
Submitted:
10 April 2024
Posted:
10 April 2024
You are already at the latest version
Abstract
Lynch syndrome (LS) is one of the most common hereditary cancer syndrome in human populations, associated with germline variants in MLH1, MSH2/EPCAM, MSH6 and PMS2 genes. The advent of next generation sequencing has proven a significant impact in germline variant detection in the causative genes; however, a large proportion of patients with clinical criteria still receive uncertain or negative results. PMS2 is the least frequent reported gene, associated with up to 15% of LS cases with late-onset disease and low penetrance phenotype; however, the proportion of PMS2-LS cases is considered to be highly underestimated. In this context, we consulted public available databases (ClinVar, LOVD) for missense and intronic PMS2 variants and performed an in silico analysis of the genomic sequence. Splicing bioinformatics tools proven effective in other genes were used to assess both the wild-type DNA sequence and reported genetic variants. Splicing alterations were predicted in silico and using GTEx short-read RNA expression data. Out of the 2384 missense variants discovered, 90% were classified with uncertain significance (VUS). 4.9% of missense variants were shown to have a potential splicing consequence (DS > 0.2) using SpliceAI. As described in the original publication, SpliceAI-visual was proven effective in annotation of short intronic variants (< 50 bp). Four short intronic variants were identified using SpliceAI-visual as potentially splicing disturbing, in spite of using a lower threshold (DS > 0.1). Moreover, we observed that Splicing Regulatory Elements (SREs) may play a fundamental role in alternative splicing in PMS2 gene, ESE motifs being overrepresented in highly expressed exons 5, 11 and 14. Computational analysis performed in our study serves as a valuable filtering step for guiding further RNA sequencing experiments. Additional functional data is necessary to evaluate the significance of our findings.
Keywords:
Lynch syndrome
; mismatch repair gene PMS2
; alternative splicing
; RNA splicing
; splicing muta-tions
; exon skipping
; splicing regulatory elements
1. Introduction
Despite the latest advancements in high-throughput sequencing technologies, PMS2 germline testing still continues to pose significant challenges in current clinical practice. Pathogenic and likely pathogenic PMS2 variants are reported in up to 15% of all individuals diagnosed with Lynch syndrome (LS, OMIM #120435), a recently acknowledged underestimation [1,2,3,4,5,6]. Supporting observations for this assertion include the high reported frequency of PMS2 defects in individuals with Constitutional Mismatch Repair Deficiency (CMMRD) syndrome (OMIM #276300), a rare pediatric cancer syndrome [1,3,7]. In heterozygous state, PMS2 pathogenic variants are associated with Lynch-related cancers, mainly colorectal and endometrial cancers [8]. However, the low penetrance phenotype and late-onset disease associated with heterozygous PMS2 variants are contributing factors that might impede accurately determining of the true prevalence of healthy carriers in population [5,9,10].
Historically, the only exonic positions regarded as critical for RNA splicing were those corresponding to acceptor and donor canonical splice sites, specifically the first and last three nucleotides of the exon [11,12]. Further research uncovered the significance of additional factors in exon recognition, such as splicing regulatory elements, chromatin structure, transcription rate, and the secondary and tertiary structure of the transcript [13,14,15]. With the recent development and implementation of in silico tools and deep learning-based algorithms [16,17], many exonic variants formerly classified as missense, nonsense, or silent are susceptible to reclassification, with some revealing a predicted impact on splicing [17,18,19,20,21]. In this scenario, the pathogenic mechanism usually involve a mixture of transcripts with abnormal splicing patterns and transcripts carrying the causative variant [22,23,24].
The vast majority of PMS2 mutations currently documented in public databases are missense, with only a minority classified as clinically significant (class 4 and 5) according to ACMG guidelines [25,26,27]. Comparing to other Lynch-related genes, PMS2 truncating variants, including nonsense, frameshift and splicing variants, are only infrequently reported as disease causing [27,28]. In other human diseases, exonic variations are linked to transcript alterations in as much as 25% of cases [29], primarily involving exon skipping, but in PMS2-associated LS this aspect remains largely unknown. In this context, our study aimed to examine the significance of alternative splicing in the processing of the PMS2 gene. To assess the impact of splicing in silico, our analysis focused on missense and short intronic germline variants documented in public clinical databases (ClinVar, LOVD) related to Lynch syndrome. We discovered genetic variants that could potentially affect splicing, along with recognizing certain limitations of the in silico software used. Bioinformatics tools, proven effective for other genes and previously employed in literature, were utilized to analyze both the wild-type DNA sequence and reported variants. To address the limitations of bioinformatics tools and provide strength to our analysis, publicly available gene expression data was complementary employed for quantifying exon expression in a tissue-specific manner.
2. Results
2.1. Acceptor Loss, the Major Mechanism for Missense Variants with Predicted Splicing Impact in SpliceAI
Within the consulted databases, a total of 2384 missense variants were documented (Figure 1). Notably, the preponderant majority of these variants fell within the category of variants of uncertain significance (VUS), comprising 90.81% of the total. Variants with conflicting interpretations constituted 6.87%, while benign/likely benign variants accounted for 1.63%, and pathogenic/likely pathogenic variants were observed in 0.67%.
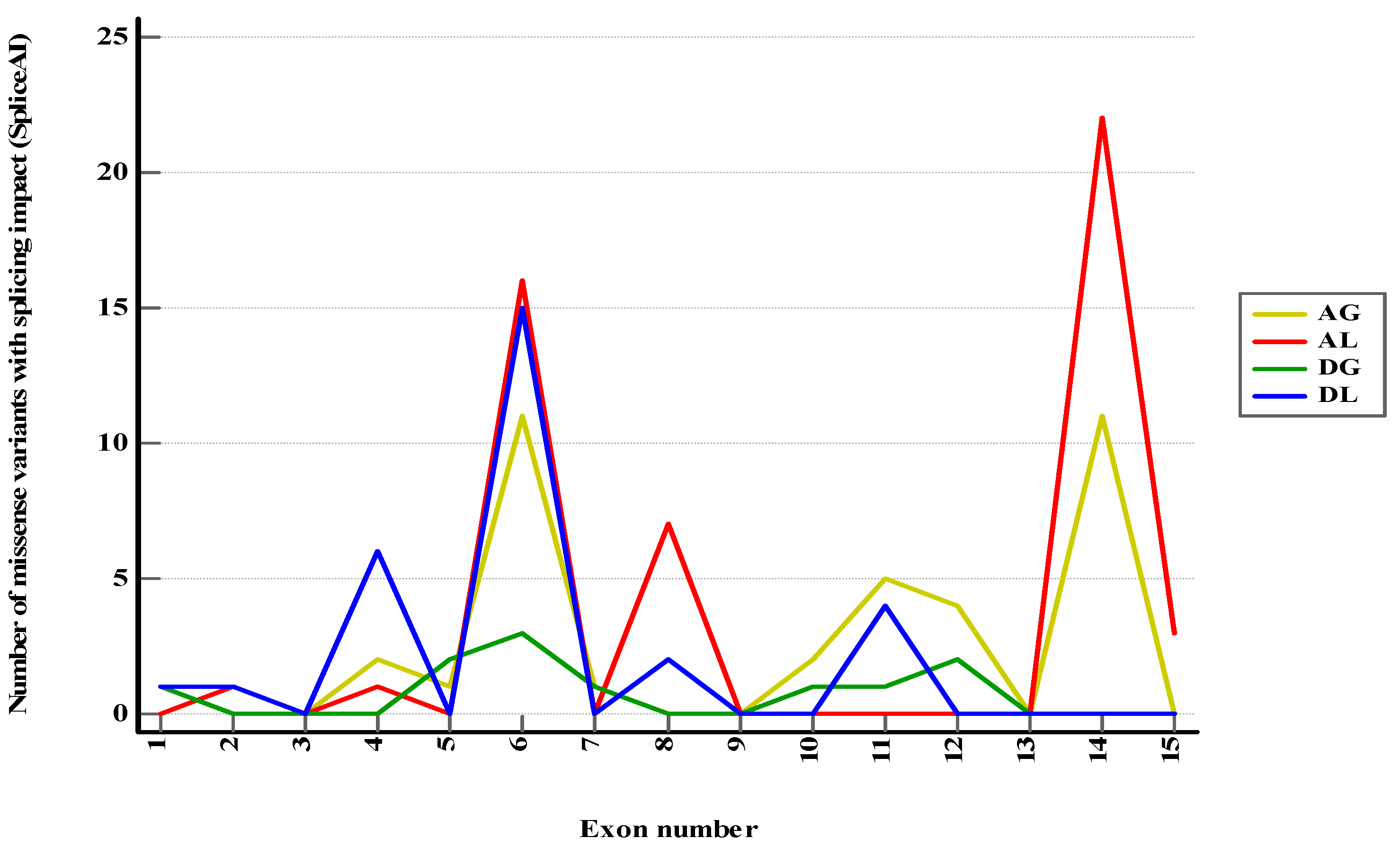
From all class 1–3 and conflicting missense variants included in the study, 117 variants (4.90%) were anticipated to exhibit at least a mild effect on RNA splicing (DS > 0.2) and 34 (1.42%) a moderate or high impact (DS > 0.5) (Figure S1). Sixteen variants of uncertain significance were reported in 3′ canonical splice sites (first exonic position), with 7 (43.75%) having a potential impact (5 of acceptor loss type and 2 of acceptor gain type). Thirty-eight variants of uncertain significance and one conflicting variant were reported in 5′ canonical splice sites (last 3 exonic positions). Among these, 14 variants (35.89%) were expected to have an impact of donor loss type. When categorized by predicted consequences, the exons most commonly linked to specific splicing mechanisms were as presented in Figure 2 and Table 1. Additionally, wild-type ESRseq scores and ΔESRseq scores corresponding to splicing variants (DS > 0.2) in exons enriched in acceptor loss (AL) and donor loss (DL) variants (Table 1) were compared to those from variants without such a prediction (Table 2, Figure S3).
Figure 1.
Distribution of reported PMS2 missense variants across exons, stratified depending on ACMG classification.
Figure 1.
Distribution of reported PMS2 missense variants across exons, stratified depending on ACMG classification.
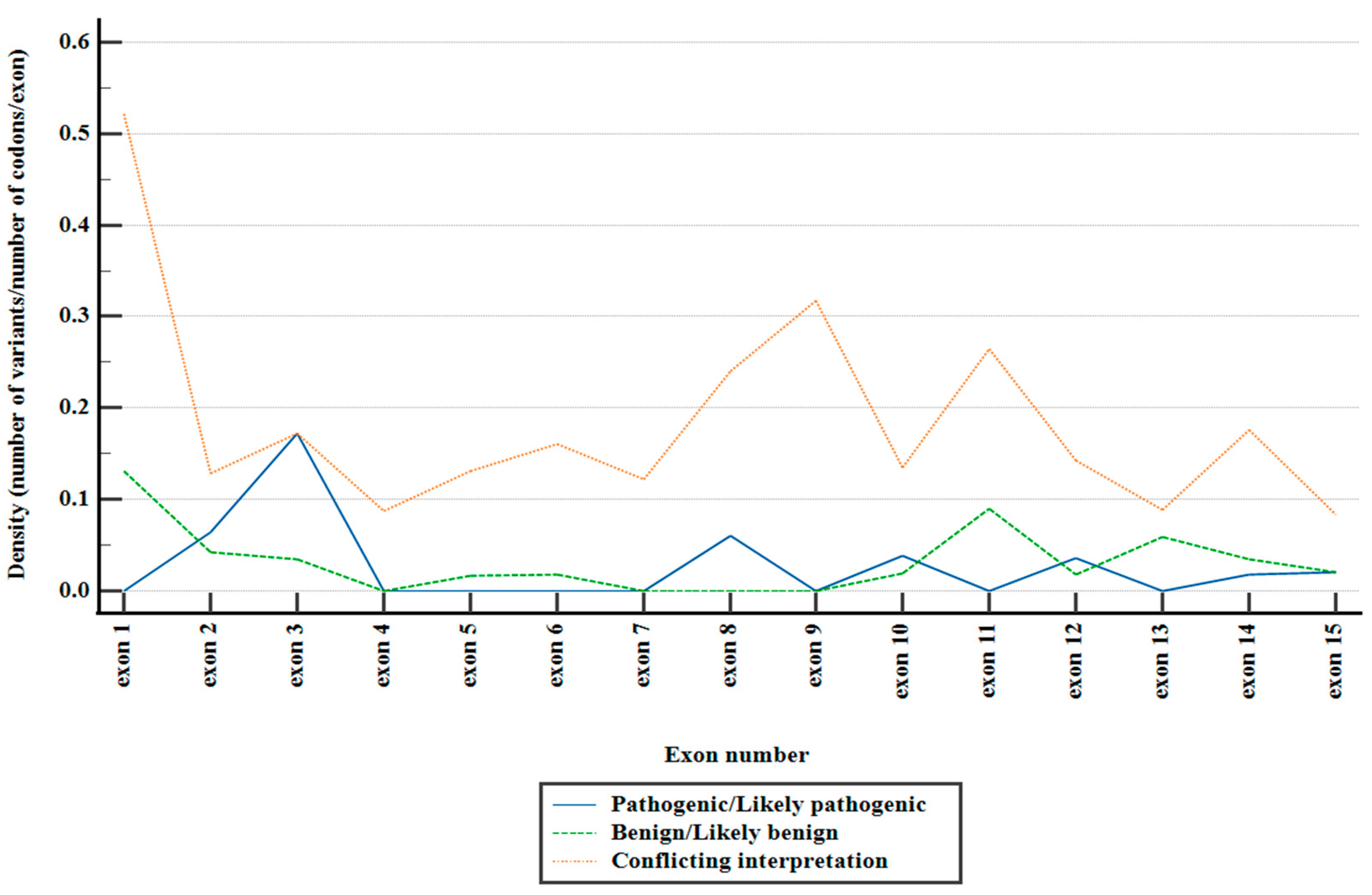
Figure 2.
Class 1–3 and conflicting PMS2 missense variants with predicted splicing impact (DS > 0.2) in SpliceAI. AG—Acceptor Gain, AL—Acceptor Loss, DG—Donor Gain, DL—Donor Loss.
Figure 2.
Class 1–3 and conflicting PMS2 missense variants with predicted splicing impact (DS > 0.2) in SpliceAI. AG—Acceptor Gain, AL—Acceptor Loss, DG—Donor Gain, DL—Donor Loss.
2.2. SpliceAI-Visual, a Valuable Prediction Tool for PMS2 Complex Short Intronic Variants
Out of 838 intronic variants reported in ClinVar, we discovered 71 non-point short genetic variants (<50 bp). Upon filtering by clinical significance, 61 (85.91%) variants had conflicting interpretation or were interpreted as class 1–3 according to ACMG criteria. Among these, 11 out of 61 variants affected the intronic canonical splice sites (the first 6 and last 3 intronic positions). Notably, 14 variants were predicted to have a potential splicing impact when we used a lower threshold (<0.2) for DS in conjunction with SpliceAI-visual. To the best of our knowledge, at the time when this manuscript was written, with one exception, no clinical or functional data were available for the mentioned variants. Additional details regarding the selected variants are available in the Table S1.
2.2.1. Canonical Splice Site Interpreted as Novel Splice Site by SpliceAI
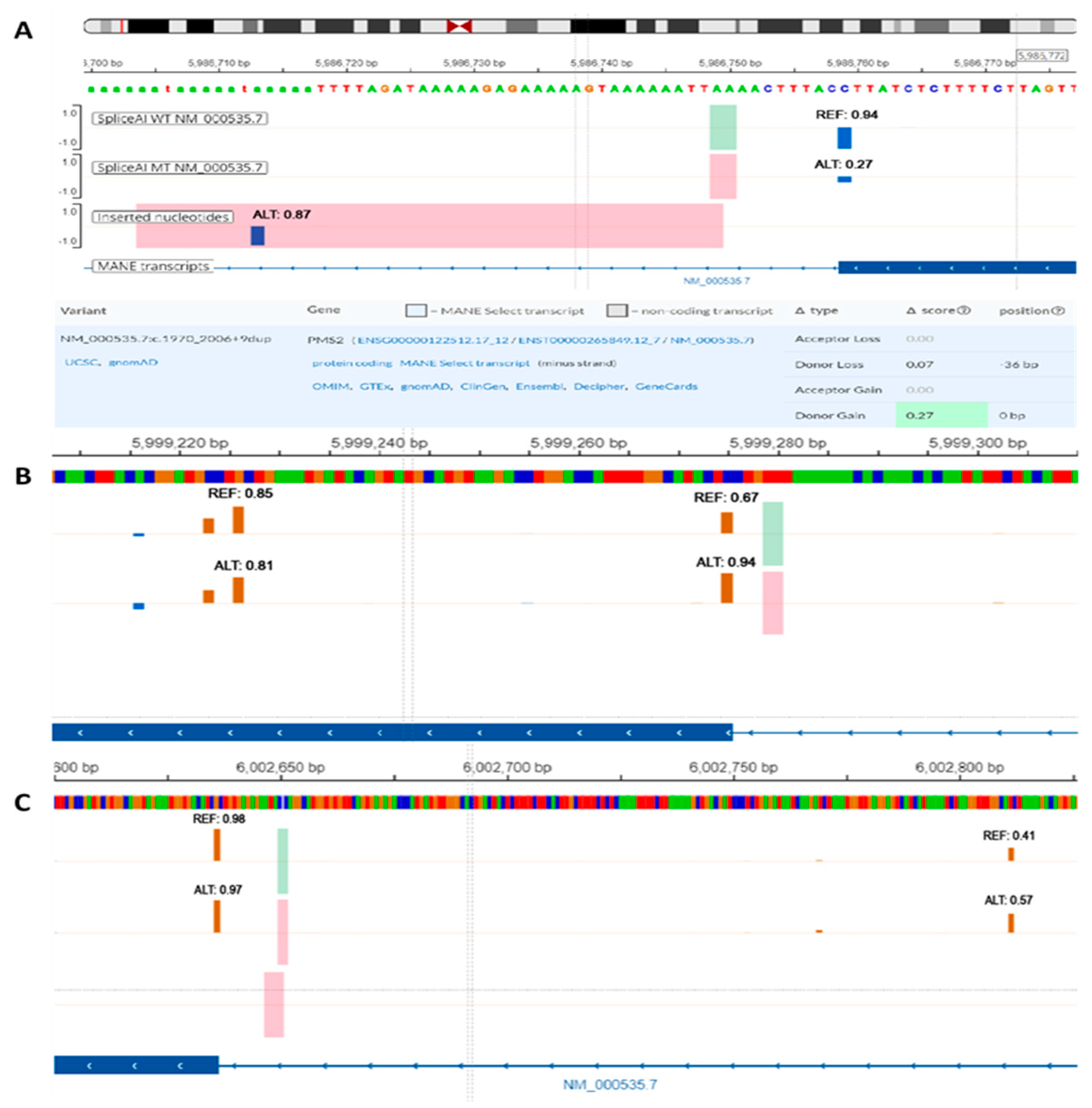
The variant of uncertain significance NM_000535.7:c.1970_2006+9dup is a 46 bp duplication that spans over the 3′ end of exon 11 and exon-intron junction (Figure 3A). In this particular case, relying solely on the DS provided by SpliceAI might suggest a mild increase in the strength of the canonical donor splice site. When the sequence was inspected using SpliceAI-visual, it was visible that SpliceAI located the original donor site in the duplicated region, showing only a slight decrease in strength (0.07). The canonical donor site was consequently interpreted as a novel splice site with DS score of 0.27. Upon further analysis, the duplicated region was anticipated to induce a frameshift and introduce a premature termination codon in the sequence. This event is expected to trigger transcript degradation via nonsense-mediated decay (NMD).
2.2.2. Variants Increasing the Strength of a Weak Canonical Splice Site
NM_000535.7:c.538-5_538-4del is a likely benign variant located in the proximity of acceptor site of exon 6 (Figure 3B). As previously described, the 5′ end of exon 6 corresponds to a relatively weak splice site (REF score: 0.67). Within the exonic sequence, there is at least one alternative cryptic acceptor splice site that is stronger than the canonical site (REF score: 0.81). While the variant DS (0.28) suggests a mild increase in strength of the acceptor splice site, the cumulative effect renders the canonical splice site stronger than cryptic sites (ALT score: 0.94). This, in turn, could consequently alter the natural proportion of Δ6 and Δ6p transcripts with potential clinical impact. Similarly, variant NM_000535.7:c.538-12dup with conflicting interpretation of pathogenicity increases the strength of the same acceptor splice site (ALT score: 0.78), despite having only a mild DS (0.12).
2.2.3. Intronic Inclusion and Premature Termination Predicted by SpliceAI-Visual
The variant NM_000535.7:c.354-18_354-15dup previously classified as likely benign, constitutes a 4 bp intronic duplication near the 3′ splice site of exon 5 (Figure 3C). SpliceAI indicates a mild acceptor gain effect (DS: 0.17) that could be easily filtered out using a standard DS cutoff value of 0.2. When using SpliceAI-visual, we observed that the variant enhances the strength of an intronic cryptic acceptor site (ALT score: 0.57). This site could compete with the canonical site and lead to intronic inclusion. Using the alternative splice site is important in this context since the variant induces a frameshift and includes a premature termination codon naturally present in the intronic sequence. This, consequently, is predicted to induce NMD and eventual transcript degradation.
Figure 3.
SpliceAI-visual predictions (IGV interface, MobiDetails) for PMS2 short intronic variants: (A) NM_000535.7:c.1970_2006+9dup, (B) NM_000535.7:c.538-5_538-4del, (C) NM_000535.7:c.354-18_354-15dup. REF—reference (wild-type) score, ALT—alternative (variant) score. Vertical blue bars signify donor sites and orange bars signify acceptor sites.
Figure 3.
SpliceAI-visual predictions (IGV interface, MobiDetails) for PMS2 short intronic variants: (A) NM_000535.7:c.1970_2006+9dup, (B) NM_000535.7:c.538-5_538-4del, (C) NM_000535.7:c.354-18_354-15dup. REF—reference (wild-type) score, ALT—alternative (variant) score. Vertical blue bars signify donor sites and orange bars signify acceptor sites.
2.3. Bioinformatics Assessment of Donor and Acceptor Splice Sites Strength
Canonical exon-intron junction motifs were evaluated using five alternative software tools (Table 3, Figure S4). As was previously indicated [37], splice sites with scores falling below the lower boundary of the 90% confidence interval (90% CI) were deemed weak. Ten exons displayed at least one putative weak splicing signal, with exons 2, 3, 4, 6, 8, and 14 consistently reported in at least three out of five predictions. Exons 1, 9, 10, 13, and 15, were lacking identifiable weak canonical splice sites.
2.4. Exonic Splicing Regulatory Elements (SREs) Predictions: ESEs, ESSs and ESS/ESE Ratio
The coding regions of all PMS2 exons, except from the first and last exons, were assessed utilizing motif matrices in ESEfinder to detect potential binding sites for SR proteins: SF2/ASF, SF2/ASF (IgM-BRCA1), SC35, SRp40 and SRp55 (Figure 4, Table S3). Significance in the analysis was attributed solely to motifs with high scores surpassing the standard thresholds recommended by developers. Exons 3, 7, 10, and 13 exhibited the lowest overall density of ESE motifs, with an isolated overrepresentation of binding sites for SRp40 in exon 10. The findings from the parallel analysis, employing both HOT-SKIP and HExoSplice, are comprehensively outlined in Table 4, Table S4.
Exons displaying consistently high predictions for ESE (exons 5, 11, 14) or ESS (exons 6, 7, 10) densities in all three SREs-specific software, were further examined based on their wild-type ESRseq scores returned by HExoSplice, in order to explore motifs distribution across exons (Figure 5, Table S5). Correlation between exonic position and ESRseq score in exon 5 (R = −0.34, p = 0.001) and exon 14 (R = 0.30, p = 0.006) was weak-moderate and statistically significant. In exon 5, ESEs were located preferentially in the first 50 bp, while ESSs were found in the latter half of the exon. This pattern was reversed in exon 14.
2.5. PMS2 Expression Data—GTEx Database and RefSeq Coding Transcripts
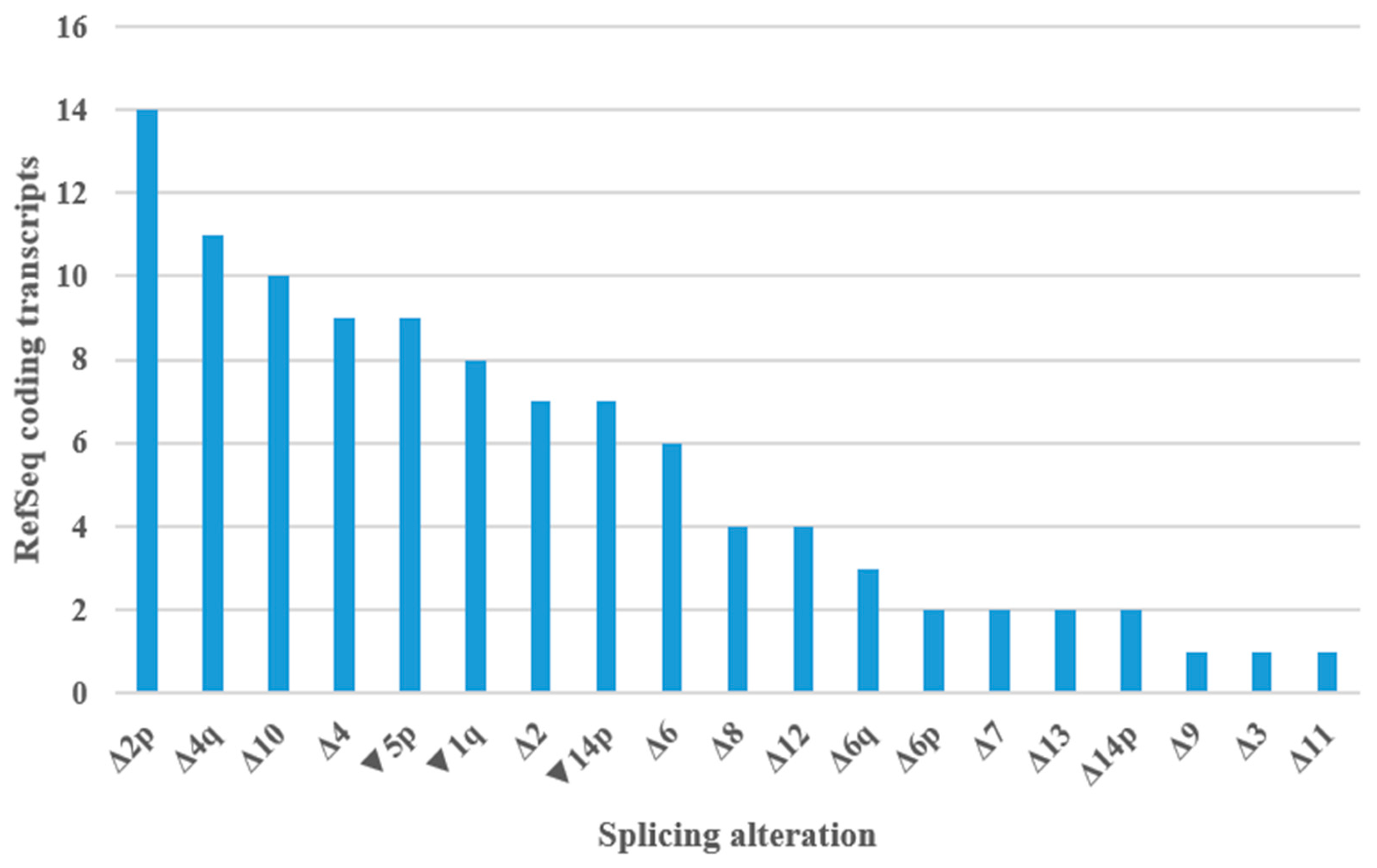
The available data regarding PMS2 gene expression revealed that exons 11, 13, and 14 had a significantly higher median count per base in all tissues included in the study (Table 5, Table S6). On the other hand, exons 1, 2, and 15 consistently exhibited low expression in almost all tissues. 60 protein coding transcripts were included in the study (Table S7, Figure S5). The most frequent splicing aberrations in studied transcripts were presented in Figure 6.
3. Discussion
3.1. Low Level of Exonic Splicing Variants in PMS2 Predicted by SpliceAI
In our study, only 4.9% of missense variants were predicted to have a significant splicing consequence (DS > 0.2) using SpliceAI. This value is lower than expected given the data available in literature which states that up to 25% of exonic disease-causing variants may disturb exon definition in mature transcripts [29]. There are several potential explanations for this result: (1) A lower performance of bioinformatics predictions in exonic positions outside the canonical splice sites [12,38]; (2) Variants situated deep in the exonic regions are less likely to influence exon inclusion [39]; (3) The DS threshold > 0.2 could impact variant classification in PMS2 gene, clinically significant variants being reportedly overseen in other genes when a standard cutoff value was used [17]; (4) Alternative splicing have a secondary role in the biology of PMS2 gene, according to what was previously reported in literature [40].
Figure 6.
Splicing alterations encountered in PMS2 RefSeq coding transcripts. Δ—complete or partial (adjacent to canonical splice sites) exonic deletion, ▼—intronic sequence inclusion (adjacent to canonical splice sites), p—acceptor site shift, q—donor site shift.
Figure 6.
Splicing alterations encountered in PMS2 RefSeq coding transcripts. Δ—complete or partial (adjacent to canonical splice sites) exonic deletion, ▼—intronic sequence inclusion (adjacent to canonical splice sites), p—acceptor site shift, q—donor site shift.
3.2. Exons Harboring Weak Canonical Splice Sites May Require Regulatory Elements for Proper Definition
Critical for exon definition, canonical splice sites may demand additional signals, such as ESEs or ESSs, when their sequence is less conserved [11,16]. Not surprisingly, in the past, other analysis of variants that disrupt SREs supported the same idea of SREs exhibiting activity predominantly in exons having splice sites of weak or moderate consensus [38]. In accordance with these observations, in our study exons 2, 4, 6, 8, 14 predicted to have weak splice sites exhibited a high frequency of at least one ESE motif in ESEfinder. Exon 3, however, although predicted with a weak 3′ splice site, had a lower than average level of ESEs in ESEfinder and HExoSplice, but a significantly higher one in HOT-SKIP, suggesting a potential for alternative splicing events [41]. Notably, the expression of exon 3 was below average in all tissues, with whole blood and medullary kidney showing significantly low expression levels. In contrast, the median read count per base in bladder tissue was above the average. When taken globally, in all exons with a weak 5′ss or 3′ss, except for exon 4, ESE or ESS motifs were overrepresented in at least one prediction. Nevertheless, in exon 4, ESEfinder detected a high density of SC35 and SF2/ASF (IgM-BRCA1) specific domains, but the overall ESEs density was above the average, reaching the upper bound of the confidence interval.
3.3. PMS2 Exons with Weak Splice Sites May Be Prone to Exon Skipping
As indicated by ESEs frequency, ESSs frequency, or the ESS/ESR ratio [35,36], exons 2, 3, 6 and 8 were predicted by at least one software as being potentially prone to exon skipping. This association is plausible, given similar in silico observations reported in the literature for other genes [39], which were subsequently confirmed in functional studies [37]. Indeed, when consulting transcripts reported to date, high number of wild type and mutant isoforms aligned with this prediction [40,42]. Bulk RNA-seq data was concordant over exons 2 and 3, with the mention of variations in exon 3 that were tissue-dependent. On the other hand, exons 6 and 8 displayed an average median read count per base across all tissues, as indicated by GTEx database. Moreover, exon 8 was shown to harbor higher than average levels of SF2/ASF sites, but the overall density of SR proteins-binding domains remained low. SpliceAI was concordant with canonical splice site software regarding exon 8, indicating a high density of acceptor loss variants, which may serve as an indirect proof of the same biological event. Indeed, Δ8_11 transcript, naturally occurring by a splice acceptor shift mechanism, was reported [40]. Other similar ‘multi-cassette’ RefSeq transcripts were curated, suggesting once again the role of alternative splicing in processing of this exon. Intriguingly, exon 4, also featuring a weak donor site, undergoes alternative splicing in certain transcripts, while exon 14 utilizes distinct acceptor sites [42], an aspect also foreseen by SpliceAI, which identifies a strong exonic cryptic acceptor site. Recently, several exonic and intronic variants associated with exon 4 skipping were reported in literature, highlighting even more the relevance of alternative splicing in PMS2-related LS [38,42,43].
Other exons potentially sensitive to skipping with no weak consensus were 7, 9, 10 and 13. Exons 7 and 10 were mentioned by all 3 tools, with a low ESEs incidence in ESEfinder and high ESSs levels in HOT-SKIP and HExoSplice. The current data indicates that expression of exon 7 is below the average in all tissues, with a significant decrease, particularly in blood. On the other hand, exon 10 is one of the most highly expressed exons across all tissues. Of note, several protein-coding RefSeq transcripts revealed, however, an isolated exclusion of this exon. Minor Δ10 transcripts were also evident in vitro assays [42]. Exon 9 is sometimes skipped in a ‘multi-cassette’ event with other exons. However, from our knowledge, there are no natural transcripts where exon 9 is skipped individually. In this case, ESRs predictions were conflictual, with ESEfinder indicating a high density of ESE motifs, while the other tools supported a low level. Similarly, exon 13 is a constitutive exon, being included and highly expressed in the majority of transcripts. This comes as no surprise, given that it encodes the MutL C terminal dimerization domain, a region known for its high conservation in the protein [44,45].
3.4. High ESE Levels Concordantly Predicted in Exons Critical for PMS2 Function
The vast majority of ESE domains in exon 5 are located next to the acceptor site in HExoSplice. Interestingly, an alternative 3’ss located proximally in the intronic sequence (and predicted by SpliceAI) was observed in at least one naturally occurring transcript. This distribution prompts questions regarding the role of ESEs in the region, particularly considering that 2 out of 5 predictions indicated a 3’ weak splice site [46]. Of note, exons 2–5 codify N-terminal ATPase domain of PMS2 (HATPase_c_3, InterPro, PF08676), which is important for maintaining mismatch repair proficiency. However, it is worth highlighting that this domain may be working in an asymmetric manner with the similar domain in MLH1, with the later appearing to play a more decisive role in this biological process [47,48,49].
By comparison, in exon 11, ESEs and ESSs hexamers are evenly distributed throughout the exon, with a region with high ESSs density and a relative ESEs depletion near the 3’ss. Exon 11, being a long exon, suggests that ESEs may play a fundamental role in facilitating the accurate selection of exonic boundaries by the splicing machinery [41]. Additionally, certain isoforms include a shorter variant of this exon due to a shift in the 5′ splice site, in contrast with the presence of a relatively strong 5′ donor consensus. Similarly, SpliceAI predicted a significant number of variants in exon 11 that could lead to donor site gain or loss. At least one transcript, Δ11q_14p, has been reported to occur via splice donor shift process [40]. An analogous event was noticed in atypical CMMRD related to a benign missense founder variant that created a novel donor splice site in intron 11, generating a 5 bp deletion frameshift at the exon 11–12 junction [50,51]. Remarkably, exon 11 is rarely spliced out in reported transcripts, which indicates a fundamental biological significance of the regional coding sequence. Upon delving into the GTEx exon expression data, we observed a consistent trend of high expression for exon 11 across all investigated tissues. When we referred to the literature, we discovered that PMS2 interacts with MLH1 in a critical area spanning amino acid residues 675 to 850 [52]. This sequence defines the C-terminus of the protein, a region that overlaps with exons 11–15, sensitive to phosphorylation and involved in regulating the degradation of PMS2 [53]. Additionally, missense variants outside this hotspot but still situated in exon 11 may impact MutLα heterodimer formation by changing the binding affinity of the protomers [54].
Within exon 14, ESSs are primarily located at the 5’ end (3’ acceptor site), whereas ESEs are complementarily positioned in the middle and 3’ end (5’ donor site). Since exon 14 was consistently predicted to have a weak acceptor site, and confirmed by wild type and mutant Δ14p transcripts [42], the high ESEs incidence may be relevant for its inclusion in mature transcripts, as it was reported in other exons with similar properties [41]. As mentioned earlier, exon 14 encodes the MutL C terminal dimerisation domain (MutL_C, InterPro, IPR014790) and, thus, critically contributes to MutLα heterodimerization [44,45]. As anticipated, the RNA expression analysis revealed, once again, a consistent pattern of elevated expression across all considered tissues.
3.5. High ESS Levels Concordantly Predicted in Exons 6, 7 and 10
Both HOT-SKIP and HExoSplice indicated potential elevated levels of exonic splicing silencers in exons 6, 7 and 10. When we visually inspected exon 6 in HExoSplice, ESSs were strikingly located in the 5’ half of the exon. In combination with ESE depletion in the same region, it could explain, at least partially, the acceptor splice shift, Δ6p and Δ6 transcripts described in literature [40,42] and present in RefSeq data. SpliceAI identified at least 2 exonic cryptic acceptor sites in the region that may play a decisive role in the process. In line with this, SpliceAI predicted a significant number of acceptor gain and acceptor loss variants in exon 6 based on the variants collected in this study. Moreover, the median ΔtESRseq score of predicted splicing variants was negative and significanty lower from other reported variants in this exon. In exon 7, ESSs are located preferentially at both 5’ and 3’ ends of the exon, middle of the exon being enriched in ESEs instead. We suspect that this particular distribution may provide a possible explanation for the observed exon 7 skipping and intron 7 inclusion in some transcripts [40]. In exon 10, ESSs are relatively evenly distributed, interspersed with ESEs, showing no specific pattern.
3.6. Limitations
The present study conducted a bioinformatics evaluation of the significance of alternative splicing in the expression of the PMS2 gene. This analysis is relevant given the numerous PMS2 variants of uncertain significance reported, that complicate the translation of DNA sequencing data into clinical practice. However, we acknowledge that the present study has several limitations, primarily related to the fact that the analysis was conducted exclusively in silico. Despite including bioinformatics tools commonly used and verified by other authors, future experimental data are still required to validate the results. Additionally, the included expression data may exhibit certain known biases [32,55]. The analyzed transcripts are solely protein-coding, excluding non-coding transcripts, which may not fully capture the diversity and complexity of alternative splicing.
4. Materials and Methods
4.1. Reference Sequence and Variant Nomenclature
PMS2 variants were described according to Human Genetic Variation Society guidelines (https://hgvs-nomenclature.org/stable/). MANE Select transcript (NM_000535.7, ENST00000265849.12) was considered the reference sequence, position c.1 corresponding to the first coding nucleotide. The examined variants were retrieved from ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) and LOVD (https://databases.lovd.nl/shared/genes/PMS2) public databases on 4 December 2023. Variants with no reported classification based on ACMG criteria [25] have been excluded from the analysis. Variant annotation was performed using Ensembl Variant Effect Predictor (VEP) (https://grch37.ensembl.org/Homo_sapiens/Tools/VEP/). For exonic regions, we focused on nonsynonymous missense variants. Truncating variants, known to be deleterious, and synonymous variants, very rarely reported in consulted databases, were excluded. Among the selected intronic variants, we specifically included short genetic variants (<50 base pairs). Complex and large variants were beyond the scope of our study.
4.2. Tissue-Specific Bulk RNA Expression Data and Public Available Transcripts
Protein-coding PMS2 transcripts under analysis were retrieved from NCBI RNA reference sequences collection (RefSeq), a public database that provides an extensive and carefully curated collection of sequences [30,31]. RefSeq transcripts available on UCSC genome browser (https://genome.ucsc.edu) were included in the analysis. To further enhance our understanding, the GTEx database was employed for quantifying exon expression using median read per base provided in a tissue-specific manner [32]. The analysis focused on tissues more commonly affected in Lynch syndrome.
4.3. Bioinformatics Analysis of Splicing Impact and Statistical Analysis
Several bioinformatics approaches were employed to assess the impact of PMS2 variants on RNA splicing. The strength analysis of canonical acceptor and donor splice sites was conducted using freely available online tools, including ESEfinder 3.0 for splice sites (https://esefinder.ahc.umn.edu/), FSplice (http://www.softberry.com/), MaxEntScan (http://hollywood.mit.edu/), NetGene2 (https://services.healthtech.dtu.dk/) and NNSplice (https://www.fruitfly.org/). The potential splicing impact of reported variants was estimated using SpliceAI, one of the most proficient deep learning-based tool reported to date [16,17,33]. Delta scores (DS) greater than 0.2 were used to screen for splice-altering variants, as outlined in the original publication [16]. To enhance the characterization of intronic variants, we utilized the SpliceAI-visual IGV interface [34] accessible on MobiDetails (https://mobidetails.iurc.montp.inserm.fr). Splicing regulatory elements (SREs) were screened across the coding regions using three alternative strategies: ESEfinder 3.0 for SR proteins (https://esefinder.ahc.umn.edu/), HOT-SKIP (https://hot-skip.img.cas.cz/) and HExoSplice (http://bioinfo.univ-rouen.fr/). Exon skipping was considered in either case based on the resulting low putative ESEs and high ESSs densities, as well as increased ESS/ESE ratios [35,36]. All statistical analyses were performed using MedCalc® Statistical Software version 22.019 (MedCalc Software Ltd, Ostend, Belgium; https://www.medcalc.org; 2024). Statistical significance was assigned for p-value < 0.05. Except where otherwise stated, a 95% confidence interval (CI) was employed. The Student’s t-test was used to statistically evaluate numerical and normally distributed values. The Mann-Whitney U test was used as a non-parametric test in non-normally distributed data.
5. Conclusions
This study highlights the potential importance of PMS2 mRNA analysis to improve the diagnostic yield in Lynch syndrome. The model could be easily extended to other genes with high amount of variants of uncertain significance. In this process, bioinformatics tools could facilitate the variant prioritization and patient selection for reflexive RNA sequencing. In silico software used underlines the frequency of splicing alterations associated to PMS2 gene, providing a potential explanation for the current underdiagnosis in PMS2-associated LS. In this regard, we identified several missense and intronic variants, candidates for functional analysis. However, some potential limitations of in silico tools occurred, thus further functional data are required to assess the biological significance of computational observations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Data extraction and bioinformatics analysis, C.V.M. and A.P.T.; writing—original draft preparation, C.V.M. and A.P.T; writing—review and editing, A.C.-E., C.V.M., M.P.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Supporting data is contained within the article and Supplementary material.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wimmer, K.; Etzler, J.; Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008, 124, 105–22. [CrossRef]
- Palomaki, G.E.; McClain, M.R.; Melillo, S.; Hampel, H.L.; Thibodeau, S.N.; EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009, 11, 42. [CrossRef]
- Goodenberger, M.L.; Thomas, B.C.; Riegert-Johnson, D.; Boland, C.R.; Plon S.E.; Clendenning, M.; et al. PMS2 monoallelic mutation carriers: the known unknown. Genet Med. 2016, 18, 13–9. [CrossRef]
- Vaughn, C.P.; Baker, C.L.; Samowitz, W.S.; Swensen, J.J.; The frequency of previously undetectable deletions involving 3’ Exons of the PMS2 gene. Genes Chromosomes Cancer. 2013, 52, 107–12. [CrossRef]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008, 135. [CrossRef]
- Yuan, L.; Chi, Y.; Chen, W.; Chen, X.; Wei, P.; Sheng, W.; et al. Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency. Int J Clin Exp Med. 2015, 8, 20988. /pmc/articles/PMC4723875/.
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.A.; Caron, O.; Colas, C.; Entz-Werle, N.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J Med Genet. 2014, 51, 355–65. [CrossRef]
- Andini, K.D.; Nielsen, M.; Suerink, M.; Helderman, N.C.; Koornstra, J.J.; Ahadova, A.; et al. PMS2-associated Lynch syndrome: Past, present and future. Front Oncol. 2023, 13, 1–12. [CrossRef]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020, 22, 15–25. [CrossRef]
- ten Broeke S.W.; Suerink, M.; Nielsen, M.; Response to Roberts et al. 2018: is breast cancer truly caused by MSH6 and PMS2 variants or is it simply due to a high prevalence of these variants in the population? Genet Med. 2019, 21, 256–7. [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R.; Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002, 3, 285–98. [CrossRef]
- Walker, L.C.; Hoya, M.; Wiggins, G.A.R.; Lindy, A.; Vincent, L.M.; Parsons, M.T.; et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Genet. 2023, 110, 1046–67. [CrossRef]
- Wang, Z.; Burge, C.B.; Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008, 14, 802–13. [CrossRef]
- Georgakopoulos-Soares, I.; Parada, G.E.; Hemberg, M. Secondary structures in RNA synthesis, splicing and translation. Comput Struct Biotechnol J. 2022, 20, 2871–84. [CrossRef]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA. 2013, 4, 49–60. [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019, 176, 535-548. [CrossRef]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; et al. SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Hum Genomics. 2023, 17. [CrossRef]
- Pagani, F.; Baralle, F.E.; Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. 2004, 5, 389–96. [CrossRef]
- Valentine, C.R. The association of nonsense codons with exon skipping. Mutat Res. 1998, 411, 87–117. [CrossRef]
- Yamaguchi, T.; Wakatsuki, T.; Kikuchi, M.; Horiguchi, S.I.; Akagi, K. The silent mutation MLH1 c.543C>T resulting in aberrant splicing can cause Lynch syndrome: a case report. Jpn J Clin Oncol. 2017, 47, 576–80. [CrossRef]
- Horton, C.; Hoang, L.; Zimmermann, H.; Young, C.; Grzybowski, J.; Durda, K.; et al. Diagnostic Outcomes of Concurrent DNA and RNA Sequencing in Individuals Undergoing Hereditary Cancer Testing. JAMA Oncol. 2023, 92656, 212–9. [CrossRef]
- Kim, E; Goren, A.; Ast, G. Alternative splicing: current perspectives. BioEssays. 2008, 30, 38–47. [CrossRef]
- Majewski, J.; Ott, J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002, 12, 1827–36. [CrossRef]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell. 2009, 136, 777–93. [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405. [CrossRef]
- Landrum, M.J.; Kattman, B.L.; ClinVar at five years: Delivering on the promise. Hum Mutat. 2018, 39, 1623–30. [CrossRef]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014, 46, 107–15. [CrossRef]
- Lagerstedt-Robinson, K.; Rohlin, A.; Aravidis, C.; Melin, B.; Nordling, M.; Stenmark-Askmalm, M.; et al. Mismatch repair gene mutation spectrum in the Swedish Lynch syndrome population. Oncol Rep. 2016, 36, 2823–35. [CrossRef]
- Lim, K.H.; Ferraris, L.; Filloux, M.E.; Raphael, B.J.; Fairbrother, W.G. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci U S A. 2011, 108, 11093–8. [CrossRef]
- Frankish, A.; Uszczynska, B.; Ritchie, G.R.S.; Gonzalez, J.M.; Pervouchine, D.; Petryszak, R.; et al. Comparison of GENCODE and RefSeq gene annotation and the impact of reference geneset on variant effect prediction. BMC Genomics. 2015, 16, 1–11. [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–45. [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013, 45, 580–5. [CrossRef]
- Jang, W.; Park, J.; Chae, H.; Kim, M. Comparison of In Silico Tools for Splice-Altering Variant Prediction Using Established Spliceogenic Variants: An End-User’s Point of View. Int J Genomics. 2022. [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; et al. Integrative Genomics Viewer. Nat Biotechnol. 2011, 29, 24. [CrossRef]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–71. [CrossRef]
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; et al. Prediction of single-nucleotide substitutions that result in exon skipping: identification of a splicing silencer in BRCA1 exon 6. Hum Mutat. 2011, 32, 436–44. [CrossRef]
- Aissat, A.; de Becdelièvre, A.; Golmard, L.; Vasseur, C.; Costa, C.; Chaoui, A.; et al. Combined computational-experimental analyses of CFTR exon strength uncover predictability of exon-skipping level. Hum Mutat. 2013, 34, 873–81. [CrossRef]
- Canson, D.; Glubb, D.; Spurdle, A.B. Variant effect on splicing regulatory elements, branchpoint usage, and pseudoexonization: Strategies to enhance bioinformatic prediction using hereditary cancer genes as exemplars. Hum Mutat. 2020, 41, 1705–21. [CrossRef]
- Tubeuf, H.; Charbonnier, C.; Soukarieh, O.; Blavier, A.; Lefebvre, A.; Dauchel, H.; et al. Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum Mutat. 2020, 41, 1811–29. [CrossRef]
- Thompson, B.A.; Martins, A.; Spurdle, A.B. A review of mismatch repair gene transcripts: issues for interpretation of mRNA splicing assays. Clin Genet. 2015, 87, 100–8. [CrossRef]
- Wu, Y.; Zhang, Y.; Zhang, J. Distribution of exonic splicing enhancer elements in human genes. Genomics. 2005, 86, 329–36.
- van der Klift, H.M.; Jansen, A.M.L.; Steenstraten, N.; Bik, E.C.; Tops, C.M.J.; Devilee, P.; et al. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol Genet Genomic Med. 2015, 3, 327. doi.org/10.1002/mgg3.145.
- Bouras, A.; Naibo, P.; Legrand, C.; Marc’hadour, F.; Ruano, E.; Grand-Masson, C.; et al. A PMS2 non-canonical splicing site variant leads to aberrant splicing in a patient suspected for lynch syndrome. Fam Cancer. 2023, 22, 303–6. [CrossRef]
- Guarné, A.; Ramon-Maiques, S.; Wolff, E.M.; Ghirlando, R.; Hu, X.; Miller, J.H.; et al. Structure of the MutL C-terminal domain: A model of intact MutL and its roles in mismatch repair. EMBO J. 2004, 23, 4134–45. [CrossRef]
- Mohd, A.B.; Palama, B.; Nelson, S.E.; Tomer, G.; Nguyen, M.; Huo, X.; et al. Truncation of the C-terminus of human MLH1 blocks intracellular stabilization of PMS2 and disrupts DNA mismatch repair. DNA Repair (Amst). 2006, 5, 347–61. [CrossRef]
- Ke, S.; Shang, S.; Kalachikov, S.M.; Morozova, I.; Yu, L.; Russo, J.J.; et al. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 2011, 21, 1360–74. [CrossRef]
- Johnson, J.R.; Erdeniz, N.; Nguyen, M.; Dudley, S.; Liskay, R.M. Conservation of Functional Asymmetry in the Mammalian MutLα ATPase. DNA Repair (Amst). 2010, 9, 1209. [CrossRef]
- Tomer, G.; Buermeyer, A.B.; Nguyen, M.M.; Michael Liskay, R. Contribution of Human Mlh1 and Pms2 ATPase Activities to DNA Mismatch Repair. J Biol Chem. 2002, 277, 21801–9. [CrossRef]
- D’Arcy, B.M.; Arrington, J.; Weisman, J.; McClellan, S.B.; Vandana, J.; Yang, Z.; et al. PMS2 variant results in loss of ATPase activity without compromising mismatch repair. Mol Genet Genomic Med. 2022, 10, 1908. [CrossRef]
- Li, L.; Hamel, N.; Baker, K.; McGuffin, M.J.; Couillard, M.; Gologan, A.; et al. A homozygous PMS2 founder mutation with an attenuated constitutional mismatch repair deficiency phenotype. J Med Genet. 2015, 52, 348–52. [CrossRef]
- Biswas, K.; Couillard, M.; Cavallone, L.; Burkett, S.; Stauffer, S.; Martin, B.K.; et al. A novel mouse model of PMS2 founder mutation that causes mismatch repair defect due to aberrant splicing. Cell Death Dis. 2021, 12. [CrossRef]
- Guerrette, S.; Acharya, S.; Fishel, R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem. 1999, 274, 6336–41. [CrossRef]
- Hinrichsen, I.; Weßbecher, I.M.; Huhn, M.; Passmann, S.; Zeuzem, S.; Plotz, G.; et al. Phosphorylation-dependent signaling controls degradation of DNA mismatch repair protein PMS2. Mol Carcinog. 2017, 56, 2663–8. [CrossRef]
- Yuan, Z.Q.; Gottlieb, B.; Beitel, L.K.; Wong, N.; Gordon, P.H.; Wang, Q.; et al. Polymorphisms and HNPCC: PMS2-MLH1 protein interactions diminished by single nucleotide polymorphisms. Hum Mutat. 2002, 19, 108–13. [CrossRef]
- Carithers, L.J.; Ardlie, K.; Barcus, M.; Branton, P.A.; Britton, A.; Buia, S.A.; et al. A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank. 2015, 13, 311–7. [CrossRef]
Figure 4.
Exonic density of SR proteins motifs identified using ESEfinder 3.0 software. Red lines represent the 95% CI for means. Exons outside the interval were noted in the graph.
Figure 4.
Exonic density of SR proteins motifs identified using ESEfinder 3.0 software. Red lines represent the 95% CI for means. Exons outside the interval were noted in the graph.
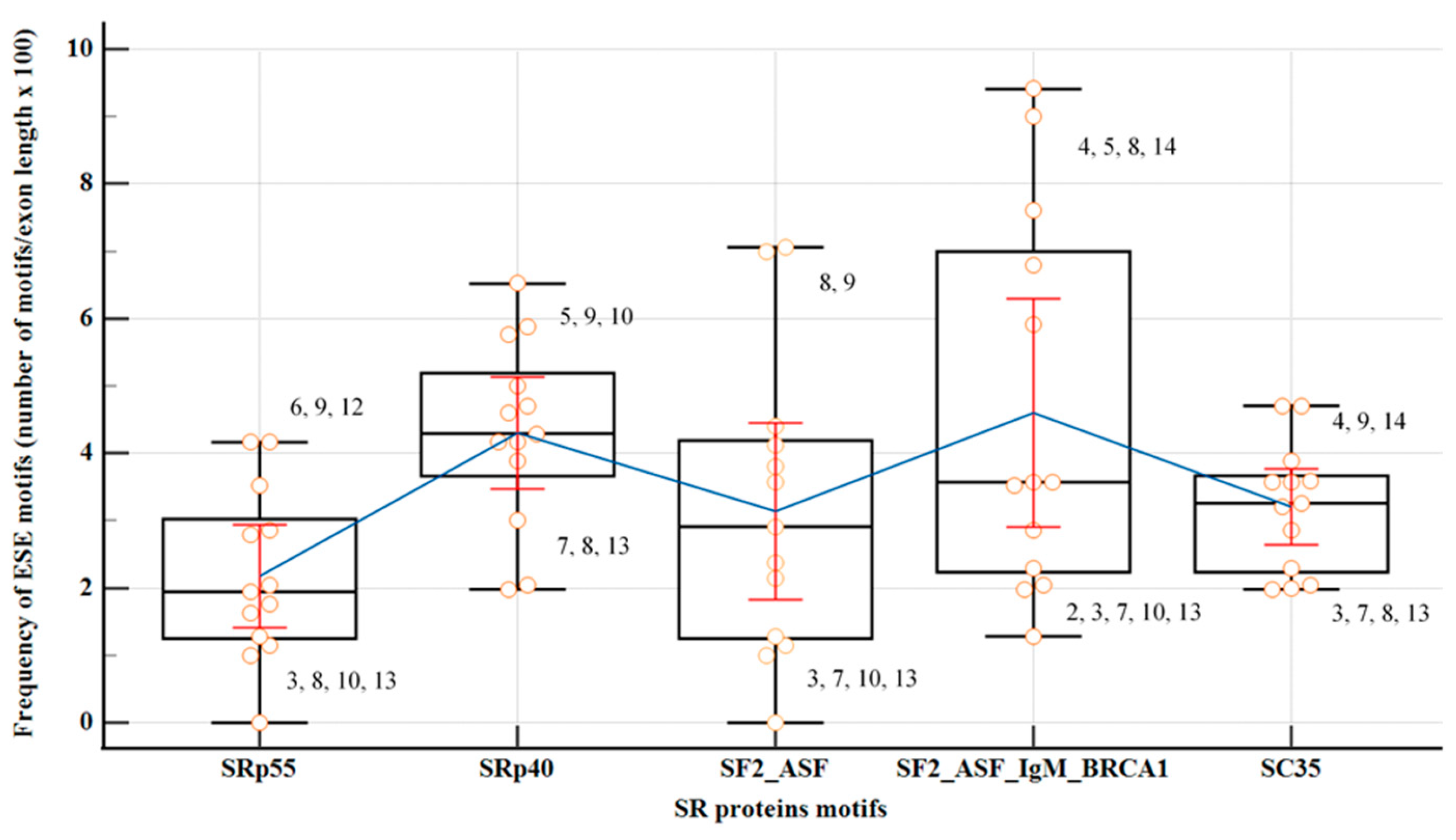
Figure 5.
Exonic distribution (HExoSplice) of SREs motifs in exons with high density of ESEs or ESSs. ESRseq scores > 0 correspond to ESE motifs and ESRseq scores < 0 represent ESS motifs.
Figure 5.
Exonic distribution (HExoSplice) of SREs motifs in exons with high density of ESEs or ESSs. ESRseq scores > 0 correspond to ESE motifs and ESRseq scores < 0 represent ESS motifs.
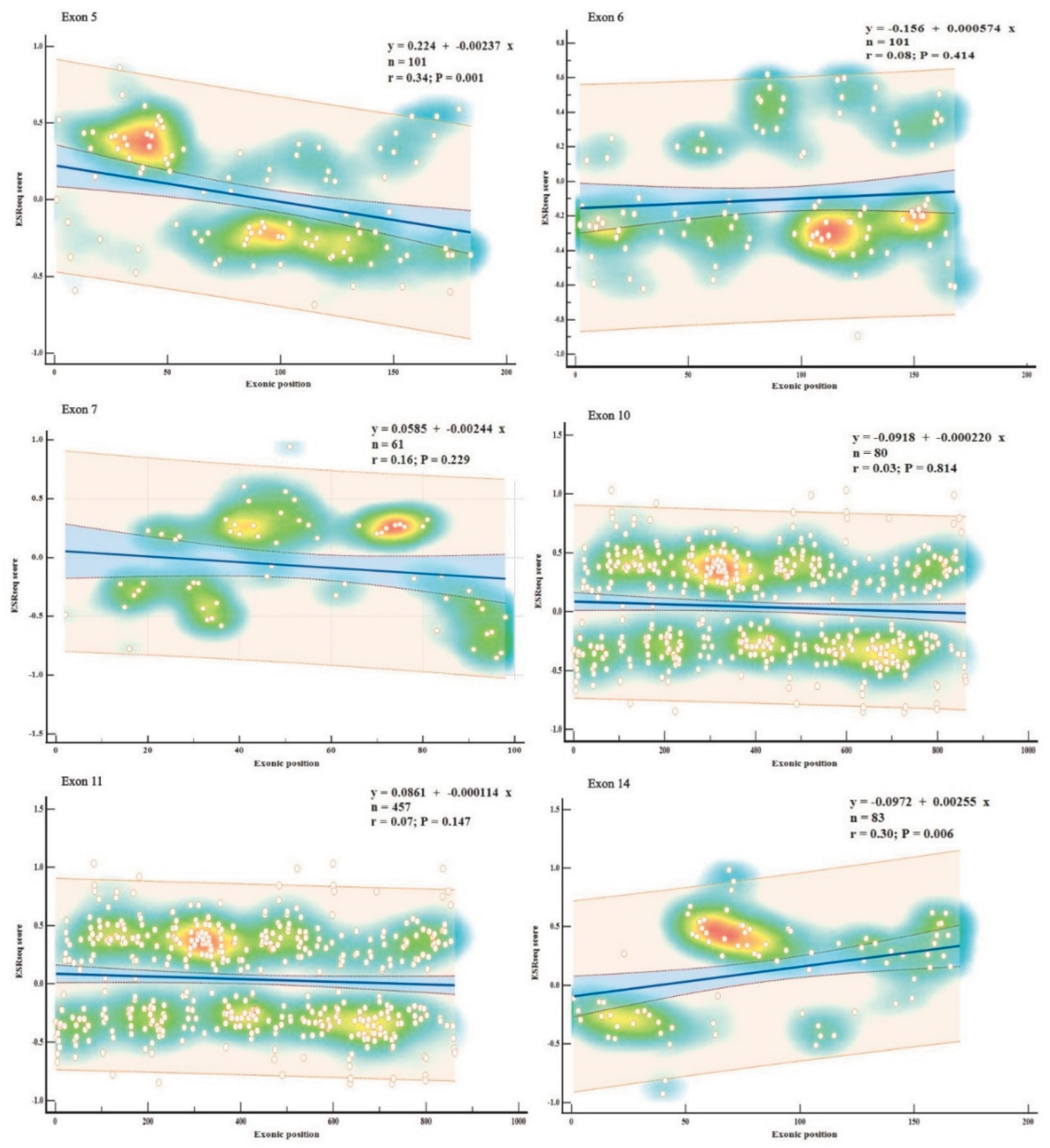

Table 1.
PMS2 exons harboring missense variants with predicted splicing impact (SpliceAI). Overrepresented exons denote exons with means above the 95% CI.
Table 1.
PMS2 exons harboring missense variants with predicted splicing impact (SpliceAI). Overrepresented exons denote exons with means above the 95% CI.
| Predicted mechanism | Total number of splicing variants | Overrepresented exons | Mean (CI 95%) |
|---|---|---|---|
| Acceptor gain | 38 (32.47%) | 6, 11, 14 | 2.53 (0.45–4.61) |
| Acceptor loss | 50 (42.73%) | 6, 8, 14 | 3.33 (0–7.05) |
| Donor gain | 11 (9.40%) | 1, 5, 6, 11 | 0.40 (0–0.80) |
| Donor loss | 29 (24.78%) | 4, 6, 11 | 1.93 (0–4.16) |
Table 2.
Wild-type ESRseq and ΔESRseq scores comparison between predicted splicing variants (DS > 0.2) and variants with no predicted splicing impact (DS < 0.2) in exons enriched in acceptor loss (AL) and donor loss (DL) variants.
Table 2.
Wild-type ESRseq and ΔESRseq scores comparison between predicted splicing variants (DS > 0.2) and variants with no predicted splicing impact (DS < 0.2) in exons enriched in acceptor loss (AL) and donor loss (DL) variants.
| Exon | ESRseq score wild type | ΔESRseq score |
|---|---|---|
| p-value | p-value | |
| Exon 4 | 0.7868 | 0.0598 |
| Exon 6 | 0.5272 | 0.0013* |
| Exon 8 | 0.1429 | 0.5495 |
| Exon 11 | 0.005 | 0.2413 |
| Exon 14 | 0.0076 | 0.8120 |
Statistically significant results (p < 0.05, Mann-Whitney U test) are highlighted in bold. *Student’s t-test.
Table 3.
Summary of PMS2 exons predicted to harbor strong and weak canonical splice sites.
| Software | Weak donor | Strong donor | Mean (CI 90%) |
Weak acceptor | Strong acceptor | Mean (CI 90%) |
|---|---|---|---|---|---|---|
| ESEfinder 3.0 | 4, 5, 7, 8, 12, 14 | 1, 2, 9, 10, 11, 13 | 5.09 (2.92–7.27) |
3, 5, 8, 11 | 4, 7, 9, 10, 13, 14 | 6.24 (4.09–8.39) |
| FSplice | 4, 6, 8, 12 | 2, 9, 10, 13, 14 | 11.38 (9.45–13.31) |
2, 3, 8, 14 | 7, 9, 10, 12, 13, 15 | 7.77 (6.13–9.40) |
| MaxEntScan | 4, 6, 8 | 2, 9, 10, 13, 14 | 8.67 (7.64–9.70) |
2, 3, 8, 14 | 6, 7, 10, 13 | 8.17 (7.01–9.32) |
| NetGene2 | 4, 7, 8 | 3, 9, 10, 12, 13, 14 | 0.68 (0.51–0.86) |
3, 5, 8, 11, 14 | 4, 6, 9, 12, 13, 15 | 0.51 (0.34–0.68) |
| NNSplice | 4, 6 | 1, 2, 3, 9, 10, 13, 14 | 0.88 (0.79–0.96) |
2, 8, 15 | 7, 9, 10, 13, 14 | 0.66 (0.50–0.83) |
Table 4.
PMS2 exons predicted to be prone to exon skipping, based on ESEs and ESSs densities and ESS/ESE ratio.
Table 4.
PMS2 exons predicted to be prone to exon skipping, based on ESEs and ESSs densities and ESS/ESE ratio.
| Software | Low ESEs density | Mean (CI 90%) |
High ESSs density | Mean (CI 90%) |
High ESS/ESE ratio | Mean (CI 90%) |
|---|---|---|---|---|---|---|
| ESEfinder 3.0 | 3, 7, 10, 13 | 17.23 (14.26–20.19) |
- | - | - | - |
| HExoSplice | 2, 3, 9, 10 | 21.96 (17.91–26.01) |
2, 6, 7, 10 | 26.65 (21.63–31.67) |
2, 6, 9, 10 | 119.86 (89.60–150.11) |
| HOT-SKIP | 8, 9 | 80.26 (75.13–85.39) |
6, 7, 10, 13 | 57.72 (52.26–63.18) |
6, 7, 9, 10, 13 | 74.03 (63.67–84.39) |
Table 5.
Mean RNA expression (median count/base) of PMS2 exons in Lynch syndrome-related tissues. Overexpressed exons denote exons with mean expression above the 95% CI. Underexpressed exons denote exons with mean expression below the 95% CI.
Table 5.
Mean RNA expression (median count/base) of PMS2 exons in Lynch syndrome-related tissues. Overexpressed exons denote exons with mean expression above the 95% CI. Underexpressed exons denote exons with mean expression below the 95% CI.
| Tissue type | Overexpressed exons | Underexpressed exons | Mean (CI 95%) |
|---|---|---|---|
| Bladder | 11, 13, 14 | 1, 2, 15 | 0.1937 (0.1502–0.2372) |
| Brain—cortex | 11, 13, 14 | 1, 2, 15 | 0.2263 (0.1767–0.2760) |
| Breast—mammary tissue | 11, 13, 14 | 1, 2, 15 | 0.1883 (0.1536–0.2230) |
| Colon—transverse | 11, 13, 14 | 1, 2, 15 | 0.1392 (0.1114–0.1671) |
| Colon—sigmoid | 11, 13, 14 | 1, 2, 15 | 0.1516 (0.1195–0.1837) |
| Kidney—medulla | 10, 11, 13, 14 | 1, 2, 3, 5 | 0.1818 (0.1309–0.2327) |
| Ovary | 10, 11, 13, 14 | 1, 2, 15 | 0.1728 (0.1371–0.2085) |
| Pancreas | 11, 13, 14 | 1, 2, 15 | 0.0618 (0.0501–0.0735) |
| Prostate | 11, 13, 14 | 1, 2, 4 | 0.1585 (0.1163–0.2007) |
| Skin—sun exposed | 10, 11, 13, 14 | 1, 2, 15 | 0.2941 (0.2360–0.3522) |
| Stomach | 11, 13, 14 | 1, 2, 15 | 0.10568 (0.0842–0.1270) |
| Uterus | 11, 13, 14 | 1, 2, 15 | 0.1714 (0.1363–0.2065) |
| Whole blood | 1, 11, 13, 14 | 2, 3, 4, 5, 7 | 0.0525 (0.0404–0.0646) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



