Submitted:
03 April 2024
Posted:
04 April 2024
You are already at the latest version
Abstract
An entomopathogenic nematode, Oscheius tipulae, was isolated from a soil sample. The identification of this species was supported by morphological and molecular markers. The nematode isolate exhibited pathogenicity against different target insects including lepidopteran, coleopteran, and dipteran insects. The virulence of this nematode was similar to that of a well-known entomopathogenic nematode, Steinernema carpocapsae, against the same insect targets. A comparative metagenomics analysis of these two nematode species predicted the existence of a combined total of 272 bacterial species in their intestines, of which 51 bacterial species were shared between the two nematode species. In particular, the common gut bacteria included several entomopathogenic bacteria including Xenorhabdus nematophila, which is known as a symbiotic bacterium to S. carpocapsae. The nematode virulence of O. tipulae to insects was enhanced by an addition of dexamethasone but suppressed by an addition of arachidonic acid, suggesting that the immune defenses of the target insects against the nematode infection is mediated by eicosanoids, which would be manipulated by the symbiotic bacteria of the nematode. Unlike S. carpocapsae, O. tipulae showed high virulence against dipteran insects including fruit flies, onion flies, and mosquitoes. O. tipulae showed particularly high control efficacies against the onion maggot, Delia platura, infesting the Welsh onion in the rhizosphere in both pot and field assays.
Keywords:
Oscheius tipulae
; Steinernema carpocapsae
; metagenomics
; virulence
; immunity
1. Introduction
Entomopathogenic nematodes (EPNs) have come to be widely accepted as some of the most optimal biological control agents to replace chemical control methods in insect pest management [1,2]. Two well-known EPNs include two independently originated families of Heterorhabditidae and Steinernematidae, which exhibit similar parasitic modes of life cycles through a convergent evolution [3]. They are typically found in the soil and search for their hosts based on characteristic behaviors wherein they act as ambushers or cruisers [4]. Ambusher nematodes implement a strategy where they sit and wait in soil in search of hosts whereas cruiser nematodes use active searching to seek out potential target insects using the host-emitted volatile organic compounds [5]. Once they are damaged by herbivores, plant roots release terpenes such as α-pinene, linalool, d-limonene, and pregeijerene, which are detected by these EPNs [6].
Once the infective juveniles (IJs) enter the host insect hemocoel through natural openings such as the mouth, anus, or spiracles, or through direct cuticle penetration, they release their symbiotic bacteria [7]. The symbiosis between the nematodes and their bacteria is species-specific, where Steinernema and Heterorhabditis nematodes respectively have Photorhabdus and Xenorhabdus bacteria in their gut [8]. The bacteria are located in a specific receptacle in the intestine near the esophagus of the nematodes, and they are released by intestinal contraction in response to insect hemolymph factor(s) [9]. In the complexes of S. carpocapsae and X. nematophila, nematode intestine localization (nil) factors A, B, and C are identified as molecular components that explain the specificity between the nematode and bacteria [10]. NilB and NilC are an outer membrane beta barrel protein and periplasmic lipoprotein, respectively [11,12], suggesting their roles in the molecular interactions with host gut epithelium.
The bacteria released in the hemocoel suppress the insect immune responses to protect the bacteria from the fatal cellular and humoral responses, with the intention of killing the insect by septicemia [13]. To suppress the immune responses, these bacteria release several secondary metabolites to inhibit phospholipase A2 (PLA2) [14]. PLA2 catalyzes phospholipids to release free fatty acids, with are typically arachidonic acid or its biosynthetic precursor in insects [15]. AA is then oxygenated to produce prostaglandins, leukotrienes, or eicosatrienoic acid, which are collectively called eicosanoids [16]. Eicosanoids mediate various immune responses in insects [17]. In this way, the inhibitory activity by the bacterial metabolites leads to host immunosuppression. In the insect cadaver, the IJs are then developed to the reproductive stage to produce progeny. After consuming all nutrients in the host insect, the nematodes develop into IJs through re-association with their specific bacteria and then emerge from the insects to look for new hosts.
Nematodes can be effective in controlling coleopteran larvae inhabiting the rhizosphere. For example, one study showed that, in peanut fields, the white grub, Holotrichia parallela, was effectively controlled by the nematodes, S. longicaudum and H. bacteriophora at 10,000 and 5,000 IJs per plant, respectively [18]. Insect pests inhabiting the aquatic environment are also targets of control by nematodes. For example, mosquitoes and tephritid flies have previously been controlled by Steinernema and Heterorhabditis nematodes [19,20].
In addition to Steinernema and Heterorhabditis, other rhabditid nematodes have been evaluated as efficient EPNs, with notable examples including Oscheius tipulae, O. carolinensis [21], O. chongmingensis [22], and O. gingeri [23]. In particular, O. tipulae has been proposed to control tephritid flies due to its high virulence in addition to its previously documented pathogenicity to a lepidopteran insect [24]. This relatively wide host range has led to the nematode, O. tipulae, being proposed an effective biological control agent against insect pests. The present study identified a strain of O. tipulae in Korea and evaluated its pathogenic spectrum and its potential to control insect pests.
2. Materials and Methods
2.1. Test Insects
Altogether, this study used three different lepidopterans (Spodoptera exigua, Maruca vitrata, and Plutella xylostella), three different dipterans (Delia platura, Drosophila melanogaster, and Aedes albopictus), and one coleopteran (Tenebrio molitor) species. The rearing conditions in the laboratory were a temperature of 25 ± 2°C with a relative humidity of 60 ± 5% and a photoperiod of 16:8 h (L:D). The larvae of S. exigua, which were collected from a field in which they were infesting Welsh onion (Allium fistulosum) in Andong, Korea, were reared on an artificial diet [25] and underwent five instars (L1-L5). For bioassay, L4 larvae were used. The larvae of P. xylostella collected from Andong, Korea were reared on fresh cabbage (Brassica rapa) and underwent four instars, in which L4 larvae were used for bioassay. The larvae of M. vitrata, which were collected from an Adzuki bean (Vigna angularis) field in Suwon, Korea, were reared on an artificial diet [26] and underwent five instars. For bioassay, L4 larvae were used. The larvae of T. molitor, which were supplied by Bio Utility (Andong, Korea), were reared on a wheat bran-based diet [27] and underwent ~20 instars [28]. For bioassay, two groups of larvae were selected based on their body length: ~10 mm for young larvae and ~25 mm for old larvae. D. melanogaster adults were purchased from Biological Nara (Seoul, Korea) and reared on an artificial diet (Hansol Tech, Yangsan, Korea). The larvae of D. platura were collected from a Welsh onion field in Andong, Korea and reared on bulb onion. The larvae of A. albopictus were donated by Professor Donghun Kim in Kyungpook National University (Sangju, Korea). First instar larvae were reared in a plastic box (12 × 8 × 5 cm) containing 1 L distilled water and provided with a fish food mini pellet (Ilsung, Daegu, Korea), while adults were maintained on 10% sugar solution using a cotton plug. Female mosquitoes were fed mouse blood as described by Choi et al. [29]. For laboratory bioassays of the dipteran insects, the penultimate larval instar (L3) was used.
2.2. Culturing Nematodes of Two Steinernema carpocapsae Strains
Two S. carpocapsae strains, Sc-AD and Sc-DR, were respectively collected from Andong and Dongrae in Korea [30]. Sc-AD was multiplied using S. exigua host while Sc-DR was cultured in T. molitor. The infection and collection of the nematodes were conducted following the method described by Park et al. [30]. Briefly, approximately 200 IJs in 500 μL distilled water were applied to Petri dishes (9 cm diameter, 3 cm height) that each contained an individual insect larva. Treated larvae were reared with artificial diet under the laboratory rearing conditions. At 5 days after the nematode infection, the emerging IJs were collected from the dead larvae using a White trap and then transferred to a 6 cm culture flask (Bibby Sterilin, Staffordshire, UK). IJs were kept in 10 ± 1°C in distilled water under darkness until being used.
2.3. Chemicals
Arachidonic acid (AA: 5,8,11,14-eicosatetraenoic acid) and dexamethasone (DEX: (11β,16α)-9-fluoro-11,17,21-trihydroxy-16-methylpregna-1,4-diene-3) were obtained from Sigma-Aldrich Korea (Seoul, Korea) and dissolved in dimethyl sulfoxide (DMSO). Chelex 100 Resin was purchased from Bio-Rad (Hercules, CA, USA) and used for DNA extraction. Luria-Bertani (LB) medium was purchased from MBcell (Seoul, Korea). Phosphate-buffered saline (PBS) solution was prepared with 100 mM phosphoric acid and adjusted to pH 7.4 with 1 N NaOH. A commercial version (Captin®, Kyungnong, Seoul, Korea) of fluxamethamide, an insecticide, was purchased from a local distributor.
2.4. Soil Samplings and EPN Collection
Soils were collected from three different field sites (SC1, SC2, and YS) located at Andong, Korea (Figure S1). The sampling sites were host to different plants, including Gingko biloba, Quercus dentata, Cucumis sativus, and Juglans regia. At each site, approximately 200-400 g of each soil sample was gathered using a hand shovel and stored in a plastic bag once a week from March through September.
Live nematodes were collected from these gathered soil samples using a modified Baermann funnel technique, as described by Lee et al. [31]. The nematode suspensions were topically applied on the larvae of T. molitor and S. exigua. Subsequent dead larvae were collected and placed onto a White trap that was kept at room temperature to facilitate the emergence of IJs [32]. The IJs were multiplied using S. exigua that were collected using the method described above.
2.5. Nematode Identification with Morphological Characteristics
For morphological characterization, 20 IJs were randomly selected from T. molitor cadavers. The nematodes were fixed using warm water (60°C) in 4% formaldehyde solution and mounted on slide glass with the addition of glycerin. A glass rod was used to prevent sample flattening while the cover glass was overlaid. Observations were made using a stereoscopic microscope (Stemi SV11, Zeiss, Jena, Germany) and a light microscope (Eclipse 80i, Nikon, Tokyo, Japan) at 50× magnification.
2.6. Nematode Identification with Molecular Characters
To begin, a sample of about 0.5 g of IJs was homogenized in 100 μL of 5% Chelex-100 (Sigma-Aldrich, Seoul, Korea) and incubated in water bath at 100°C for 10 min. After centrifugation at 12,500 × g for 5 min, the extracted DNA was transferred to a new tube and stored at -20°C. A rRNA gene covering two internal transcribed spaces (ITS-1 and ITS-2) was amplified using universal primers: 5ʹ-TTGATTACGTCCCTTGCCCTT-3ʹ and 5ʹ-TTTCACTCGCCGTTACTAAGG-3ʹ [33]. The PCR mixture consisted of 5 μL of 10× PCR buffer, 15 μL of deionized water, 1 μL of dNTP, 1 μL of Taq polymerase, 1 μL of forward primer, 1 μL of reverse primer, and 1 μL of template DNA (~100 ng). PCR comprised of 35 amplification cycles, where each cycle was programmed to consist of 1 min at 94°C for denaturation, 1 min at 46°C for annealing, and 1 min at 72°C for extension. After cloning into pCR2.1 cloning vector, the PCR product was bidirectionally sequenced with M13 forward and reverse universal primers by Macrogen sequencing company (Seoul, Korea). The obtained nucleotide sequences were analyzed using the BlastN program from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov) to match the most similar sequences of specific species. Phylogenetic tree analysis was conducted using a Neighbor- Joining method with MEGA6 [34]. Bootstrap values on the branches were estimated with 1,000 repetitions.
2.7. Nematode Virulence Test
The insecticidal activity of the nematode isolate was determined by applying different concentrations (0, 100, 200, 400 IJs/larva) to filter paper in a 9-cm diameter Petri dish containing 10 larvae. The experimental unit (i.e., each Petri dish) was replicated three times per nematode concentration. For mosquito bioassay, L3 larvae of A. albopictus were bathed in 4 mL of distilled water and then placed in 8 mL plate well (4 cm in diameter). Four concentrations of nematodes were tested (0, 50, 100, and 200 IJs/mL). Distilled water was used for a control. For this bioassay, three lepidopteran species (S. exigua, M. vitrata, and P. xylostella) at L4 instar and D. melanogaster at L3 instar were used. Further, the larvae of a coleopteran insect (T. molitor) with approximately 25 mm body length were used in the assay. Treated larvae were provided with diets and kept in the previously described rearing conditions. Mortality was assessed at 4 days after treatment (DAT).
2.8. Nematode Life Cycle Assessment
The life cycle of the nematode isolate was examined using M. vitrata larvae as hosts. Each L4 larva was infected with 50 IJs per larva as described above. The treated insects were dissected daily for 10 days. Each time point was replicated with three different infected larvae and each nematode life stage was counted. The time to emergence, i.e., the number of days after infection to the moment of IJ release from the cadaver, was determined on the White traps through daily observation until the first infective juveniles started to come out of the cadavers.
2.9. Isolation of the Nematode-Associated Bacteria
The bacterial extraction from the nematodes was conducted following the method described by Loulou et al. [35]. Briefly, approximately 200 IJs were washed three times with sterile PBS and incubated in 1% sodium hypochlorite (Yakuri Pure Chemicals, Kyoto, Japan) in PBS for 5 min with gentle agitation. After removing the incubation solution, the nematode sample was incubated again with 1% sodium hypochlorite. After several washes with sterile PBS, the nematodes were sonicated with a power strength of 100 for 1 min using a ultrasonicator (MS73, Bandelin Sonoplus, Heinrichstraße, Berlin, Germany). After centrifugation at 650 × g for 10 min at 4°C, the supernatant was used for bacterial culture or metagenomics analysis. For bacterial culture, the extracts were plated on LB agar plates for 24-48 h at 28°C. The resulting colonies were then classified into three types based on the colony shape: fuzzy, small, and big. These colonies were used for subsequent virulence testing.
2.10. Bacterial Virulence Assessment
To assess the insecticidal activities of the isolated bacteria, freshly overnight cultured bacterial cells were washed three times with sterilized PBS through centrifugation at 8,000 rpm for 2 min at 4°C, and bacterial pellets were resuspended in PBS. After counting the bacterial cells, the different bacterial concentrations (0, 101, 102, 103, 104, and 105 colony-forming unit (cfu)/larva) were prepared through a serial dilution and injected into the L5 larvae of M. vitrata. One microliter of bacterial solution was injected into the larvae hemocoel using a 10 μL micro syringe (Hamilton, Reno, NV, USA). Ten surface-sterilized L5 larvae were injected per bacterial concentration. The control larvae were injected with PBS buffer. The larvae were placed in sterile plastic boxes and kept in the dark at 25°C; insect mortality was then determined at 72 h post-injection. All assays were replicated three times.
2.11. Metagenomics Analysis of the Nematode-Associated Bacteria
Three different nematodes were used in this metagenomics analysis: Sc-DR, Sc-AD, and O. tipulae (Ot). The bacteria were extracted while following the method described above. The DNA was amplified for the V3-V4 region of the 16S rRNA gene with the Herculase II Fusion DNA Polymerase kit using 27-F (5ʹ-TTGATTACGTCCCTTGCCCTT-3ʹ and 1492-R (5ʹ-TTTCACTCGCCGTTACTAAGG-3ʹ). The library preparation was done using the DNA Polymerase Nextera XT kit, which is suitable for working with small genomes. Indexed libraries were then joined at equimolar concentrations, and the product was sequenced with a MiSeq device, and it was set to sequence the PCR product 300 base pairs from both ends. The MiSeq benchtop sequencer output is over 100,000 reads with a length of 300 and a total of more than 30 million bases. Upon completion of this sequencing, Illumina MiSeq raw data were sorted by sample utilizing the index sequences, and paired-end FASTQ files were generated for each sample. According to Martin [36], Cutadapt v3.2 was used to eliminate sequencing adapter and primer sequences. The DADA2 package in R v4.0.3 was used to rectify errors in the amplicon sequencing process [37]. Briefly, paired-end reads with expected errors exceeding 2 were excluded. Post-preprocessing, an error model was constructed for each batch to eliminate sample noise. The corrected paired-end sequences were merged into a single sequence, and chimera sequences were filtered out using DADA2’s consensus method, which ultimately resulted in the formation of Amplicon Sequence Variants (ASVs). ASVs shorter than 350 bp were removed. Moreover, for microbial community comparison analysis, subsampling was utilized for normalization via QIIME v1.9 [38,39] based on the minimum read count across all samples. Each ASV sequence was taxonomically annotated by BLAST+ v2.9.0 against the reference database (NCBI 16S Microbial DB), where taxonomy information was assigned based on the organism with the highest similarity. Utilizing the abundance and taxonomy data of ASVs, various comparative analyses of microbial communities were conducted using QIIME, and species diversity and evenness within microbial communities were evaluated using Shannon and Simpson index [40].
2.12. Statistical Analysis
The means and variances of the treatments were analyzed by one-way ANOVA by PROC GLM of the SAS program [41]. All the data were plotted in terms of mean ± standard error using Sigma Plot (Systat Software, Inc., Point Richmond, CA, USA) and GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). The least squared difference (LSD) test was used to compare the means with a Type I error of 0.05.
3. Results
3.1. Identification of an EPN, Oscheius tipulae
A nematode population killing bait insect was isolated from soil collections in Andong, Korea (Figure 1). The soil samples under a walnut—J. regia—tree in site SC1 were rich in the nematode population. Using a White trap, thousands of infective juveniles (IJs) were collected from a dead coleopteran insect, T. molitor (Figure 1A). Male and female adults was examined for morphological identification (Figure 1B). Compared to other EPN genera (Steinernema and Heterorhabditis), the isolate was similar (indicated with ‘+’) to Oscheius in the six morphological characters (Figure 1C). Within Oscheius, the four morphological characters (indicated with ‘+’) of the isolate matched those of O. tipulae (Figure 1D). For molecular identification, ITS1 and ITS2 of the isolates, which respectively covered partial 18S and 28S rRNA regions, were sequenced (Figure 2A). A phylogenetic analysis indicates that the isolate was clustered with other O. tipulae isolates and distinct from other EPNs such as Steinernema and Heterorhabditis spp. (Figure 2B). Thus, both the morphological and molecular characteristics of the isolate indicate that the isolate is O. tipulae.
3.2. Biological Characteristics of O. tipulae
To determine the developmental periods of different life stages of O. tipulae, the nematodes were isolated from the infected host at every 24 h. The nematodes spent four larval instars (L1-L4) for about four days and became adults in T. molitor (Figure 3A). After 7 days post-infection (pi), IJs (= L3 stage) began to come out of the dead host. In total, ~10,000 IJs in M. vitrata and ~6,000 IJs in T. molitor emerged from each larva for 3 days (Figure 3B).
Long-term storage of the IJs of O. tipulae would be necessary to apply this nematode to control insect pests. Two parameters for storage were assessed by analyzing storage temperature and preservative. The IJs were kept in low temperatures and monitored in terms of survival (Figure 3C). After two weeks, the IJs exhibited a significant (P < 0.05) decrease in survival at 20°C. However, low temperatures such as 10 °C and 15°C kept more larvae alive while low temperatures such as 0 °C and 5°C were found to be detrimental to nematode survival. The optimal temperature to preserve the IJs was 10°C. To increase the survival at 10°C, glycerol was added to the distilled water, but it did not significantly increase the survival compared to IJs in the distilled water (Figure 3D). PBS was not found to be favorable for IJ nematode storage.
3.3. Pathogenicity of O. tipulae against Different Insect Taxa
The nematode isolate of O. tipulae was analyzed in its pathogenic host range (Figure 4). The IJs were applied to the diets of four different insects, and these groups showed significantly different mortalities compared to the controls (Figure 4A). With an increase in the treated IJ concentration, the virulence of O. tipulae increased in a dose-dependent manner against the four different insects. However, there was a variation in the virulence among the different insects, where a coleopteran T. molitor was highly susceptible compared to lepidopteran (S. exigua, P. xylostella, and M. vitrata) insects (Figure 4B). At 400 IJs/larva, the highest mortality was observed in T. molitor. The speed-to-kill of the nematodes was measured in a median lethal time (LT50), and the different species showed relatively similar rates, aside from P. xylostella (Figure 4C). The nematode also showed its preference against larvae to pupae and adults in T. molitor (Figure 4D).
3.4. Entomopathogenic Bacterial Isolates Derived from O. tipulae
To analyze the entomopathogenicity, the bacteria were isolated from the IJs of O. tipulae (Figure 5A). In particular, the bacteria were isolated from surface-sterilized nematodes. Three different colony forms of big (‘B’), small (‘S’), and fuzzy (‘F’) shapes were evident. These bacteria were injected into larvae of M. vitrata at 103 cfu per larva, and they exhibited pathogenicity. When their virulence was measured by LD50, the fussy shape colony was highly virulent compared to other colony forms (Table 1). These three bacterial colonies were identified by their 16S rRNA sequences (Figure 5B). The fussy colony was Serratia marsescens while the two round shape bacteria were S. nematodiphila and Klebsiella pneumonia with identities exceeding 96%.
To test how the entomopathogenicity of O. tipulae was affected by the bacterial virulence, the immunity induced by eicosanoid in response to the bacterial infection was assessed (Figure 5C). Dexamethasone (DEX) has previously been shown to inhibit PLA2 to shut down eicosanoid biosynthesis in insects [42]. The nematode virulence was found to be highly enhanced by the injection of DEX in M. vitrata or T. molitor larvae. By contrast, the injection of arachidonic acid (AA), which is the catalytic product of PLA2, suppressed the nematode virulence in both larval species. These results suggest that entomopathogenic bacteria play a role in nematode virulence.
3.5. Metagenomics Analysis of the Bacteria Associated with IJs of O. tipulae
The identification of the entomopathogenic bacteria derived from the IJs of O. tipulae suggested the additional presence of the entomopathogens. To test this hypothesis, the bacteria derived from the IJs were analyzed using a metagenomics approach and compared with the bacterial fauna of the other entomopathogenic nematode, S. carpocapsae (Figure 6). Two different strains (Sc-AD and Sc-DR) of S. carpocapsae were multiplied on different insect hosts: Sc-AD from S. exigua insect host and Sc-DR from T. molitor insect host. For reference, O. tipulae were multiplied using T. molitor insect host. The metagenomics analysis showed that 276 bacterial species were ultimately predicted from these three nematode isolates (Figure 6A) but different among the three bacterial isolates (Table S1). They also showed variations in bacterial composition (Figure 6B). Compared to O. tipulae, both S. carpocapsae isolates had higher bacterial diversity, as measured by Shannon and Gini-Simpson indices (Figure 6C). The total numbers of bacterial species were different even within the same species, where Sc-AD had more than two-fold the bacterial species of Sc-DR. More than 10% bacterial species were commonly shared among the three nematode isolates (Table S2). Surprisingly, the common bacterial fauna included Xenorhabdus nematophila, which is known to be associated with S. carpocapsae in a species-specific manner [42]. This suggests that the IJs of O. tipulae possess the entomopathogenic bacteria. By contrast, the metagenomics analysis showed that the IJs of O. tipulae possess 32 unique bacteria including K. pneumonia (Table S3), which showed entomopathogenicity in our bacterial isolation (see above). The entomopathogenic bacteria predicted from the metagenomics analysis showed that at least seven species that were found in O. tipulae were shared with those of S. carpocapsae aside from K. pneumonia.
3.6. Virulence of O. tipulae against Dipteran Insect Pests
To apply O. tipulae to insect pest control, its virulence was assessed against dipteran insect pests living in humid conditions, which is the where nematodes inhabit (Figure 7). When the virulence of O. tipulae was compared with that of S. carpocapsae, there was little difference in the virulence against lepidopteran and coleopteran insects (Figure 7A). However, O. tipulae were found to be more potent than S. carpocapsae against dipteran insects. Among three dipteran insects used in this virulence test, the fruit fly (D. melanogaster) and the onion fly (D. platura) were more susceptible to O. tipulae compared to a mosquito (A. albopictus) (Figure 7B). The susceptible flies suffered from dose-dependent mortality of the nematodes.
3.7. Control Efficacy of O. tipulae against D. platura
The nematode, O. tipulae, was further analyzed for its efficacy in controlling against D. platura, which is an agricultural insect pest that damages young Welsh onion at the rhizosphere [43]. In a pot assay, the seedlings were infected with D. platura and had different concentrations of IJs applied (Figure 8A). Significant (P < 0.05) control efficacies were observed above 100 IJs per plant. Dose-dependent control efficacy was shown, with high control efficacy above 80% observed at 1,000 IJs per plant. This IJ dose was applied to Welsh onions cultivated in a greenhouse (Figure 8B). A chemical control using fluxamethamide (FLX) insecticide was used as a positive control. FLX treatment showed fast control efficacy even at 3 days after treatment. At 7 days after treatment, O. tipulae showed 85% control efficacy whereas the chemical insecticide treatment showed 95% control efficacy.
4. Discussion
Entomopathogenic nematodes have been successfully used to control insect pests. Although there is substantial nematode species diversity, which has been estimated at ~30,000 described species [44], two families of EPNs have been the main objects of focus since the first proposal [45]: Steinernematidae and Heterorhabditidae. This study isolated a non-model EPN species and analyzed its application in the control of insect pests.
An EPN isolate from a soil sample killed lepidopteran and coleopteran insects and was found to multiply in insect cadavers. It was identified as Oscheius tipulae based on analyses of both morphological and molecular characters. The species was named as such because it was first isolated from a dipteran insect, Tipula paludosa [46]. Our phylogenetic analysis using ITS sequences showed that O. tipulae was clustered with other Oscheius spp. and Heterorhabditis spp. and separated from Steinernema spp. This supports the nematode classification [47] that classified two separate suborders by clustering Oscheius and Heterorhabditis into Rhabditina while Steinernema was classified into Tylenchina in the Order Rhabditida.
The infective juveniles (= 3rd instar) developed into adults in 4-5 days. O. tipulae has previously been shown to have a fast developmental rate in in vitro culture, where it develops 3-4 days at room temperature after four juvenile stages [48]. Some delay in the parasitic life cycle might consist of elapsed time taken during the infection stage, such as penetration to insect hemocoel and suppression of host immune defense.
The insect-killing activity of O. tipulae was unusual because nematodes have been known to be bacterivorous and used as animals in genetic research due to their close relationship with a well-known Caenorhabditis elegans and its easy culture [49,50]. The insecticidal activity of O. tipulae was assessed in a Mediterranean fly, Ceratitis capitata, in which the nematodes killed all the immature stages of the flies, especially with high susceptibility of pupae, even at a few IJs per pupa [24]. This study suggested that O. tipulae could be used as a biological control agent against C. capitata flies. In Oscheius, about 30 species are recorded, of which more than 13 are known to kill insects [51]. O. tipulae appear to occur in a wide range of soil environments. A survey of EPNs using White trap baited with Galleria mellonella collected O. tipulae from agricultural soils in Brazil in addition to Heterorhabditis and Steinernema [52]. Our current study showed that O. tipulae was obtained from a non-agricultural area and showed a wide host range, including lepidopteran, coleopteran, and dipteran insects, for its parasitic life.
Suppression of host immunity was required for the insecticidal activity of O. tipulae. Eicosanoids mediate various immune responses in insects against virus, bacterial, and fungal infections [17]. PLA2 catalyzes the biosynthetic pathway of the eicosanoids by releasing arachidonic acid (AA) as the biosynthetic substrate [53]. Dexamethasone treatment significantly increased the nematode insecticidal activity whereas AA treatment suppressed the activity. This suggests that eicosanoids play a crucial role in defending against nematode infection. For EPNs such as Heterorhabditis and Steinernema, their symbiotic bacteria suppress the target insect immune responses by inhibiting PLA2 [14]. The enzyme activity is inhibited by the secondary metabolites released by the nematode-specific symbiotic bacteria, Photorhabdus and Xenorhabdus [54]. These findings led us to examine the bacteria associated with O. tipulae: Three different bacteria were isolated from the surface-sterilized IJs of O. tipulae and showed insecticidal activities. These bacteria were identified to be S. marsescens, S. nematodiphila, and K. pneumonia from 16S rRNA sequences. They were also predicted from the metagenomics analysis of O. tipulae. Surprisingly, the metagenomics analysis also suggested that X. nematophila, which is specific to S. carpocapsae, is associated with O. tipulae. Our previous work showed that X. nematophila produces at least eight different secondary metabolites, inhibiting PLA2 [55]. These findings suggest that the insecticidal activity of O. tipulae may be caused by its associated bacteria. However, it is too premature to conclude that X. nematophila is associated with O. tipulae until the bacteria can be isolated. Our current metagenomics analysis showed that two strains of the same species, S. carpocapsae, exhibited a bacterial fauna that was presumably attributable to their insect hosts. Even though there were differences in the two strains of S. carpocapsae, O. tipulae had lower species diversity than S. carpocapsae. The relatively low bacterial species of O. tipulae occurred in both entomopathogenic and non-entomopathogenic bacteria from the metagenomics analysis.
The use of EPNs has been regarded as one of the highly promising biological control strategies due to their capacity to efficiently control different insect pests with a well-developed mass production technique [56]. Our current study applied the nematode isolate identified as O. tipulae to control an onion fly, Delia platura, of which larvae damages the seedlings of Welsh onions, particularly at the rhizosphere [57]. O. tipulae gave significant insecticidal activities to other dipteran insects, including a mosquito, A. albopictus. This mosquito showed high resistance to the other EPN, S. carpocapsae, which did not show more than 40% mosquitocidal activity. The resistance of A. albopictus to S. carpocapsae might be a cellular immune defense of the mosquito in exhibiting hemocytic encapsulation around the IJs infected to the mosquito hemocoel [58]. This then leads to the question of why O. tipulae can evade or suppress the mosquito immune responses. It could be attributable to be the immunosuppressive activity of the nematode-associated bacteria by releasing secondary metabolites. This needs to be explored in a subsequent study. Our current study also showed that O. tipulae demonstrated outstanding control efficacy to Drosophila melanogaster. These results suggest that O. tipulae exhibits a wide range of pathogenic spectrum against various insect pests, which is a useful characteristic for its application to biological control. Our current study also showed the potential for the long-term storage of O. tipulae using 10°C in distilled water. The storability may be enhanced by formulating IJs in alginate gel to increase survival without impairing infectivity [59].
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: A map indicating soil sampling sites in Andong, Korea; Table S1: List of the predicted bacterial species of three nematode isolates according to a metagenomics analysis: O. tipulae (Ot), S. carpocapsae Dongrae (Sc-DR), and S. carpocapsae Andong (Sc-AD); Table S2: List of the common bacteria shared by three nematode isolates according to a metagenomics analysis: O. tipulae (Ot), S. carpocapsae Dongrae (Sc-DR), and S. carpocapsae Andong (Sc-AD); Table S3: List of 32 bacterial species uniquely detected in the IJs of O. tipulae.
Author Contributions
Conceptualization, Y.K., E.A., M.E.; Methodology, Y.K., E, A. M.E., J.K., G.J.; formal analysis, E.A., M.E., J.K., G.J.; Data curation and software, E.A., M.E., J.K., G.J.; Investigation, Y.K. E.A., M.E., J.K., G.J.; Funding acquisition, Y.K.; supervision, Y.K.; writing—original draft, Y.K., E.A; writing—review & editing, Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant (No. 2022R1A2B5B03001792) from the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning, Republic of Korea. This study was also funded by a research grant from Andong National University.
Data Availability Statement
The data presented in this study is available in the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Akhurst, R.J. Neoaplectana species: specificity of association with bacteria of the genus Xenorhabdus. Exp. Parasitol. 1983, 55, 258–263. [CrossRef]
- Shapiro-Ilan, D.I.; Han, R.; Dolinksi, C. Entomopathogenic nematode production and application technology. J. Nematol. 2012, 44, 206–217.
- Trejo-Meléndez, V.J.; Ibarra-Rendón, J.; Contreras-Garduño, J. The evolution of entomopathogeny in nematodes. Ecol. Evol. 2024, 14, e10966. [CrossRef]
- Bal, H.K.; Grewal, P.S. Lateral dispersal and foraging behavior of entomopathogenic nematodes in the absence and presence of mobile and non-mobile hosts. PLoS ONE. 2015, 10, e0129887. [CrossRef]
- Zhang, X.; Li, L.; Kesner, L.; Robert, C.A.M. Chemical host-seeking cues of entomopathogenic nematodes. Curr. Opin. Insect Sci. 2021, 44, 72–81. [CrossRef]
- Frankenstein, D.; Luu, M.S.; Luna-Ayala, J.; Willett, D.S.; Filgueiras, C.S. Soil moisture conditions alter behavior of entomopathogenic nematodes. J. Sci. Food Agric. 2024. [CrossRef]
- Kaya, H.K.; Gaugler, R. Entomopathogenic nematodes. Annu. Rev. Entomol. 1993, 38, 181–206.
- Chaston, J.M.; Suen, G.; Tucker, S.L.; Andersen, A.W.; Bhasin, A.; Bode, E.; Bode, H.B.; Brachmann, A.O.; Cowles, C.E.; Cowles, K.N.; et al. The entomopathogenic bacterial endosymbionts Xenorhabdus and Photorhabdus: convergent lifestyles from divergent genomes. PLoS ONE. 2011, 6, e27909. [CrossRef]
- Snyder, H., Stock, S.P.; Kim, S.K.; Flores-Lara, Y.; Forst, S. New insights into the colonization and release processes of Xenorhabdus nematophila and the morphology and ultrastructure of the bacterial receptacle of its nematode host, Steinernema carpocapsae. Appl. Environ. Microbiol. 2007, 73, 5338–5346.
- Heungens, K.; Cowles, C.E.; Goodrich-Blair, H. Identification of Xenorhabdus nematophila genes required for mutualistic colonization of Steinernema carpocapsae nematodes. Mol. Microbiol. 2002, 45, 1337–1353. [CrossRef]
- Cowles, C.E.; Goodrich-Blair, H. Characterization of a lipoprotein, NilC, required by Xenorhabdus nematophila for mutualism with its nematode host. Mol. Microbiol. 2004, 54, 464–477. [CrossRef]
- Bhasin, A.; Chaston, J.M.; Goodrich-Blair, H. Mutational analyses reveal overall topology and functional regions of NilB, a bacterial outer membrane protein required for host association in a model of animal-microbe mutualism. J. Bacteriol. 2012, 194, 1763–1776. [CrossRef]
- Eleftherianos, I.; Shokal, U.; Yadav, S.; Kenney, E.; Maldonado, T. Insect immunity to entomopathogenic nematodes and their mutualistic bacteria. Curr. Top. Microbiol. Immunol. 2017, 402, 123–156.
- Kim, Y.; Ji, D.; Cho, S.; Park, Y. Two groups of entomopathogenic bacteria, Photorhabdus and Xenorhabdus, share an inhibitory action against phospholipase A2 to induce host immunodepression. J. Invertebr. Pathol. 2005, 89, 258–264. [CrossRef]
- Hasan, M.A., Ahmed, S., Mollah, M.M.I., Lee., Kim, Y. Variation in pathogenicity of different strains of Xenorhabdus nematophila; Differential immunosuppressive activities and secondary metabolite production. J. Invertebr. Pathol. 2019, 166, 107221.
- Kim, Y.; Stanley, D. Eicosanoid signaling in insect immunology: new genes and unresolved issues. Genes 2021, 12, 211. [CrossRef]
- Kim, Y.; Ahmed, S.; Stanley, D.; An, C. Eicosanoid-mediated immunity in insects. Dev. Comp. Immunol. 2018, 83, 130–143. [CrossRef]
- Guo, W.; Yan, X.; Zhao, C.; Han, R. Efficacy of entomopathogenic Steinernema and Heterorhabditis nematodes against white grubs (Coleoptera: Scarabaeidae) in peanut fields. J. Econ. Entomol. 2013, 106, 1112–1117. [CrossRef]
- Cardoso, D.D.O.; Gomes, V.M.; Dolinski, C.; Souza, R.M. Potential of entomopathogenic nematodes as biocontrol agents of immature stages of Aedes aegypti. Nematoda 2015, 2, e092015. [CrossRef]
- Godjo, A.; Zadji, L.; Decraemer, W.; Willems, A.; Afouda, L. Pathogenicity of indigenous entomopathogenic nematodes from Benin against mango fruit fly (Bactrocera dorsalis) under laboratory conditions. Biol. Control. 2018, 117, 68–77. [CrossRef]
- Ye, W.; Foye, S.; MacGuidwin, A.E.; Stephan, S. Incidence of Oscheius onirici (Nematoda: Rhabditidae), a potentially entomopathogenic nematode from the marshlands of Wisconsin, USA. J. Nematol. 2018, 50, 1–9. [CrossRef]
- Zhang, C.; Liu, J.; Xu, M.; Sun, J.; Yang, S.; An, X.; Gao, G.; Lin, M.; Lai, R.; He, Z.; Wu, Y.; Zhang, K. Heterorhabditidoides chongmingensis gen. nov., sp. nov. (Rhabditida: Rhabditidae), a novel member of the entomopathogenic nematodes. J. invertebr. Pathol. 2008, 98, 153–168. [CrossRef]
- Pervez, R.; Eapen, S.J.; Devasahayam, S.; Jacob, T.K. A new species of entomopathogenic nematode Oscheius gingeri sp. n. (Nematoda: Rhabditidae) from ginger rhizosphere. Arch. Phytopathol. Plant Proc. 2013, 46, 526–535.
- Loulou, A.; M’saad, G. M.; Muller, A.; Bhat, A.H.; Abolafia, J.; Machado, R.; Kallel, S. Potential of Oscheius tipulae nematodes as biological control agents against Ceratitis capitata. PLoS ONE 2022, 17, 0269106. [CrossRef]
- Goh, H.G.; Lee, S.G.; Lee, B.P.; Choi, K.M.; Kim, J.H. Simple mass-rearing of beet armyworm, Spodoptera exigua (Hübner) (Lepidoptera: Noctuidae), on an artificial diet. Korean J. Appl. Entomol. 1990, 29, 180–183.
- Jung, J.K.; Seo, B.Y.; Cho, J.R.; Kwon, Y.H.; Kim, G.H. Occurrence of lepidopteran insect pests and injury aspects in Adzuki bean fields. Korean J. Appl. Entomol. 2009, 48, 29–35. [CrossRef]
- Michael, P.; Susanne, S-E.; Silvia, A.; Stefan, H.; Cornelia, H.; Erwin, S.; Wolfgang, K. Wheat bran-based biorefinery: composition of wheat bran and strategies of functionalization. Food Sci. Technol. 2014, 56, 211–221.
- Kröncke, N.; Wittke, S.; Steinmann, N.; Benning, R. Analysis of the composition of different instars of tenebrio molitor larvae using near-infrared reflectance spectroscopy for prediction of amino and fatty acid content. Insects 2023, 14, 310. [CrossRef]
- Choi, D.; Al Baki, M.A.; Ahmed, S.; Kim, Y. Aspirin inhibition of prostaglandin synthesis impairs mosquito egg development. Cells 2022, 11, 4092. [CrossRef]
- Park, Y.; Kim, Y.; Yi, Y. Identification and characterization of a symbiotic bacterium associated Steinernema carpocapsae in Korea. J. Asia-Pac. Entomol. 1999, 2, 105–111. [CrossRef]
- Lee, S.; Kim, Y.; Han. S. An improved collecting method of the infective juveniles of the entomopathogenic nematode, Steinernema carpocapsae Weiser. Korean J. Soil Zool. 2000, 5, 97100.
- Kaya, H. K.; Stock, S. P. Techniques in insect nematology. In: Lacey, L.A. (Ed.), Manual of Techniques in Insect Pathology. Academic Press, San Diego, 1997; pp. 281–324.
- Vrain, T.C.; Wakarchuk, D.A.; Levesque, A.C.; Hamilton, R.I. Intraspecific rDNA restriction fragment length polymorphism in the Xiphinema americanum group. Fund. Appl. Nematol. 1992, 5, 563–574.
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729.
- Loulou, A.; Mastore, M.; Caramella, S.; Bhat, A.H.; Brivio, M.F.; Machado, R.A.; Kallel, S. Entomopathogenic potential of bacteria associated with soil-borne nematodes and insect immune responses to their infection. PLoS ONE 2023, 18, 0280675. [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K., Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; Huttley, G.A. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336.
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome. 2018, 6, 1–17. doi:10.1186/s40168-018-0470-z.
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [CrossRef]
- SAS Institute, Inc. SAS/STAT User’s Guide; SAS Institute: Cary, NC, USA, 1989.
- Park, Y.; Kim, Y. Eicosanoids rescue Spodoptera exigua infected with Xenorhabdus nematophilus, the symbiotic bacteria to the entomopathogenic nematode Steinenerma carpocapsae. J. Insect Physiol. 2000, 46, 1469–1476.
- Filgueiras, C.C.; Shields, E.J.; Nault, B.A.; Willett, D.S. Entomopathogenic nematodes for field control of onion maggot (Delia antiqua) and compatibility with seed treatments. Insects 2023, 14, 623. [CrossRef]
- Hodda, M. Phylum Nematoda: trends in species descriptions, the documentation of diversity, systematics, and the species concept. Zootaxa 2022, 5114, 290–317. [CrossRef]
- Gaugler, R., Kaya, H.K. Entomopathogenic namtodes in biological control. 1990, pp. 365. CRC Press, Boca Raton, FL, USA.
- Lam, A.B.Q.; Webster, J.M. Morphology and biology of Panagrolaimus tipulae n. sp. (Panagrolaimidae) and Rhabditis (Rhabditella) tipulae n. sp. (Rhabditidae), from leatherjacket larvae, Tipula paludosa (Diptera: Tipulidae). Nematol. 1971, 17, 201–212. [CrossRef]
- de Ley, P.; Blaxter, M. Systematic position and phylogeny. In: The biology of nematodes (Ed. Lee, D.L.), Taylor & Francis Press, NY, USA, 2002; pp. 1–30.
- Winter, C.E. The yolk polypeptides of a free-living rhabditid nematode. Comp. Biochem. Physiol. 1992, 103B, 189–196. [CrossRef]
- Félix M.A.; Vierstraete, A.; Vanfleteren, J. Three biological species related to Rhabditis (Oscheius) pseudodolichura Körner in Osche, 1952. J. Nematol. 2001, 33, 104–109.
- Besnard, F.; Koutsovoulos, G.; Dieudonné, S.; Blaxter, M.; Félix, M.A. Toward universal forward genetics: using a draft genome sequence of the nematode Oscheius tipulae to identify mutations affecting vulva development. Genetics 2017, 206, 1747–1761. [CrossRef]
- Tabassum, K.A.; Shahina, F.; Nasira, K.; Erum, Y. Description of six new species of Oscheius Andrassy, 1976 (Nematoda: Rhabditida) from Pakistan with a key and diagnostic compendium to species of the genus. Pakistan J. Nematol. 2016, 34, 109–161.
- de Brida, A.L.; Rosa, J.M.; Oliveira, C.M.; Castro, B.M.; Serrão, J.E.; Zanuncio; J.C.; Leite, L.G.; Wilcken, S.R. Entomopathogenic nematodes in agricultural areas in Brazil. Sci. Rep. 2017, 7, 45254. [CrossRef]
- Dennis, E.; Cao, J.; Hsu, Y. H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185.
- Shi, Y.M.; Hirschmann, M.; Shi, Y.N.; Ahmed, S., Abebew, D.; Tobias, N.J., Grün, P.; Crames, J.J.; Pöschel, L.; Kuttenlochner, W.; et al. Global analysis of biosynthetic gene clusters reveals conserved and unique natural products in entomopathogenic nematode-symbiotic bacteria. Nat. Chem. 2022, 14, 1–21. [CrossRef]
- Shapiro-Ilan, D.I.; Morales-Ramos, J.A.; Rojas, M.G. In vivo production of entomopathogenic nematodes. Methods Mol. Biol. 2016, 1477, 137–158.
- Seo, S.; Lee, S.; Hong, Y.; Kim, Y. Phospholipase A2 inhibitors synthesized by two entomopathogenic bacteria, Xenorhabdus nematophila and Photorhabdus temperata subsp. temperata. Appl. Environ. Microbiol. 2012, 78, 3816–3823.
- Abdisa, E.; Park, H.; Kwon, J.; Jin, G.; Esmaeily, M.; Kim, Y. Enhancement of an entomopathogenic fungal virulence against the seedcorn maggot, Delia platura, by suppressing immune responses with a bacterial culture broth of Photorhabdus temperata subsp. temperata. Arch. Insect Biochem. Physiol. 2024, 115, e22103.
- Liu, W.T.; Chen, T.L.; Hou, R.F.; Chen, C.C.; Tu, W.C. The invasion and encapsulation of the entomopathogenic nematode, Steinernema abbasi, in Aedes albopictus (Diptera: Culicidae) larvae. Insects 2020, 11, 832. [CrossRef]
- Lalitha, K.; Nithya, K.; Bharathi, B.G.; Venkatesan, S.; Shivakumar, M.S. Long-term storage does not affect the infectivity of entomopathogenic nematodes on insect hosts. Appl. Microbiol. Biotechnol. 2023, 107, 419–431. [CrossRef]
- Park, Y.; Kim, K.; Kim, Y. A pathogenic bacterium, Enterococcus faecalis, to the beet armyworm, Spodoptera exigua. J. Asia Pac. Entomol. 2002, 5, 221–225.
- Torres-Barragan, A., Suazo, A., Buhler, W.G., Cardoza, Y.J. Studies on the entomopathogenicity and bacterial associates of the nematode Oscheius carolinensis. Biol. Control. 2011, 59, 123–129. [CrossRef]
- Ruiu, L.; Marche, M.G.; Mura, M.E.; Tarasco, E. Involvement of a novel Pseudomonas protegens strain associated with entomopathogenic nematode infective juveniles in insect pathogenesis. Pest Manag. Sci. 2022, 78, 5437–5443. [CrossRef]
- Matthijs, S.; Laus, G.; Meyer, J.M.; Abbaspour-Tehrani, K.; Schäfer, M.; Budzikiewicz, H.; Cornelis, P. Siderophore-mediated iron acquisition in the entomopathogenic bacterium Pseudomonas entomophila L48 and its close relative Pseudomonas putida KT2440. Biometal. 2009, 22, 951–964. [CrossRef]
- Xu, M.; Cai, H.; Zhang, K. Pathogenesis of entomopathogenic nematode symbiotic bacterial strain Serratia nematodiphila DR186 against fruit flies. J. Nanjing Agric. Univ. 2017, 40, 827–834.
- Houard, J.; Aumelas, A.; Noël, T.; Givaudan, A.; Fitton-Ouhabi, V.; Villain-Guillot, P.; Gualtieri, M. Cabanillasin, a new antifungal metabolite, produced by entomopathogenic Xenorhabdus cabanillasii JM26. J. Antibiot. 2013, 66, 617–620. [CrossRef]
- Zhou, Q.; Grundmann, F.; Kaiser, M.; Schiell, M.; Gaudriault, S.; Batzer, A.; Kurz, M.; Bode, H.B. Structure and biosynthesis of xenoamicins from entomopathogenic Xenorhabdus. Chem. 2013, 19, 16772–16779. [CrossRef]
Figure 1.
Identification of a nematode isolate killing insects using morphological characteristics. (A) Release of infective juveniles (IJs) from the dead T. molitor larva. The right panel shows an enlarged picture of the square region in the left panel as well as IJs. (B) Morphological traits of male and female specimens: TL for tail length, RL for rectum length, ovary, BDM for body diameter, BL for body length, and SL for stylet length. (C) Genus identification of the nematode isolate using the morphological characters. Each measurement used 10 nematode samples and was reported as mean ± SD. (D) Species identification of the nematode isolate using the morphological characteristics. ‘+’ and ‘-’ in the parenthesis indicate being identical and non-identical with the nematode isolate with respect to each morphological characteristic.
Figure 1.
Identification of a nematode isolate killing insects using morphological characteristics. (A) Release of infective juveniles (IJs) from the dead T. molitor larva. The right panel shows an enlarged picture of the square region in the left panel as well as IJs. (B) Morphological traits of male and female specimens: TL for tail length, RL for rectum length, ovary, BDM for body diameter, BL for body length, and SL for stylet length. (C) Genus identification of the nematode isolate using the morphological characters. Each measurement used 10 nematode samples and was reported as mean ± SD. (D) Species identification of the nematode isolate using the morphological characteristics. ‘+’ and ‘-’ in the parenthesis indicate being identical and non-identical with the nematode isolate with respect to each morphological characteristic.
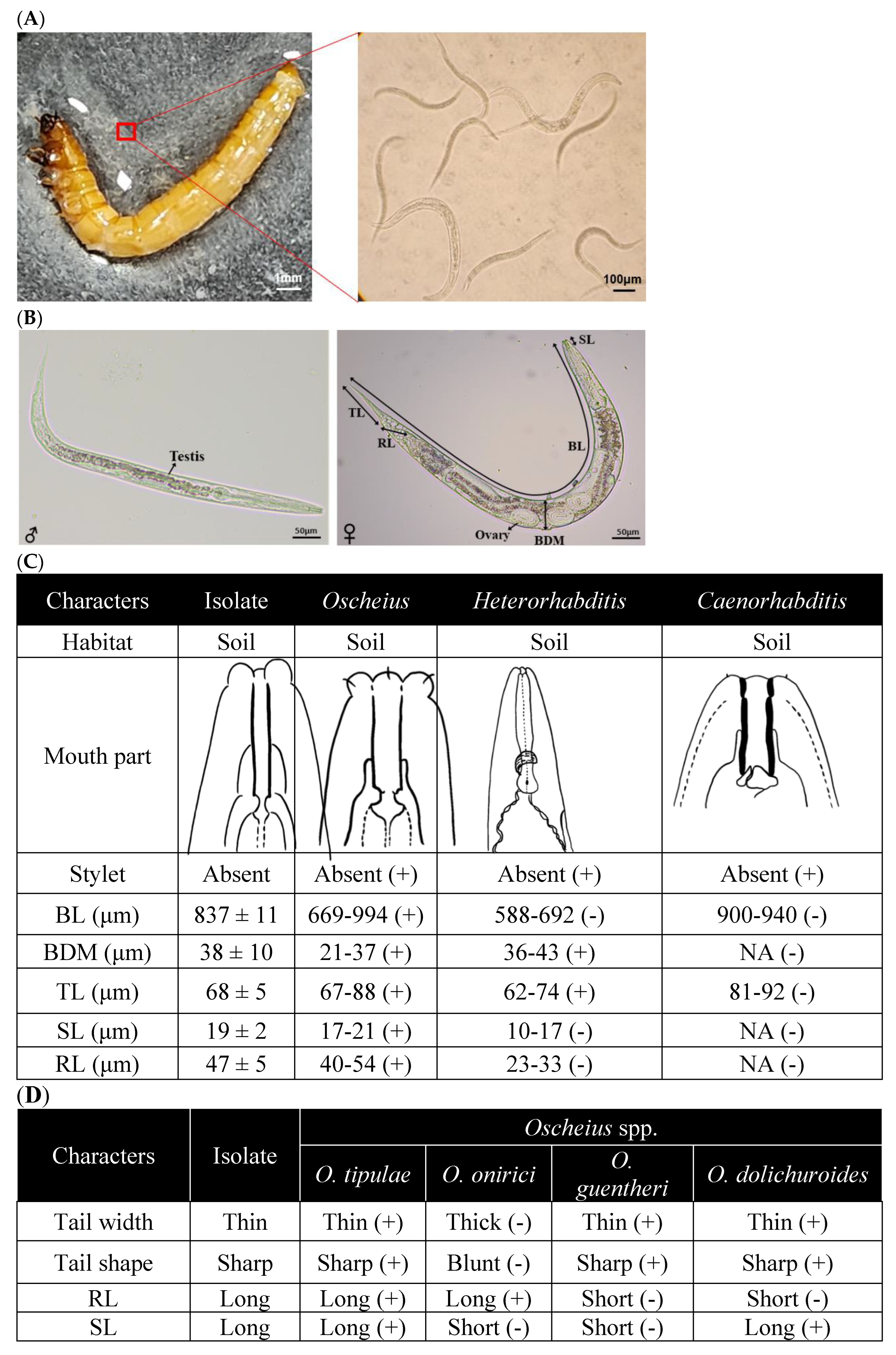
Figure 2.
Molecular identification of a nematode isolate killing insects using ITS sequence. (A) Nucleotide sequence of internal transcribed spacer (‘ITS’) region of the nematode isolate. The sequence was submitted with the GenBank access number of PP358559. With sequence comparison of the ITS sequences of Heterorhabditis spp. (Adams et al., 1998), the following putative rDNA regions were identified: partial 18S rDNA (1-161), ITS-1 (162-470), 5.8S rDNA (470-600), ITS-2 (600-843), and partial 28S rDNA (843-929). (B) Phylogenetic analysis of the ITS sequence of the nematode isolate with those of the closely related species. The tree was constructed with MEGA.11 based on Neighbor-Joining analysis, in which the bootstrap values were obtained with 1,000 repetitions.
Figure 2.
Molecular identification of a nematode isolate killing insects using ITS sequence. (A) Nucleotide sequence of internal transcribed spacer (‘ITS’) region of the nematode isolate. The sequence was submitted with the GenBank access number of PP358559. With sequence comparison of the ITS sequences of Heterorhabditis spp. (Adams et al., 1998), the following putative rDNA regions were identified: partial 18S rDNA (1-161), ITS-1 (162-470), 5.8S rDNA (470-600), ITS-2 (600-843), and partial 28S rDNA (843-929). (B) Phylogenetic analysis of the ITS sequence of the nematode isolate with those of the closely related species. The tree was constructed with MEGA.11 based on Neighbor-Joining analysis, in which the bootstrap values were obtained with 1,000 repetitions.
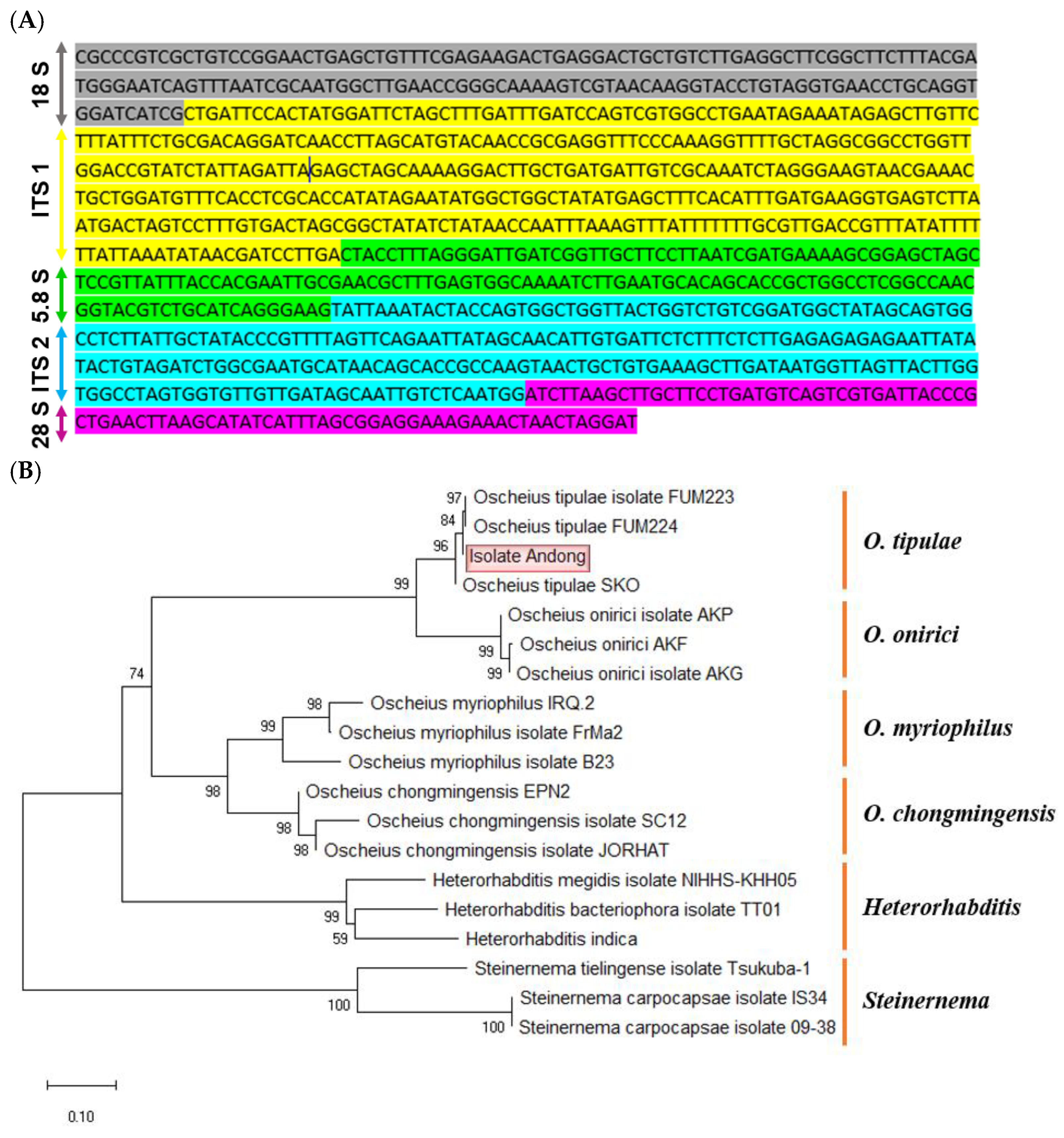
Figure 3.
Biological characteristics of O. tipulae. (A) Parasitic life cycle of O. tipulae in M. vitrata larvae. Parasitized host larvae with O. tipulae IJs (= L3) were kept at room temperature and dissected every day to assess the nematode development of different larval instars (L1-L4). Each measurement consists of three replications (= three parasitized larvae). Relative proportions were calculated by the ratios of individual numbers among different developmental stages. (B) Comparison of IJ yield between M. vitrata (‘Mv’) and T. molitor (‘Tm’) larvae. IJs were collected using a White trap for 3 days after the first IJ emergence from the cadaver. Each measurement consisted of three replications, and the error bar indicates the standard deviation. The different letters above the standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test). (C) Survival analysis of IJs at different temperatures and preservatives for a storage period of 30 days. The different letters following the survival means at 30 days indicate significant differences among means at Type I error = 0.05 (LSD test).
Figure 3.
Biological characteristics of O. tipulae. (A) Parasitic life cycle of O. tipulae in M. vitrata larvae. Parasitized host larvae with O. tipulae IJs (= L3) were kept at room temperature and dissected every day to assess the nematode development of different larval instars (L1-L4). Each measurement consists of three replications (= three parasitized larvae). Relative proportions were calculated by the ratios of individual numbers among different developmental stages. (B) Comparison of IJ yield between M. vitrata (‘Mv’) and T. molitor (‘Tm’) larvae. IJs were collected using a White trap for 3 days after the first IJ emergence from the cadaver. Each measurement consisted of three replications, and the error bar indicates the standard deviation. The different letters above the standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test). (C) Survival analysis of IJs at different temperatures and preservatives for a storage period of 30 days. The different letters following the survival means at 30 days indicate significant differences among means at Type I error = 0.05 (LSD test).
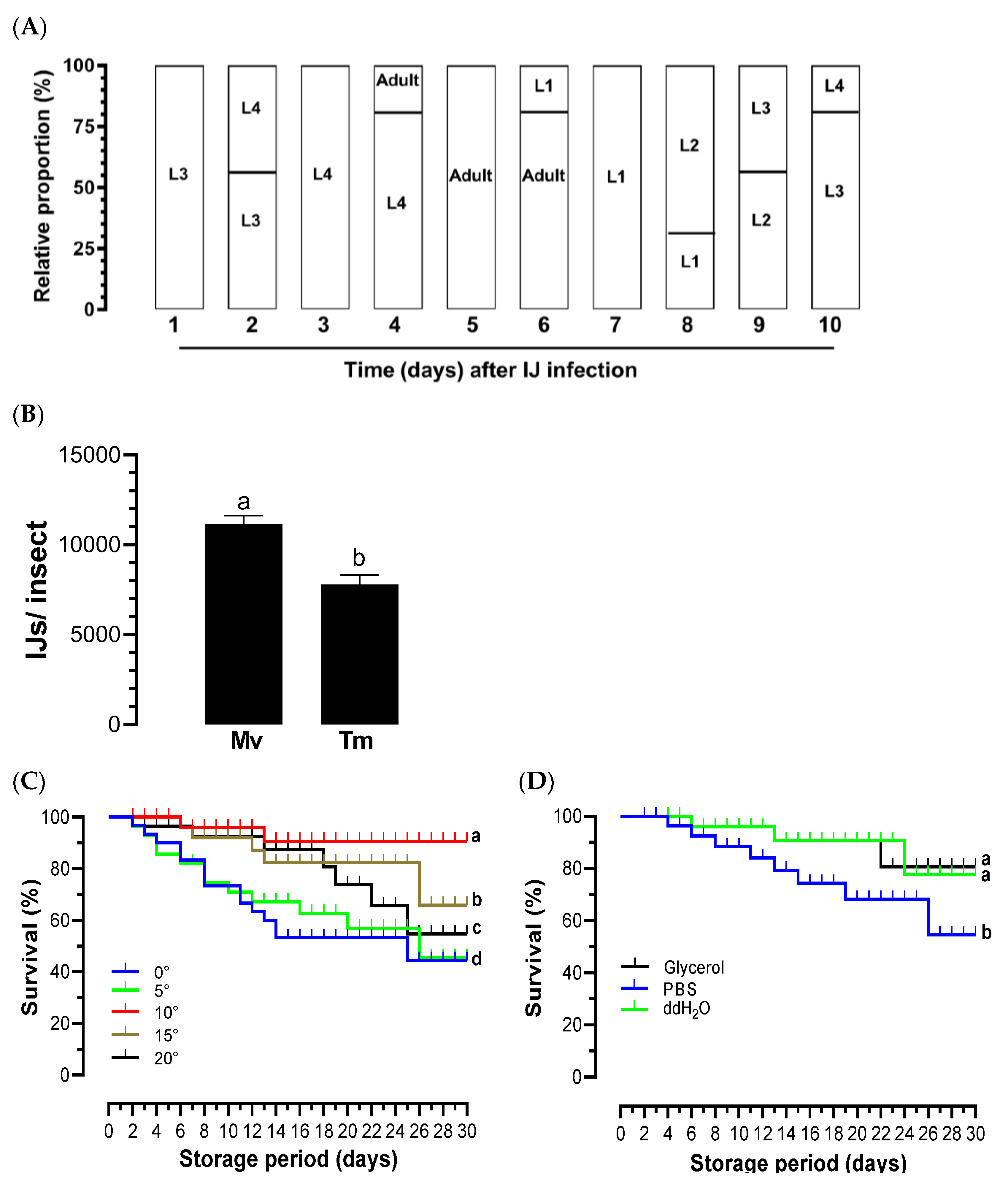
Figure 4.
Insecticidal spectrum of the infective juveniles (IJs) of O. tipulae. (A) Virulence of O. tipulae against larvae of four different insects: T. molitor (‘Tm’), S. exigua (‘Se’), P. xylostella (‘Px’), and M. vitrata (‘Mv’). The experimental unit was a Petri dish containing 10 larvae, and each one was replicated three times. Mortality was recorded at 4 days after treatment (‘DAT’). (B) Medium lethal time (LT50) at a concentration of 400 IJs per larva. (C) Pathogenicity of O. tipulae against different developmental stages of T. molitor at 400 IJs per larva. Different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test).
Figure 4.
Insecticidal spectrum of the infective juveniles (IJs) of O. tipulae. (A) Virulence of O. tipulae against larvae of four different insects: T. molitor (‘Tm’), S. exigua (‘Se’), P. xylostella (‘Px’), and M. vitrata (‘Mv’). The experimental unit was a Petri dish containing 10 larvae, and each one was replicated three times. Mortality was recorded at 4 days after treatment (‘DAT’). (B) Medium lethal time (LT50) at a concentration of 400 IJs per larva. (C) Pathogenicity of O. tipulae against different developmental stages of T. molitor at 400 IJs per larva. Different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test).
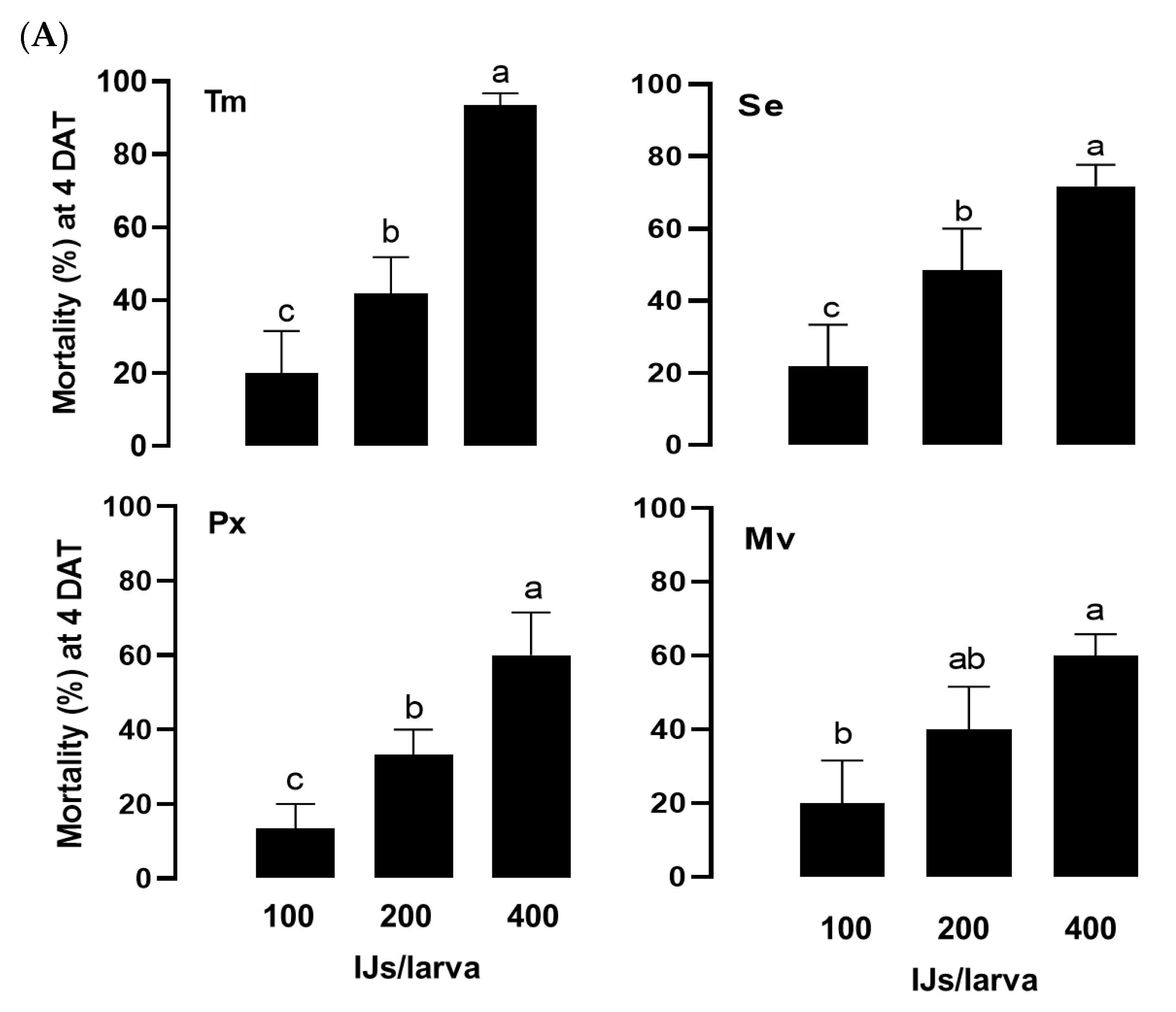
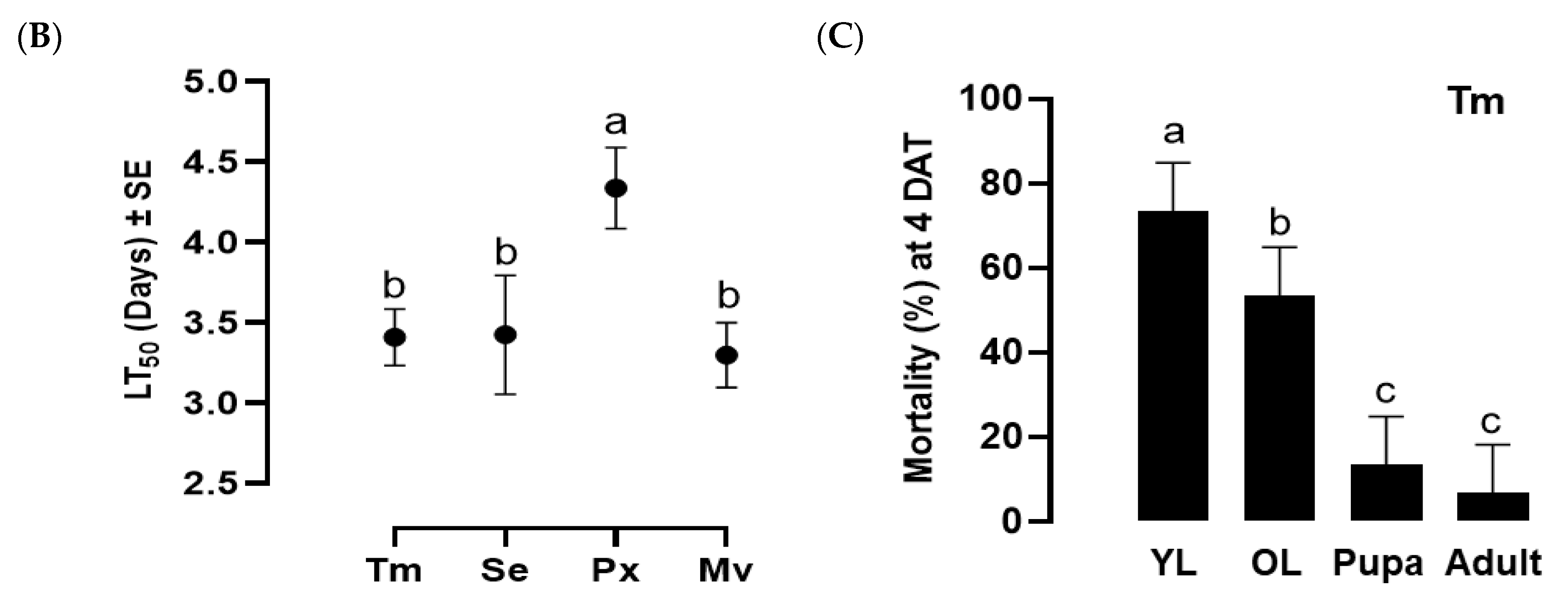
Figure 5.
Entomopathogenic bacteria isolated from O. tipulae (‘Ot’). (A) Virulence of three different colonies of small (‘S’), big (‘B’), or fuzzy (‘F’) shapes against M. vitrata larvae. The bacteria (103 cfu/larva) were injected into the larvae of M. vitrata. The experimental unit was a Petri dish containing 10 larvae and each unit was replicated three times. Mortality was recorded at 48 h after the treatment. (B) Identification of the bacterial colonies using 16S rRNA sequence. (C) Influence of eicosanoids on the virulence of the nematode against two insect hosts: M. vitrata (‘Mv’) and T. molitor (‘Tm’). Ot was treated at 400 IJs per larva to the two insects. Dexamethasone (‘DEX’) or arachidonic acid (‘AA’) was injected into the larvae at 1 μg per larva. The experimental unit was a Petri dish containing 10 larvae, and each unit was replicated three times. Mortality was recorded at 4 days after the nematode treatment (‘DAT’). The different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test).
Figure 5.
Entomopathogenic bacteria isolated from O. tipulae (‘Ot’). (A) Virulence of three different colonies of small (‘S’), big (‘B’), or fuzzy (‘F’) shapes against M. vitrata larvae. The bacteria (103 cfu/larva) were injected into the larvae of M. vitrata. The experimental unit was a Petri dish containing 10 larvae and each unit was replicated three times. Mortality was recorded at 48 h after the treatment. (B) Identification of the bacterial colonies using 16S rRNA sequence. (C) Influence of eicosanoids on the virulence of the nematode against two insect hosts: M. vitrata (‘Mv’) and T. molitor (‘Tm’). Ot was treated at 400 IJs per larva to the two insects. Dexamethasone (‘DEX’) or arachidonic acid (‘AA’) was injected into the larvae at 1 μg per larva. The experimental unit was a Petri dish containing 10 larvae, and each unit was replicated three times. Mortality was recorded at 4 days after the nematode treatment (‘DAT’). The different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test).
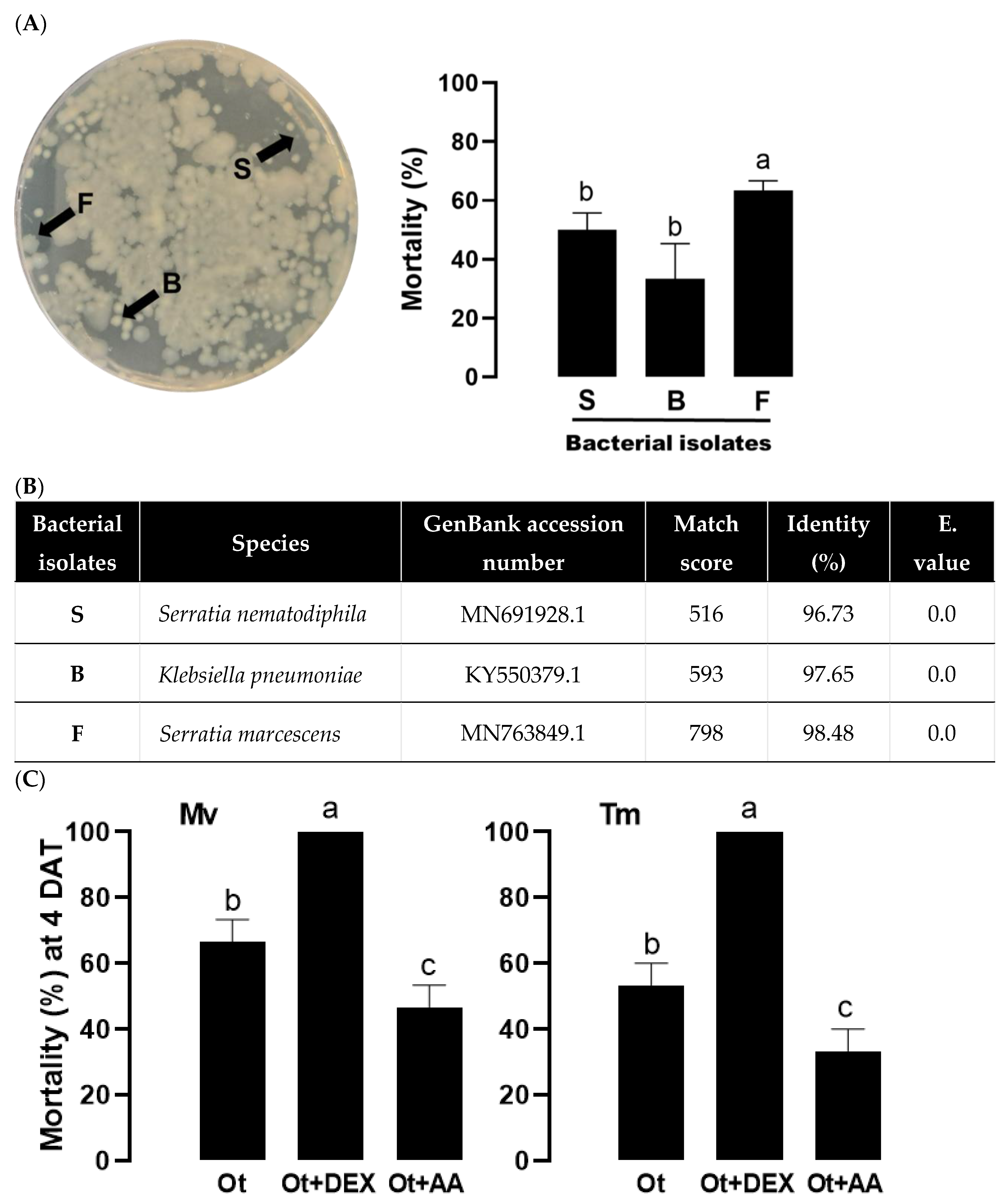
Figure 6.
Metagenomics analysis of the gut bacteria isolated from the nematodes of O. tipulae (‘Ot’) and two S. carpocapsae (‘Sc-AD’ and ‘Sc-DR’). (A) Comparison of bacterial diversity based on Amplicon Sequence Variants (ASVs). The figures within the bars indicate the total numbers of the bacterial species based on ASVs. (B) Relative bacterial abundance in taxonomic composition at phylum level. (C) Comparative analysis of bacterial diversity on two community indices: Shannon index and Gini-Simpson index. (D) Comparison of the entomopathogenic bacteria identified from the nematodes. Presence and absence are respectively denoted by + and -.
Figure 6.
Metagenomics analysis of the gut bacteria isolated from the nematodes of O. tipulae (‘Ot’) and two S. carpocapsae (‘Sc-AD’ and ‘Sc-DR’). (A) Comparison of bacterial diversity based on Amplicon Sequence Variants (ASVs). The figures within the bars indicate the total numbers of the bacterial species based on ASVs. (B) Relative bacterial abundance in taxonomic composition at phylum level. (C) Comparative analysis of bacterial diversity on two community indices: Shannon index and Gini-Simpson index. (D) Comparison of the entomopathogenic bacteria identified from the nematodes. Presence and absence are respectively denoted by + and -.
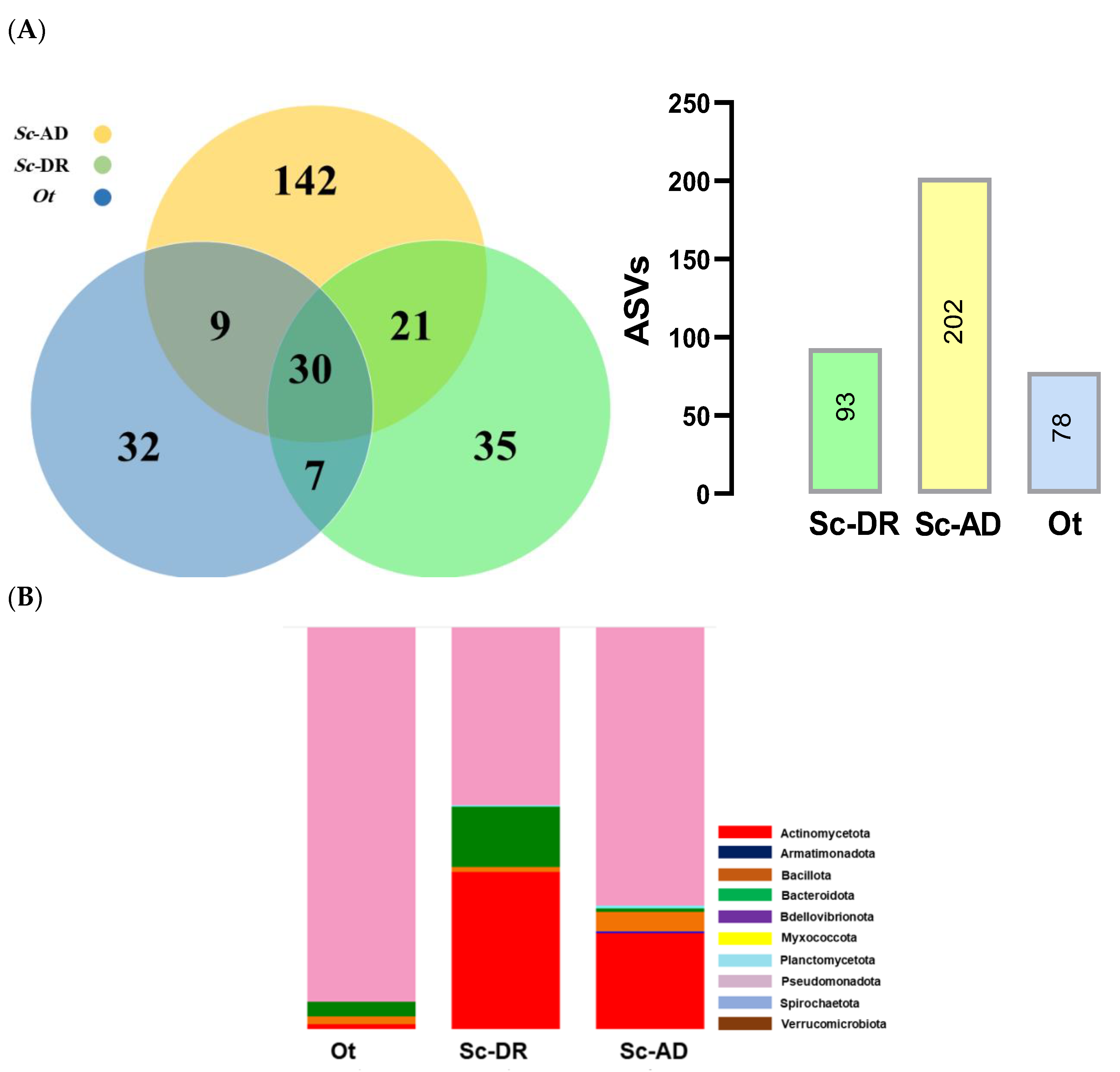
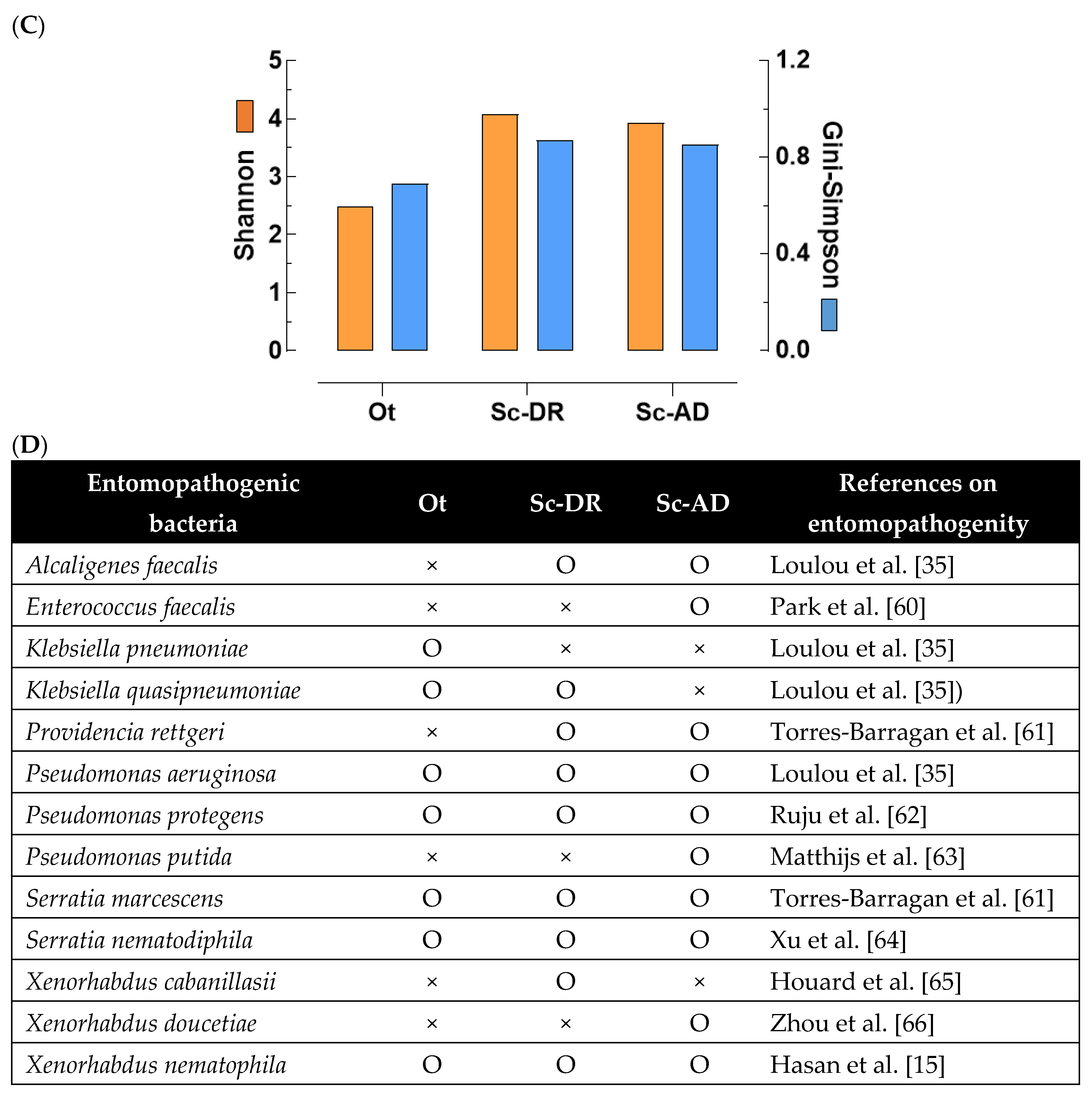
Figure 7.
High virulence of O. tipulae against dipteran insect pests of D. platura (‘Dp’), D. melanogaster (‘Dm’), and A. albopictus (‘Aa’). (A) Comparative virulence analysis of the nematodes of O. tipulae (‘Ot’) and two S. carpocapsae (‘Sc-AD’ and ‘Sc-DR’) against different insect hosts including lepidopteran (‘Lep’), coleopteran (‘Col’), and dipteran (‘Dip’) species. The Lep and Col insects were M. vitrata (‘Mv’) and T. molitor (‘Tm’) larvae, respectively. All larvae were treated at 400 IJs per larva. The experimental unit was a Petri dish (each well of a 24-well plate) containing 10 larvae, and each unit was replicated three times. Mortality was recorded at 4 days after the nematode treatment (‘DAT’). Asterisks above the standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test). ‘ns’ represents no significant difference. (B) Ot virulence against three Dip insects. Each treatment used 10 larvae and was replicated three times. Different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test). Photos indicate the emerging IJs from the cadavers.
Figure 7.
High virulence of O. tipulae against dipteran insect pests of D. platura (‘Dp’), D. melanogaster (‘Dm’), and A. albopictus (‘Aa’). (A) Comparative virulence analysis of the nematodes of O. tipulae (‘Ot’) and two S. carpocapsae (‘Sc-AD’ and ‘Sc-DR’) against different insect hosts including lepidopteran (‘Lep’), coleopteran (‘Col’), and dipteran (‘Dip’) species. The Lep and Col insects were M. vitrata (‘Mv’) and T. molitor (‘Tm’) larvae, respectively. All larvae were treated at 400 IJs per larva. The experimental unit was a Petri dish (each well of a 24-well plate) containing 10 larvae, and each unit was replicated three times. Mortality was recorded at 4 days after the nematode treatment (‘DAT’). Asterisks above the standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test). ‘ns’ represents no significant difference. (B) Ot virulence against three Dip insects. Each treatment used 10 larvae and was replicated three times. Different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test). Photos indicate the emerging IJs from the cadavers.
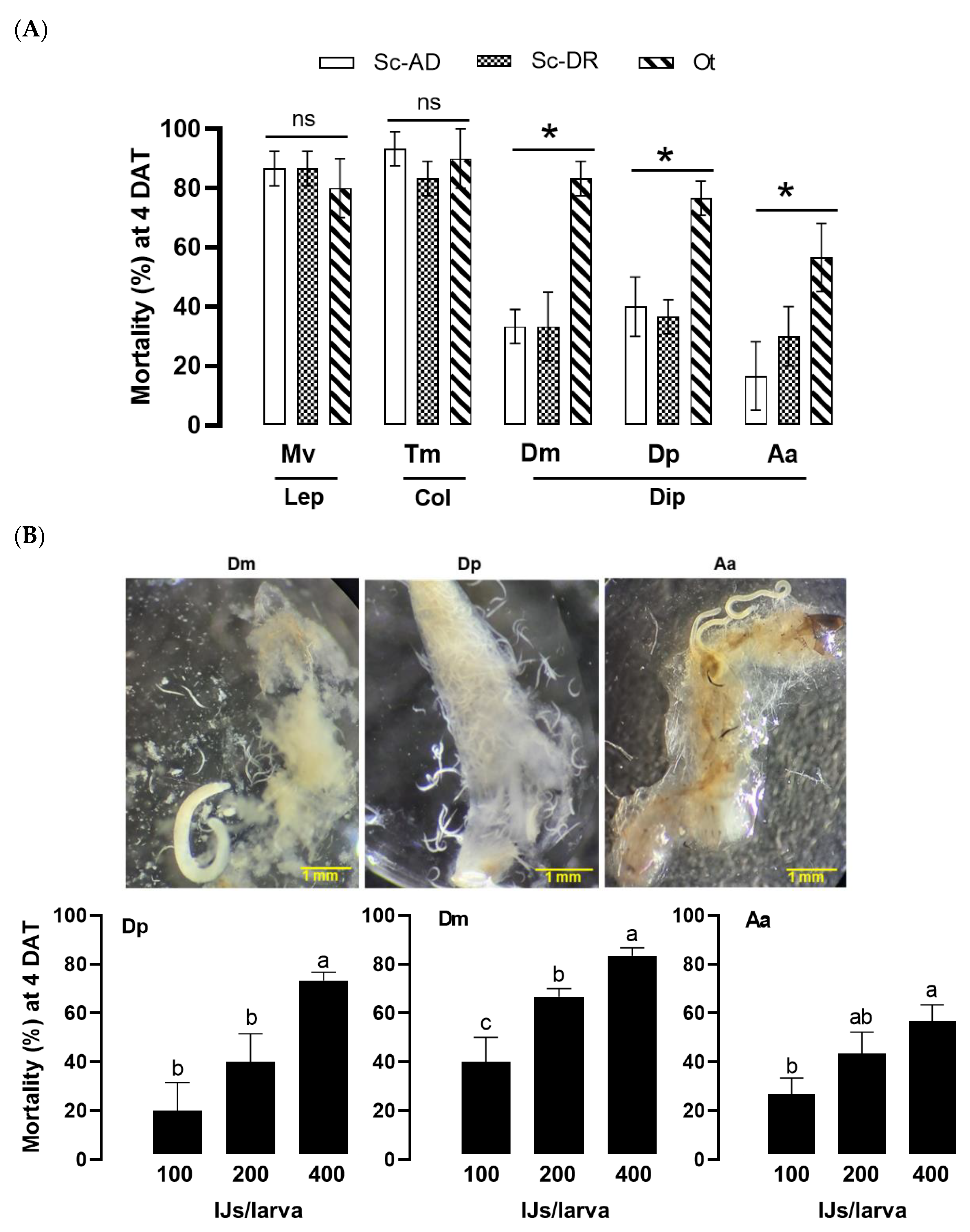
Figure 8.
Control efficacies of O. tipulae (‘Ot’) against D. platura in pot (A) and field (B) assays. In pot assay, each Welsh onion was grown in a pot and inoculated with 30 nematodes. Different concentrations of the nematodes were applied to the pot. The nematode mortality was recorded at six days after treatment (‘DAT’). In the field assay, each onion plant infested by D. platura was drenched with the nematode suspension containing 1,000 IJs. Fluxametamide (Flx, 500 ppm) was treated as a positive control in the experiment. Control efficacy was estimated by comparing survival IJ numbers in untreated and treated plants. The different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test).
Figure 8.
Control efficacies of O. tipulae (‘Ot’) against D. platura in pot (A) and field (B) assays. In pot assay, each Welsh onion was grown in a pot and inoculated with 30 nematodes. Different concentrations of the nematodes were applied to the pot. The nematode mortality was recorded at six days after treatment (‘DAT’). In the field assay, each onion plant infested by D. platura was drenched with the nematode suspension containing 1,000 IJs. Fluxametamide (Flx, 500 ppm) was treated as a positive control in the experiment. Control efficacy was estimated by comparing survival IJ numbers in untreated and treated plants. The different letters above standard deviation bars indicate significant differences among means at Type I error = 0.05 (LSD test).
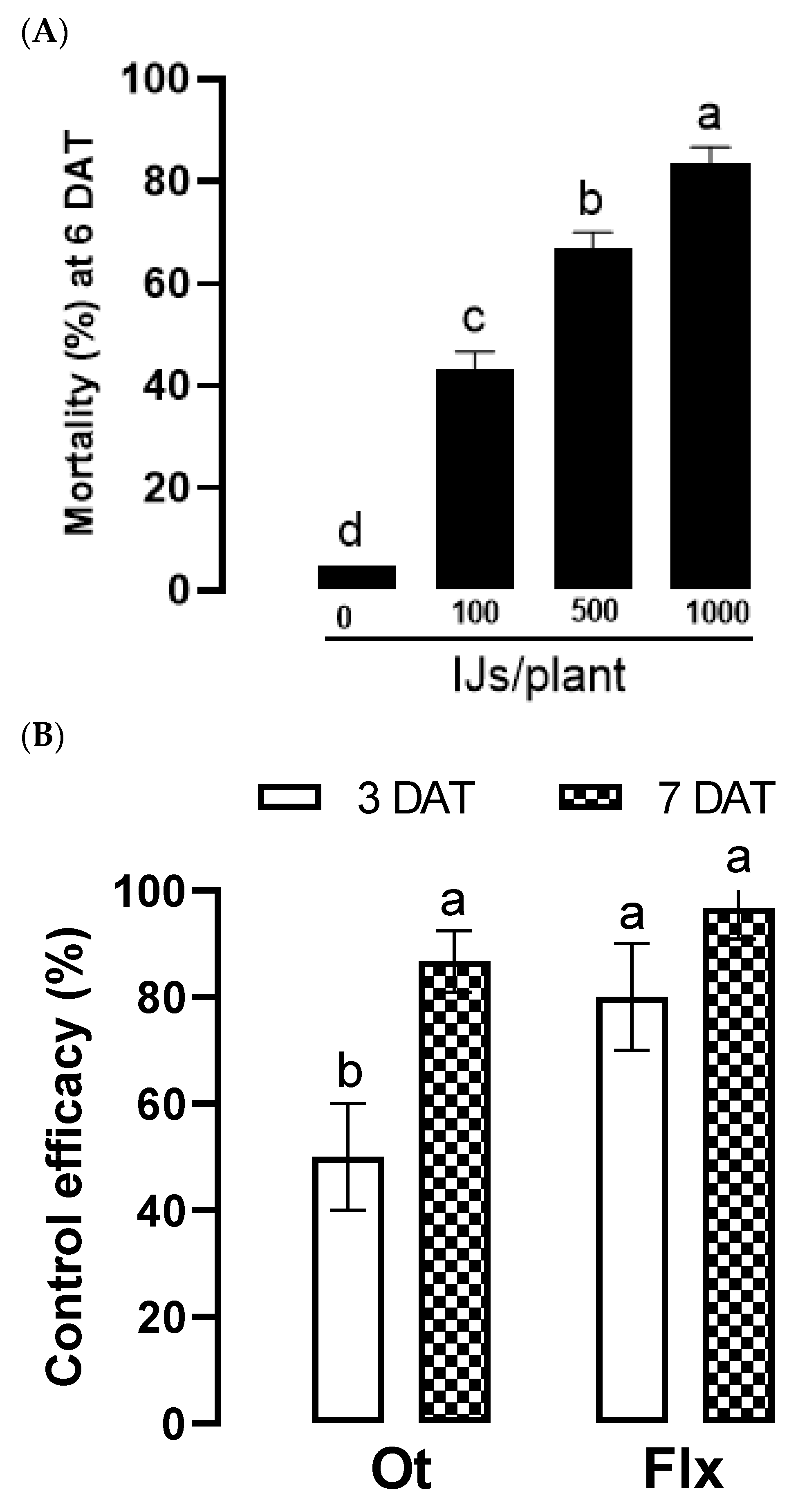

Table 1.
Virulence of the three bacterial colonies isolated from O. tipulae against L5 larvae of M. vitrata. The virulence was estimated using the median lethal dose (LD50). Each bioassay used 10 larvae at the L5 stage and was replicated three times.
Table 1.
Virulence of the three bacterial colonies isolated from O. tipulae against L5 larvae of M. vitrata. The virulence was estimated using the median lethal dose (LD50). Each bioassay used 10 larvae at the L5 stage and was replicated three times.
| Colony shape | Number of larvae used for bioassay |
LD50 (95% CI) cfu/larva |
Slope ± SE | X2 |
| Fuzzy | 50 | 249.3 (89.1-698.6) |
0.49 ± 0.1 | 8.37 |
| Small | 50 | 1,649.7 (667-4,350.4) |
0.47 ± 0.1 | 8.94 |
| Big | 50 | 8,313.9 (3,564.4-2,4477.0) |
0.53 ± 0.2 | 7.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



