
Submitted:
14 April 2024
Posted:
15 April 2024
You are already at the latest version
Abstract
Effects of the enzyme N-acetylgalactosamine-4-sulfatase (Arylsulfatase B, ARSB), which removes the 4-sulfate group at the non-reducing end of chondroitin 4-sulfate, on the expression of PD-L1 were determined and the underlying mechanism of PD-L1 expression elucidated. Initial experiments in human melanoma cells (A375) showed PD-L1 expression increased from 357 ± 31 to 796 ± 50 pg/mg protein (p
Keywords:
arylsulfatase B
; chondroitin sulfate
; HDAC3
; galectin-3
; c-Jun
1. Introduction
Agents to modify checkpoint inhibition by PD-L1 (programmed death ligand 1) and its immune cell receptor (PD-1) are widely used to treat malignant melanoma and other malignancies. However, response to treatment is unpredictable and may not be sustained [1,2,3,4,5]. Treatment with exogenous activated Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase), the enzyme that removes 4-sulfate groups at the non-reducing end from chondroitin 4-sulfate (C4S), was recently shown to reduce tumor progression and improve the survival of C57BL/6J mice with B16F10 subcutaneous melanomas [6]. The potential impact of ARSB on PD-L1 expression and on PD-L1-mediated checkpoint therapy was unknown.
In prior work, decline in ARSB was associated with increasing invasiveness of human melanoma cell lines, as well as with more aggressive human prostate and colonic cancers and in association with malignant mammary cells [7,8,9,10,11]. As ARSB decreases, chondroitin 4-sulfation increases, leading to increased binding of SHP2 (PTP11; non-receptor tyrosine phosphatase) and reduced binding of galectin-3 with C4S [11,12,13]. Subsequent effects attributed to decline in SHP2 availability include increases in phospho-ERK1/2 and phospho-p38 MAPK and transcriptional effects, including by hypermethylation of the DKK3 promoter and reduced expression of DKK (Dickkopf WNT pathway signaling inhibitor) 3, leading to activation of Wnt signaling in prostate cells [14]. Other transcriptional events following silencing of ARSB and increased availability of galectin-3 include the increased expression of CSPG4 and CHST15 in melanoma cells [6,7], increased expression of versican in prostate epithelial cells [13], and increased expression of Wnt9A in colonic epithelial cells [15]. The transcriptional events initiated by changes in SHP2 or galectin-3 binding with C4S, which follow either decline or increase in ARSB activity, exerted profound effects on vital cell processes. The relationship between these ARSB-C4S-initiated transcriptional mechanisms and immune-mediated effects on melanoma tumor proliferation had not been addressed previously.
Initial experiments showed that silencing ARSB in melanoma cells increased the expression of PD-L1 mRNA and protein and, inversely, treatment by exogenous ARSB reduced PD-L1 expression in human and mouse melanoma cells, as well as in human prostate cell lines [16]. The experiments detailed in this report were performed to clarify the mechanism by which ARSB regulated PD-L1 expression in the subcutaneous B16F10 mouse melanomas, in human A375 melanoma cells, and in normal human melanocytes.
Reports in the literature have presented several different transcriptional mechanisms affecting PD-L1 expression, including epigenetic mechanisms involving methylations and histone deacetylations, as well as transcription factors [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. HDACs 3, 6, and 8 have been implicated in effects on histone acetylation in melanoma [18,24,30,31,32,34,35]. Changes in ARSB or chondroitin-4 sulfate had not previously been associated with regulation of histone acetylation and HDACs. The findings which follow support the impact of ARSB and chondroitin 4-sulfation on galectin-3, c-jun, HDAC3, and H3 on the regulation of PD-L1 expression in melanoma and suggest that attention to ARSB may help to inform treatment decisions about checkpoint inhibition.
2. Results
2.1. Exogenous ARSB Reduces and ARSB siRNA Increases PD-L1 Expression
PD-L1 was measured in tumor tissue from B16F10 subcutaneous melanomas in C57Bl/6J mice which were treated by local injection of exogenous ARSB (rhARSB 0.2 mg/kg on days 2, 7, and 14 following tumor inoculation. PD-L1 protein and mRNA expression declined compared to saline treated controls (Figure 1A,1B). Measurements of PD-L1 protein and mRNA were performed following ARSB silencing or exposure to rhARSB (1 ng/ml x 24h) in cultured normal melanocytes (Figure 1C,1D) and in A375 cells (Figure 1E,1F). Consistently, ARSB silencing increased PD-L1 expression and PD-L1 expression declined following exogenous ARSB (p<0.0001, p<0.0001; unpaired t-test, two-tailed, unequal variance). Images of A375 cells confirm decline in PD-L1 immunostaining following exogenous ARSB (Figure 1G) compared to untreated control (Figure 1H). Also, marked increase in PD-L1 immunostaining is evident following ARSB silencing (Figure 1I), compared to control siRNA (Figure 1J).
The reduction of PD-L1 expression following exogenous ARSB in the normal melanocytes (Figure 1K) and the A375 cells (Figure 1L) was inhibited by silencing of the IGF2 receptor (IGF2R), but not by the mannose-6-phosphate receptor (M6PR). This suggests that exogenous ARSB acts through a calcium-independent receptor in these cell lines [39].
2.2. Exogenous ARSB Reduces Free Galectin-3 Due to Increased Binding with Chondroitin 4-Sulfate
The mechanism by which PD-L1 expression was regulated by ARSB was evaluated in the following experiments, based on prior findings of changes in free galectin-3, due to ARSB-induced alteration in C4S-galectin-3 binding [11,13,15,40]. Free galectin-3 was reduced in the melanoma tissue of the C57BL/6J mice (Figure 2A), following treatment with exogenous ARSB (p<0.0001). This is attributable to increased binding of galectin-3 with chondroitin 4-sulfate (C4S) (Figure 2B) after treatment with bioactive, exogenous ARSB (p<0.0001) and is consistent with previous findings showing decline in galectin-3 binding to more sulfated C4S present when ARSB activity is less. Experiments in A375 cells (Figure 2C) and in normal human melanocytes (M0) (Figure 2D) indicate increases in free galectin-3 when ARSB is silenced (p<0.0001) and declines following exposure to exogenous galectin-3 (p<0.0001). Galectin-3 was effectively silenced by siRNA in the M0 and A375 cells (Figure 2C,2D).
In contrast to the observed binding of galectin-3 with chondroitin 4-sulfate, the quantity of PD-L1 which co-immunoprecipitated with chondroitin sulfate (CS) in the A375 cells was negligible (0.10 pg/mg CS), with no significant difference following ARSB siRNA or exogenous ARSB.
2.3. Impact of Galectin-3 on Phospho-(Thr183/Tyr185)-JNK and Nuclear c-jun
Galectin-3 is known to enhance the activation of AP-1 [13,41], so the impact of exogenous ARSB on DNA-bound c-Jun and on c-Jun N-terminal kinase 1 (JNK) was addressed in the melanoma tumor tissue and in the normal melanocyte (M0) and A375 cells. In the tumor tissue, both phospho-(Thr185/Tyr185)-JNK and nuclear c-jun decreased following treatment with exogenous ARSB (Figure 3A,3B) (p<0.0001, p<0.0001). In the B16F10 tumors, phospho-JNK declined by ~41% and nuclear c-jun by ~45%. In the A375 (Figure 3C,3D) and normal melanocyte cell lines (Figure 3E,3F), galectin-3 silencing blocked the ARSB siRNA-induced increases in phospho-JNK and c-Jun. Exogenous rhARSB significantly reduced phospho-JNK and c-Jun in the cell lines (p<0.0001, p<0.0001). The c-Jun mimetic peptide cJP and the c-Jun inhibitory peptide JIP1 both inhibited control siRNA and ARSB siRNA-induced increases in DNA-bound c-Jun in the A375 cells. PHPS1, a small molecule chemical inhibitor of the SHP2 (PTPN11), did not inhibit the ARSB siRNA-induced increase in phospho-JNK or DNA-bound c-Jun in the M0 and A375 cells. The IGF2R siRNA almost completely reversed the rhARSB-induced decline in DNA-bound c-Jun in the A375 (Figure 3G) and M0 (Figure 3H) cells.
2.4. HDAC3 Activity and Expression Modulated by ARSB and Galectin-3
Reports of mechanisms linking c-Jun and HDAC3 in the regulation of transcription [25,42,43] instigated experiments to address the impact of exogenous ARSB and ARSB silencing on HDAC3 expression and activity. In the subcutaneous mouse melanoma tissue, HDAC3 activity increased following exogenous ARSB (p<0.0001) (Figure 4A), in contrast to the decline in free galectin-3 (Figure 2B). In the A375 and M0 cells, HDAC3 activity decreased when ARSB was silenced and increased following exogenous ARSB (Figure 4B,4D). mRNA expression of HDAC decreased in the A375 cells when ARSB was silenced (Figure 4C). The c-Jun mimetic peptide cJP increased the baseline HDAC3 activity and expression. Galectin-3 silencing partially reversed the ARSB silencing associated decline in HDAC3; in contrast, PHPS1 had no effect. The HDAC3 inhibitor RGFP966 completely blocked the HDAC3 activity in the cells.
2.5. Regulation of PD-L1 Promoter by H3 Acetylation
To determine if the effects on HDAC3 affected H3 acetylation of the PD-L1 promoter, the promoter was probed for H3 acetylation by ChIP assay using two sets of primers following immunoprecipitation of DNA oligonucleotides by acetyl-H3 antibody using A375 cell extracts treated by IgG control, control siRNA, ARSB siRNA, or ARSB siRNA and cJP (Figure 5A). Differential effects are apparent, with reduced band intensity of PCR product following ARSB siRNA and increased intensity following ARSB siRNA with cJP (Figure 5A), as shown by optical density (Figure 5B). % Input confirms reduced acetyl-H3 binding to both promoter regions following ARSB silencing (Figure 5C,5D). These results indicate open chromatin following ARSB knockdown and decline in HDAC3, leading to enhanced transcription of PD-L1.
2.6. Impact of β-D-xyloside on Mediators of PD-L1 Expression
The impact of treatment by methyl-β-D-xylopyranoside (BDX) on free galectin-3 (Figure 6A), nuclear c-Jun (Figure 6B), HDAC3 activity (Figure 6C), and PD-L1 (Figure 6D) was examined in the A375 cells. BDX acts to inhibit construction of functional proteoglycans, by impairing attachment of chondroitin sulfate chains to the core protein of proteoglycans [44]. The experiments indicate some additional effect on free galectin-3, nuclear c-Jun, HDAC3 activity, and PD-L1 when ARSB is silenced and cells are treated with BDX.
2.7. Overall Pathway of PD-L1 Expression by ARSB
The pathway by which decline in ARSB increases PD-L1 expression and exogenous ARSB reduces PD-L1 expression involves galectin-3, nuclear c-Jun, HDAC3, and acetyl-H3, leading to effects on the responsiveness of the PD-L1 promoter. The impact of ARSB siRNA, exogenous ARSB, and inhibitors (including galectin-3, cJP, JIP1, RGFP966, and potential inhibitors (including PHPS1 and JQ1) on PD-L1 protein and mRNA in A375 and M0 cells is presented (Figure 7A-7E). The overall pathway is presented schematically in Figure 8.
3. Discussion
Initial measurements of programmed death ligand 1 (PD-L1) in human prostate cell lines, including metastatic PC-3 cells, normal prostate epithelial cells, and normal prostate stromal cells, showed that PD-L1 mRNA expression increased to 1.8 times, 2.2 times, and 1.6 times the baseline level when ARSB was silenced (Supplementary Figure 1a). Other measurements indicated that in mononuclear cells of participants with prediabetes in a dietary study of carrageenan withdrawal [45], PD-L1 expression declined by 62% in participants on the no-carrageenan diet over a 12-week period, as ARSB activity increased by 34% (Supplementary Figure 1b). These findings indicated significant impact of ARSB on PD-L1 expression in other human cells and are consistent with the findings in this report about the effects of both ARSB knockdown and exogenous, bioactive ARSB on PD-L1 expression through an epigenetic mechanism.
PD-L1 has emerged as a major focus in immune-mediated control of tumor proliferation. The PD-1 – PD-L1 binding between tumor-invading immune cells and tumor cells has been elucidated as a checkpoint which impedes other, potentially more cytotoxic, immune cell-tumor cell interactions from proceeding. Many excellent clinical responses arise from treatment by checkpoint inhibitors using antibodies directed at either PD-1 or PD-L1; however, responses may not endure, some patients do not respond, and treatment toxicity may be severe [1,2,3,4,5]. Recognition that exogenous ARSB reduces PD-L1 expression may help to focus interest and treatment on the impact of chondroitin sulfate- and sulfatase-mediated effects in cancer biology and on regulation of immune-mediated effects.
Recent work has demonstrated that exogenous, bioactive ARSB reduces progression and improves survival of mice with B16F10 subcutaneous melanomas is provocative [6], and further evaluation of the potential therapeutic benefit of exogenous ARSB treatment is of interest. Treatment benefit from rhARSB occurred at a dose of 0.2 mg/kg SQ on days 2, 7, and 14 post tumor inoculation, with no evidence of toxicity. Recombinant human ARSB is used successfully in enzyme replacement therapy of the congenital deficiency of ARSB, Mucopolysaccharidosis VI, by administration of a weekly dose of 1.0 mg/kg IV [46].
In cultured prostate stem and epithelial cells, effects of ARSB on signaling events, including increase in phospho-JNK, were attributed to enhanced binding of SHP2 with C4S when ARSB activity was reduced, leading to sustained phosphorylation of vital signaling molecules [12,13,14,15]. In the current experiments, the SHP2 inhibitor PHPS1 had no impact on PD-L1 expression, and did not affect nuclear c-Jun, phospho-JNK, or HDAC activity. The pathway by which phospho-JNK is modified in the melanoma cells appears to be independent of SHP2 and is not yet clarified. The epigenetic mechanism in the melanoma cells affecting PD-L1 expression resembles the previously observed mechanisms by which ARSB and chondroitin 4-sulfation affect galectin-3 and AP-1, as shown with transcriptional effects on HIF-1α and versican [11,13]. The impact of ARSB on chondroitin 4-sulfation and, thereby, on binding of galectin-3 and restriction of free galectin-3 leads to exogenous ARSB acting as an inhibitor of galectin-3. Inhibition of galectin-3 is under investigation as a treatment approach to interfere with PD-1-PD-L1 interaction in melanoma and other malignancies [47,48,49,50]. Additional, detailed investigations are required to elucidate further how ARSB-induced changes in C4S impact on galectin-3, c-Jun, HDACs, and histones, potentially regulating multiple transcriptional events sites and malignant progression.
4. Materials and Methods
4.1. Cell Culture
A375 human melanoma cells (CRL-1619, ATCC, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; ATCC), and 1% penicillin-streptomycin (ATCC). The cells were maintained at 37°C in a humidified, 5% CO2 environment with media exchange every 2 days. Confluent cells in T-25 flasks were harvested by EDTA-trypsin (ATCC), and sub-cultured. Primary normal melanocytes were cultured in Airway Epithelial cell basal medium (ATCC, Manassas, VA) with melanocyte growth kit (ATCC). The cells were maintained at 37°C in a humidified, 5% CO2 environment with media exchange every 3 days. Confluent cells in T-25 flasks were harvested by trypsin for primary cells (ATCC) and sub-cultured. B16F10 mouse melanoma cells were purchased (ATCC), and cells were cultured in DMEM supplemented with 10% FBS, and 1% penicillin-streptomycin. Cells were screened for pathogens by IDEXX BioAnalytics (Columbia, MO). Cells were maintained at 37°C in a humidified, 5% CO2 environment with media exchange every 2 days, and confluent cells in T-25 flasks were harvested by EDTA-trypsin, and sub-cultured.
4.2. Animal Procedures
Eight-week-old female C57BL/6J mice (n = 40) were purchased (Jackson Laboratories, Bar Harbor, Maine, USA) and housed in the Veterinary Medicine Unit at the Jesse Brown VA Medical Center (JBVAMC, Chicago, IL, USA) [6]. Principles of laboratory animal care were followed, and all procedures were approved by the AALAC accredited Animal Care Committee of the JBVAMC. Mice were fed a standard diet and maintained in groups of three in a cage with routine light–dark cycles. Mice were inoculated subcutaneously with 2.5 x 105 B16F10 mouse melanoma cells (ATCC) in 100 μl of normal saline. Treatment by injection of recombinant, human, bioactive ARSB, which was expressed in E. coli (R&D Systems, Minneapolis, MN, USA) or normal saline control injection was started 48 hours following tumor inoculation. Recombinant ARSB was diluted in sterile, normal saline and injected subcutaneously around the tumor with a 25-gauge needle at a dose of 0.2 mg/kg body weight on days 2, 7, and 14 [6]. Body weight and tumor volume were measured using calipers, and the volume was expressed in cm3 [0.5 × L × W2] (L=long diameter; W=short diameter of the tumor).
4.3. Treatment of A375 Human Melanoma Cells by Exogenous ARSB, siRNAs, and Other Agents
A375 cells were treated by exogenous, bioactive rhARSB (1 ng/ml x 24h; R&D, Biotechne). ARSB, galectin-3, mannose-6 phosphate receptor, and IGFII receptor were silenced by specific siRNA in the A375 cells at 70% confluence using standard procedures and verified siRNA (Oncogene, Thermofisher s7218, s8375). Media were exchanged after 24h, and cell treatments were initiated. Treatments were for 24h, unless indicated otherwise, and included:
cJP (400 μm with 2h pre-incubation and total 26h exposure; #1989, R&D Systems, Bio-Techne, Minneapolis, MN, USA), a cell-permeable c-Jun mimetic peptide, [36]; JIP-1 (10 μm with 2h pre-incubation and total 26h exposure; #1565, Tocris, Bio-Techne), an inhibitor of activated JNK based on interacting protein-1 [37]; RGFP966 (5 µM, SelleckChem, Houston, TX, USA), an HDAC3 inhibitor; PHPS1 (phenylhydrazonopyrazolone sulfonate or PTPN11, 30 μM; Sigma-Aldrich, St. Louis, MO, USA), a chemical inhibitor of SHP2, the tyrosine-protein phosphatase non-receptor type 11; JQ1 (10 µM, MedChem Express, Monmouth Junction, NJ, USA), a thienotriazolodiazepine and a potent inhibitor of the BET family of bromodomain proteins [38]; and methyl β-D-xylopyranoside (1 mM, #M5878, Sigma-Aldrich).
4.4. Treated and Control Cells Were Harvested and Frozen at -80°C for Subsequent Analysis
Small interfering (si) RNAs for ARSB (Qiagen, Germantown, MD, USA), galectin-3 (Qiagen), mannose-6-phosphate receptor (#s8375. Thermofisher) and IGF2R (#s7218, Thermofisher) were validated by QRT-PCR to confirm effective inhibition. The siRNA sequences for ARSB (NM_000046) silencing were: sense 5′- GGGUAUGGUCUCUAGGCA - 3′ and antisense: 5′- UUGCCUAGAGACCAUACCC - 3′. The sequence of the DNA template for human galectin-3 silencing (Hs_ LGALS3_9) was: 5΄ - ATGATGTTGCCTTCCACTTTA - 3′. A375 cells and normal melanocytes were grown to ~60% confluence, then silenced by adding 0.6 μl of 20 μM siRNA (150 ng), mixed with 100 μl of serum-free medium and 12 μl of HiPerfect Transfection Reagent (Qiagen).
4.5. Immunohistochemistry of PD-L1 in A375 Cells
A-375 human melanoma cells were plated in compartment slides and grown to 70–80 % confluence. Cells were treated with ARSB siRNA (Qiagen) or control siRNA (Qiagen) for 24 h or by recombinant human ARSB (1 ng/ml x 24 h). Preparations were then fixed for 2 h with 2 % paraformaldehyde and washed in 1×-PBS containing 1 mM calcium chloride (pH 7.4) for 5 min. Cells were then incubated with 5 % normal goat serum (#501972, Invitrogen, Thermo Fisher Scientific, Inc., Hanover Park, IL) for blocking and permeabilized with 0.08 % saponin in 1×-PBS/Ca++/saponin. Slides were incubated overnight with PD-L1 antibody (#13684, Cell Signaling, Danvers, MA, USA) at 4 °C, washed four times in 1×-PBS/Ca++/saponin, and then stained with secondary antibody rabbit anti-mouse FITC IgG (1:100, ab8517, Abcam) for 1 h. Slides were washed four times in 1×-PBS/Ca++/saponin and coverslipped using ProLong™ Gold anti-fade reagent with DAPI (P36941, Invitrogen) for nuclear staining. The fluorochromes were scanned using EVOS M5000 Imaging System (Thermo Fisher Scientific) at 20x. The captured TIFF images were exported for analysis and reproduction.
4.6. Arylsulfatase B (ARSB) Activity Assay
ARSB measurements were performed using a fluorometric assay, following a standard protocol with 20 μl of homogenate and 80 μl of assay buffer (0.05 M Na acetate buffer with 20 mM barium sulfate pH 5.6 at 37°C) with 100 μl of substrate (5mM 4-methylumbelliferyl sulfate in fresh assay buffer) in a black microplate, as previously reported [11].
4.7. Measurement of Total Sulfated Glycosaminoglycans (sGAG) and Chondroitin-4-sulfate
Total sulfated glycosaminoglycans (sGAG) were measured in the cell extracts by sulfated GAG assay (Blyscan™, Biocolor Ltd, Newtownabbey, Northern Ireland), as previously described [2]. Chondroitin sulfate (CS) or chondroitin 4-sulfate (C4S) in the samples was determined following immunoprecipitation by dynabeads (Life Technologies, Carlsbad, CA, USA) coated with a total CS antibody (CS-56, Abcam, Waltham, MA, USA) or C4S antibody (LY111, Amsbio, Cambridge, MA, USA). Beads were mixed with samples and incubated, and the immunoprecipitated CS molecules were eluted and subjected to the Blyscan sulfated GAG assay, as above.
4.8. Total Sulfotransferase Activity
Total Sulfotransferase activity was determined using the Universal Sulfotransferase Activity kit (R&D Systems, Minneapolis, MN, USA). The activity results were normalized using the total cellular protein and expressed as percentage of the control value.
4.9. ELISAs for Galectin-3, phospho-(Thr183/Tyr185)-JNK, PD-L1
Galectin-3 in the cell lysates was determined by a sandwich ELISA kit (R&D Systems) for human galectin-3. The wells of a microtiter plate were coated with specific anti-galectin-3 monoclonal antibody, and nonspecific sites were blocked by a blocking buffer with 1% bovine serum albumin (BSA). Sample galectin-3 captured into the microtiter wells was detected by biotin-conjugated secondary galectin-3 antibody and streptavidin-horseradish peroxidase (HRP). Hydrogen peroxide-tetramethylbenzidine (TMB) chromogenic substrate was used to develop the color, and color intensity was measured at 450 nm in an ELISA plate reader (FLUOstar, BMG Labtech, Inc., Cary, NC, USA). The galectin-3 concentrations were extrapolated from a standard curve, and sample values were normalized using total protein content. Galectin-3 in mouse tumor tissues was determined by a similar mouse sandwich ELISA kit (R&D Systems). Galectin-3 was also measured following immunoprecipitation of mouse tumor treated and control tissue lysates with the C4S antibody. Chondroitin 4-sulfate was immunoprecipitated from the cell lysates, as previously described, and the immunoprecipitate was eluted with dye-free elution buffer and subjected to mouse galectin-3 ELISA.
Cell extracts were prepared from both treated and control cells in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA; 9803S). Tumor tissues from treated and untreated animals were homogenized for the measurement of phospho-(Thr183/Tyr185)-JNK (c-Jun N-terminal kinase 1). Phospho-JNK was measured in cell and tissue samples using a DuoSet sandwich ELISA kit (R&D Systems). Samples and standards were added to the wells of the microtiter plate precoated with a capture antibody to human JNK. Phospho-JNK in the lysates was captured by the coated antibody on the plate and detected with biotinylated antibody to phospho-JNK. Streptavidin-HRP and hydrogen peroxide/TMB substrate were used to develop color which was read at 450 nm in a plate reader (FLUOstar). Phospho-JNK concentrations in the samples were extrapolated from a curve derived using known standards and expressed as % control.
Cell extracts were prepared from both treated and untreated control cells in cell lysis buffer. Malignant tumor issues from treated and untreated mice were homogenized. PD-L1 was measured in cell and tissue samples using a DuoSet sandwich ELISA kit (R&D Systems). Samples and standards were added to the wells of the microtiter plate precoated with a capture antibody to human/mouse PD-L1. PD-L1 in the lysates was captured by the coated antibody on the plate and detected with biotinylated antibody to PD-L1. Streptavidin-HRP and hydrogen peroxide/TMB substrate were used to develop color proportional to the bound HRP activity. The reaction was stopped, and the optical density of the color was read at 450 nm in a plate reader (FLUOstar). PD-L1 concentrations in the samples were extrapolated from a curve derived using known standards. PD-L1 was also measured by ELISA (Abcam ab278124) following immunoprecipitation with chondroitin sulfate antibody (CS-56, Abcam), which reacts with C4S and chondroitin 6-sulfate.
4.10. Oligonucleotide-based ELISA to Detect Nuclear c-Jun
Oligonucleotide binding assay (TransAM Kit, Active Motif, Carlsbad, CA) was used to detect nuclear c-Jun in the rhARSB-treated and control mouse melanoma tissue, in A375 melanoma cells and in normal melanocyte cells. Nuclear extracts were prepared using a nuclear extract preparation kit (Active Motif, Carlsbad, CA, USA), and were added to the wells of a 96-well microtiter plate, pre-coated with the AP-1 consensus oligonucleotide sequence (5′-TGAGTCA-3′). The bound c-Jun was recognized by a specific c-Jun primary antibody and finally detected by HRP-conjugated secondary antibody. Color was developed, using hydrogen peroxide/TMB substrate, and was proportional to the activity of the bound HRP. The reaction was stopped by acid stop solution, and the optical density of the color was read at 450 nm in a plate reader (FLUOstar). Sample values were normalized by total cell protein and expressed as percent of untreated control.
4.11. HDAC3 Activity and Expression
Histone Deacetylase (HDAC)3 in the tissue and cell samples was determined following immunoprecipitation with a specific HDAC3 antibody (Rabbit mAb #85057, Cell Signaling Technology, Inc., Danvers, MA 01923). Dynabeads (Life Technologies, Carlsbad, CA) were coated with specific HDAC3 (D201K) antibody, and beads were mixed with samples, incubated, and immunoprecipitated. Immunoprecipitated HDAC3 molecules were eluted and subjected to HDAC Activity Assay (ab156064, Abcam, Cambridge, MA, USA). Assay buffer and substrate peptide were added to the wells of a black microtiter plate. Reactions were initiated by adding 5 μl buffer for the no enzyme control assay or the enzyme sample to each well, mixed thoroughly, and incubated for 20 minutes at room temperature (RT). 20 μL of stop solution were added to each well of the microtiter plate and mixed thoroughly, before adding 5 μL of developer to each well and mixed thoroughly. Following incubation for 20 min at RT, fluorescence intensity was read at Ex/Em = 350 – 380 nm / 440 – 460 nm. HDAC3 activity was expressed as % control.
4.12. mRNA Expression of HDAC3 and PDL1
Total RNA was prepared from treated and control cells using an RNeasy Mini Kit (Qiagen, Germantown, MD, USA). Equal amounts of purified RNAs from the control and treated cells were reverse-transcribed and amplified using Brilliant SYBR Green QRT-PCR Master Mix (BIO-RAD, Hercules, CA, USA). Human β-actin was used as an internal control. QRT-PCR was performed using the following specific primers-
PDL1 human (NM_014143) forward: 5′-TTTACTGTCACGGTTCCCAAG-3′ and reverse: 5′-GCTGAACCTTCAGGTCTTCCT-3′;
PDL1 mouse (NM_021893) forward: 5′-AGTTTGTGGCAGGAGAGGAG-3′ and reverse: 5′-CTGGTTGATTTTGCGGTATG-3′.
HDAC3 human (NM_003880) forward: 5′-GAGTTCTGCTCGCGTTACACAG-3′ and reverse: 5′-CGTTGACATAGCAGACAGAG-3′;
HDAC3 mouse (NM_010411) forward: 5′-AACCTCATCGCCTGGCATTGAC-3′ and reverse: 5′-GTAGTCCTCAGAGAAGCGG-3′;
4.13. Chromatin Immunoprecipitation (ChIP) Assay for Histone 3-Acetylation
Chromatin immunoprecipitation assays were performed using the Acetyl-Histone H3 Immunoprecipitation (ChIP) Assay Kit (#17–245, MilliporeSigma, Burlington, MA, USA). A375 melanoma cells were fixed with 1% formaldehyde for 10 min at room temperature. This was followed by shearing of chromatin by sonication on ice to obtain DNA lengths between 200 and 1000 base pairs. Soluble chromatin fragments of 200 to 1000 bp in length were incubated with 5 μg of anti-acetyl-histone H3 antibodies at 4°C overnight. Normal rabbit IgG was used as a negative control for validating the ChIP assay. Protein–DNA complexes were precipitated by protein A/G-coupled magnetic beads. DNA was purified from the immunoprecipitated complexes by reversal of cross-linking and followed by proteinase K treatment. Real-time RT-PCR was performed using SYBR Green QRT-PCR master mix (BIORAD). Two sets of ChIP primers covering 1800 bp upstream of the human PD-L1 gene start codon and designed by NCBI-Blast software were: primer 1 (− 1178 bp to − 1117 bp), forward 5′-GCT GGG CCC AAA CCC TAT T−3′ and reverse 5′-TTT GGC AGG AGC ATG GAG TT-3′; primer 2 (− 455 bp to − 356 bp), forward 5′-ATG GGT CTG CTG CTG ACT TT-3′ and reverse 5′-GGC GTC CCC CTT TCTGAT AA-3′ [25]. The ChIP qPCR result was calculated using the ΔΔCt method. Briefly, each ChIP fractions’ Ct value was normalized to the input DNA fraction and expressed as % Input. Band intensity was compared among the IgG, treated, and control samples on a 1.5% agarose gel, and densitometric analysis was carried out using ImageJ software.
4.14. Statistical Analysis
Data presented are the mean ± SD of at least six independent experiments. Statistical significance was determined by unpaired t-tests, two-tailed, corrected for unequal variance using Microsoft Excel or Prizm 10 software (GraphPad, La Jolla, CA, USA), unless stated otherwise. In the figures, **** represents p<0.0001, *** represents p≤0.001, ** represents p≤ 0.01, and * is for p≤ 0.05. The top of the bar is the mean value and horizontal bars indicate one standard deviation. Each dot represents an independent experiment.
5. Conclusion
This is the first demonstration of epigenetic effects of exogenous, bioactive ARSB, which was previously shown to reduce tumor volume and improve survival of mice with subcutaneous B16F10 melanomas. Treatment of melanoma cells and B16F10 melanomas by rhARSB led to increases in HDAC3 and reduced PD-L1 expression. The ARSB-mediated pathway involves effects on chondroitin 4-sulfate, galectin-3, nuclear c-Jun, HDAC3, and H3 acetylation at the PD-L1 promoter. Effects of rhARSB were inhibited by Insulin Growth Factor-2 Receptor knockdown. Findings indicate that ARSB may be an effective treatment of malignant melanoma and may influence the outcome of checkpoint inhibitor therapy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
SB: Conceptualization, Data curation, Formal analysis; Investigation; Writing original draft; IO: Investigation, Methodology; JKT: Conceptualization, Funding acquisition, Investigation, Methodology, Project Administration, Resources, Supervision, Validation, Writing-original draft and review and editing.
Data Availability
Data are available by communication with JKT.
Acknowledgments
The facilities of the VA and VA Merit Review grant to JKT supported the research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the VA.
References
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Pilon-Thomas S, Mackay A, Vohra N, Mulé JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol. 2010, 184, 3442–3449. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino V, Pizzimenti C, Franchina M, Pepe L, Russotto F, Tralongo P, Micali MG, Militi GB, Lentini M. Programmed cell death ligand 1 immunohistochemical expression and cutaneous melanoma: A controversial relationship. Int J Mol Sci. 2024, 25, 676. [Google Scholar] [CrossRef] [PubMed]
- Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, Okazaki T, Tokura Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010, 116, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Massi D, Brusa D, Merelli B, Ciano M, Audrito V, Serra S, Buonincontri R, Baroni G, Nassini R, Minocci D, Cattaneo L, Tamborini E, Carobbio A, Rulli E, Deaglio S, Mandalà M. PD-L1 marks a subset of melanomas with a shorter overall survival and distinct genetic and morphological characteristics. Ann Oncol. 2014, 25, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya S, O-Sullivan I, Tu J, Chen Z, Tobacman JK. Exogenous recombinant N-acetylgalactosamine-4-sulfatase (Arylsulfatase B; ARSB) inhibits progression of B16F10 cutaneous melanomas and modulates cell signaling. Biochim Biophys Acta Mol Basis Dis. 2024, 1870, 166913. [Google Scholar]
- Bhattacharyya S, Feferman L, Terai K, Dudek AZ, Tobacman JK. Decline in arylsulfatase B leads to increased invasiveness of melanoma cells. Oncotarget. 2017, 17, 4169–4180. [Google Scholar]
- Feferman L, Bhattacharyya S, Deaton R, Gann P, Guzman G, Kajdacsy-Balla A, Tobacman JK. Arylsulfatase B (N-acetylgalactosamine-4-sulfatase): potential role as a biomarker in prostate cancer. Prostate Cancer Prostatic Dis. 2013, 16, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya S, Tobacman JK. Arylsulfatase B regulates colonic epithelial cell migration by effects on MMP9 expression and RhoA activation. Clin Exp Metastasis. 2009, 26, 535–545. [Google Scholar] [CrossRef]
- Bhattacharyya S, Tobacman JK. Steroid sulfatase, arylsulfatases A and B, galactose-6-sulfatase, and iduronate sulfatase in mammary cells and effects of sulfated and non-sulfated estrogens on sulfatase activity. J Steroid Biochem Mol Biol. 2007, 103, 20–34. [Google Scholar] [CrossRef]
- Tobacman JK, Bhattacharyya S. Profound impact of decline in N-acetylgalactosamine-4-sulfatase (Arylsulfatase B) on molecular pathophysiology and human diseases. Int J Mol Sci. 2022, 23, 13146. [Google Scholar] [CrossRef]
- Bhattacharyya S, Feferman L, Han X, Ouyang Y, Zhang F, Linhardt RJ, Tobacman JK. Decline in arylsulfatase B expression increases EGFR expression by inhibiting the protein-tyrosine phosphatase SHP2 and activating JNK in prostate cells. J Biol Chem. 2018, 293, 11076–11087. [Google Scholar] [CrossRef]
- Bhattacharyya S, Feferman L, Tobacman JK. Arylsulfatase B regulates versican expression by galectin-3 and AP-1 mediated transcriptional effects. Oncogene. 2014, 33, 5467–5476. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya S, Feferman L, Tobacman JK. Chondroitin sulfatases differentially regulate Wnt signaling in prostate stem cells through effects on SHP2, phospho-ERK1/2, and Dickkopf Wnt signaling pathway inhibitor (DKK3). Oncotarget. 2017, 8, 100242–100260. [Google Scholar] [CrossRef]
- Bhattacharyya S, Feferman L, Tobacman JK. Increased expression of colonic Wnt9A through Sp1-mediated transcriptional effects involving arylsulfatase B, chondroitin 4-sulfate, and galectin-3. J Biol Chem. 2014, 289, 17564–17575. [Google Scholar] [CrossRef]
- Tobacman JK, Bhattacharyya B, Feferman L. Decline in Arylsulfatase B (ARSB) increases PD-L1 expression in melanoma, hepatic, prostate, and mononuclear cells [abstract]. Cancer Res 2020, 80, 4699. [Google Scholar] [CrossRef]
- Woods DM, Sodré AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. Cancer Immunol Res. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. 2015, 3, 1375–1385.
- Wang YF, Liu F, Sherwin S, Farrelly M, Yan XG, Croft A, Liu T, Jin L, Zhang XD, Jiang CC. Cooperativity of HOXA5 and STAT3 Is critical for HDAC8 inhibition-mediated transcriptional activation of PD-L1 in human melanoma cells. J Invest Dermatol. 2018, 138, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Booth L, Roberts JL, Poklepovic A, Kirkwood J, Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget. 2017, 8, 83155–83170. [Google Scholar] [CrossRef]
- Chatterjee A, Rodger EJ, Ahn A, Stockwell PA, Parry M, Motwani J, Gallagher SJ, Shklovskaya E, Tiffen J, Eccles MR, Hersey P. Marked Global DNA hypomethylation Is associated with constitutive PD-L1 expression in melanoma. iScience. 2018, 4, 312–325. [Google Scholar] [CrossRef]
- Gallagher SJ, Tiffen JC, Hersey P. Histone modifications, modifiers and readers in melanoma resistance to targeted and immune therapy. Cancers (Basel). 2015, 7, 1959–1982. [Google Scholar] [CrossRef]
- Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, Jochems C, Clavijo PE, Allen CT, Hodge JW, Tsang KY, Schlom J, Gameiro SR. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. Oncoimmunology. 2018, 7, e1466018. [Google Scholar] [CrossRef] [PubMed]
- Darvin P, Sasidharan Nair V, Elkord E. PD-L1 expression in human breast cancer stem cells is epigenetically regulated through posttranslational histone modifications. J Oncol. 2019, 2019, 3958908. [Google Scholar]
- Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, Woan KV, Lee C, Cheng F, Deng S, Smalley KSM, Montecino M, Kozikowski A, Pinilla-Ibarz J, Sarnaik A, Seto E, Weber J, Sotomayor EM, Villagra A. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. 2016, 10, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Wang H, Fu C, Du J, Wang H, He R, Yin X, Li H, Li X, Wang H, Li K, Zheng L, Liu Z, Qiu Y. Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells. J Exp Clin Cancer Res. 2020, 39, 29. [Google Scholar] [CrossRef] [PubMed]
- Deng S, Hu Q, Zhang H, Yang F, Peng C, Huang C. HDAC3 inhibition upregulates PD-L1 expression in B-cell lymphomas and augments the efficacy of anti-PD-L1 therapy. Mol Cancer Ther. 2019, 18, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Li X, Su X, Liu R, Pan Y, Fang J, Cao L, Feng C, Shang Q, Chen Y, Shao C, Shi Y. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene. 2021, 40, 1836–1850. [Google Scholar] [CrossRef] [PubMed]
- Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, Yang H, Singh S, Ying HY, Chu CS, Ma MCJ, Toska E, Alig S, Durant M, de Stanchina E, Ghosh S, Mottok A, Nastoupil L, Neelapu SS, Weigert O, Inghirami G, Baselga J, Younes A, Yee C, Dogan A, Scheinberg DA, Roeder RG, Melnick AM, Green MR. Selective inhibition of HDAC3 targets synthetic vulnerabilities and activates immune surveillance in lymphoma. Cancer Discov. 2020, 10, 440–459. [Google Scholar] [CrossRef]
- Elleback E, Khan S, Bastholt L, Schmidt H, Haslund CA, Donis M, Svane IM. PD-L1 is a biomarker of real-world clinical outcomes for anti-CTLA-4 plus anti-PD-1 or anti-PD-1 monotherapy in metastatic melanoma. Eur J Cancer 2023, 198, 113476. [Google Scholar]
- .Peng X, Li L, Chen J, Ren Y, Liu J, Yu Z, Cao H, Chen J. Discovery of novel histone deacetylase 6 (HDAC6) inhibitors with enhanced antitumor immunity of anti-PD-L1 immunotherapy in melanoma. J Med Chem. 2022, 65, 2434–2457. [Google Scholar] [CrossRef]
- Peng X, Yu Z, Surineni G, Deng B, Zhang M, Li C, Sun Z, Pan W, Liu Y, Liu S, Yu B, Chen J. Discovery of novel benzohydroxamate-based histone deacetylase 6 (HDAC6) inhibitors with the ability to potentiate anti-PD-L1 immunotherapy in melanoma. J Enzyme Inhib Med Chem. 2023, 38, 2201408. [Google Scholar] [CrossRef] [PubMed]
- Wang YF, Liu F, Sherwin S, Farrelly M, Yan XG, Croft A, Liu T, Jin L, Zhang XD, Jiang CC. Cooperativity of HOXA5 and STAT3 is critical for HDAC8 inhibition-mediated transcriptional activation of PD-L1 in human melanoma cells. J Invest Dermatol. 2018, 138, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Yeon M, Kim Y, Jung HS, Jeoung D. Histone deacetylase inhibitors to overcome resistance to targeted and immuno therapy in metastatic melanoma. Front Cell Dev Biol. 2020, 8, 486. [Google Scholar]
- Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, Kim J, Powers J, Gracia-Hernandez M, Oliveira V, Cheng F, Chen J, Barinka C, Pinilla-Ibarz J, Lee NH, Kozikowski A, Villagra A. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci Rep. 2019, 9, 6136. [Google Scholar] [CrossRef] [PubMed]
- Han J, Xu X, Liu Z, Li Z, Wu Y, Zuo D. Recent advances of molecular mechanisms of regulating PD-L1 expression in melanoma. Int Immunopharmacol. 2020, 88, 106971. [Google Scholar] [CrossRef] [PubMed]
- Holzberg D, Knight CG, Dittrich-Breiholz O, Schneider H, Dörrie A, Hoffmann E, Resch K, Kracht M. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN δ domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J Biol Chem 2003, 278, 40213–40223. [Google Scholar] [CrossRef] [PubMed]
- Barr R K, Kendrick T S, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002, 277, 10987–10997. [Google Scholar] [CrossRef] [PubMed]
- Zhang Z, Zhang Q, Xie J, Zhong Z, Deng C. Enzyme-responsive micellar JQ1 induces enhanced BET protein inhibition and immunotherapy of malignant tumors. Biomater Sci. 2021, 9, 6915–6926. [Google Scholar] [CrossRef]
- Dahms NM, Lobel P, Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem. 1989, 264, 12115–12118. [Google Scholar] [CrossRef]
- Iwaki J, Hirabayashi J. Evaluation of galectin binding by frontal affinity chromatography (FAC). Methods Mol Biol. 2015, 1207, 63–74. [Google Scholar]
- Song S, Byrd JC, Mazurek N, Liu K, Koo JS, Bresalier RS. Galectin-3 modulates MUC2 mucin expression in human colon cancer cells at the level of transcription via AP-1 activation. Gastroenterology. 2005, 129, 1581–1591. [Google Scholar] [CrossRef]
- Wolter S, Doerrie A, Weber A, Schneider H, Hoffmann E, von der Ohe J, Bakiri L, Wagner EF, Resch K, Kracht M. c-Jun controls histone modifications, NF-kappaB recruitment, and RNA polymerase II function to activate the ccl2 gene. Mol Cell Biol. 2008, 28, 4407–4423. [Google Scholar] [CrossRef] [PubMed]
- Weiss C, Schneider S, Wagner EF, Zhang X, Seto E, Bohmann D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO J. 2003, 22, 3686–3695. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, NB. Regulation of chondroitin sulfate synthesis. Effect of beta-xylosides on synthesis of chondroitin sulfate proteoglycan, chondroitin sulfate chains, and core protein. J Biol Chem. 1977, 252, 6316–6321. [Google Scholar] [CrossRef] [PubMed]
- Feferman L, Bhattacharyya S, Oates E, Haggerty N, Wang T, Varady K, Tobacman JK. Carrageenan-free diet shows improved glucose tolerance and insulin signaling in prediabetes: A randomized, pilot clinical trial. J Diabetes Res. 2020, 2020, 8267980. [Google Scholar]
- D'Avanzo F, Zanetti A, De Filippis C, Tomanin R. Mucopolysaccharidosis Type VI, an updated overview of the disease. Int J Mol Sci. 2021, 22, 13456. [Google Scholar] [CrossRef] [PubMed]
- Guo Y, Shen R, Yang K, Wang Y, Song H, Liu X, Cheng X, Wu R, Song Y, Wang D. RNF8 enhances the sensitivity of PD-L1 inhibitor against melanoma through ubiquitination of galectin-3 in stroma. Cell Death Discov. 2023, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Curti BD, Koguchi Y, Leidner RS, Rolig AS, Sturgill ER, Sun Z, Wu Y, Rajamanickam V, Bernard B, Hilgart-Martiszus I, Fountain CB, Morris G, Iwamoto N, Shimada T, Chang S, Traber PG, Zomer E, Horton JR, Shlevin H, Redmond WL. Enhancing clinical and immunological effects of anti-PD-1 with belapectin, a galectin-3 inhibitor. J Immunother Cancer. 2021, 9, e002371. [Google Scholar] [CrossRef] [PubMed]
- Mabbitt J, Holyer ID, Roper JA, Nilsson UJ, Zetterberg FR, Vuong L, Mackinnon AC, Pedersen A, Slack RJ. Resistance to anti-PD-1/anti-PD-L1: galectin-3 inhibition with GB1211 reverses galectin-3-induced blockade of pembrolizumab and atezolizumab binding to PD-1/PD-L1. Front Immunol. 2023, 14, 1250559. [Google Scholar] [CrossRef]
- Derosiers N, Aguilar W, DeGaramo DA, Posey AD Jr. Sweet immune checkpoint targets to enhance T cell therapy. J Immunol. 2022, 208, 278–285. [Google Scholar] [CrossRef]
Figure 1.
PD-L1 expression and protein in B16F10 subcutaneous melanomas, normal melanocytes, and A375 melanoma cells following ARSB silencing and exposure to exogenous ARSB. A,B. In the mouse melanomas, PD-L1 protein (n=9) and mRNA expression (n=6) are reduced following treatment with exogenous, bioactive ARSB (p<0.0001). C,D. In normal human melanocytes, ARSB silencing increases protein and mRNA expression of PD-L1 (p<0.0001, n=9). E,F. Similarly, in the malignant A375 cells, PD-L1 protein and mRNA are increased by ARSB silencing and reduced by rhARSB (p<0.0001, n=9). The baseline PD-L1 value is greater in the malignant than in the normal cells. G-J. Immunohistochemistry of cultured A375 cells stained for PD-L1 demonstrates decline following rhARSB (G) and marked increase following ARSB siRNA (I), compared to control (H) and control siRNA (J). K,L. In the normal melanocytes and the A375 cells, silencing of the mannose-6-phosphate receptor (M6PR) by siRNA did not block the effect of exogenous ARSB on PD-L1 expression. In contrast, silencing of the calcium-independent insulin growth factor 2 receptor (IGF2R) by siRNA blocked almost completely the rhARSB-induced decline in PD-L1. P-values are <0.0001, determined by unpaired t-test, two-tailed, with unequal variance and n=>6. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; IGF2R=insulin growth factor 2 receptor; M6PR = mannose-6-phosphate receptor; PD-L1=programmed death ligand-1; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
Figure 1.
PD-L1 expression and protein in B16F10 subcutaneous melanomas, normal melanocytes, and A375 melanoma cells following ARSB silencing and exposure to exogenous ARSB. A,B. In the mouse melanomas, PD-L1 protein (n=9) and mRNA expression (n=6) are reduced following treatment with exogenous, bioactive ARSB (p<0.0001). C,D. In normal human melanocytes, ARSB silencing increases protein and mRNA expression of PD-L1 (p<0.0001, n=9). E,F. Similarly, in the malignant A375 cells, PD-L1 protein and mRNA are increased by ARSB silencing and reduced by rhARSB (p<0.0001, n=9). The baseline PD-L1 value is greater in the malignant than in the normal cells. G-J. Immunohistochemistry of cultured A375 cells stained for PD-L1 demonstrates decline following rhARSB (G) and marked increase following ARSB siRNA (I), compared to control (H) and control siRNA (J). K,L. In the normal melanocytes and the A375 cells, silencing of the mannose-6-phosphate receptor (M6PR) by siRNA did not block the effect of exogenous ARSB on PD-L1 expression. In contrast, silencing of the calcium-independent insulin growth factor 2 receptor (IGF2R) by siRNA blocked almost completely the rhARSB-induced decline in PD-L1. P-values are <0.0001, determined by unpaired t-test, two-tailed, with unequal variance and n=>6. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; IGF2R=insulin growth factor 2 receptor; M6PR = mannose-6-phosphate receptor; PD-L1=programmed death ligand-1; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
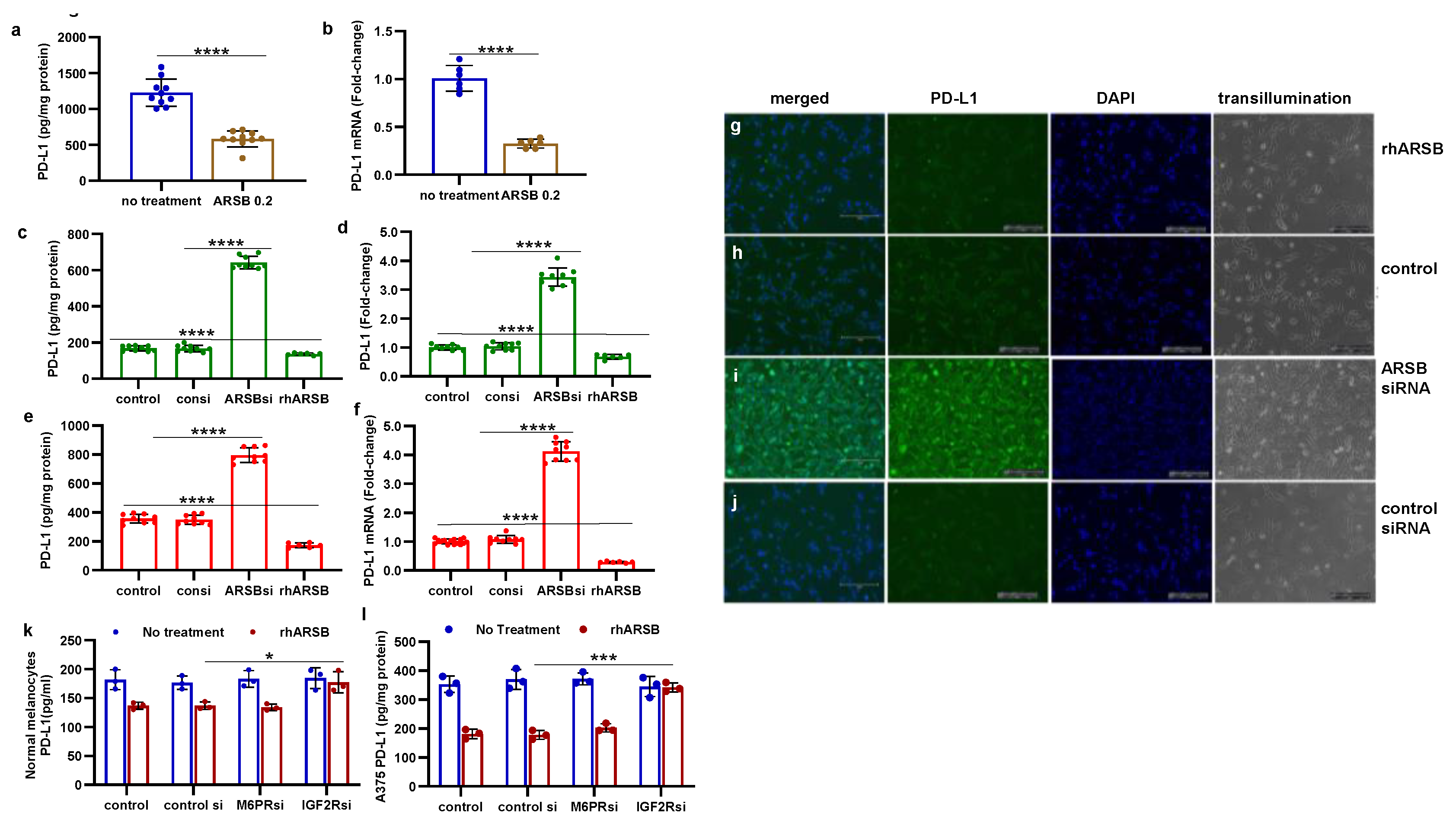
Figure 2.
Inverse effects of silencing ARSB and rhARSB on galectin-3 due to altered binding with chondroitin 4-sulfate. A. In the mouse melanoma tissue, treatment by recombinant ARSB reduced the free galectin-3, as measured by ELISA (p<0.0001, n=6). B. The decline in free galectin-3 is attributed to increased binding of galectin-3 with chondroitin 4-sulfate, which was detected following immunoprecipitation of tumor tissue with C4S antibody. C. In the A375 melanoma cells, free galectin-3 increased when ARSB was silenced and declined following exogenous ARSB (p<0.0001), consistent with altered binding with C4S following decline or increase in ARSB activity. Galectin-3 siRNA effectively reduced galectin-3 protein. D. Similarly, in the normal human melanocytes, ARSB siRNA led to increase in free galectin-3 and exogenous ARSB led to decline (p<0.0001). P-values are <0.0001, determined by unpaired t-test, two-tailed, with unequal variance and n>6 for all determinations. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
Figure 2.
Inverse effects of silencing ARSB and rhARSB on galectin-3 due to altered binding with chondroitin 4-sulfate. A. In the mouse melanoma tissue, treatment by recombinant ARSB reduced the free galectin-3, as measured by ELISA (p<0.0001, n=6). B. The decline in free galectin-3 is attributed to increased binding of galectin-3 with chondroitin 4-sulfate, which was detected following immunoprecipitation of tumor tissue with C4S antibody. C. In the A375 melanoma cells, free galectin-3 increased when ARSB was silenced and declined following exogenous ARSB (p<0.0001), consistent with altered binding with C4S following decline or increase in ARSB activity. Galectin-3 siRNA effectively reduced galectin-3 protein. D. Similarly, in the normal human melanocytes, ARSB siRNA led to increase in free galectin-3 and exogenous ARSB led to decline (p<0.0001). P-values are <0.0001, determined by unpaired t-test, two-tailed, with unequal variance and n>6 for all determinations. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
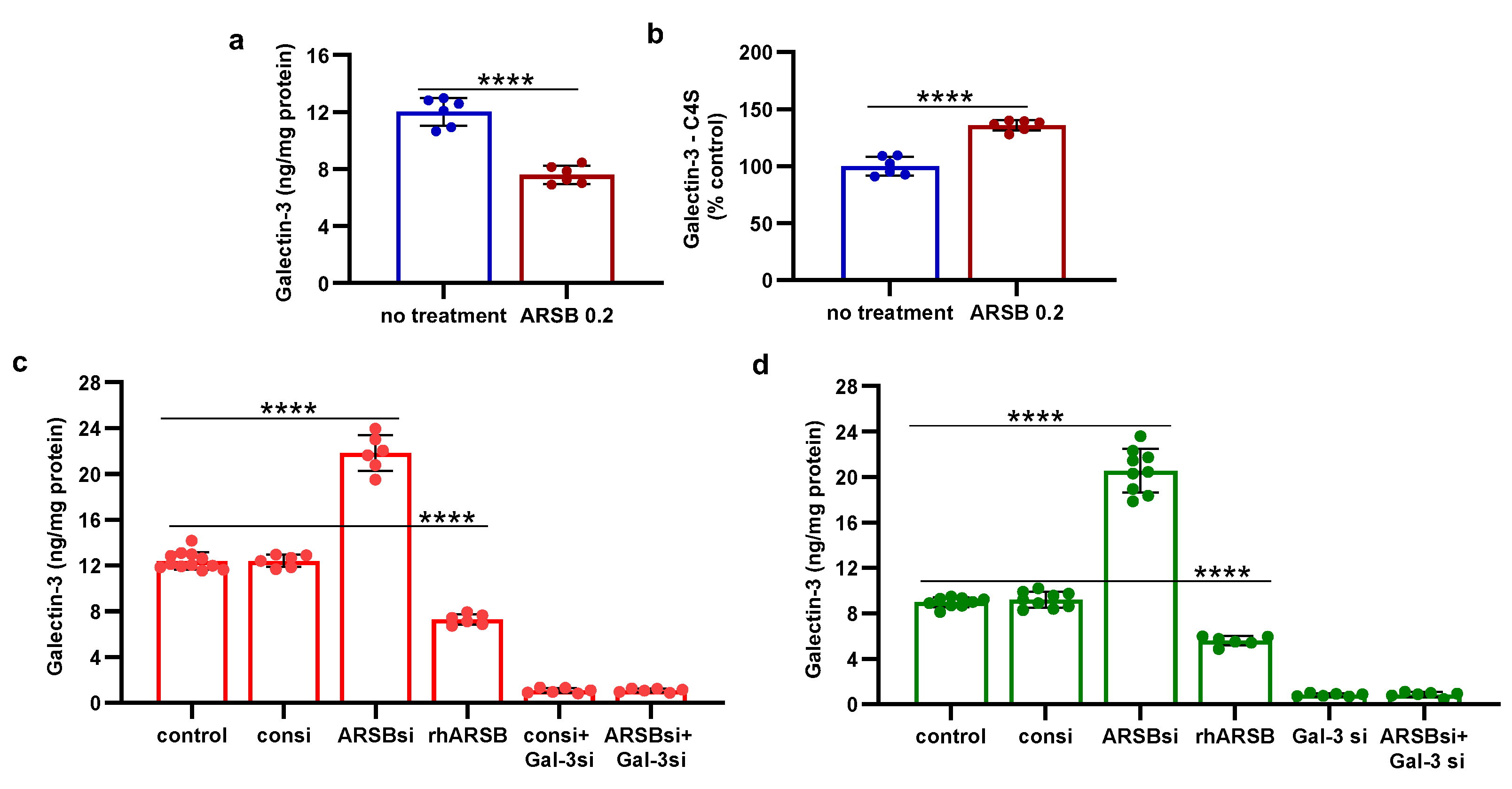
Figure 3.
Inverse effects of ARSB siRNA and rhARSB on phospho-JNK and nuclear c-jun. A,B. In the mouse melanoma tissue, phospho-(Thr183/Tyr185)-JNK and nuclear c-Jun declined following exogenous ARSB (p<0.0001, n=6). C,D. In the A375 cells, ARSB siRNA increased phospho-JNK (p<0.0001, n=9) and nuclear c-Jun (p<0.0001, n=9).. In contrast, rhARSB reduced both phospho-JNK (p<0.0001, n=6) and c-Jun (p<0.0001, n=6). The SHP-2 inhibitor PHPS1 did not reduce the effect of ARSB siRNA. In contrast, galectin-3 siRNA significantly reduced the ARSB si-induced increases. JIP1, a JNK-selective inhibitory peptide, and cJP, a c-Jun mimetic peptide, both completely blocked the increases caused by ARSB siRNA. E,F. Similarly, in the normal melanocytes, phospho-JNK and DNA-bound c-Jun were affected by ARSB siRNA and recombinant ARSB (p<0.0001, n=6). Galectin-3 silencing blocked the effect of ARSB siRNA. G,H. In the A375 cells and the normal melanocytes, the effect of exogenous ARSB on DNA-bound c-Jun was inhibited by the IGF2R siRNA (p=0.005, p=0.002; n=3, n=3), but not by M6PR silencing. P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; IGF2R=insulin growth factor 2 receptor; JIP1=JNK-selective inhibitory peptide; cJP=c-Jun mimetic peptide; JNK=c-Jun N-terminal kinase 1; M6PR = mannose-6-phosphate receptor; PD-L1=programmed death ligand-1; PHPS1=SHP2 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
Figure 3.
Inverse effects of ARSB siRNA and rhARSB on phospho-JNK and nuclear c-jun. A,B. In the mouse melanoma tissue, phospho-(Thr183/Tyr185)-JNK and nuclear c-Jun declined following exogenous ARSB (p<0.0001, n=6). C,D. In the A375 cells, ARSB siRNA increased phospho-JNK (p<0.0001, n=9) and nuclear c-Jun (p<0.0001, n=9).. In contrast, rhARSB reduced both phospho-JNK (p<0.0001, n=6) and c-Jun (p<0.0001, n=6). The SHP-2 inhibitor PHPS1 did not reduce the effect of ARSB siRNA. In contrast, galectin-3 siRNA significantly reduced the ARSB si-induced increases. JIP1, a JNK-selective inhibitory peptide, and cJP, a c-Jun mimetic peptide, both completely blocked the increases caused by ARSB siRNA. E,F. Similarly, in the normal melanocytes, phospho-JNK and DNA-bound c-Jun were affected by ARSB siRNA and recombinant ARSB (p<0.0001, n=6). Galectin-3 silencing blocked the effect of ARSB siRNA. G,H. In the A375 cells and the normal melanocytes, the effect of exogenous ARSB on DNA-bound c-Jun was inhibited by the IGF2R siRNA (p=0.005, p=0.002; n=3, n=3), but not by M6PR silencing. P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; IGF2R=insulin growth factor 2 receptor; JIP1=JNK-selective inhibitory peptide; cJP=c-Jun mimetic peptide; JNK=c-Jun N-terminal kinase 1; M6PR = mannose-6-phosphate receptor; PD-L1=programmed death ligand-1; PHPS1=SHP2 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
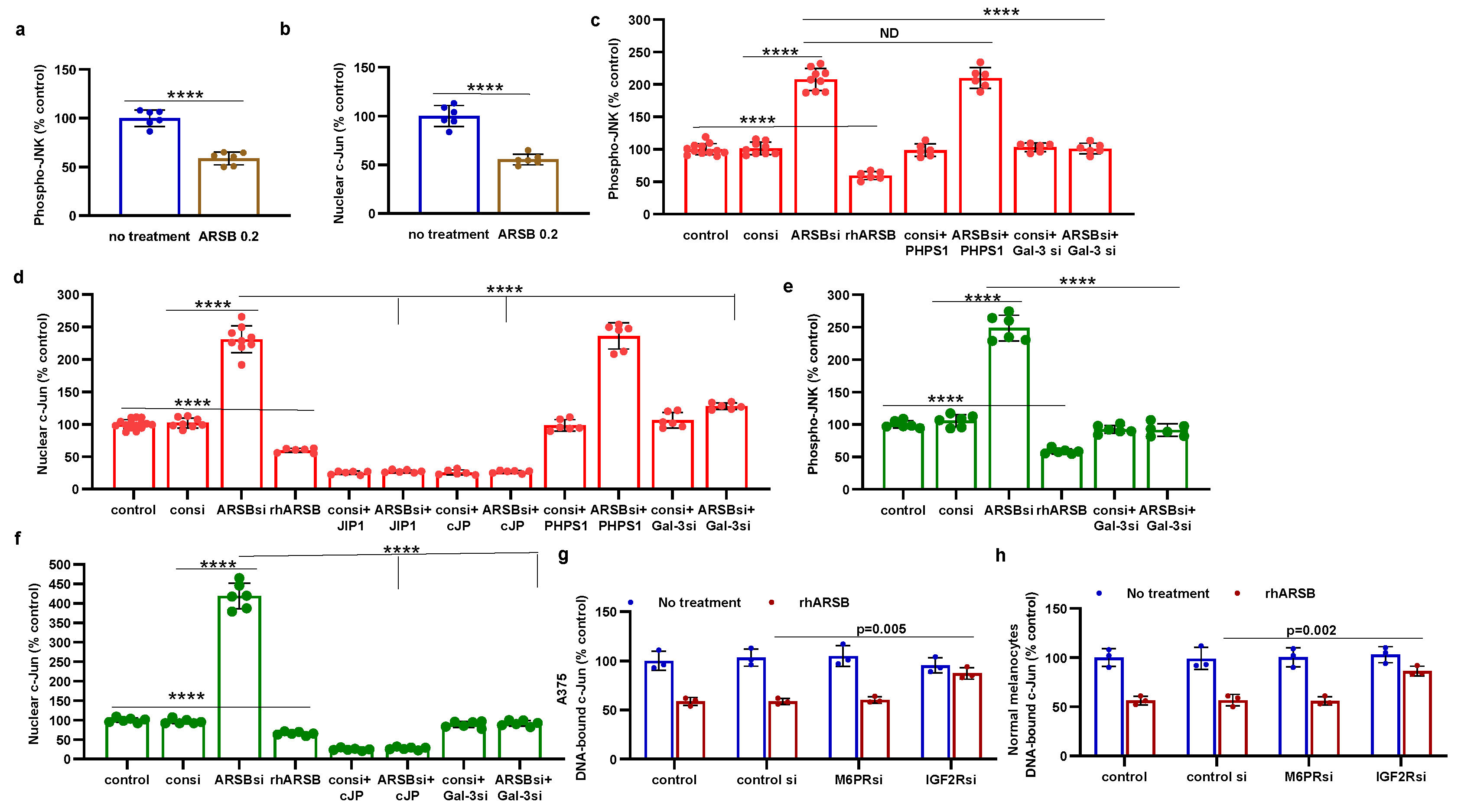
Figure 4.
HDAC3 activity and expression are increased following exogenous ARSB A. In the B16F10 tumors, histone deacetylase (HDAC)3 activity was increased by over 50% following local treatment by exogenous ARSB (0.2 mg/kg x 3 doses) (p<0.001, n=9). B. In the A375 melanoma cells, ARSB silencing reduced (p<0.0001, n=9) and rhARSB increased (p<0.001, n=6) the HDAC3 activity. Treatment by c-Jun mimetic peptide (cJP) markedly increased the HDAC3 activity to 200% of the control level. The SHP2 inhibitor PHPS1 did not affect the ARSB siRNA induced decline, but galectin-3 silencing restored the HDAC-3 activity to near baseline (p<0.001, n=6). C. HDAC3 expression was reduced by ARSB siRNA (p<0.0001, n=6) and normalized by galectin-3 silencing (p<0.0001, n=6). HDAC3 mRNA was increased to 2.3 ± 0.3 times baseline following treatment by cJP. D. In the normal human melanocytes, similar effects of ARSB siRNA, cJP, and galectin-3 siRNA are apparentas in the malignant A375 cells (p<0.0001). RGFP699 completely blocked the HDAC3 activity. P-values are determined by unpaired t-test, two-tailed, with unequal variance, and n>6. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; cJP=c-Jun mimetic peptide; RGFP=RGFP966=HDAC3 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
Figure 4.
HDAC3 activity and expression are increased following exogenous ARSB A. In the B16F10 tumors, histone deacetylase (HDAC)3 activity was increased by over 50% following local treatment by exogenous ARSB (0.2 mg/kg x 3 doses) (p<0.001, n=9). B. In the A375 melanoma cells, ARSB silencing reduced (p<0.0001, n=9) and rhARSB increased (p<0.001, n=6) the HDAC3 activity. Treatment by c-Jun mimetic peptide (cJP) markedly increased the HDAC3 activity to 200% of the control level. The SHP2 inhibitor PHPS1 did not affect the ARSB siRNA induced decline, but galectin-3 silencing restored the HDAC-3 activity to near baseline (p<0.001, n=6). C. HDAC3 expression was reduced by ARSB siRNA (p<0.0001, n=6) and normalized by galectin-3 silencing (p<0.0001, n=6). HDAC3 mRNA was increased to 2.3 ± 0.3 times baseline following treatment by cJP. D. In the normal human melanocytes, similar effects of ARSB siRNA, cJP, and galectin-3 siRNA are apparentas in the malignant A375 cells (p<0.0001). RGFP699 completely blocked the HDAC3 activity. P-values are determined by unpaired t-test, two-tailed, with unequal variance, and n>6. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; cJP=c-Jun mimetic peptide; RGFP=RGFP966=HDAC3 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
Figure 5.
Histone-3 acetylation at PD-L1 promoter follows ARSB silencing in A375 cells. A. A375 melanoma cells, including cells treated by ARSB siRNA, control siRNA, ARSB siRNA + cJP, and IgG negative control, were fixed with 1% formaldehyde, chromatin was sheared, and soluble chromatin fragments were incubated with acetyl-H3 antibodies. Protein–DNA complexes were precipitated, DNA was immunoprecipitated by reversal of cross-linking, and QRT-PCR was performed using two sets of primers upstream of the PD-L1 start codon. Band intensity is diminished when ARSB was silenced, consistent with open chromatin and increased expression of PD-L1. Treatment of the cells ARSB siRNA and cJP normalized the band intensity. B. Densitometry of the bands quantifies the observed differences in intensity using two treated cell samples. C,D. Primer pair 1 yields slightly greater % DNA input than primer pair 2. Both show similar effects of ARSB siRNA and ARSB siRNA + cJP. P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; cJP=c-Jun mimetic peptide].
Figure 5.
Histone-3 acetylation at PD-L1 promoter follows ARSB silencing in A375 cells. A. A375 melanoma cells, including cells treated by ARSB siRNA, control siRNA, ARSB siRNA + cJP, and IgG negative control, were fixed with 1% formaldehyde, chromatin was sheared, and soluble chromatin fragments were incubated with acetyl-H3 antibodies. Protein–DNA complexes were precipitated, DNA was immunoprecipitated by reversal of cross-linking, and QRT-PCR was performed using two sets of primers upstream of the PD-L1 start codon. Band intensity is diminished when ARSB was silenced, consistent with open chromatin and increased expression of PD-L1. Treatment of the cells ARSB siRNA and cJP normalized the band intensity. B. Densitometry of the bands quantifies the observed differences in intensity using two treated cell samples. C,D. Primer pair 1 yields slightly greater % DNA input than primer pair 2. Both show similar effects of ARSB siRNA and ARSB siRNA + cJP. P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; cJP=c-Jun mimetic peptide].
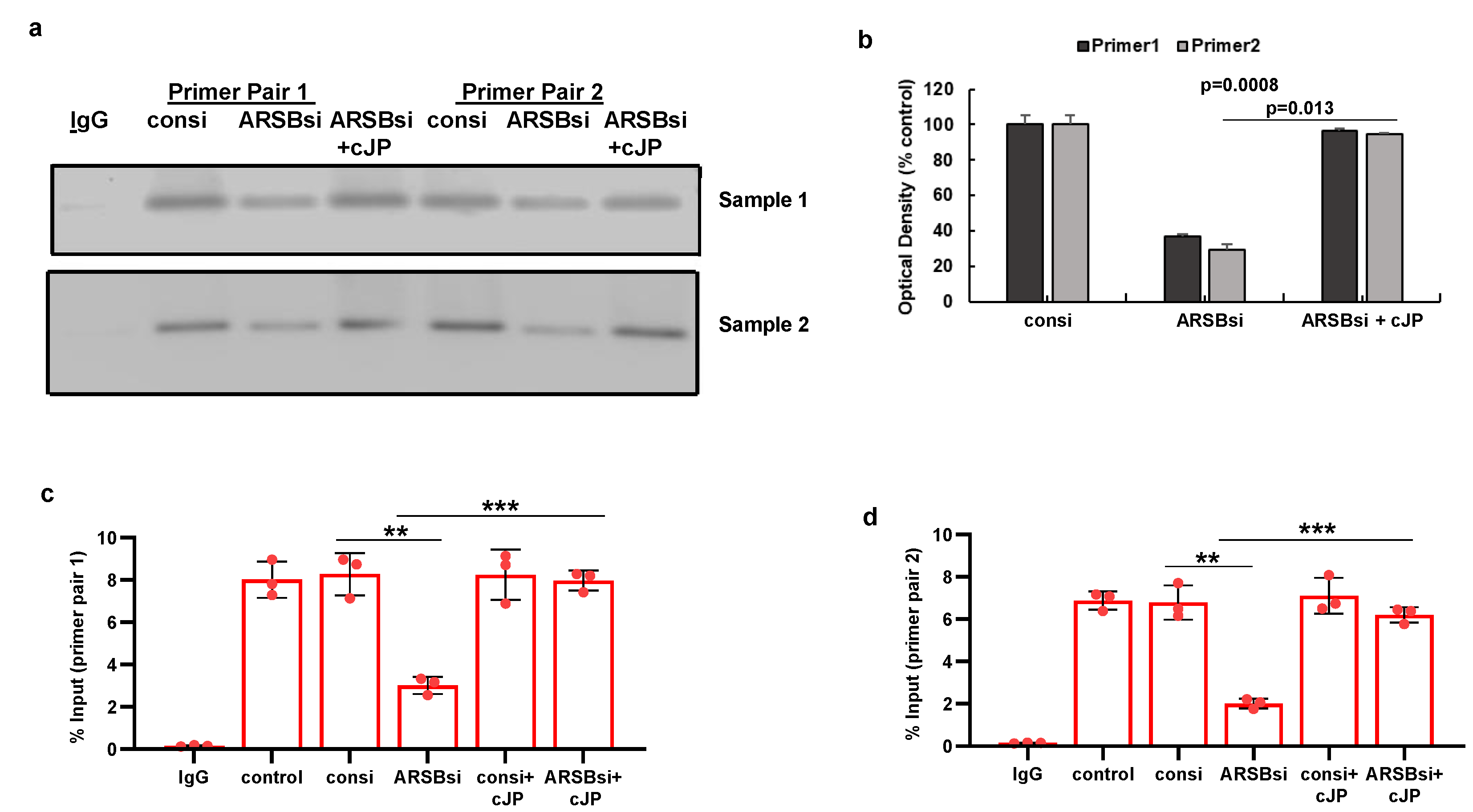
Figure 6.
Impact of methyl-β-D-xylopyranoside (BDX) on mediators of PD-L1 expression. A. Methyl-β-D-xylopyranoside (BDX), an inhibitor of chondroitin sulfate proteoglycan biosynthesis, increases free galectin-3 in the A375 cells (p<0.001, n=3). The combination of ARSB siRNA and BDX has slightly greater effect than ARSB siRNA alone (p<0.05, n=3). B. Nuclear c-Jun is increased by BDX (p<0.001, n=3) and ARSB siRNA (p<0.001, n=3). cJP inhibits the effects of BDX and ARSB siRNA. C. HDAC3 activity is reduced by both ARSB siRNA (p=0.002, n=3) and BDX (p=0.0002, n=3), and further reduced by their combination (p=0.002, p=0.003). These effects are reversed by cJP. D. PD-L1 expression is increased by both ARSB siRNA and BDX, and these increases are inhibited by cJP. P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; BDX=methyl-β-D-xylopyranoside; consi=control siRNA; cJP=c-Jun mimetic peptide; HDAC=histone deacetylase; PD-L1=programmed death ligand-1; PHPS1=SHP2 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose].
Figure 6.
Impact of methyl-β-D-xylopyranoside (BDX) on mediators of PD-L1 expression. A. Methyl-β-D-xylopyranoside (BDX), an inhibitor of chondroitin sulfate proteoglycan biosynthesis, increases free galectin-3 in the A375 cells (p<0.001, n=3). The combination of ARSB siRNA and BDX has slightly greater effect than ARSB siRNA alone (p<0.05, n=3). B. Nuclear c-Jun is increased by BDX (p<0.001, n=3) and ARSB siRNA (p<0.001, n=3). cJP inhibits the effects of BDX and ARSB siRNA. C. HDAC3 activity is reduced by both ARSB siRNA (p=0.002, n=3) and BDX (p=0.0002, n=3), and further reduced by their combination (p=0.002, p=0.003). These effects are reversed by cJP. D. PD-L1 expression is increased by both ARSB siRNA and BDX, and these increases are inhibited by cJP. P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; BDX=methyl-β-D-xylopyranoside; consi=control siRNA; cJP=c-Jun mimetic peptide; HDAC=histone deacetylase; PD-L1=programmed death ligand-1; PHPS1=SHP2 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose].
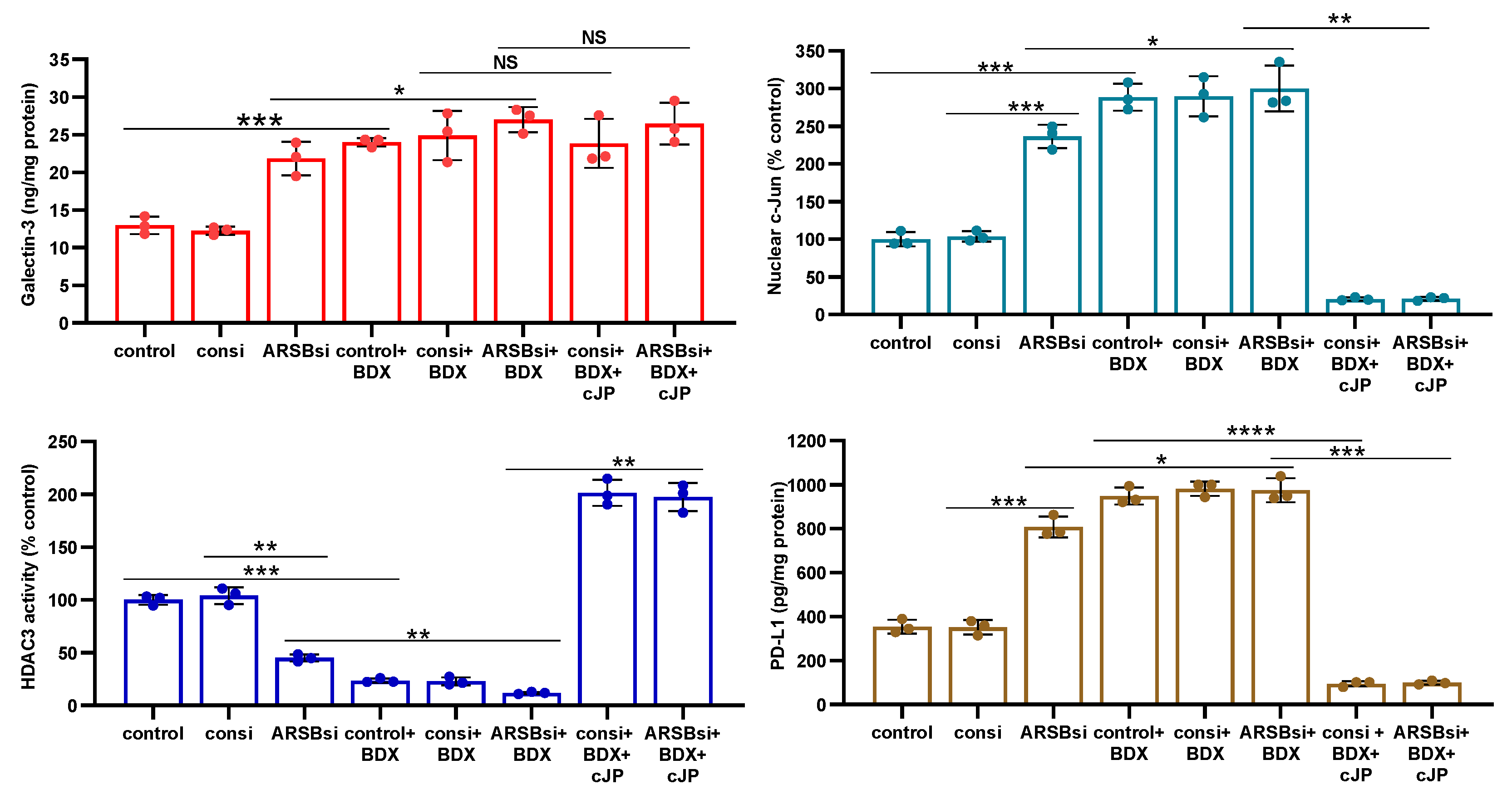
Figure 7.
Effects of inhibition of c-jun, galectin-3, and HDAC3 on ARSB-mediated changes in PD-L1 expression in A375 melanoma cells and normal melanocytes. A. In the A375 cells, PD-L1 protein is markedly increased by ARSB siRNA, and this increase is inhibited by cJP and galectin-3 siRNA. PHPS1 does not affect the PD-L1 expression. The HDAC3 inhibitor, RGFP966, further increases the ARSB siRNA-induced increase in PD-L1. B. In normal melanocytes, similar effects of ARSB siRNA, cJP, galectin-3 siRNA, and RGFP are observed. The total production of PD-L1 is less than in the A375 cells (642 ± 34 pg/mg protein vs. 796.± 50 pg/mg protein; p<0.0001, n=9). C,D. Similar effects are observed in PD-L1 mRNA expression in the A375 malignant cells and the normal melanocytes (p<0.0001). Galectin-3 siRNA inhibits the effect of ARSB silencing, consistent with dependence on galectin-3 for the observed effects. E. JQ1, the inhibitor of the BET family of bromodomain proteins, blocks the effects of ARSB siRNA and RGFP on PD-L1 expression (p<0.0001). P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; cJP=c-Jun mimetic peptide; JQ1=inhibitor of the BET family of bromodomain proteins; PD-L1=programmed death ligand-1; PHPS1=SHP2 inhibitor; RGFP=RGFP966=HDAC3 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
Figure 7.
Effects of inhibition of c-jun, galectin-3, and HDAC3 on ARSB-mediated changes in PD-L1 expression in A375 melanoma cells and normal melanocytes. A. In the A375 cells, PD-L1 protein is markedly increased by ARSB siRNA, and this increase is inhibited by cJP and galectin-3 siRNA. PHPS1 does not affect the PD-L1 expression. The HDAC3 inhibitor, RGFP966, further increases the ARSB siRNA-induced increase in PD-L1. B. In normal melanocytes, similar effects of ARSB siRNA, cJP, galectin-3 siRNA, and RGFP are observed. The total production of PD-L1 is less than in the A375 cells (642 ± 34 pg/mg protein vs. 796.± 50 pg/mg protein; p<0.0001, n=9). C,D. Similar effects are observed in PD-L1 mRNA expression in the A375 malignant cells and the normal melanocytes (p<0.0001). Galectin-3 siRNA inhibits the effect of ARSB silencing, consistent with dependence on galectin-3 for the observed effects. E. JQ1, the inhibitor of the BET family of bromodomain proteins, blocks the effects of ARSB siRNA and RGFP on PD-L1 expression (p<0.0001). P-values are determined by unpaired t-test, two-tailed, with unequal variance. [ARSB=arylsulfatase B=N-acetylgalactosamine-4-sulfatase; consi=control siRNA; gal-3=galectin-3; cJP=c-Jun mimetic peptide; JQ1=inhibitor of the BET family of bromodomain proteins; PD-L1=programmed death ligand-1; PHPS1=SHP2 inhibitor; RGFP=RGFP966=HDAC3 inhibitor; rh=recombinant human; si=small interfering siRNA; 0.2=0.2 mg/kg treatment dose] .
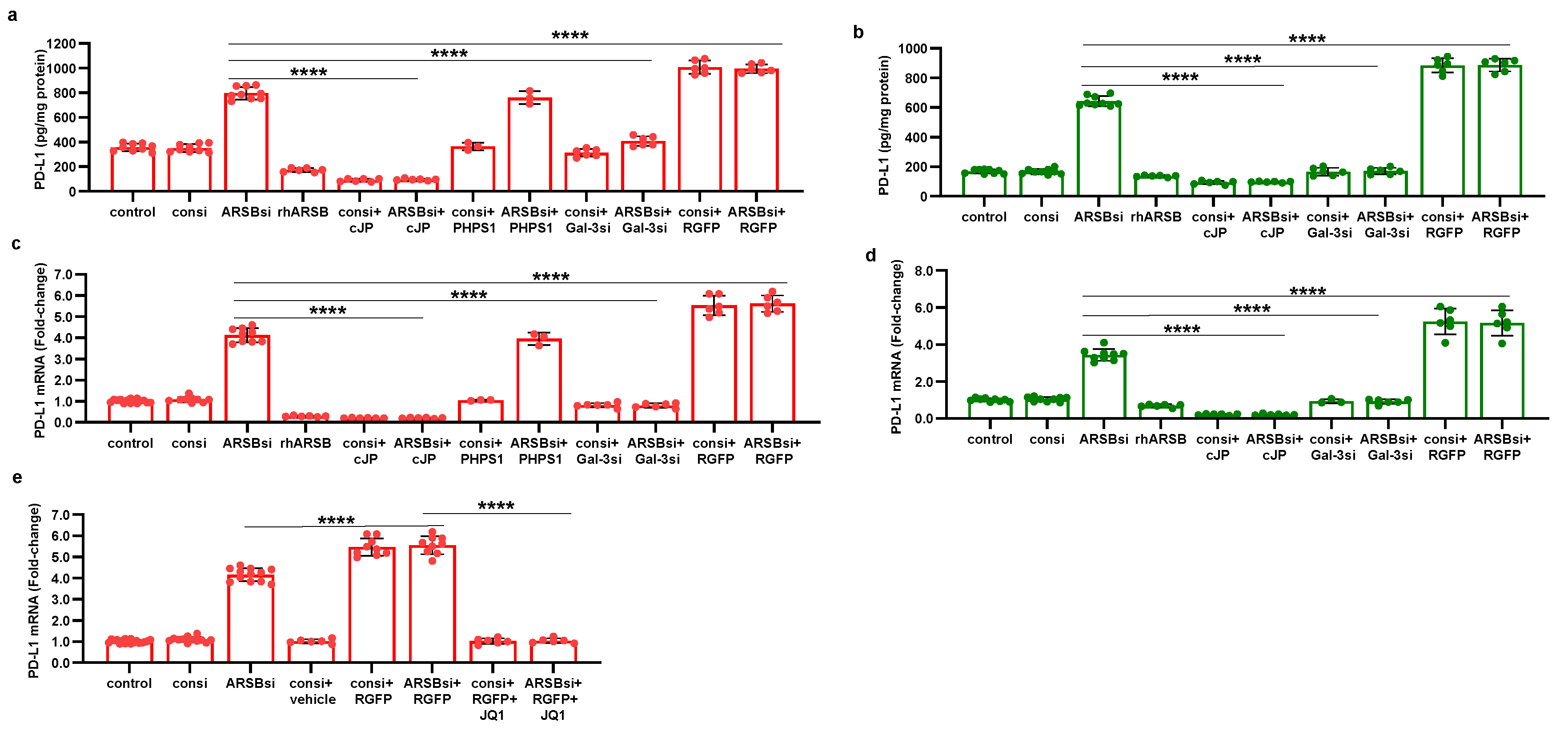
Figure 8.
Schematic of overall pathway. Following exposure to recombinant human ARSB, either in the A375 melanoma cells, normal melanocytes, or mouse subcutaneous B16F10 melanoma, the sulfate group of chondroit. in 4-sulfate is removed and galectin-3 binding with C4S is enhanced. This leads to decline in the free galectin-3 and reduced availability to undergo nuclear translocation and to impact on c-Jun DNA binding, which is then reduced. Reciprocal effects lead to increase in HDAC3, reduced acetyl-H3, increased availability of histone lysines to bind with DNA, leading to closed chromatin, and reduced expression of PD-L1. When ARSB is silenced, opposite effects occur, due to increased availability of galectin-3 and increased effects of c-jun, reduced HDAC3 activity, reduced binding of histone lysines to promoter DNA, leading to open chromatin, and enhanced PD-L1 expression.
Figure 8.
Schematic of overall pathway. Following exposure to recombinant human ARSB, either in the A375 melanoma cells, normal melanocytes, or mouse subcutaneous B16F10 melanoma, the sulfate group of chondroit. in 4-sulfate is removed and galectin-3 binding with C4S is enhanced. This leads to decline in the free galectin-3 and reduced availability to undergo nuclear translocation and to impact on c-Jun DNA binding, which is then reduced. Reciprocal effects lead to increase in HDAC3, reduced acetyl-H3, increased availability of histone lysines to bind with DNA, leading to closed chromatin, and reduced expression of PD-L1. When ARSB is silenced, opposite effects occur, due to increased availability of galectin-3 and increased effects of c-jun, reduced HDAC3 activity, reduced binding of histone lysines to promoter DNA, leading to open chromatin, and enhanced PD-L1 expression.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



