
Submitted:
03 April 2024
Posted:
04 April 2024
You are already at the latest version
Abstract
Most neurodegenerative diseases, including Alzheimer's disease, ischemic stroke, subarachnoid hemorrhage, and intracerebral hemorrhage are associated with inflammation. Tumor necrosis factor (TNF) is a pleiotropic pro-inflammatory cytokine that regulates cerebral infarction in stroke pathology, and its action is influenced by the bioavailability of its membrane-bound receptors, TNFR1 and TNFR2, and microglial activation. During the initial onset of these diseases, the soluble variant of the cytokine presents with prolonged and excessive activation of TNFR1, resulting in cell death and long-term neurological impairments. Therapeutic interventions for neurodegenerative diseases have targeted TNF to limit the onset of neuroinflammation. First-generation therapeutics have been demonstrated to inhibit membrane-bound TNF to TNFR2 receptor binding, resulting in severe side effects such as infections and cancer. As such, second-generation drugs, including XPro1595, have been developed to selectively inhibit soluble TNF and impede the effects of TNFR1 while still allowing for TNFR2 activation. Early results in murine TBI models demonstrate reduced glial reactivity by 50%, reduced dendritic degeneration by 30%, increased plasticity by 15%, and improved functional outcomes by 20% post-TBI. This scoping review of TNF receptors in various neurodegenerative diseases seeks to evaluate current and future therapeutic strategies as well as highlight potential strategies to eliminate confounding variables present in the current literature.
Keywords:
cerebroprotection
; ischemia
; tumor necrosis factor
1. Introduction
Cytokines are part of a network of innate and acquired immune responses to brain injury and disease. Tumor necrosis factor (TNF) is a well-studied cytokine that contributes to neuroinflammation in a range of conditions including subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), and ischemic stroke (IS). Despite being recognized as a pro-inflammatory cytokine, TNF signaling is linked to diverse outcomes such as cell survival and proliferation. This multifaceted impact is attributed to the distinct downstream effects of TNF receptors, where TNF receptor 1 (TNFR1) activation yields detrimental effects, while TNF receptor 2 (TNRF2) activation can result in beneficial effects. This comprehensive review aims to elucidate the complexities and crosstalk inherent in TNF signaling as well as analyze current therapeutic interventions designed to limit the threat of excessive inflammation and neuronal cell death associated with TNFR1 activation1. Since TNF can signal in its membrane-bound and soluble conformations, current therapeutic methods include neutralizing antibodies, TNFα converting enzyme (TACE) antagonists, and soluble receptor2–4. Moreover, this review demonstrates the potential of novel treatments such as Xpro1595 and evaluates certain limitations in the current research paradigm. Overall, this review highlights the potential of selectively targeting TNFR1 signaling pathways as a novel therapeutic approach. It also emphasizes the critical need for future research utilizing appropriate models to ensure the accuracy and replicability of results in human trials.
2. Materials and Methods
The literature in this review included peer-reviewed publications published between the years 1989 to 2022. The online databases utilized in this literature review included Embase, PubMed, Google Scholar, and Biogen Pipeline patent US20150239951A1. Pertinent search filters included TNF, receptors, neuroinflammation, ischemia, stroke, and cerebral hemorrhage. Selected literature consisted of peer-reviewed publications written in English, spanning preclinical and clinical case studies, cross-sectional studies, retrospective studies, meta-analyses, and systematic literature reviews. Exclusion criteria comprised non-peer-reviewed articles, conference abstracts, editorials, letters, and commentaries. Additionally, studies beyond the specified time period, not in English, solely focusing on cell lines, lacking clear methodological details, or not directly addressing TNF and its receptors in neurodegenerative diseases and injuries were excluded. These criteria were applied to ensure the inclusion of relevant and high-quality studies aligned with the review’s objectives. The collected data were tabulated into clinical and preclinical reviews. Data variables gathered from each experimental publication, when applicable, included the species tested, population size, average age, sex predominance, study duration, drug administration, anatomical outcomes, and functional outcomes.
3. TNF Receptors
The majority of known cellular responses to TNF are mediated by either TNF receptors 1 (TNFR1) or 2 (TNFR2). Existing literature has focused on TNFR1 interactions due to its higher binding affinity and variable responses compared to TNFR2. TNFR1, expressed in all cell types, can be activated by both membrane-bound TNF (tmTNF) and soluble TNF (sTNF) [1]. TNFR2 is expressed primarily on endothelial cells and predominantly binds to tmTNF. While both variants contribute to cerebral-protection, TNFR2 has demonstrated some capacity in inflammatory reactions as well [2]. Despite this, most cells express both forms of the receptor, and each lead to unique responses [3].
3.1. TNFR1
To initiate a signal transduction event, homotrimeric TNF must bind to the cysteine-rich extracellular domains of membrane-bound TNFR homodimers [4]. The binding process can be inhibited by soluble variants of TNF receptors (sTNFR), which compete with membrane-bound receptors for ligands or directly bind to the N-terminal pre-ligand binding assembly domain, preventing the formation of active receptor trimers [5]. Upon binding to its ligand, TNFR1 homotrimerizes and interacts with intracellular signaling proteins through its death domain (Figure 1). This leads to the formation of complex I, consisting of TNFR1-associated death domain protein (TRADD), receptor-interacting protein kinase-1 (RIPK1), and TNF receptor-associated factor 2 (TRAF2) or TRAF5. Complex I can activate JUN NH2-terminal kinase (JNK), p38, and nuclear factor κB (NFκB) transcription by parallel recruitment of TAK1 and the IKK complex [4]. TAK1 activates MAP kinases (MAPK) such as JNK, p38, and ERK, as well as ceramide/sphingomyelinase pathways by recruiting cellular inhibitor apoptosis proteins 1 and 2 (cIAP1/2). The IKK complex, comprising IKKα, IKKβ, and NFκB essential modulators (NEMO), promotes Jak1, Jak2, STAT3, and STAT5 activation, freeing RelA/p50 for nuclear translocation and the transcription of NFκB inflammatory genes [6]. These pathways can induce cytoprotective or apoptotic effects depending on the duration and intensity of stimulation. However, canonical activation of NFκB via RelA/p50 is typically proinflammatory in nature [4].
Moreover, internalization of the activated TNFR1 complex I results in the dissociation of some adaptor proteins, leading to the binding of the FAS-associated death domain (FADD), recruitment of pro-caspase 8, and formation of executioner caspases, ultimately resulting in cell death [7]. The structure of this new complex, denoted as complex 2, is subject to conflicting evidence, as it may retain proteins such as RIPK1, TRADD, or TRAF2, and gain FLIP-c [6,7,8]. Figure 1 depicts one proposed structure of complex II [4].
Activation of caspase 8 leads to the degradation of RIPK1 and RIPK3. However, insufficient concentrations of caspase can result in the accumulation of RIPK1 and RIPK3 in a fibrous structure that promotes programmed necrosis under certain conditions [9]. In chronic inflammatory conditions, TNFR1 has also been shown to inhibit TNFR2 signaling by activating TACE through the iRhom2 and p38 pathways [10]. As TNFR2 is only capable of activation via membrane-bound TNF, TACE cleavage of TNF results in decreased TNFR2 activation.
3.2. TNFR2
In contrast to TNFR1, TNFR2 lacks a death domain but can directly interact with TNF receptor-associated factors 1 and 2 or 3 (TRAF1 and TRAF2/3) using an amino acid motif near the C-terminus (Figure 1). The recruitment of TRAF2/3 to the activated complex results in the production of NF-κB-inducing kinase (NIK), which is typically bound to TRAF2/3 or cIAP1/2 in an inhibitory conformation [4]. NIK is capable of non-canonical NFκB activation, leading to the activation of IKKα and subsequent nuclear translocation of RelB/p52, responsible for the transcription of genes relevant to cell survival and proliferation [11,12,13]. This non-canonical activation generally has a longer duration compared to canonical NFκB activation via TNFR1.
Despite lacking a death domain, TNFR2 has also demonstrated the capability to canonically activate NFκB. The exact mechanism is unknown, but activation is still dependent upon TAK1 and IKKβ (Figure 1) [14]. Dissociation of the TRAF1/2 complex can also bind to cIAP1 and be transported to the endoplasmic reticulum, triggering stress and promoting cell death [6]. Significant crosstalk exists between TNFR2 and TNFR1, as the activation of either complex utilizes common proteins. For instance, TNFR2 activation depletes available TRAF2, inhibiting the formation of the TNFR1 complex and promoting non-canonical NFκB activation, as well as the activation of apoptotic factors [4].
4. Inflammatory Response to Stroke
Stroke ranks as the fifth leading cause of death and the primary cause of disability in the United States [15]. At a rate of 40 stroke incidents occurring every second, there are 800,000 new strokes annually in the United, resulting in prolonged cognitive and physiological dysfunction[16]. A stroke is commonly defined as a vascular neurological deficiency lasting longer than 24 hours, and can be classified as ischemic or hemorrhagic based on the mechanism, with ischemic stroke constituting 85% of all acute stroke incidents [16,17].
Ischemic stroke (IS), also referred to as a cerebrovascular accident, occurs due to the occlusion of cerebral vasculature, blocking blood flow and oxygen to brain regions and causing the breakdown of the blood-brain barrier (BBB)[18]. The lack of blood flow suspends the membrane potentials of neurons and glial cells, interrupting energy-dependent mechanisms and ion pumps in cell membranes, leading to the release of harmful extracellular components and cell death [19]. After an IS, the body’s natural response involves releasing proinflammatory cytokines such as interleukins IL-1 and IL-6, along with TNF. These cytokines play a crucial role in regulating the size of cerebral infarctions caused by IS, and their levels increase following focal ischemic injury [20,21]. It is crucial to document the modulation of these cytokines over time to understand the mechanism, targets upon their release, and identifying specific treatments at various time points. Existing evidence, utilizing reverse transcription PCR, indicates an 8-fold increase of TNF mRNA-expressing microglial cells and microglia-macrophages as early as 4 hours following neuronal ischemia [22]. TNF mRNA levels typically rise early, at the 4-hour mark post focal ischemia, compared to IL, which instead demonstrates a late increase at 96 hours [22,23].
The occlusion of blood flow to regions in the brain leads to a measurable area of lethally damaged and hypoxic tissue known as the infarct core, surrounded by a reversibly damaged tissue zone that is known as the penumbra [20,24]. Clinical aspects of infarction examined in experimentation and clinical studies include infarct volume, location, and quantity at variable locations. Many studies have explored the measured expression and concentration of TNF mRNA and cytokines concerning these factors to implement therapies controlling the concentration of these highly regulated and pleiotropic ligands. Higher levels of TNF following IS have been suggested to correlate with larger infarct volumes. In a study involving transgenic rats overexpressing the murine TNF gene and subjected to Middle Cerebral Artery Occlusion (MCAO), the transgenic rats exhibited a higher infarct volume at 24 hours (p≤0.05) and 3 days (p≤0.01). Within the first 10 min of MCAO, cortical perfusion was reduced in the transgenic rats (p≤0.05), leading to increased cellular death and DNA denaturation [25]. Preclinical studies have also demonstrated an increase in the concentration of microglia surrounding the penumbra in a time-dependent manner, starting at 12 hours up to 1 day (p≤0.01) and 3 days (p≤0.005) following transient MCAO [18]. NFκB was also evaluated in this study and was observed to be activated 12 hours after global ischemia in both wild-type and TNF-deficient mice [18]. With increasing levels of TNF under most experimental conditions correlated with cell death and larger infarct volumes, potential therapies for limiting the long-term cognitive and physiological effects of IS involve introducing specific receptors that will bind to and reduce the toxic inflammatory effects [1,26]. Table 1 and Table 2 present pre-clinical studies analyzed regarding the effects of TNF stimulation and current clinical studies regarding TNF activation in the context of stroke, respectively.
4.1. sTNFRs and Stroke
Soluble receptors, investigated for their potential to prevent cytokines from binding to membrane receptors and inhibit their biological activity, offer advantages such as high ligand specificity and low immunogenicity [58]. However, they may exhibit lower affinities and half-lives as compared to membrane receptors, making the therapeutic response of sTNFR dependent on TNF concentration as well as TNFR1 and TNFR2 [59,60].
In stroke therapy, soluble cytokine receptors serve as natural antagonists. The increase in sTNFR is achieved through multiple pathways triggered by TNF, interleukins, transforming growth factors, and interferons. Proteolytic release of TNFR1 is regulated mainly by metalloprotease ADAM8, while TNFR2 release is mediated by TACE in a process known as ectodomain shedding [1]. Studies monitoring TNF regulation in CNS diseases, including stroke, have shown that ADAM8 mRNA expression increases in affected regions proportionally to TNF levels. To measure the effect of sTNFR1 levels on neuronal survival, a comparative analysis of ADAM8 deficient neurons and wild-type neurons was conducted in the presence of exogenous TNF. ADAM8 deficiency increases neuronal sensitivity to varying TNF concentrations, suggesting that shedding of TNFR1 has a protective effect on primary neuron survival [39].
Experimenters have explored alternative methods of anti-cytokine therapy to protect against ischemic damage, including TNF binding protein (TNF-BP) and monoclonal antibodies (mAb). TNF-BP, formed by the proteolytic cleavage of membrane TNF receptors, demonstrated therapeutic capability to modulate TNF toxicity [61,62]. TNF itself induces the release of TNF-BP, self-modulating the toxic effects related to increased concentrations. In experiments examining TNF upregulation in the brain following ischemic injury, TNF-BP administration immediately or 60 minutes after MCAO led to smaller infarct volumes, with animals receiving TNF-BP showing a 25% reduction in infarct volume up to 2 weeks following treatment compared to control groups [33,63,64,65]. Studies have investigated the protective role of mAb in the context of TNF upregulation in the brain following ischemic injury. Rats were administered exogenous TNF before transient or permanent MCAO. Despite TNF injection leading to increased infarct size, prior mAb injection significantly reduced hemispheric infarct size by 8%. Treatment with mAb prior to exogenous TNF also alleviated cognitive deterioration and mitigated neurological scores in the TNF groups [32].
5. TNF as a Therapeutic Target
The release of TNF by microglia and a subsequent pattern of increased sTNFR suggests an anti-inflammatory role of these receptors as they scavenge and decrease the serum concentration of this cytokine. Currently, two commonly used therapeutic options targeting TNF are etanercept and infliximab. These drugs share similar mechanisms of action, aiming to decrease inflammation caused by the pro-inflammatory release of TNF. Etanercept, marketed as Enbrel®, functions as an artificial sTNFR and consists of two TNFR2 receptors joined to the Fc portion of human IgG as demonstrated in Figure 2 [66]. It exhibits significantly greater binding efficiency and half-life compared to sTNFR2 (Table 3). The protective role of soluble receptors in physiological and neurological outcomes has been examined through controlled preclinical experiments. Rats subjected to fluid percussion injury (FPI) were treated with etanercept every 12 hours for 3 days. In the etanercept-administered groups, there was a significant decrease in cerebral ischemia area (p<.01), microglial production of TNF (p<.01), and neurological and motor dysfunction (p<.05) [67]. Additionally, etanercept therapy led to a significant reduction in glial scar formation, a major inhibitor of neuronal regeneration. This study suggests etanercept attenuates microglia-associated TNF expression without changes to astrocyte or neuron expression following TBI [67,68]. In a retrospective study of patients with chronic neurological deficits post-TBI, etanercept demonstrated clinical improvements in motor function, spasticity, walking, cognition, sensation, aphasia, and pain, even when administered 115 to 160 months after traumatic brain injury84.
Additional TNF-binding drugs include infliximab, a chimeric monoclonal anti-TNF antibody that has been used to reduce inflammation associated with a variety of chronic diseases and conditions [69,70]. Marketed as Remicade®, infliximab functions to target TNF, attenuating inflammation by binding to Fc receptors to decrease the activation of monocyte and caspase-3-induced oxidative stress [71,72]. While infliximab has been widely used for chronic diseases, its potential therapeutic role in neuroinflammation, like etanercept, warrants exploration. However, limitations associated with TNF inhibitors, such as etanercept or infliximab, have prompted research for novel TNF blockers. While Etanercept offers therapeutic benefits, unintended side effects have been observed, leading to decreased long-term functional outcomes. The drug’s inability to differentiate between sTNF and tmTNF poses challenges, inhibiting baseline activity and causing adverse effects [69]. Exogenous TNF blockers, including etanercept, may increase the risk of serious infections and cancer according to a meta-analysis of clinical trials comparing TNF-binding medications like etanercept and infliximab [73]. Etanercept, in placebo-controlled trials, demonstrated a 35% infection rate over the therapy course, associated with serious bacterial, viral, and fungal infections [69]. Infliximab has similar associations and a close link to tuberculosis development, often reflecting latent infection reactivation. Give the limitations and potential side effects of current TNF blockers like etanercept and infliximab, a second-generation drug, XPro1595, has been developed to address these issues and enhance patient outcomes.
5.1. Proposed Second-Generation Therapies
A novel sTNF inhibitor, XPro1595, has demonstrated success in inflammatory disease trials, notably for its ability to penetrate the BBB [74,75]. Also known as INB03, XPro1595 inhibits TNFR1 activation by forming a non-viable heterotrimer with sTNF as shown in Figure 2 [76]. Xpro1595 mimics the monomeric proteins in sTNF homotrimers and freely exchanges native proteins in a 1:2 or 2:1 ratio. XPro1595 is dominant-negative mutant of TNF, with specific mutations (A145R and Y87H) at the TNFR binding site, selectively inhibiting sTNF activity at TNFR1 receptors [77]. As mentioned earlier, TNFR1 is responsible for downstream neuroinflammatory effects and subsequent cognitive impairments. Unlike other TNF inhibitors, XPro1595 selectively inhibits the effects of TNFR1, ensuring continued TNFR2 function, crucial for neurogenesis and myelination. The newly formed XPro1595 heterotrimer does not interfere with the affinity of transmembrane TNF for TNFR1 or TNFR2. Allowing the continued function of TNFR2 while blocking TNFR1 differentiates XPro1595 from other TNF inhibitors and contributes to the drug’s increased effectiveness. To enhance its properties, XPro1595 has been modified via PEGylation, resulting in XENP1595, with increased half-life and reduced immunogenicity [77].
In preclinical studies, XPro1595 administration in mice subcutaneously 1-hour post-TBI reduced decreased glial activity, prevented dendritic degeneration, and improved spatial learning and memory [75]. Similar beneficial results have been demonstrated in focal cerebral ischemia models where topical Xpro1595 decreased infarct volumes 1 and 3 days post MCAO, outperforming Etanercept [40]. Additionally, XPro1595 treatment has shown promise in mitigating the effects of obesogenic diets in a preclinical model of Alzheimer’s disease and increased BBB permeability, attenuating inflammation and improving cognitive function [78].
A clinical trial evaluating XPro1595 treatment in patients with Alzheimer’s disease was completed in 2021. The open-label dose-identification study of XPro1595 in Alzheimer’s disease patient aimed to evaluate the drug’s safety, efficacy in reducing neuroinflammation, and impact on cognitive function. Study results are yet to be published but are available in online videos through the study sponsor’s website [79]. Results of the phase 1b study showed that weekly treatment with XPro1595 for 3 months in patients with signs of neuroinflammation demonstrated decreased white-matter free water and CSF inflammatory proteins as well as increased axonal fiber density and remyelination [79]. Prompted by these initial results, two phase II studies are currently recruiting with the same primary outcomes in a larger patient population to assess the long-term safety, tolerability, and efficacy of XPro1595.
Proof of concept trials for novel antibody-drug conjugates such as ABBV-3373 may be a potential alternative to current-generation anti-inflammatory therapies such as adalimumab. Composed of a glucocorticoid receptor modulator conjugated with an anti-TNF monoclonal antibody, ABBV-3373 was able to significantly reduce DAS28 (CRP) scores in rheumatoid arthritis patients with greater efficacy when compared to adalimumab (p<.022) [80]. It should be noted that the study lacked sufficient sample size to draw statistical conclusions past 12 weeks and that patients treated with ABBV-3373 received a placebo past 12 weeks but demonstrated similar responses to the 24-week adalimumab regimen. These results warrant continuing research and the potential application of antibody-drug conjugates in a cerebroprotective role.
6. Conclusions
This review investigates the role of TNF and potential therapeutic targets in cerebrovascular and neurodegenerative disease. TNF, a major inflammatory cytokine, plays a crucial role in neuroinflammation associated with these conditions. It can signal through its membrane-bound form or the homotrimeric sTNF, formed by cleaving tmTNF from the cell membrane via the proteolytic enzyme TACE. TNF activates two receptors, TNFR1 and TNFR2, with promoting detrimental effects, including cell death and increased inflammation, and TNFR2 demonstrating beneficial effects, such as enhanced neuronal tissue growth and CNS autoimmunity. Selectively targeting the sTNF and TNFR1 signaling pathway may facilitate new mechanisms to combat neuroinflammation.
The generation of sTNF and sTNFRs involved the proteolytic cleavage of tmTNF and TNFR1/2, respectively. Metalloproteinases, particularly ADAM8, preferentially cleave TNFR1 without affecting TNFR2, providing a potential therapeutic avenue to modulate TNFR1 signaling. Studies suggest a negative feedback mechanism in which TNF signaling leads to ADAM8 activation and therefore TNFR1 shedding. TNFR2 appears to be mainly cleaved through TACE, and further research is required to explore similar feedback mechanisms in sTNFR2 production.
The myriad of chemical and biological reactions that TNF can activate makes it an ideal target for therapeutic interventions. Several drugs, including etanercept and infliximab, have been developed to target TNF signaling. However, these drugs can cause unwanted side effects due to the indiscriminate inhibition of TNFR2 activation. The second-generation TNF inhibitor XPro1595 has been designed to selectively neutralize sTNF without affecting tmTNF or TNFR2, aiming to maintain pathways of neurogenesis and innate immunity [81]. Early results in mice with TBI demonstrated positive effects on dendritic plasticity, reduced neurodegeneration, and glial reactivity [75]. Clinical trials of the drug have demonstrated decreased neuroinflammation and neurodegeneration in Alzheimer’s Disease patients and the ability of XPro1595 to cross the BBB has aided in attenuating inflammatory biomarkers in other neurodegenerative diseases [79].
Limitations in using mice models for TNF inhibitors include the unresponsiveness of mouse TNF to human TNF inhibitors [82]. Humanized mouse models expressing human TNF (hTNF) or human TNFR (hTNFR) are utilized to study the molecular mechanisms of these cytokines [83]. However, overexpression of hTNF can result in spontaneous autoimmune diseases, prompting the use low-copy or tissue-specific models [83]. Humanized mice with hTNFR2 LoxP sites have been developed to address these challenges [84]. Recently, there has been a rise in various humanized mouse models used to study the TNF/TNFR1/2 mechanism [85,86]. Future research should leverage these humanized mouse models to test TNF inhibitor drugs, ensuring accuracy and replicability in human trials.
Author Contributions
Conceptualization, R.A., K.E., E.B., A.M., and B.L.; methodology, R.A., K.E., and E.B; validation, A.M., and B.L.; formal analysis, R.A., K.E., and E.B; investigation, R.A., K.E., E.B., A.M., and B.L.; writing—original draft preparation, R.A., K.E., and E.B; writing—review and editing, R.A., K.E., A.M., and B.L.; visualization, R.A., A.M., and B.L.; supervision, A.M., and B.L.; project administration, R.A., B.L.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Gregory AP, Dendrou CA, Attfield KE; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature. 2012, 488, 508. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia LA, Goeddel D V. Two TNF receptors. Immunol Today. 1992, 13, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Gough P, Myles IA. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Published online, 1975. [CrossRef]
- Chan FKM, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A Domain in TNF Receptors That Mediates Ligand-Independent Receptor Assembly and Signaling. Science (1979). 2000, 288, 2351–2354. [Google Scholar] [CrossRef]
- Ruiz A, Palacios Y, Garcia I, Chavez-Galan L, Nieuwenhuizen NE. Molecular Sciences Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections. Molecular Sciences Transmembrane TNF and Its Receptors TNFR1 and TNFR2 in Mycobacterial Infections. Published online 2021. [CrossRef]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008, 133, 693–703. [Google Scholar] [CrossRef]
- Li J, McQuade T, Siemer AB; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Green LA, Njoku V, Mund J; et al. Endogenous Transmembrane TNF-Alpha Protects Against Premature Senescence in Endothelial Colony Forming Cells. Circ Res. 2016, 118, 1512–1524. [Google Scholar] [CrossRef]
- Rauert H, Wicovsky A, Müller N; et al. Membrane Tumor Necrosis Factor (TNF) Induces p100 Processing via TNF Receptor-2 (TNFR2). J Biol Chem. 2010, 285, 7394. [Google Scholar] [CrossRef]
- Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel ULM. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JPY. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Emmerich CH, Ordureau A, Strickson S; et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A. 2013, 110, 15247–15252. [Google Scholar] [CrossRef] [PubMed]
- George MG, Fischer L, Koroshetz W; et al. CDC Grand Rounds: Public Health Strategies to Prevent and Treat Strokes. MMWR Morb Mortal Wkly Rep. 2019, 66, 479–481. [Google Scholar] [CrossRef]
- Shatri G, Senst B. Acute Stroke. StatPearls. Published online August 17, 2023. Accessed February 18, 2024. https://www.ncbi.nlm.nih.gov/books/NBK535369/.
- Onwuekwe I, Ezeala-Adikaibe B. Ischemic Stroke and Neuroprotection. Ann Med Health Sci Res. 2012, 2, 186. [Google Scholar] [CrossRef] [PubMed]
- Chen AQ, Fang Z, Chen XL; et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. [CrossRef]
- Joshi I, Andrew RD. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J Neurophysiol. 2001, 85, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012, 32, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008; 5. [CrossRef]
- Lambertsen KL, Meldgaard M, Ladeby R, Finsen B. A quantitative study of microglial-macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005, 25, 119–135. [Google Scholar] [CrossRef]
- Hill JK, Gunion-Rinker L, Kulhanek D; et al. Temporal modulation of cytokine expression following focal cerebral ischemia in mice. Brain Res. 1999, 820, 45–54. [Google Scholar] [CrossRef]
- Meschia JF, Brott T. Ischaemic stroke. Eur J Neurol. 2018, 25, 35–40. [Google Scholar] [CrossRef]
- Creed Pettigrew L, Kindy MS, Scheff S; et al. Focal cerebral ischemia in the TNFalpha-transgenic rat. Published online, 2008. [CrossRef]
- 26. Maddahi A, Kruse LS, Chen QW, Edvinsson L. The role of tumor necrosis factor-α and TNF-α receptors in cerebral arteries following cerebral ischemia in rat. J Neuroinflammation. 2011; 8. [CrossRef]
- 27. Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. Published online. [CrossRef]
- Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A. 1998, 95, 570–575. [Google Scholar] [CrossRef] [PubMed]
- King MD, Alleyne CH, Dhandapani KM. TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice. Neurosci Lett. 2013, 542, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Murakami Y, Saito K, Hara A; et al. Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J Neurochem. 2005, 93, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Ma L, Jiang Y, Dong YN, Gao J, Du B, Liu DW. Anti-TNF-alpha antibody attenuates subarachnoid hemorrhage-induced apoptosis in the hypothalamus by inhibiting the activation of Erk. Neuropsychiatr Dis Treat. 2018, 14, 525. [Google Scholar] [CrossRef] [PubMed]
- Belarbi K, Jopson T, Tweedie D; et al. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J Neuroinflammation. 2012; 9. [CrossRef]
- Low PC, Manzanero S, Mohannak N; et al. PI3Kδ inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nature Communications 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Barone FC, Arvin B, White RF; et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997, 28, 1233–1244. [Google Scholar] [CrossRef]
- Nawashiro H, Martin D, Hallenbeck JM. Neuroprotective effects of TNF binding protein in focal cerebral ischemia. Brain Res. 1997, 778, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Sumbria RK, Boado RJ, Pardridge WM. Combination stroke therapy in the mouse with blood-brain barrier penetrating IgG-GDNF and IgG-TNF decoy receptor fusion proteins. Brain Res. 2013, 1507, 91–96. [Google Scholar] [CrossRef]
- Li J, Zhang J, Zhang Y; et al. TRAF2 protects against cerebral ischemia-induced brain injury by suppressing necroptosis. Cell Death Dis. 2019; 10. [CrossRef]
- Aoki T, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol Commun. 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. Journal of Cerebral Blood Flow and Metabolism. 1998, 18, 1283–1287. [Google Scholar] [CrossRef]
- Bartsch JW, Wildeboer D, Koller G; et al. Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease-disintegrin ADAM8: Evidence for a protease-regulated feedback loop in neuroprotection. J Neurosci. 2010, 30, 12210–12218. [Google Scholar] [CrossRef]
- Yli-Karjanmaa M, Clausen BH, Degn M; et al. Topical administration of a soluble TNF inhibitor reduces infarct volume after focal cerebral ischemia in mice. Front Genet. 2019, 10, 781. [Google Scholar] [CrossRef]
- Nawashiro H, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J Cereb Blood Flow Metab. 1997, 17, 229–232. [Google Scholar] [CrossRef]
- Zhang YY, Huang NN, Zhao YX; et al. Elevated Tumor Necrosis Factor-a-induced Protein 8-like 2 mRNA from Peripheral Blood Mononuclear Cells in Patients with Acute Ischemic Stroke. Int J Med Sci. 2018, 15, 1713. [Google Scholar] [CrossRef] [PubMed]
- Emsley HCA, Smith CJ, Gavin CM; et al. Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007, 7, 1–12. [Google Scholar] [CrossRef]
- Tufekci KU, Vurgun U, Yigitaslan O; et al. Follow-up Analysis of Serum TNF-Related Apoptosis-Inducing Ligand Protein and mRNA Expression in Peripheral Blood Mononuclear Cells from Patients with Ischemic Stroke. Front Neurol. 2018; 9. [CrossRef]
- Cui G, Wang H, Li R; et al. Polymorphism of tumor necrosis factor alpha (TNF-alpha) gene promoter, circulating TNF-alpha level, and cardiovascular risk factor for ischemic stroke. Published online 2012. Accessed February 23, 2024. http://www.jneuroinflammation.com/content/9/1/235.
- 47. Lasek-Bal A, Jedrzejowska-Szypulka H, Student S; et al. The importance of selected markers of inflammation and blood-brain barrier damage for short-term ischemic stroke prognosis. J Physiol Pharmacol. 2019; 70. [CrossRef]
- Elneihoum AM, Falke P, Axelsson L, Lundberg E, Lindgärde F, Ohlsson K. Leukocyte Activation Detected by Increased Plasma Levels of Inflammatory Mediators in Patients With Ischemic Cerebrovascular Diseases. Stroke. 1996, 27, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Svensson EH, Söderholm M, Abul-Kasim K, Engström G. Tumor Necrosis Factor Receptor 1 and 2 Are Associated With Risk of Intracerebral Hemorrhage. Stroke. 2017, 48, 2710–2715. [Google Scholar] [CrossRef] [PubMed]
- Christensen H, Boysen G, Christensen E, Johannesen HH, Bendtzen K. Plasma cytokines in acute stroke. J Stroke Cerebrovasc Dis. 2002, 11, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Fragata I, Bustamante A, Penalba A; et al. Venous and arterial TNF-R1 predicts outcome and complications in acute subarachnoid hemorrhage. Neurocrit Care. 2019, 31, 107–115. [Google Scholar] [CrossRef]
- De Torres R, Mancha F, Bustamante A, Canhao P, Fragata I, Montaner J. Usefulness of TNFR1 as biomarker of intracranial aneurysm in patients with spontaneous subarachnoid hemorrhage. Future Sci OA. 2020, 6. [Google Scholar] [CrossRef]
- Witkowska AM, Borawska MH, Socha K, Kochanowicz J, Mariak Z, Konopka M. TNF-α and sICAM-1 in intracranial aneurismal rupture. Arch Immunol Ther Exp (Warsz). 2009, 57, 137. [Google Scholar] [CrossRef] [PubMed]
- Beeftink MMA, Ruigrok YM, Rinkel GJE, Van Den Bergh WM. Relation of serum TNF-α and TNF-α genotype with delayed cerebral ischemia and outcome in subarachnoid hemorrhage. Neurocrit Care. 2011, 15, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Lv S yin, Wu Q, Liu J peng; et al. Levels of Interleukin-1β, Interleukin-18, and Tumor Necrosis Factor-α in Cerebrospinal Fluid of Aneurysmal Subarachnoid Hemorrhage Patients May Be Predictors of Early Brain Injury and Clinical Prognosis. World Neurosurg. 2018, 111, e362–e373. [Google Scholar] [CrossRef] [PubMed]
- Hanafy KA, Grobelny B, Fernandez L; et al. Brain interstitial fluid TNF-α after subarachnoid hemorrhage. J Neurol Sci. 2010, 291, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Wu W, Guan Y, Zhao G; et al. Elevated IL-6 and TNF-α Levels in Cerebrospinal Fluid of Subarachnoid Hemorrhage Patients. Mol Neurobiol. 2016, 53, 3277–3285. [Google Scholar] [CrossRef] [PubMed]
- Fragata I, Bustamante A, Penalba A; et al. TNF-R1 Correlates with Cerebral Perfusion and Acute Ischemia Following Subarachnoid Hemorrhage. Neurocrit Care. 2020, 33, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Botran R, Crespo FA, Sun X. Soluble cytokine receptors in biological therapy. Expert Opin Biol Ther. 2002, 2, 585–605. [Google Scholar] [CrossRef]
- Jones SA, Rose-John S. The role of soluble receptors in cytokine biology: The agonistic properties of the sIL-6R/IL-6 complex. Biochim Biophys Acta Mol Cell Res. 2002, 1592, 251–263. [Google Scholar] [CrossRef]
- Mohler,’ KM, Torrance DS, Smith CA; et al. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. The Journal of Immunology. 1993, 151, 1548–1561. [Google Scholar] [CrossRef]
- Olsson I, Gatanaga T, Gullberg U, Lantz M, Granger GA. Tumour necrosis factor (TNF) binding proteins (soluble TNF receptor forms) with possible roles in inflammation and malignancy. Eur Cytokine Netw. 1993, 4, 169–180. [Google Scholar]
- Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci Lett. 1996, 218, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Lantz M, Malik S, Slevin ML, Olsson I. Infusion of tumor necrosis factor (TNF) causes an increase in circulating TNF-binding protein in humans. Cytokine. 1990, 2, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Engelmann H, Aderka D, Rubinstein M, Rotman D, Wallach D. A tumor necrosis factor-binding protein purified to homogeneity from human urine protects cells from tumor necrosis factor toxicity. Journal of Biological Chemistry. 1989, 264, 11974–11980. [Google Scholar] [CrossRef]
- Seckinger P, Isaaz S, Dayer JM. Purification and biologic characterization of a specific tumor necrosis factor α inhibitor. Journal of Biological Chemistry. 1989, 264, 11966–11973. [Google Scholar] [CrossRef]
- Famenini S, Sako EY, Butler DC, Wu JJ. Etanercept. Moderate to Severe Psoriasis: Fourth Edition. Published online July 24, 2023, 191–198. [CrossRef]
- Chio CC, Chang CH, Wang CC; et al. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-α. BMC Neurosci. 2013, 14. [Google Scholar] [CrossRef]
- Chio CC, Lin JW, Chang MW; et al. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem. 2010, 115, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatolog Treat. 2004, 15, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Remicade (Infliximab) : Gastroenterology Nursing. Accessed February 23, 2024. https://journals.lww.com/gastroenterologynursing/citation/1998/11000/remicade__infliximab_.8.aspx.
- Zhou Y, Fan R, Botchway BOA, Zhang Y, Liu X. Infliximab Can Improve Traumatic Brain Injury by Suppressing the Tumor Necrosis Factor Alpha Pathway. Mol Neurobiol. 2021, 58, 2803–2811. [Google Scholar] [CrossRef] [PubMed]
- Pergel A, Tümkaya L, Çolakoğlu MK; et al. Effects of infliximab against carbon tetrachloride-induced intestinal injury via lipid peroxidation and apoptosis. Hum Exp Toxicol. 2019, 38, 1275–1282. [Google Scholar] [CrossRef]
- Li J, Zhang Z, Wu X, Zhou J, Meng D, Zhu P. Risk of Adverse Events After Anti-TNF Treatment for Inflammatory Rheumatological Disease. A Meta-Analysis. Front Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Karamita M, Barnum C, Möbius W; et al. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 2017, 2. [Google Scholar] [CrossRef]
- Larson K, Damon M, Randhi R, Nixon-Lee N, Dixon KJ. Selective inhibition of soluble TNF using XPro1595 improves hippocampal pathology to promote improved neurological recovery following traumatic brain injury in mice. CNS Neurol Disord Drug Targets. 2022, 22, 1378–1390. [Google Scholar] [CrossRef]
- Steed PM, Tansey MG, Zalevsky J; et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science. 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Fischer R, Kontermann RE, Pfizenmaier K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front Cell Dev Biol. 2020, 8. [Google Scholar] [CrossRef]
- MacPherson KP, Sompol P, Kannarkat GT; et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed]
- INmune Bio Announces First Patient Dosed in Phase 2 XPro1595 Trial for Treatment of Neuroinflammation as a Cause of Alzheimer’s disease. Accessed February 23, 2024. https://www.inmunebio.com/index.php/newsroom/2022-news/inmune-bio-announces-first-patient-dosed-in-phase-2-xpro1595-trial-for-treatment-of-neuroinflammation-as-a-cause-of-alzheimers-disease?highlight=WyJ4cHJvMTU5NSJd.
- Buttgereit F, Aelion J, Rojkovich B; et al. Efficacy and Safety of ABBV-3373, a Novel Anti-Tumor Necrosis Factor Glucocorticoid Receptor Modulator Antibody-Drug Conjugate, in Adults with Moderate-to-Severe Rheumatoid Arthritis Despite Methotrexate Therapy: A Randomized, Double-Blind, Active-Controlled Proof-of-Concept Phase IIa Trial. Arthritis Rheumatol. 2023, 75, 879–889. [Google Scholar] [CrossRef]
- Randhi R, Damon M, Dixon KJ. Selective inhibition of soluble TNF using XPro1595 relieves pain and attenuates cerulein-induced pathology in mice. BMC Gastroenterol. 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Winsauer C, Kruglov AA, Chashchina AA, Drutskaya MS, Nedospasov SA. Cellular sources of pathogenic and protective TNF and experimental strategies based on utilization of TNF humanized mice. Cytokine Growth Factor Rev. 2014, 25, 115–123. [Google Scholar] [CrossRef]
- Gogoleva VS, Atretkhany KSN, Dygay AP, Yurakova TR, Drutskaya MS, Nedospasov SA. Current Perspectives on the Role of TNF in Hematopoiesis Using Mice With Humanization of TNF/LT System. Front Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Atretkhany KSN, Mufazalov IA, Dunst J; et al. Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc Natl Acad Sci U S A. 2018, 115, 13051–13056. [Google Scholar] [CrossRef]
- Williams SK, Fairless R, Maier O; et al. Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis. Sci Rep. 2018, 8. [Google Scholar] [CrossRef]
- Dong Y, Fischer R, Naudé PJW; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci U S A. 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of TNFR1 and TNFR2 pathways. Current research indicates that TNFR1 primarily promotes cell death events through its TRADD complex whereas TNFR2 signaling primarily promotes cellular survival through non-canonical NFκB activation. Note that the process by which TNFR2 activates TAK1 is currently unknown. Furthermore, adequate and inadequate caspases refer to the concentration of Caspase 8 relative to RIPK1 and RIPK3, with adequate caspases resulting in the complete degradation of RIPK1 and 3 leading to cell death and inadequate caspases resulting in the accumulation of RIPK1 and 3 and necrosis. Created with BioRender.com.
Figure 1.
Illustration of TNFR1 and TNFR2 pathways. Current research indicates that TNFR1 primarily promotes cell death events through its TRADD complex whereas TNFR2 signaling primarily promotes cellular survival through non-canonical NFκB activation. Note that the process by which TNFR2 activates TAK1 is currently unknown. Furthermore, adequate and inadequate caspases refer to the concentration of Caspase 8 relative to RIPK1 and RIPK3, with adequate caspases resulting in the complete degradation of RIPK1 and 3 leading to cell death and inadequate caspases resulting in the accumulation of RIPK1 and 3 and necrosis. Created with BioRender.com.
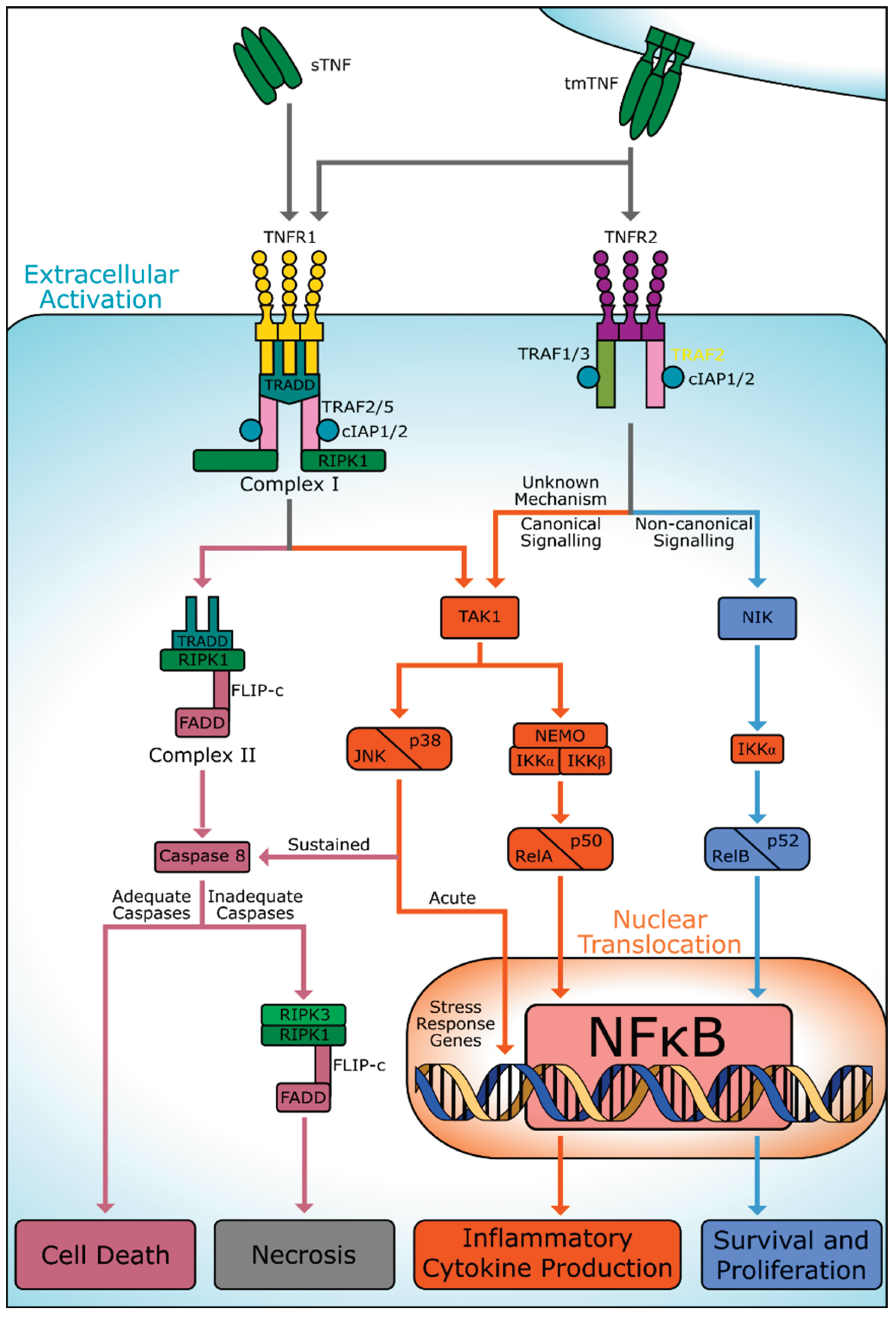
Figure 2.
Illustration of various sTNF signaling inhibitors. Infliximab, Etanercept, and sTNRF1 prevent the interaction of sTNF and tmTNF homotrimers with their respective receptors. XPro1595 prevents the formation of only the sTNF homotrimer, thereby inhibiting only TNFR1 activation. Created with BioRender.com.
Figure 2.
Illustration of various sTNF signaling inhibitors. Infliximab, Etanercept, and sTNRF1 prevent the interaction of sTNF and tmTNF homotrimers with their respective receptors. XPro1595 prevents the formation of only the sTNF homotrimer, thereby inhibiting only TNFR1 activation. Created with BioRender.com.
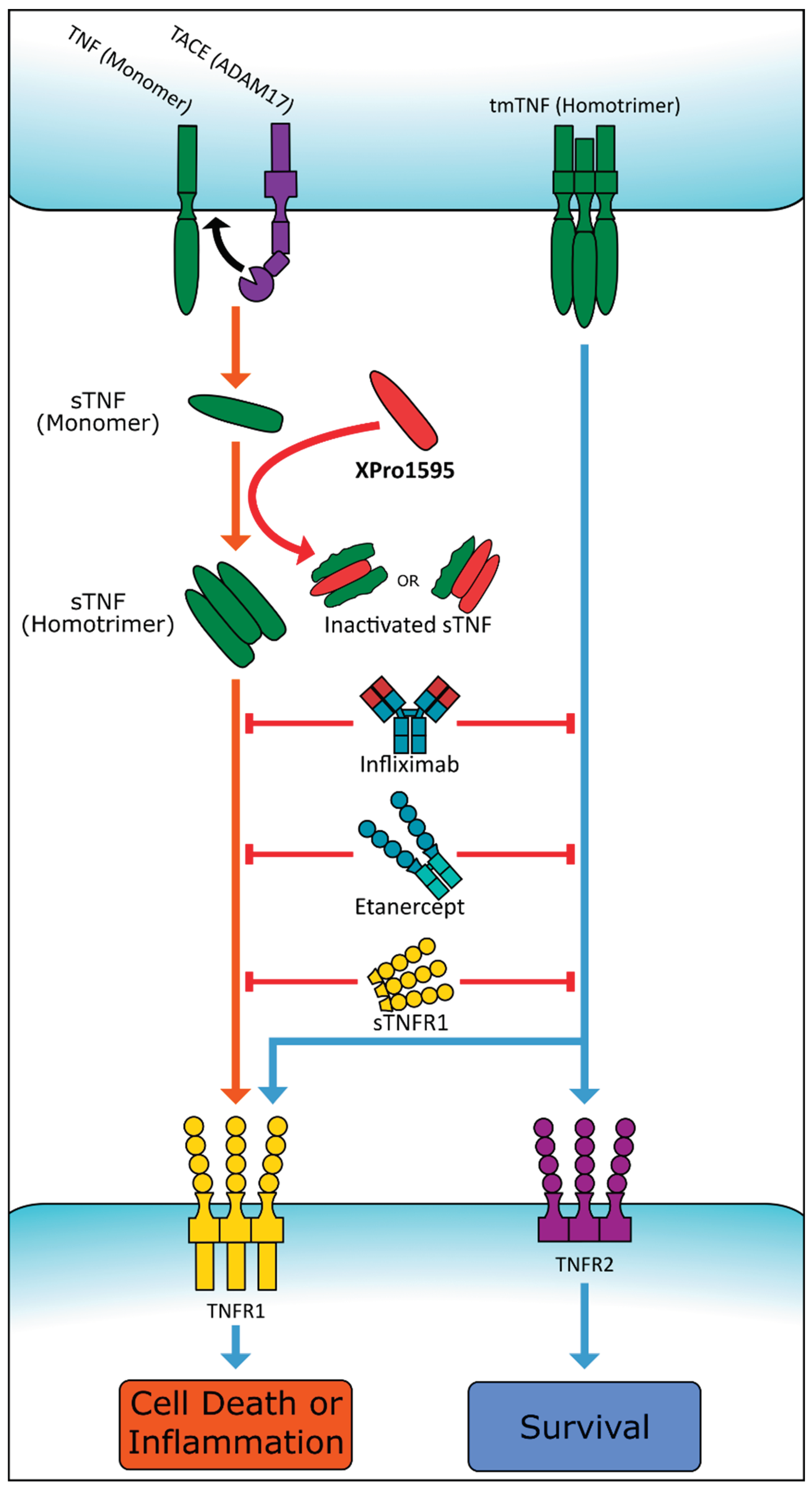

Table 1.
Pre-Clinical Studies Analyzed Regarding the Effects of TNF Stimulation.
| Reference | Age, Weight, Species Tested, Population Size, Sex Predominance, Duration of Study | Disease Studied & Model | Drugs/Treatments | Anatomical Outcomes | Functional Outcomes | |
|---|---|---|---|---|---|---|
| Chen et al. (2019)[18] Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke |
250-300gSprague-Dawley ratsn=ND100% M7d-duration | tMCAO (2h): intraluminal vascular occlusion Mice sacrifice: 1-7d post-ischemia |
Microglial cells transfected with 10nM siRNA for 24h. Infliximab (anti-TNF) administered by tail vein injection (5mg/kg) at 1d, 2d, and 3d post-reperfusion. 7d survival. | Levels of TNF post-tMCAO consistent with double staining results of rat brain tissue sections.TNF ELISA analysis shows decreased microglia-secreted TNF after oxygen-glucose deprivation/reoxygenation (n=6; p<.001 vs. control group p<.0001). Infliximab dramatically reduced microglia migration. | Neurological severity score results revealed that multiple doses of infliximab improved neurological function 3d after tMCAO (p<.05). | |
| King et al. (2013)[27] TNF-alpha receptor antagonist, R-7050, improves neurological outcomes following intracerebral hemorrhage in mice |
8-10wkCD-1 micen=32100% M72h-duration | ICH (collagenase model): 0.5mm diameter hole drilled over parietal cortex. 26-gauge Hamilton syringe, loaded with 0.04U of bacterial type IV collagenase in 0.5μL saline lowered 3mm into left striatum. Syringe depressed at 450nL/min. | R-7050 (6–18mg/kg) administered via IP route at the time of injury or up to 2h post-ICH. | R-7050 maintains BBB integrity post-ICH. increased Evans blue brain tissue in sham-operated mice from 12.2±1.5μg to 47.2±5.8μg at 24h post-ICH (p<.01 vs. sham). Decreased Evans blue brain tissue extravasation in R-7050 (6mg/kg) to 28.7±5.9μg and 30.3±1.9μg when administered at 0.5h or 2h post-ICH, respectively (p<.05 and p<.01 vs. ICH). TNF elevated in ICH rats. | R-7050 resulted in protective effect during first 3d post-ICH, as compared to placebo, with complete decrease in neurological deficits (p<.05). | |
| Murakami et al. (2005)[28] Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus |
10-15wk WT C57BL/6J mice n=ND100% M7d-duration |
Transient Global ischemia: bilateral common carotid arteries occluded by aneurysm clips (75g pressure) for 20min. Mice sacrificed 12h post-ischemia. |
ND | Hippocampal TNF mRNA levels increased (sham: 0.16±0.064 vs. ischemia: 0.459±0.096; p<.01) 3h after recirculation from endogenous brain cells, then decreased to sham-operated levels by 24h. Increase of TNF mRNA again at 36h (sham: 0.160±0.064 vs. ischemia: 0.760±0.092; p<.01). Up-regulated TNF protein level in the hippocampus only at 6h post-ischemia (sham: 0.946±0.150 vs. ischemia: 6.534±2.646; p<.05). | ND | |
| Ma et al. (2018)[29] Anti-TNF-alpha antibody attenuates subarachnoid hemorrhage-induced apoptosis in the hypothalamus by inhibiting the activation of Erk |
250-350gWistar Ratsn=153100% M7d-duration | SAH: fresh non-heparinized blood (0.3mL) from femoral artery injected into cisterna magna at 0.05 mL/min. | U0126 (dissolved in PBS; 250 ng/μL, 10μL per rat) microinfused into left lateral cerebral ventricle 30min before SAH at 0.5μL/min. | U0126 injection blocked SAH-induced TNF increase (p=.024).U0126 micro infusion decreased mRNA levels in hypothalamus (p=.001).Apoptotic and anti-apoptotic gene expression levels increase post-SAH. U0126 decreased Erk phosphorylation. | Decreased anxiety-like behaviors. PBS-injected SAH rats showed decreased total traveled distance compared to control group at 2d and 7d post-SAH. U0126 SAH rats spent less time in the center of the open field both at 2d (p=.003) and 7d (p=.026) post-surgery. | |
| Pettigrew et al. (2008)[25] Focal cerebral ischemia in the TNF alpha-transgenic rat | 250-350gSprague-Dawley, carried the TNF genen=ND100% M7d-duration | Focal cerebral ischemia: Zea Longa suture-occlusion of MCA for 1h. Cortical perfusion measured by LDF. Reperfusion at 3 or 24h. |
ND | TNF-Tg rats had greater infarct volume than non-Tg control at 24h (p≤.05) and 7d (p≤.01).TNF-Tg rats had decreased cortical perfusion within 10min of MCAO (p≤.05). Neural cellular apoptosis increased in transgenic animals with elevated caspase-3 activity (p≤.05) and DNA fragmentation (p≤.001) at 24h. | TNF showed a dose-dependent adverse effect on motor function. | |
| Belarbi et al. (2012)[30] TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation |
3moF344 rats (Charles River Labs) n=28100% M43d-duration | Chronic neuroinflammation via LPS-induced sustained microglia activation. | Artificial cerebrospinal fluid (aCSF; n=11) or lipopolysaccharide (LPS; n=17) loaded into an osmotic minipump at 0.25μl/h; 28d delivery into the fourth brain ventricle. | LPS-vehicle rats had increased TNF mRNA levels (129.97±9.09%; p<.05) and increased TNFR2 expression (124.91±6.25%; p<.05) when compared to aCSF-vehicle rats.DT treatment returned TNF mRNA to control levels (102.18±8.90%; p<.05 vs. LPS-vehicle). | Neuroinflammation decreased novel place recognition, spatial learning, and memory function, but not novel object recognition (aCSF-vehicle: p=.0016; aCSF-DT: p=.0124; LPS-vehicle: p<.0001; LPS-DT: p=.0094).DT treatment restored cognitive function in LPS-infused rats. All treatment groups had similar swim velocity (p=.6878) and similar escape latency to locate the platform (p=.7775). | |
| Low et al. (2014)[31] PI3Kδ inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model | 12-22wkp110d kinase-dead PI3Kδ mice, C57BL/6n=ND100% M20wk-duration | tMCAO (1h): internal carotid artery occluded with a clipReperfusion: 24h to 72h | 40 mg/kg CAL-101 by 25mL infusion into the femoral vein, 15min before ischemia/reperfusion treatment, or 3h and 6h after reperfusion. | PI3Kδ controls intracellular TNF trafficking in macrophages and is prospective target to limit neuroinflammation. p110δ mice had decreased infarct volume than tMCAO WT (p<.005). PI3Kδ inhibition protects from ischemia. Brain PI3Kδ drives ischemia-induced leukocyte infiltration. | p110 mice had decreased brain damage and neurological deficit at 72h post-ischemia/reperfusion than WT mice. | |
| TNFR1 | Barone et al. (1997)[32] Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury | 280-340gSpontaneously Hypertensive Rat (SHR) n=ND100% M3d-duration | IS: induced by permanent or tMCAO for 80m or 160m Reperfusion: 24h |
TNF (2.5 or 25pmol) 24h before pMCAO or tMCAO.mAb (60pmol) or sTNFR1 (0.7nmol) administered ICV 30m before and after tMCAO at 3h and 6h. | Infarct size increase seen in injections of:- 2.5pmol TNF 24h pre-pMCAO (p<.05)- 25pmol TNF (p<.01) (greater than 2.5pmol TNF pre-pMCAO)- tMCAO (80 and 160min) 2.5pmol TNF (p<.05).Reduced focal ischemic injury seen in treatment before and during 24h of focal ischemia, with repeated ICV mAb or sTNFR1 (p<.05). | Neurological deficits not decreased by blocking TNF. |
| Nawashiro et al. (1997)[33] Neuroprotective effects of TNF binding protein in focal cerebral ischemia | 22-26gBALB/Cn=67100% M2wk-duration | Permanent MCAO: anesthetized mice dura and arachnoid opened and left MCAO performed by electrocoagulation. | Treatment groups topically given 3mg/kg TNF-BP or vehicle immediately or 60m post MCAO. | All treatments resulted in reduced infarct volumes, with TNF-BP treatment most potent (p<.001). Significant neuroprotection when TNF-BP administered 60m post-MCAO. In mice treated with TNF-BP, 3/7 showed no DNA fragmentation. | ND | |
| Sumbria et al. (2013)[34] Combination stroke therapy in the mouse with blood-brain barrier penetrating IgG-GDNF and IgG-TNF decoy receptor fusion proteins | 25-33gC57Bl/6J micen=38100% M7d-duration | tMCAO (1h): electrocoagulation of ECA branchesReperfusion: 23h or 7d | After 1h MCAO, 100μl total volume of either saline, rGDNF (170μg/kg), GDNF (1mg/kg), or GDNF and TNFR (1mg/kg each) intravenously injected via tail vein at 45min post arterial occlusion, after 1h MCAO. | Combined GDNF and TNFR fusion protein treatment reduced hemispheric, cortical, and subcortical stroke volumes. Decreased hemispheric, cortical, and subcortical stroke volume present at 7d post-injury. | ND | |
| TNFR2 | Li et al. (2019)[35] TRAF2 protects against cerebral ischemia-induced brain injury by suppressing necroptosis | 25-30gICR micen=86100% M48h-duration | tMCAO (1h): Ligation carried out 1cm to internal and external cervical vascular branch. Reperfusion at 24h. |
2 injections of negative control shRNA lentivirus or TRAF2 shRNA lentivirus (1μl 5×108 TU/ml) injected into ipsilateral striatum of the mice 2wk before MCAO. | TRAF2 levels increased in the ipsilateral cortex 24h post-reperfusion.TRAF2 knockdown increased infarct volumes, cell death, and neuroinflammation. Post-ischemic induction of TRAF2 protected microglia and neurons against necroptotic cell death. | ND |
| TNF, TNFR1, TNFR2 | Aoki et al. (2014)[36] Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation | 7wkSprague-DawleyTNFR1−/− micen=122100% M5mo-duration | SAH by intracranial aneurysm ligation of the left carotid artery and systemic hypertension induced by salt overloading and left renal artery ligation. | ND | Intracranial artery TNF increase in rats with advanced stage IA 3mo post-aneurysm.Increased TACE activity compared to control rats. suggesting TNF production up- TNFR1-deficient mice had decreased IA when compared to WT mice (p=.012). | ND |
| Maddahi et al. (2011)[37] The role of tumor necrosis factor-α and TNF-α receptors in cerebral arteries following cerebral ischemia in rat | MCAO: 300-350g SAH: 300-350gWistar-Hanover rats n=24100% M48h-duration |
MCAO: Blood flow in the right MCA blocked by an intraluminal occluderSAH: suture to perforate ICA. Protein expression evaluated after 48h. |
ND | MCAO: TNF, TNF-R1, and TNF-R2 protein expression in increased compared to control group, P< .05, <.01, and <.05, respectively. SAH: TNF, TNF-R1, and TNF-R2 protein expression increased compared to control, P<.05, <.01, and <.05, respectively. |
ND | |
| Gary et al. (1998)[38] Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor | 3mo25-30gC57BL/6n=129100% M24h-duration | tMCAO (1h): Middle cerebral artery blocked by rounded nylon thread.Reperfusion: 24h | Kainic acid (.3µg in 0.5 µL) injected into the dorsal hippocampus. | WT and p75−/− (TNFR2) mice had similar infarct sizes. p55−/− (TNFR1) and p55/p75−/−mice had increased infarct sizes when compared to WT and p75−/− mice. | No change in brain structure or behavioral test performance in p55/p75 −/− mice when compared to WT. | |
| sTNFR1 and sTNFR2 | Bartsch et al. (2010)[39] Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease-disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection | wr/+ Adam8+/+wr/+ Adam8−/−wr/wr Adam8+/−wr/wr Adam8−/−n=15055d-duration | Motor Neuron Disease (CNS) | NA | 7 to 10 times increased expression of ADAM8 mRNA after administration of 100U/ml TNF. Administration of ≥500U/ml of TNF resulted in decline in ADAM8 mRNA levels. Concentration of sTNFR1 significantly increased in standard mice, but not in homozygous ADAM8 deficient. | ADAM8-deficient Wobbler mice showed more dramatic decline in the forelimb force at P18 and complete loss of force at approximately day 24 after birth. |
| Yli-Karjanmaa et al. (2019)[40] Topical administration of a soluble TNF inhibitor reduces infarct volume after focal cerebral ischemia in mice |
7-8wkC57BL/6 n=56100% MND | MCAO by electrocoagulation | Topical (2.5 mg/ml for 3d) or ICV (1.25mg) at 1μl/h 30m post-pMCAO with saline, XPro1595, or etanercept post-pMCAO. Topical XPro1595 concentration in brain tissue 1d post-pMCAO: 630,300±160,000pg/mg. ICV XPro1595 concentration: 69,400±51,300pg/mg. | Topical XPro1595 decreased infarct volume at 1d and 3d post-pMCAO. TNF mRNA expression increased 1d and 3d post-pMCAO in mice treated topically with XPro1595 when compared to saline and etanercept mice. Topical etanercept showed no effect. | No difference in grip strength test for ICV Etanercept and ICV XPro1595. | |
| Nawashiro et al. (1997)[41] Inhibition of tumor necrosis factor and amelioration of brain infarction in mice | 22-26gBALB/C micen=38100% M24h-duration | MCAO by electrocoagulation | sTNFR1 linked to polyethylene glycol (TNF-BP) immediately after MCAO, 0.3 or 3mg/kg body weight of TNF-BP (1μl or 10μl) applied topically. |
26% reduction in brain damage volume in animals that received 3mg/kg of TNF-BP, 10% in those given 0.3mg/kg of TNF-BP, 15% in those injected with 3mg/kg of TNF-BP IP, and 20% in animals administered 3mg/kg of TNF-BP IV. | NA |
aSAH, aneurysmal subarachnoid hemorrhage; BBB, blood-brain barrier; CI, cerebral infarction; CSF, cerbrospinal fluid; CVA, cerebrovascular accident; CCI, Charlson Comorbidity Index; CHI, closed head injury; CTA, computed tomography angiogram; CCI, cortical controlled impact d, day; DCI, delayed cerebral ischemia; DT, 3,6′-dithiol thalidomide; F, female; FPI, fluid percussion injury; g, gram; h, hour; ICH, intracerebral hemorrhage; IS, ischemic stroke; LFP, lateral fluid percussion; LPS, lipopolysaccharide; M, male; MCA, middle cerebral artery; MCAO, middle cerebral artery occlusion; ms, millisecond; min, minute; mRS, modified Rankin Scale; mAB, monoclonal antibody; mo, month; NIHSS, National Institute of Health Stroke Scale; ND, not discussed; OR, odds ratio; SD, Sprague Dawley rats; SAH, subarachnoid hemorrhage; TACE, tumor necrosis factor-α converting enzyme; tMCAO, transient middle cerebral artery occlusion; TBI, traumatic brain injury; TNF-BP, tumor necrosis factor-binding protein; TRAIL, tumor necrosis factor related apoptosis inducing ligand; wk, week; WT, wildtype; yr, year.
Table 2.
Current Clinical Studies Regarding TNF Activation in the Context of Stroke.
| Reference | Mean Age, Population size, Sex, Duration, Location | Exclusion Criteria | Anatomical Outcomes | Functional Outcomes | |
|---|---|---|---|---|---|
| IS | Zhang et al. (2018)[42] Elevated tumor necrosis factor-a-induced protein 8-like 2 mRNA from peripheral blood mononuclear cells in patients with acute ischemic stroke |
IS: 68.00yr n=182 49.4% M Ctrl: 64.50yr n=40 42.5% M 8mo-duration Shandong, China |
Patients with TIA, greater than 24h from stroke onset, hemorrhage stroke, severe infections, or malignant tumors. | mRNA levels of TNF in peripheral blood mononuclear cells elevated compared controls [3.74 (2.40-5.48) vs 2.16 (1.68-3.69); p<.001]. TNF mRNA median levels lower in survivals than non-survivals [TNF, 3.42 (2.20-4.86) vs 5.67 (4.67-7.72); p<.001]. | After 3mo follow-up, 33 AIS patients had died. |
| Emsley et al. (2007)[43] Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production |
IS: 69.6±13.0yr n=36 67% M Ctrl: 68.7±12.6yr n=36 67% M 12mo-duration Salford, England |
Patients with decreased symptoms since onset, an indeterminant onset of symptoms, or evidence of active malignancy. | Plasma sTNFR1 concentration was correlated with infarct volume in the first week (r=.62, p=.001), 3mo (r=.59, p<.001), and 1yr (r=.57, p=.001). | 14 AIS patients died by 12mo. Causes of death: index stroke: 8 recurrent stroke:1 pulmonary embolism: 1 left ventricular failure secondary to myocardial infarction: 1 sepsis: 3 Barthel Index (BI) for Activities of Daily Living (ADL) at 3mo (p=.001) and 1yr (p<.001). |
|
| Tufekci et al. (2018)[44] Follow-up analysis of serum TNF-related apoptosis-inducing ligand protein and mRNA expression in peripheral blood mononuclear cells from patients with ischemic stroke |
IS: 67.35yr n=95 58.6% M Ctrl: 69.7yr n=95 39.4% M 6mo-duration Izmir, Turkey |
Inclusion: Patients admitted to hospital within 24h of stroke onset with no history of previous acute ischemic stroke. | Lower TRAIL levels of stroke patients than healthy control (p<.0001). Serum TRAIL levels were increased 1mo after stroke onset (p=.002). Stroke patients had higher TRAIL mRNA expression as compared to the controls (p<.0001). | ND | |
| Cui et al. (2012)[45] Polymorphism of tumor necrosis factor alpha (TNF-alpha) gene promoter, circulating TNF-alpha level, and cardiovascular risk factor for ischemic stroke |
IS: n=961 Ctrl: n=821 ND 5yr-duration Wuhan, China |
Non-Chinese Han populations. | Higher serum TNF levels in IS than in control (SMD=2.33, 95%CI=1.85-2.81). | ND | |
| Lasek-Bal et al. (2019)[46] The importance of selected markers of inflammation and blood-brain barrier damage for short-term ischemic stroke prognosis |
73.11±11.48yr n=138 46.04% M 30d-duration University of Silesia |
No history of a previous stroke, admission greater than 24h after symptoms, pre-stroke status on mRS less than 1 point. | Patients with higher NIHSS scores showed higher TNF concentrations p=.00218. Median NIHSS score of all patients is 3. Data suggests TNF has an unfavorable effect on stroke infarct volume. | Patients with good functional status on 30d, despite poor initial status, had higher TNF levels. Median mRS score is 2. Influence of TNF on neurological improvement compared to other cytokines (p<.04). High TNF concentration on the first day of stroke correlates with severe status. |
|
| Elneihoum et al. (1996)[47] Leukocyte activation detected by increased plasma levels of inflammatory mediators in patients with ischemic cerebrovascular diseases |
IS: 70yr n=72 59.7% M TIA: 69yr n=48 54.2% M Ctrl: 68yr n=35 57.1% M ND Malmo, Sweden |
Patients with ICH, renal deficit, acute infection, or vascular ischemia. | Increased TNFR1 levels in stroke group than in control (3.1 μg/L vs. 2.1 μg/L, p<.04). Increased TNF in 18% of patient group and 17% of control group. Correlation between TNFR1 and TNF plasma levels (p<.0006). |
ND | |
| ICH | Svensson et al. (2017)[48] Tumor necrosis factor receptor 1 and 2 are associated with risk of intracerebral hemorrhage |
ICH: 62yr n=220 48% M Ctrl: 62yr n=244 49% M 48mo-duration Malmö, Sweden |
Missing plasma samples, laboratory errors, or missing blood pressure information. | Patients who developed ICH during follow-up had higher concentrations of TNFR1/2 (TNFR1: OR, 2.28; 95% CI p=.006; TNFR2: OR, 1.77; p=.008). | TNFR1 and TNFR2 associated with increased fatal ICH (TNFR1: OR, 4.42; TNFR2: OR, 2.90) and with poor mRS results. |
| Christensen et al. (2002)[49] Plasma cytokines in acute stroke |
ICH: 74yr n=17 54% M CI: n=162 3mo-duration Copenhagen, Denmark |
Within 24h of stroke onset. | Positive correlation between sTNFR1&2 levels and age (p<.001). Plasma levels in pg/mL of cytokines and soluble cytokine receptors on inclusion/3mo: sTNFR1 =1.306/1.469 (p=.044) sTNFR2 =2.307/2.805 (p<.001). Higher levels of sTNFR1&2 correlated to an unfavorable outcome at 3mo. |
Increased levels of sTNFR1 and sTNFR2 correlated to bladder cancer, colon cancer, disseminated terminal cancer without identified primary cancer, and urinary tract infection in treatment at 3mo. 3mo fatality rate was 11.2%. | |
| SAH | Fragata et al. (2019)[50] Venous and arterial TNF-R1 predicts outcome and complications in acute subarachnoid hemorrhage |
56.7±16.1yr n=58 39.7% M 6mo-duration ND |
Inclusion: Patients admitted in the first 72h of SAH symptoms. Exclusion: Patients in very poor clinical condition. |
Correlation of arterial and venous levels of TNFR1 (p<.001). No association of TNFR1 with DCI. Cut-off for arterial TNFR1 of 1523.7pg/mL had 75% sensitivity/66% specificity for the prediction of hydrocephalus. | Patients with high venous TNFR1 are correlated with poor outcomes in SAH by GCS and Fisher scales (OR 8.74; p=.018). |
| de Torres et al. (2019)[51] Usefulness of TNFR1 as biomarker of intracranial aneurysm in patients with spontaneous subarachnoid hemorrhage |
56.7±16.1yr n=58 37.9% M 6mo-duration Seville, Spain |
Older than 18yr, within the first 72h of acute SAH, imaging studies performed within the first 72h of SAH. | Venous TNFR1 levels greater than 1658pg/ml had 46.3% sensitivity/94.1% specificity for aneurysms and is independent predictor for its presence [OR=12.03 (1.13-128.16); p=.039]. | Higher clinical and radiological severity in the aneurysm group, both on the HH grade (3 vs 1; p=.022) and on the Fisher scale (4 vs 3; p=.019). | |
| Witkowska et al. (2009)[52] TNF-alpha and sICAM-1 in intracranial aneurismal rupture |
SAH: 56yr n=27 51.8% M Ctrl: 55yr n=17 72h-duration Poland |
Diagnosed with intracranial aneurismal after 72h of ER arrival. | Concentrations of TNF in patients with SAH were 12.42 ± 9.70 pg/ml, and 11.29 ± 8.80 pg/ml in control. Increased TNF levels in the CSF observed 4-10d after SAH. | All GCS scores between 11 and 15. 74% of patients presented with HH grades 0–1. 81% of patients presented with GOS scores between 3 and 5. | |
| Beeftink et al. (2011)[53] Relation of serum TNF-α and TNF-α genotype with delayed cerebral ischemia and outcome in subarachnoid hemorrhage |
TNF mutation: 57yr n=31 26% M Ctrl: 57yr n=67 30% M 3mo-duration Utrecht, The Netherlands |
ND | High-serum TNF levels during days 0–12 showed an adjusted HR of 0.6 for DCI. Non-WT TNF genotype individuals exhibited decreased serum TNF levels with an HR of 0.4 for DCI. | Lower risk of DCI for patients with higher values of TNF. Patients with the variant allele tended to have a lower serum TNF. Patients with good outcomes and control patients had low TNF levels. | |
| Lv et al. (2018)[54] Levels of interleukin-1β, interleukin-18, and tumor necrosis factor-α in cerebrospinal fluid of aneurysmal subarachnoid hemorrhage patients may be predictors of early brain injury and clinical prognosis |
55yr n=81 42% M 6mo-duration Nanjing, China |
Patients who had previously experienced central nervous system diseases or are on aspirin/anti-inflammatory analgesics. | TNF in the CSF of aSAH patients with WFNS grades 4 and 5 were higher than those of patients with WFNS grades 1 and 3 (p<.0001). TNF in the CSF of aSAH patients with Fisher grades 3 and 4 were higher than those of patients with Fisher grades 1 and 2 (p<.01). | TNF (1-3d, r=.4982; 4-6d, r=.5470, and 7-9d, r=.4757) was correlated with mRS (p<.001). Patients with a poor outcome had higher TNF levels than those with good outcomes. | |
| Hanafy et al. (2010)[55] Brain interstitial fluid TNF-alpha after subarachnoid hemorrhage |
48yr n=14 29% M 72h-duration New York, NY |
Patients without cerebral micro dialysis or at least 24h of recoverable data. | Brain interstitial fluid glucose was associated with TNF (p=.018). Aneurysm size was a strong predictor of the TNF response (p<.001). | ND | |
| Wu et al. (2016)[56] Elevated IL-6 and TNF-α levels in cerebrospinal fluid of subarachnoid hemorrhage patients |
SAH: 57.9yr n=57 57.9% M Ctrl: 59.2yr n=65 53.8% M 48h-duration Jilin Univ, China |
Patients with infection, heart disease, malignant tumor, and hematonosis. |
CSF TNF levels of SAH patients and controls were 49.68±7.02 and 12.47±2.15pg/mL, respectively (p<.001). CSF TNF levels in Caucasian SAH patients were higher than corresponding controls (p<.001). | Higher TNF levels in CSF of SAH patients (HH 1-4) compared to healthy controls (p<.05). HH grade 2 SAH patients showed higher TNF levels in CSF compared to HH grade 1 (p=.008). | |
| Fragata et al. (2020)[57] TNF-R1 correlates with cerebral perfusion and acute ischemia following subarachnoid hemorrhage |
54.6yr n=41 48.8% M 18mo-duration Lisbon, Portugal |
Patients with renal insufficiency, unknown time of SAH onset, pregnant women, or in poor clinical condition (GCS 3). Hospital admission 72h after. |
Higher arterial TNF-R1 levels correlated with trends of lower cerebral blood flow (r = -0.262, p = 0.069) and cerebral blood volume (r = -0.24, p = 0.096). TNFR1 levels independent predictors of poor outcomes. |
ND |
aSAH, aneurysmal subarachnoid hemorrhage; BBB, blood-brain barrier; CI, cerebral infarction; CSF, cerbrospinal fluid; CVA, cerebrovascular accident; CCI, Charlson Comorbidity Index; CHI, closed head injury; CTA, computed tomography angiogram; CCI, cortical controlled impact d, day; DCI, delayed cerebral ischemia; DT, 3,6′-dithiol thalidomide; F, female; FPI, fluid percussion injury; g, gram; h, hour; ICH, intracerebral hemorrhage; IS, ischemic stroke; LFP, lateral fluid percussion; LPS, lipopolysaccharide; M, male; MCA, middle cerebral artery; MCAO, middle cerebral artery occlusion; ms, millisecond; min, minute; mRS, modified Rankin Scale; mAB, monoclonal antibody; mo, month; NIHSS, National Institute of Health Stroke Scale; ND, not discussed; OR, odds ratio; SAH, subarachnoid hemorrhage; tMCAO, transient middle cerebral artery occlusion; TBI, traumatic brain injury; TNF-BP, tumor necrosis factor-binding protein; TRAIL, tumor necrosis factor related apoptosis inducing ligand; wk, week; WT, wildtype; yr, year.
Table 3.
Brief Overall Comparison of Known Therapeutic Treatments: Range of XPro1595 molecular weight accounts for PEGylation in some studies.
Table 3.
Brief Overall Comparison of Known Therapeutic Treatments: Range of XPro1595 molecular weight accounts for PEGylation in some studies.
| XPro1595 | Etanercept | sTNFR | |
|---|---|---|---|
| MW (kDa) | 7-20 | 105 | 75 |
| Half-life (h) | ~9 | 68 | 2 |
| Ligands | sTNF monomer/dimer | sTNF trimer, TNF | |
| IC50 (ng/mL) | 12.5±4.0 | 10.8±1.8 | 3500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



