalian target of rapamycin; NCOA: selective cargo receptor nuclear receptor coactivator; NF-kB: nuclear factor kappa beta; NLRP: NOD-like receptor family pyrin domain containing; NO: nitric oxide; NOX: NADPH oxidase; NQO1: NAD(P)H dehydrogenase (quinone); Nrf2: nuclear factor erythroid 2-related factor 2; PERK: protein kinase R-like endoplasmic reticulum kinase; PM: particulate matter; PPAR: peroxisome proliferator-activated receptor; ROS: reactive oxygen species; Smad: mothers against decapentaplegic homolog; SOCS: suppressor of cytokine signaling; SOD: superoxide dismutase; STAT: signal transducer and activator of transcription; TGF: transforming growth factor; TNF: tumor necrosis factor; TRPM: transient receptor potential cation channel, subfamily M; TXNIP: thioredoxin-interacting protein; TXNRD1: thioredoxin reductase; VEGF: vascular endothelial growth factor; XO: xanthine oxidase.
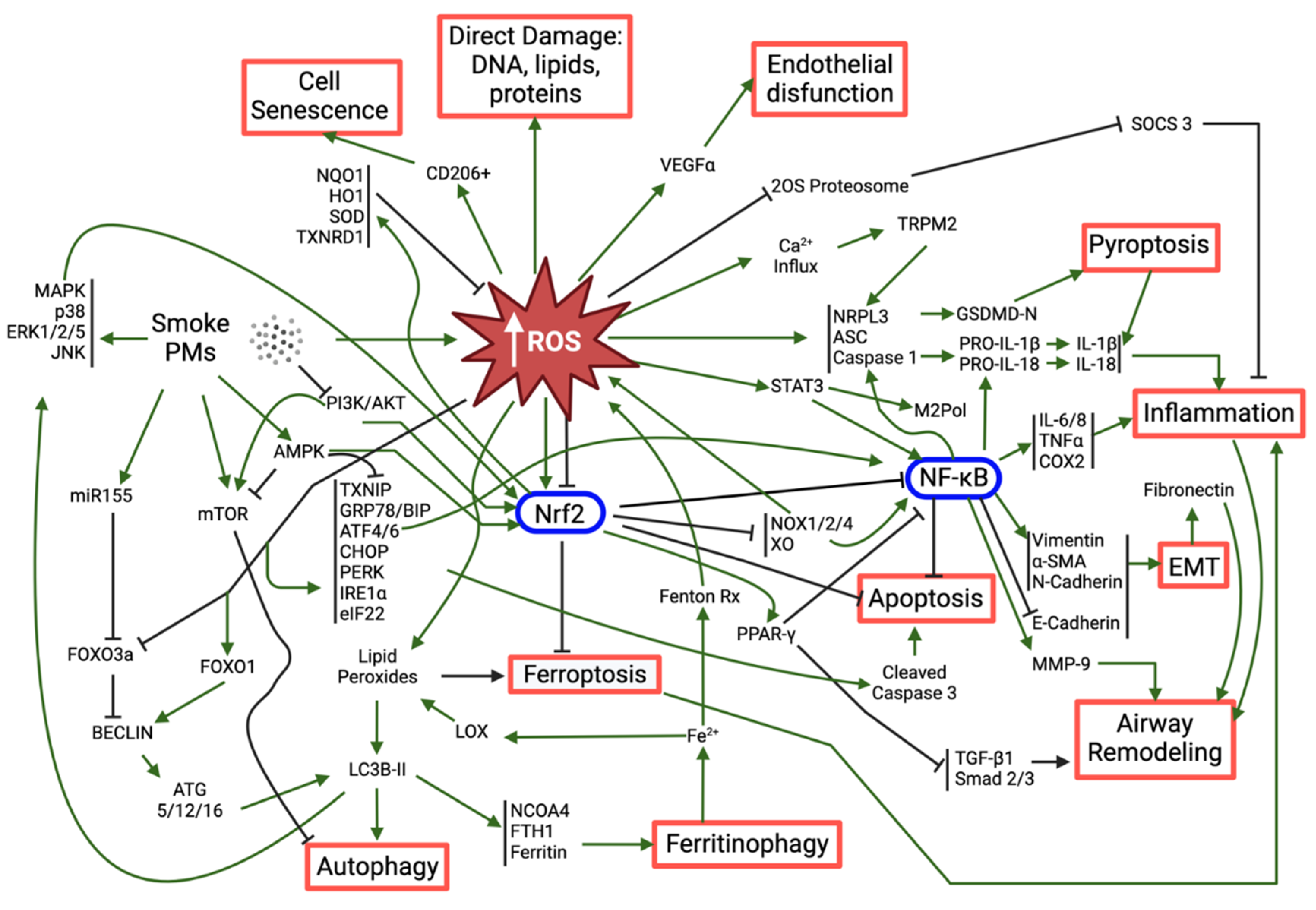
Figure 4.
Interconnection between oxidative stress, antioxidant system, DNA damage repair, and epigenetic factors. It visually depicts the impact of oxidative stress on cellular components, the role of antioxidant systems and DNA repair mechanisms, and the bidirectional relationship between oxidative stress and epigenetic modifications in regulation cellular homeostasis. Created with BioRender.com. Abbreviations: HAT: Histone acetyltransferase; HDACs: Histone deacetylases; HMT: Histone methyltransferases; HDM: Histone demethylases; DNMT: DNA methyltransferases; TET: ten-eleven translocation methylcytosine dioxygenases; miRNA: microRNA; lncRNA: long-noncoding RNA; circRNA: circular RNA.
Figure 4.
Interconnection between oxidative stress, antioxidant system, DNA damage repair, and epigenetic factors. It visually depicts the impact of oxidative stress on cellular components, the role of antioxidant systems and DNA repair mechanisms, and the bidirectional relationship between oxidative stress and epigenetic modifications in regulation cellular homeostasis. Created with BioRender.com. Abbreviations: HAT: Histone acetyltransferase; HDACs: Histone deacetylases; HMT: Histone methyltransferases; HDM: Histone demethylases; DNMT: DNA methyltransferases; TET: ten-eleven translocation methylcytosine dioxygenases; miRNA: microRNA; lncRNA: long-noncoding RNA; circRNA: circular RNA.
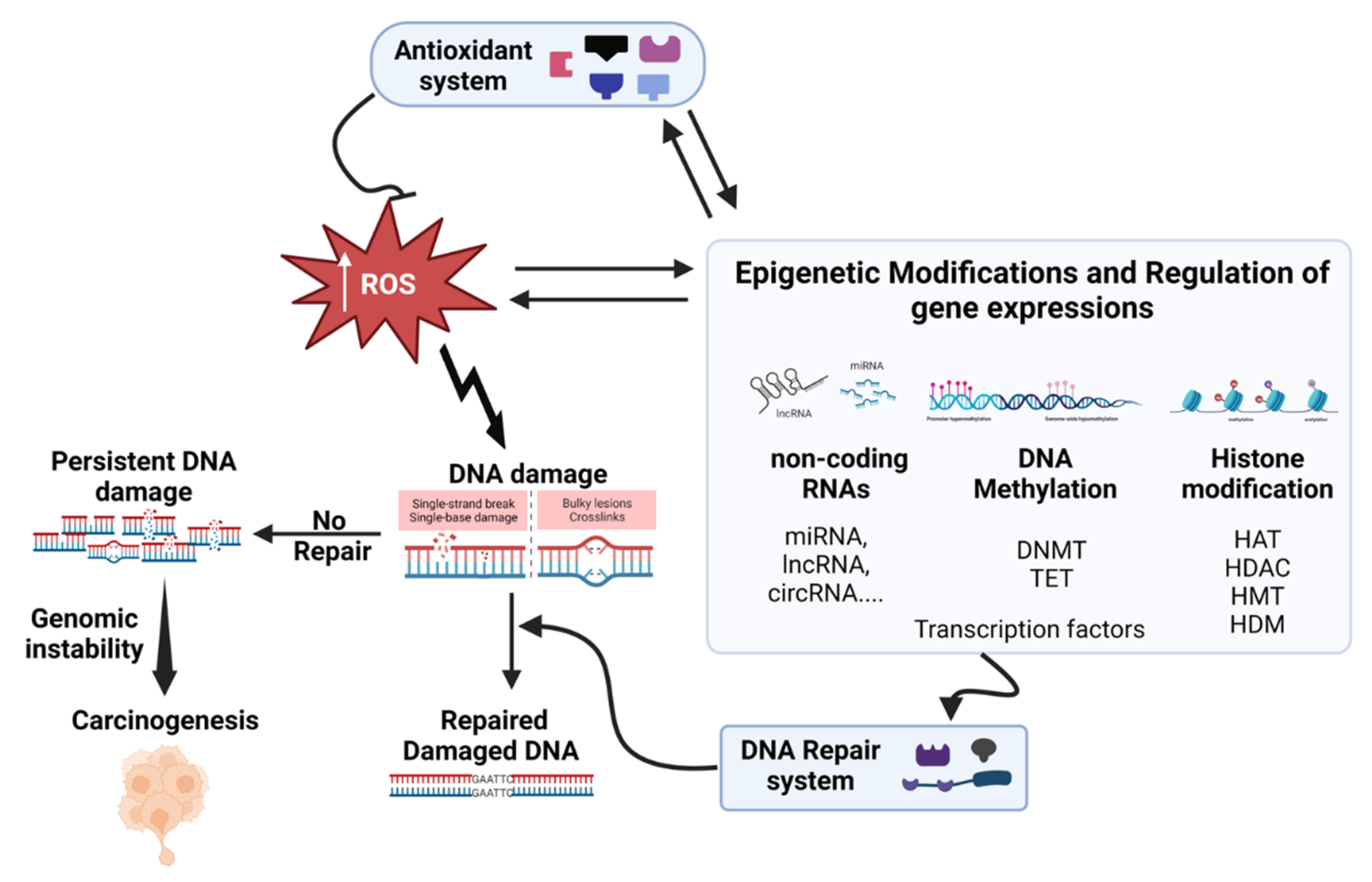

Table 1.
Characteristics of inhaled particulate matter based on their average aerodynamic size.
| PM size | Average aero-dynamic size | Surface area per particle (µm2) | Particles fitting in a 10 µm diameter sphere | Equivalent area vs a 10 µm diameter sphere (µm2) | Main sources | Main components |
|---|---|---|---|---|---|---|
| PM10 | 10 µm | 314 | 1 | 314 | Soil, industry, traffic, construction | Soil products, salts and oxides, biological components (i.e. bacteria and fungi) |
| PM2.5 | 2.5 µm | 19.63 | 64 | 1256 | Fuel combustion, traffic (gasoline and diesel engines) | Black carbon particles, PAHs and other organic compounds |
| PM0.1 | 0.1 µm | 0.0314 | 1 *106 | 31400 | Cigarette smoke, fuel combustion, waste incineration, wear off of materials containing NPs | Carbon derived nanomaterials, metal derived nanomaterials |
Table 2.
Main oxidative stress-related effects triggered by particulate matter, reported in the latest in vitro lung studies (2020.01.01-2023.07.13). Studies are organized in alphabetical order of tested cell line.
Table 2.
Main oxidative stress-related effects triggered by particulate matter, reported in the latest in vitro lung studies (2020.01.01-2023.07.13). Studies are organized in alphabetical order of tested cell line.
| Cells/ cell line | Type of PM, conc., time of exposure, condition | Main oxidative stress-related effects | Reference |
|---|---|---|---|
| A549 | CSE, 3 and 6%, 12h, SUB | - LC3B-II, protein carbonylation, translocation of ADAR1 from nucleus to cytosol - ADAR1 (but not mRNA, so it’s post-transcriptional), CYP1A1, RNA editing levels of AhR, SOD act. |
(Takizawa et al., 2020) |
| A549 | CSE (commercial), 0.25 & 1mg/mL, 24h, SUB | - IL-6, IL-8, MCP-1, CCL5, CYBA, SOD, GPx, CAT, NOX, Nrf2, ATG5, ATG12, ATG16, beclin-1, LC3B-II/LC3B-I, autophagosome formation, FOXO1, nuclear FOXO3a - FOXO3a, mTOR No change in cell viability, ANXV+ or Pi+ cells (= no necrosis, no apoptosis) |
(Bagam et al., 2021) |
| A549 | PM10 SRM 1648a water-soluble fraction, 400mg/mL, 24h, SUB | - MDA, NO, MEK5, ERK5, p-ERK5, Nrf2, HO-1 - cell viability, SOD act., CAT, GSH |
(Xue et al., 2021b) |
| A549 | PM SRM 1648a, 25-200mg/cm2 (119-950mg/mL), 24h, SUB | - ROS, p-AMPKa, Sestrin2 (oxidative stress suppressor), IL-8, TNF-a, COX-2 - cell viability, mitochondrial function |
(So et al., 2022) |
| A549 | PM SRM 1649b Organic extractable fraction, 100mg/mL, 24h, SUB | - wound healing, cell migration, vimentin, fibronectin, ETS-1, p-p65 NF-κb - E-cadherin |
(Chen et al., 2020) |
| A549 | PM2.5 (Water-soluble fraction in simulated lung fluid), 50-200mg/mL, 24h, SUB | - LDH, DNA damage, proline expression - cell viability, TAC |
(Barzgar et al., 2023) |
| A549 | PM2.5 (brake-derived) w/ ¹ Cu conc., 50-500mg/mL, 48h, SUB |
- ROS, % apoptotic cells, MitoMP, IL-8, IL-1a, IL-6, TNF-a, HO-1 - Cell viability, Bcl-2 |
(Figliuzzi et al., 2020) |
| A549 | PM2.5 Urban vs industrial, 80mg/mL, 24h, SUB | - ROS, TNF-a (non-pollution), IL-6 (industrial) - Cell viability, NOQ1 |
(Pang et al., 2020) |
| A549 | PM2.5, 80mg/mL, 24h, SUB | - ROS, IL-6, TNF-a, LDH - Cell viability (significant but not relevant) |
(Li et al., 2022) |
| A549+HUVEC on chip | PM2.5, 100mg/mL, 24h, SUB | - ROS, IL-1a, IL-1β, IL-6, INF-a, % apoptotic cells, BIP, PERK, p-eIF2a, CHOP, caspase-3 | (Guan et al., 2021) |
| A549, SD-1 |
PM2.5, 100mg/mL, 12h, SUB | - ROS, Ca2+, IL-1b, IL-6, TNF-a, NLRP3, caspase-1, TRPM2 | (Wang et al., 2020) |
| A549, RAW 264.7 |
PM, 50mg/mL, 24h, SUB | - ROS, NO, O2-, IL-6, TNF-a, cells in G2/M, % apoptotic cells - viability |
(Guerra e Oliveira et al., 2022) |
| A549+ diff THP-1 | Cow stable dust, 25-100mg/mL, 18h, SUB | - ROS, IL-6, TNF-a, cells in G1/G0 - metabolic act, cell in S-G2/M |
(Martikainen et al., 2021) |
| A549, BEAS-2B |
CSE, 3 (A549) 1.38% (BEAS-2B), 48h, SUB | - p-NF-kb/NF-kb, vimentin, N-cadherin, a-SMA - Cell viability, Nrf2, SIRT1, p-b-catenin/b-catenin, E-cadherin |
(Saha et al., 2022) |
| BEAS-2B | 3¹ functionalized carbon black vs carbon black (PM2.5), 1.56-25mg/mL, 24h, SUB | - IL-1b, IL-6, protein carbonylation - Cell viability, SOD2, Nrf2 |
(Housseiny et al., 2020) |
| BEAS-2B | CSE, 8%, 24h, SUB |
- ROS, MDA, ERK p-p38 MAPK, IL-6, TNF-a, MMP-9, mitochondrial fission factor - SOD and GPx act, OPA1 |
(Yang et al., 2021) |
| BEAS-2B | CSE, 5%, 24h, SUB |
- ROS, apoptotic cells, Bax, cleaved caspase-3/caspase-3, cleaved PARP/ PARP, MDA, TNFa, IL-6, IL-1b - cell viability, Bcl-2, SOD, GSH-Px, ANXA1, FRP2, pAMPK/AMPK |
(Yu and Zhang, 2022) |
| BEAS-2B | CSE, 5%, 24h, SUB | - ROS, MDA, Nrf2, HO-1, NQO1, TRIM25, caspase-1, LDH, NLRP3, GSDMD-N, IL-1b, IL-18 - cell viability, SOD-1, SOD-2, SOD-3, Keap-1 |
(Tian et al., 2021) |
| BEAS-2B | CSE (1%, 7days) & PM10 (SRM 1648 100mg/mL, 24h) alone vs combined, SUB |
- ROS (combined exposure), LDH (not CSE), MDA, IL-6, IL-8, p-ERK, p-JNK, Nrf2, IL-1β, IL-6, IL-8, TNF-α, MCP-1, CXCL-1, HO-1, NQO1 - Cell viability (not CSE), GSH, TXN |
(Son et al., 2020) |
| BEAS-2B | PM SRM 1649b, 200μg/mL, 24h, SUB | - ROS, IL-6, IL-8, p-IκBα/IκBα, p-p65/p65 NF-κb, Nrf2, HO-1, NQO1 - Keap1 |
(Wang et al., 2022b) |
| BEAS-2B | PM SRM 1649b, 200mg/mL, 24h, SUB | - ROS, Pi+ cells, NLRP3, ASC, GSDM-N/GSDMD, cleaved caspase-1/caspase-1, caspase-1 act., LDH release, mature IL-1b/IL-1b, mature IL-18/IL-18, Nrf2 (total + nuclear), NQO1, HO-1, p-Akt/Akt - cell viability |
(Liu et al., 2022b) |
| BEAS-2B | PM2.5 (SRM 2786), 20μg/cm2, 36h, SUB | - Lipid ROS, ROS, MitoMP, Mitochondrial ROS, NADP+/NADPH, COX2, MDA, IL-6, IL-8, TNF-a, Fe2+ accumulation, LC3B-II, NCOA4, FTH1 - cell viability, GPX4, GSH, GPx, Nrf2, PPAR-γ |
(Wang et al., 2022c) |
| BEAS-2B | PM2.5 (China) soluble extract, 300μg/mL (~94 μg/cm2), up to 24h, SUB | - ROS, IL-1b, IL-6, IL-8, GM-CSF, cleaved PARP, cleaved caspase-3, Bax, %apoptotic cells, COX2, p-p65 NF-κb, p-ERK, p-p38 MAPK/ERK, p-JNK - cell viability, ZO-1, E-cadherin, Bcl-2, GSH act, p-mTOR |
(Zhao et al., 2020) |
| BEAS-2B | PM2.5, 25-200mg/mL, 24h, SUB | - Nrf2, NF-kb, IL-1, IL-6, IL-8, a-SMA - Cell viability (lower in direct exp), E-cadherin |
(Wang et al., 2022d) |
| BEAS-2B | PM2.5-0.3 vs organic extractable & non-extractable fractions, 12mgEq. PM/cm2, 6-48h, SUB | - ROS, Nrf2, Nrf2 binding act, Keap-1, NQO1, HO, SOD, GSSG/GSH, DNA damage protein carbonylation, 8-isoprostane, TNF-a, IL-6, IL-8, MCP-1, caspase 3/7, caspase 8, caspase 9 - cell viability, ATG5, Beclin, LC3B-II |
(Badran et al., 2020) |
| BEAS-2B, WL-38, primary rat alveolar macrophages |
PM2.5, 70mg/mL, 24h SUB | BEAS-2B: - ROS, apoptosis rate, collagen I/III, a-SMA, TGF-b1, p-Smad2 - cell viability WL-38: - ROS, apoptosis rate, collagen I/III, a-SMA, TGF-b1, p-Smad2 - cell viability Alveolar macrophages: - ROS, apoptosis rate, M2 phenotype, mTORC1, TIPE2 - cell viability, M1 phenotype |
(Liu et al., 2022a) |
| BEAS-2B, Primary mouse tracheal epithelial cells |
PM2.5, 100mg/mL, 24h, SUB | - ROS, MDA, miR-155 - SOD, GPx, FOXO3a, SOD2, CAT |
(Li et al., 2021) |
| BEAS-2B, primary human small airway epithelial cells |
Polycarbonate (PC) vs polyurethane (PU) incinerated thermoplastics & derivatives w/ 3% carbon nanotubes (CNT), 0.6 or 1.2mg/cm2, 48h, ND | - ROS (only for PC-CNT and results in DNA damage), LDH, CYP1 act, cells in G2 - viability, cells in G1, MitoMP |
(Coyle et al., 2020) |
| BEAS-2B, NHBE cells | Poultry organic dust extract, 0.25%, up to 24h, SUB | - ROS, mitoROS, pro-IL-1b, IL-8, IL-6, PTGS2, ICAM-1, p-p65 NF-κb, p-STAT-3 - p47phox (indicates NOX2 activation) |
(Meganathan et al., 2022) |
| BEAS-2B, THP-1 |
Organic dust extract, 5%, 24h, SUB | BEAS-2B: - ROS, RNS, Nrf2, IL-1β, IL-6, IL-8, IL-10 THP-1: - ROS, RNS, iNOS, Nrf2, Trl2, Trl4, IL-6, IL-8, NF-kb |
(Shrestha et al., 2021) |
| 16-HBE | CSE, 5%, 24h, SUB | - ROS, LDH, IL-1b, IL-18, Pi+ cells, caspase-1 act, NLRP3 - GSDMD |
(Zhang et al., 2021) |
| 16-HBE | PM2.5 (China); 67.5, 116.9, 202.5mg/mL; 4 & 24h, SUB | - ROS, LDH, MDA, HO-1, DNA damage - Cell viability, GSH |
(Niu et al., 2020a) |
| 16-HBE14o-, NuLi-1 | SRM 2585 (Organic extract of house dust), 0.2mg/mL, SUB | - ROS, mitochondrial dysfunction - TEER |
(Marques dos Santos et al., 2022) |
| HBECs | PM SRM 1649b, 300μg/mL, 24h, SUB | - ROS, IL-6, IL-1a, IL-1b, COX2, p-p65/p65 NF-κb - MitoMP |
(Zeng et al., 2022) |
| HBECs | PM SRM 1649b, 200mg/mL, 24h, SUB |
- ROS, ATF4, BIP/GRP78, CHOP, ATF6, cleaved caspase-3, NLRP3, a, GSDMD-N, IL-1β, caspase-1, IL-18, IL-6, IL-8, apoptotic and necrotic cells, Nrf2 (total and nuclear), HO-1, NQO1. | (Shi et al., 2023) |
| HBECs | CSE, 2%, 48h, SUB | - ROS, apoptosis rate, IL-8, IL-6, TNF-a, cleaved caspase-3, p-NF-kb, Keap-1, BIP/GRP78, p-PERK, p-IRE1a, ATF6, ATF4, CHOP, NOX1, NOX2, NOX4, XO, Keap-1 - Cell viability, HO-1, NQO-1, SOD, GCLM, Nrf-2 |
(Wang et al., 2022a) |
| HBSM | CSE, 2.5% 24h, SUB |
- proliferation rate, BrdU incorporation (into newly synthesized DNA of actively proliferating cells), cyclin D1, a-SMA, p-SMAD2, p-SMAD3, TGF-b1 - PPAR- γ |
(Pan et al., 2021) |
| J774A.1 | CSE, 0.5%, 24h, SUB | - ROS, NO - Cell viability |
(da Silva Araújo et al., 2020) |
| L-132 | CSE, 10%, 24h, SUB | - TXNIP, NLRP3, mitoROS, LDH release - cell viability, mitophagy (mitochondria clearance) |
(Mahalanobish et al., 2020) |
| MH-S | CSE, 3%, 1h, SUB | - ROS, EVs conc, vesicular (not intracellular) SOCS3 - 20S proteasome act. |
(Haggadone et al., 2020) |
| MLE-12 | PM2.5, 100mg/mL, 24h, SUB | - a-SMA, Txnip, p-mTOR - cell viability, E-cadherin, Txnrd1 |
(Zhongyin et al., 2022) |
| NCI-H292 | CSE, 10%, 48h, SUB | - IL-8, TNF-a, MMP-9, STAT3, JAK1, JAK2 - SOD, TIMP-1, PPAR |
(Haoran et al., 2020) |
| NCI-H292, HPAEC |
PM2.5, 10mg/cm2, up to 24h, SUB |
- ROS (sub-urban), HO-1, SOD-2, IL-8 in both cell types but higher in endothelial | (Crobeddu et al., 2020) |
| NCI-H460 | PM10, 400mg/mL, 12h, SUB | - ROS - cell viability |
(Lee et al., 2022a) |
| NHBE | CS diluted in clean air, 0.5-4L/min, 40’/day * 3x/week * 4weeks followed by a 20day-recovery phase (RP), ALI |
- HO-1 (back to basal after RP), IL-1β, IL-1 receptor antagonist, IL-6, IL-8, G-CSF, RANTES, CK6, involucrin, TEER, PPARg - GSH/GSSG (acute exposure), IL-7, MCP-1, MMP-1, MMP-2, MMP-3, MMP-7, MMP-10, MMP-13, MUC5AC, MUC5B, CCSP, cilliated cells, goblet cells, number of cilia, cilia lenght, cilia beating frequency, |
(Xiong et al., 2021a) |
| Primary human bronchial epithelial cells | DEP alone (12.5 μg/cm2, 3’/day x3days) vs single combined exposure w/ NO2 (0.1ppm) and w/ SO2 (0.2ppm), aerosol | -Alone: -IL-6, IL-8, TNF-a, GSTA1, HO, SOD3 - IL-8, MMP-9 Combined: -TNF-a, GSTA1, SOD3, MMP-9 |
(Upadhyay et al., 2022a) |
| Primary human CCR6+Th17 cells | CSE, 5%, 48h, SUB | - ROS, SA-b gal+ cells, p16INK4a + cells, VEGFa, p-ERK+ cells, HO-1, NQO1 | (Baskara et al., 2020) |
| Primary rat alveolar epithelial cells | CSE (Heat-not-burn), 20% vs CSE (conventional), 10%, up to 24h, SUB |
-Nrf2, HO-1, GSTA1, GSTA3, NQO1 | (Ito et al., 2020) |
| Rat ATII cells, NR8383 | PM2.5, 50mg/mL, 24h, SUB | - ROS, IL-6, TNF-a, apoptosis or necrosis Data related to immunomodulation is normalized to PM2.5 making it not possible to understand the effects of PM2.5 relative to control. |
(Gao et al., 2021) |
| RAW 264.7 | PMET720 (common stainless-steel wire) aerosols collected @50 or 60 psi, up to 200mg/mL, 24h, SUB vs. GMA-SS, MMA-SS welding particles |
PMET720(60) @200mg/mL: -LDH, NF-kb (>3.12ug/mL) - Cell viability PMET720(50&60) @100mg/mL: - ROS, NF-kb (>3.12ug/mL) |
(Kodali et al., 2022) |
| RAW 264.7 | PM2.5, 400mg/mL, 24h, SUB | - ROS, MDA, NLRP3, NF-kb, Bax, apoptotic rate, caspase-1, caspase-3, GSDMD, IL-1β, %cells in G2 - Bcl-2, SOD act., %cells in G1 |
(Ren et al., 2022) |
| RAW 264.7 | PM (China) urban aerossol, 30mg/cm2, 24h, SUB | - ROS, TNF-a, IL-1b, IL-6, MIP-2 | (Tanaka et al., 2022) |
| diff U937 (as alveolar macrophages), HMC3 (microglia) | DPM SRM 2975, 25mg/mL, 24h, SUB Conditioned serum, 48h, SUB |
-U937: - ROS, H2O2, MCP-1, IL-1b, IL-6, IL-8, TNF-a HMC3: - ROS, H2O2, IL-6, IL-8, IL-1b, TNF-a, CD-14 activation |
(Pradhan et al., 2023) |
A more comprehensive version of this table can be found as supplementary material, including information on tested antioxidative treatments and whether the results have been verified in vivo, as well. Abbreviations in the table: Act: activity; ADAR: adenosine deaminase acting on RNA; AhR: aryl hydrocarbon receptor; a-SMA: alpha smooth muscle actin; AKT: protein kinase B; ANX: annexin; AMPK: 5' adenosine monophosphate-activated protein kinase; ASC: Apoptosis-associated speck-like protein containing a caspase recruitment domain; ATF: activating transcription factor; ATG: autophagy-related; BAX: Bcl-2-associated protein X; Bcl: B-cell lymphoma; BrdU: bromodeoxyuridine; CAT: catalase; CCL: CC chemokine ligand; CCR6+: CC chemokine receptor; CCSP: club-cell secretory protein; CD: cluster of differentiation; CHOP: CCAAT/enhancer-binding protein homologous protein; CK: keratin; COX: cyclooxygenase; CS: cigarette smoke; CSE: cigarette smoke extract; CXCL: CXC chemokine ligand; CYBA: Cytochrome b-245 light chain; CYP: cytochrome P450; DEP: diesel exhaust particles; diff: differentiated; DPM: diesel particulate matter; eIF: eukaryotic initiation factor; ERK: extracellular signal-regulated kinase; ETS: E26 transformation-specific sequence; EV: extracellular vesicle; FOXO: forkhead box protein class O; FRP: N-formyl peptide receptor; FTH: ferritin heavy chain; GCLM: glutamate-cysteine ligase regulatory subunit; GM-CSF: granulocyte-macrophage colony-stimulating factor; GPx: glutathione peroxidase; GPX4: phospholipid hydroperoxide glutathione peroxidase; GRP78: 78 kDa glucose-regulated protein; G-CSF: granulocyte colony-stimulating factor; GMA-SS: gas metal arc- stainless steel; GSDMD: gasdermin D; GSH: reduced glutathione; GSSG: oxidized glutathione; GST: glutathione S-transferase; HO: heme oxygenase; IC: inhibitory concentration; ICAM: intracellular adhesion molecule; IkB: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor; IL: interleukin; iNOS: inducible nitric oxide synthase; IRE: inositol-requiring enzyme; JAK: Janus kinase; JNK: c-Jun N-terminal kinases; Keap: Kelch-like ECH-associated protein; LC3B: microtubule-associated proteins 1A/1B light chain; LDH: lactate dehydrogenase; MAPK: mitogen-activated protein kinase; MCP: monocyte chemoattractant protein; MDA: malondialdehyde; MEK: mitogen-activated protein kinase kinase; MIP: macrophage inflammatory protein; miR: microRNA; mito: mitochondrial; MitoMP: mitochondrial membrane potential; MMA-SS: manual metal arc stainless steel; MMP: matrix metallopeptidase; mTOR: mammalian target of rapamycin; MUC: mucin; NCOA: selective cargo receptor nuclear receptor coactivator; ND: not disclosed; NF-kb: nuclear factor kappa beta; NLRP: NOD-like receptor family pyrin domain containing; NO: nitric oxide; NOX: NADPH oxidase; NQO1: NAD(P)H dehydrogenase (quinone); Nrf2: nuclear factor erythroid 2-related factor 2; OPA: dynamin-like 120 kDa protein, mitochondrial; p: phosphorylated; p47-phox: Neutrophil cytosol factor 1; PARP: poly [ADP-ribose] polymerase; PERK: protein kinase R-like endoplasmic reticulum kinase; Pi: propidium iodide; PM: particulate matter; PMET: common consumable stainless-steel wire; PPAR: peroxisome proliferator-activated receptor; PTGS: prostaglandin-endoperoxide synthase; ROS: reactive oxygen species; SA-bgal: senescence-associated b galactosidase; SIRT: sirtuin; Smad: mothers against decapentaplegic homolog; SOCS: suppressor of cytokine signaling; SOD: superoxide dismutase; SRM: standard reference material; STAT: signal transducer and activator of transcription; SUB: submerged; TAC: total antioxidant capacity; TEER: transepithelial electrical resistance; TGF: transforming growth factor; Th17: T-helper lymphocyte; TIMP: tissue inhibitor metalloproteinase; TIPE: TNF alpha induced protein 8 like; TNF: tumor necrosis factor; TRIM: tripartite motif-containing protein; Trl: toll-like receptor; TRPM: transient receptor potential cation channel, subfamily M; TXN: thioredoxin; TXNIP: thioredoxin-interacting protein; TXNRD1: thioredoxin reductase; VEGF: vascular endothelial growth factor; XO: xanthine oxidase; ZO-1: zonula occludens 1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



