
Submitted:
03 April 2024
Posted:
03 April 2024
You are already at the latest version
Abstract
Analysing the human body by direct observation can lead to misinterpretation of underlying trends, whereas in vitro organ models facilitate their understanding. Likewise, in vitro organ models serve as valuable tools for assessing the toxicity of compounds. However, none of the existing models had the features we considered critical for toxicity assessment, so we developed an in vitro liver model for toxicity assessment. We used four different types of primary liver cells: Hepatic sinusoidal endothelial cells, hepatic stellate cells, Kupffer cells and hepatocytes. We cultured them in different combinations of composition and volume of cell medium, hepatocyte proportion of total cells, and addition of extracellular matrix. We added rifampicin, ibuprofen and 5-fluorouracil to this model and observed the microanatomy and physiology changes for a week with preclinical and clinical instruments. Among the different model configurations, we selected the feature combination of the in vitro model that had similar biomarker values to those measured by the clinical instruments used in clinical diagnostics. When we exposed the selected model configuration to rifampicin, ibuprofen and 5-fluorouracil, the viability of Kupffer cells and liver sinusoidal endothelial cells was significantly lower than in the untreated samples. In contrast, we observed negligible viability of hepatocytes compared to the untreated samples. We have built an in vitro liver model that resembles the liver microenvironment and we have analysed it with clinical instrumentation to facilitate the data translation. In this sense, we have established that Kupffer and LSEC cells are suitable candidates for the search for clinical diagnostic markers of liver function.
Keywords:
liver in vitro model
; in vitro toxicity
; translational research
; novel biomarkers
1. Introduction
The most direct way to analyse the human body it’s from its origin. Advances in various fields (e.g., proteomics, imaging, scanners) make it possible to study the human body, but there is too much “noise” to understand the data from observations [1,2,3]. In contrast, in in vitro organ models make it easier to understand the underlying trends because there are considerably fewer variables, which also makes the models incomplete [4,5]. Each model has its advantages and disadvantages, and considering the purpose of the study, researchers select the most appropriate model for their studies [6]. In the beginning, the models were oversimplified by using a single cell line, and the models consistently had paradoxical results [7,8,9,10]. However, in recent years, more comprehensive preclinical models have been developed [11,12]. Therefore, we can consider organ models as the first step toward a better understanding of the processes/mechanisms in the human body.
Assuming that an organ model recreates the in vivo microenvironment accurately, any toxicity assessment in such model should be accurate. Under these assumptions, numerous new organ models have been developed to test the safety of drugs [13,14,15]. Two dimensional hepatocyte models have the advantage that there are relevant and primary human hepatocytes are recognised to be the ‘gold standard’ for in vitro hepatotoxic assays [13]. Yet, they have a short lifespan, and they lack other cell types present in the liver that are essential for the liver normal functioning [16]. The liver organ-on-a-chip (Ooc) models showed promising results [15], but they are designed with a limited number of cells and low cell density in comparison to the in vivo environment [16]. The spheroid liver models have a greater cell number than the Ooc models and resemble most closely the in vivo liver microenvironment [14]. But the cells in the middle of the spheroid lack nourishment and the cell density is lower than the in vivo microenvironment [16]. Thus, there are many different liver in vitro models, but none of the existing models had the characteristics that we considered critical for toxicity testing.
In this sense, we designed an in vitro liver model based on the characteristics previously proposed by Madorran et al. [16]. In addition, we used preclinical and clinical instruments to measure the physiology and anatomy of the built model to favour the comparison between the values we measured in the model with the clinical data from the literature (clinical cases). Under this premises, we were able to build a cell-based model that have similar values to that observe in clinical cases, therefore, building a liver model that recreated the in vivo liver microenvironment. At the same time, we uncovered promising targets that may be used as liver function early markers in clinical diagnostics.
2. Materials and Methods
2.1. Cell Culture Medium
We used two different cell culture mediums to culture the cells alternatively:
- i
- Williams E colorless medium (Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, USA). L-glutamine (2 mM, Sigma), penicillin (100 U mL-1, Sigma), and streptomycin (1 mgmL-1, Fluka, Buchs, Switzerland) were also added for optimal cell growth.
- ii
- A medium based on Williams E’s colorless medium, which we will refer to from now on as Hep medium (Supplement Table S1). This medium was supplemented with additional amino acid, fatty acids, vitamins and insulin. We use Maxgel, a commercial extra cellular matrix (ECM) (Merck KGaA, Darmstadt, Germany).
2.2. Cells
The liver model was built coculturing hepatocytes from Lonza (Switzerland) and nonparenchymal liver cells (NPC) from (ZEN-BIO, USA); liver sinusoidal endothelial cells (LSEC), human stellate cells (HSC), Kupffer cells. NPC and hepatocytes were grown separately in a 25 cm2 flask (NUNC, Denmark) in a controlled environment at 37º C and 5% CO2 and later seeded together in a 96-well microplate (NUNC, Denmark) to build the liver models.
We seeded the cells in two different seeding arrangements. In the first configuration, we seeded 10,000 hepatocytes and 2500 NPC in each well (80% hepatocytes of total cells). In the second seeding arrangement, we seeded 10,000 hepatocytes and 6,600 NPC (60% hepatocytes of total cells).
In the following experimental setup (when exposing the model to hepatotoxic drugs), more cells were seeded in each sample: 20000 hepatocytes and 5000 NPC cells.
2.3. Toxic Agents
Rifampicin (RIF), ibuprofen (IBU) and 5-fluorouracil (5-FU) (Merck KGaA, Darmstadt, Germany) were added to the two cell culture media at the following final concentrations: 50 µmol/l RIF, 1 mmol/l IBU or 500 µmol/l 5-FU.
2.4. Analytical Techniques
Three different analytical techniques were used to evaluate the physiology and anatomy of the model: the Zeiss Axiovert 40CFL inverted microscope (Zeiss, Germany), the Cobas C111 biochemical analyser (Roche, Switzerland) and the Imagestream MK2 (ISX) imaging flow cytometer (Luminex, USA).
2.5. Statistical Analysis
All statistical analyses in this study were performed using R program. We determined any statistical difference among the feature influence in the viability or the biomarker values of the samples with the Wilcoxon signed-ranked test. We evaluated the differences in viability and biomarkers between the in vitro liver model samples treated with hepatotoxic drugs with the ANOVA/Tukey HSD test.
3. Results
We observed the cell morphology, cell viability and clinical biochemical markers in the liver model we built with different features. We tested different feature combinations: we cultured the cells with different cell culture volumes (65 µL and 85 µL), different cell culture media composition (Williams E medium and Hep medium), different hepatocyte density (80% and 60%), and the addition of ECM (some samples with and some without it) (Figure 1a).
3.1. Cell Morphology
We observed two important cell arrangements under the inverted microscope. The cells cultured with 65 µL of the medium clustered in the centre of the well (Figure 2a). The cells cultured with 85 µL of medium formed colonies on the entire surface of the well (Figure 2b). We didn’t observe any morphological differences between the samples cultured with or without ECM (Figure 2c). It is noteworthy that the cells form similar structures to that observed in liver spheroid models (Figure 2c).
3.2. Cell Viability Assessment
The models with the highest hepatocyte density (80%) had the highest cell viability (Figure 3). On the contrary, the models with the lowest hepatocyte density (60%) had the lowest cell viability (Figure 3).
The formulation of cell medium also influences the viability of the models. Cells cultured with Hep medium had significantly higher viabilities than cells cultured with William E (Figure 3).
3.3. Clinical Biochemistry
The value of each liver marker was subtracted from the liver values measured in both cell culture media (without the cells). These values are summarized in Supplementary Table S2. We analyzed alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), glucose, triglycerides, and albumin in the pooled samples using a Cobas C111 (Roche, Switzerland) [17].
We observed a significant difference in liver marker values between the two cell culture media (Figure 4a, red and brown boxplots). Significantly higher albumin levels were measured in the samples cultured with Hep medium. In addition, the cells cultured in this medium had a higher net triglyceride release. In contrast, cells cultured with Williams E medium had a net uptake of triglycerides. The glucose dynamics were significantly different (p= 0.00024) and had an opposite effect to the triglyceride dynamics.
We did not observe any significant differences between the rest of the samples (Figure 4a). However, the normalisation of the values of each biomarker with the cell number of the corresponding sample revealed significant differences (Figure 4b). The normalisation showed a significantly lower ALP value in the samples cultured with Hep medium (Figure 4b). It also showed a significantly lower ALP value in samples with higher hepatocyte density.
3.4. Testing the Selected Model with RIF, IBU, 5-FU
3.4.1. Morphology Assessment
There were no visible morphologic differences between groups in the first 5 days (Figure 5). Longer incubation periods should be performed to observe possible morphological changes. We did not performed longer incubations since the focus of our study was to culture cell-based liver model for 5 days.
3.4.2. Viability Assessment
We didn’t observe any significant difference in the viability of the samples treated with 5-FU in respect to the untreated samples (Figure 6a). However, when analysing the viability of each cell type of the sample, we observed that Kupffer cells had the lowest viability (Figure 6b). In contrast, exposure to 5-FU induced the LSEC to proliferate during the first week (Figure 6b). However, we observed a high cell death ratio on day 5 (Table 1). So, continuous exposure of 5-FU to LSEC cells may reduce their viability.
We observed an ambivalent effect on the viability of cells exposed to IBU (Figure 6b). On the one hand, we observed that the viability of Kupffer and HSC cells was lower when the samples were exposed to IBU compared to the untreated samples (Figure 6b). On the other hand, we observed a higher viability of LSEC cells exposed to IBU (Figure 6b).
3.4.3. Clinical Biochemistry
We observed a significant decrease in the ALP value for all treated samples compared to untreated samples (Figure 7). We also observed a lower albumin content in treated samples, but the decrease was significant only in samples treated with IBU and RIF.
We also observed that samples exposed to IBU had a significantly higher net release of glucose than the untreated sample (Figure 7). On the contrary, there was a net uptake of glucose and triglycerides in cells treated with RIF (Figure 7).
ALP, ALT, AST, GGT were similar in all the samples, no significant effect was observed when treating the model with RIF, IBU nor 5-FU (Figure 7).
4. Discussion
Our goal was to build an in vitro liver model to resemble the liver microenvironment and allow clinical marker usage. To that end, we used primary liver cells: HSC, LSEC, Kupffer, and hepatocytes, because they reportedly retain the characteristics of the original cells, unlike cancer cells or immortalized cell lines [6]. We also used different combinations of cell medium volume and formulation, ECM addition, and cell type proportion to configure a liver model. In this sense, we evaluate the influence of each feature in the liver model.
The influence of cell culture media
We observed that cells cultured with a medium rich on supplements (Hep medium) increased the viability of the cells (Figure 3a), which is concordant with previous studies [18,19]. The addition of insulin to the medium increased the glucose uptake by the cells (Figure 4), as described in the literature [20,21,22]. In contrast, the net release of triglycerides observed in cells cultured with the same media may be due to the higher amino acid content of the media [23]. The opposing dynamics of glucose and triglycerides (Figure 4a) are consistent with in vivo observations [23]. The addition of amino acids in the Hep medium increased the albumin synthesis significantly (Figure 4a), which concurs with the literature [24].
The influence of cell culture volume
In the initial phase, the higher cell density and interaction in samples cultured with 65 µl of cell medium (due to the cell arrangement seen in Figure 2a) may increase cell proliferation [25]. Yet, later necrosis of the cells may occur due to nutrient deficiency [6,26]. In this sense, and considering the lobule arrangement of the liver [27], the cells cultured with 85 µl more closely resemble the in vivo situation (Figure 2b), scattered colonies resembling lobule-like arrangement). There are many different types of stimulus that induce cell migration [28]. But, considering that the different migrations we observed were related to cell density, we associate the migration to chemotaxis [29]. This complex process needs to be studied in much more detail to better understand the observed migrations.
Furthermore, the clinical instrumentation is designed to measure the markers at certain range of concentration. However, the concentration of the molecules of interest is different in in vitro models and in vivo environment. Thus, lowering the volume to increase the cell number to cell volume ratio enabled the use of clinical instrumentation.
The influence of ECM
The addition of ECM did not affect the morphology of cells in the model (Figure 2c). On the contrary, the addition of ECM had a noticeable impact on the viability of the cells. ECM induced the proliferation of hepatocytes (as previously observed by Wang Y. et al. [30]) but decreased the viability of HSC cells. The designed model had HSC cells responsible for synthesising ECM in the liver [31]. Thus, ECM production by the existing HSCs was sufficient [31], and the additional ECM coating did not affect any of the biomarker values.
The influence of the proportion of the cell types
Higher hepatocyte density (80%) induced cell proliferation (Figure 3), which was previously observed [25]. This samples (80 % hepatocyte density) had less variable glucose content (Figure 4a), as expected since hepatocytes are the liver cells that regulate glucose levels [23]. The samples with higher hepatocyte density also had higher albumin content (Figure 4a), as albumin is only synthesised in hepatocytes [32].
After evaluating the influence of each feature in the model, we selected the feature combination that most faithfully recreated the liver microenvironment and expose the liver model to 50 µmol/l RIF, 1 mmol/l IBU or 500 µmol/l 5-FU. The concentrations chosen are based on previous studies by other authors [33,34,35], analysing cell proliferation, IC50, EC50, gene expression, CYP activities and data from our own experiments.
Exposing the liver model to 5-FU
5-FU is a widely used chemotherapeutic drug and one of the most commonly utilized drugs for the treatment of various types of cancers because it inhibits thymidylate synthase [36]. The hepatotoxicity of this drug is well documented [34,36,37,38], thus, is interesting as a toxic agent for liver models. The samples exposed to 5-FU had the lowest viability of all the evaluated samples (Figure 6a) because only 5-FU was cytotoxic to hepatocytes (Figure 6b). Since hepatocytes account for the vast majority of the population, any change in their cell number induces a higher effect in the total cell number of the sample. Yet, the highest cytotoxic effect was evaluated in Kupffer cells. In contrast, 5-FU induced the proliferation of LSEC cells during the first week, for which there are no previous data in literature). However, given the high cell death observed on day 5 (Table 1), continuous exposure to 5-FU may also induce cell death in LSEC cells [39]. Inhibition of thymidylate synthase and accumulation of toxic by-products from 5-FU catabolism (fluorocitrate for instance) may induce this delayed toxic effect [40]. We also observed that adding 5-FU to the model decreased albumin synthesis. This is more likely due to the reduction of hepatocyte number [41].
Exposing the liver model to IBU
IBU is a nonsteroidal anti-inflammatory drug and one of the most used drugs worldwide [42]. There are many animal studies that observed the hepatotoxicity of this drug [43,44,45,46], and considering its exposure [42], it is an interesting candidate for the evaluation of our model. The addition of IBU had an ambivalent effect on the cell viability of the samples. On the one hand, IBU significantly decreased the viability of Kupffer and HSC cells (Figure 6b). On the other hand, samples with added IBU had twice as many LSEC cells as the untreated samples (Table 1). The proliferation of LSEC was also observed in a previous study [47], yet the mechanisms involved are unknown to date. As a result of both trends (cytotoxicity and proliferation), samples exposed to IBU had similar viability to the untreated samples, but the cell composition of the liver model changed (Figure 6b). Yet, the most significant biological response induced by IBU was related to glucose dynamics. The samples treated with IBU had a significantly higher net glucose release than untreated samples. And these higher values were not the result of greater net triglyceride uptake. Thus, the influence of IBU on gluconeogenesis and glycogenolysis was also observed in vivo [43,48,49]and could be the cause of this observation. Notably, there was no albumin content in samples treated with IBU, which may be caused due to the higher net glucose release (and lower availability for albumin synthesis) [41].
Exposing the liver model to RIF
RIF is a widely used antibiotic to treat tuberculosis and other bacterial infections. Treatment with RIF is effective, but is known to induce drug-metabolizing enzymes in the liver [50]. Thus, it is an interesting compound to evaluate the toxic assessment of our model. The addition of RIF significantly reduced the viability of Kupffer cells, like in the rest of the treated samples (Figure 6b). On the contrary, RIF addition had a very distinct effect on the glucose and triglyceride dynamics. Both markers had a significantly higher net uptake by the cells compared to untreated samples (Figure 7). This is in line with different studies reporting fatty acid accumulation upon RIF treatment [51]. Moreover, this observation could also indicate an increase in FA accumulation within cells [23]. This evaluation is consistent with previous studies in which upregulation of the free fatty acid transporter was observed in HepG2 cells treated with 10 µM of RIF [51]. The albumin synthesis was significantly reduced when exposing the cells to RIF, maybe due to the above mentioned changes in the molecular pathways involved in glucose and triglyceride dynamics [24]. Although further investigation is needed to corroborate the latter.
Evaluation of the liver model
The model has sufficient cell density to be statistically relevant, unlike Ooc models [52] and it has a cell arrangement similar to that in spheroids, which reinforces the native physiology of the cells [14]. It is visible under the microscope, allowing observation of detailed cell morphology. It can also be analysed with clinical instruments, which facilitates comparison with clinical data. In addition, the model enables comprehensive cell analysis with preclinical instruments. Most importantly, the observations in the model suggest that it resembles the liver microenvironment. The liver regulates the dynamics of triglycerides and glucose, which are in an inverse relationship [23,49] and these conditions are met in the model we developed (Figure 4a). Furthermore, the addition of hepatotoxic drugs to the model resulted in similar physiological responses as in vivo. Albumin synthesis is a key element in any liver model because albumin production is one of the liver’s main functions [24]. In this sense, there are changes in albumin synthesis associated with various pathologies [24,53]. And in our model, we observed the influence on albumin synthesis by the hepatotoxic drugs (Figure 7).
In the tested liver models, we did not observe any correlation between transaminase values and the viability of the cells. But the influence of transaminase levels in the liver is not clear in clinical studies either. High liver transaminases are found in patients whose liver is proliferating (after the liver recession) [54]. But various studies have measured similar levels in patients with liver diseases [55,56,57,58,59]. In addition, clinical studies have observed patients with abnormal levels who did not have liver disease [58,59]. These observations have led other authors to search for alternative markers in recent years [60]. In this sense, this model may aid in the search for alternative markers, which is a rising concern in recent years [61,62,63]. Moreover, this model may shed further light on some specific pathology related features, since it allows a more concrete analysis using preclinical evaluation methods besides the use of clinical instrumentation. To that end, our study may uncover an underlying trend that was unclear earlier. We observed that all hepatotoxic drugs had a greater impact on the viability of LSEC and Kupffer cells than on HSC and hepatocytes. Previous studies observed transcriptomic shifts between healthy and cirrhotic liver disease (CLD) scenarios [64,65]on LSEC cells. But they did not focus on the viability of both cell types. This finding are supported by the function of both cells, since LSEC and Kupffer cells are the liver cells responsible for xenobiotic uptake [66]. Thus, LSEC and Kupffer cells are the liver cells first affected by exogenous agents [66]. Therefore, we can use this model (or similar) to monitor molecules related to LSEC and Kupffer injury.
4.1. Limitations of the Model
In this experimental setup we observed the cells in the model with an inverted microscope, but we are working on the next experimental setup where we will observe the model with a confocal microscope. The use of a confocal microscope for further studies should allow us a more detailed study of the anatomy of the cells in this model.
The use of clinical instrumentation to evaluate the model was troublesome due to the scarcity of information on the use of clinical instruments in an in vitro liver model. In this sense, more replicates under similar conditions should be performed to increase confidence in the observed trends. Special focus should be drawn to molecules present in LSEC and Kupffer cells in the model when exposing it to toxic compounds. If these molecules were also present in the peripheral circulation, they may be used as markers of liver health.
With the current settings, the model is not a suitable for pharmacokinetic studies, but key changes in its configuration may adequate it to such studies. The introduction of a rocker or even perfusion is possible and would enable pharmacokinetic studies.
4.2. Highlights
- Our in vitro liver model had similar biomarker levels to those observed in vivo, confirming the representativeness of the model.
- The biological effects elicited by the selected drugs had similar trends to those observed in clinical studies.
- This new approach facilitates the translation between basic and clinical research.
- LSEC and Kupffer had a more pronounced biological effect (viability) than the other cell types when exposed to hepatotoxic drugs.
5. Conclusions
The developed model allows a comprehensive evaluation of the toxicity of substances, since is possible to observed with preclinical and clinical instruments. Moreover, when exposing the model to hepatotoxic drugs we observed similar values to the data available from clinical diagnostics. Thus, this should aid to a better comparison between the data from preclinical and clinical observations. At the same time, it contributes to a better understanding of the physiology and anatomy of the human body. In this sense, a possible underlying physiological trend can be observed in the evaluation of toxicity, since LSEC and Kupffer cells were the only cell types that showed a measurable biological response. Thus, both cell types are reasonable candidates as clinical diagnostic markers of liver function, and we recommend further studies to substantiate our findings..
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Hep medium formulation; Table S2: Summary of liver markers in the two cell culture media used for liver model culturing.
Author Contributions
Conceptualization, E.M.; methodology, E.M.; software, E.M.; validation, E.M.; formal analysis, E.M.; investigation, E.M.; resources, E.M.; data curation, E.M.; writing—original draft preparation, E.M.; writing—review and editing, E.M, L.K.S.; M.R. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
The research leading to these results has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no 311820 (ECsafeSEAFOOD).
Data Availability Statement
Authors agree to make data and materials supporting the results or analyses presented in their paper available upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dominguez, D.C.; Lopes, R.; Torres, M.L. Proteomics: clinical applications. Clinical laboratory science : journal of the American Society for Medical Technology 2007, 20, 245–8. [Google Scholar]
- Schwenk, J.M.; Omenn, G.S.; Sun, Z.; Campbell, D.S.; Baker, M.S.; Overall, C.M.; Aebersold, R.; Moritz, R.L.; Deutsch, E.W. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J Proteome Res 2017, 16, 4299–4310. [Google Scholar] [CrossRef]
- Edge, R.; Ford, C. CADTH Rapid Response Reports. In Clinical Decision Support Systems for Appropriate Medical Imaging: Clinical Evidence and Cost-Effectiveness, Canadian Agency for Drugs and Technologies in Health Copyright © 2019 Canadian Agency for Drugs and Technologies in Health.: Ottawa (ON), 2019.
- Oltvai, Z.N.; Barabási, A.-L.; Jeong, H.; Tombor, B.; Albert, R. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar]
- Maresova, P.; Klimova, B.; Kuca, K. ; Legislation, regulation and policies issues of orphan drugs in developed countries from 2010 to 2016. Journal of Applied Biomedicine 2018. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Shafagh, R.Z.; Hendriks, D.F.G.; Ingelman-Sundberg, M. 3D Primary Hepatocyte Culture Systems for Analyses of Liver Diseases, Drug Metabolism, and Toxicity: Emerging Culture Paradigms and Applications. Biotechnol J 2019, 14, e1800347. [Google Scholar] [CrossRef]
- Ouedraogo, M.; Nguyen, A.T.; Duez, P. Methods Applied to the In Vitro Primary Toxicology Testing of Natural Products: State of the Art, Strengths, and Limits. 2014.
- Kharasch, E.D.; Whittington, D.; Hoffer, C.; Krudys, K.; Craig, K.; Vicini, P.; Sheffels, P.; Lalovic, B. Paradoxical role of cytochrome P450 3A in the bioactivation and clinical effects of levo-alpha-acetylmethadol: importance of clinical investigations to validate in vitro drug metabolism studies. Clinical pharmacokinetics 2005, 44, 731–51. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P. Paradoxical echinocandin activity: a limited in vitro phenomenon? Medical mycology 2009, 47 Suppl 1, S369–75. [Google Scholar] [CrossRef]
- Yang, P.; Zhao, Y.; Zhao, L.; Yuan, J.; Chen, Y.; Varghese, Z. Paradoxical effect of rapamycin on inflammatory stress-induced insulin resistance in vitro and in vivo. Nature Publishing Group 2015, 1–13. [Google Scholar] [CrossRef]
- Lee, M.; Nam, K.T.; Kim, J.; Lim, S.E.; Yeon, S.H.; Lee, B.; Lee, J.Y.; Lim, K.M. Evaluation of ocular irritancy of coal-tar dyes used in cosmetics employing reconstructed human cornea-like epithelium and short time exposure tests. Food Chem Toxicol 2017, 108, 236–243. [Google Scholar] [CrossRef]
- OECD, Test No. 492B: Reconstructed Human Cornea-like Epithelium (RHCE) Test Method for Eye Hazard Identification. 2022.
- Polidoro, M.A.; Ferrari, E.; Marzorati, S.; Lleo, A.; Rasponi, M. Experimental liver models: From cell culture techniques to microfluidic organs-on-chip. Liver Int 2021, 41, 1744–1761. [Google Scholar] [CrossRef]
- Brooks, A.; Liang, X.; Zhang, Y.; Zhao, C.X.; Roberts, M.S.; Wang, H.; Zhang, L.; Crawford, D.H.G. Liver organoid as a 3D in vitro model for drug validation and toxicity assessment. Pharmacol Res 2021, 169, 105608. [Google Scholar] [CrossRef]
- Messelmani, T.; Morisseau, L.; Sakai, Y.; Legallais, C.; Le Goff, A.; Leclerc, E.; Jellali, R. Liver organ-on-chip models for toxicity studies and risk assessment. Lab Chip 2022, 22, 2423–2450. [Google Scholar] [CrossRef]
- Madorran, E.; Stozer, A.; Bevc, S.; Maver, U. In vitro toxicity model: Upgrades to bridge the gap between preclinical and clinical research. Bosn J Basic Med Sci 2019. [Google Scholar] [CrossRef]
- Bowling, J.L.; Katayev, A. An Evaluation of the Roche Cobas c 111. Laboratory Medicine 2010, 41, 398–402. [Google Scholar] [CrossRef]
- Yuan, H.X.; Xiong, Y.; Guan, K.L. Nutrient sensing, metabolism, and cell growth control. Mol Cell 2013, 49, 379–87. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metabolism 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Boron, W.F.; Boulpaep, E.L. Medical Physiology. Proceedings of the Royal Society of Medicine 2012, 68, 3487. [Google Scholar]
- Boron, W.F.; Boulpaep, E.L. Medical physiology: a cellular and molecular approach; Saunders Elsevier: Philadelphia, PA, 2012. [Google Scholar]
- Mescher, A.L. Junqueira’s Basic Histology: Text and Atlas. 16th Edition ed.; McGraw Hill: New York, 2021. [Google Scholar]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr Physiol 2017, 8, 1–8. [Google Scholar]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J Gen Med 2016, 9, 229–255. [Google Scholar] [CrossRef]
- Chinnici, C.M.; Miceli, V.; Pampalone, M.; Lo Nigro, A.; Amico, G.; Conaldi, P.G. In vitro evidences of epithelial to mesenchymal transition in low cell-density cultured human fetal hepatocytes. Biochem Biophys Res Commun 2017, 490, 472–479. [Google Scholar] [CrossRef]
- Carotti, S.; Morini, S.; Carpino, G.; Gaudio, E. Liver Histology. In Liver Diseases: A Multidisciplinary Textbook, Radu-Ionita, F.; Pyrsopoulos, N.T., Jinga, M., Tintoiu, I.C., Sun, Z., Bontas, E., Eds.; Eds. Springer International Publishing: Cham, 2020; pp. 17–28. [Google Scholar]
- Saxena, R.; Theise, N.D.; Crawford, J.M. Microanatomy of the human liver-exploring the hidden interfaces. Hepatology 1999, 30, 1339–46. [Google Scholar] [CrossRef]
- Trepat, X.; Chen, Z.; Jacobson, K. Cell migration. Compr Physiol 2012, 2, 2369–92. [Google Scholar]
- SenGupta, S.; Parent, C.A.; Bear, J.E. The principles of directed cell migration. Nature Reviews Molecular Cell Biology 2021, 22, 529–547. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, M.H.; Shirahama, H.; Lee, J.H.; Ng, S.S.; Glenn, J.S.; Cho, N.J. ECM proteins in a microporous scaffold influence hepatocyte morphology, function, and gene expression. Sci Rep 2016, 6, 37427. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Moman, R.N.; Gupta, N.; Varacallo, M. Physiology, Albumin. In StatPearls, StatPearls Publishing Copyright © 2020; StatPearls Publishing LLC.: Treasure Island (FL), 2020. [Google Scholar]
- Akrami, H.; Moradi, B.; Borzabadi Farahani, D.; Mehdizadeh, K. Ibuprofen reduces cell proliferation through inhibiting Wnt/β catenin signaling pathway in gastric cancer stem cells. Cell Biol Int 2018, 42, 949–958. [Google Scholar] [CrossRef]
- Sommer, J.; Mahli, A.; Freese, K.; Schiergens, T.S.; Kuecuekoktay, F.S.; Teufel, A.; Thasler, W.E.; Müller, M.; Bosserhoff, A.K.; Hellerbrand, C. Analysis of molecular mechanisms of 5-fluorouracil-induced steatosis and inflammation in vitro and in mice. Oncotarget 2017, 8, 13059–13072. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology 2012, 28, 69–87. [Google Scholar] [CrossRef]
- Diseases, B.M.N.I. o. D. a. D. a. K.; LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]-fluorouracil. In livertox.nih.gov: 2018.
- Alessandrino, F.; Qin, L.; Cruz, G.; Sahu, S.; Rosenthal, M.H.; Meyerhardt, J.A.; Shinagare, A.B. 5-Fluorouracil induced liver toxicity in patients with colorectal cancer: role of computed tomography texture analysis as a potential biomarker. Abdominal Radiology 2019, 44, 3099–3106. [Google Scholar] [CrossRef]
- da Silva, M.C.; Fabiano, L.C.; da Costa Salomão, K.C.; de Freitas, P.L.Z.; Neves, C.Q.; Borges, S.C.; de Souza Carvalho, M. d. G.; Breithaupt-Faloppa, A.C.; de Thomaz, A.A.; dos Santos, A.M.; Buttow, N.C. A Rodent Model of Human-Dose-Equivalent 5-Fluorouracil: Toxicity in the Liver, Kidneys, and Lungs. Antioxidants 2023, 12, 1005. [Google Scholar] [CrossRef]
- Eggert, T.; Greten, T.F. Tumor regulation of the tissue environment in the liver. Pharmacol Ther 2017, 173, 47–57. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda (MD), 2012.
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Frontiers in physiology 2014, 5, 299. [Google Scholar] [CrossRef]
- Diseases, B.M.N.I. o. D. a. D. a. K.; LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]- ibuprofen. In livertox.nih.gov: 2018.
- de Souza, C.O.; Kurauti, M.A.; de Fatima Silva, F.; de Morais, H.; Curi, R.; Hirabara, S.M.; Rosa Neto, J.C.; de Souza, H.M. Celecoxib and Ibuprofen Restore the ATP Content and the Gluconeogenesis Activity in the Liver of Walker-256 Tumor-Bearing Rats. Cellular Physiology and Biochemistry 2015, 36, 1659–1669. [Google Scholar] [CrossRef]
- Hartung, T.; Daston, G. Are In Vitro Tests Suitable for Regulatory Use? 2009, 111, 233–237. [Google Scholar] [CrossRef]
- Ilic, S.; Drmic, D.; Zarkovic, K.; Kolenc, D.; Brcic, L.; Radic, B.; Djuzel, V.; Blagaic, A.B.; Romic, Z.; Dzidic, S.; Kalogjera, L.; Seiwerth, S.; Sikiric, P. Ibuprofen hepatic encephalopathy, hepatomegaly, gastric lesion and gastric pentadecapeptide BPC 157 in rats. European Journal of Pharmacology 2011, 667, 322–329. [Google Scholar] [CrossRef]
- Bendele, A.M.; Hulman, J.F.; White, S.; Brodhecker, C.; Bendele, R.A. Hepatocellular proliferation in ibuprofen-treated mice. Toxicol Pathol 1993, 21, 15–20. [Google Scholar] [CrossRef]
- Maher, J.J. Cell-specific expression of hepatocyte growth factor in liver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride. J Clin Invest 1993, 91, 2244–52. [Google Scholar] [CrossRef]
- Brass, E.P.; Garrity, M.J. Effect of nonsteroidal anti-inflammatory drugs on glycogenolysis in isolated hepatocytes. Br J Pharmacol 1985, 86, 491–6. [Google Scholar] [CrossRef]
- Klover, P.J.; Mooney, R.A. Hepatocytes: critical for glucose homeostasis. The International Journal of Biochemistry & Cell Biology 2004, 36, 753–758. [Google Scholar]
- Diseases, B.M.N.I. o. D. a. D. a. K.; LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]- rifampicin. In livertox.nih.gov: 2018.
- Zhang, J.; Wei, Y.; Hu, B.; Huang, M.; Xie, W.; Zhai, Y. Activation of human stearoyl-coenzyme A desaturase 1 contributes to the lipogenic effect of PXR in HepG2 cells. PLoS One 2013, 8, e67959. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: recent breakthroughs and future prospects. BioMedical Engineering OnLine 2020, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Soeters, P.B.; Wolfe, R.R.; Shenkin, A. Hypoalbuminemia: Pathogenesis and Clinical Significance. JPEN J Parenter Enteral Nutr 2019, 43, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Siu, J.; McCall, J.; Connor, S. Systematic review of pathophysiological changes following hepatic resection. HPB (Oxford) 2014, 16, 407–21. [Google Scholar] [CrossRef] [PubMed]
- Moriles, K.E.; Azer, S.A. Alanine Amino Transferase. In StatPearls, StatPearls Publishing Copyright © 2020; StatPearls Publishing LLC.: Treasure Island (FL), 2020. [Google Scholar]
- Aulbach, A.D.; Amuzie, C.J. Chapter 17 - Biomarkers in Nonclinical Drug Development. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development (Second Edition), Faqi, A.S.; Ed. Academic Press: Boston, 2017; pp. 447–471. [Google Scholar]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Lilford, R.J.; Bentham, L.; Girling, A.; Litchfield, I.; Lancashire, R.; Armstrong, D.; Jones, R.; Marteau, T.; Neuberger, J.; Gill, P.; Cramb, R.; Olliff, S.; Arnold, D.; Khan, K.; Armstrong, M.J.; Houlihan, D.D.; Newsome, P.N.; Chilton, P.J.; Moons, K.; Altman, D. Birmingham and Lambeth Liver Evaluation Testing Strategies (BALLETS): a prospective cohort study. Health Technol Assess 2013, 17, 1–307. [Google Scholar] [CrossRef] [PubMed]
- Lala, V.; Goyal, A.; Bansal, P.; Minter, D.A. Liver Function Tests. In StatPearls, StatPearls Publishing Copyright © 2020; StatPearls Publishing LLC.: Treasure Island (FL), 2020. [Google Scholar]
- Lehmann-Werman, R.; Magenheim, J.; Moss, J.; Neiman, D.; Abraham, O.; Piyanzin, S.; Zemmour, H.; Fox, I.; Dor, T.; Grompe, M.; Landesberg, G.; Loza, B.L.; Shaked, A.; Olthoff, K.; Glaser, B.; Shemer, R.; Dor, Y. Monitoring liver damage using hepatocyte-specific methylation markers in cell-free circulating DNA. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Arechederra, M.; Argemí, J.; Fernández-Barrena, M.G.; Avila, M.A. Loss of liver function in chronic liver disease: An identity crisis. J Hepatol 2023, 78, 401–414. [Google Scholar] [CrossRef]
- Lurie, Y.; Webb, M.; Cytter-Kuint, R.; Shteingart, S.; Lederkremer, G.Z. Non-invasive diagnosis of liver fibrosis and cirrhosis. World J Gastroenterol 2015, 21, 11567–83. [Google Scholar] [CrossRef]
- Arechederra, M.; Recalde, M.; Gárate-Rascón, M.; Fernández-Barrena, M.G.; Ávila, M.A.; Berasain, C. Epigenetic Biomarkers for the Diagnosis and Treatment of Liver Disease. Cancers 2021, 13, 1265. [Google Scholar] [CrossRef]
- Holland, C.H.; Ramirez Flores, R.O.; Myllys, M.; Hassan, R.; Edlund, K.; Hofmann, U.; Marchan, R.; Cadenas, C.; Reinders, J.; Hoehme, S.; Seddek, A.L.; Dooley, S.; Keitel, V.; Godoy, P.; Begher-Tibbe, B.; Trautwein, C.; Rupp, C.; Mueller, S.; Longerich, T.; Hengstler, J.G.; Saez-Rodriguez, J.; Ghallab, A. Transcriptomic Cross-Species Analysis of Chronic Liver Disease Reveals Consistent Regulation Between Humans and Mice. Hepatol Commun 2022, 6, 161–177. [Google Scholar] [CrossRef]
- Gallon, J.; Coto-Llerena, M.; Ercan, C.; Bianco, G.; Paradiso, V.; Nuciforo, S.; Taha-Melitz, S.; Meier, M.A.; Boldanova, T.; Pérez-Del-Pulgar, S.; Rodríguez-Tajes, S.; von Flüe, M.; Soysal, S.D.; Kollmar, O.; Llovet, J.M.; Villanueva, A.; Terracciano, L.M.; Heim, M.H.; Ng, C.K.Y.; Piscuoglio, S. Epigenetic priming in chronic liver disease impacts the transcriptional and genetic landscapes of hepatocellular carcinoma. Mol Oncol 2022, 16, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, S.; Larsen, A.K.; McCourt, P.; Smedsrød, B.; Sørensen, K.K. The Scavenger Function of Liver Sinusoidal Endothelial Cells in Health and Disease. Frontiers in physiology 2021, 12, 757469. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic presentation of the experimental protocol. a) Cells were cultured with different cell culture medium formulation and volume, ECM, and cell percentage of the different cell types. b) Table summarizing the samples with their feature combinations (each combination was cultured in parallel). c) All the combinations were analysed with the inverted microscope, the biochemical analyser and ISX. d) The feature combination that most resembled the liver microenvironment was selected, and e) was exposed to IBU, RIF and 5-FU. f) The same analysing methods were used to determine the toxic effect of the drugs in the liver model with the selected feature combination.
Figure 1.
Schematic presentation of the experimental protocol. a) Cells were cultured with different cell culture medium formulation and volume, ECM, and cell percentage of the different cell types. b) Table summarizing the samples with their feature combinations (each combination was cultured in parallel). c) All the combinations were analysed with the inverted microscope, the biochemical analyser and ISX. d) The feature combination that most resembled the liver microenvironment was selected, and e) was exposed to IBU, RIF and 5-FU. f) The same analysing methods were used to determine the toxic effect of the drugs in the liver model with the selected feature combination.
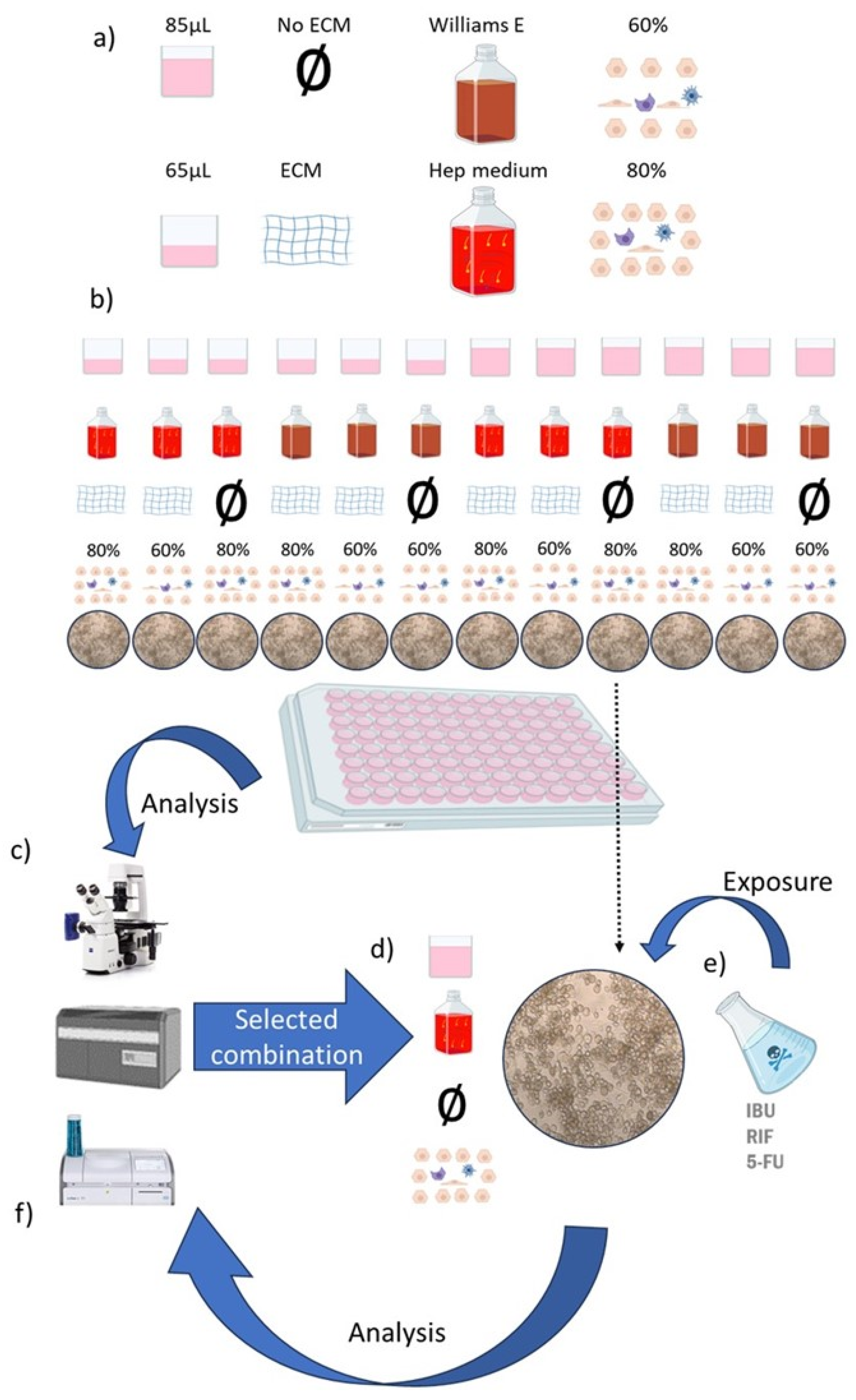
Figure 2.
Observations of cell arrangement and morphology with the inverted microscope. a) A sample cultured with 65 µL in which the cells have migrated towards the centre (5x magnification). b) A sample cultured with 85 µL in which the cells have formed scattered colonies (5x magnification). In both cases, a) and b), we observed the transition of the cells from day 1 to day 5 in a 96-well microplate. c) On the left, cells cultured with ECM and on the right, cells cultured without ECM (20x magnification).
Figure 2.
Observations of cell arrangement and morphology with the inverted microscope. a) A sample cultured with 65 µL in which the cells have migrated towards the centre (5x magnification). b) A sample cultured with 85 µL in which the cells have formed scattered colonies (5x magnification). In both cases, a) and b), we observed the transition of the cells from day 1 to day 5 in a 96-well microplate. c) On the left, cells cultured with ECM and on the right, cells cultured without ECM (20x magnification).

Figure 3.
Viability of cells concerning the different combinations of features (p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***).
Figure 3.
Viability of cells concerning the different combinations of features (p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***).
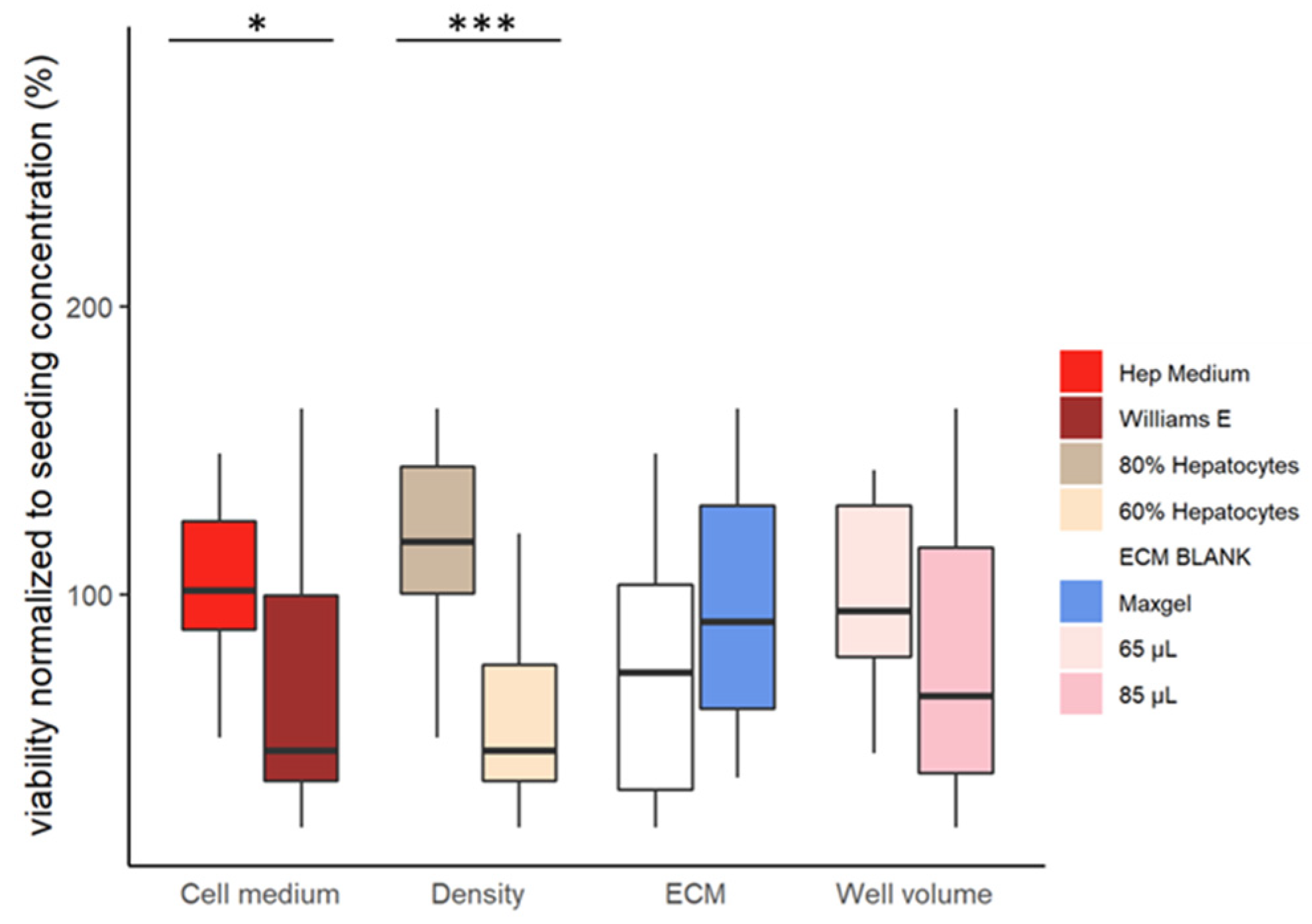
Figure 4.
Values of the liver markers in the various liver models built with different feature combinations. a) Liver marker concentrations. b) Liver marker values normalised to cell number (molecule biomarker per cell). (p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***). We analysed alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), glucose, triglycerides, and albumin.
Figure 4.
Values of the liver markers in the various liver models built with different feature combinations. a) Liver marker concentrations. b) Liver marker values normalised to cell number (molecule biomarker per cell). (p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***). We analysed alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), glucose, triglycerides, and albumin.
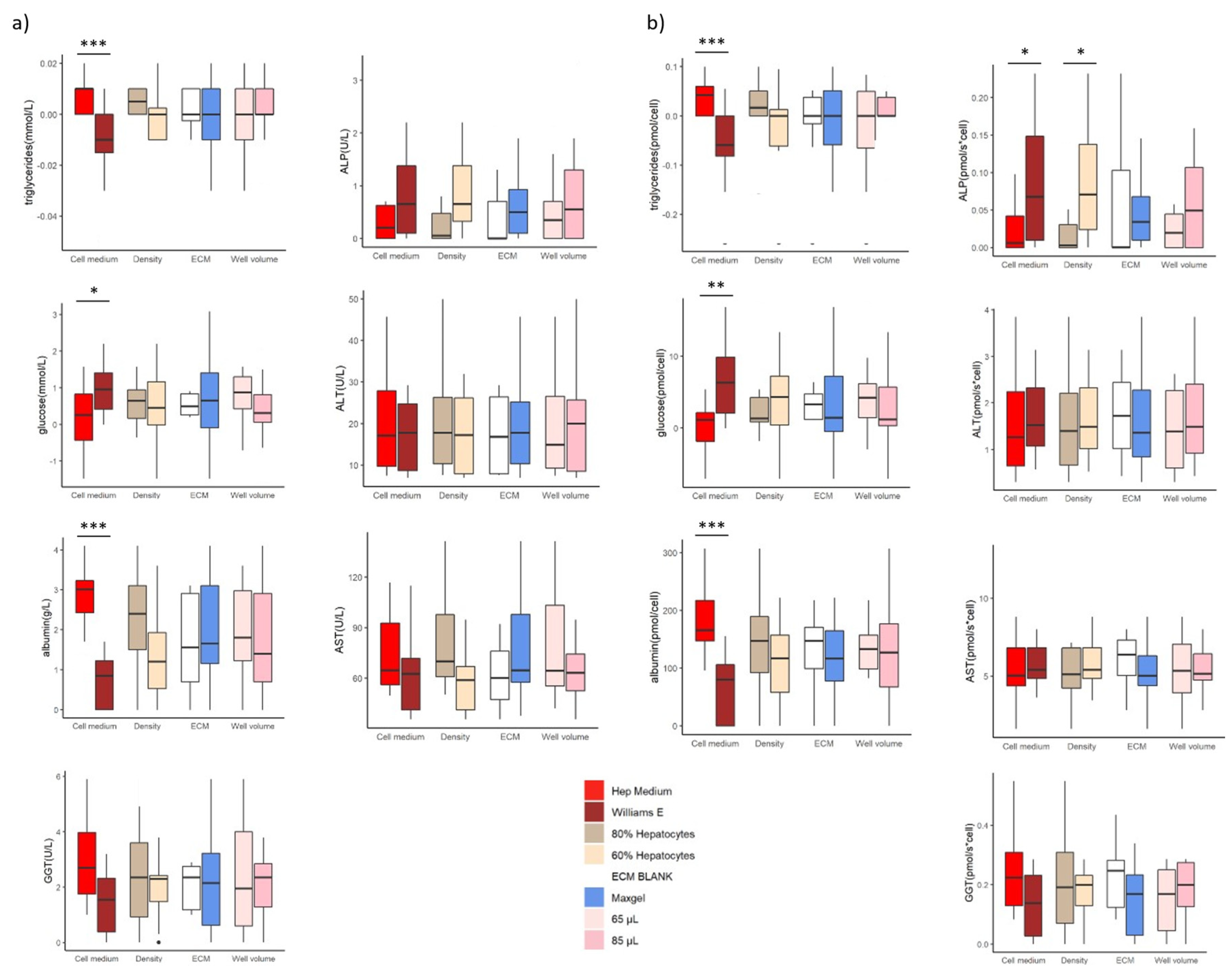
Figure 5.
Evaluation of cell morphology with the inverted microscope (20x magnification) on each cell treatment. a) Untreated, b) RIF, c) IBU, d) 5- FU.
Figure 5.
Evaluation of cell morphology with the inverted microscope (20x magnification) on each cell treatment. a) Untreated, b) RIF, c) IBU, d) 5- FU.

Figure 6.
Viability of samples treated with RIF, IBU, and 5-FU. a) Viability of samples relative to the initial seeding number. b) Viability of each cell type normalised to the untreated sample(p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***). Rifampicin (RIF), ibuprofen (IBU) and 5-fluorouracil (5-FU), liver sinusoidal endothelial cells (LSEC), human stellate cells (HSC).
Figure 6.
Viability of samples treated with RIF, IBU, and 5-FU. a) Viability of samples relative to the initial seeding number. b) Viability of each cell type normalised to the untreated sample(p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***). Rifampicin (RIF), ibuprofen (IBU) and 5-fluorouracil (5-FU), liver sinusoidal endothelial cells (LSEC), human stellate cells (HSC).
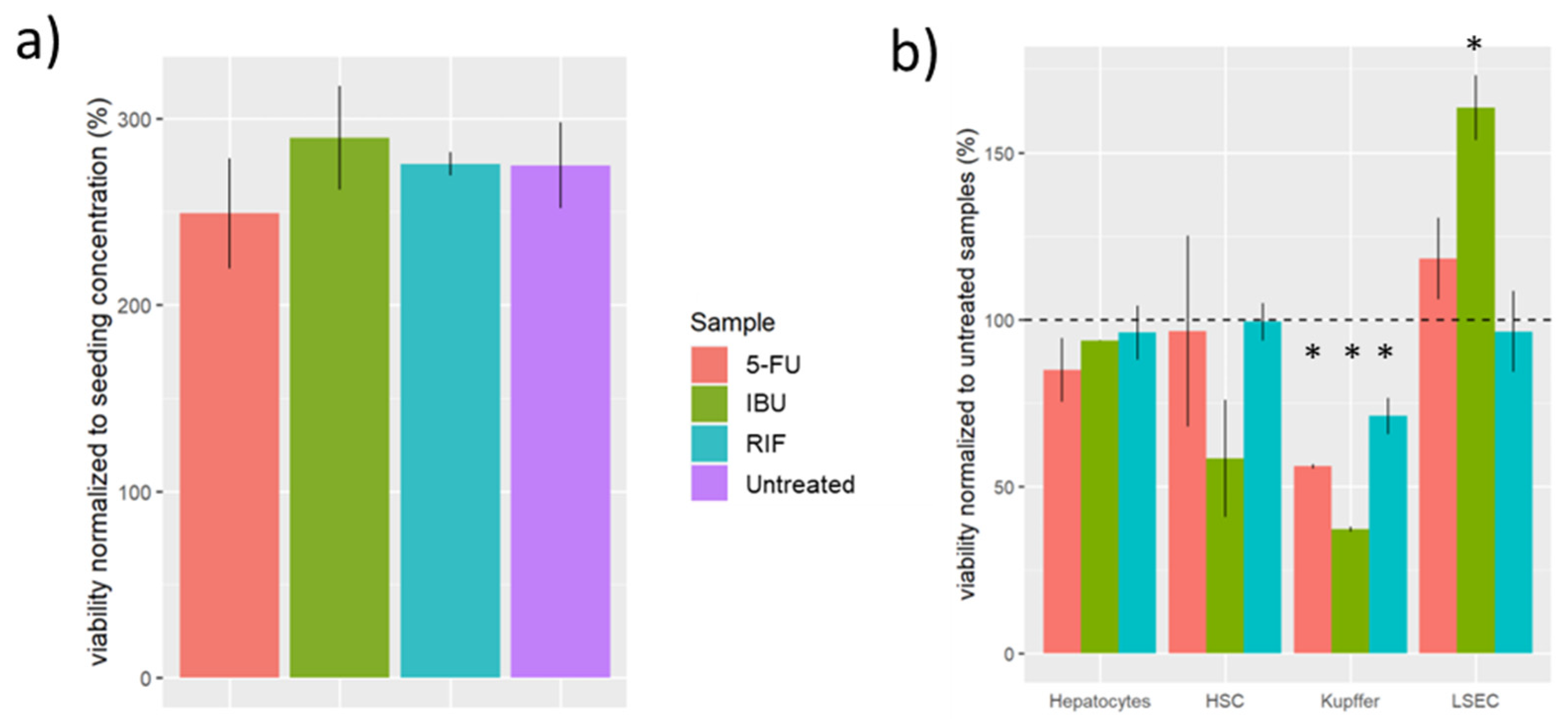
Figure 7.
Clinical markers of samples treated with RIF, IBU, and 5-FU. (p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***). Rifampicin (RIF), ibuprofen (IBU) and 5-fluorouracil (5-FU), liver sinusoidal endothelial cells (LSEC), human stellate cells (HSC).
Figure 7.
Clinical markers of samples treated with RIF, IBU, and 5-FU. (p-value > 0.05, < 0.05 *, <0.01 **, < 0.001 ***). Rifampicin (RIF), ibuprofen (IBU) and 5-fluorouracil (5-FU), liver sinusoidal endothelial cells (LSEC), human stellate cells (HSC).
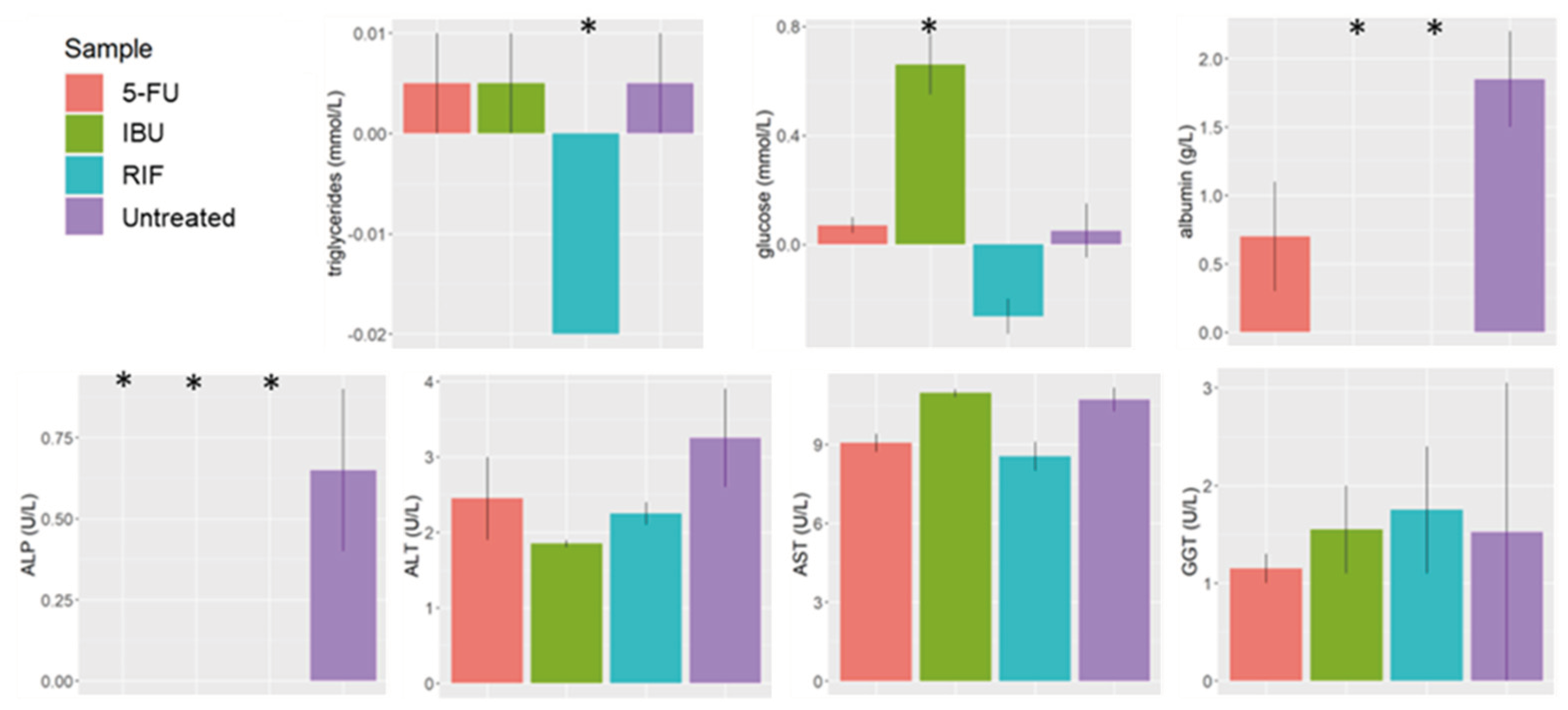

Table 1.
Cell death ratio and population share of each cell type 5 days after exposing the cell-based liver model to hepatotoxic drugs.
Table 1.
Cell death ratio and population share of each cell type 5 days after exposing the cell-based liver model to hepatotoxic drugs.
| Group | Share of the population (%) | Cell death ratio (%) | Share of the population (%) | Cell death ratio (%) |
|---|---|---|---|---|
| LSEC | HSC | |||
| Control | 4%±1 | 2%±0 | 5%±1 | 2%±1 |
| 5-FU | 6%±0 | 3%±0 | 5%±1 | 5%±1 |
| IBU | 8%±1 | 2%±1 | 3%±1 | 5%±1 |
| RIF | 5%±1 | 3%±1 | 5%±0 | 3%±0 |
| Kupffer | Hepatocytes | |||
| Control | 4%±0 | 3%±1 | 88%±1 | 3%±1 |
| 5-FU | 2%±0 | 12%±0 | 87%±1 | 4%±0 |
| IBU | 2%±0 | 12%±2 | 88%±1 | 2%±1 |
| RIF | 3%±0 | 8%±0 | 88%±0 | 2%±0 |
Rifampicin (RIF), ibuprofen (IBU) and 5-fluorouracil (5-FU), liver sinusoidal endothelial cells (LSEC), human stellate cells (HSC).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



