
Submitted:
06 April 2024
Posted:
08 April 2024
You are already at the latest version
Abstract
Respiratory syncytial virus (RSV) infection is a major cause of severe respiratory disease in infants and young children worldwide. Two vaccines targeting the elderly and pregnant women have been recently approved employing the prefusion form of the RSV-fusion protein (F). However, there are currently no vaccines for infants. Studies have shown a fusion-inactive prefusogenic F form is significantly more immunogenic and produces higher antibody titers to both the prefusion and postfusion structures of the F protein. Here we have developed RSV virus-like particles (RSV-VLPs) consisting of prefusogenic F, RSV glycoprotein and matrix proteins, produced them using the baculovirus expression system, and studied their protective efficacy in Balb/c mice. Morphology and successful assembly of VLPs were confirmed by transmission electron microscopy and western blot. Mice immunized with VLPs developed high levels of serum IgG and neutralizing antibodies as compared with mice immunized with inactivated virus. The VLP vaccine also induced higher levels of IFNγ and IL4, elicited limited proliferation of CD4+ and CD8+ T cells but higher cytotoxic-T-lymphocyte responses. VLP immunization abolished lung pathology in the mice after RSV challenge. Overall, our results indicate that RSV-VLPs consisting of prefusogenic F, glycoprotein and matrix proteins are a potential vaccine candidate against RSV.
Keywords:
respiratory syncytial virus (RSV)
; virus-like particles (VLPs)
; prefusogenic F
; glycoprotein
; matrix protein
; vaccine
1. Introduction
Human respiratory syncytial virus (RSV) is a negative-strand, enveloped RNA virus of the Pneumoviridae family and is one of the leading causes of lower respiratory tract infection (LRTI) in young children and older adult populations. The disease burden is particularly high in developing countries with between 2.95 to 3.35 million hospitalizations and 50,000–70,000 deaths in young children [1,2]. Children under 5 years of age are most susceptible for RSV-related deaths, with the vast majority (>90%) in developing countries [3,4]. The RSV disease burden is also substantial in older adults with over 1 million infections, 300,000 hospitalizations, and over 10,000 in-hospital deaths worldwide [3,5]. Palivizumab and nirsevimab are the only licensed prophylaxis for prevention of RSV in high-risk newborns [6]. However, in many countries these monoclonal antibodies are not yet available. Prohibitive costs and need for multiple doses in the first few months of life limit their use in the high-risk populations and make them unaffordable for use in low- and middle-income countries [7,8].
The RSV genome contains more than 15,000 nucleotides coding for 11 known proteins: 2 non-structural and 9 structural proteins, with glycoprotein (G), fusion protein (F) and small hydrophobic (SH) protein being the surface proteins [9]. The F protein is a major component of the virus envelope [10], and is responsible for viral fusion with the membrane of the host cells [11]. The sequence of the F protein is highly conserved among RSV isolates, and antibodies to the RSV-A F protein provide protection against both A and B strains of RSV [12,13]. In contrast, the sequence of the G protein is variable, and 2 serotypes (A and B strains) showed different antigenicity with strain-specific antibody responses [14]. The G protein helps in attaching the virus to host cells. Both of these proteins elicit potent neutralizing antibodies and provide protection against both serotypes [15,16]. Additionally, the matrix (M) protein serves as the inner envelope protein and plays a central role in viral assembly [17,18]. The M protein was shown to enhance CD8+ T cell response in mice upon infection [19,20].
Despite the large health impact of RSV infections and extensive efforts for vaccine development, a safe and effective vaccine remained unavailable for decades. The development was hindered for many years by safety concerns arising from the first RSV candidate vaccine trial in the 1960s where two immunized children enrolled in a clinical trial died due to enhanced RSV-associated pneumonia [21]. The reasons behind the occurrence of enhanced disease were conformation changes in the protein epitopes because of formalin treatment, activation of innate immune cells that resulted in induction of a Th2-biased cellular immune response and high titers of non-neutralizing antibodies [22,23,24]. Since then, different vaccine strategies have been explored for preventing severe RSV infection: protein vaccines that use different RSV protein subunits, VLPs, virus vectors expressing RSV proteins or live vaccines that included attenuated RSV strains [25].
The F protein of RSV is metastable and undergoes significant rearrangements from the prefusion (preF) to a stable postfusion (postF) structure with neutralizing epitopes on intermediate structures [26,27]. Many preF-based particulate and non-particulate vaccines are under development. Recently, the world’s first preF-based vaccine, Arexvy, from GSK, was approved for use in older adults. Another preF-based RSV vaccine, Abrysvo, from Pfizer, for pregnant women has been approved in the USA and Europe [28]. The F protein is expressed as a single polypeptide (F0), that is cleaved into F1 and F2 subunits by furin proteases at two sites. This cleavage is required for fusion of the virus with the host cell membrane and transformation of the preF to the postF form [29]. Patel et. al. (2019) developed a stable F protein form that is partially cleaved with mutations in the furin cleavage site II of the fusion protein, called prefusogenic F (preFg). Immunogenicity studies using the preFg have shown it to be more immunogenic than preF and producing higher titers of neutralizing antibodies than both preF and postF structures to the most potent antigenic sites Ø and V of the fusion protein and the p27 domain [30,31].
Often, protein-based vaccines induce limited antibody responses due to their non-particulate nature. For this reason, we and others have developed VLP-based RSV vaccines. VLPs are complexes of multiple copies of proteins that assemble to form particles which mimic the virus structure but lack the viral genetic material. Due to their highly repetitive display of antigenic epitopes, VLPs are more immunogenic and are processed by antigen presenting cells (APCs) much more efficiently than purified proteins [32]. Previously, RSV-VLPs were made using F0 or preF with or without G proteins [33]. More recently, VLPs were developed using M or M2-1 together with preF and G [34,35,36]. These preF-based RSV-VLP were immunogenic and protective.
To further improve RSV-specific antibody and cellular immune response, we decided to combine the highly immunogenic preFg form with the VLP technology. In this study, we describe the development and characterization of VLPs consisting of preFg together with G and M proteins. The immunogenicity of these VLPs was assessed in mice with in-depth analysis of the evoked humoral and cellular immune responses. A challenge study was performed to determine the protective potential of the vaccine.
2. Materials and Methods
2.1. Cells and Live Virus Stock Preparation
HEp-2 cells (ATCC) were grown and maintained in Minimum Essential Medium (MEM) containing 10% fetal bovine serum (FBS), and 100 I.U/ml penicillin/streptomycin (all reagents from Gibco). The RSV-A2 (VR-1540, ATCC) strain was propagated by infecting HEp-2 cells. ExpiSf9 cells (Gibco) were used to make VLPs. These cells are a non-engineered derivative of the Spodoptera frugiperda Sf9 cells (Gibco) and are maintained in suspension employing a shaker platform in serum-free ExpiSf CD medium (Gibco) in non-baffled, plain bottom conical flasks and incubated at 27 ⁰C under non-humidified, non-CO2 cell culture condition.
2.2. Construction and Generation of Recombinant Baculoviruses Expressing RSV PreFg, RSV G and M Protein
For obtaining plasmids with RSV M and G gene sequences, viral RNA was extracted using a viral RNA extraction kit (Qiagen). Reverse transcription was performed on extracted viral RNA using the One-Step RT-PCR kit (Invitrogen) with M or G gene-specific forward oligonucleotide primers, given in Table 1. The M and G genes were then PCR-amplified from the RSV complementary DNA (cDNA) by use of the primer pairs indicated in Table 1 and cloned into the transfer vector, pFastBac1 (Gibco) with BamHI/XhoI sites, resulting in plasmids pFastBac1-M and pFastBac1-G. Recombinant clones were confirmed by restriction digestion and sequencing. For the PreFg gene, a codon-optimized construct was synthetically cloned into the pFastBac1 vector, named pFastBac1-PreFg, by GeneArt (Thermo Fisher) also with overlapping BamHI and XhoI sites. For the construction of PreFg, a mutation in the furin cleavage site II (KKQKQQ to KKRKRR) and a 10 amino acid deletion in the fusion domain (F137 – S146) of F protein was introduced as described in Patel et. al. 2020 [30]. The protein-encoding plasmids were amplified employing DH5α E. coli cells (Gibco).
To generate recombinant baculoviruses (rBVs) containing the three genes, the Bac-to-Bac baculovirus expression system (Gibco) was used as per manufacturer instructions. Briefly, cloned pFastBac1 plasmids were individually transformed into DH10Bac E. coli containing a baculovirus shuttle vector (bacmid), to create expression bacmids containing the inserted RSV genes. Expression bacmid clones were screened with blue/white screening. After confirmation, the expression bacmid DNAs were extracted and used for transfection of ExpiSf9 cells using Cellfectin II reagent (Gibco) to generate rBVs expressing M, G and PreFg gene as per the manufacturers protocol.
2.3. Protein Expression and Confirmation
Expression and secretion of proteins was confirmed by execution of indirect ELISAs on cell supernatant. Briefly, cell supernatants from the transfected ExpiSf9 cells were diluted ten-fold in carbonate-bicarbonate buffer, pH 9.2, and coated on ELISA wells. Coated wells were incubated overnight at 4 ⁰C and washed five times using phosphate buffer saline (PBS) containing 0.05% tween 20 (Sigma-Aldrich). Plates were blocked for 1 hr at 37 ⁰C using PBS containing 10% FBS after and washed five times with wash buffer. The washing protocol was kept constant for all further washing steps. One hundred microliters of mouse anti-RSV immune serum and serum from naïve mice were diluted (1:100 dilution) in sample diluent buffer (PBS containing 10% FBS and 0.1% tween 20) was added to plates and incubated for 1 hr at 37 ⁰C. RSV positive sera was acquired by infecting mice intranasally thrice with 40µl of 106 TCID50/ml of RSV-A2 at 2-week intervals. After incubation, wells were washed, and 100 µl 1:20,000 diluted anti-mouse-IgG-HRP (SouthernBiotech) conjugate in PBS with 10% FBS and 0.1% Tween 20 was added to the wells. After incubation for 30 min at 37 ⁰C, 100 µl tetramethylbenzidine (TMB) substrate containing hydrogen peroxide (Clinical Science Products Inc.) was added and the reaction was stopped after 10 min using 50 µl of 2N H2SO4 (Sisco Research Laboratories). Plates were read at 450 nm with a filter of 655 nm.
2.4. Production and Characterization of RSV-VLPs
For production of RSV-VLPs, ExpiSf9 cells (5 × 106 cells/mL, ≥ 90% viability) were cultured in presence of ExpiSf enhancer as per the manufacturers protocol. After 16-18 hrs, cells were co-infected with the three rBVs expressing PreFg, G and M proteins at a ratio of 1:1:1 (v/v). Cell culture supernatant collected on day 5 post-infection was centrifuged at 4000g for 30 minutes at 4 ⁰C for removal of cell debris. Clarified cell supernatant was layered on 20% sucrose in PBS and centrifuged in a SW-32Ti rotor for 3 hrs at 150,000g at 4°C. The pellet was resuspended in PBS and further purified on discontinuous 20%-30%-35%-40%-50%-60% sucrose gradients by centrifugation at 100,000g for 3 hrs at 4 ⁰C in a SW-55Ti rotor. 25 0.2ml fractions were collected from the top and total protein content by Folin-Lowry assay and RSV specificity by indirect ELISA of each fraction was determined. The VLP bands between 30%-35% were collected and dialysed against PBS to remove sucrose. The VLPs were characterized by western blot and transmission electron microscopy (National Institute of Virology, India). For western blot analysis, mouse anti-RSV serum was used. Phospholipid determination was conducted using VLPs dialyzed against PBS. Briefly, the standards were made using a known amount of sodium phosphate. Samples and standard were treated with 70% perchloric acid (Sigma-Aldrich) for 45 min at 180 ⁰C followed by addition of molybdate reagent (Sigma-Aldrich). Colour was developed using ascorbic acid and samples were read at 812 nm [37].
2.5. Preparation of Inactivated RSV
Briefly, HEp-2 cells were seeded in 175cm2 multilayer flasks using MEM containing 10% FBS, 100 I.U/ml penicillin and streptomycin. After 70% confluency was reached, cells were washed with PBS (HiMedia) and infected with 0.01 MOI RSV in MEM without FBS. On day 7, the cell supernatant containing detached cells was collected from the cell culture flasks and centrifuged to remove cell debris. Beta-propiolactone (BPL) inactivated RSV (BPL-RSV) was prepared by treating RSV-containing cell supernatant with a final concentration of 0.1% (v/v) BPL (HiMedia) for 24 hrs at 4 ⁰C under gentle shaking [38]. Formalin-inactivated RSV (FI-RSV) was prepared by incubating RSV-containing cell supernatant for 3 days with formalin at a final dilution of 1:4000 at 37 ⁰C [23]. Inactivated viruses were purified by ultracentrifugation at 100,000g at 4 ⁰C for 3 hrs and the pellets were resuspended in PBS. Purity of inactivated RSV preparations were checked by SDS-gel electrophoresis and protein concentration was determined using the Folin-Lowry assay.
2.6. Immunization and Challenge of Mice
In vivo experiments in mice were conducted after the approval by the institutional animal ethics committee of Bharati Vidyapeeth Medical College, Bharati Vidyapeeth (Deemed to be University), Pune. Female Balb/c mice (Advanced Centre for Treatment, Research and Education in Cancer, India) aged 6-8 weeks were used for immunization studies. All procedures on mice were executed under isoflurane anesthesia. Mice (6 mice per group) were intramuscularly immunized twice with a 3-week interval with 5 µg BPL-RSV, 5 µg FI-RSV, or VLPs at a dose of 5 or 10 µg. For intramuscular (i.m.) immunization, 50 µl solution was administered in the hind limb. Non-immunized or immunized mice were challenged with 40 µl of 106 TCID50/ml RSV seven days after the second dose. For challenge, the 40 µl was solution containing RSV was slowly administered equally in both nostrils. Mice were observed daily for weight loss. Mice were sacrificed 7 days after the second immunization dose or 4 days after challenge by cervical dislocation under the influence of anesthesia.
2.7. Sample Collection and Processing
Blood samples were collected by retro-orbital puncture 21 days after the first dose and 7 days after the second dose using capillary tubes. A piece of lung was collected in tissue cassettes to study pathology induced after the virus challenge. Lungs fixed in 10% formalin were sectioned and stained using hematoxylin and eosinophil staining (H&E). Lung pathology was scored on alveolar infiltrates, bronchitis and vasculitis. Spleens were collected individually in complete Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 100 I.U/ml penicillin/streptomycin, 0.1% beta-mercaptoethanol (BME) and 10% FBS and stored on ice until processed. Briefly, the spleens were processed individually by dissociating using sterile syringe plungers and passing the suspension through a 70 µm mesh (Becton Dickinson) employing IMDM complete medium. Subsequently, erythrocytes were lysed by incubating with 1 ml ACK lysis buffer (Gibco) for 5 min on ice. The cells were washed with IMDM complete medium, counted and brought to appropriate concentrations.
2.8. ELISA
Antibodies reacting with VLP, F, G or M protein (total IgG titre, IgG1, and IgG2a) were determined in mouse sera by enzyme-linked immunosorbent assay (ELISA). Briefly, ELISA plates were coated with 300 ng purified VLP or 150 ng RSV proteins (Sino Biologicals) per well in 100 µl carbonate-bicarbonate buffer, pH 9.2 overnight at 4 ⁰C. Plates were washed three times using PBS containing 0.05% tween 20 (PBST). Plates were blocked for 1 hr at 37 ⁰C using PBS containing 5% FBS and washed three times with wash buffer. The washing protocol was kept constant for all further washing steps. IgG1 and IgG2a concentrations were determined using calibration curves made by coating of ELISA plates with 0.1 μg anti IgG (Southern Biotech) at 4 ⁰C, overnight. Following extensive washing, increasing concentrations of 100 μl of IgG1 or IgG2a (Southern Biotech) were added to the plates. Diluted serum was added to the plates and incubated for 1 hr at 37 ⁰C. Serum samples were double diluted for IgG titre determination. Next, wells were washed and conjugated antibodies were added (anti-mouse-IgG HRP, anti-mouse-IgG2a HRP, or anti-mouse-IgG1 HRP; all from SouthernBiotech) to the respective wells and the plates were. incubated for 1 hr at 37 ⁰C. After washing, tetramethylbenzidine (TMB) substrate containing hydrogen peroxide (Clinical Science Products Inc.) was added and the reaction was stopped after 10 min using 2N H2SO4 (Sisco Research Laboratories). Plates were read at 450 nm with a filter of 655 nm using Synergy HTX reader and Gen5 software (BioTek).
2.9. Micro Neutralization Test
Mice sera were complement-inactivated at 56 ⁰C for 30 minutes. The serum samples were 1:4 diluted in MEM with 2% FBS and subsequently double diluted in the same medium in 96-well cell culture plates. Live virus was diluted in 2% MEM with 2% FBS to obtain titer of 100 TCID50/ml and added to the diluted serum. The virus-serum mixture was incubated at 37 ⁰C for 1 hr. After incubation, 2x104 HEp-2 cells in MEM with 2% FBS were added followed by incubation for 7 days at 37 ⁰C. Cells were fixed using 3.7% formaldehyde in PBS for 30 min, washed with PBS three times and stained with 1% crystal violet. Plates were observed for cell detachment and neutralizing 50 (NT50) antibody titre was calculated by the Reed-Münch method [39].
2.10. RSV-Specific Antibody-Secreting Cells
To quantitate anti-RSV IgG and IgA antibody-secreting cells (ASCs) elicited by immunization, a dual colour enzyme-linked immunosorbent spot assay (ELISpot) was performed using splenocytes. Briefly, MultiScreen-HA filter plates (Millipore) were coated with 1 µg purified VLPs in 100 µl PBS overnight at 4 ⁰C. Later, plates were blocked using complete IMDM complete medium for two hrs at 37 ⁰C. 1x106 splenocyte cells were added and the plates were incubated overnight in IMDM complete medium. After 24 hrs, cells were discarded, plates were washed with PBST and a mixture of anti-mouse IgG-HRP and anti-mouse IgA-AP (SouthernBiotech) at dilutions of 1:2000 in PBST was added to the plates. After incubation at 37 ⁰C, IgG-ASC were developed using amino-ethylcarbazole (AEC) substrate containing hydrogen peroxide (Sigma) followed by staining for IgA-ASC employing 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) substrate for detection of IgA ASC. The reaction was stopped by washing the plates with commercially available mineral water. Plates were scanned on the S6 Ultra-M2 system (Cellular Technology Ltd.) and spots were counted using the ImmunoSpot® Double Colour Enzymatic software (v7.0.26).
2.11. IFNγ and IL4 ELISpot
IFNγ and IL4 ELISpot was performed on splenocytes using a dual colour ELISpot kit as per manufacturer instructions (Cellular Technology Ltd.). 1x106 splenocytes were added to PVDF plates coated with anti-mouse IFNγ and anti-mouse IL4 antibodies in complete IMDM medium supplemented with 10% FBS and 0.1% BME. Cells were cultured without stimulant or were stimulated with 1 µg purified VLPs for 18 hrs. Subsequently, cells were discarded, plates were washed with PBST and cytokine-specific detection antibodies were added. Blue spots specific for IL4 and red spots specific for IFNγ were developed to measure cytokine-specific spot forming cells (SFC). Plates were scanned on the S6 Ultra-M2 system (Cellular Technology Ltd.) and spots were counted using the ImmunoSpot® Double Colour Enzymatic software (v7.0.26).
2.12. Cytokine Response after Immunization
Inflammatory (IFNγ, TNFα, IL6, IL2, IL17A) and anti-inflammatory cytokines (IL4, IL10) secreted by splenocytes were determined by cytometric bead array (Becton Dickinson) as per manufacturer protocol. To this end, 1x106 splenocytes from each mouse were cultured in 96 well U-bottom plates in complete IMDM medium for 72 hrs and either stimulated with 1 µg purified VLPs or non-stimulated. After incubation, the supernatants were collected and used for cytokine determination.
2.13. Flow Cytometry
Splenocytes were used to determine the CD4 and CD8 T cell response after immunization. 1×106 splenocytes were seeded into U-bottom 96-well plates in complete IMDM media, stimulated with 1 µg purified VLPs and the plates were incubated at 37 ⁰C in a humidified incubator with 5% CO2 for 48 hrs. After 48 hrs, cytotoxic T lymphocytes (CTLs) and proliferating T cells were evaluated by flow cytometry by staining with antibodies (see Table 2). Data from stained cells was acquired on the CytoFLEX LX system (Beckman Coulter) and analysed using CytExpert software (v2.5).
2.14. Statistical Analysis
Graphpad Prism v10.0.0 (Graphpad Software) was used to perform statistical analysis. Either a two-tailed t test or one-way ANOVA test was used to compare differences between various groups. p-values below 0.05 were considered statistically significant. * represents p < 0.05, ** represents p < 0.01, *** represents p < 0.001 and **** represents p < 0.0001.
3. Results
3.1. Generation of Transfer Vectors, Expression Bacmids and Protein Expression
RSV M and G genes were amplified by PCR with primers containing sites for restriction enzymes BamHI and XhoI and cloned into pFastBac1 transfer vectors. Insertion of the M and G gene into the vector was confirmed by sequencing of the plasmids. The pFastBac1 transfer vector containing the PreFg gene was constructed by GeneArt and encodes changes in amino acid sequences as mentioned in Materials and Methods above. All three transfer vectors were confirmed for gene insertion by restriction digestion with BamHI and XhoI (Figure 1a). Expression bacmids for the three genes were made by recombination into parent bacmid in DH10Bac E. coli. Production and secretion of PreFg, G and M proteins by the ExpiSf9 cells transfected with the individual bacmids confirmed by an indirect ELISA of the cell supernatant (Figure 1b).
3.2. Production and Characterization of VLPs
Cell culture supernatant from infected ExpiSf9 cells was clarified by centrifugation and RSV-VLPs were purified by sucrose density ultracentrifugation (Figure 2a). 25 fractions from the discontinuous sucrose gradient were collected from the top of the gradient. Total protein content and RSV-specific reactivity were measured for each fraction by indirect ELISA using anti-RSV antibody-positive mice sera. Protein content and ELISA reactivity coincided at fractions 8-10 (30%-35% sucrose) indicating the presence of an RSV antibody-reactive structures (Figure 2a). A drop in protein content and ELISA-OD values was observed with in fractions with a sucrose content from 35% to 60%. Western blot using RSV-reactive mouse sera of the pooled fractions 8-10 confirmed the presence of PreFg, G and M proteins in the VLPs (Figure 2b). A PreFg protein band at ~63 kDa is consistent with the size described by Patel et. al. 2020 [31]. The matrix protein band (~28 kDa) and the glycoprotein band (~70 kDa) produced by baculovirus-infected cells had the expected molecular weights described previously [40,41]. TEM images of the purified VLPs revealed spherical particles with a size of 70-100 nm (Figure 2c). Western blot analysis and TEM of the purified VLPs confirms the incorporation of preFg, G and M proteins within the VLP structure. The presence of phosphate indicated that a lipid membrane is indeed an integral part of the formed VLPs (Figure 2d) [42].
3.3. RSV-VLPs Induce Potent Humoral Immune Responses after Immunization
In vivo studies were conducted to determine the immunogenicity of the VLPs. The immune response elicited by BPL-RSV and FI-RSV were the comparators. Of significance, the mice immunized with RSV-VLPs (5 or 10 µg) generated significantly higher VLP-specific IgG titers than the mice that received BPL- or formalin-inactivated RSV (Figure 3a). Similarly, IgG responses against RSV fusion (RSV-F, Figure 3b) and matrix (RSV-M, Figure 3d) proteins were higher in the VLP-immunized mice. Surprisingly, the response against glycoprotein, RSV-G, was not enhanced for the VLP vaccine (p value 0.07 to 0.88) (Figure 3c). B cell ELISpot using splenocytes from immunized mice was conducted to determine the numbers of antigen-specific ASCs. The VLPs induced similar numbers of ASCs as the inactivated RSV vaccines (Figure 3e). Determination of neutralizing antibody titers highlighted that the mice immunized with either 5 or 10 µg VLPs generated significantly higher levels of neutralizing antibodies than mice immunized with either of the inactivated RSV vaccines (Figure 3f).
3.4. IgG Subclass Immune Responses after Immunization
Antibody subtype responses against RSV-VLPs, F, and G proteins were characterized by measuring IgG2a and IgG1 subclass antibodies, the hallmark antibodies for Th1 and Th2 immunity respectively. VLP-immunized mice produced robust VLP- and F-specific IgG2a (Figure 4a,b) and IgG1 (Figure 4d,e) antibodies, and these responses were higher than those in mice immunized with inactivated RSV. However, this increase was not observed for G-specific antibodies. Similar to total IgG, the use of 10 µg VLPs for immunization did not increase the induction of either of the antibody subtypes against any of the proteins, as compared to the 5 µg dose (Figure 4a–f). Immunization with VLPs elicited an IgG1-dominant response (Figure 4e–f). Calculation of the IgG1/IgG2a ratio highlighted that VLPs gave rise to a skewed Th2 response but the skewing was less prominent than observed in the FI-RSV group (Figure 4g–i).
3.5. IFNγ- and IL4-Producing T Cells after Immunization
To further characterize the immune response generated after immunization, we quantified Th1 cytokine (IFNγ) and Th2 cytokine (IL4) from in vitro stimulated or non-stimulated splenocytes. Spots in non-stimulated wells highlighted immunization with either of the antigens induced significantly higher number of IFNγ or IL4 SFCs than in naïve mice. The number of SFCs was further increased after overnight stimulation (Figure 5c). Immunization with VLPs induced highest numbers of IFNγ SFCs upon stimulation compared to immunization with inactivated RSV virus or mock immunization (Figure 5a). The number of IL4 SFCs were similar in all immunized groups after stimulation (Figure 5b). Similar observations were noticed for secreted IFNγ and IL4 in supernatants of the restimulated and non-stimulated splenocytes (p values 0.11 to 0.90) (Figure 5d,e).
3.6. Inflammatory and Anti-Inflammatory Cytokine Responses after Immunization
We next estimated other cytokines that are shown to modulate the adaptive immune response. Non-stimulated splenocytes obtained from VLP-immunized mice showed enhanced secretion of TNFα (Figure 6a) and IL6 (Figure 6b). Only splenocytes from mice immunized with VLP induced significantly higher secretion of TNFα, IL6 and IL2 compared to the control group upon stimulation. Notably, IL2 secretion increased almost five-fold upon stimulation of the splenocytes (Figure 6c). Secretion of IL17A was similar across the different groups (Figure 6d). Similar to inflammatory cytokines, immunization with the VLPs showed significantly higher secretion of IL10 compared to the control group in non-stimulated and stimulated cultures (Figure 6e).
3.7. Memory T Cell Responses after Immunization
The ability of T cells to proliferate and develop memory T cells after vaccination is essential for protection and sustainability of the adaptive immune responses. For this assessment, we investigated the ability of the RSV-VLPs to induce proliferation of total CD4+ and CD8+ T cells as well as effector memory cells. The proliferation status was assessed by measuring Ki67 expression. Immunization with either vaccine resulted in the proliferation of CD4+ and CD8+ T cells (Figure 7a,b). Although no significant differences were observed in the total numbers of CD4+ and CD8+ T cells among the groups (data not shown), immunization with 10µg VLPs induced proliferation among CD8+ T cells (CD8+Ki67+, Figure 7b) as compared to the other groups. However, the proliferation of CD4+ T cells (CD4+Ki67+, Figure 7a) was not enhanced even after immunization with 10µg VLP. The levels of proliferating CD4+ and CD8+ effector memory T cells (CD4+CD44+CD62L-Ki67+ and CD8+CD44+CD62L-Ki67+, Figure 7c,d) in mice immunized with RSV-VLPs were similar to the levels found in other immunized groups. Overall, these results suggest that the performance of RSV-VLPs in inducing T cell proliferation is similar to that of inactivated RSV but can be enhanced with an increase in dose.
3.8. Cytotoxic CD8+ T Cell Response after Immunization
CTLs are known to mediate viral clearance through targeted killing of infected cells by releasing granzymeB. Increased expression of the degranulation marker CD107a on CD8+ T cells is directly proportional to cytotoxic activity. Therefore, we next evaluated the CTL response after immunization. Immunization with VLPs enhanced the number of granzymeB+CD8+ (Figure 8a) and CD107a+CD8+ T cells (Figure 8b) compared to immunization with inactivated RSV. This enhancement was not dependent on the VLP dose. An increase in dual granzymeB+CD107a+CD8+ T cells was also seen only after immunization with the VLPs (Figure 8d) and the increase was significantly higher as compared to that in the other immunizations groups. Surprisingly, no enhancement of IFNγ+CD8+ T cells was observed after immunization with VLPs (Figure 8c).
3.9. Protection from Live RSV after Challenge in Immunized Mice
To further investigate the possibility of enhanced respiratory disease (ERD) in the immunized mice, we examined lung pathology upon challenge infection (Figure 9a–e). The lungs of mice infected with live virus had mild congested vascular tissue in the lung parenchyma, mild alveolar pathological changes with alveolitis with mononuclear inflammatory cellular infiltration in the alveolar parenchyma (Figure 9b). Minimal pathology was seen in mice that received BPL-RSV (Figure 9c). However, mice immunized with FI-RSV showed signs of enhanced inflammation such as alveolitis and infiltrates in both the peribronchial and perivascular areas (Figure 9d). In contrast, the lungs of the mice that received RSV-VLP (Figure 9e) showed no signs of lung pathology and were very similar to the lungs of naïve mice (Figure 9a). Overall, mice immunized with FI-RSV showed ERD while immunization with VLPs prevented lung inflammation after RSV challenge (Figure 9f).
4. Discussion
In the current study, we aimed to develop RSV-VLPs consisting of PreFg, G and M protein as a vaccine candidate against RSV and study its protective efficacy in mice. The RSV-VLPs were produced by coinfection of ExpiSf9 cells with recombinant baculoviruses encoding the respective proteins. Immunogenicity studies demonstrated that these VLPs induced stronger humoral and cellular immune responses and provided better protection against lung inflammation upon challenge than inactivated RSV vaccines.
We wish to emphasize here that the VLPs developed by us contain the preFg and G proteins which possess most of the neutralizing epitopes of RSV [15]. The reasons for using preFg F protein in our study are (i) preFg does not get converted to the postF conformation, (ii) consists of the p27 protein and (iii) preFg is shown to be more immunogenic than the preF form [31]. Co-infection with two or more rBVs for simultaneous expression of three proteins is shown to give rise to VLPs that are released from cells similar to native RSV [27,36]. Similarly, we employed baculovirus encoding preFg and co-infected the cells with baculoviruses encoding G and M proteins to obtain the VLPs.
Although it was reported earlier that the M protein of RSV might lead to pleomorphic structures and may interfere with the formation of VLPs in absence of phosphoprotein [43], using M protein of RSV we were able to generate VLPs that resemble the structure of native RSV particles. In our study, we used ExpiSf9 cells, which are a non-engineered, suspension culture derivative of Sf9 insect cells. As per our expectations, the VLPs formed after co-infection of ExpiSf9 cells with baculoviruses encoding preFg, G and M consisted of all the three proteins. The structure of the VLPs was spherical having size between 70-100 nm. After co-infection and further downstream purification, we found substantial yield of approximately 1mg/litre of purified VLP. The VLPs possessed a lipid bilayer as observed by TEM and phosphate determination. The presence of lipids is adventitious because the VLPs can be associated with adjuvants that have lipophilic tails viz. MPLA [44,45]. The presence of lipid bilayer has also been visualized in the TEM images in other studies wherein VLPs were developed using baculoviruses encoding the preF and G protein [46,47].
Our study also demonstrated that immunization of mice with the VLPs induced robust humoral and cellular responses after two doses and conferred protective immunity against the virus challenge. Most importantly, no lung pathology or enhance respiratory disease was recorded. The VLPs showed higher antibody induction against all the three included proteins than the inactivated-RSV immunized mice. These results further confirm that the VLPs indeed consisted of the F, G and M proteins. The number of ASCs induced by the VLPs was comparable to that induced by the inactivated-RSV immunized mice. However, the higher amount of antibodies produced by a similar number of ASCs highlights the superior quality of VLP-induced ASCs. The neutralization potential of antibodies against preF is well known [48]. Additionally, various animal studies have documented protection by antibodies against G protein [49,50]. Immunization with VLPs induced antibodies against both F and G proteins indicative of additive neutralization effect [51].
Different immunoglobulin subtypes display differences in their ability to mediate effector responses [52]. Therefore, the IgG antibody subtypes were studied to understand the influence of the VLPs on their induction after immunization. In our study, the VLPs induced both IgG2a and IgG1 subtypes, though the response was IgG1 dominated pointing to a Th2 skewed response. Previous studies employing inactivated RSV or RSV-virosomes have shown induction of balanced IgG2a/IgG1 or skewed Th2 response upon intramuscular immunization [38,53,54]. The induction of IgG2a may reflect the role of humoral response in assisting the effective virus clearance without inducing pathology [55,56].
The antibody subtype responses are strongly influenced by the cytokines released by the CD4 T helper (Th) cells. Th1 cells are shown to promote IgG2a response due to the predominant release of IFNγ while Th2 cells are shown to induce IgG1 antibody with the help of IL4 [57,58]. In our study, the secretion of IFNγ and IL4 upon in vitro stimulation of the splenocytes with VLPs were also in line with the IgG subtype results. Although the VLPs induced higher IFNγ than FI-RSV, the number of IL4-producing cells were higher than IFNγ-producing cells which may have resulted in skewing of antibody response towards IgG1. Along with these cytokines, TNFα, IL10 and IL6 secretion was also found to be higher in the VLP immunized mice which may have contributed further to enhanced IgG1 antibody response [59]. IFNγ can promote the production of TNFα and can synergize with this cytokine in its actions on responder cells. Increased secretion of IL6 and IL2 after in vivo stimulation in our study suggests that the VLPs might be capable of promoting follicular T helper cells response leading to memory B cells production [60,61]. The increased IL10 production indicated capability of VLPs to promote Treg response [62,63]. Overall, the VLPs induced cytokines that are required to enhance the antigen-specific immune response.
In addition, VLPs induced higher granzymeB+, CD107a+ and dual positive granzymeB+CD107a+ CD8+ T cells than inactivated vaccines. Along with neutralizing antibodies, the induction of CD8+ T cells-mediated immunity may have promoted rapid virus clearance from the lungs of mice immunized with VLPs thereby decreasing pathology [64,65,66]. Similar results were reported for CD8+ T cell epitope vaccine against RSV [67]. It may be summarized that both CD8+ T cells together with neutralizing antibodies contributed towards the protection of the VLP-immunized mice.
Memory CD4+ T cells are the key players in promoting proliferation of memory B cells and contribute decisively to protection [68]. They also rapidly proliferate during recall response and facilitate the rapid production of antibodies. RSV vaccine-induced enhanced disease was shown to be orchestrated by memory CD4+ T cells [69]. Similarly, memory CD8+ T cells proliferate rapidly and perform their effector function of killing RSV-infected cells [67]. It has been well established that CD8+ T cells provide protection during acute RSV infection by mediating viral clearance [70,71]. We observed that mice immunized with VLPs developed memory CD4 and CD8 T cell responses that are further enhanced with an increased VLP dose. These observations strongly suggest that the VLPs developed by us have an intrinsic property of promoting memory T cell response.
Even though RSV has been recognized as an important pathogen of concern for the children, no vaccine is registered for the use in children. Formalin inactivated RSV vaccine has been abandoned because of the detrimental outcome of a previous clinical trial [4]. Although, other F protein-based particulate vaccines are immunogenic, the presence of metastable F makes their usage unadvisable [49,53,72]. Importantly, preF-based protein subunit RSV-vaccines are approved for use in elderly and pregnant women and approval may be extended to children. However, purified proteins are inferior to particulate-based vaccines in eliciting broader immunity [35,73,74]. Our study demonstrates that VLPs-based on the preFg and preF along with G and M proteins could be an attractive approach. Head-to-head comparison studies are needed to prove the superiority of these stabilized F protein-based VLPs over preF, preFg or G proteins-based vaccines. In conclusion, we provide strong evidence for preFg VLPs as to be a vaccine candidate that needs to be explored further. The presence of a lipid bilayer might allow to further improve the immunogenicity of the VLPs by inclusion of adjuvants with lipophilic tails and thereby enabling stimulation of a single APC with both adjuvant and antigen.
Author Contributions
Conceptualization, H.P.P., V.A.A. and A.L.W.H.; methodology, A.S.M. and H.P.P.; investigation, A.S.M., A.K.M. and H.P.P.; formal analysis, A.S.M. and H.P.P.; resources, H.P.P.; writing—original draft preparation, A.S.M. and H.P.P.; writing—review and editing, A.S.M. and H.P.P.; supervision, A.L.W.H, V.A.A. and H.P.P.; funding acquisition, H.P.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the DBT-Wellcome India Alliance (Reference number – IA/E/17/1/503651).
Institutional Review Board Statement
The in vivo study in mice was conducted after the approval from the Institutional Animal Ethics Committee of Bharati Vidyapeeth (Deemed to be University), Pune, India (Proposal number BVDUMC/819/2022/002/017).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data can be provided after request to the corresponding authors.
Acknowledgments
The authors thank Dr. Atanu Basu from the National Institute of Virology, Pune, India for kindly providing us with the transmission electron microscopy images of the VLPs. We also acknowledge Sanika Ghayal and Sawani Karandikar for their assistance with the animal experiments.
Conflicts of Interest
Patent application of Virus-like particles for respiratory syncytial virus and method of use is submitted at Controller General of Patents, Designs and Trade Marks, India. Application reference number: 202121036717.
References
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Children Younger than 5 Years in 2019: A Systematic Analysis. The Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Antillón, M.; Li, X.; Willem, L.; Bilcke, J.; Investigators, R.; Jit, M.; Beutels, P. The Age Profile of Respiratory Syncytial Virus Burden in Preschool Children of Low- and Middle-Income Countries: A Semi-Parametric, Meta-Regression Approach. PLoS Med 2023, 20, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Young Children in 2015: A Systematic Review and Modelling Study. The Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Willem, L.; Antillon, M.; Bilcke, J.; Jit, M.; Beutels, P. Health and Economic Burden of Respiratory Syncytial Virus (RSV) Disease and the Cost-Effectiveness of Potential Interventions against RSV among Children under 5 Years in 72 Gavi-Eligible Countries. BMC Med 2020, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, A.C.; Bont, L.J. Respiratory Syncytial Virus Infection and Novel Interventions. Nat Rev Microbiol 2023, 21, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Nirsevimab: First Approval. Drugs 2023, 83, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Wittenauer, R.; Pecenka, C.; Baral, R. Cost of Childhood RSV Management and Cost-Effectiveness of RSV Interventions: A Systematic Review from a Low- and Middle-Income Country Perspective. BMC Med 2023, 21, 121. [Google Scholar] [CrossRef] [PubMed]
- Mezei, A.; Cohen, J.; Renwick, M.J.; Atwell, J.; Portnoy, A. Mathematical Modelling of Respiratory Syncytial Virus (RSV) in Low- and Middle-Income Countries: A Systematic Review. Epidemics 2021, 35, 100444. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory Syncytial Virus—A Comprehensive Review. Clin Rev Allergy Immunol 2013, 45, 331–379. [Google Scholar] [CrossRef]
- Swanson, K.A.; Balabanis, K.; Xie, Y.; Aggarwal, Y.; Palomo, C.; Mas, V.; Metrick, C.; Yang, H.; Shaw, C.A.; Melero, J.A.; et al. A Monomeric Uncleaved Respiratory Syncytial Virus F Antigen Retains Prefusion-Specific Neutralizing Epitopes. J Virol 2014, 88, 11802–11810. [Google Scholar] [CrossRef]
- Tripp, R.A.; Mejias, A.; Ramilo, O. Host Gene Expression and Respiratory Syncytial Virus Infection. Curr Top Microbiol Immunol 2013, 372, 193–209. [Google Scholar] [CrossRef]
- Walsh, E.E. Humoral, Mucosal, and Cellular Immune Response to Topical Immunization with a Subunit Respiratory Syncytial Virus Vaccine. J Infect Dis 1994, 170, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Dennis, V.A.; Carter, C.L.; Pillai, S.R.; Jefferson, A.; Sahi, S.V.; Moore, E.G. Immunogenicity and Efficacy of Recombinant RSV-F Vaccine in a Mouse Model. Vaccine 2007, 25, 6211–6223. [Google Scholar] [CrossRef] [PubMed]
- Sullender, W.M. Respiratory Syncytial Virus Genetic and Antigenic Diversity. Clin Microbiol Rev 2000, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and Function of Respiratory Syncytial Virus Surface Glycoproteins. Curr Top Microbiol Immunol 2013, 372, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Rainho-Tomko, J.N.; Pavot, V.; Kishko, M.; Swanson, K.; Edwards, D.; Yoon, H.; Lanza, L.; Alamares-Sapuay, J.; Osei-Bonsu, R.; Mundle, S.T.; et al. Immunogenicity and Protective Efficacy of RSV G Central Conserved Domain Vaccine with a Prefusion Nanoparticle. NPJ Vaccines 2022, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Ghildyal, R.; Ho, A.; Jans, D.A. Central Role of the Respiratory Syncytial Virus Matrix Protein in Infection. FEMS Microbiol Rev 2006, 30, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.; Bierre, M.; Veyrié, L.; Richard, C.-A.; Eleouet, J.-F.; Muriaux, D.; Bajorek, M. Selective Targeting and Clustering of Phosphatidylserine Lipids by RSV M Protein Is Critical for Virus Particle Production. Journal of Biological Chemistry 2023, 299, 105323. [Google Scholar] [CrossRef]
- Rutigliano, J.A.; Ruckwardt, T.J.; Martin, J.E.; Graham, B.S. Relative Dominance of Epitope-Specific CD8+ T Cell Responses in an F1 Hybrid Mouse Model of Respiratory Syncytial Virus Infection. Virology 2007, 362, 314. [Google Scholar] [CrossRef]
- Rutigliano, J.A.; Rock, M.T.; Johnson, A.K.; Crowe, J.E.; Graham, B.S. Identification of an H-2Db-Restricted CD8+ Cytotoxic T Lymphocyte Epitope in the Matrix Protein of Respiratory Syncytial Virus. Virology 2005, 337, 335–343. [Google Scholar] [CrossRef]
- Acosta, P.L.; Caballero, M.T.; Polack, F.P. Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease. Clinical and Vaccine Immunology 2016, 23, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Mejias, A.; Rodríguez-Fernández, R.; Oliva, S.; Peeples, M.E.; Ramilo, O. The Journey to an RSV Vaccine. Ann Allergy Asthma Immunol 2020, 125, 36. [Google Scholar] [CrossRef] [PubMed]
- Waris, M.E.; Tsou, C.; Erdman, D.D.; Zaki, S.R.; Anderson, L.J. Respiratory Syncytial Virus Infection in BALB/c Mice Previously Immunized with Formalin-Inactivated Virus Induces Enhanced Pulmonary Inflammatory Response with a Predominant Th2-Like Cytokine Pattern. J Virol 1996, 70, 2852–2860. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.F.; Coviello, S.; Monsalvo, A.C.; Melendi, G.A.; Hernandez, J.Z.; Batalle, J.P.; Diaz, L.; Trento, A.; Chang, H.-Y.; Mitzner, W.; et al. Lack of Antibody Affinity Maturation Due to Poor Toll-like Receptor Stimulation Leads to Enhanced Respiratory Syncytial Virus Disease. Nat Med 2009, 15, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mazur, N.I.; Higgins, D.; Nunes, M.C.; Melero, J.A.; Langedijk, A.C.; Horsley, N.; Buchholz, U.J.; Openshaw, P.J.; McLellan, J.S.; Englund, J.A.; et al. The Respiratory Syncytial Virus Vaccine Landscape: Lessons from the Graveyard and Promising Candidates. Lancet Infect Dis 2018, 18, e295–e311. [Google Scholar] [CrossRef] [PubMed]
- Cullen, L.M.; Schmidt, M.R.; Morrison, T.G. The Importance of RSV F Protein Conformation in VLPs in Stimulation of Neutralizing Antibody Titers in Mice Previously Infected with RSV. Hum Vaccin Immunother 2017, 13, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Qin, H.; Lei, L.; Lou, W.; Li, R.; Pan, Z. Virus-like Particles Containing a Prefusion-Stabilized F Protein Induce a Balanced Immune Response and Confer Protection against Respiratory Syncytial Virus Infection in Mice. Front Immunol 2022, 13, 1054005. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, P. First RSV Vaccine Approvals. Lancet Microbe 2023, 4, e577. [Google Scholar] [CrossRef] [PubMed]
- Gonzá Lez-Reyes, L.; Begoñ, A.; Ruiz-Argü Ello, M.; García-Barreno, B.; Calder, L.; Ló Pez, J.A.; Albar, J.P.; Skehel, J.J.; Wiley, D.C.; Melero, J.A. Cleavage of the Human Respiratory Syncytial Virus Fusion Protein at Two Distinct Sites Is Required for Activation of Membrane Fusion. Proc Natl Acad Sci U S A 2001, 98, 9859–9864. [Google Scholar] [CrossRef]
- Patel, N.; Tian, J.H.; Flores, R.; Jacobson, K.; Walker, M.; Portnoff, A.; Gueber-Xabier, M.; Massare, M.J.; Glenn, G.; Ellingsworth, L.; et al. Flexible RSV Prefusogenic Fusion Glycoprotein Exposes Multiple Neutralizing Epitopes That May Collectively Contribute to Protective Immunity. Vaccines (Basel) 2020, 8, 1–20. [Google Scholar] [CrossRef]
- Patel, N.; Massare, M.J.; Tian, J.H.; Guebre-Xabier, M.; Lu, H.; Zhou, H.; Maynard, E.; Scott, D.; Ellingsworth, L.; Glenn, G.; et al. Respiratory Syncytial Virus Prefusogenic Fusion (F) Protein Nanoparticle Vaccine: Structure, Antigenic Profile, Immunogenicity, and Protection. Vaccine 2019, 37, 6112–6124. [Google Scholar] [CrossRef] [PubMed]
- Grgacic, E.V.L.; Anderson, D.A. Virus-like Particles: Passport to Immune Recognition. Methods 2006, 40, 60–65. [Google Scholar] [CrossRef] [PubMed]
- McGinnes Cullen, L.; Luo, B.; Wen, Z.; Zhang, L.; Durr, E.; Morrison, T.G. The Respiratory Syncytial Virus (RSV) G Protein Enhances the Immune Responses to the RSV F Protein in an Enveloped Virus-Like Particle Vaccine Candidate. J Virol 2023, 97, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.; Yang, J.E.; Chen, X.; Jadhao, S.J.; Wright, E.R.; Anderson, L.J. Two RSV Platforms for G, F, or G+F Proteins VLPs. Viruses 2020, 12, 906. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Chu, K.-B.; Kim, M.-J.; Mao, J.; Eom, G.-D.; Yoon, K.-W.; Ahmed, M.A.; Quan, F.-S. Virus-like Particle Vaccine Expressing the Respiratory Syncytial Virus Pre-Fusion and G Proteins Confers Protection against RSV Challenge Infection. Pharmaceutics 2023, 15, 782. [Google Scholar] [CrossRef]
- Lee, S.-H.; Chu, K.-B.; Kim, M.-J.; Quan, F.-S. Virus-Like Particles Assembled Using Respiratory Syncytial Virus Matrix Protein Elicit Protective Immunity in Mice. Infect Drug Resist 2023, 16, 6099–6110. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. Assay of Inorganic Phosphate, Total Phosphate and Phosphatases. 1966; pp. 115–118.
- Shafique, M.; Wilschut, J.; de Haan, A. Induction of Mucosal and Systemic Immunity against Respiratory Syncytial Virus by Inactivated Virus Supplemented with TLR9 and NOD2 Ligands. Vaccine 2012, 30, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.A. Determination of 50% Endpoint Titer Using a Simple Formula. World J Virol 2016, 5, 85–86. [Google Scholar] [CrossRef]
- Ward, C.; Maselko, M.; Lupfer, C.; Prescott, M.; Pastey, M.K. Interaction of the Human Respiratory Syncytial Virus Matrix Protein with Cellular Adaptor Protein Complex 3 Plays a Critical Role in Trafficking. PLoS ONE 2017, 12, e184629. [Google Scholar] [CrossRef]
- Kim, S.; Chang, J. Baculovirus-Based Vaccine Displaying Respiratory Syncytial Virus Glycoprotein Induces Protective Immunity against RSV Infection without Vaccine-Enhanced Disease. Immune Netw 2012, 12, 8–17. [Google Scholar] [CrossRef]
- McCurdy, L.H.; Graham, B.S. Role of Plasma Membrane Lipid Microdomains in Respiratory Syncytial Virus Filament Formation. J Virol 2003, 77, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Bajorek, M.; Galloux, M.; Richard, C.-A.; Szekely, O.; Rosenzweig, R.; Sizun, C.; Eleouet, J.-F. Tetramerization of Phosphoprotein Is Essential for Respiratory Syncytial Virus Budding While Its N-Terminal Region Mediates Direct Interactions with the Matrix Protein. J Virol 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.; Murugappan, S.; ter Veer, W.; Meijerhof, T.; de Haan, A.; Frijlink, H.W.; Wilschut, J.; Hinrichs, W.L.J.; Huckriede, A. Evaluation of Monophosphoryl Lipid A as Adjuvant for Pulmonary Delivered Influenza Vaccine. Journal of Controlled Release 2013, 174, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Gosavi, M.; Kulkarni-Munje, A.; Patil, H.P. Dual Pattern Recognition Receptor Ligands CL401, CL413, and CL429 as Adjuvants for Inactivated Chikungunya Virus. Virology 2023, 585, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chu, K.; Kim, M.; Mao, J.; Eom, G.; Yoon, K.; Ahmed, A.; Quan, F. Virus-like Particle Vaccine Expressing the Respiratory Syncytial Virus Pre-Fusion and G Proteins Confers Protection against RSV Challenge Infection. Pharmaceutics 2023, 15, 782. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Chu, K.B.; Lee, S.H.; Mao, J.; Eom, G.D.; Yoon, K.W.; Moon, E.K.; Quan, F.S. Assessing the Protection Elicited by Virus-like Particles Expressing the RSV Pre-Fusion F and Tandem Repeated G Proteins against RSV RA2 Line19F Infection in Mice. Respir Res 2024, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.C.G.; Pletneva, L.M.; McGinnes-Cullen, L.; Otoa, R.O.; Patel, M.C.; Fernando, L.R.; Boukhvalova, M.S.; Morrison, T.G. Efficacy of a Respiratory Syncytial Virus Vaccine Candidate in a Maternal Immunization Model. Nat Commun 2018, 9. [Google Scholar] [CrossRef]
- Quan, F.S.; Kim, Y.; Lee, S.; Yi, H.; Kang, S.M.; Bozja, J.; Moore, M.L.; Compans, R.W. Viruslike Particle Vaccine Induces Protection against Respiratory Syncytial Virus Infection in Mice. Journal of Infectious Diseases 2011, 204, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, H.C.; Murray, J.; Juarez, M.G.; Nangle, S.J.; DuBois, R.M.; Tripp, R.A. Immunogenicity and Protective Efficacy of an RSV G S177Q Central Conserved Domain Nanoparticle Vaccine. Front Immunol 2023, 14, 1215323. [Google Scholar] [CrossRef]
- Su, C.; Zhong, Y.; Zhao, G.; Hou, J.; Zhang, S.; Wang, B. RSV Pre-Fusion F Protein Enhances the G Protein Antibody and Anti-Infectious Responses. NPJ Vaccines 2022, 7, 1–11. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Immunology: Divergent Immunoglobulin G Subclass Activity through Selective Fc Receptor Binding. Science (1979) 2005, 310, 1510–1512. [Google Scholar] [CrossRef]
- Shafique, M.; Meijerhof, T.; Wilschut, J.; de Haan, A. Evaluation of an Intranasal Virosomal Vaccine against Respiratory Syncytial Virus in Mice: Effect of TLR2 and NOD2 Ligands on Induction of Systemic and Mucosal Immune Responses. PLoS ONE 2013, 8, e61287. [Google Scholar] [CrossRef]
- Kamphuis, T.; Meijerhof, T.; Stegmann, T.; Lederhofer, J.; Wilschut, J.; de Haan, A. Immunogenicity and Protective Capacity of a Virosomal Respiratory Syncytial Virus Vaccine Adjuvanted with Monophosphoryl Lipid a in Mice. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Coutelier, J.P.; Van Der Logt, J.T.M.; Heessen, F.W.A.; Warnier, G.; Van Snick, J. IgG2a Restriction of Murine Antibodies Elicited by Viral Infections. Journal of Experimental Medicine 1987, 165, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.C.; McKeon, R.M.; Brackin, M.N.; Miller, L.A.; Keating, R.; Brown, S.A.; Makarova, N.; Perez, D.R.; MacDonald, G.H.; McCullers, J.A. Distinct Contributions of Vaccine-Induced Immunoglobulin G1 (IgG1) and IgG2a Antibodies to Protective Immunity against Influenza. Clinical and Vaccine Immunology 2006, 13, 981–990. [Google Scholar] [CrossRef]
- Bossie, A.; Vitetta, E.S. IFN-γ Enhances Secretion of IgG2a from IgG2a-Committed LPS-Stimulated Murine B Cells: Implications for the Role of IFN-γ in Class Switching. Cell Immunol 1991, 135, 95–104. [Google Scholar] [CrossRef] [PubMed]
- MOON, H.B.; SEVERINSON, E.; HEUSSER, C.; JOHANSSON, S.G.O.; MÖLLER, G.; PERSSON, U. Regulation of IgG1 and IgE Synthesis by Interleukin 4 in Mouse B Cells. Scand J Immunol 1989, 30, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding Roles for CD4+ T Cells in Immunity to Viruses. Nat Rev Immunol 2012, 12, 136–148. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Ditoro, D.; Winstead, C.; Pham, D.; Witte, S.; Andargachew, R.; Singer, J.R.; Wilson, C.G.; Zindl, C.L.; Luther, R.J.; Silberger, D.J.; et al. Differential IL-2 Expression Defines Developmental Fates of Follicular versus Nonfollicular Helper T Cells. Science (1979) 2018, 361. [Google Scholar] [CrossRef]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding Roles for CD4+ T Cells in Immunity to Viruses. Nat Rev Immunol 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, X.; Shen, G.; Wang, W.; Li, J.; Zhao, J.; Wei, Y.Q.; Edwards, C.K. Interleukin-10 Deficiency Impairs Regulatory T Cell-Derived Neuropilin-1 Functions and Promotes Th1 and Th17 Immunity. Sci Rep 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.E.; Varga, S.M. The CD8 T Cell Response to Respiratory Virus Infections. Front Immunol 2018, 9, 357939. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.E.; Meyerholz, D.K.; Varga, S.M. Pre-Existing Neutralizing Antibodies Prevent CD8 T Cell-Mediated Immunopathology Following Respiratory Syncytial Virus Infection. Mucosal Immunol 2020, 13, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Woodland, D.L.; Hogan, R.J.; Zhong, W. Cellular Immunity and Memory to Respiratory Virus Infections. Immunol Res 2001, 24, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Stokes, K.L.; Currier, M.G.; Sakamoto, K.; Lukacs, N.W.; Celis, E.; Moore, M.L. Vaccine-Elicited CD8+ T Cells Protect against Respiratory Syncytial Virus Strain A2-Line19F-Induced Pathogenesis in BALB/c Mice. J Virol 2012, 86, 13016. [Google Scholar] [CrossRef]
- Gray, J.I.; Westerhof, L.M.; MacLeod, M.K.L. The Roles of Resident, Central and Effector Memory CD4 T-Cells in Protective Immunity Following Infection or Vaccination. Immunology 2018, 154, 574–581. [Google Scholar] [CrossRef]
- Knudson, C.J.; Hartwig, S.M.; Meyerholz, D.K.; Varga, S.M. RSV Vaccine-Enhanced Disease Is Orchestrated by the Combined Actions of Distinct CD4 T Cell Subsets. PLoS Pathog 2015, 11, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Uddback, I.; Kohlmeier, J.E.; Christensen, J.P.; Thomsen, A.R. Vaccine Induced Memory CD8+ T Cells Efficiently Prevent Viral Transmission from the Respiratory Tract. Front Immunol 2023, 14. [Google Scholar] [CrossRef]
- Retamal-Díaz, A.; Covián, C.; Pacheco, G.A.; Castiglione-Matamala, A.T.; Bueno, S.M.; González, P.A.; Kalergis, A.M. Contribution of Resident Memory CD8+ T Cells to Protective Immunity Against Respiratory Syncytial Virus and Their Impact on Vaccine Design. Pathogens 2019, 8, 147. [Google Scholar] [CrossRef]
- Walpita, P.; Johns, L.M.; Tandon, R.; Moore, M.L. Mammalian Cell-Derived Respiratory Syncytial Virus-Like Particles Protect the Lower as Well as the Upper Respiratory Tract. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Vartak, A.; Sucheck, S.J. Recent Advances in Subunit Vaccine Carriers. Vaccines (Basel) 2016, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M.; Hu, J. The Integrated Consideration of Vaccine Platforms, Adjuvants, and Delivery Routes for Successful Vaccine Development. Vaccines (Basel) 2023, 11, 695. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cloning and expression of preFg, G and M proteins. (a) pFastBac1 transfer vectors carrying the RSV PreFg (lane 1), G (lane 2) and M (lane 3) gene were cut with BamHI and XhoI to confirm correct insertion of the genes in the vectors. (b) Expression of the PreFg, G and M proteins by ExpiSf9 cells infected with the respective rBVs confirmed by indirect ELISA using sera negative and positive for anti-RSV antibodies.
Figure 1.
Cloning and expression of preFg, G and M proteins. (a) pFastBac1 transfer vectors carrying the RSV PreFg (lane 1), G (lane 2) and M (lane 3) gene were cut with BamHI and XhoI to confirm correct insertion of the genes in the vectors. (b) Expression of the PreFg, G and M proteins by ExpiSf9 cells infected with the respective rBVs confirmed by indirect ELISA using sera negative and positive for anti-RSV antibodies.
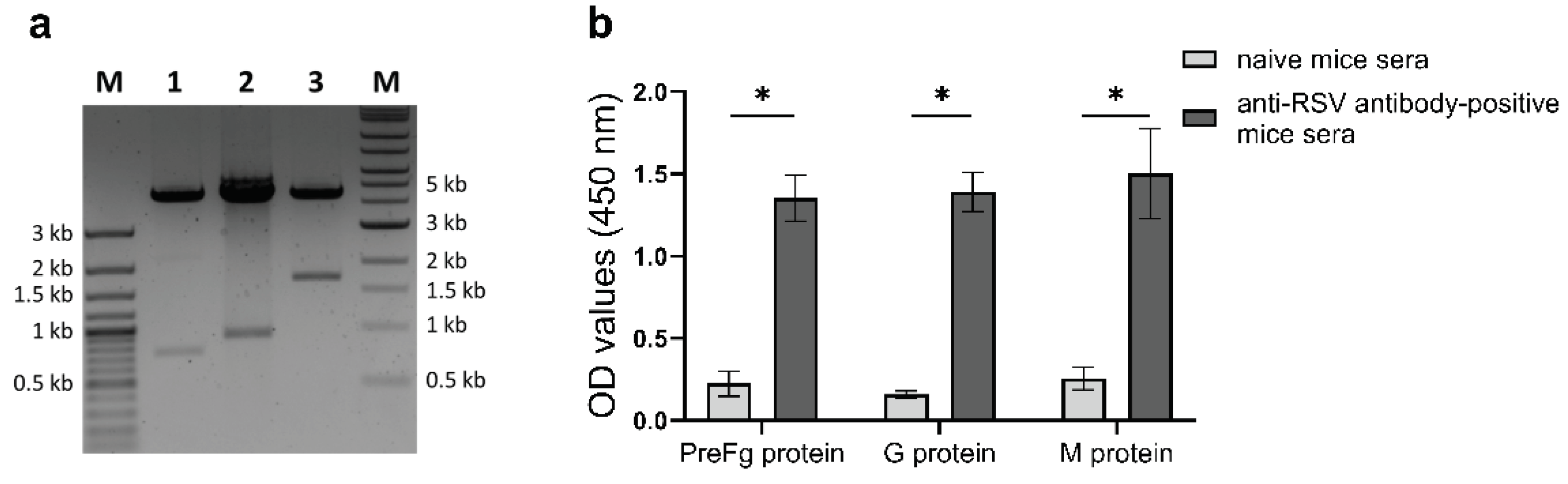
Figure 2.
Purification and characterization of RSV-VLPs. (a) Cell culture supernatant of the ExpiSf9 cells coinfected with PreFg, G and M-encoding rBVs was harvested and RSV-VLPs were purified on a 20%-60% sucrose gradient by ultracentrifugation at 100,000g for 3 hrs at 4 ⁰C as described in materials and methods. A total of 25 fractions were collected and total protein content and RSV reactivity by indirect ELISA of each fraction was determined. (b) Western blot analysis of the dialysed fractions 8-10, corresponding to 30%-35% sucrose gradient showing purified RSV-VLPs containing the three proteins, PreFg and G and M. (c) The same pooled fractions were visualized by transmission electron microscopy to confirm the formation of RSV-VLPs. Scale bar equals 200nm. (d) The phospholipid content of 5µg purified RSV-VLP and inactivated BPL-RSV was also determined.
Figure 2.
Purification and characterization of RSV-VLPs. (a) Cell culture supernatant of the ExpiSf9 cells coinfected with PreFg, G and M-encoding rBVs was harvested and RSV-VLPs were purified on a 20%-60% sucrose gradient by ultracentrifugation at 100,000g for 3 hrs at 4 ⁰C as described in materials and methods. A total of 25 fractions were collected and total protein content and RSV reactivity by indirect ELISA of each fraction was determined. (b) Western blot analysis of the dialysed fractions 8-10, corresponding to 30%-35% sucrose gradient showing purified RSV-VLPs containing the three proteins, PreFg and G and M. (c) The same pooled fractions were visualized by transmission electron microscopy to confirm the formation of RSV-VLPs. Scale bar equals 200nm. (d) The phospholipid content of 5µg purified RSV-VLP and inactivated BPL-RSV was also determined.
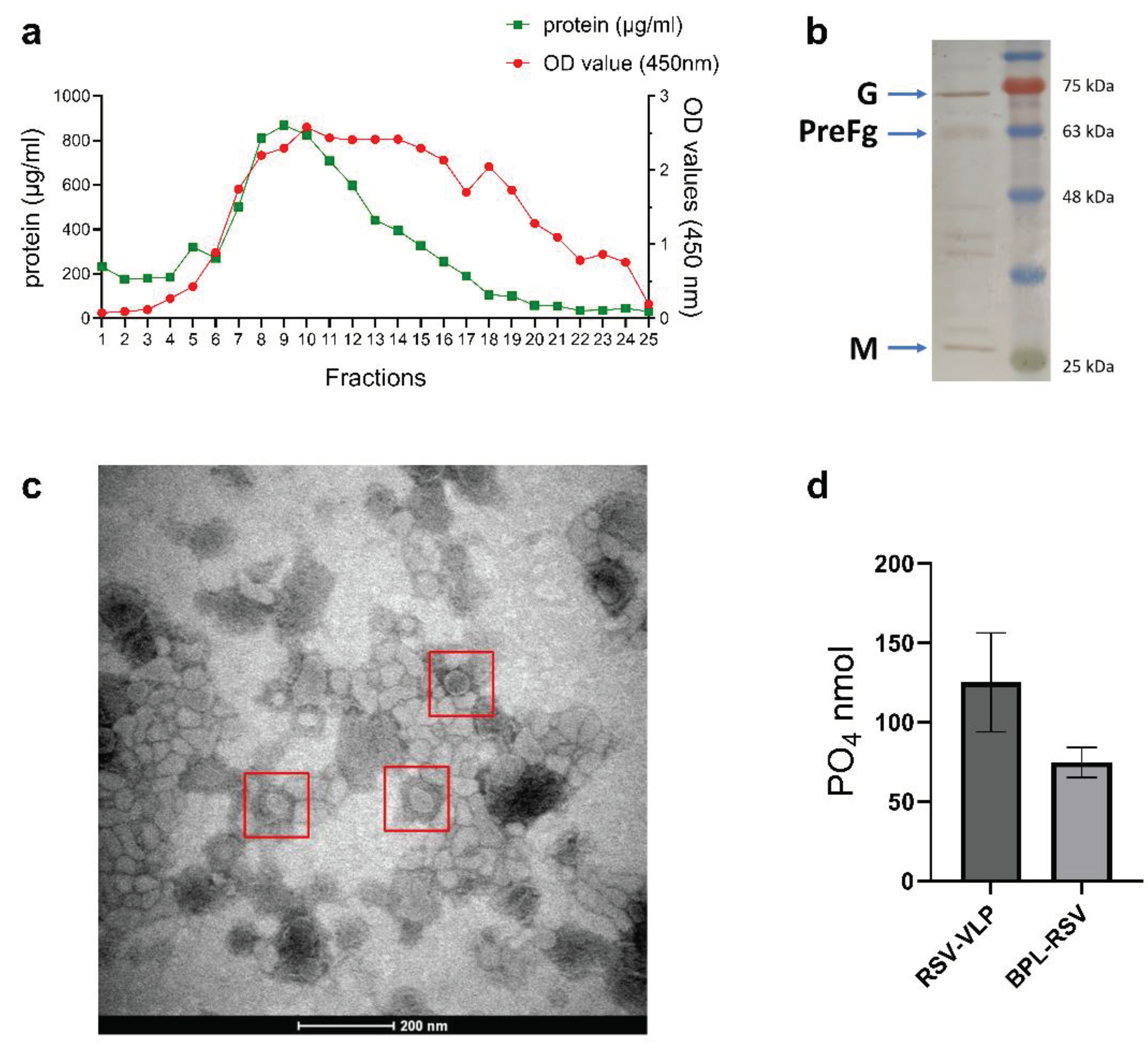
Figure 3.
Humoral response after immunization with RSV-VLPs. Mice (n=6) were immunized twice on days 1 and 21 with 5µg BPL-RSV, FI-RSV or 5 or 10 µg RSV-VLP. Control mice remained unimmunized. Blood was collected 21 days after the first dose and 7 days after the second dose (day 28) upon sacrifice. Antibody responses after immunization were determined by measuring (a) the total IgG titre against RSV and ELISA reactivity of the collected sera against (b) RSV-F, (c) RSV-G and (d) RSV-M proteins. (e) ASCs producing RSV-specific IgG or IgA from splenocytes were measured by ELISpot. HEp-2 cells were used to determine (f) neutralizing antibody levels in sera.
Figure 3.
Humoral response after immunization with RSV-VLPs. Mice (n=6) were immunized twice on days 1 and 21 with 5µg BPL-RSV, FI-RSV or 5 or 10 µg RSV-VLP. Control mice remained unimmunized. Blood was collected 21 days after the first dose and 7 days after the second dose (day 28) upon sacrifice. Antibody responses after immunization were determined by measuring (a) the total IgG titre against RSV and ELISA reactivity of the collected sera against (b) RSV-F, (c) RSV-G and (d) RSV-M proteins. (e) ASCs producing RSV-specific IgG or IgA from splenocytes were measured by ELISpot. HEp-2 cells were used to determine (f) neutralizing antibody levels in sera.
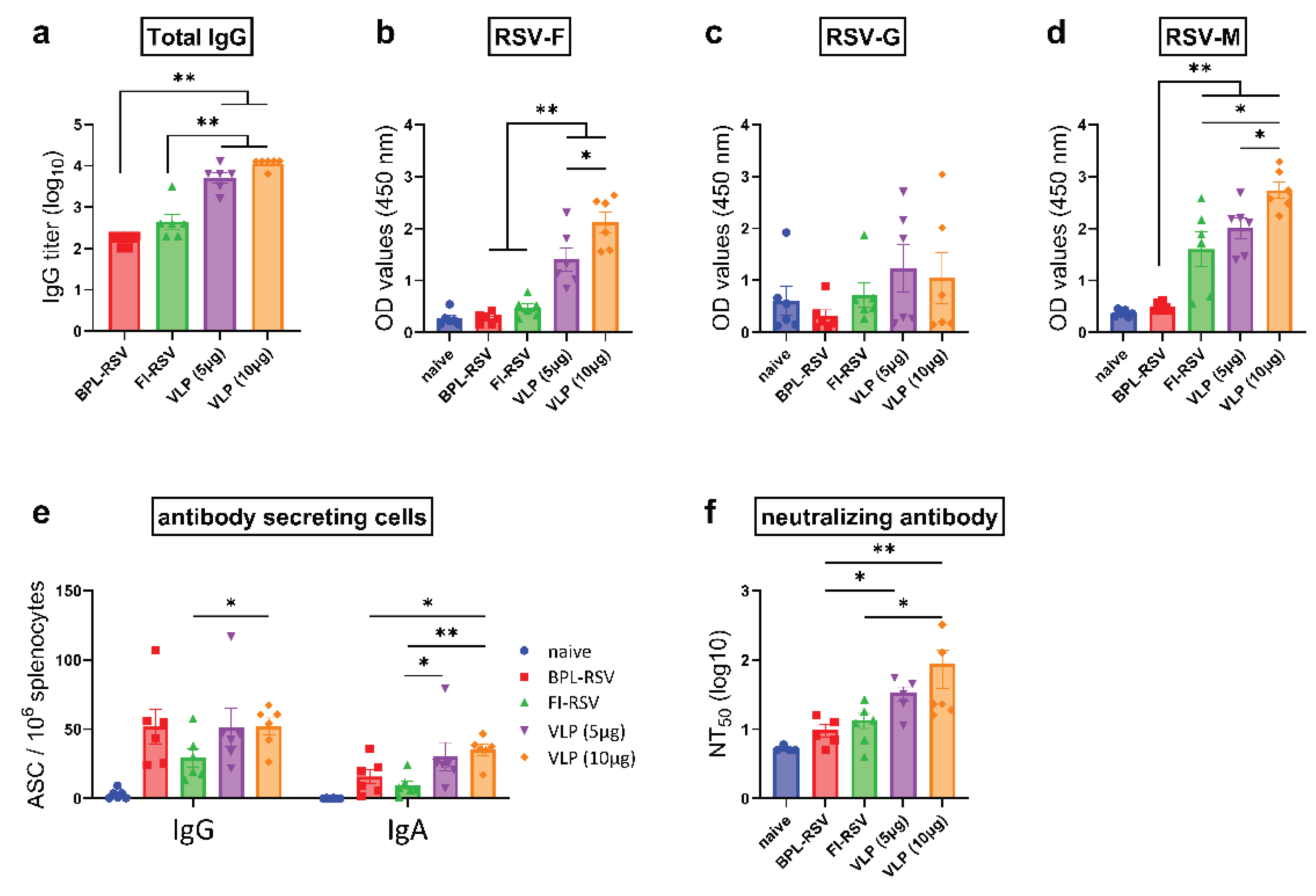
Figure 4.
Antibody subtype response after immunization with RSV-VLP. Mice were vaccinated with two doses as described previously. Sera obtained from the mice were analysed using for IgG subtypes using RSV-VLPs, RSV-F and RSV-G for coating. (a-c) IgG2a and (d-f) IgG1 response were evaluated by ELISA using serum obtained from the blood collected from mice on day 28. The ratios of IgG1 to IgG2a to (g) RSV-VLPs, (h) RSV-F and (i) RSV-G were calculated for individual mice.
Figure 4.
Antibody subtype response after immunization with RSV-VLP. Mice were vaccinated with two doses as described previously. Sera obtained from the mice were analysed using for IgG subtypes using RSV-VLPs, RSV-F and RSV-G for coating. (a-c) IgG2a and (d-f) IgG1 response were evaluated by ELISA using serum obtained from the blood collected from mice on day 28. The ratios of IgG1 to IgG2a to (g) RSV-VLPs, (h) RSV-F and (i) RSV-G were calculated for individual mice.
Figure 5.
IFNγ and IL4 responses after immunization. Mice were sacrificed 7 days post the second vaccination (day 28) and spleens were collected. Splenocytes were left untreated or stimulated with RSV-VLPs for 18 hours to determine (a) IFNγ and (b) IL-4 SFC. (c) Representative images of stimulated and non-stimulated wells of each immunization group are shown for IFNγ/IL-4 dual color ELISpot. Blue and red spots are specific for IL4 and IFNγ, respectively. Similarly, splenocytes were cultured for 72 hrs in the presence or absence of VLPs to determine the amount of secreted (d) IFNγ and (e) IL-4. Open and filled symbols indicate cytokine responses from the non-stimulated and stimulated splenocytes respectively.
Figure 5.
IFNγ and IL4 responses after immunization. Mice were sacrificed 7 days post the second vaccination (day 28) and spleens were collected. Splenocytes were left untreated or stimulated with RSV-VLPs for 18 hours to determine (a) IFNγ and (b) IL-4 SFC. (c) Representative images of stimulated and non-stimulated wells of each immunization group are shown for IFNγ/IL-4 dual color ELISpot. Blue and red spots are specific for IL4 and IFNγ, respectively. Similarly, splenocytes were cultured for 72 hrs in the presence or absence of VLPs to determine the amount of secreted (d) IFNγ and (e) IL-4. Open and filled symbols indicate cytokine responses from the non-stimulated and stimulated splenocytes respectively.
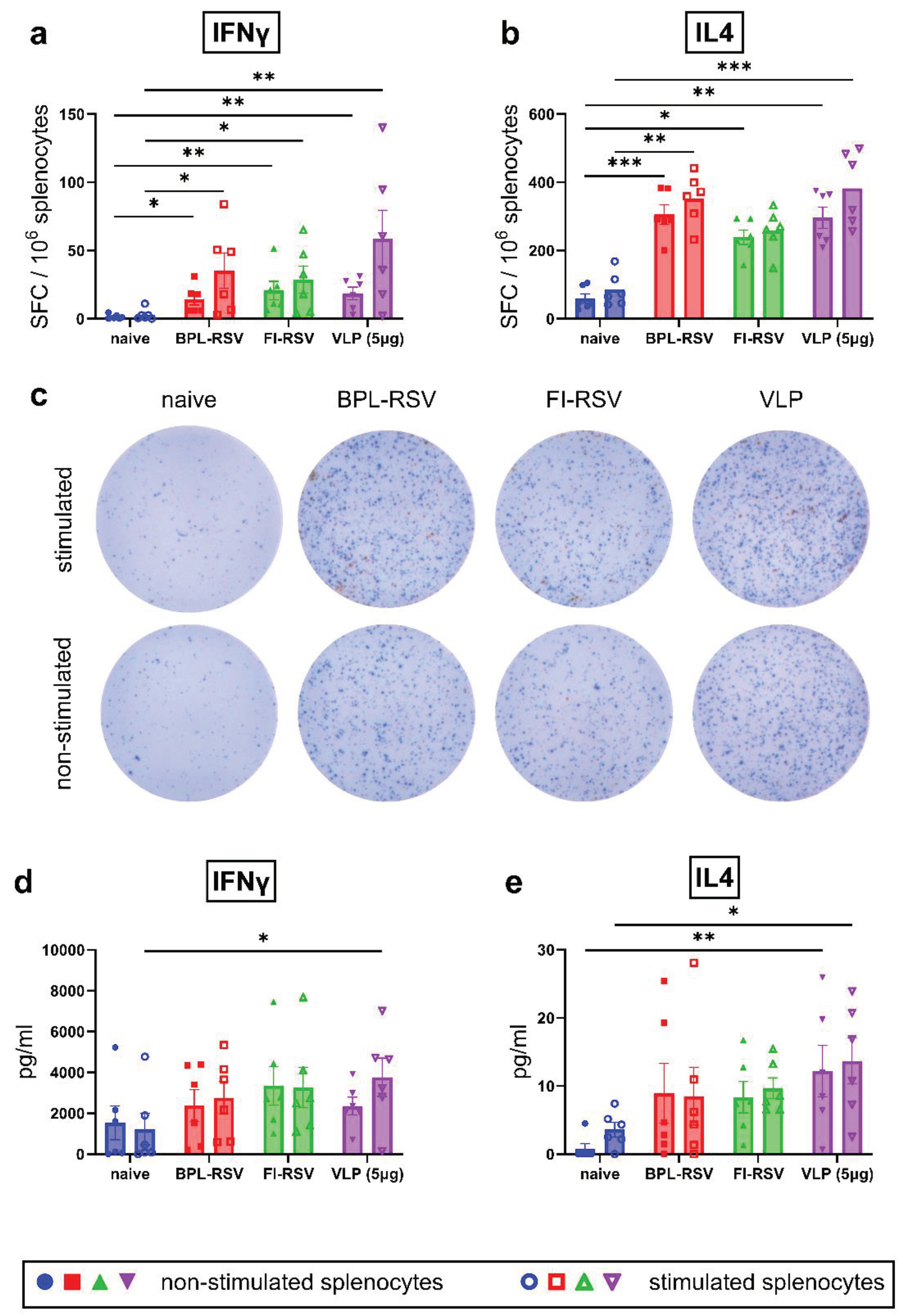
Figure 6.
Inflammatory and non-inflammatory cytokine responses after immunization. Mice were sacrificed 7 days post second vaccination (day 28) and splenocytes were cultured in the presence or absence of RSV-VLPs for 72 hours to determine secretion of (a) TNFα, (b) IL6, (c) IL17A, (d) IL10 and (e) IL2. Antigen-specific secretion (hollow dots) of cytokines was determined by subtracting values of the secreted cytokine of non-stimulated wells from the stimulated wells for each mouse. Antigen mediated response (solid dots) of cytokines was determined from the supernatant of unstimulated wells. The quantity of cytokines released from the unstimulated splenocytes was subtracted from cytokines released from the stimulated splenocytes to determine antigen specific cytokine response. Antigen mediated response which is a measure of in vivo stimulation due to the second immunization dose was determined by evaluating cytokine levels from unstimulated splenocytes culture.
Figure 6.
Inflammatory and non-inflammatory cytokine responses after immunization. Mice were sacrificed 7 days post second vaccination (day 28) and splenocytes were cultured in the presence or absence of RSV-VLPs for 72 hours to determine secretion of (a) TNFα, (b) IL6, (c) IL17A, (d) IL10 and (e) IL2. Antigen-specific secretion (hollow dots) of cytokines was determined by subtracting values of the secreted cytokine of non-stimulated wells from the stimulated wells for each mouse. Antigen mediated response (solid dots) of cytokines was determined from the supernatant of unstimulated wells. The quantity of cytokines released from the unstimulated splenocytes was subtracted from cytokines released from the stimulated splenocytes to determine antigen specific cytokine response. Antigen mediated response which is a measure of in vivo stimulation due to the second immunization dose was determined by evaluating cytokine levels from unstimulated splenocytes culture.
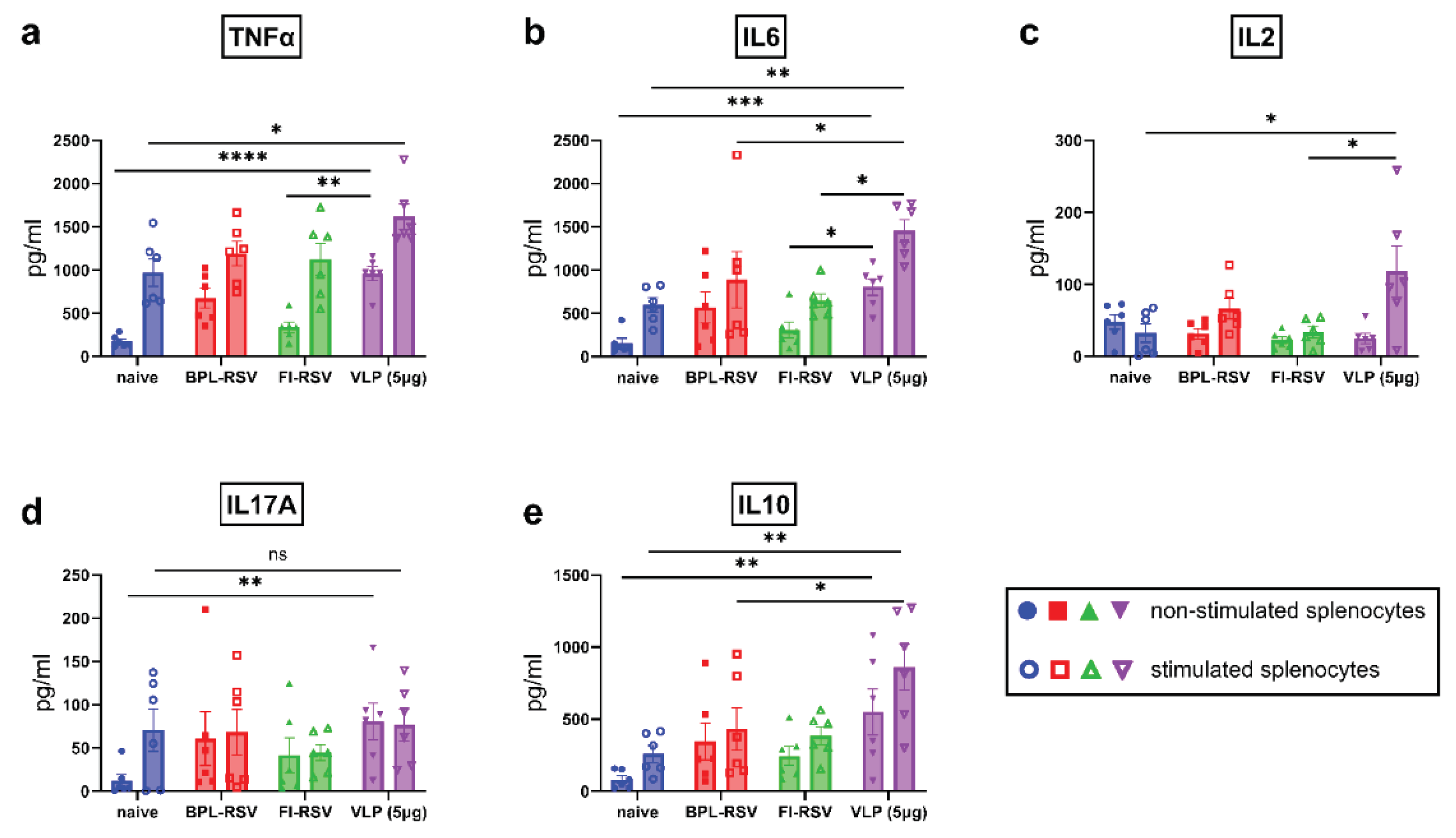
Figure 7.
Proliferating T cell responses after immunization. The proliferating CD4 and CD8 responses were evaluated using splenocytes of immunized mice that were sacrificed 7 days after the second vaccination. Ki67 was used as a marker of proliferation. The percentage of total (a) CD4+ and (b) CD8+ proliferating cells as well as proliferating effector memory (c) CD4+ and (d) CD8+ T cells was determined employing flowcytometry. (e) Gating strategy to determine levels of proliferating CD4+ and CD8+ T cells is depicted.
Figure 7.
Proliferating T cell responses after immunization. The proliferating CD4 and CD8 responses were evaluated using splenocytes of immunized mice that were sacrificed 7 days after the second vaccination. Ki67 was used as a marker of proliferation. The percentage of total (a) CD4+ and (b) CD8+ proliferating cells as well as proliferating effector memory (c) CD4+ and (d) CD8+ T cells was determined employing flowcytometry. (e) Gating strategy to determine levels of proliferating CD4+ and CD8+ T cells is depicted.
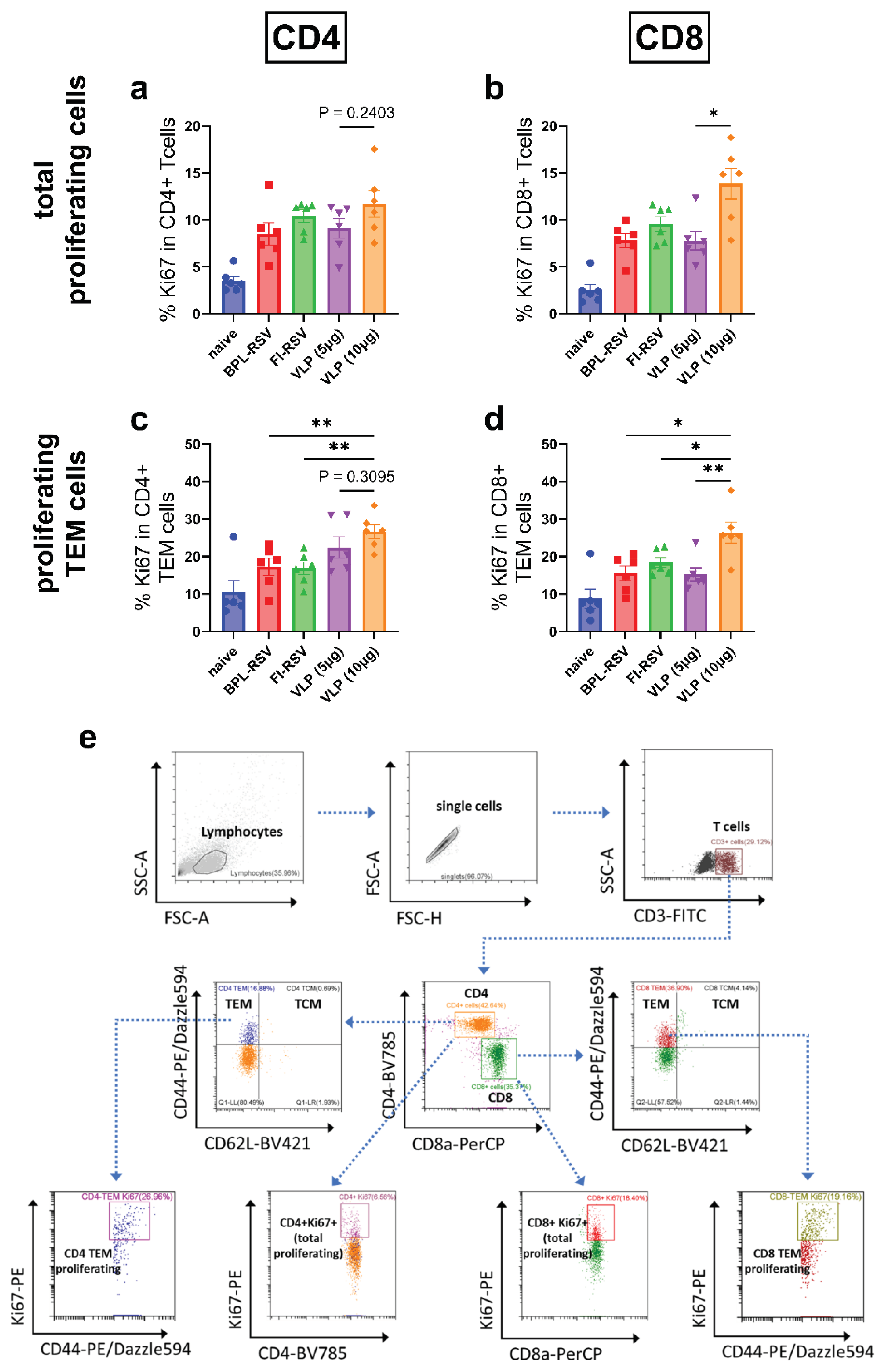
Figure 8.
Cytotoxic T cell responses after immunization. CTL responses were evaluated using splenocytes of immunized mice that were sacrificed 7 days after the second immunization. The percentage of (a) granzyme B+, (b) CD107a+, (c) IFNγ+ and (d) CD107a+granzyme B+ -expressing CD8+ T cells was determined employing flowcytometry. (e) Gating strategy and representative images showing the percentages of granzyme B-, CD107a- and IFNγ-positive cells from the immunized groups.
Figure 8.
Cytotoxic T cell responses after immunization. CTL responses were evaluated using splenocytes of immunized mice that were sacrificed 7 days after the second immunization. The percentage of (a) granzyme B+, (b) CD107a+, (c) IFNγ+ and (d) CD107a+granzyme B+ -expressing CD8+ T cells was determined employing flowcytometry. (e) Gating strategy and representative images showing the percentages of granzyme B-, CD107a- and IFNγ-positive cells from the immunized groups.
Figure 9.
Lung pathology in mice challenged with live RSV after immunization. Mice were immunized twice and subsequently challenged with live RSV. The lungs were harvested 4 days post challenge, fixed, sectioned and stained with H&E to evaluate RSV-mediated enhanced respiratory disease. Representative images of lungs from (a) naive, (b) non-immunized and challenged, (c) BPL-RSV-, (d) FI-RSV- and (e) RSV-VLP-immunized and challenged mice. (f) Lung pathology score was calculated after analyzing the lung sections from each mouse.
Figure 9.
Lung pathology in mice challenged with live RSV after immunization. Mice were immunized twice and subsequently challenged with live RSV. The lungs were harvested 4 days post challenge, fixed, sectioned and stained with H&E to evaluate RSV-mediated enhanced respiratory disease. Representative images of lungs from (a) naive, (b) non-immunized and challenged, (c) BPL-RSV-, (d) FI-RSV- and (e) RSV-VLP-immunized and challenged mice. (f) Lung pathology score was calculated after analyzing the lung sections from each mouse.
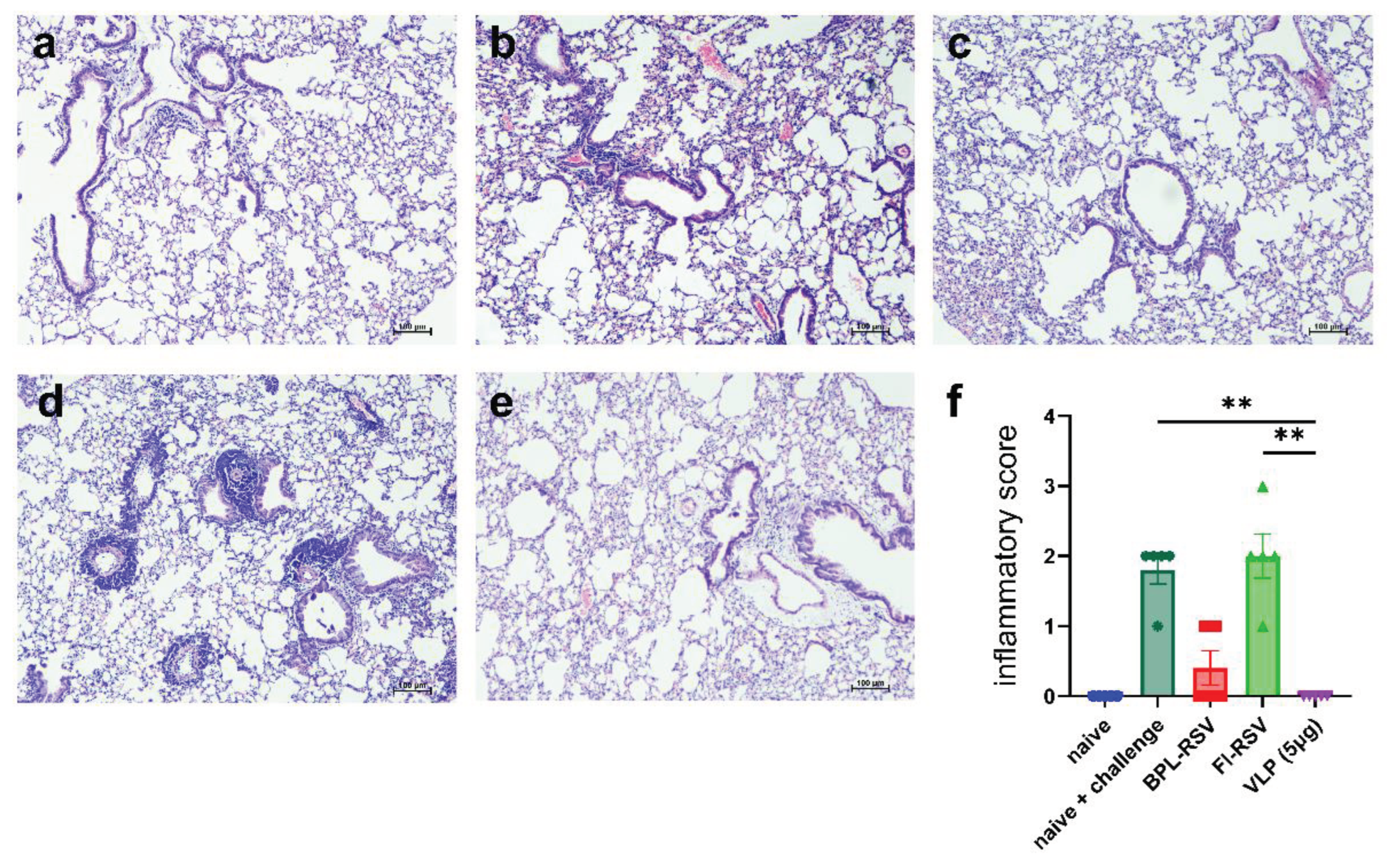

Table 1.
Primer sequences of RSV M and G genes used. Sequences of BamHI and XhoI restriction enzymes are underlined in the forward and reverse primers respectively.
Table 1.
Primer sequences of RSV M and G genes used. Sequences of BamHI and XhoI restriction enzymes are underlined in the forward and reverse primers respectively.
| Target | Primer | Sequence (5′-3′) |
|---|---|---|
| M | Forward | CGCGGATCCATGGAAACATACGTGAACAAGCTTC |
| Reverse | CCGCTCGAGAGTAACTACTGGCGTGGTGTG | |
| G | Forward | CGCGGATCCATGCAAACATGTCCAAAAACAAG |
| Reverse | CCGCTCGAGAGTAACTACTGGCGTGGTGTG |
Table 2.
Antibodies used for CTL and T cell proliferation evaluation.
| Antibody | Clone | Source |
|---|---|---|
| CTL panel | ||
| CD3-FITC | 17A2 | BioLegend |
| CD8a-PerCP | 53-6.7 | BioLegend |
| CD107a-PE | 1D4B | BioLegend |
| Granzyme B-PE/Dazzle594 | QA16A02 | BioLegend |
| IFNγ-PECy7 | XMG1.2 | BioLegend |
| T cell proliferation panel | ||
| CD3-FITC | 17A2 | BioLegend |
| CD8a-PerCP | 53-6.7 | BioLegend |
| CD4-BV785 | RM4-5 | BioLegend |
| CD44-PE/Dazzle594 | IM7 | BioLegend |
| CD62L-BV421 | MEL-14 | BioLegend |
| Ki67-PE | 11F6 | BioLegend |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



