
Submitted:
09 April 2024
Posted:
10 April 2024
You are already at the latest version
Abstract
RAMOSA1 (RA1) is a Cys2-His2-type (C2H2) zinc finger transcription factor that controls plant meristem fate and identity and has played an important role in maize domestication. Despite its importance, the origin of RA1 is unknown and the evolution in plants is partially understood. In this paper, we present a well resolved phylogeny based on 73 amino acid sequences from 48 embryophyte species. The recovered tree topology indicates that, during grass evolution, RA1 arose from two consecutive SUPERMAN duplications resulting in three distinct grass sequence lineages: RA1-like A, RA1-like B, and RA1; however, most of these copies have unknown functions. Our findings indicate that RA1 and RA1-like play roles in the nucleus despite lacking a traditional nuclear localization signal. Here we report that copies diversified their coding region and, with it, their protein structure, suggesting different patterns of DNA binding and protein-protein interaction. In addition, each of the retained copies diversified regulatory elements along their promoter regions, indicating differences in their upstream regulation. Taken together, we propose that the RA1 and RA1-like gene families in grasses may have undergone subfunctionalization and neofunctionalization enabled by gene duplication.
Keywords:
zinc finger
; embryophyte
; phylogeny
; paralogs
; collinearity
; nuclear localization
; secondary structure
; divergent motifs
; promoter
1. Introduction
RAMOSA1 (RA1) is a C2H2 zinc finger protein first cloned and studied in Zea mays (maize), a major crop species [1,2]. The RA1 is a small protein (175 aa.) that is localized in the nucleus despite lacking a traditional nuclear localization signal [3]. The coding sequence of RA1 is characterized by having a unique zinc finger domain and two well characterized ethylene-responsive element binding factor-associated amphiphilic repression (EAR) motifs (xLxLxLx) towards the C-terminal. In ra1 mutants, long indeterminate branches replace short determinate branches in both the male and female maize inflorescences suggesting that RA1 controls meristem fate and identity. Protein-protein interaction in yeast showed that RA1 interacts with RAMOSA1 ENHANCER LOCUS2 (REL2), a co-repressor homolog of Arabidopsis thaliana (Arabidopsis) TOPLESS (TPL), to repress the expression of target genes [4]. Indeed, it is well documented that RA1 and REL2 interact in vitro and in vivo via the two EAR motifs of RA1 with the Lyssencephaly type1-like homology, LISH (CTHL), region of REL2 [4]. Although RA1 was originally suggested as a strong repressor of transcription, chromatin immunoprecipitation (CHIP-seq) experiments in maize showed that it mainly behaves as a promoter of gene expression that controls several regulatory and developmental pathways [5]. In addition, recent studies suggest that cis-acting regulatory elements upstream of RA1 are a key factor in modulating branch meristem determinacy affecting ear and tassel morphology in maize and grass inflorescence architecture [6]. Indeed, previous studies have found evidence that RA1 was a selected locus during maize domestication [7].
So far, the origin and evolution of RA1 in plants is uncertain. In terms of sequence similarity, RA1 is similar to the Arabidopsis SUPERMAN (SUP) protein [2,8]. Within the functional context, SUP is involved during floral development preventing the initiation of supernumerary stamens, while RA1 has a central role in inflorescence development and does not seem to intervene in floral development [2,8]. The overexpression of RA1 (35S::RA1) in Arabidopsis sup5 mutants fails to restore the number of stamens in the flower [9]. Likewise, the 35S::RA1 transgenic plants of Arabidopsis generate pleiotropic effects in the plant, such as an increase in the size of the reproductive organs due to cell expansion [10]. These results indicate that the role of RA1 differs from SUP. In particular, within grasses, RA1 was cloned in maize and its closest relatives, but it is absent in the genomes of other grass species of the BOP clade (such as rice, wheat, and oat) [1,11].
Given the (1) importance of RA1 during maize inflorescence development and domestication, (2) its central role as a regulator of various plant development pathways, and (3) fragmented knowledge that we have about the evolution and role of the RA1 in other cereals, we decided to deepen the studies in RA1 to increase our understanding on the protein evolutionary history. For that, we reconstructed the phylogeny of RA1 in embryophyte to identify homologs and paralogs protein sequences. Additionally, phylogeny reconstruction results were analyzed considering genome and chromosome collinearity. Homologs and paralogs of RA1 sequences were comparatively characterized with a focus on: conserved/divergent motifs along coding and promoter regions, subcellular protein localization, protein secondary structure and conformational plasticity. We found a complex pattern of gene duplication followed by different patterns of gene copy retention and losses. Retained copies showed partial conservation of the coding sequences and secondary structure, as well as cis-elements in the promoter regions suggesting diversification of protein functionality and upstream regulation. The resulting information will be useful to further explore the mode of action of these proteins in the future.
2. Materials and Methods
2.1. Data Sets
Protein sequences were retrieved from Phytozome Database v13 [12], National Center for Biotechnology Information [13] and Gramene Released 66 [14] in March 2023 (Datasheet 1 available at Mendeley Digital Repository, https://doi: 10.17632/m45fk8hxs4.1, Table S1). Three annotated protein sequences of A. thaliana (SUP, AT3G23130), Setaria viridis (XP_034583085), and Z. mays (NP_001143449) were used as templates for a BLASTp search in each of the 50 studied embryophyte genomes: Amborella trichopoda (AmTr), Anana comosus (Ac), Aquilegia coerulea (AqCo), A. thaliana (At), Brachypodium distachyon (Bd), Brachypodium sylvaticum (Bs), Brachypodium stacei (BrSt), Brassica rapa (Br), Carica papaya (Cp), Capsella rubella (Cr), Cenchrus americanus (Ca), Ceratodon purpureus (CePu), Citrus clementina (Cc), Dioscorea alata (Da), Eleusine coracana (Ec), Hordeum vulgare (Hv), Joinvillea ascendens (Ja), Leersia perrieri (Lp), Marchantia polymorpha (Mp), Medicago truncatula (Mt), Miscanthus sinensis (Ms), Olea europaea (Oe), Oryza barthii (OrBa), Oryza brachyantha (Ob), Oryza glaberrima (Og), Oryza glumipatula (OrGl), Oryza meridionalis (Om), Oryza nivara (On), Oryza punctata (Op), Oryza rufipogon (Or), Oryza sativa (rice) (Os), Panicum hallii (Ph), Panicum virgatum (Pv), Pharus latifolius (Pl), Physcomitrella patens (Pp), Populus trichocarpa (Pt), Prunus persica (PrPe), Ricinus communis (Rc), Selaginella moellendorffii (Sm), Setaria viridis (Sv), Sorghum bicolor (Sb), Solanum tuberosum (St), Theobroma cacao (Tc), Thinopyrum intermedium (Ti), Thuja plicata (Tp), Urochloa fusca (Uf), Vaccinium darrowii (Vd), Zea luxurians (Zl), Z. mays (Zm), Zizania palustris (Zp). Duplicated sequences were pruned and raw datasets were scanned using Multiple EM for motif elicitation (MEME) search with MEME v5.5.2 [15]. The search was set up to a maximum of ten motifs and default settings. Based on MEME exploratory scans, sequences containing an incomplete zinc finger motif and/or truncated C-terminal EAR motif were discarded. Protein sequences were aligned with the online version of MAFFT using an E-INS-i strategy [16]. The alignments were manually inspected using MEGA v.10.2.4 [17]. The final aligned dataset contained a total of 73 sequences is presented in Figure S1.
2.2. Phylogeny
Phylogenetic trees were built with MrBayes 3.2.6 [18] via the CIPRES Science Gateway [19] using a fixed substitution model and run for 15 million generations (sampling every 1000 generations). Four Markov chains were run simultaneously in two independent runs starting with a random tree until the convergence diagnostic (standard deviation of split sequences) dropped below 0.01. The first 3.75 million generations were discarded as burn-in (25 %) and the rest of the samples from the two replicates were combined. A majority rule consensus of 22 502 trees was constructed and visualized using Mesquite [20]. Further tree edition was performed using Inkscape 1.2.1. To search for chromosomal collinearity (synteny) among duplicated genes we used the GENESPACE Synteny Viewer available at Phytozome Database v13 [12,21] using A. thaliana TAIR10, O. sativa v7.0, S. viridis v2.1, Z. mays Zm-B73-REFERENCE-NAM-5.0.55, and Z. mays RefGen_V4 as reference genomes.
2.3. Conserved Domain Analysis
To identify conserved motifs, 35 amino acid sequences from 16 grass species were scanned with MEME using MEME v5.5.1 [22]. The search was set to 15 domains from 6 to 50 amino acids long and default settings. For each lineage, a box scheme was presented graphically to show the distribution and frequency of occurrence of their specific motifs.
2.4. Plant Growth Conditions
Nicotiana benthamiana (tabacco) seeds were grown directly on soil at 22–24 °C and 120 µE (µEinstein = µmol m−2 s−1) in a growth chamber under long-day conditions (16 hours of light and 8 hours of darkness). Setaria viridis accession A10 seeds were grown in a growth chamber at 24-28 °C and 300 µE under long-day conditions. Zea mays cultivar B73 seeds were grown in a greenhouse (Instituto de Agrobiotecnología del Litoral) at 25-28°C and 800-1000 µE under long-day conditions.
2.5. Genetic Constructs
The open reading frame of RA1 homologs from Z. mays and S. viridis and paralogs of S. viridis were amplified by PCR using Taq Pegasus DNA polymerase (Productos Bio-Lógicos PBL SA, Buenos Aires, Argentina) with specific primers carrying restriction sites (Table S2). Amplicons were digested with the corresponding enzymes (Promega Corporation, Madison, WI, US) and ligated into the entry vector pENTR3C (Invitrogen) using T4 DNA ligase (Invitrogen, Carlsbad, CA, US). For the subcellular localization analyses, each sequence-verified entry vector clone was sub-cloned into the pFK247 destination vector by site-specific recombination using Gateway LR Clonase II Enzyme Mix (Invitrogen) [23]. The pFK247 vector is derived from the pGreen vector series in which a 35S CaMV promoter drives the expression of an N-terminal fusion protein with Green Fluorescent Protein (GFP) [24].
2.6. Transient Transformation
Tabacco leaves were agro-infiltrated with Agrobacterium tumefaciens LBA4404 cells carrying the appropriate translational fusion construct described above. In order to suppress gene silencing, A. tumefaciens cells that express the tomato bushy stunt virus p19 protein [25] were used in the co-infiltration method, as previously reported by Belda-Palazón et al. [26]. For that, an equal mixture of Agrobacterium strains containing the appropriate translational fusion constructs and the p19 plasmid was prepared for co-infiltration. For transient transformation, the abaxial side of four weeks old N. benthamiana leaves were co-infiltrated with a needleless syringe, as previously described [27]. After infiltration, the plants were exposed to light for one hour and moved into a dark chamber overnight. Seventy-two hours after infiltration, samples were collected at the beginning of the photoperiod and used for visualization under confocal laser scanning microscopy. Fluorescence of at least two transformed leaves from three plants of similar age was analyzed with confocal microscope. The experiments were repeated at least two times for each construct.
2.7. Nuclei Staining
The reagent 4′ ,6-diamidino-2-phenylindole (DAPI; Sigma, St. Louis, MO) was used to stain the nuclei. Before fluorescence confocal microscopy analysis, the agro-infiltrated N. benthamiana leaves were removed from the plant and cut into one inch diameter circles; these were incubated in distilled water supplemented with 1µg/ml DAPI for 5 to 45 min. Then, leaf sections were mounted on a microscope slide and covered with distilled water for observation through the leaf abaxial side.
2.8. Fluorescence Microscopy
Digitized confocal images were acquired at 1024 x 1024 pixel resolution with a ×20 dry objective on a TCS ST8 inverted confocal laser scanning microscope (Leica Microsystems, Germany) by using a 488 nm excitation line laser for GFP and spectral detection was made at 497 and 537 nm for GFP. For DAPI laser excitation the wavelength was set to 405 nm and detection was made at 410–492 nm. Image plates were assembled with InkScape 1.2.2.
2.9. Molecular Modeling
2.10. Molecular Dynamic Simulations
Molecular dynamics studies in solution were carried out using the GROMACS 2021.5 package [30] in conjunction with the AMBER99SB-ILDN force field [31]. The molecular dynamics protocol was performed for the zinc finger domain regions of SUP (aa: 42-78) and RA1 (aa: 32-83) proteins, to characterize their structure and the dynamics. The initial structure of the SUP zinc finger was taken as one of the Nuclear Magnetic Resonance (NMR) determined model results. The initial structure used to start the simulation of the RA1 zinc finger was taken as the AlphaFold prediction for this region of the protein [32]. In the case of the simulation starting from the AlphaFold model, the length of the simulated peptide has been enlarged to avoid disturbing the secondary structure features predicted by the initial model of RA1.
The simulation protocol was the same for each simulation and it consisted in: (1) total energy minimization of the initial system using the steepest descent energy minimization algorithm using a tolerance of 100 kJ/mol, (2) molecular dynamic simulations with position restraint of the protein heavy atoms for 2 ns, in order to allow the water molecules to rearrange around the protein, (3) simulated annealing with position restraint of the zinc finger heavy atoms, increasing the temperature from 300 K to 400 K during 200 ps, equilibrating the temperature for 600 ps and finally gradually cool the system back to 300 K during 4.2 ns (5 ns in total), in order to enhance conformational sampling, and (4) molecular dynamics production run at a temperature of 300 K for 1 μs. In all cases, the proteins were inserted into an octahedral box of simple point charge (SPC) water molecules. The minimum distance between the protein and the simulation box was 1.1 nm, to avoid any inappropriate behavior of the water molecules. The timestep of 2 fs was used for the integration of the equations of motions. The Berendsen thermostat and barostat were used to couple the systems to a temperature of 300 K (with coupling time constant 0.1 ps) and a pressure of 1 bar (with coupling time constant 2.0 ps) baths respectively. The particle mesh Ewald method [33] was used to treat long-range Coulombic interactions. The LINCS algorithm [34] was used to constrain bond lengths of protein atoms and SETTLE for the water molecules [35]. Van der Waals forces were considered up to distances of 1.2 nm and Coulomb interactions were truncated at 1.2 nm. The Root-Mean-Square Deviation (RMSD) and the Root-Mean-Square Fluctuation (RMSF) of backbone atoms were calculated during the 1μs for the RMSD and final 0.2 μs of the molecular dynamics trajectory. The central structure during the last 200 ns (defined as the frame structure with the smallest average RMSD from all other frames in this range) was used as reference for fitting the structures in the calculation of the RMSD and the RMSF.
2.11. Analysis of Promoter cis-Acting Regulatory Elements in Grasses
To identify conserved regulatory motifs on the promoter regions, we downloaded 2kb upstream genomic sequences from the translation initiation site of RA1, RA1-like A, and RA1-like B of 16 grass species. Downloaded sequences were screened to search for codified genes using FGENESH (http://www.softberry.com/berry.phtml) [36]. When additional genes were identified, retrieved sequences were shortened accordingly. Retrieved sequences were organized in eight different datasets: (a) RA1 Andropogoneae dataset, comprising RA1 promoter sequences from four Andropogoneae species; (b) RA1 Paniceae dataset, comprising RA1 promoter sequences from four Paniceae species; (c) RA1-like A Andropogoneae dataset, comprising RA1-like A promoter sequences from three Andropogoneae species; (d) RA1-like A Paniceae dataset, comprising RA1-like A promoter sequences from four Paniceae species; (e) RA1-like A BOP dataset, comprising RA1-like A promoter sequences from five species of BOP clade; (f) RA1-like B Andropogoneae dataset, comprising RA1-like B promoter sequences from three Andropogoneae species; (g) RA1-like B Paniceae dataset, comprising RA1-like B promoter sequences from five Paniceae species; and (h) RA1-like B BOP dataset, comprising RA1-like B promoter sequences from six species of BOP clade. Potential cis-acting regulatory elements were determined against the JASPAR CORE (2022) non redundant database from plants [37] using XSTREME [38]. Each dataset was scanned for motifs ranging from 6 to 30 characters and default settings. We considered motifs with E-value < 0.05.
3. Results
3.1. Phylogenetic Analysis
3.1.1. SUP Evolution and the Origin of RA1
The BLASTp searches recovered SUP sequences along most of the embryophyte species, except from M. polymorpha and S. moellendorffii. To identify the origin of RA1 and understand its evolution, we reconstructed a phylogenetic tree with amino acid sequences obtained from genomes of grasses and other embryophytes. For that, we used SUP sequences from non-seeds plants (P. patens and C. purpureus) as outgroups. The recovered tree topology and the presence of a single copy sequence of the grass P. latifolia indicates that RA1 originated from two successive duplications of SUP that occurred at the split of the BOP and PACMAD clade (Figure 1). As a result of these duplications, three lineages of sequences were generated, one of them includes RA1 sequences and the other two are here named arbitrarily RA1-like A and RA1-like B.
3.1.2. Evolution of RA1 in Grasses
To study the relationship between RA1 and RA1-like more closely, we reconstructed a new phylogeny with amino acid sequences obtained from 16 genomes of grass species using as outgroup the SUP sequence from the monocot J. ascendence (Figure 2a, Figures S2 and S3). The obtained tree topology indicates that: (1) RA1 was originated after a second duplication around the split of the BOP and PACMAD clades, (2) RA1 is sister to the RA1-like B proteins, (3) RA1 was loss in the BOP clade and, currently, is present in the PACMAD clade, (4) RA1-like A and RA1-like B are found in species of both BOP and PACMAD clades, and (5) species specific duplications and loss of some copies of RA1 and RA1-like suggest a complex evolution of these molecules during the diversification of the grasses (Figure 2a, Figure S4).
Genome annotations indicate that SUP is on Chr3:8242256…8243372 of Arabidopsis genome, whereas RA1, RA1-like A, and RA1-like B are on maize chromosomes Chr7:114958642…114959398, Chr5:68312034…68312578, and Chr10:103427449…103428364, respectively. Chromosomal collinearity visualization maps indicate that Arabidopsis chromosome 3 lacks synteny with maize chromosomes 7, 5 and 10 (Figure S5). In contrast, RA1 and RA1-like genes are placed in syntenic chromosome blocks among maize, Setaria, and rice (Figure S6). These results suggest that RA1 and RA1-like are syntenic paralogs.
3.2. Conserved Motifs Analysis among RA1 and RA1-Like Coding Sequences
To discover specific features that characterize the coding region of each lineage, we performed a motif analysis (Figure 2b). In total, 15 motifs were identified (Figure S7 and Figure S8). Some of them are lineage specific (for instance, Motif 7 [WPPPQVRS], Motif 8 [PPPNPNPSCTVLDL], Motif 11 [FPWPPQ], Motif 12 [VVCSCSST], and Motif 13 [MESRSAARAGDQQH]) and others are shared by lineages, such as Motif 3 [ARAPJPNLNYSPPHPA] which is present in RA1-like A and RA1-like B sequences, whereas Motif 4 [APPVVYSFFSLAASA] is shared by RA1 and RA1-like B sequences (Figure 2b, Figure S7). All of the sequences have in common the presence of one C2H2 zinc finger domain (Motif 1 [SSSSSYTCGYCKREFRSAQALGGHMNVHRRDRARLRHGQSP]) (Figure 2b; Figure S9). In particular, sequences from RA1 lineage have the variant QGLGGH in the zinc finger domain as it was previously reported [2] (Figure 2c). In addition, Motif 2 (GDGAEEGLDLELRLG) appeared two to three times towards the C-terminal of all the sequences. Our analysis identified two Motif 2 along the C-terminal of the RA1-like sequences, whereas three Motif 2 were detected on most of the RA1 sequences of the PACMAD clade (Figure 2b, Figure S9). In addition, we found that the number of Leu (L) and the distances between the Motifs 2 vary among clades (Figure 2b, Figure S9). According to literature, Motif 2 is a putative EAR-like repressor motif [2,39,40,41,42,43].
3.3. Subcellular Localization
To determine the in vivo subcellular localization of the RA1 and RA1-like proteins of maize and S. viridis, we generated a N-terminal translational fusion with GFP. The 35S:GFP::RA1 and 35S:GFP::RA1-like constructs were analyzed by a transient expression system in agro-infiltrated tabacco young leaves. The fluorescence was observed through a laser scanning confocal microscope. DAPI staining was used to visualize nuclei localization. As shown in Figure 3, the RA1 and RA1-like proteins are localized in the nuclei of tobacco epidermal cells.
3.4. Molecular Modeling
3.4.1. Proteins Secondary Structure and Conformational Plasticity
The secondary structure properties of the SUP/RA1 members are poorly characterized [44,45]. Only the zinc finger C2H2-type of SUP has had its secondary structure experimentally solved by NMR [45]. So, to gain perspective on protein structure among SUP, RA1, and RA1-like, disordered regions and secondary structures were predicted using the DisoPred 3.1 tool [29] and PsiPred server [28,46], respectively (Figure 4, Figure S10, and Table S4). Overall, the N-terminal region of SUP, RA1, and RA1-like is predicted as disordered with protein binding affinities (Figure 4, Figure S10, and Table S4). Such a disordered region is followed by a region defined as a coil that continues with the C2H2-type domain. The structural predictions suggest the zinc finger to be formed by a β-sheet (two consecutive β-strands) followed by an α-helix (Table S4). In general, the C-terminal of RA1 and RA1-like is presented as a disordered region with protein binding affinities. In addition, we found stabilized ordered segments, such as two to three α-helix. These ordered segments are mostly correlated with the allocation of Motif 2 (Figure 4, Figure S10, and Table S4).
3.4.2. Zinc Finger Molecular Dynamics Simulation
The zinc finger of SUP (1NJQ) is a structure resolved by NMR (20 models), which has 75% sequence identity with the zinc finger of RA1 (Figure S11a). There is no experimental structure of RA1. Given differences in sequence identity between SUP and RA1 we performed 1μs Molecular dynamics simulations of each protein zinc finger domain to evaluate differences in the structural stability of these variants. The RMSDs of these simulations as a function of simulated time (Figure S11b) show small departures from their initial values (average RMSD values lower than 0.5Å). This fact is associated with the robustness of the structure of the zinc finger domain of both proteins and the proximity of the initial models to the equilibrated structures. We assumed that the simulated time in the period [0.8μs, 1.0μs] is adequate to calculate equilibrium properties of the systems since the RMSDs of the simulations of both proteins show small fluctuations without any appreciable definite tendency.
Analysis of the time evolution of the secondary structure of SUP and RA1 peptides showed the classical pattern of a zinc finger with a β-sheet composed of two strands (Y47T48-F55E56 and Y46T47-F54E55, in SUP and RA1 respectively) and a turn stabilized by H-bonds (S50F51C52 and G49Y50C51, for SUP and RA1), followed by an α-helix (A59:H69 and A58:R72, for SUP and RA1) (Figure 5a, Table S4). The α-helix structural motif is four residues longer for the case of RA1 (an additional turn). Beyond the α-helix structural motif, the SUP peptide presents a turn of a 310-helix (D72:R75) (Figure 5a, Table S4). We observed an additional structure with fluctuating character 310-helix, α-helix, and turns in RA1 peptide (I76:Y80) (Figure 5a, Table S4).
Previous work suggests that the change of Ala (A) to Gly (G) in the QALGGH motif of the RA1 zinc finger could lead to an enhanced mobility of the residues in the α-helix [2]. However, such a hypothesis has not been tested yet. Here, we perform a comparative RMSF analysis between RA1 and SUP to test whether the G affects the structure dynamic of the zinc finger α-helix (Figure 5b). The RMSF of the SUP and RA1 amino acids were calculated in the equilibrium interval of the simulations, for the backbone atoms of each residue. We found that, at this particular site, the amino acids of RA1 show smaller fluctuations than the A of SUP (Figure 5b). One could argue that the effect of the substitution of A by G may not be local; however, we found that the RA1 amino acids show smaller fluctuations along all the extensions of the canonical zinc finger domain. The existence of a longer α-helix motif in the RA1 peptide, followed by the additional helix pattern, causes the region of small fluctuations or relative rigidity of the peptide to extend well beyond the canonical zinc finger motif (Figure 5b). These results suggest that the substitution of A for G does not affect the tridimensional structural mobility of the zinc finger domain.
3.5. Cis-Acting Regulatory Elements Analysis among RA1 and RA1-Like Promoter Sequences
When the promoter of RA1 and RA1-like genes of 16 grass species were examined, a large number of regulatory motifs were identified and grouped into 33 types of elements, according to the corresponding transcription factor (TF) family (Figure 6, Table S5). Conserved sequences containing binding motifs for well-known transcriptional regulators were recognized. We identified TF motifs involved in different biological processes related to: (1) seed germination, such as embryo development, somatic embryogenesis, and positive regulation of cell population proliferation; (2) plant development, such as regulation of secondary shoot formation, leaf development, flower development, and root development; (3) response to abiotic stress, such as response to salt stress, response to light stimulus, and response to water; and (4) response to biotic stress, such as defense response to bacterium (Table S6). In addition, we identified TF binding sites related to positive or negative regulation of hormone signaling pathways, hormone biosynthetic processes, as well as auxin, gibberellin, abscisic acid, and ethylene responses (Table S6).
The conserved regulatory motifs harbored eight putative TF binding sites for RA1-like A lineage, 22 putative TF binding sites for RA1-like B lineage, and 13 putative TF binding sites for RA1 lineage. We found two conserved motifs shared by RA1-like and RA1 promoter sequences: (1) ABI-motifs are present among at least 24 of 35 promoter sequences analyzed; (2) WRKY-motifs were identified in 12 of 35 promoter sequences.
RA1-like A shares: (1) TCP motifs with the promoter sequences of RA1-like B of the PACMAD clade, except for the ones of Andropogoneae, and (2) DREB1E motifs with Andropogoneae promoter sequences of RA1.
RA1-like B has five motifs in common with RA1: (1) ARF-motifs are present in all RA1-like B and RA1 promoter sequences of the Andropogoneae tribe; (2) AGL-motifs are well conserved in RA1-like B promoter sequences, except in the Andropogoneae tribe, while AGL-motifs are restricted to the Andropogoneae tribe in RA1 lineage; (3) MYB-motifs were identified in RA1-like B sequences of the PACMAD and RA1 promoter sequences from the Andropogoneae tribe; (4-5) MNB1A-motif and ERF-motifs were identified among RA1-like B promoter sequences of the PACMAD clade, except for the ones of Andropogoneae, and RA1 promoter sequences from the Andropogoneae tribe.
We found that each species lineage copy is characterized by unique cis-elements. The RA1-like A lineage has conserved BPC-motifs outside the Andropogoneae tribe and a RA1 binding motif in the Andropogoneae tribe. TGA9-motif, GRP9-motif, and HHO3-motif were identified only in promoter sequences from PACMAD clade species outside the Andropogoneae tribe. The RA1-like B lineage is characterized by the presence of NAC-motifs on all promoter sequences analyzed, except for the one of T. intermedium. OsRR22-motif, BZIP48-motif, and GATA20-motif are restricted to the BOP clade. Between species from the PACMAD clade, bHLH130-motif, ANL2-motif, and an unknown motif were identified in the Andropogoneae tribe, while PLT1-motif, FUS3-motif, ABF2-motif, Zm00001d027846-motif, ATHB-12-motif, AHL20-motif, and DOF3.6-motif were identified outside the Andropogoneae tribe. Finally, the promoter sequences of the RA1 lineage are characterized by the presence of: (1) SPL14-motif, TB1-motif, CDF5-motif, and O2-motif sequences in the Andropogoneae tribe, and (2) SMZ-motif outside the Andropogoneae tribe.
4. Discussion
RAMOSA1 is a small C2H2 zinc finger transcription factor that has an unquestionable role during maize inflorescence development and domestication [1,4,5]. Indeed, RA1 is the core of the RAMOSA circuit that regulates the fate of axillary meristem determining the final form of the maize inflorescence architecture. Despite its remarkable role, so far, the origin and the evolution of RA1 remained uncertain. It has been mentioned that SUP may be the Arabidopsis homolog of RA1 based on sequence similarity; however, it has been documented that SUP and RA1 have different functions [1,8,47]. To link the fragmented, and sometimes conflicting, knowledge on the evolution and functionality of SUP/RA1 proteins, in this work we present: (1) a solid phylogenetic framework to understand the origin and evolution of RA1 in embryophytes, (2) a detailed SUP/RA1 homology clustering among model and non-model embryophyte species, and (3) new insights on the RA1 gene features and protein structure evolution.
We identified that SUP was present, at least, from the early diversification of embryophytes (land plants). Based on such results we reconstructed the evolutionary history of SUP/RA1 in embryophytes. The phylogeny presented here suggests that RA1 arose from two successive duplications of SUP, one around the base of Poaceae and the other at the diversification of the BOP and PACMAD clades. The first duplication around the origin of the grasses gave rise to the RA1-like A lineage that is sister to the paralogs RA1-like B and RA1 lineages which arose later during the split of the BOP and PACMAD clades. The phylogeny is also supported here by genome synteny studies. Indeed, previous literature and the collinearity analysis presented in this work suggest that the first duplication correlates with the rho WGD of grasses (at ~100 million years ago) and the second duplication may correlate with GD that occurred at the base of BOP/PACMAD separation (at ~80 million years ago) [48,49,50]. Interestingly, RA1 and RA1-like paralogs genes are exclusive of grasses and show different patterns of retentions and losses as is usually observed after genome duplication events [48,49,50]. It is well documented that differential gene loss or subfunctionalization and neofunctionalization of retained copies, as the case presented here, may promote morphological, physiological, and ecological diversification, in particular, in grasses [51,52].
The nuclear localization of RA1 is well described [3]; however, the localization of RA1-like proteins is unknown. To generate knowledge on the general role of such proteins we performed protein subcellular localization studies. We observed that RA1 and RA1-like localized in the nucleus besides lacking a classical nuclear localization signal [3]. So far, our results indicate that RA1 and RA1-like proteins may move to the nucleus to regulate gene expression like most of the C2H2 zinc finger proteins.
Duplication of SUP/RA1 correlates with changes in the coding region, secondary structure, and diversification of the binding properties of their promoter regions.
Evolution of RA1 and RA1-like amino acid sequences. We found conserved and divergent motifs along the coding region of RA1 and RA1-like studied sequences. All of the sequences studied in this work presented the C2H2-type zinc finger with the motif QALGGH, except for the variant of QGLGGH in the RA1 sequences. Overall, we observed conserved motifs between RA1-like A and RA1-like B and between RA1-like B and RA1 sequences as well as specific motifs that characterize each lineage. Some of these conserved motifs were identified in regions predicted as disordered structures with binding protein affinities, suggesting that RA1 and RA1-like have diversified their function through variations in protein-protein interaction.
Interestingly, we identified motifs linked to positive and negative regulators of the transcription as was previously identified in SUP [53]. Negative elements at the N-terminal of the zinc finger domain (Motif 1) might affect nucleosome positioning in the core promoter region, as was shown in SUP [53]. Also, the region around the C-terminal (Motif 2) could be a target site for methylation and silencing the gene expression [53,54]. Likewise, results of ChIP-seq and genome-wide analysis of RA1 occupancy showed that RA1 can repress and also activate genes associated with nucleic acid-related processes, such as chromatin, and TFs implicated in cell specification with final effects on inflorescence development [5].
In particular, in the present study, Motif 2 has been identified as a putative EAR-like repressor motif (xLxLxLx; [39,41,42,43,55,56,57]). Indeed, SUP has been characterized as a repressor protein whose repression activity falls on the unique C-terminal EAR motif [40]; however, RA1 was described as a repressor protein bearing two EAR motifs: an EAR motif towards the C-terminal of the protein as in SUP [2] and a second one close to the C-terminal of the zinc finger domain [4]. It was documented that both EAR repressor motifs of RA1 are involved in the interaction with REL2 (a co-repressor homolog to TPL of Arabidopsis) which, in turn, regulates their target genes promoting the formation of short braches bearing paired spikelets in maize [4]. Similarly, more recently, it has been documented that SUP acts as a repressor of B class genes in the 4th whorl during flower development by interacting with TPL via its single EAR motif located at the C-terminal region of the protein [57]. Indeed, a site-directed mutagenesis of the SUP EAR motif demonstrated that the removal of a L will abolish the interaction between SUP and TPL [57]. Interestingly, among RA1 sequences, we found natural L mutations in Motif 2 located next to the zinc finger domain in sequences of PACMAD species, but the Andropogoneae. In addition, results presented in this work indicate that most RA1 and RA1-like proteins also carry an additional putative EAR motif in between. So far, experiments on the role of the third putative EAR motif observed in RA1 and RA1-like were not carried out yet. Then, RA1 sequences of the Andropogoneae members may have up to three putative EAR motifs. The phylogeny presented here supports the hypothesis of an increment in the number of putative EAR motifs during the evolution of SUP/RA1. Interestingly, it is well documented that a greater number of L along the EAR, a greater number of EAR motifs along the amino acid sequences, and regions that border EAR motifs are equally important for stabilizing binding to repressor proteins such as TPL [43]. These results suggest that, given the differences in the number and conservation of L residues of the EAR motifs, RA1 may have evolved towards an increased strength of repression activity through more stable binding to its co-repressor counterpart.
Changes in secondary structures. Studies on how folding information is distributed along a protein sequence provide information on the formation of different secondary structures, such as α-helices and β-sheets, and on binding properties [58]. Secondary structure predictions carried on in this work suggest that SUP, RA1, and RA1-like mainly differ towards the C-terminal. Such differences are correlated with the presence of one to three Motif 2, identified here as putative EAR motifs. Interestingly, EAR motifs were, previously, described as important motifs for the interaction with co-repressors [2,43,58].
Most zinc finger proteins of plants bind DNA via a short α-helix containing the highly conserved QALGGH sequence [60,61]. Interestingly, RA1 orthologs are characterized by having the QGLGGH sequence instead [2]. It has been suggested that G may act as a helix-relaxing residue that confers unique functional attributes [2]. Our analysis identified that the SUP zinc finger domain secondary structure consists of a very well-defined ββα motif, as it was experimentally determined [45]. The same structure was predicted for the RA1 zinc finger domain, suggesting that RA1 binds DNA in a similar way to all other C2H2 zinc finger proteins structurally characterized [45,61,62,63,64,65,66,67,68,69]. The RMSF studies presented in this work showed that the change of A to G in the QALGGH motif of the RA1 zinc finger does not affect the mobility of the helix region as it was suggested in previous work [2].
On the other hand, the zinc finger is one of the major structural motifs involved in eukaryotic protein-DNA interaction [59,70]. Structural studies on the C2H2 zinc finger have revealed that three positions (determinant residues) in the helical region of the zinc finger participate in major interactions with sequences in target DNA. In animals, the three amino acid residues of the finger that specifically make contacts with the DNA bases occupy the helix positions +2 (Arg), +3 (Asn), and +6 (Arg) and, in plants, two of them correspond to the sequence QALGGH [55,61,71,72,73,74,75]. In this context, in SUP the QALGGH sequence occupies positions 2–7 of the helix, that is, it includes all three residues (+2, +3, and +6) [45]. In SUP, position 2 is occupied by a Gln (Q) residue, position 3 by an A, and position 6 by a G residue [45]. Even though Q and more infrequently A residues are reported to act as base determinants, it is difficult to conceive that G can bind a base of the nucleotide due to the lack of a side chain [45]. Indeed, it has been proposed that the binding of SUP is performed through residues at relative positions -1 (S), 2 (Q), and 3 (A) [45]. The G residues at relative positions 5 and 6 could allow the helix to draw closer to the DNA and the residues at the C-terminal of the zinc finger helix, relative positions 9 (N), 10 (V), and 12 (R), may bind the DNA, as in the case of EPF2-5 and EPF2-7 of Petunia [76]. For the case of RA1 residue at relative position 3 is a G therefore the proposal of positions -1, 2, and 3 as base determinants can be ruled out. Nevertheless, it is still possible that G plays a passive role in the approximation of the α-helix to the major groove of DNA allowing that other determinant residues upstream in the structure can bind the DNA bases [45]. In the same way, taking the alternative proposal of Isernia et al. [45], the determinant residues would be 9 (N), 10 (I), and 12 (R), near the zinc finger α-helix C-terminal. The variability of residues in this zinc finger sequence among zinc finger proteins could be related to specificities for different and unique base triplet sequences, generating a huge diversity of target sequences [61,75]. To explore these possibilities additional experiments are needed that, however, are beyond the scope of this work.
Changes in promoter binding properties. Regulatory motifs in promoter regions serve as recognition sites for TFs that promote the initiation of transcription as well as specific regulation of gene expression [36]. In general, most of the cis-acting elements identified in all promoters suggest that these proteins are related to plant growth and development, but via different pathways or interacting with diverse partners.
The analysis of regulatory elements in the potential promoters of RA1 and RA1-like genes identified binding motifs for TFs involved in seed germination (ABI3, [77]; ABI5, [78]; FUS3, [79]), plant development (BPC5, BPC6, [80]; TB1, [81]; ARF4, [82]; ARF16, [83]; RA1, [2]; SMZ, TGA9, [84]; bHLH130, [85]; SPL14, [86]; ATHB12, [87]; AGL27, [88]; AGL42, [89]; CDF5, [90]; ARALYDRAFT_897773, also known as TCP4, [91]; ARALYDRAFT_496250, also known as TCP5, [92]; GRF9, [93]; ANL2, [94]; PLT1, [95]; HHO3, [96]; ABF2, [97]; DOF3.6, [98]), response to abiotic stress (DREB1E, [99]; ERF008, [100]; ERF055, [101]; ERF115, [102]; NAC020, [103]; NAC045, [104]; NAC092, [105]; OsRR22, [106]), and response to biotic stress (WRKY62, [107]; WRKY75, [108]; MYB73, [109]).
The conserved promoter sites found in the RA1 sequences in this study are consistent with the findings of Strable et al. [6]. The authors had identified several highly conserved motifs in the RA1 promoter sequences of Andropogoneae species, such as ARFs-motifs, which were also identified in the present study. Also, our findings support the hypothesis that RA1 plays a role in the transition to flowering, the integration of both developmental and environmental cues [5], as well as the positive or negative regulation of gene expression in specific organs and at certain stages of development [53]. Additionally, our results suggest that these sites may also be involved in the regulation of hormonal pathways. In line with this, previous RNA-seq and Chip-seq studies also indicate that RA1 may be involved in the biosynthesis and signaling of gibberellic acid and linked to auxin pathways [5].
In summary, the phylogenetic reconstruction presented in this work suggests that RA1 arose from two successive SUP duplications during the origin of the grass family and the diversification of grass species. This gave rise to three different grass sequence lineages, namely RA1-like A, RA1-like B, and RA1, most of which have unknown functions. These results indicate that SUP and RA1 are paralog sequences. The phylogenetic distance and duplication events that separated SUP and RA1 may explain the different roles reported for these proteins. It is interesting to note that, most of the studied species retained RA1 and RA1-like proteins in their genomes, indicating their functional importance.
In this report, we have discovered that RA1 and RA1-like have diversified their coding region, which may have led to variations in their protein structure. This suggests that there may be differences in their DNA binding patterns and protein-protein interactions among copies. Additionally, each of the conserved copies have diversified regulatory elements in their promoter regions, indicating differences in their upstream regulation. Overall, we have found that RA1 and RA1-like are involved in different pathways of plant growth and development. Therefore, we propose that gene duplication has enabled subfunctionalization and neofunctionalization in the RA1 and RA1-like gene families in grasses.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Multiple sequence alignment of 73 C2H2 zinc finger amino acid sequences from 48 embryophyte species; Figura S2: RA1 evolution in grasses. Majority rule consensus tree (N=22502 trees) of RA1 TF in grasses generated by Bayesian inference using 54 amino acid sequences. JaSUP was used as outgroup; Figure S3: Resolved phylogenetic tree between RA1 and RA1-like B. Majority rule consensus tree (N=22502 trees) of RA1 and RA1-like B TFs in grasses generated by Bayesian inference using 33 amino acid sequences. BdRA1-A and ZmRA1-A were used as outgroups; Figure S4: Scheme representing the presence or absence of RA1 and RA1-like proteins in grass species. Colored circle represents the presence of the protein; Figure S5: Chromosome synteny between A. thaliana and Z. mays. (a) Analysis of synteny between genomes. Colored lines link collinearity blocks that represent syntenic regions identified by GENESPACE. (b) Gene alignment between chromosome 3 of A. thaliana and chromosome 1 of Z. mays; Figure S6: Chromosome synteny between O. sativa, Z. mays, and S. viridis. (a) Analysis of synteny between O. sativa and Z. mays genomes. Colored lines link collinearity blocks that represent syntenic regions identified by GENESPACE. (b) Analysis of synteny between O. sativa and S. viridis genomes. Colored lines link collinearity blocks that represent syntenic regions identified by GENESPACE. (c) Analysis of synteny between S. viridis and Z. mays genomes. Colored lines link collinearity blocks that represent syntenic regions identified by GENESPACE; Figure S7: Schematic distribution of conserved motifs in RA1 and RA1-like proteins identified by MEME. The fifteen motifs are represented with different colors. Motif 1 represents the conserved zinc finger domain. Motif 2 represents the conserved putative EAR domain; Figure S8: Motif consensus sequences identified by MEME. The 35 amino acid C2H2 sequences from 16 grass species were used to identify conserved motifs among them. Fifteen motifs were identified. Motif 1 represents the conserved zinc finger domain. Motif 2 represents the conserved putative EAR motif; Figure S9: Multiple sequence alignment of 35 C2H2 zinc finger amino acid sequences from 16 grass species. Black box indicates the zinc finger domain (Motif 1). Gray boxes indicate the putative EAR motifs (Motif 2). Dashed gray box indicates the putative EAR motif partially conserved among these proteins. Dotted gray box indicates a motif composed by only two Leu, similar to an EAR motif (EAR-like). Solid gray box indicates the EAR motif towards the C-terminal. Black arrowhead indicates the amino acid position changed (A -> G) in the zinc finger domain; Figure S10: Proteins conformational plasticity. Relative disorder levels of the structures are measured in a range of 0 to 1.0, following the algorithms used by DisoPred 3.1 server. Levels above 0.5 (dashed line) indicate disordered regions. The segment corresponding to the zinc finger (ZF) domain is marked in the figure; Figure S11: Zinc finger dynamic simulation. (a) Local sequence alignment of RA1 and SUP zinc finger domain as determined by Smith-Waterman algorithm (residue identity 75%). (b) Time evolution of the RMSD of the backbone atoms of the RA1 (above) and SUP (below) peptides in the simulated interval 0.0 to 1.0 μs. The structure with the smallest RMSD in the interval 0.0 to 1.0 μs of each simulation has been used as reference for fitting the structures of each peptide. The vertical gray dashed line at 0.8 μs marks the time that has been assumed as the beginning of the thermodynamic equilibrium interval for properties calculations. Table S1: Protein data information from embryophyte genomes; Table S2: Primers and enzymes used in each genetic construction; Table S3: Functional characterized proteins information; Table S4: Proteins conformational plasticity and secondary structures; Table S5: Cis-acting regulatory elements analysis; Table S6: TF motifs associated a different biological processes.
Author Contributions
Conceptualization, C.B. and R.R.; methodology, C.B., F.H., D.R., A.G., S.H. and RR; experiments, C.B., F.H., D.R., A.G., S.H. and RR; formal analysis, C.B., F.H., D.R., A.G., S.H. and RR.; writing—original draft preparation, C.B., F.H., D.R., and RR; writing—review and editing, C.B., D.R., and RR; funding acquisition, R.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Universidad Nacional del Litoral (CAID+D 2020 - 50620190100039LI to R.R.), Fondo para la Investigación Científica y Tecnológica (FONCYT, PICT StartUp-2020-029 to R.R.; FONCYT, PICT-2021-I-A-00756 to R.R.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in Mendeley Digital Repository at doi: 10.17632/m45fk8hxs4.1.
Acknowledgments
We thank Santiago Prochetto for sharing his phylogenetic reconstruction knowledge and providing constructive comments on an early version of the manuscript. We also thank members of the Development and Evolution Lab (LED, IAL) and the Instituto de Agrobiotecnología del Litoral (UNL-CONICET) for helpful discussions. This work used computational resources from CCAD-UNC, which is part of SNCAD-MinCyT, Argentina.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martienssen, R.; Vollbrecht, E. Nucleotide Sequences Enconding Ramosa 1 Gene and Methods of Use for Same. WO2001090343A2. 2001. [Google Scholar]
- Vollbrecht, E.; Springer, P.S.; Goh, L.; Buckler IV, E.S.; Martienssen, R. Architecture of Floral Branch Systems in Maize and Related Grasses. Nature 2005, 436, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. Study of RAMOSA1 Function during Maize Inflorescence Development. Ph.D. Thesis, Iowa State University, Ames, IA, USA, 2011. [Google Scholar]
- Gallavotti, A.; Long, J.A.; Stanfield, S.; Yang, X.; Jackson, D.; Vollbrecht, E.; Schmidt, R.J. The Control of Axillary Meristem Fate in the Maize Ramosa Pathway. Developmen, 2010, 137, 2849–2856. [Google Scholar]
- Eveland, A.L.; Goldshmidt, A.; Pautler, M.; Morohashi, K.; Liseron-Monfils, C.; Lewis, M.W.; Kumari, S.; Hirag, S.; Yang, F.; Unger-Wallace, E.; et al. Regulatory Modules Controlling Maize Inflorescence Architecture. Genome Res. 2014, 24, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Strable, J.; Unger-Wallace, E.; Raygoza, A.A.; Briggs, S.; Vollbrecht, E. Interspecies Transfer of RAMOSA1 Orthologs and Promoter Cis Sequences Impacts Maize Inflorescence Architecture. Plant Physiol. 2023, 191, 1084–1101. [Google Scholar] [CrossRef] [PubMed]
- Sigmon, B.; Vollbrecht, E. Evidence of Selection at the Ramosa1 Locus during Maize Domestication. Mol. Ecol. 2010, 19, 1296–1311. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Sakai, H.; Jack, T.; Weigel, D.; Mayer, U.; Meyerowitz, E.M. SUPERMAN, a Regulator of Floral Homeotic Genes in Arabidopsis. Development 1992, 114, 599–615. [Google Scholar] [CrossRef]
- Cassani, E.; Landoni, M.; Pilu, R. Characterization of the Ra1 Maize Gene Involved in Inflorescence Architecture. Sex. Plant Reprod. 2006, 19, 145–150. [Google Scholar] [CrossRef]
- Landoni, M.; Cassani, E.; Pilu, R. Arabidopsis Thaliana Plants Overexpressing Ramosa1 Maize Gene Show an Increase in Organ Size Due to Cell Expansion. Sex. Plant Reprod. 2007, 20, 191–198. [Google Scholar] [CrossRef]
- Estep, M.C.; Vela Diaz, D.M.; Zhong, J.; Kellogg, E.A. Eleven Diverse Nuclear-Encoded Phylogenetic Markers for the Subfamily Panicoideae (Poaceae). Am. J. Bot. 2012, 99, 443–446. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A Comparative Platform for Green Plant Genomics. Nucleic Acids Res. 2012, 40, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Tello-Ruiz, M.K.; Jaiswal, P.; Ware, D. Gramene: A Resource for Comparative Analysis of Plants Genomes and Pathways; Springer US: New York, NY, USA, 2022. [Google Scholar]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE : Tools for Motif Discovery and Searching. Nucleic Acids Research 2009, 37, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop, New Orleans, LA, USA, 14 November 2010; Volume 14, pp. 1–6. [Google Scholar]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Evolution 2008, 62, 1103–1118. [Google Scholar]
- Lovell, J.T.; Sreedasyam, A.; Schranz, M.E.; Wilson, M.; Carlson, J.W.; Harkess, A.; Emms, D.; Goodstein, D.M. GENESPACE Tracks Regions of Interest and Gene Copy Number Variation across Multiple Genomes. eLife 2022, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.; Elkan, C. Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Hartley, J.L.; Temple, G.F.; Brasch, M.A. DNA Cloning Using in Vitro Site-Specific Recombination. Genome Res, 2000, 10, 1788–1795. [Google Scholar] [CrossRef]
- Hellens, R.P.; Anne Edwards, E.; Leyland, N.R.; Bean, S.; Mullineaux, P.M. PGreen: A Versatile and Flexible Binary Ti Vector for Agrobacterium-Mediated Plant Transformation. Plant Mol. Biol. 2000, 42, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O.; Rivas, S.; Mestre, P.; Baulcombe, D. An Enhanced Transient Expression System in Plants Based on Suppression of Gene Silencing by the P19 Protein of Tomato Bushy Stunt Virus. Plant J. 2003, 33, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Belda-Palazón, B.; Ruiz, L.; Martí, E.; Tárraga, S.; Tiburcio, A.F.; Culiáñez, F.; Farràs, R.; Carrasco, P.; Ferrando, A. Aminopropyltransferases Involved in Polyamine Biosynthesis Localize Preferentially in the Nucleus of Plant Cells. PLoS ONE 2012, 7, e46907. [Google Scholar] [CrossRef] [PubMed]
- Felippes, F.F. De; Weigel, D. Transient Assays for the Analysis of MiRNA Processing and Function. In Plant MicroRNAs; Meyers, B.C., Green, P.J., Eds.; Humana Press: Totowa, NJ, USA, 2009. [Google Scholar]
- Jones, D.T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise Disordered Region Predictions with Annotated Protein-Binding Activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Solovyev, V.V.; Shahmuradov, I.A.; Salamov, A.A. Computational Biology of Transcription Factor Binding; Humana Press: Totowa, NJ, USA, 2010; Volume 674. [Google Scholar]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Berhanu Lemma, R.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Perez, N.M.; et al. JASPAR 2022: The 9th Release of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2021, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L. XSTREME: Comprehensive Motif Analysis of Biological Sequence Datasets. bioRxiv, 4587. [Google Scholar]
- Ohta, M.; Matsui, K.; Hiratsu, K.; Shinshi, H.; Ohme-Takagi, M. Repression Domains of Class II ERF Transcriptional Repressors Share an Essential Motif for Active Repression. Plant Cell 2001, 13, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Hiratsu, K.; Ohta, M.; Matsui, K.; Ohme-Takagi, M. The SUPERMAN Protein Is an Active Repressor Whose Carboxy-Terminal Repression Domain Is Required for the Development of Normal Flowers. FEBS Lett. 2002, 514, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Hiratsu, K.; Mitsuda, N.; Matsui, K.; Ohme-Takagi, M. Identification of the Minimal Repression Domain of SUPERMAN Shows That the DLELRL Hexapeptide Is Both Necessary and Sufficient for Repression of Transcription in Arabidopsis. Biochem. Biophys. Res. Commun. 2004, 321, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.B.; Hagen, G.; Guilfoyle, T.J. Aux/IAA Proteins Contain a Potent Transcriptional Repression Domain. Plant Cell 2004, 16, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Ma, H.; Gu, X.; Thelen, A.; Brunzelle, J.S.; Li, J.; Xu, H.E.; Melcher, K. Structural Basis for Recognition of Diverse Transcriptional Repressors by the TOPLESS Family of Corepressors. Sci. Adv. 2015, 1, e1500107. [Google Scholar] [CrossRef] [PubMed]
- Dathan, N.; Zaccaro, L.; Esposito, S.; Insernia, C.; Omichinski, J.G.; Riccio, A.; Pedone, C.; Di Blasio, B.; Fattorusso, R.; Pedone, P.V. The Arabidopsis SUPERMAN Protein Is Able to Specifically Bind DNA through Its Single Cys2-His2 Zinc Finger Motif. Nucleic Acids Res. 2002, 30, 4945–4951. [Google Scholar] [CrossRef]
- Isernia, C.; Bucci, E.; Leone, M.; Zaccaro, L.; Di Lello, P.; Digilio, G.; Esposito, S.; Saviano, M.; Di Blasio, B.; Pedone, C.; et al. NMR Structure of the Single QALGGH Zinc Finger Domain from the Arabidopsis Thaliana SUPERMAN Protein. ChemBioChem 2003, 4, 171–180. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 Years On. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Sakai, H.; Medrano, L.J.; Meyerowitz, E.M. Role of SUPERMAN in Maintaining Araidopsis Floral Whorl Boundaries. Nature 1995, 378, 199–202. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Patterns in Grass Genome Evolution. Curr. Opin. Plant Biol., 2007, 10, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Marcussen, T.; Meseguer, A.S.; Fjellheim, S. The Grass Subfamily Pooideae: Cretaceous–Palaeocene Origin and Climate-Driven Cenozoic Diversification. Glob. Ecol. Biogeogr. 2019, 28, 1168–1182. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, L.; Columbus, J.T.; Hu, Y.; Zhao, Y.; Tang, L.; Guo, Z.; Chen, W.; McKain, M.; Bartlett, M.; et al. A Well-Supported Nuclear Phylogeny of Poaceae and Implications for the Evolution of C4 Photosynthesis. Mol. Plant 2022, 15, 755–777. [Google Scholar] [CrossRef] [PubMed]
- Preston, J.C.; Kellogg, E.A. Reconstructing the Evolutionary History of Paralogous APETALA1/FRUITFULL- like Genes in Grasses (Poaceae). Genetics 2006, 174, 421–437. [Google Scholar] [CrossRef] [PubMed]
- McKain, M.R.; Tang, H.; McNeal, J.R.; Ayyampalayam, S.; Davis, J.I.; Depamphilis, C.W.; Givnish, T.J.; Chris Pires, J.; Stevenson, D.W.; Leebens-Mack, J.H. A Phylogenomic Assessment of Ancient Polyploidy and Genome Evolution across the Poales. Genome Biol. Evol. 2016, 8, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Sakai, H.; Meyerowitz, E.M. Whorl-Specific Expression of the SUPERMAN Gene of Arabidopsis Is Mediated by Cis Elements in the Transcribed Region. Curr. Biol. 2003, 13, 1524–1530. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Locus-Specific Control of Asymmetric and CpNpG Methylation by the DRM and CMT3 Methyltransferase Genes. Proc. Natl. Acad. Sci. USA 2002, 99, 16491–16498. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.I.; Sakamoto, A.; Kobayashi, A.; Rybka, Z.; Kanno, Y.; Nakagawa, H.; Nishino, T.; Takatsuji, H. CYs2/His2 Zinc-Finger Protein Family of Petunia: Evolution and General Mechanism of Target-Sequence Recognition. Nucleic Acids Res. 1998, 26, 608–615. [Google Scholar] [CrossRef]
- Kagale, S.; Links, M.G.; Rozwadowski, K. Genome-Wide Analysis of Ethylene-Responsive Element Binding Factor-Associated Amphiphilic Repression Motif-Containing Transcriptional Regulators in Arabidopsis. Plant Physiol. 2010, 152, 1109–1134. [Google Scholar] [CrossRef]
- Chow, V.; Kirzinger, M.W.; Kagale, S. Lend Me Your EARs: A Systematic Review of the Broad Functions of EAR Motif-Containing Transcriptional Repressors in Plants. Genes 2023, 14, 270. [Google Scholar] [CrossRef]
- Terao, J. SUPERMAN, the Guardian of Floral Organ Gene Expression in Arabidopsis thaliana. Master’s Thesis, American University, Washington, DC, USA, 2019. [Google Scholar]
- Berg, J.M.; Godwin, H.A. Lessons from zinc-binding peptides. Annu. Rev. Biophys. Biomol. Struct. 1997, 26:1, 357–371.
- Takatsuji, H. Zinc-Finger Transcription Factors in Plants. Cell. Mol. Life Sci. 1998, 54, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Takatsuji, H. Zinc-Finger Proteins: The Classical Zinc Finger Emerges in Contemporary Plant Science. Plant Mol. Biol. 1999, 39, 1073–1078. [Google Scholar] [CrossRef]
- Lee, M.I.N.S.; Gippert, G.P.; Soman, K.V.; Case, D.A.; Wright, P.E. Three-Dimensional Solution Structure of a Single Zinc Finger DNA-Binding Domain. Science 1989, 245, 635–637. [Google Scholar] [CrossRef]
- Kochoyan, M.; Keutmannt, H.T.; Weiss, M.A. Architectural Rules of the Zinc-Finger Motif: Comparative Two-Dimensional NMR Studies of Native and “Aromatic-Swap” Domains Define. Proc. Natl. Acad. Sci. USA 1991, 88, 8455–8459. [Google Scholar] [CrossRef]
- Michael, S.F.; Kilfoil, V.J.; Schmidt, M.H.; Amann, B.T.; Berg, J.M. Metal Binding and Folding Properties of a Minimalist Cys2His2 Zinc Finger Peptide. Proc. Natl. Acad. Sci. USA 1992, 89, 4796–4800. [Google Scholar] [CrossRef] [PubMed]
- Fairall, L.; Schwabe, J.W.R.; Chapman, L.; Finch, J.T.; Rhodes, D. The Crystal Structure of a Two Zinc-Finger Peptide Reveals an Extension to the Rules for Zinc-Finger/DNA Recognition. Nature 1993, 366, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Elrod-Erickson, M.; Rould, M.A.; Nekludova, L.; Pabo, C.O. Zif268 Protein-DNA Complex Refined at 1.6 Å: A Model System for Understanding Zinc Finger-DNA Interactions. Structure 1996, 4, 1171–1180. [Google Scholar] [CrossRef]
- Houbaviy, H.B.; Usheva, A.; Shenk, T.; Burley, S.K. Cocrystal Structure of YY1 Bound to the Adeno-Associated Virus P5 Initiator. Proc. Natl. Acad. Sci. USA 1996, 93, 13577–13582. [Google Scholar] [CrossRef]
- Omichinski, J.G.; Pedone, P.V.; Felsenfeld, G.; Gronenborn, A.M.; Clore, G.M. The Solution Structure of a Specific GAGA Factor-DNA Complex Reveals a Modular Binding Mode. Nat. Struct. Biol. 1997, 4, 122–132. [Google Scholar] [CrossRef]
- Bowers, P.M.; Schaufler, L.E.; Klevit, R.E. A Folding Transition and Novel Zinc Finger Accessory Domain in the Transcription Factor ADR1. Nat. Struct. Biol. 1999, 6, 478–485. [Google Scholar] [PubMed]
- Berg, J.M. Zinc Finger Domains: From Predictions to Design. Acc. Chem. Res. 1995, 28, 14–19. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Pabo, C. Zinc Finger-DNA Recognition: Crystal Structure of a Zif268-DNA Complex at 2.1 A. Science 1991, 252, 809–817. [Google Scholar] [CrossRef]
- Jacobs, G.H. Determination of the Base Recognition Positions of Zinc Fingers from Sequence Analysis. EMBO J. 1992, 11, 4507–4517. [Google Scholar] [CrossRef]
- Omichinski, J.G.; Clore, G.M.; Schaad, O.; Felsenfeld, G.; Trainor, C.; Appella, E.; Stahl, S.J.; Gronenborn, A.M. NMR Structure of a Specific DNA Complex of Zn-Containing DNA Binding Domain of GATA-1. Science 1993, 261, 438–446. [Google Scholar] [CrossRef]
- Takatsuji, H.; Nakamura, N.; Katsumoto, Y. A New Family of Zinc Finger Proteins in Petunia: Structure, DNA Sequence Recognition, and Floral Organ-Specific Expression. Plant Cell 1994, 6, 947–958. [Google Scholar] [PubMed]
- Ciftci-Yilmaz, S.; Mittler, R. The Zinc Finger Network of Plants. Cell. Mol. Life Sci. 2008, 65, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Takatsuji, H. A Single Amino Acid Determines the Specificity for the Target Sequence of Two Zinc-Finger Proteins in Plants The EPF Family Is a Group of DNA-Binding Proteins with Two Canonical Cys 2 / His 2 Zinc- Expressed in a Floral Organ-Specific Manner That Is Corre. Biochem. Biophys. Res. Commun. 1996, 223, 219–223. [Google Scholar] [CrossRef]
- Turquetti-Moraes, D.K.; Moharana, K.C.; Almeida-Silva, F.; Pedrosa-Silva, F.; Venancio, T.M. Integrating Omics Approaches to Discover and Prioritize Candidate Genes Involved in Oil Biosynthesis in Soybean. Gene 2022, 808. [Google Scholar] [CrossRef]
- Finkelstein, R.R. Mutations at Two New Arabidopsis ABA Response Loci Are Similar to the Abi3 Mutations. Plant J. 1994, 5, 765–771. [Google Scholar] [CrossRef]
- Bäumlein, H.; Miséra, S.; Luerßen, H.; Kölle, K.; Horstmann, C.; Wobus, U.; Müller, A.J. The FUS3 Gene of Arabidopsis Thaliana Is a Regulator of Gene Expression during Late Embryogenesis. Plant J. 1994, 6, 379–387. [Google Scholar] [CrossRef]
- Monfared, M.M.; Simon, M.K.; Meister, R.J.; Roig-Villanova, I.; Kooiker, M.; Colombo, L.; Fletcher, J.C.; Gasser, C.S. Overlapping and Antagonistic Activities of BASIC PENTACYSTEINE Genes Affect a Range of Developmental Processes in Arabidopsis. Plant J. 2011, 66, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Burnham, C.R.; Yagyu, P. Linkage Relations of Teosinte Branched. Maize Genet. Coop. Newsl. 1961. [Google Scholar]
- Pekker, I.; Alvarez, J.P.; Eshed, Y. Auxin Response Factors Mediate Arabidopsis Organ Asymmetry via Modulation of KANADI Activity. Plant Cell 2005, 17, 2899–2910. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Wang, L.J.; Mao, Y.B.; Cai, W.J.; Xue, H.W.; Chen, X.Y. Control of Root Cap Formation by MicroRNA-Targeted Auxin Response Factors in Arabidopsis. Plant Cell 2005, 2005 17, 2204–2216. [Google Scholar] [CrossRef]
- Murmu, J.; Bush, M.J.; de Long, C.; Li, S.; Xu, M.; Khan, M.; Malcolmson, C.; Fobert, P.R.; Zachgo, S.; Hepworth, S.R. Arabidopsis Basic Leucine-Zipper Transcription Factors TGA9 and TGA10 Interact with Floral Glutaredoxins ROXY1 and ROXY2 and Are Redundantly Required for Anther Development. Plant Physiol. 2010, 154, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Song, Y.H.; Josephson-Day, A.R.; Miller, R.J.; Breton, G.; Olmstead, R.G.; Imaizumi, T. FLOWERING BHLH Transcriptional Activators Control Expression of the Photoperiodic Flowering Regulator CONSTANS in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 3582–3587. [Google Scholar] [CrossRef]
- Miura, K.; Ikeda, M.; Matsubara, A.; Song, X.J.; Ito, M.; Asano, K.; Matsuoka, M.; Kitano, H.; Ashikari, M. OsSPL14 Promotes Panicle Branching and Higher Grain Productivity in Rice. Nat. Genet. 2010, 42, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Son, O.; Hur, Y.S.; Kim, Y.K.; Lee, H.J.; Kim, S.; Kim, M.R.; Nam, K.H.; Lee, M.S.; Kim, B.Y.; Park, J.; et al. ATHB12, an ABA-Inducible Homeodomain-Leucine Zipper (HD-Zip) Protein of Arabidopsis, Negatively Regulates the Growth of the Inflorescence Stem by Decreasing the Expression of a Gibberellin 20-Oxidase Gene. Plant Cell Physiol. 2010, 51, 1537–1547. [Google Scholar] [CrossRef]
- Scortecci, K.C.; Michaels, S.D.; Amasino, R.M. Identification of a MADS-Box Gene, FLOWERING LOCUS M, That Represses Flowering. Plant J. 2001, 26, 229–236. [Google Scholar] [CrossRef]
- Dorca-Fornell, C.; Gregis, V.; Grandi, V.; Coupland, G.; Colombo, L.; Kater, M.M. The Arabidopsis SOC1-like Genes AGL42, AGL71 and AGL72 Promote Flowering in the Shoot Apical and Axillary Meristems. Plant J. 2011, 67, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; Panigrahi, K.C.S.; Gissot, L.; Sauerbrunn, N.; Rühl, M.; Jarillo, J.A.; Coupland, G. Arabidopsis DOF Transcription Factors Act Redundantly to Reduce CONSTANS Expression and Are Essential for a Photoperiodic Flowering Response. Dev. Cell 2009, 17, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Palatnik, J.F.; Allen, E.; Wu, X.; Schommer, C.; Schwab, R.; Carrington, J.C.; Weigel, D. Control of Leaf Morphogenesis by MicroRNAs. Nature 2003, 425, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, L.; Wang, W.; Tian, P.; Wang, W.; Wang, K.; Gao, Z.; Liu, S.; Zhang, Y.; Irish, V.F.; et al. TCP5 Controls Leaf Margin Development by Regulating KNOX and BEL-like Transcription Factors in Arabidopsis. J. Exp. Bot. 2021, 72, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Omidbakhshfard, M.A.; Fujikura, U.; Olas, J.J.; Xue, G.P.; Balazadeh, S.; Mueller-Roeber, B. GROWTH-REGULATING FACTOR 9 Negatively Regulates Arabidopsis Leaf Growth by Controlling ORG3 and Restricting Cell Proliferation in Leaf Primordia. PLoS Genet. 2018, 14, e1007484. [Google Scholar] [CrossRef] [PubMed]
- Kubo, H.; Peeters, A.J.M.; Aarts, M.G.M.; Pereira, A.; Koornneef, M. ANTHOCYANINLESS2, a Homeobox Gene Affecting Anthocyanin Distribution and Root Development in Arabidopsis. Plant Cell 1999, 11, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Aida, M.; Beis, D.; Heidstra, R.; Willemsen, V.; Blilou, I.; Galinha, C.; Nussaume, L.; Noh, Y.S.; Amasino, R.; Scheres, B. The PLETHORA Genes Mediate Patterning of the Arabidopsis Root Stem Cell Niche. Cell 2004, 119, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, H.; Wu, C.; Feng, J.; Liu, X.; Qin, H.; Wang, D. Overexpressing HRS1 Confers Hypersensitivity to Low Phosphate-Elicited Inhibition of Primary Root Growth in Arabidopsis Thaliana. J. Integr. Plant Biol. 2009, 51, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, J.Y.; Cho, D.I.; Park, J.H.; Soo, Y.K. ABF2, an ABRE-Binding BZIP Factor, Is an Essential Component of Glucose Signaling and Its Overexpression Affects Multiple Stress Tolerance. Plant J. 2004, 40, 75–87. [Google Scholar] [CrossRef]
- Kang, H.G.; Singh, K.B. Characterization of Salicylic Acid-Responsive, Arabidopsis Dof Domain Proteins: Overexpression of OBP3 Leads to Growth Defects. Plant J. 2000, 21, 329–339. [Google Scholar] [CrossRef]
- Akhtar, M.; Jaiswal, A.; Taj, G.; Jaiswal, J.P.; Qureshi, M.I.; Singh, N.K. DREB1/CBF Transcription Factors: Their Structure, Function and Role in Abiotic Stress Tolerance in Plants. J. Genet. 2012, 91, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.Y.; DeFalco, T.A.; Moeder, W.; Li, B.; Gong, Y.; Liu, X.M.; Taniguchi, M.; Lumba, S.; Toh, S.; Shan, L.; et al. Arabidopsis ETHYLENE RESPONSE FACTOR 8 (ERF8) Has Dual Functions in ABA Signaling and Immunity. BMC Plant Biol. 2018, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sheerin, D.J.; von Roepenack-Lahaye, E.; Stahl, M.; Hiltbrunner, A. The Phytochrome Interacting Proteins ERF55 and ERF58 Repress Light-Induced Seed Germination in Arabidopsis Thaliana. Nat. Commun. 2022, 13, 1656. [Google Scholar] [CrossRef]
- Heyman, J.; Cools, T.; Canher, B.; Shavialenka, S.; Traas, J.; Vercauteren, I.; Van Den Daele, H.; Persiau, G.; De Jaeger, G.; Sugimoto, K.; et al. The Heterodimeric Transcription Factor Complex ERF115-PAT1 Grants Regeneration Competence. Nat. Plants 2016, 2016 2, 16165. [Google Scholar] [CrossRef]
- Hao, Y.J.; Wei, W.; Song, Q.X.; Chen, H.W.; Zhang, Y.Q.; Wang, F.; Zou, H.F.; Lei, G.; Tian, A.G.; Zhang, W.K.; et al. Soybean NAC Transcription Factors Promote Abiotic Stress Tolerance and Lateral Root Formation in Transgenic Plants. Plant J. 2011, 68, 302–313. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, B.; Lu, G.; Han, B. Overexpression of a NAC Transcription Factor Enhances Rice Drought and Salt Tolerance. Biochem. Biophys. Res. Commun. 2009, 379, 985–989. [Google Scholar] [CrossRef]
- Balazadeh, S.; Siddiqui, H.; Allu, A.D.; Matallana-Ramirez, L.P.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Köhler, B.; Mueller-Roeber, B. A Gene Regulatory Network Controlled by the NAC Transcription Factor ANAC092/AtNAC2/ORE1 during Salt-Promoted Senescence. Plant J. 2010, 62, 250–264. [Google Scholar] [CrossRef]
- Takagi, H.; Tamiru, M.; Abe, A.; Yoshida, K.; Uemura, A.; Yaegashi, H.; Obara, T.; Oikawa, K.; Utsushi, H.; Kanzaki, E.; et al. MutMap Accelerates Breeding of a Salt-Tolerant Rice Cultivar. Nat. Biotechnol. 2015, 33, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Lai, Z.; Fan, B.; Chen, Z. Arabidopsis WRKY38 and WRKY62 Transcription Factors Interact with Histone Deacetylase 19 in Basal Defense. Plant Cell 2008, 20, 2357–2371. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Lin, G.; Wang, A.; Wang, Z.; Lu, G. Overexpression of AtWRKY28 and AtWRKY75 in Arabidopsis Enhances Resistance to Oxalic Acid and Sclerotinia Sclerotiorum. Plant Cell Rep. 2013, 32, 1589–1599. [Google Scholar] [CrossRef]
- Jia, J.; XinG, J. hong; Dong, J. gao; Han, J. min; Liu, J. sheng. Functional Analysis of MYB73 of Arabidopsis Thaliana Against Bipolaris Oryzae. Agric. Sci. China 2011, 10, 721–727. [Google Scholar] [CrossRef]
Figure 1.
SUP evolution and the origin of RA1. Mayority rule consensus tree (N=22502 trees) of RA1 transcription factor in embryophytes generated by Bayesian inference using 73 peptide sequences (Figure S1 and Table S1). Black dots indicate Bayesian Posterior Probability (PP) = [0.9 to 1]. Green arrows indicate gene duplication events. Black asterisk points out proteins with known function (Table S2).
Figure 1.
SUP evolution and the origin of RA1. Mayority rule consensus tree (N=22502 trees) of RA1 transcription factor in embryophytes generated by Bayesian inference using 73 peptide sequences (Figure S1 and Table S1). Black dots indicate Bayesian Posterior Probability (PP) = [0.9 to 1]. Green arrows indicate gene duplication events. Black asterisk points out proteins with known function (Table S2).

Figure 2.
Evolution of RA1 in grasses. (a) Mayority rule consensus tree (N=22502 trees) of RA1 and RA1-like transcription factors in grass species generated by Bayesian inference using 35 amino acid sequences (Figure S2, Figure S3 and Table S1). Each clade is identified by a color. Black dots indicate Bayesian Posterior Probability (PP) = [0.9 to 1]. Black asterisk points out proteins with known function (Table S3). (b) Motif distribution patterns on RA1 and RA1-like amino acid sequences in grass species. Colored boxes represent motif occurrence (Figure S7 and Figure S8). (c) Sequence representation logo of Motif 1 obtained from the multiple sequence alignment (Figure S9). Black arrowhead indicates the amino acid variant (A->G) in Motif 1.
Figure 2.
Evolution of RA1 in grasses. (a) Mayority rule consensus tree (N=22502 trees) of RA1 and RA1-like transcription factors in grass species generated by Bayesian inference using 35 amino acid sequences (Figure S2, Figure S3 and Table S1). Each clade is identified by a color. Black dots indicate Bayesian Posterior Probability (PP) = [0.9 to 1]. Black asterisk points out proteins with known function (Table S3). (b) Motif distribution patterns on RA1 and RA1-like amino acid sequences in grass species. Colored boxes represent motif occurrence (Figure S7 and Figure S8). (c) Sequence representation logo of Motif 1 obtained from the multiple sequence alignment (Figure S9). Black arrowhead indicates the amino acid variant (A->G) in Motif 1.
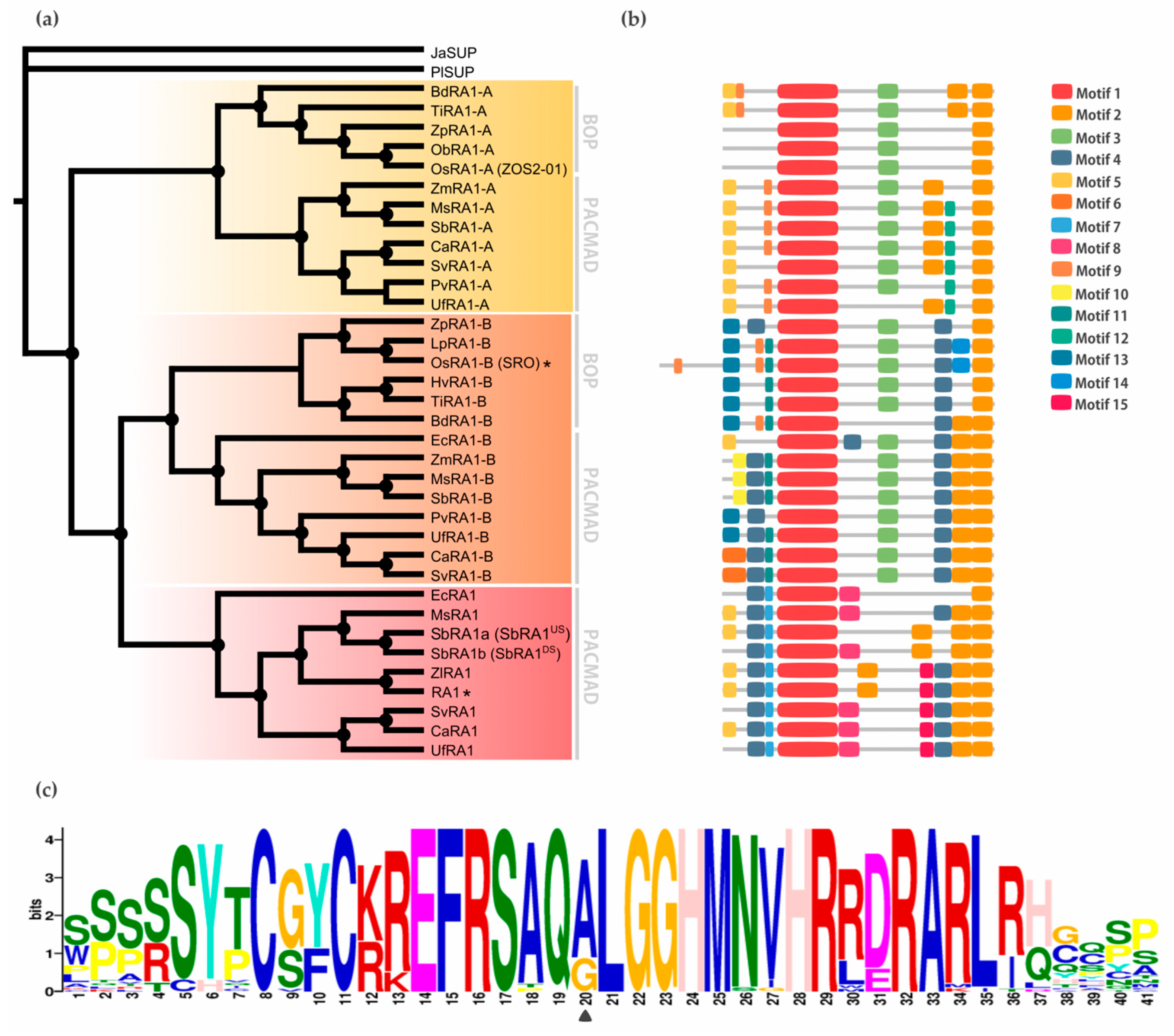
Figure 3.
Sub-cellular localization analysis of RA1 and RA1-like proteins. Sub-cellular localization of the RA1-GFP, SvRA1-GFP, SvRA1-like A-GFP, and SvRA1-like B-GFP constructs in tobacco leaf epidermal cells. Green color is GFP protein signal. Blue color represents DAPI stained nucleus.
Figure 3.
Sub-cellular localization analysis of RA1 and RA1-like proteins. Sub-cellular localization of the RA1-GFP, SvRA1-GFP, SvRA1-like A-GFP, and SvRA1-like B-GFP constructs in tobacco leaf epidermal cells. Green color is GFP protein signal. Blue color represents DAPI stained nucleus.
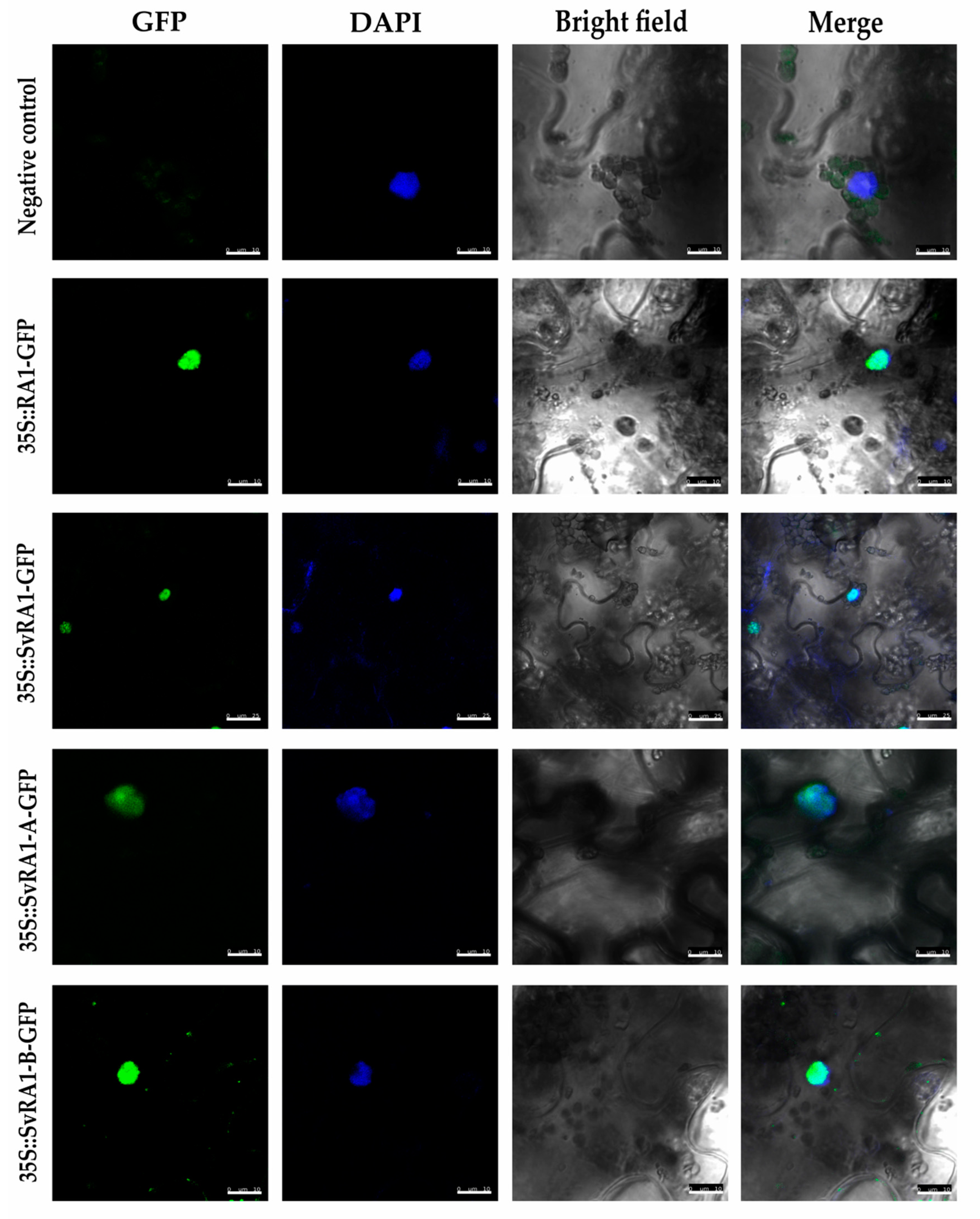
Figure 4.
Proteins secondary structure and conformational plasticity. (a) Secondary structure prediction of SUP, ZmRA1-like A, ZmRA1-like B, and RA1 proteins obtained by PsiPred server. α-helix corresponding to the zinc finger domain is represented in orange. α-helix corresponding to Motif 2, putative EAR motif, is represented in violet. Other α-helices are represented in pink. β-strands are represented in yellow. (b) Relative disorder levels of the structures measured in a range of 0 to 1.0 (Figure S10 and Table S4). Levels above 0.5 (dashed line) are considered disordered regions. Abbreviations: ZF, zinc finger domain; EAR, EAR repressor motifs.
Figure 4.
Proteins secondary structure and conformational plasticity. (a) Secondary structure prediction of SUP, ZmRA1-like A, ZmRA1-like B, and RA1 proteins obtained by PsiPred server. α-helix corresponding to the zinc finger domain is represented in orange. α-helix corresponding to Motif 2, putative EAR motif, is represented in violet. Other α-helices are represented in pink. β-strands are represented in yellow. (b) Relative disorder levels of the structures measured in a range of 0 to 1.0 (Figure S10 and Table S4). Levels above 0.5 (dashed line) are considered disordered regions. Abbreviations: ZF, zinc finger domain; EAR, EAR repressor motifs.
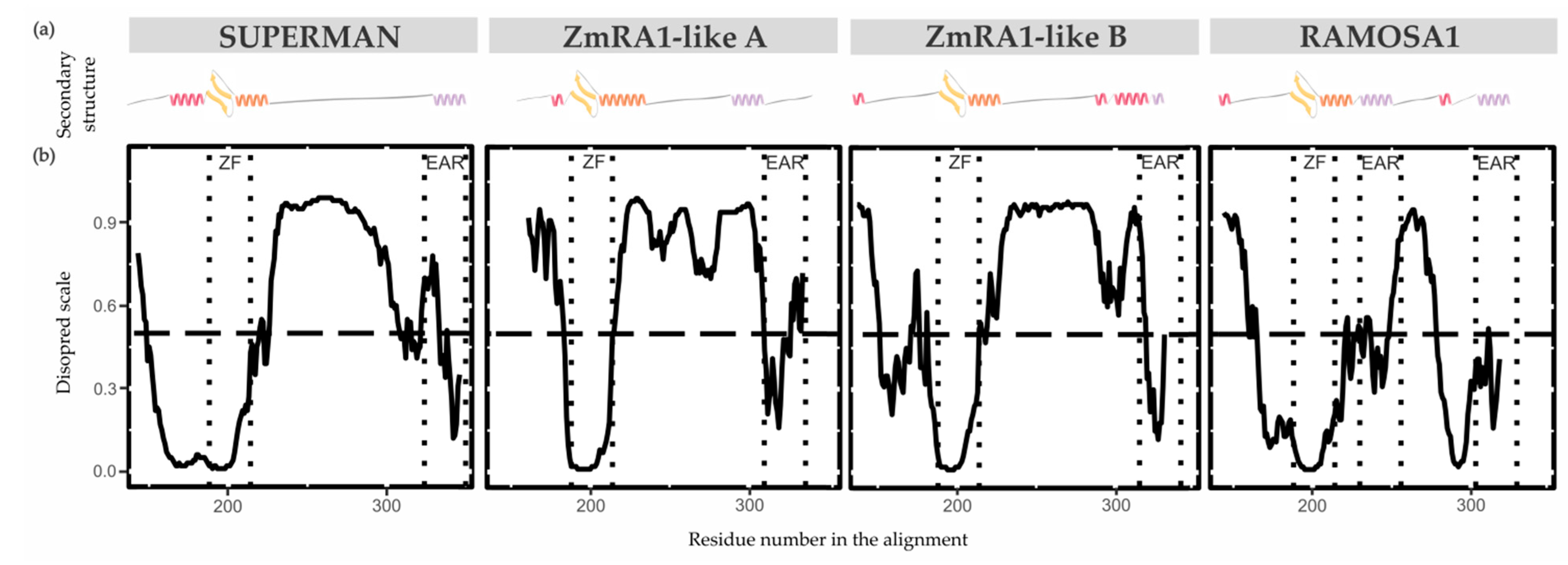
Figure 5.
Zinc finger molecular dynamics simulation. (a) Time evolution of the secondary structure of SUP and RA1 proteins as determined by the DSSP algorithm using the simulated structures, in the thermodynamics equilibrium interval (0.8 to 1.0 μs). At the side of the graph, the amino acid sequences are quoted with their residue number in the proteins. In the center of the graph, the relative position of each residue referenced to the first one of the α-helix N-terminal is indicated. (b) RMSF of the backbone atoms of each amino acid sequence in the equilibrium interval (0.8 to 1.0 μs) taking as reference for fitting the structures the one with smallest RMSD in the same period.
Figure 5.
Zinc finger molecular dynamics simulation. (a) Time evolution of the secondary structure of SUP and RA1 proteins as determined by the DSSP algorithm using the simulated structures, in the thermodynamics equilibrium interval (0.8 to 1.0 μs). At the side of the graph, the amino acid sequences are quoted with their residue number in the proteins. In the center of the graph, the relative position of each residue referenced to the first one of the α-helix N-terminal is indicated. (b) RMSF of the backbone atoms of each amino acid sequence in the equilibrium interval (0.8 to 1.0 μs) taking as reference for fitting the structures the one with smallest RMSD in the same period.
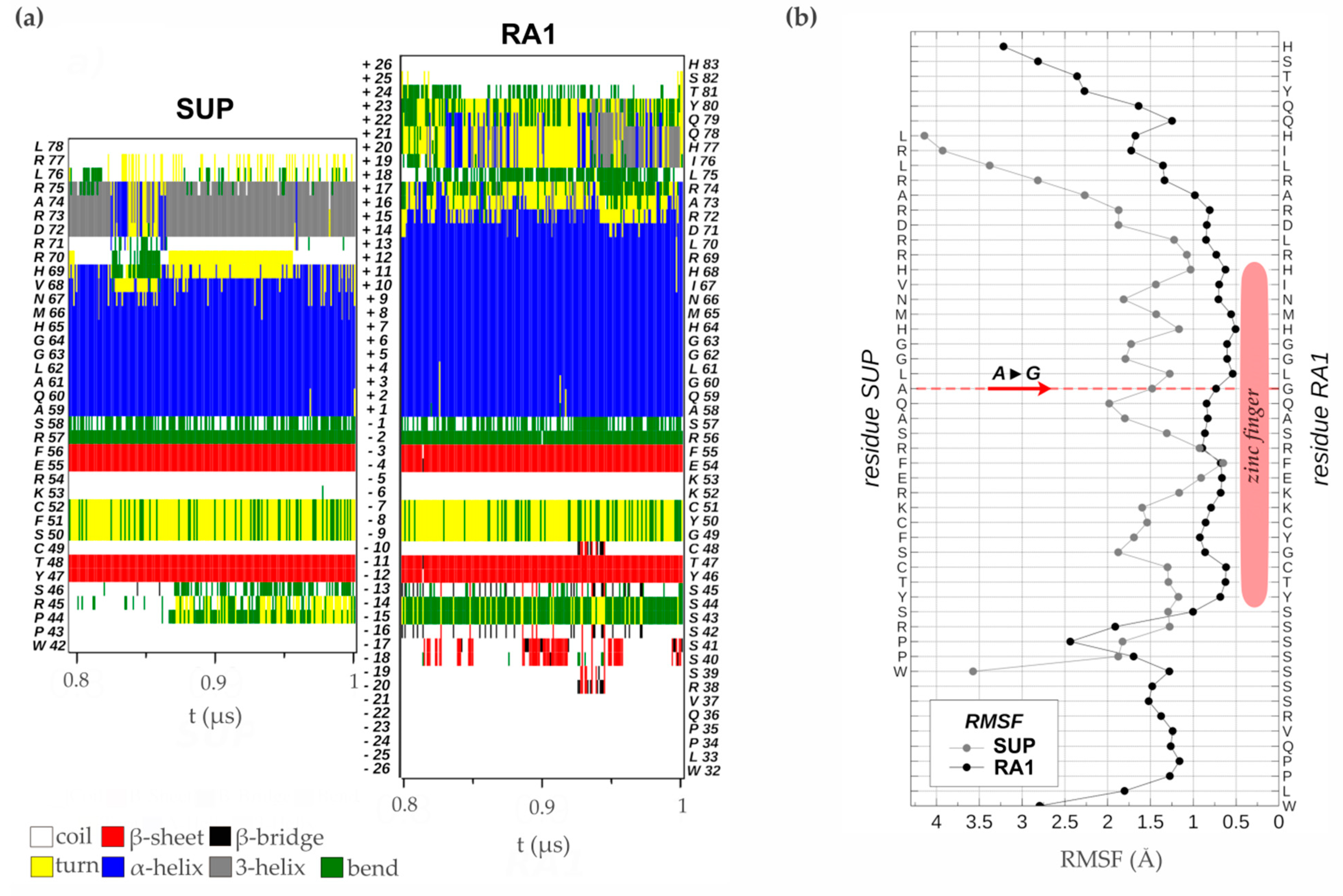
Figure 6.
Noncoding cis-elements identified in predicted promoter regions of RA1 and RA1-like sequences from grass species. Colored circles represent presence of conserved motifs in the promoter. Motif consensus sequences are showed in Table S5.
Figure 6.
Noncoding cis-elements identified in predicted promoter regions of RA1 and RA1-like sequences from grass species. Colored circles represent presence of conserved motifs in the promoter. Motif consensus sequences are showed in Table S5.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



