
Submitted:
04 April 2024
Posted:
05 April 2024
You are already at the latest version
Abstract
Hepatocellular carcinoma (HCC) development is associated with altered modifications in DNA methylation, changing transcriptional regulation. Emerging evidence indicates that DNA methyltransferase 1 (DNMT1) plays a key role in carcinogenesis process. This study aimed to investigate how pirfenidone (PFD) modifies this pathway and the effect generated by the association between c-Myc expression and DNMT1 activation. Rats F344 were used for HCC development using 50 mg/kg of diethylnitrosamine (DEN) and 25 mg/kg of 2-aminofluorene (2-AAF). HCC/PFD group received simultaneous doses of 300 mg/kg of PFD. All treatments lasted 12 weeks. On the other hand, HepG2 cells were used to evaluate the effects of PFD in restoring DNA methylation in the presence of the inhibitor 5-Aza. Histopathological, biochemical, immunohistochemical, and western blot analysis were carried out and our findings showed that PFD treatment reduced the amount and size of tumors along with decreased Glipican-3, β-catenin, and c-Myc expression in nuclear fractions. Also, this treatment improved lipid metabolism by modulating PPARγ and SREBP1 signaling. Interestingly, PFD augmented DNMT1 and DNMT3a protein expression, which restores global methylation, both in our in vivo and in vitro models. In conclusion, our results suggest that PFD could slow down HCC development by controlling DNA methylation.
Keywords:
Hepatocellular carcinoma
; DNMT1
; DNMT3a
; c-Myc
; β-catenin
1. Introduction
Hepatocellular carcinoma (HCC) represents the most frequent primary liver cancer and the third leading cause of death related to this illness worldwide [1]. Several etiological factors are associated with HCC genesis, i.e., HBV and HCV viral infection, diseases linked with metabolic disorders, and excessive alcohol intake, which are amongst the most frequent [2]. On the other hand, HCC development is associated with an increase in oxidative, inflammatory, fibrogenic, and proliferative events and an increase in growth and cellular death [3]. Mutations in specific genes involved in these processes mediate many of these processes. In recent years, interest has grown in studying the effects of the environment on the expression of critical genes involved in the genesis of various diseases, including cancer. More importantly, understanding the expression of a given gene when altered and without modifying its nucleotide sequence has become a key issue in this new era of pharmaceutics and epigenomics [4].
Epigenetic modifications depend on environmental factors that favor changes in a particular gene expression, while no alterations in its DNA sequence occur [5]. Two types of epigenetic modifications related to the initiation and development of cancer are known: DNA methylation and histone modifications. DNA methylation mainly occurs on the cytosine ring, specifically CpG islands, driven by DNA methyltransferases (DNMTs) activity. Global hypomethylation of the genome is a common feature in many cancers, including HCC. This widespread hypomethylation can contribute to genomic instability and the activation of other oncogenes. Primarily, methylation of the gene promoter region causes transcriptional repression, while methylation in the gene body promotes gene expression in various cancers, including HCC [4].
On the other hand, experimental models are an adequate strategy for evaluating the efficacy and safety of new drugs [6]. Castro-Gil et al. used an HCC animal model administering diethylnitrosamine (DEN) and 2-acetylaminofluorene (2AAF) to rats. This method limited weight gain and increased hepatomegaly and liver changes. Also, increased levels of liver cancer markers like γ-GTP, prostaglandin reductase 1, and GSTP1 were found. Importantly, this experimental model allowed the study of HCC development from its earliest stages to its final stage [7].
Pirfenidone (PFD) is a drug with important pharmacological properties; evidence from basic and clinical studies has shown that PFD has antifibrotic, antioxidant, and anti-inflammatory effects [8]. In in vitro models, PFD can inhibit proliferation, promote apoptosis of HepG2 cells [9], and regulate SIRT1-related mechanisms [10]. Silva-Gómez et al. demonstrated that PFD efficacy is involved in preventing HCC genesis via induction of p50 nuclear translocation, modifying the p65/p50 ratio in favor of p50, and knocking down IL-6, TNF-α, and COX-2 expression. In the early stages of experimental HCC PFD also changed the expression of p53, the activation of caspase-3p17, and the cleavage of PARP-1 [11].
Previous studies have reported that overexpression levels of oncogenes such as c-Myc, cyclinD1, ß-catenin, and tumor suppressor genes such as p53, E-cadherin, DLC-1, and pRb are downregulated to different degrees during the development of HCC. Particularly, c-Myc is associated with hypomethylation, while p53 has been associated with hypermethylation in its promoter sequence [12].
Our main goal in this study was to elucidate whether PFD regulates the formation of DNMT1/3a complexes and restores global DNA methylation, which might be related to HCC progression.
2. Material and Methods
2.1. Experimental Animals
Male Fischer-344 rats were provided by UPEA-Bioterio at CUCS, “Universidad de Guadalajara”, México and housed in accordance with the guidelines of Universidad de Guadalajara under the approval number of the bioethics and research committees CI-03020. Animals were housed under a 12-hour light/dark cycle and constant temperature of 25°C ± 2 °C and received humane care.
2.2. Animals and Experimental Design
Hepatocarcinogenesis model implemented by Castro-Gil et al. [7] was developed. Eighteen rats of the same weight (180 g) were randomized into three groups: No treatment (NT), administered with the vehicle (0.5% carboxymethyl cellulose, p.o. (CMC); hepatocellular carcinoma (HCC); rats was injected weekly with DEN (50 mg/kg/i.p. Sigma Aldrich, St. Louis, MO, USA) plus 2AAF (25 mg/kg/p.o. Sigma Aldrich); and HCC/PFD group, administered with the same treatments of HCC group, plus PFD 300 mg/kg/day, p.o. (Tecoland, Irvine, CA, USA) Noteworthy, this protocol was carried out for 12 weeks (Figure 1A) instead of 4 weeks as previously described (11). Finally, all animals were euthanized by administering isoflurane (SOFLORAN®VET, PISA. Guadalajara, Jal, MX).
2.3. Biochemical Determination of ɣ-GTP and ALT
Alanine aminotransferase (ALT), and gamma-glutamyl transpeptidase γ-GTP) activities were measured in rat plasma as described in supplementary material [11]
2.4. Histologic Assessment of Liver Sections
Liver samples from all animals were fixed with 10% formaldehyde for 24 hours. They were subsequently embedded in paraffin. Sections were sliced into 4μm-thick and mounted in glass slides. For the different stains performed, tissues were deparaffinized at 60 °C overnight in xylene and rehydrated steps in graded ethanol solutions. HCC diagnosis was histologically confirmed by Glypican-3 (GPC3, GENETEX, Irvine, CA, USA), and all HCC tumor tissues were assessed by hematoxylin and eosin staining. Fibrosis was evaluated with Masson trichrome. A certified pathologist analyzed liver histology. All experiments were performed according to standard procedures.
2.5. Cell Culture and Treatments
To perform in vitro assays, HepG2 cells (ATCC. HB-8065, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle medium (DEMEM; Invitrogen Life Technologies-GIBCO, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen Life Technologies-GIBCO), 100 U/mL penicillin, and 100g/mL streptomycin (GIBCO. Life Technologies) in a humidified atmosphere of 5% CO2 in air at 37°C. Cells were seeded at a density of 3×105 cells per ml. Treatments were performed after 8 h of fetal bovine serum starving. Then, cells were incubated with the inhibitor of DNMTs, 5-aza-2′-deoxycytidine (5-Aza) (0.05 mM) (Santa Cruz Biotechnology, Dallas, TX, USA), Rosiglitazone (Ros) (0.03 mM) (Santa Cruz Biotechnology, Dallas, TX, USA), and PFD (0.5 mM) for 48 h to evaluate effect on DNMTs and proteins expression (See supplementary Table S1). DMSO (0.1μM) was used as control.
2.6. Proteins Extraction and Western Blot Assay
Samples of normal and HCC tissue or HepG2 cell cultures were extracted using nuclear and cytoplasmic extraction reagents, containing a protease inhibitor and a phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA). The extracts were collected and centrifuged at 17,000 rpm at 4 °C for 10 minutes (GYROZEN 1730MR, Gimpo, Korea). Mini-Bradford determined protein concentrations. Proteins were boiled for 10 min in Laemmli Sample Buffer 2X (Bio-Rad), separated with SDS-PAGE, and transferred to PVDF membranes (Bio-Rad). The membranes were blocked with 10% non-fat milk in phosphate-buffered saline (PBS) containing 0.1% Tween-20 (Bio-Rad) at 4°C overnight. Multiple membranes were probed with primary antibodies (see Supplementary Table S1) overnight at 4°C and then incubated with secondary antibodies for 2h at room temperature. Bands of interest were visualized using BioRad ChemiDoc™ XRS+ System software (Bio-Rad).
2.7. Immunofluorescence
Immunofluorescence for DNMT1, DNMT3a, DNMT3b, and 5mC in tissue and HepG2 cells was performed as described by Silva-Gomez et al. [11]. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Images were collected using an epifluorescence microscope OLYMPUS BX51 (Shinjuku, Tokyo, Japan) and analyzed using Image-ProPlus 6.0 (OLYMPUS).
2.8. Global Methylation Assessment in genomic DNA
Total DNA was extracted from liver tissue using the commercial kit QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) following provider instructions. DNA was quantified using NanoDrop, and 100 ng of DNA was used to measure the percentage of total methylation using MethylFlash Global DNA Methylation (5mC) ELISA (EpiGentek, Farmingdale, NY, USA) and compared against a standard curve. Briefly, wells were pretreated to generate a high affinity for DNA. Subsequently, a capture antibody (5mC) specific against 5-methylcytosine was included. Finally, a developing solution that binds to the capture antibody was added, allowing colorimetric detection at 450nm. The amount of methylated DNA was proportional to the intensity of the optical density obtained, and compared to the calibration curve using the following formula:
5mC% = (sample od – negative control od)/ (slope x ng DNA) x100%
2.9. Dot Blot of Global DNA Methylation
Dot Blot was used to determine global DNA methylation through 5mc detection in cells and liver tissue. DNA was denatured by incubation with 0.1 M of NaOH for 10 min at 95°C; to prevent annealing DNA was kept on ice. DNA solution was neutralized by 1 M of NH4OAc for 1 min. 30 µg in 3 µl of DNA was dot-blotted for 30 min at 80 °C in a nitrocellulose membrane (Bio-Rad). The membranes were blocked with 5 % BSA (Sigma-Aldrich) for 1 h/TBS-T (Bio-Rad) at room temperature and were incubated with a mouse anti-5mC monoclonal antibody and IgG antibody as an isotype control. Finally, membranes were washed for 5 min three times in TBS-T and were visualized using Bio-Rad ChemiDoc™ XRS+ System software.
2.11. Protein-Protein Interaction Analysis through the STRING Platform
For the construction of the protein-protein interaction network, particularly between PPARγ and DNMTs, the STRING tool v11.5 (https://string-db.org/) was used. This tool allows the integration of all known possible IPPs. The information of proteins to be studied is introduced to the server, STRING searches for all interactions between the corresponding molecules and generates a network with nodes (proteins) and edges (interaction).
2.12. Molecular Docking Protein-Protein
A crystal structure of both proteins was downloaded from Protein Data Bank (https://www.rcsb.org/). PPARɣ (5U46) and DNMT1 (3PTA). Subsequently, to predict the possible interaction between both proteins, the PDB codes of each protein were entered into the HawkDock server to perform molecular docking. The HawkDock server determines the best positions and protein-protein docking sites through HawkRank and ATTRAC, and MM/GBSA allows the identification of the key residues that determine the interaction.
2.13. Statistical Analysis
All data is expressed as the mean values ± SD. ANOVA followed by Tukey’s was used to test statistical significance between groups as appropriate. Graphs and statistical analysis were generated using GraphPad Prism 10.0 software (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant when p<0.05.
All assays and analyses were performed in triplicate, as shown in Supplementary Figures S2 to S8.
3. Results
3.1. Liver Damage Caused by Chemicals is Prevented by Pirfenidone
Carcinogenic damage induced during twelve weeks by the administration of DEN and 2-AAF, caused a decrease in body weight in animals of HCC group (Figure 1B); in addition, in this same group, severe morphological alterations in liver tissue and neoplastic nodules development were observed (Figure 1C). Also, hepatomegaly observed in HCC group was corroborated by quantifying the liver weight/body weight ratio, which increased significantly in damage group rats versus NT group (p<0.0001) (Figure 1D,E). Regarding serum markers of liver damage, γ-GTP and ALT, activity of both enzymes increased significantly, corroborating the deterioration of liver function induced in the HCC group (Figure 1F,G). Noteworthy, the concomitant administration of PFD favored the recovery of body weight from the eighth week of the experimental procedure, prevented the macroscopic and microscopic alterations observed in the damaged group, and prevented the increase in the activity of liver enzymes.
All data of the morphological evaluation of liver tissue are shown in Table 1 and Table 2. It is important to mention that in HCC group livers, as well as in livers of rats administered with PFD, the formation of nodules is observed; however, in the HCC/PFD group, nodules number and their size was significantly lower than in the HCC group (less than 1 mm (*), Figure 1C.
3.2. Pirfenidone Slows Down the Expression of Carcinogenic Markers, Reduces Fibrosis, and Decreases Damage in Liver Architecture
To demonstrate the PFD effect on microscopic alterations in liver parenchyma, H&E and Masson’s trichrome stains were performed. Additionally, IHC examined GPC3 as a marker of liver neoplastic lesions. Figure 2A shows microscopic changes in the HCC group suggestive of malignancy, such as abundant anaplastic hepatocytes, hyperchromic nuclei, and multinucleated cells with alterations in the nuclear-cytoplasmic ratio (asterisk), in addition to abundant foci of steatosis (yellow arrows) and a marked appearance of ductular reaction in the portal vasculature. On the other hand, PFD administration prevented these histological changes.
Moreover, in Figure 2C, extensive areas of fibrosis (positive turquoise blue) in the liver of rats with HCC were observed. In addition, greater sinusoidal capillarization and a high infiltration of inflammatory cells were detected. On the contrary, the administration of PFD prevented the accumulation of extracellular matrix by 20% versus damage group (Figure 2D; p<0.001) and averted inflammatory cells infiltration. Figure 2G shows the IHC for GPC3 detection in liver tissues from HCC group where the expression of this surface oncoprotein has significantly increased. Remarkably, PFD prevented the expression of GPC3 in approximately 50% (Figure 2F, p<0.0001).
A cellular fractional process was performed to analyze the expression of cytoplasmic and nuclear proteins. Figure 2G displays representative western blots of the expression of β-catenin. A significant decrease in expression of this protein is observed in HCC rats compared to NT group, while the HCC/PFD group did not present significant changes (Figure 2H). On the other hand, in Figure 2I, it is observed that in the nuclear fraction, there is an increase in β-catenin and c-Myc in the HCC group; however, treatment with PFD significantly reduced the expression of both proteins in the cell nucleus (Figure 2J,K).
3.3. Pirfenidone Regulates Expression and Translocation of PPARɣ and PPARγ2.
To determine PPARα expression and localization, along with two isoforms of PPARγ, cytoplasmic and nuclear protein extracts were analyzed by western blot Figure 3 shows that PFD significantly increases PPARα expression in both, cytoplasm (Figure 3A,B; p < 0.001) and nucleus (Figure 3E,F; p < 0.001), compared with HCC group. In addition, this drug stimulates PPARγ expression in both, cytoplasmic (Figure 3A,C; p < 0.001) and nuclear fractions (Figure 3E,G: p<0.05), versus damage group. Contrary to the above, PPARγ2 isoform expression exhibited a decrease in both, cytoplasmic (Figure 3A,D; p < 0.0001) and nuclear fraction (Figure 3E,H: p < 0.00001), in groups treated with PFD, compared with HCC group. PPARγ2 is overexpressed in the liver and adipose tissue of animals developing fatty liver disease, it is also known that this transcription factor regulates SREBP1 protein function involved in the lipogenic response. Figure 3I (3J and 3K; p < 0.0001) shows that in the HCC group, the expression and phosphorylation of SREBP1 increased, specifically in serine 372, however in the group treated with PFD, levels of this protein maintained their basal expression and phosphorylation.
3.4. Pirfenidone Reverses Global DNA Hypomethylation Through Regulation of DNMT1.
Nuclear extracts were used for the analysis of different DNMT isoforms, as well as for understanding of scaffold protein’s role, UHRF1, and PCNA. In our study, we analyzed PCNA as a clamp protein that facilitates DNMT1 function on hemimethylated DNA. Figure 4A shows representative Western blots of the mentioned proteins. In HCC group, a decrease in DNMT1, DNMT3a, and UHRF1 nuclear expression, but not DNMT3b, is observed; in addition, a slight increase in DNMT1 acetylated form (DNMT1Ac; p<0.05). However, in PFD-treated group, a significant increase in DNMT1 expression (p<0.001), DNMT3a (p<0.0001), and UHRF1(p<0.0001), as well as DNMT1Ac (p<0.00001) was observed.
On the other hand, 5-methylcytosine (5mC) modification is the most frequent form of DNA methylation. To evaluate this mark, double immunofluorescence was carried out to detect the expression and localization of DNMT1 and modification of 5mC in liver tissue of experimental animals. In Figure 4H, the DNMT1 and 5mC signals present in the cellular nucleus of samples NT and HCC/PFD groups are observed. The first is distinguished in the green channel, while the second is in the red channel.
To corroborate the results obtained in the in vivo model, we carried out an in-situ assay, analyzing the global DNA methylation by dot-blot using a monoclonal antibody directed against 5mC, mouse IgG as isotype control and staining with methylene blue to control total DNA loading. Figure 4J,K, respectively, show that global DNA hypomethylation ensued in animals from HCC group. This response is prevented by PFD, which can maintain this global DNA methylation in a similar way to NT group (p<0.001). Additionally, to corroborate the result obtained in the dot blot analysis, we performed an ELISA Methyl Flash Global DNA Methylation (5-mC) assay. In Figure 4L, it was observed that in HCC/PDF group, there was an increase in global DNA methylation versus HCC group (p<0.001).
3.5. Pirfenidone Regulates DNMT1 Expression and Prevents Global DNA Hypomethylation In Vitro.
HepG2 cell line was employed in in vitro studies to support the in vivo analyses. DNMT inhibitor, 5-Aza, and the PPARγ activator, rosiglitazone, were administered to cells for 48 hours, Subsequently, the nuclear extracts were purified and analyzed. Figure 5A,B demonstrate that 5-Aza administration reduces DNMT1, DNMT3A, and DNMT3bexpression, as well as DNMT1 acetylation (p<0.0001). In contrast, post-treatment with PFD markedly elevated the expression of scaffolding proteins, PCNA and UHRF1, as well as DNMT1 and DNMT3a (p<0.0001), while DNMT3b showed a modest but significant increase (p<0.001). On the other hand, post-treatment with rosiglitazone did not significantly affect the expression of proteins of interest (Figure 5A).
The expression patterns of oncoproteins c-Myc and β-catenin in nucleus were also looked at, along with the anticancer marker, p53. Representative WBs are shown in Figure 5C,D. Treatment with 5-Aza decreased p53 expression; however, after combined 5-Aza/PFD treatment, an increase in p53 expression is observed (p<0.0001). Conversely, combined treatment with 5-Aza/PFD prevented increased c-Myc and β-catenin oncogenes expression induced by 5-Aza treatment. p<0.05). Also, rosiglitazone treatment enhanced p53 protein expression and reduced b-catenin and c-Myc levels in nuclear fraction (Figure 5C). These data suggest that PFD can effectively suppress genes implicated in carcinogenesis, which reduces cell viability and, as the MTT experiment showed, lowers rates of cell proliferation (Supplementary Figure S1).
To corroborate the effect of PFD on DNMTs, we performed an immunofluorescence assay. Figure 5E shows in green channel the nuclear expression of DNMT1, DNMT3a, and DNMT3b in cells without treatment and in cells treated with PFD for 48 hours. The signal intensity decreased significantly in cells incubated with 5-Aza for 48 h in a comparable way with the red signal of 5mC. Remarkably, in cells treated with PFD, and after 5-Aza treatment, a significant increase in DNMT1, DNMT3a and 5mC signals was observed.
Finally, a dot-blot assay of the total genomic DNA extracted from HepG2 cells with the different treatments was performed. Figure 5F,G show that 5mC spot on the dot blot is decreased in cells that were treated with 5-Aza. However, when post-treatment with PFD is performed, DNA methylation escalated to its basal levels.
3.6. In Silico Analysis Reveals that PPARγ Complexes with DNMT1.
We performed an in-silico examination employing STRING platform to determine potential interactions of PPARγ with various isoforms of DNMTs, PCNA, and UHRF1 proteins. Figure 6A shows that PPARγ interacts with DNMT1 and DNMT3a, and indirectly with PCNA and UHRF. Finally, we made representative diagrams of interaction between PPARγ and PFD, highlighting their junction with the ligand-binding domain (LBD); in addition, a second diagram represents DNMT1 structure, highlighting main domains of this protein (Figure 6B).
To complete the in-silico analysis, we obtained from PDB the three-dimensional structures of PPARγ (5U46), BAH1, BAH2 domains, and catalytic domain of DNMT1 (3PTA) (Figure 6C). The HawkDock server was used to determine interaction of both proteins. Figure 6D shows the interaction of N-terminal domain of PPARγ with BAH domain of DNMT1, causing a change in the conformation of catalytic domain. Finally, ATTRAC algorithm was used to identify amino acids with highest binding free energy, and it was verified by means of MM/GBSA (Figure 6E).
4. Discussion
HCC is a gradual damage process generated by deregulation of gene expression and aberrant molecular signaling, which trigger histopathological and physiological alterations in the liver. Additionally, epigenetic alterations contribute to the inception of this pathology development. DNA methylation is the most studied epigenetic modification, and the most consistent in various types of human cancer, including HCC [12,13].
PPARs role in HCC remains controversial. PPARα high expression may inhibit tumor cell growth by preventing cell division and initiating cell death via IκBα and NF-κB signaling pathways, as reported by several authors [14]. Additionally, PPARα overexpression was linked to extended survival times in HCC patients [15]. Moreover, the involvement of PPARα in NOX1-mediated angiogenesis has been disclosed. NOX1 presence decreases PPARα activity, whereas its absence increases its expression, preventing endothelial cell migration and angiogenesis [16]. Our findings indicate that PFD treatment increased PPARα expression, mostly in cytoplasmic fractions, improving lipid metabolism and slowing HCC development.
On the other hand, PPARγ are transcription factors that play an important role in HCC development. However, the available evidence is not yet conclusive, nor is it entirely clear whether the ligands of this molecule promote or prevent the tumorigenic process. In different cell lines, and in human HCC, increased expression of PPARγ plays an important role in moderately and poorly differentiated tumors [17]; although the mechanisms are not yet well understood, it has been suggested that the use of antagonists blocking PPARγ activity, promote cell arrest and death through apoptosis of HCC cells [18]. In a recent study conducted by our research group, an increase in PPARγ expression at cytoplasmic and nuclear levels in liver tissue of patients with HCC was observed. Furthermore, observations derived from an in-silico assay, allowed us to conclude that PFD is an agonist that interacts with PPARγ ligand-binding domain (LBD). Its mechanism of action was elucidated in vitro, using the HepG2 cell line, and an in vivo model of chemical hepatocarcinogenesis at 30 days. In these studies, PFD treatment induced PPARγ nuclear translocation, increased IkB-α expression, p53, and caspase 3-p17 activity; in addition, it prevented the inflammatory process by blocking the activity of NF-κB p65, that is, it intervened in antitumor and anti-inflammatory mechanisms characteristic of this disease [11].
Our results indicate that PFD treatment (300 mg/kg) prevented weight loss, maintained normal liver enzyme activity, and delayed tumor formation (Figure 1). There is evidence that the use of PPAR agonists, such as pioglitazone (10 mg/kg), is beneficial in preventing HCC development and reducing the formation of macroscopic tumor nodules [19].
In this work, we have also demonstrated that animals treated with PFD, maintained intact hepatic cytoarchitecture, prevented synthesis and accumulation of extracellular matrix fibers, and decreased GPC3 expression significantly. Regarding this, Capurro et al. found that non-glycosylated GPC3 protein forms a complex with Wnt-β-catenin pathway proteins, they also demonstrated that glycosaminoglycan chains are not required to stimulate Wnt signaling and hepatocellular carcinoma growth [20]. Also, Hu et al. demonstrated that curcumin treatment inhibits Wnt/-β-catenin signaling in addition to downregulating GPC3 expression, in vitro experiments confirmed that silencing the expression of GPC3 potentiates the effects of curcumin on Wnt/β-catenin signaling; while in vivo experiments showed that this drug inhibits cell proliferation and induces apoptosis in HepG2 cells [21]. Our results indicate that PFD can decrease GPC3 membrane expression, preventing β-catenin translocation to the nucleus, and reducing c-Myc oncogene expression (Figure 2A,I).
Several evidence have postulated the antifibrogenic mechanism exerted by PFD. Lv et al. observed that PFD reduced pulmonary fibrosis, in vitro and in vivo, by regulating the Wnt/GSK-3β/β-catenin pathway and the TGF-β1/Smad2/3 signaling pathway [22]. More recently, our research group postulated that the molecular interaction of PFD with PPARγ and its effects on the redistribution and signaling of β-catenin in the HepG2 line might be responsible for this molecular pathway [23]
DNA methylation, primarily carried out by DNMT1, DNMT3a, and DNMT3b, plays a crucial role in epigenetic reprogramming. DNA methyltransferase inhibitors, such as 5-azacitidine, can greatly improve the efficiency of reprogramming and change gene expression, which can affect p53-mediated cell fate decisions like apoptosis or proliferation [12]. Also, abnormal Wnt/β-catenin activation is linked to DNA methylation and affects tumor suppressor genes like E-cadherin. This is because Wnt/β-catenin activation helps to keep tumors cell stem-like in liver and other types of cancer. It is important to note that abnormal DNA methylation plays a substantial role in cancer growth by changing genes related to the Wnt/β-catenin pathway and changes in its translated process. Furthermore, evidence suggests a regulatory link between c-Myc activity and DNMTs, specifically DNMT1 and DNMT3a [12]. Elevated c-Myc levels have been linked to increased DNA methylation activity by DNMT3a. The connection between c-Myc and DNMT3a is important for c-Myc specific methylation [12,13], which controls transcription of many genes, including those that regulate cell cycle. Additionally, it has been proposed that c-Myc proto-oncogene overexpression can induce DNA damage by generating reactive oxygen species (ROS), promoting oncogenesis [12]. Although DNMT1 and p53 are involved in distinct cellular processes, there can be interactions between different cellular pathways. Several research has demonstrated that p53 regulates DNMT1 synthesis, and alterations to p53 function, could impact DNA methylation patterns. Cancer development can be linked to molecular alterations through both, DNMT1 and p53 dysregulation [24].
In humans, tumors show differential changes in DNA methylation patterns, and hypermethylation of gene-specific promoters has been documented, though genome-wide hypomethylation may also occur [25,26,27,28,29]. It is obvious then, that a strong controversy exists regarding this issue. In early stages of hepatocarcinoma, an overexpression of DNMT1, DNMT3a, and DNMT3b is suggested, as well as an increase in methylation of specific genes in patients with HCC [28,29,30,31]. Along these lines, this response is observed in experimental models of early HCC [32,33]. These findings suggest that abnormal DNA methylation predicts poor survival in patients with this disease. Conversely to these facts, it has been established that in normal cells, heterochromatin is highly methylated, therefore transcriptional activity is epigenetically silenced; however, in several types of cancer, global hypomethylation is observed, for example, methylation of LINE-1 (long interspersed nucleotide elements) maintains genomic stability and integrity. Loss of methylation increases genomic instability and increases the likelihood of mitotic recombination, leading to tumor development [31]. Also, in a study by Eden A et al., DNA hypomethylation is proposed to favor cancer promotion by altering chromosome stability. This research group also states that further characterization of the relationship between DNA methylation, chromatin composition, and structure is required to understand how hypomethylation affects DNA structure and integrity in cancer [35].
Our results indicate that in HCC group there is a decrease in the expression of DNMT1 and DNMT3a, this response correlated with global DNA hypomethylation, while PFD treatment induced overexpression of both isoforms of DNMTs, and the scaffolding protein UHRF1, which could explain, at least in part, the reversal of global DNA hypomethylation caused by carcinogenic damage. Additionally, 5-Aza is a DNA demethylating drug that inhibits DNMT activity by forming covalent adducts with the catalytic region of the enzyme and DNMT1 degradation as a result. [36]. Also, in vitro studies have demonstrated the reactivation of tumor suppressor genes, including p53 [24]. In this work, we found that 5-Aza administration reduces global DNA methylation, and DNMT1 and DNMT3a expression; nonetheless, PFD reduces 5-azacitidine effects on both DNMTs (Figure 5). A study conducted by Nishimori H et al. observed that there is an increase in PCNA expression in HCC patients [37]. Interestingly, our findings reveal that PFD treatment reduces the expression of this protein, while in HCC group, it remains increased (Figure 4A). On the other hand, in our in vitro assay, 5-Aza treatment in HepG2 cells reduces PCNA expression. A similar response was observed by Tikoo K. et al. [38]. This group postulates that 5-Aza incorporation into DNA causes direct cytotoxicity and antiproliferative effects on tumor cells. Our results show that treatment with 5-Aza/PFD maintains basal levels of PCNA, however, it is necessary to carry out additional experiments to clearly understand PFD effects on PCNA.
Yu J et al. were the first to suggest that the loss of a PPARγ allele increases the development of HCC. They postulated that PPARγ suppresses tumor cell growth by reducing cell proliferation and inducing G2/M phase arrest and apoptosis. Thus, PPARγ acts as a tumor suppressor gene in the liver [39]. Likewise, Lee YK et al. demonstrated that PPARγ2 is expressed in liver tissue, specifically in hepatocytes, and its expression level correlates with the expression and phosphorylation of SREBP1, leading to fat accumulation induced by pathological conditions, such as obesity and diabetes, promoting HCC development [40].
Our results showed that PFD treatment induced PPARγ translocation to the nucleus, and correlated with c-Myc expression inhibition (Figure 3), we also showed that in HCC group, PPARγ2 is overexpressed both in cytoplasm and nuclear fractions, contributing to lipid accumulation mediated by SREBP-1c, however, PFD treatment significantly reduced PPARγ2 expression and SREBP-1c (Ser372) phosphorylation, reducing hepatic steatosis present in liver tissue from animals of HCC group (Figure 3I,J.
Pazienza et al. showed that PPARγ and DNMT1 play an important role in the development of carcinogenesis, having the same expression patterns in cell lines derived from human pancreatic cancer [41]. Recently, Ceccarelli et al. postulated that activation of PPARγ with eicosatetraenoic acid promotes physical interaction with DNMT1 and HDAC1 in CpG island of Hic-1 gene [42].
Furthermore, Sharma A et al. evaluated in an in-silico model the potential role of PPARγ-DNMT1 interaction via PPAR-binding elements (PPRE) [43]. This evidence indicates that is possible that PFD activates PPARγ facilitating the formation of complexes with BAH1 domain of DNMT1, thus increasing the activity of this enzyme. Here, we performed a molecular interaction analysis through STRING demonstrating that PPARγ interacts with DNMT1 and DNMT3a.
5. Conclusions
Our results show that PFD treatment slows down the development of experimental hepatocarcinogenic damage. This effect takes place through several pharmacodynamic mechanisms including an increase in PPARγ nuclear translocation and its expression, a decrease in tumor markers expression, and, on the other hand, acting as a strong epigenetic regulator through the possible formation of PPARγ-DNMT1 complex, preventing DNA hypomethylation observed in HCC. Based on the results and the preventative nature of the experimental model used in this work, we suggest further investigation into the molecular mechanisms of PFD and its potential application as an adjuvant therapy in treating HCC. Figure 7 is a schematic representation of epigenetic mechanisms by which PFD can prevent the development of HCC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
H. O. M-R., Conceptualization, Investigation, Methodology, Formal analysis, Writing – original draft, Visualization. L. F. S-S., Investigation, Conceptualization, Methodology, Formal analysis, Writing – original draft, Visualization. M. G-M., Investigation, Methodology, Writing – review & editing. S. A-O., Investigation, Formal analysis, Visualization. F C-C., Investigation, Formal analysis. C. F-G., Visualization. A. S-R., Methodology, Formal analysis. J.G-B., Methodology, Formal analysis. M. A-L., Visualization. A. S., Writing – review & editing, Visualization. J A-B., Conceptualization, Writing – review & editing, Supervision, Funding acquisition. H. C.M-R., Conceptualization, Methodology, Investigation, Visualization, Re-sources, Writing – review & editing, Supervision, Funding acquisition.
Funding
This project was founded by Programa de Fortalecimiento de Institutos, Centros y Laboratorios de Investigación 2022, awarded to J. A. B.: Fondo para Proyectos de Impulso a la Investigación (PIN 2020- I) to H.C.M-R. Fondo CONACyT Ciencia Básica y/o Ciencia de Frontera, Modalidad: Paradigmas y Controversias de la Ciencia 2022, number project: 320341 to J.A.B. The funding sources had no role in the design of the study, collection, analysis, and interpretation of data, or writing of the manuscript.
Institutional Review Board Statement
The study was carried out in compliance with the ARRIVE guidelines. All mouse assays were following the Animal Research Reporting In vivo Experiments guidelines and accomplished according to guidelines for the care and use of laboratory animals published by the US National Institutes of Health (NIH, publication No. 85-23, revised 1996). All the experiments were performed using male Fischer-344 rats (spp. Rattus norvegicus) after the approval of the Institute Animal Ethics Committee (CI-03020) Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara. Consent to participate is not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank CONAHCYT for the scholarship granted to Hipolito Otoniel Miranda-Roblero and Liliana Faridi Saavedra-Salazar to obtain his PhD degree.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Ahmed, T.S.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Med. 2019, 55, 1–22. [Google Scholar] [CrossRef]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’alessandro, R. Inflammatory Mechanisms of Hcc Development. Cancers (Basel). 2020, 12. [Google Scholar] [CrossRef]
- Fernández-Barrena, M.G.; Arechederra, M.; Colyn, L.; Berasain, C.; Avila, M.A. Epigenetics in Hepatocellular Carcinoma Development and Therapy: The Tip of the Iceberg. JHEP Reports. 2020, 2, 100167. [Google Scholar] [CrossRef]
- Braghini, M.R.; Lo Re, O.; Romito, I.; Fernandez-Barrena, M.G.; Barbaro, B.; Pomella, S.; Rota, R.; Vinciguerra, M.; Avila, M.A.; Alisi, A. Epigenetic Remodelling in Human Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 1–21. [Google Scholar] [CrossRef]
- Galicia-Moreno, M.; Silva-Gomez, J.A.; Lucano-Landeros, S.; Santos, A.; Monroy-Ramirez, H.C.; Armendariz-Borunda, J. Liver Cancer: Therapeutic Challenges and the Importance of Experimental Models. Can. J. Gastroenterol. Hepatol. 2021, 2021. [Google Scholar] [CrossRef]
- Castro-Gil, M.P.; Sánchez-Rodríguez, R.; Torres-Mena, J.E.; López-Torres, C.D.; Quintanar-Jurado, V.; Gabiño-López, N.B.; Villa-Treviño, S.; del-Pozo-Jauner, L.; Arellanes-Robledo, J.; Pérez-Carreón, J.I. Enrichment of Progenitor Cells by 2-Acetylaminofluorene Accelerates Liver Carcinogenesis Induced by Diethylnitrosamine in Vivo. Mol. Carcinog. 2021, 60, 377–390. [Google Scholar] [CrossRef]
- Lopez-de la Mora, D.A.; Sanchez-Roque, C.; Montoya-Buelna, M.; Sanchez-Enriquez, S.; Lucano-Landeros, S.; Macias-Barragan, J.; Armendariz-Borunda, J. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015, 12, 840–847. [Google Scholar] [CrossRef]
- Zou, W.J.; Huang, Z.; Jiang, T.P.; Shen, Y.P.; Zhao, A.S.; Zhou, S.; Zhang, S. Pirfenidone Inhibits Proliferation and Promotes Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the Wnt/β-Catenin Signaling Pathway. Med. Sci. Monit. 2017, 23, 6107–6113. [Google Scholar] [CrossRef]
- Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Meza-Rios, A.; Garcia-Bañuelos, J.; Vera-Cruz, J.; Gutiérrez-Cuevas, J.; Silva-Gomez, J.; Staels, B.; Dominguez-Rosales, J.; Galicia-Moreno, M.; et al. Pirfenidone Is an Agonistic Ligand for PPARα and Improves NASH by Activation of SIRT1/LKB1/PAMPK. Hepatol. Commun. 2020, 4, 434–449. [Google Scholar] [CrossRef]
- Silva-Gomez, J.A.; Galicia-Moreno, M.; Sandoval-Rodriguez, A.; Miranda-Roblero, H.O.; Lucano-Landeros, S.; Santos, A.; Monroy-Ramirez, H.C.; Armendariz-Borunda, J. Hepatocarcinogenesis Prevention by Pirfenidone Is Pparγ Mediated and Involves Modification of Nuclear Nf-Kb P65/P50 Ratio. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular Pathogenesis of Human Hepatocellular Carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Nagasaka, T.; Nishimura, T.; Ikai, I.; Richard, C.; Goel, A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2010, 47, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chu, E.S.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.; Yu, J. ; Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway. Oncotarget. 2014, 30, 8330–40. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.B.; Cai, S.H.; Liu, L.L.; Yang, X.; Yun, J.P. Decreased expression of peroxisome proliferator-activated receptor alpha indicates unfavorable outcomes in hepatocellular carcinoma. Cancer Manag Res. 2018, 26, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS One. 2011, 6, e14665. [Google Scholar] [CrossRef]
- Schaefer, K.L.; Wada, K.; Takahashi, H.; Matsuhashi, N.; Ohnishi, S.; Wolfe, M.M.; Turner, J.R.; Nakajima, A.; Borkan, S.C.; Saubermann, L.J. Peroxisome Proliferator-Activated Receptor γ Inhibition Prevents Adhesion to the Extracellular Matrix and Induces Anoikis in Hepatocellular Carcinoma Cells. Cancer Res. 2005, 65, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Sakisaka, S.; Harada, M.; Takagi, T.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Sasatomi, K.; Kimura, R.; Hashimoto, O.; et al. Involvement of P21WAF1/Cip1, P27Kip1, and P18INK4c in Troglitazone-Induced Cell-Cycle Arrest in Human Hepatoma Cell Lines. Hepatology. 2001, 33, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ghoshal, S.; Sojoodi, M.; Arora, G.; Masia, R.; Erstad, D.J.; Lanuti, M.; Hoshida, Y.; Baumert, T.F.; Tanabe, K.K.; et al. Pioglitazone Reduces Hepatocellular Carcinoma Development in Two Rodent Models of Cirrhosis. J. Gastrointest. Surg. 2019, 23, 101–111. [Google Scholar] [CrossRef]
- Capurro, M.I.; Xiang, Y.Y.; Lobe, C.; Filmus, J. Glypican-3 Promotes the Growth of Hepatocellular Carcinoma by Stimulating Canonical Wnt Signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef]
- Hu, P.; Ke, C.; Guo, X.; Ren, P.; Tong, Y.; Luo, S.; He, Y.; Wei, Z.; Cheng, B.; Li, R.; et al. Both Glypican-3/Wnt/β-Catenin Signaling Pathway and Autophagy Contributed to the Inhibitory Effect of Curcumin on Hepatocellular Carcinoma. Dig. Liver Dis. 2019, 51, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Wang, J.; Xu, C.; Huang, X.; Ruan, Z.; Dai, Y. Pirfenidone Alleviates Pulmonary Fibrosis in Vitro and in Vivo through Regulating Wnt/GSK-3β/β-Catenin and TGF-Β1/Smad2/3 Signaling Pathways. Mol. Med. 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Monroy-Ramírez, H.C.; Silva-Gómez, J.A.; Galicia-Moreno, M.; Santos-García, A.; Armendáriz-Borunda, J. Analysis of the Molecular Interaction of Pirfenidone with PPAR-Gamma and Effects on the Beta-Catenine Pathway in HEPG2 Line. Ann. Hepatol. 2020, 19, 9. [Google Scholar] [CrossRef]
- Georgia, S.; Kanji, M.; Bhushan, A. DNMT1 Represses P53 to Maintain Progenitor Cell Survival during Pancreatic Organogenesis. Genes Dev. 2013, 27, 372–377. [Google Scholar] [CrossRef]
- Feinberg, A.P. The Epigenetics of Cancer Etiology. Semin. Cancer Biol. 2004, 14, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.J.; Baylin, S.B. The Epigenomic of Cancer. Cell. 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA Hypomethylation in Cancer Cells. Epigenomics. 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Wu, H.C.; Yazici, H.; Yu, M.W.; Lee, P.H.; Santella, R.M. Global Hypomethylation in Hepatocellular Carcinoma and Its Relationship to Aflatoxin B1 Exposure. World J. Hepatol. 2012, 4, 169–175. [Google Scholar] [CrossRef]
- Oh, B.; Kim, H.; Park, H.; Shim, Y.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int J Mol Med. 2007, 20, 65–73. [Google Scholar] [CrossRef]
- Arai, E.; Kanai, Y. DNA Methylation Profiles in Precancerous Tissue and Cancers: Carcinogenetic Risk Estimation and Prognostication Based on DNA Methylation Status. Epigenomics. 2010, 2, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Simile, M.M.; Satta, G.; Seddaiu, M.A.; Daino, L.; Pinna, G.; Vinci Gaspa, M.A.L.; Feo, F. Comparative Effects of L-Methionine, S-Adenosyl-L-Methionine and 5′-Methylthioadenosine on the Growth of Preneoplastic Lesions and DNA Methylation in Rat Liver during the Early Stages of Hepatocarcinogenesis. Anticancer Res. 1991, 11, 1617–1624. [Google Scholar] [PubMed]
- Valencia Antúnez, C.A.; Chayeb, L.T.; Rodriguez-Segura, M.Á.; López Álvarez, G.S.; Garcia-Cuéllar, C.M.; Treviño, S.V. DNA Methyltransferases 3a and 3b Are Differentially Expressed in the Early Stages of a Rat Liver Carcinogenesis Model. Oncol. Rep. 2014, 32, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Article, R.; Floden, A. , Combs, C.; Article, R. DNA methylation of cancer genome. Birth Defects Res C Embryo Today. 2008, 23, 1–7. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal Instability and Tumors Promoted by DNA Hypomethylation. Science. 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of Action of the DNA Methyltransferase Inhibitors Azacytidine and Decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, H.; Tsukishiro, T.; Nambu, S.; Okada, K.; Shimizu, Y.; Miyabayashi, C.; Higuchi, K.; Watanabe, A. Analyses of proliferating cell nuclear antigen-positive cells in hepatocellular carcinoma: comparisons with clinical findings. J Gastroenterol Hepatol. 1994, 9, 425–32. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, K.; Ali, I.Y.; Gupta, J.; Gupta, C. 5-Azacytidine prevents cisplatin induced nephrotoxicity and potentiates anticancer activity of cisplatin by involving inhibition of metallothionein, pAKT and DNMT1 expression in chemical induced cancer rats. Toxicol Lett. 2009, 19, 158–66. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Shen, B.; Chu, E.S.H.; Teoh, N.; Cheung, K.F.; Wu, C.W.; Wang, S.; Lam, C.N.Y.; Feng, H.; Zhao, J.; et al. Inhibitory Role of Peroxisome Proliferator-Activated Receptor Gamma in Hepatocarcinogenesis in Mice and in Vitro. Hepatology. 2010, 51, 2008–2019. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic Lipid Homeostasis by Peroxisome Proliferator-Activated Receptor Gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Pazienza, V.; Tavano, F.; Benegiamo, G.; Vinciguerra, M.; Burbaci, F.P.; Copetti, M.; Di Mola, F.F.; Andriulli, A.; Di Sebastiano, P. Correlations among PPARγ, DNMT1, and DNMT3B Expression Levels and Pancreatic Cancer. PPAR Res. 2012, 2012. [Google Scholar] [CrossRef]
- Ceccarelli, V.; Ronchetti, S.; Marchetti, M.C.; Calvitti, M.; Riccardi, C.; Grignani, F.; Vecchini, A. Molecular Mechanisms Underlying Eicosapentaenoic Acid Inhibition of HDAC1 and DNMT Expression and Activity in Carcinoma Cells. Biochim. Biophys. Acta - Gene Regul. Mech. 2020, 1863, 194481. [Google Scholar] [CrossRef]
- Sharma, A.; Tobar-Tosse, F.; Dakal, T.C.; Liu, H.; Biswas, A.; Menon, A.; Paruchuri, A.; Katsonis, P.; Lichtarge, O.; Gromiha, M.M.; et al. Ppar-Responsive Elements Enriched with Alu Repeats May Contribute to Distinctive Pparγ–Dnmt1 Interactions in the Genome. Cancers (Basel). 2021, 13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Liver damage caused by chemicals is prevented by PFD. (A) Established experimental design; NT, non-treated group; HCC, hepatocellular carcinoma group injected weekly with DEN (50 mg/kg/i. p.) plus 2AAF (25 mg/kg/p. o); HCC/PFD group, HCC treatment plus 300 mg/kg PFD from week 0. All experimental groups were euthanized after 12 weeks. (B) Weekly logging of body weight. (C) Representative images of livers after 12 weeks of treatment. Greater size and number of dysplastic nodules were observed in HCC group than in the HCC/PFD group (white asterisks). (D) Graph of the liver weight at the end of treatment. (E) The ratio of liver weight to body weight of animals in each study group. (F) Serum gamma-glutamyl transferase (GTP) assay. (G) Serum alanine transaminase (ALT) assay. Data are presented as mean ± SD using ANOVA followed by Tukey’s multiple comparison test. ns, not significantly difference, *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001.
Figure 1.
Liver damage caused by chemicals is prevented by PFD. (A) Established experimental design; NT, non-treated group; HCC, hepatocellular carcinoma group injected weekly with DEN (50 mg/kg/i. p.) plus 2AAF (25 mg/kg/p. o); HCC/PFD group, HCC treatment plus 300 mg/kg PFD from week 0. All experimental groups were euthanized after 12 weeks. (B) Weekly logging of body weight. (C) Representative images of livers after 12 weeks of treatment. Greater size and number of dysplastic nodules were observed in HCC group than in the HCC/PFD group (white asterisks). (D) Graph of the liver weight at the end of treatment. (E) The ratio of liver weight to body weight of animals in each study group. (F) Serum gamma-glutamyl transferase (GTP) assay. (G) Serum alanine transaminase (ALT) assay. Data are presented as mean ± SD using ANOVA followed by Tukey’s multiple comparison test. ns, not significantly difference, *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001.
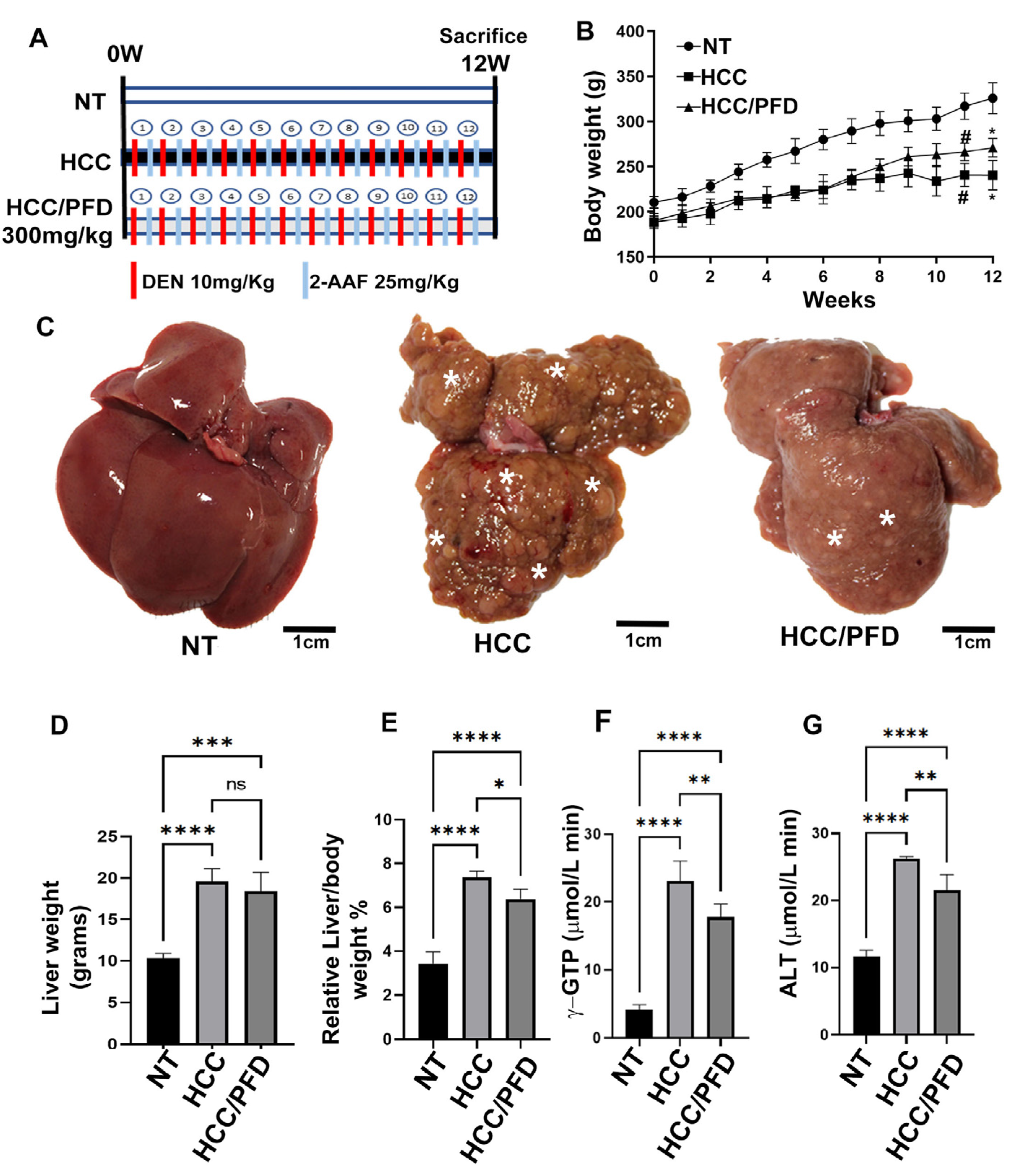
Figure 2.
PFD prevented alteration of hepatic architecture, fibrosis, and neoplastic lesions. (A) Photomicrograph representative of H&E staining (H&E) of groups at 12 weeks. Deformed portal tracts and thickened hepatic plaques are evident in HCC (asterisks). (B) Quantification of atypical hepatocytes. Cells with many nuclei and changes in the nuclear-cytoplasmic ratio (asterisk), in addition to numerous steatosis sites (yellow arrows). (C) Masson’s trichrome staining (MT). (D) Quantification of the percentage of collagen fibers deposited in the liver tissues of the different groups. (E) Representative expression of GPC3. Hepatic GPC-3 was analyzed in tissue by immunohistochemistry using primary anti-rabbit GPC-3 antibody. (F) Percentage of areas positive for GPC-3. (G) Representative western blot of cytoplasmic β-catenin. (H) Quantification of β-catenin expression. (I) Western blotting representative of the nuclear fraction of β-catenin and c-Myc. J) Quantification of β-catenin nuclear expression. (K) Quantification of c-Myc nuclear expression. Significantly different at **p < 0.001, ***, p < 0.0001 and ****p<0.00001.
Figure 2.
PFD prevented alteration of hepatic architecture, fibrosis, and neoplastic lesions. (A) Photomicrograph representative of H&E staining (H&E) of groups at 12 weeks. Deformed portal tracts and thickened hepatic plaques are evident in HCC (asterisks). (B) Quantification of atypical hepatocytes. Cells with many nuclei and changes in the nuclear-cytoplasmic ratio (asterisk), in addition to numerous steatosis sites (yellow arrows). (C) Masson’s trichrome staining (MT). (D) Quantification of the percentage of collagen fibers deposited in the liver tissues of the different groups. (E) Representative expression of GPC3. Hepatic GPC-3 was analyzed in tissue by immunohistochemistry using primary anti-rabbit GPC-3 antibody. (F) Percentage of areas positive for GPC-3. (G) Representative western blot of cytoplasmic β-catenin. (H) Quantification of β-catenin expression. (I) Western blotting representative of the nuclear fraction of β-catenin and c-Myc. J) Quantification of β-catenin nuclear expression. (K) Quantification of c-Myc nuclear expression. Significantly different at **p < 0.001, ***, p < 0.0001 and ****p<0.00001.
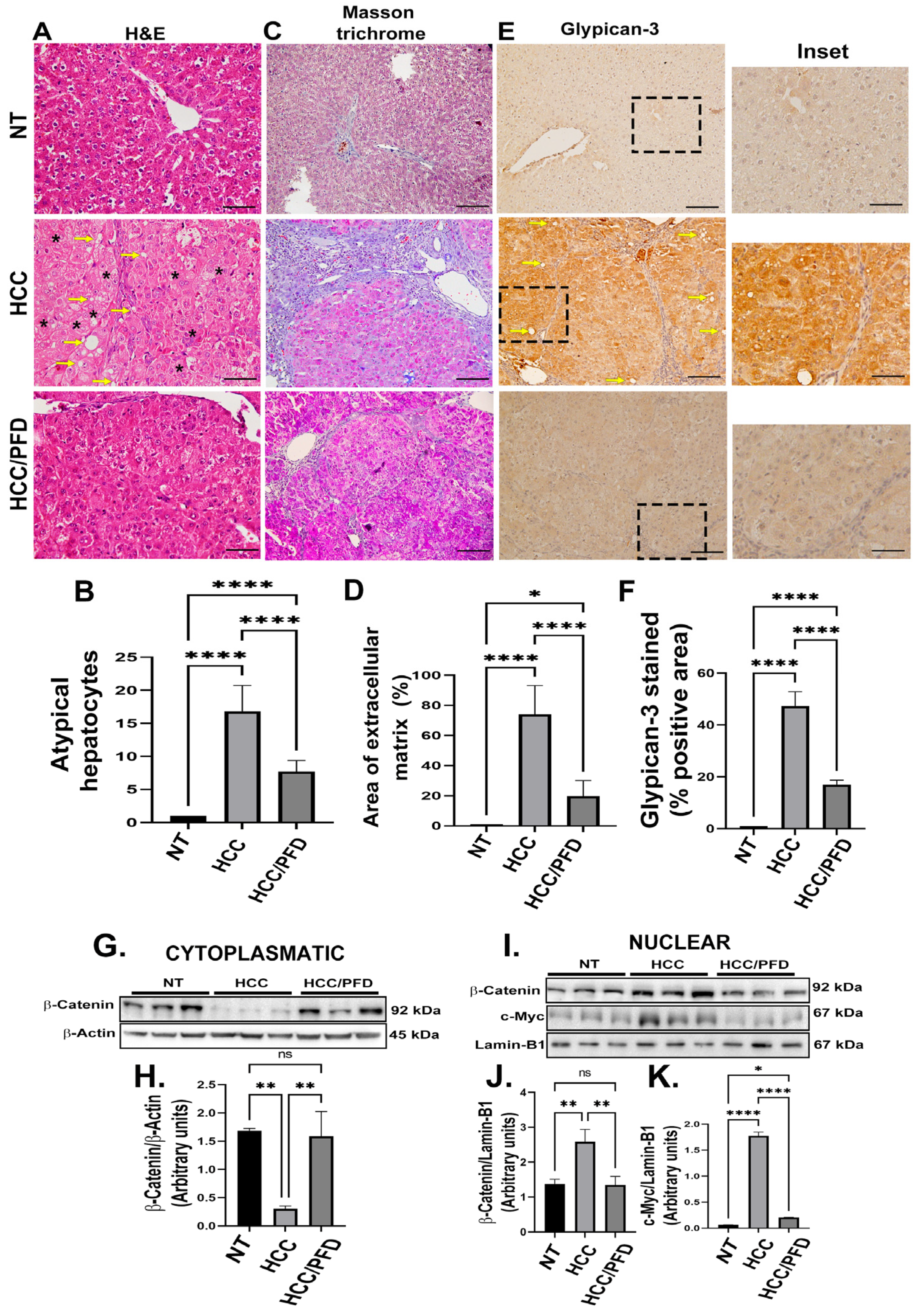
Figure 3.
PFD modulates the expression and subcellular localization of PPAR isoforms and SREBP1. (A) Western blot representative of the cytoplasmic expression of different PPAR isoforms. (B) Densitometry determination of PPARα expression, (C) PPARɣ and (D) PPARɣ2. (E) Western blotting representative of the nuclear expression of different isoforms of PPARs. (F) Densitometry determination of the nuclear expression of PPARα (G) PPARɣ and (H) PPARɣ2. (I) Western blot representative of total and phosphorylated SREBP expression. (J) Graph of the determination of SREBP expression and (K) pSREBP-1c (ser372). The results are shown as the mean ± standard deviation (SD) of triplicate assays. One-way ANOVA and Tukey’s post-hoc tests were performed. Significantly different at *p<0.05, **p < 0.001, *** p < 0.0001 and ****p<0.00001.
Figure 3.
PFD modulates the expression and subcellular localization of PPAR isoforms and SREBP1. (A) Western blot representative of the cytoplasmic expression of different PPAR isoforms. (B) Densitometry determination of PPARα expression, (C) PPARɣ and (D) PPARɣ2. (E) Western blotting representative of the nuclear expression of different isoforms of PPARs. (F) Densitometry determination of the nuclear expression of PPARα (G) PPARɣ and (H) PPARɣ2. (I) Western blot representative of total and phosphorylated SREBP expression. (J) Graph of the determination of SREBP expression and (K) pSREBP-1c (ser372). The results are shown as the mean ± standard deviation (SD) of triplicate assays. One-way ANOVA and Tukey’s post-hoc tests were performed. Significantly different at *p<0.05, **p < 0.001, *** p < 0.0001 and ****p<0.00001.
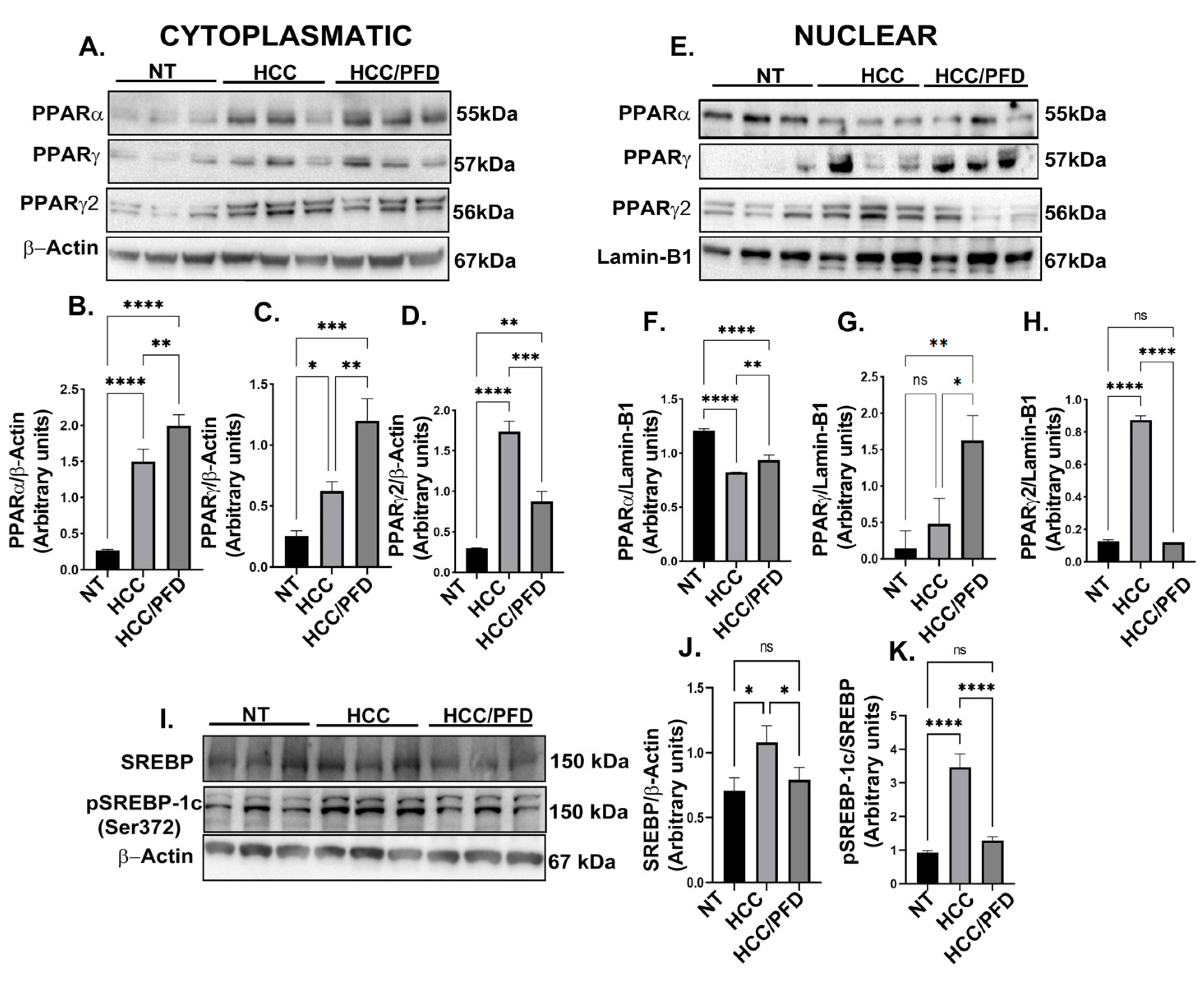
Figure 4.
Pirfenidone modulates the expression of enzymes that modify DNA and promote global methylation. (A) Representative western blots of DNMT1, DNMT1Ac, DNMT3a, DNMT3b, UHRF1, and PCNA. (B) Graphs showing the relative expression levels of DNMT1, (C) DNMT1Ac, (D) DNMT3a, (E) DNMT3b, (F) UHRF1, and (G) PCNA. (H) Representative images of the nuclear localization of DNMT1 and 5-mC in liver tissues. Nuclei were stained with DAPI (blue), DNMT1 (green) and 5-Methylcytosine (5-mC) (red). Images were captured using an epifluorescence microscope. White asterisks indicate positivity to the different markers analyzed. (I) Graph showing the number of positive hepatocytes for DNMT1. (J) Dot blot representative of global DNA methylation through the detection of 5mC. (K) Quantification of densitometry results of the relative levels of 5mC. (L) Determination of overall percentage of methylated DNA. One-way ANOVA and Tukey’s post-hoc tests were performed. Significantly different at *p<0.05, **p < 0.001, ***p< 0.0001, ****p<0.00001.
Figure 4.
Pirfenidone modulates the expression of enzymes that modify DNA and promote global methylation. (A) Representative western blots of DNMT1, DNMT1Ac, DNMT3a, DNMT3b, UHRF1, and PCNA. (B) Graphs showing the relative expression levels of DNMT1, (C) DNMT1Ac, (D) DNMT3a, (E) DNMT3b, (F) UHRF1, and (G) PCNA. (H) Representative images of the nuclear localization of DNMT1 and 5-mC in liver tissues. Nuclei were stained with DAPI (blue), DNMT1 (green) and 5-Methylcytosine (5-mC) (red). Images were captured using an epifluorescence microscope. White asterisks indicate positivity to the different markers analyzed. (I) Graph showing the number of positive hepatocytes for DNMT1. (J) Dot blot representative of global DNA methylation through the detection of 5mC. (K) Quantification of densitometry results of the relative levels of 5mC. (L) Determination of overall percentage of methylated DNA. One-way ANOVA and Tukey’s post-hoc tests were performed. Significantly different at *p<0.05, **p < 0.001, ***p< 0.0001, ****p<0.00001.
Figure 5.
In vitro, pirfenidone affects the synthesis of enzymes altering DNA global methylation (A) Representative western blots for DNMT1, acDNMT1, DNMT3a, DNMT3b, UHRF1, and PCNA. (B) Relative expression of DNMT1, acDNMT1, DNMT3a, DNMT3b, UHRF1 and PCNA. Lamin-B1 was used as a loading control. (C) Representative western blots for p53, β-Catenin and c-Myc. (D) Relative expression of p53, β-Catenin and c-Myc. Lamin-B1 was used as a loading control. (E) HepG2 cell nuclei were stained with DNMT1, DNMT3a and DNMT3B (green) and 5-Methylcytosine (red). White asterisks indicate positivity to the different markers analyzed. (F) Dot-blot representative of global DNA methylation through 5mC detection. (J) quantification for 5mC relative levels, methylene blue was used as a DNA loading control. The results are shown as the mean ± standard deviation (SD) of triplicate assays. One-way ANOVA and Tukey’s post-hoc tests were performed. Significantly different at * p<0.05, **p < 0.001, *** p < 0.0001 and ****p<0.00001.
Figure 5.
In vitro, pirfenidone affects the synthesis of enzymes altering DNA global methylation (A) Representative western blots for DNMT1, acDNMT1, DNMT3a, DNMT3b, UHRF1, and PCNA. (B) Relative expression of DNMT1, acDNMT1, DNMT3a, DNMT3b, UHRF1 and PCNA. Lamin-B1 was used as a loading control. (C) Representative western blots for p53, β-Catenin and c-Myc. (D) Relative expression of p53, β-Catenin and c-Myc. Lamin-B1 was used as a loading control. (E) HepG2 cell nuclei were stained with DNMT1, DNMT3a and DNMT3B (green) and 5-Methylcytosine (red). White asterisks indicate positivity to the different markers analyzed. (F) Dot-blot representative of global DNA methylation through 5mC detection. (J) quantification for 5mC relative levels, methylene blue was used as a DNA loading control. The results are shown as the mean ± standard deviation (SD) of triplicate assays. One-way ANOVA and Tukey’s post-hoc tests were performed. Significantly different at * p<0.05, **p < 0.001, *** p < 0.0001 and ****p<0.00001.

Figure 6.
Analysis of the protein-protein interaction between PPARɣ and DNMT1. (A) STRING database was used to generate a comprehensive protein-protein interaction (PPI) network between PPARɣ and DNMT1. (B) The interaction between PFD and PPARɣ is depicted, along with a linear representation of PPARɣ and DNMT1 and their corresponding structural domains. (C) The 3D structures of PPARɣ (5U46) and DNMT1 (3PTA) obtained from the Protein Data Bank (https://www.rcsb.org/). (D) The high-scoring docking poses from the docking simulation of PPARγ-DNMT1 proteins are shown, with PPARγ (cyan blue) serving as a possible receptor and DNMT1 (green) as a possible ligand. Interacting amino acids are highlighted in red. (E) Amino acid residues and free energy that allow for the interaction between PPARγ and DNMT1 are depicted.
Figure 6.
Analysis of the protein-protein interaction between PPARɣ and DNMT1. (A) STRING database was used to generate a comprehensive protein-protein interaction (PPI) network between PPARɣ and DNMT1. (B) The interaction between PFD and PPARɣ is depicted, along with a linear representation of PPARɣ and DNMT1 and their corresponding structural domains. (C) The 3D structures of PPARɣ (5U46) and DNMT1 (3PTA) obtained from the Protein Data Bank (https://www.rcsb.org/). (D) The high-scoring docking poses from the docking simulation of PPARγ-DNMT1 proteins are shown, with PPARγ (cyan blue) serving as a possible receptor and DNMT1 (green) as a possible ligand. Interacting amino acids are highlighted in red. (E) Amino acid residues and free energy that allow for the interaction between PPARγ and DNMT1 are depicted.
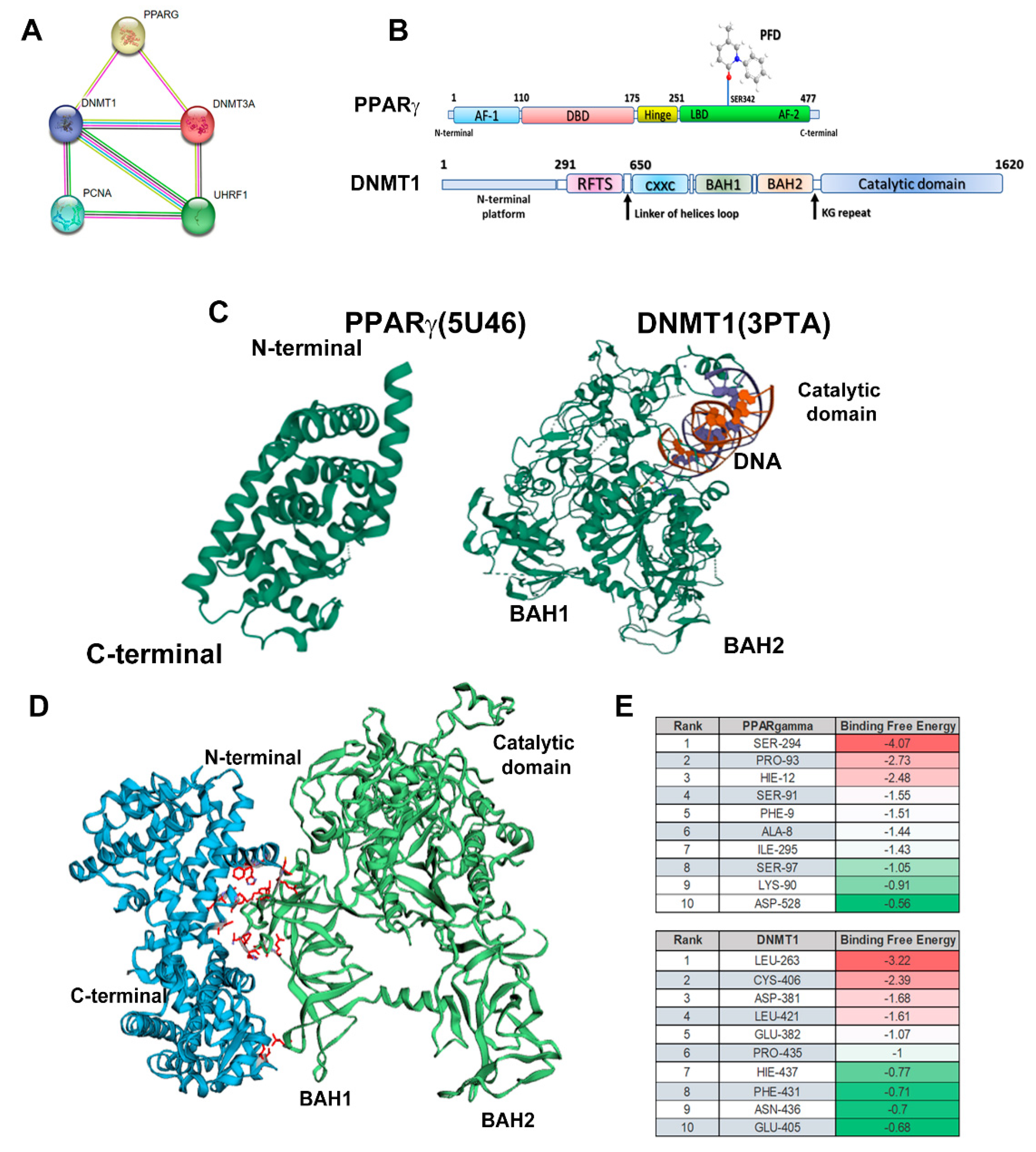
Figure 7.
Modulation of epigenetic markers induced during HCC is the proposed mechanism exerted by PFD. Left panel: Molecular mechanisms activated during HCC growth: β-catenin crosses into the nucleus, facilitating c-Myc oncogene transcription. Right panel: PFD is a PPARγ ligand/agonist that alters DNMT1 and DNMT3a function, promoting DNA hypermethylation, and reducing c-Myc expression. These mechanisms together could suppress aberrant cell division leading to HCC.
Figure 7.
Modulation of epigenetic markers induced during HCC is the proposed mechanism exerted by PFD. Left panel: Molecular mechanisms activated during HCC growth: β-catenin crosses into the nucleus, facilitating c-Myc oncogene transcription. Right panel: PFD is a PPARγ ligand/agonist that alters DNMT1 and DNMT3a function, promoting DNA hypermethylation, and reducing c-Myc expression. These mechanisms together could suppress aberrant cell division leading to HCC.
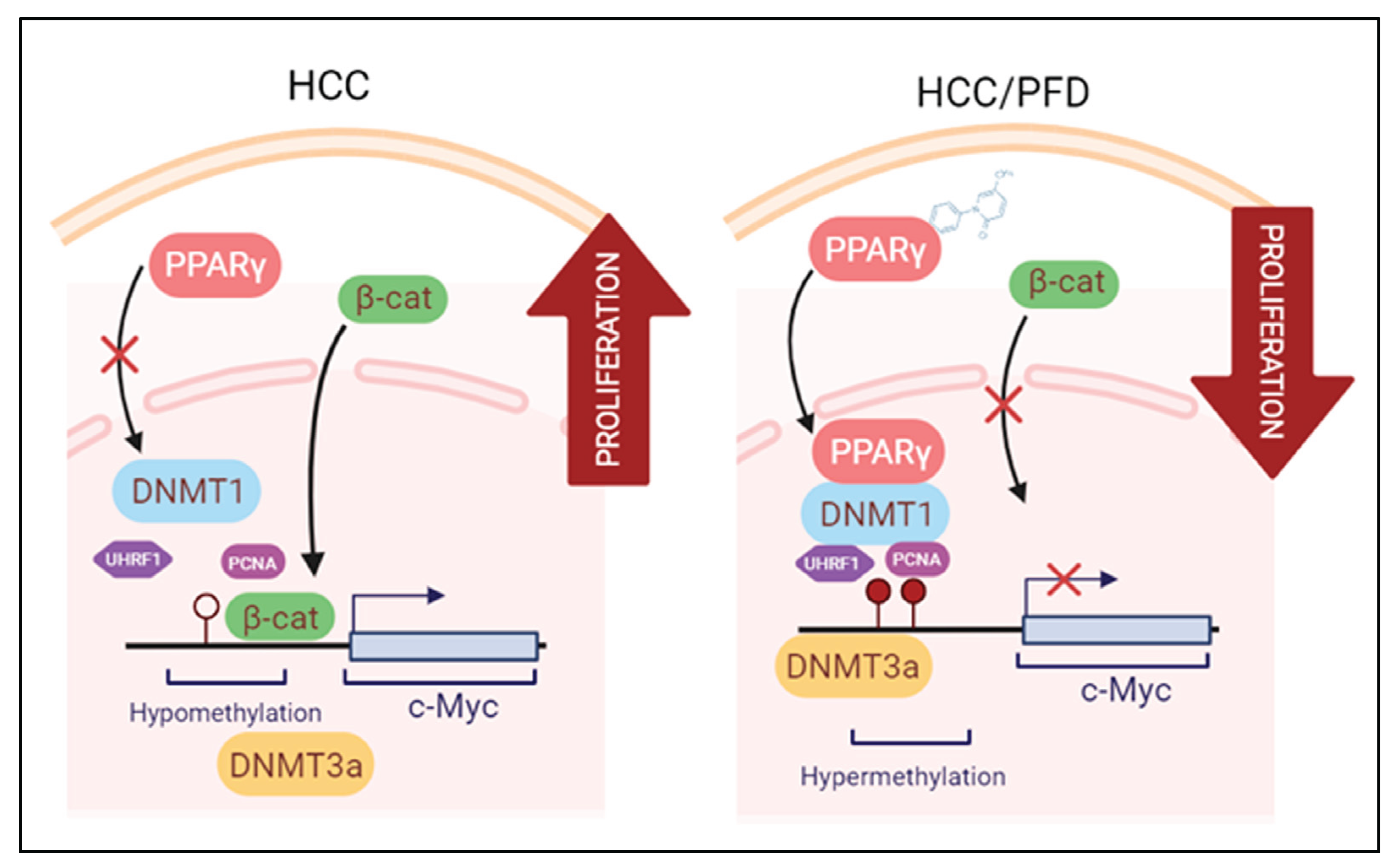

Table 1.
Effects of PFD on number of hepatocellular nodules in rats.
| Groups | No of rats with nodules / total rats | Nodule incidence (%) | Total no. of nodules | Average no. of nodules/nodule bearing liver (nodule multiplicity) | Nodules relative to size (% of total no.) | ||
| ≥3 mm | <3 to >1mm | ≤1 mm | |||||
| NT | 0/0 | 0 | 0 | 0 | 0 | 0 | 0 |
| HCC | 6/6 | 100 | 424 | 106±36.36* | 12 | 30 | 58 |
| HCC/PFD | 6/6 | 90 | 227 | 57±12.66* | 8 | 15 | 76 |
Table 2.
Histologic alterations induced at 12 weeks.
| Histological parameter | NT | HCC | HCC/PFD |
| Hyperplasia | - | + | + |
| Dysplasia | - | + | - |
| Cancer cell | - | + (MD) | +(WD) |
| Cellular infiltration | - | 1 | 0 |
| Fibrosis | - | 3 | 2 |
| Oval cells | - | 3 | 1 |
| Ballooning degeneration | - | 3 | 1 |
| Steatosis | - | ++ | - |
| Cholestasis | - | 0 | 0 |
| Mallory bodies | - | + | - |
| Lobular inflammation | - | 1 | 0 |
| Periportal bile ducts proliferation | - | 1 | 0 |
Description of histopathological scoring of the different groups: NT, non-treated; HCC, hepatocellular carcinoma group; HCC/PFD, HCC plus pirfenidone group. MD, moderate-differentiated HCC; WD, well-differentiated HCC. 0, none, 1-mild, 2 -moderate, 3-large, +, present, and − absent.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



