
Submitted:
16 April 2024
Posted:
16 April 2024
You are already at the latest version
Abstract
Background: Progression of metabolic dysfunction associated steatotic liver disease (MASLD) to steatohepatitis (MASH) is driven by stress-inducing lipids that promote liver inflammation and fibrosis. MASH can lead to cirrhosis and hepatocellular carcinoma. We showed coordinated defenses regulated by transcription factors, nuclear factor erythroid 2 related factor-1 (Nrf1) and -2 (Nrf2), protect against hepatic lipid stress. Here, we investigated protective effects of hepatocyte Nrf1 and Nrf2 against MASLD-induced liver fibrosis and tumorigenesis.
Methods: Using mice fed MASH diet for 24-52 weeks, we examined MASLD in mice with hepatocyte specific Nrf1, Nrf2, or combined deletion, and compared this to control. In a separate study, mice received weekly injections of carbon tetrachloride to induce liver fibrosis. From week 16-24, mice were treated with Nrf2 activating drug bardoxolone, hepatocyte overexpression of human NRF1 (hNRF1), or both, and groups were compared to control.
Results: Hepatocyte Nrf2 deficiency had no effect. Hepatocyte Nrf1 and combined deficiency caused MASH but only hepatocyte Nrf1 deficiency in male mice increased tumor number. Bardoxolone reduced liver steatosis, fibrosis, inflammation, and proliferation, and this effect when combined with hNRF1 was greater than bardoxolone alone.
Conclusion: Physiologic Nrf1 delays MASLD progression, Nrf2-induction alleviates MASH, and combined enhancement synergistically protects against steatosis.
Keywords:
Nrf2
; Nrf1
; HCC
; liver fibrosis
; mash
; masld
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD; aka. non-alcoholic fatty liver disease) is prevalent in 37% of the global population and has a wide clinicopathological spectrum (1-3). The most common case is asymptomatic simple steatosis whereas ~20% of people with MASLD progress to metabolic dysfunction associated steatohepatitis (MASH), characterized by liver steatosis, inflammation, and fibrosis. People with MASH have higher risk for cirrhosis and hepatocellular carcinoma (HCC), and so treatments that delay or reverse MASH onset and its progression to more severe disease stages are in need (4-8). A major factor driving MASH is the accumulation of hepatotoxic lipids that impose stress by causing reactive oxygen species (ROS) production, organelle damage and dysfunction, and inflammatory signaling which in turn promote fibrosis via activating stellate cells (4, 8-11). As this is a pivotal point for adverse effects and risk factor for mortality and severe comorbidities (5, 12), alleviating or resolving lipid-induced stress in the liver may be critical to improve outcomes for patients with MASH.
One strategy to reduce hepatic lipid stress is by enhancing endogenous defense programs. But which can be targeted to accomplish this feat is unclear. Here, we focus on the homologous transcription factors, nuclear factor erythroid 2 related factor-1 (Nrf1) and -2 (Nrf2), which are known to regulate expression of cytoprotective defenses (13, 14). Actions by Nrf1 and/or Nrf2, determined using gene deficient mice, have been shown to influence cholesterol, triglyceride, and glucose metabolism, defend against ROS, proteotoxicity, and impaired autophagy (13-23), and protect against steatohepatitis (17, 19, 20, 23), consistent with the effect of polymorphisms linked to the genes encoding NRF1 (NFE2L1) and NRF2 (NFE2L2) in people with obesity (24). Thus, enhancing Nrf1 and/or Nrf2 activity may mitigate MASH and prevent liver disease progression.
While there is therapeutic potential in targeting these factors, there are also incompletely understood aspects regarding similarities and differences for their roles in liver health and disease. One known difference is that newly synthesized Nrf1 is anchored to the endoplasmic reticulum, undergoes p97-dependent turnover by the proteasome, and is sensitive to insufficient proteasome activity (14, 25-27), whereas newly synthesized Nrf2 is localized to cytosol, forms a heterodimeric complex with kelch-like ECH-associated protein 1, and is sensitive to ROS (13). Another is that while they have common targets, they also have distinct gene targets (16, 28-32). Most important though is regarding a discrepant role they may play in HCC. Nrf1 has been found to repress tumor growth in HCC cell models (33-35) and in aged mice with hepatic Nrf1 deficiency (23). In contrast, while transient pharmacological induction of Nrf2 can improve liver health (36), the role of Nrf2 on HCC formation in transgenic mouse models has been conflicting (37-39), and analyses of HCC promoting mutations in humans suggest chronic Nrf2 induction is pro-tumorigenic (40-42). Hence, while enhancing Nrf1 activity seems to be protective throughout MASLD stages, there appears to be a ‘double-edged sword’ effect for Nrf2 dictated by mode and duration of activation. Therefore, further investigation is needed regarding the role of Nrf1 and Nrf2 in MASH and its progression to gain sufficient clarity for strategic development of appropriate therapies.
Recently, we demonstrated that Nrf1 and Nrf2 complementarily regulate the expression of hepatocyte defenses to protect against the stress of MASH-associated hepatic cholesterol overload (16). High fat, fructose, and cholesterol (HFFC) diet fed mice with a coinciding 1 to 3 weeks of hepatocyte deficiency for Nrf1 and Nrf2, but not either alone, exhibited severe MASH due to increased hepatic lipid storage, altered bile acid metabolism, and oxidative damage, whereas Nrf1 deficiency alone was sufficient to increase hepatic inflammation. Moreover, Nrf1 and Nrf2 were each required for 2 week treatment with the Nrf2-activating drug bardoxolone to reduce steatosis and inflammation in mice chronically fed HFFC diet for 24 weeks, and combining this with hepatocyte overexpression of human NRF1 potentiated such effects in mice fed HFFC diet for 16 weeks. This indicated that enhancing the actions of both Nrf1 and Nrf2 in hepatocytes can reduce MASH. However, the study durations were insufficient to evaluate the impact hepatocyte Nrf1 and Nrf2 activity may have on more severe stages of MASLD, such as cirrhosis and HCC. Here, using HFFC diet fed conditions lasting up to 52 weeks in duration or combining HFFC diet with the liver fibrosis-inducing agent carbon tetrachloride and modulating the activity of Nrf1 and/or Nrf2 in differing disease stages, we investigate whether Nrf1 and Nrf2 synergistically alleviate fibrosis in MASH and prevent HCC. Our results confirm the beneficial effect of bardoxolone to alleviate liver inflammation and fibrosis, establish a robust synergistic role for Nrf1 and Nrf2 to reduce hepatic lipid storage, and demonstrate that hepatocyte Nrf1 plays a greater physiological role than Nrf2 in preventing HCC.
2. Results
2.1. Effect of HFFC Diet on Liver in Mice with Flox Alleles for Nrf1, Nrf2, or Both
Our initial goal was to investigate the impact of hepatocyte deficiency for Nrf1 (Nfe2l1), Nrf2 (Nfe2l1), or both on the progression of diet-induced MASH, using Nfe2l1flox/flox mice, Nfe2l2flox/flox mice, and Nfe2l1flox/flox; Nfe2l2flox/flox mice respectively, as described previously (15, 16, 21). While there are several dietary approaches (8, 43, 44) with each having advantages and limitations, we selected to employ chronic feeding with diet enriched with high saturated fat, fructose, and cholesterol (HFFC; known as Gubra Amylin diet). HFFC diet has been shown to induce several histological, transcriptional, and metabolic features translationally relevant to clinical MASH (45). Cohorts of male and female mice from each line were fed 1% cholesterol containing HFFC diet or control diet for 24 weeks. At either week 16 or 22, these mice were infected with adeno-associated virus exhibiting hepatocyte expression of green fluorescent protein (AAV-GFP) or Cre recombinase (AAV-CRE). As we have shown (15, 16, 21), AAV-CRE causes hepatocyte specific loss-of-function effect for the respective flox gene whereas AAV-GFP serves as a control. At week 24, tissues were collected for assessment. With these experiments, we assessed the effect of HFFC compared to control diet as well as the impact of hepatic gene deficiency for a short term of ~7 days (i.e., infected at week 22) or prolonged duration of ~7 weeks (i.e., infected at week 16).
To determine the extent to which the flox lines were prone to developing diet-induced MASH, we pooled them and compared MASH-related parameters in AAV-GFP infected mice fed HFFC diet to AAV-GFP infected mice fed control diet. As expected, chronic HFFC diet increased the body weight and liver-to-body weight ratio in males and females (Figure 1A), coinciding with increased hepatic triglyceride and cholesterol (Figure 1B). Likewise, transcriptional analyses of livers using quantitative polymerase chain reaction (qPCR) revealed HFFC diet intake increased expression of MASH markers involved in liver inflammation (Ccl2, Tnfrsf12a), fibrogenesis (Col1a1, Col3a1) and cell proliferation (Ccnb1) (Figure 1C). To further elucidate the effect of HFFC diet on MASH development, histological examination of liver sections was done (Figure 1D and 1E). We used hematoxylin and eosin (H&E) staining to assess steatosis and inflammation, immunohistochemical (IHC) staining of Ki-67 to assess hepatocyte proliferation (46), and IHC staining of p62 as a marker of liver damage, chronic steatohepatitis, and risk for hepatocellular carcinoma (HCC) onset and progression (38, 47-49). Compared to control, HFFC diet increased hepatic steatosis and inflammation as well as proliferation and p62 accumulation (Figure 1D,E). Thus, consistent with others (45), chronic HFFC diet intake induced MASH within a 24-week time frame, confirming that using this diet on these mouse lines is suitable for investigating the role of Nrf1 and Nrf2 in the progression of MASH to cirrhosis and HCC.
2.2. Effect of Hepatocyte Deficiency for Nrf1, Nrf2 or Both in Mice Chronically Fed HFFC Diet
Recently, we showed short-term (i.e., 10-21 days) hepatocyte Nrf1 and Nrf2 deficiency, but not single gene deficiency, resulted in hepatic lipid accumulation, oxidative damage, and inflammation in mice fed HFFC diet for only 17-28 days (16). However, the effect on lipid levels did not occur in short-term gene deficient mice that already had chronic HFFC diet-induced MASH, while other aspects related to liver inflammation did still occur. Here, we investigated the impact of hepatocyte Nrf1, Nrf2, or combined deficiency for ~7 days or for ~7 weeks on MASH progression. Using the experiment described above and, in this case, pooling male and female samples of each flox line, we compared MASH-related parameters in AAV-CRE infected mice fed HFFC diet to AAV-GFP infected mice fed HFFC diet (Figure 2) as well as the AAV-CRE infected mice fed control diet to AAV-GFP infected mice fed control diet (Figure S1).
Compared to respective control, HFFC diet fed mice with ~7 days hepatocyte deficiency for Nrf1, Nrf2, or both had no difference in the level of steatosis and hepatic lipids (Figure 2A,B). However, Nrf1 deficiency increased liver inflammation (Figure 2A) and expression of MASH-related genes Ccl2, Ccnb1, Col3a1, and Tnfrsf12a (Figure 2C). Likewise, combined deficiency increased liver inflammation (Figure 2A) and expression of Ccnb1, Col3a1, Ihh, and Tnfrsf12a (Figure 2C). In contrast, Nrf2 deficiency had no effect. In the control diet fed mice, Nrf2 deficiency remained nonconsequential, while the effect of Nrf1 and combined deficiency was exacerbated, as demonstrated by hepatic steatosis as well as significant or trending increases in hepatic triglyceride and expression of Ccl2, Ccnb1, Col1a1, Col3a1, Ihh, and Tnfrsf12a (Figure S1A–C). We then evaluated whether prolonged ~7-week deficiency for Nrf1, Nrf2, or both resulted in signs of major adverse liver outcomes reflecting the progression of MASH to HCC. Using IHC, we stained for Ki67 to assess hepatocyte proliferation, and p62 to assess liver damage and HCC risk. As expected, Nrf2 deficiency had no effect, but interestingly, while mice with Nrf1 deficiency had increased Ki67 positive nuclei irrespective of being fed HFFC or control diet, this did not occur in mice with combined deficiency (Figure 2D and Figure S1D). Moreover, Nrf1 deficiency increased p62 positive nuclei, whereas this occurred only for mice with combined deficiency when fed HFFC diet (Figure 2D and Figure S1D). Taken together, results from the ~7-day deletion model demonstrate a predominant physiologic role for Nrf1 in counteracting liver inflammation, whereas results from the ~7-week deletion model suggest Nrf1 counteracts MASH progression to HCC, possibly via influencing pro-tumorigenic actions of Nrf2.
2.3. Nrf1 Deficiency Enhances Liver Tumorigenesis in Male Mice with MASH
As Nrf1 deficiency for ~7-days to ~7-weeks corresponded with signature features of liver disease progression and this was distinct from Nrf2, we next examined their roles in protecting against MASH progression to liver tumorigenesis. Male and female Nfe2l1flox/flox mice, Nfe2l2flox/flox mice, and Nfe2l1flox/flox; Nfe2l2flox/flox were fed 2% cholesterol enriched HFFC diet for 52 weeks. On week 16, 32 and 48, mice were infected with AAV-CRE to induce and maintain hepatocyte deficiency of Nrf1, Nrf2, or both respectively (Figure 3A). Control mice were infected with AAV-GFP. Thus, we examined whether ~35 weeks of hepatocyte deficiency in mice with pre-existing and persisting MASH impacted HCC tumor onset and growth.
At the study endpoint, livers were collected to assess the tumor incidence and abundance. Males and females were done separately due to apparent sex-dependent differences in incidence, as there was an average of 56% incidence in AAV-GFP infected male mice and 27% in AAV-GFP infected female mice (Figure 3B). Tumor incidence in female mice with hepatocyte deficiency for Nrf1, Nrf2, or both was not obviously different (i.e., <30%) than controls, and this was also the case for male mice with Nrf2 deficiency and those with combined deficiency (Figure 3C). In contrast, incidence in males with Nrf1 deficiency was 100% and >30% greater than control, and this also corresponded with more tumors per liver (Figure 3C). Interestingly though, tumors per liver was similar to control in males with combined deficiency, indicating tumorigenic effects of Nrf1 deficiency depended on Nrf2 activity. However, the size of the largest tumors was not greater in hepatocyte Nrf1 deficient liver (Figure 3D), indicating tumorigenic effects were due to increased tumor initiation, not tumor growth. Taken together, these results suggest that physiologic Nrf1 protects against MASH progression to HCC by suppressing tumor initiation, and that this may involve functional interactions with Nrf2 activity.
2.4. Effect of Nrf2-Inducing Drug Bardoxolone and Nrf1 Overexpression on MASH-Linked Fibrosis
Previously (16), we showed both Nrf1 and Nrf2 were required for the beneficial effects of 2 week treatment with the Nrf2 activating drug bardoxolone on MASH in 24 week HFFC diet fed mice, and combining 1 week bardoxolone treatment with hepatocyte overexpression of human NRF1 (hNRF1) exhibited partial synergistic effect in mice fed HFFC diet for 16 weeks. Using similar methods to induce Nrf1 and Nrf2 activity, here we investigated whether more prolonged induction of Nrf1, Nrf2, or both may alleviate MASH and fibrosis. As illustrated in Figure 4A, male and female C57bl/6J mice were fed 2% cholesterol enriched HFFC diet for 24 weeks. To induce liver fibrosis, mice were injected with hepatocyte damaging agent carbon tetrachloride (50) once per week for the first 15 weeks. At week 16 and 20, mice were infected with AAV-hNRF1 to induce and maintain increased hepatocyte Nrf1 activity or with AAV-GFP, as a control. Also, from week 16-24 mice were injected three times per week with bardoxolone or vehicle control. At week 24, liver was collected for analysis of the four groups: AAV-GFP + vehicle, AAV-GFP + bardoxolone, AAV-hNRF1 + vehicle, and AAV-hNRF1 + bardoxolone.
To ensure each treatment had the intended effect, we assessed expression of Nrf1 and Nrf2 target genes, Psmc2 and Gstm1 respectively, which we demonstrated previously (16). Compared to AAV-GFP + vehicle, livers expressing hNRF1 had increased Psmc2 expression, whereas livers treated with bardoxolone had increased Gstm1 expression (Figure S2A), thus validating the model. Then, we performed histological analysis via H&E staining to assess steatosis and inflammation and Masson Trichrome staining to assess liver fibrosis (Figure 4B,C). Compared to AAV-GFP + vehicle, AAV-hNRF1 + bardoxolone reduced steatosis, and this was greater than AAV-GFP + bardoxolone and AAV-hNRF1 + vehicle. In contrast, AAV-hNRF1 + bardoxolone reduced liver inflammation (Figure 4B) and fibrosis (Figure 4C) compared to the AAV-GFP + vehicle and to AAV-hNRF1 + vehicle, but not compared to AAV-GFP + bardoxolone.
Interestingly, while evaluating H&E-stained sections, the AAV-hNRF1 + bardoxolone group had a noticeable population of oval shaped cells reminiscent of liver progenitor cells, which are known to sequester in the canals of Hering and to differentiate into functional hepatocytes in conditions of chronic liver injury (51-53). As oval cells have been found to repopulate liver with functionally active hepatocytes following chronic liver injury (54), we sought to quantify whether this population was affected by Nrf1 and Nrf2 activity. To do this, we measured expression of cytokeratin 19 (Krt19), a marker of LPC activation, while also measuring known markers of liver inflammation (Emr1, Tnfrsf12a, Il1B), fibrosis (Ihh, Col1a1, and Col3a1), and proliferation (Ccnd1). Compared to AAV-GFP + vehicle control, AAV-hNRF1 + bardoxolone reduced expression of genes involved in liver inflammation, proliferation, and some involved in fibrosis, which in some cases was also reduced compared to AAV-hNRF1 + vehicle but not to AAV-GFP + bardoxolone. In contrast, AAV-hNRF1 + bardoxolone increased Krt19 expression, and this was also increased compared to AAV-GFP + bardoxolone but not to AAV-hNRF1 + vehicle. Taken together, these results indicate combined induction of Nrf1 and Nrf2 synergistically reduce hepatic lipid stores in MASH-linked fibrosis, that the Nrf2-inducing drug bardoxolone is sufficient to reduce liver inflammation, proliferation, and fibrosis whereas actions by Nrf1 may enhance repair and functional cell repopulation by progenitor cells in chronically damaged liver.
3. Discussion
MASH and its progression to cirrhosis and HCC has emerged as a global health burden (2, 4, 6, 7, 12). Effective therapeutic strategies are in need, but this is hindered by incomplete understanding of how to counteract the pathologic mechanisms. Stress caused by hepatoxic lipid accumulation is recognized as a central factor (4, 8-11), and we propose enhancing endogenous defense programs controlled by transcription factors Nrf1 and Nrf2 may alleviate this stress to mitigate MASH and its progression. Here, we explored this possibility using a validated model of diet-induced MASH on transgenic mice amenable to adult-onset loss-of-function for hepatocyte Nrf1, Nrf2, or both and by enhancing the actions of Nrf1, Nrf2, or both in wild type mice with MASH-linked fibrosis.
Consistent with work by us and others (16, 17, 19, 20, 22, 23), we found physiologic actions by hepatocyte Nrf1 were critical for mitigating liver inflammation. Some effects were independent of MASH-inducing diet, as they occurred and were, in many cases, even greater in mice fed control diet. These findings reinforce that Nrf1 plays a major role in liver homeostasis. On the other hand, physiologic actions by hepatocyte Nrf2 were not critical and, instead, may even be contributory to pathology that arises in Nrf1 deficiency, such as liver tumor development. MASH-induced HCC is a major challenge and still poorly understood (8), and a role for Nrf1 in preventing this process is possible. Xu et al. (23) found liver specific Nrf1 deletion in utero caused liver tumors when mice reached the adult age of 1 year. Here, we used a similar approach except hepatocyte Nrf1 was not deleted until mice were well into adulthood and had been fed HFFC diet for at least 16 weeks. Tumor number was elevated in male mice with Nrf1 deficiency, and interestingly this effect was lost in mice deficient for Nrf1 and Nrf2. Thus, Nrf1 may be able to mitigate pro-tumorigenic effects of chronic Nrf2 activation that have been identified by others (38-42).
Interestingly, while loss of physiologic Nrf2 activity had little effect, the opposite occurred when using the Nrf2-activating agent, bardoxolone. Combined induction of Nrf2 via bardoxolone and Nrf1 via hNRF1 expression reduced steatosis, inflammation, proliferation, and fibrosis. Most of these effects were attributable mainly to bardoxolone, as only reduced steatosis was amplified by combined treatment. Caution with interpretation is necessary, as bardoxolone could elicit these effects by impacting non-hepatic tissue. Still, these results raise two interesting questions. First, what is the mechanism by which bardoxolone and AAV-hNRF1 synergize to reduce hepatic lipids? Second, assuming actions of bardoxolone were due to hepatocyte Nrf2, what drives the disconnect between an apparently dispensable physiologic Nrf2 with an apparently potent Nrf2 inducing drug. Future work should aim to address the underlying mechanism of this discrepancy.
Consistent with our previous work (16), functional Nrf1 was important for preventing the accumulation of the liver damage and HCC marker p62, but it is unknown how this effect develops. Another interesting finding was that Nrf1, and not Nrf2, induction using AAV-hNRF1 increased expression of liver progenitor cell marker Krt19, and that LPCs were abundant in AAV-hNRF1 conditions. This may indicate Nrf1 has a role in liver repair and in resisting liver damage. Investigating Nrf1 in other conditions of experimental liver damage may provide more clarity. In conclusion, we identified distinct and complementary roles for hepatocyte NRF1 and NRF2 in protecting against MASLD progression and in promoting hepatocyte regeneration in liver with MASH-linked fibrosis. Hence, therapies targeting defense programs controlled by Nrf1 and Nrf2 may prove to be an effective approach for improving the outcomes of patients with MASLD.
4. Materials and Methods
4.1. Animal Used in Study
Mouse handling, husbandry, and experiments were performed with approval from University of Saskatchewan's Animal Care Committee. In loss-of-function studies, male and female mice on C57bl/6J background were housed at 21°C on a 12-hour light/dark cycle and provided ad libitum access to food and water. Mice were fed a control diet containing 10% fat (lard & soybean oil), 10% sucrose, 0% fructose, and 0% cholesterol (Research diets, catalog# D09100304) or MASH inducing high fat, fructose, and cholesterol (HFFC) diet (catalog# D19021910) containing 40% fat (75% palm oil / 11% lard / 14% soybean oil), 10% sucrose, 20% fructose and 1% or 2% cholesterol (Research diets, catalog# D19021910 or D09100310), as indicated in the text and figures. Mice with flox alleles for genes encoding Nrf1 (nfe2l1flox/flox), Nrf2 (nfe2l2flox/flox), or both (nfe2l1flox/flox; nfe2l2flox/flox) were generated, as in previous (15, 16, 21, 22). Diet feeding ranged from 24 to 52 weeks, as indicated. To delete Nrf1, Nrf2, or both in hepatocytes, recombination of flox alleles to remove gene elements was done by retroorbital infection of mice, while under isoflurane anesthesia, with 2.0 x 1011 particles of liver targeting serotype 8 adeno-associated virus expressing Cre recombinase via hepatocyte-specific thyroxine binding globulin promoter (AAV-CRE), as previous (15, 16, 21). Controls received virus expressing green fluorescent protein (AAV-GFP). AAV-CRE (AAV8.TBG.PI.Cre.rBG) and AAV-GFP (AAV8.TBG.PI.eGFP.WPRE.bGH) viruses were acquired from the University of Pennsylvania Vector Biocore.
In the grain-of-function study, C57bl/6J mice were purchased from Jackson Laboratory (www.jax.org/strain/000664) and fed HFFC diet throughout. Liver fibrosis was induced using a method similar to Tsuchida et al (50). Liver toxin carbon tetrachloride (CCL4; Sigma-Aldrich, catalog# 319961) was diluted 1:10 in corn oil (Sigma, catalog# C8267) and intraperitoneally (IP) injected to mice at a dose of 0.32 μg/g (0.2 μl/g) body weight once per week for 15 weeks. To enhance hepatocyte Nrf1 activity, mice were injected with the same AAV as above, but encoding for human NRF1 (AAV-hNRF1) expression transcript (NCBI: NM_003204.2), as previous (16). Littermate controls received virus expressing green fluorescent protein (AAV-GFP). AAV-hNRF1 (AAV8.TBG.hNRF1) was acquired from University of Pennsylvania Vector Biocore. To enhance hepatocyte Nrf2 activity, mice were injected IP with 3 mg/kg bardoxolone methyl (bardoxolone) (Selleckchem, catalog# S6647). Controls received vehicle (10% DMSO in PBS). Altogether, mice were infected with AAV-hNRF1 or AAV-GFP at week 16 and 20, while bardoxolone or vehicle was administered three times a week for 8 weeks, starting at week 16.
4.2. Tissue Collection and Liver Histological Analysis
At indicated endpoints, mice were anesthetized via 3 % isoflurane at an oxygen flow rate of 1 L/minute. Blood was collected into a 26-gauge 1 ml syringe needle via intracardiac puncture and then placed into a 1.5 ml tube containing ethylenediaminetetraacetic acid (EDTA, 5 mM final concentration) and placed on ice. Later, plasma was separated via 2000 g centrifugation at 4°C for 10 minutes. Immediately after collecting blood, vasculature of anesthetized mice was flushed with 20 ml of phosphate buffered saline that was at room temperature. Liver was isolated and weighed and then divided into portions that were snap frozen and stored at -80 ºC. Another portion of the liver (left lobe) was used for histological analysis. The left lobe was fixed in 10 % neutral buffered formalin, embedded in wax, sectioned at 5 µm thickness, and stained with Hematoxylin and Eosin (H&E) and Masson Trichrome (MT). Images were captured using an Aperio Scanscope CS image analysis System and Aperio Imagescope viewing software (version 12).
To assess levels of steatosis and inflammation of H&E-stained liver sections, a blinded analyst scored three separate fields for each sample, in accordance with guidelines by Liang et al. (55). For MT fibrosis scoring, the analyst scored six separate fields for each liver sample according to the NASH CRN Scoring System. For anti-ki-67 staining and anti-p62, paraffin embedded 5 µm sections were incubated with anti-ki-67 antibody (1:1000) or anti-p62 antibody (1:250) overnight at 4°C, then they were incubated with peroxidase labelled secondary antibody for 30 minutes, stained with diaminobenzidine (DAB), and counterstained with hematoxylin. 3 fields per sample were visualized and analyzed with ImageJ software to determine the percentage of stained area.
4.3. Analysis of Liver Cholesterol and Triglyceride
As previous (16), 50-100 mg of frozen liver was homogenized in 2 ml hexane: isopropanol (v: v, 3:2). Homogenate was heated to 75 ºC for 5 minutes and centrifuged at 13,000 g for 10 minutes and then lipid-containing supernatant was transferred to a new tube. Supernatant was dried overnight at 55 ºC and resuspended in isopropanol: NP-40 (v: v, 9.9:0.1). Triglyceride was measured using Infinity Triglyceride Reagent from ThermoFisher Scientific (catalog #TR22421). To measure liver cholesterol, an aliquot of the isopropanol:NP-40 suspension was dried again at 55 ºC, and then resuspended in a cholesterol suspension buffer, a component of the Amplex Red Cholesterol Assay Kit from Invitrogen (catalog #A12216) used to measure cholesterol.
4.4. Gene Expression Analysis
25-50 mg of liver was homogenized in TRIzol Reagent ThermoFisher Scientific, catalog #15596018) and RNA isolated according to manufacturer instructions. Complementary DNA (cDNA) was synthesized via Maxima H Minus First Strand cDNA Synthesis Kit with dsDNase from ThermoFisher Scientific (catalog #K1672). qPCR was performed on a CFX384 Real-Time PCR Detection System using PowerUpTM SYBRTM Green Master Mix SYBR from ThermoFisher Scientific (catalog #A25742). Cycle thresholds were normalized to ribosomal protein, large, P0 levels (36B4) and displayed as relative expression. Primers used to detect gene expression is in Supplemental Table S1.
4.5. Tumor Volume Measurement
Tumor dimensions were taken using a Vernier caliper and volume was calculated using the formula: V= (4/3*(3.14159) X (Length/2) X (Width/2)2) as described (56).
4.6. Statistical Analysis
Data analysis was done using GraphPad PRISM (version 9.3.1). Male and female mice results were combined for analysis unless otherwise indicated in text or figures. Data are presented as mean ± SEM, with individual data points shown as indicated in figures. Significant differences were determined using multiple t test (two-tailed unpaired), one way analysis of variance (ANOVA) or two-way ANOVA, as indicated in figure legends. The significance level was set at p < 0.05, after adjustment for multiple comparisons as necessary.
Supplemental Materials
The following supporting information can be downloaded at Preprints.org: Supplemental Figure S1 and S2 as well as Supplemental Table S1.
Author Contributions
Conceptualization, M.G.A. and S.B.W.; methodology, M.G.A. and L.L.; formal analysis, M.G.A. and S.B.W.; investigation, M.G.A. and S.B.W.; data curation, M.G.A. and S.B.W.; writing - original draft preparation, M.G.A.; writing - review and editing, S.B.W.; visualization, M.G.A. and S.B.W.; supervision, S.B.W.; funding acquisition, S.B.W. All authors have read and agreed to the published version of the manuscript.
Funding Acknowledgments
This research was funded by a Project Grant (PJT-174988) to S.B.W. from the Canadian Institutes of Health Research. M.G.A. was supported by a James Regan Cardiology Research scholarship from University of Saskatchewan’s College of Medicine. S.B.W. was supported by a National New Investigator Award and McDonald Scholarship from Heart and Stroke Foundation of Canada.
Data Availability Statement
Data corresponding to figures have been provided in the Mendeley repository (###).
Declaration of conflict of interest
All authors agree to the submission of the manuscript and affirm that the material submitted for publication has not been reported before and is not under consideration for publication elsewhere. The authors have no conflicts of interest to disclose.
References
- Le MH, Yeo YH, Li X, Li J, Zou B, Wu Y, Ye Q; et al. 2019 Global NAFLD Prevalence: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2022;20:2809-2817 e2828. [CrossRef]
- Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, Colombo M; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol 2018;69:896-904. [CrossRef]
- Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020;158:1999-2014 e1991. [CrossRef]
- Diehl AM, Day C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N Engl J Med 2017;377:2063-2072. [CrossRef]
- Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, Hultcrantz R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015;61:1547-1554. [CrossRef]
- Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010;51:1972-1978. [CrossRef]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J; et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11-20. [CrossRef]
- Akl MG, Widenmaier SB. Immunometabolic factors contributing to obesity-linked hepatocellular carcinoma. Front Cell Dev Biol 2022;10:1089124. [CrossRef]
- Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021;184:2537-2564. [CrossRef]
- Horn CL, Morales AL, Savard C, Farrell GC, Ioannou GN. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol Commun 2022;6:12-35. [CrossRef]
- Carvalho-Gontijo R, Han C, Zhang L, Zhang V, Hosseini M, Mekeel K, Schnabl B; et al. Metabolic Injury of Hepatocytes Promotes Progression of NAFLD and AALD. Semin Liver Dis 2022;42:233-249. [CrossRef]
- Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R, Kechagias S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol 2017;67:1265-1273. [CrossRef]
- Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev 2018;98:1169-1203. [CrossRef]
- Zhang Y, Xiang Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem J 2016;473:961-1000. [CrossRef]
- Akl MG, Baccetto R, Stebbings BM, Li L, Widenmaier SB. Euglycemia is affected by stress defense factor hepatocyte NRF1, but not NRF2. Biochem Biophys Res Commun 2023;668:96-103. [CrossRef]
- Akl MG, Li L, Baccetto R, Phanse S, Zhang Q, Trites MJ, McDonald S; et al. Complementary gene regulation by NRF1 and NRF2 protects against hepatic cholesterol overload. Cell Rep 2023;42:112399. [CrossRef]
- Hirotsu Y, Hataya N, Katsuoka F, Yamamoto M. NF-E2-related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC-1beta genes. Mol Cell Biol 2012;32:2760-2770.
- Kamisako T, Tanaka Y, Kishino Y, Ikeda T, Yamamoto K, Masuda S, Ogawa H. Role of Nrf2 in the alteration of cholesterol and bile acid metabolism-related gene expression by dietary cholesterol in high fat-fed mice. J Clin Biochem Nutr 2014;54:90-94. [CrossRef]
- Okada K, Warabi E, Sugimoto H, Horie M, Gotoh N, Tokushige K, Hashimoto E; et al. Deletion of Nrf2 leads to rapid progression of steatohepatitis in mice fed atherogenic plus high-fat diet. J Gastroenterol 2013;48:620-632. [CrossRef]
- Sugimoto H, Okada K, Shoda J, Warabi E, Ishige K, Ueda T, Taguchi K; et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol 2010;298:G283-294. [CrossRef]
- Trites MJ, Stebbings BM, Aoki H, Phanse S, Akl MG, Li L, Babu M; et al. HDL functionality is dependent on hepatocyte stress defense factors Nrf1 and Nrf2. Front Physiol 2023;14:1212785. [CrossRef]
- Widenmaier SB, Snyder NA, Nguyen TB, Arduini A, Lee GY, Arruda AP, Saksi J; et al. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017;171:1094-1109 e1015. [CrossRef]
- Xu Z, Chen L, Leung L, Yen TS, Lee C, Chan JY. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci U S A 2005;102:4120-4125. [CrossRef]
- Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, Tsai PC; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017;541:81-86. [CrossRef]
- Radhakrishnan SK, den Besten W, Deshaies RJ. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014;3:e01856. [CrossRef]
- Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 2010;38:17-28. [CrossRef]
- Bartelt A, Widenmaier SB. Proteostasis in thermogenesis and obesity. Biol Chem 2020;401:1019-1030. [CrossRef]
- Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem 2008;283:33554-33562. [CrossRef]
- Wufuer R, Fan Z, Liu K, Zhang Y. Differential Yet Integral Contributions of Nrf1 and Nrf2 in the Human HepG2 Cells on Antioxidant Cytoprotective Response against Tert-Butylhydroquinone as a Pro-Oxidative Stressor. Antioxidants (Basel) 2021;10. [CrossRef]
- Wufuer R, Fan Z, Yuan J, Zheng Z, Hu S, Sun G, Zhang Y. Distinct Roles of Nrf1 and Nrf2 in Monitoring the Reductive Stress Response to Dithiothreitol (DTT). Antioxidants (Basel) 2022;11. [CrossRef]
- Wufuer R, Liu K, Feng J, Wang M, Hu S, Chen F, Lin S; et al. Distinct mechanisms by which Nrf1 and Nrf2 as drug targets contribute to the anticancer efficacy of cisplatin on hepatoma cells. Free Radic Biol Med 2024;213:488-511. [CrossRef]
- Katsuoka F, Otsuki A, Hatanaka N, Okuyama H, Yamamoto M. Target Gene Diversity of the Nrf1-MafG Transcription Factor Revealed by a Tethered Heterodimer. Mol Cell Biol 2022;42:e0052021. [CrossRef]
- Chen J, Wang M, Xiang Y, Ru X, Ren Y, Liu X, Qiu L; et al. Nrf1 Is Endowed with a Dominant Tumor-Repressing Effect onto the Wnt/beta-Catenin-Dependent and Wnt/beta-Catenin-Independent Signaling Networks in the Human Liver Cancer. Oxid Med Cell Longev 2020;2020:5138539.
- Ren Y, Qiu L, Lu F, Ru X, Li S, Xiang Y, Yu S; et al. TALENs-directed knockout of the full-length transcription factor Nrf1alpha that represses malignant behaviour of human hepatocellular carcinoma (HepG2) cells. Sci Rep 2016;6:23775. [CrossRef]
- Wang M, Ren Y, Hu S, Liu K, Qiu L, Zhang Y. TCF11 Has a Potent Tumor-Repressing Effect Than Its Prototypic Nrf1alpha by Definition of Both Similar Yet Different Regulatory Profiles, With a Striking Disparity From Nrf2. Front Oncol 2021;11:707032.
- Sharma RS, Harrison DJ, Kisielewski D, Cassidy DM, McNeilly AD, Gallagher JR, Walsh SV; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell Mol Gastroenterol Hepatol 2018;5:367-398. [CrossRef]
- Mohs A, Otto T, Schneider KM, Peltzer M, Boekschoten M, Holland CH, Hudert CA; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J Hepatol 2021;74:638-648. [CrossRef]
- Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, Zhong Z; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016;29:935-948. [CrossRef]
- He F, Antonucci L, Yamachika S, Zhang Z, Taniguchi K, Umemura A, Hatzivassiliou G; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J Hepatol 2020;72:1182-1195. [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327-1341 e1323.
- Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, Tanaka H; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet 2016;48:500-509. [CrossRef]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505-511. [CrossRef]
- Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab 2019;29:18-26.
- Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol 2018;68:230-237. [CrossRef]
- Hansen HH, HM AE, Oro D, Evers SS, Heeboll S, Eriksen PL, Thomsen KL; et al. Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol 2020;20:210. [CrossRef]
- Assy N, Minuk GY. Liver regeneration: Methods for monitoring and their applications. J Hepatol 1997;26:945-952. [CrossRef]
- Basaranoglu M, Turhan N, Sonsuz A, Basaranoglu G. Mallory-Denk Bodies in chronic hepatitis. World J Gastroenterol 2011;17:2172-2177.
- Stumptner C, Fuchsbichler A, Zatloukal K, Denk H. In vitro production of Mallory bodies and intracellular hyaline bodies: The central role of sequestosome 1/p62. Hepatology 2007;46:851-860.
- Taniguchi K, Yamachika S, He F, Karin M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett 2016;590:2375-2397. [CrossRef]
- Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 2018;69:385-395. [CrossRef]
- Espanol-Suner R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, Jacquemin P; et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012;143:1564-1575 e1567. [CrossRef]
- Van Hul N, Lanthier N, Espanol Suner R, Abarca Quinones J, van Rooijen N, Leclercq I. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am J Pathol 2011;179:1839-1850.
- Zhang L, Theise N, Chua M, Reid LM. The stem cell niche of human livers: Symmetry between development and regeneration. Hepatology 2008;48:1598-1607. [CrossRef]
- Shafritz DA, Dabeva MD. Liver stem cells and model systems for liver repopulation. J Hepatol 2002;36:552-564. [CrossRef]
- Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, Havekes LM; et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014;9:e115922. [CrossRef]
- Sapi J, Kovacs L, Drexler DA, Kocsis P, Gajari D, Sapi Z. Tumor Volume Estimation and Quasi-Continuous Administration for Most Effective Bevacizumab Therapy. PLoS ONE 2015;10:e0142190.
Figure 1.
HFFC diet promotes weight gain and MASH. Mice were fed control diet or diet enriched with high fat, fructose, and 1% cholesterol (HFFC) for 24 weeks. A-B) % change in body weight relative and % liver to body weight ratio (A) and levels of triglyceride and cholesterol in liver (B), comparing diets (n = 11-17). C) Liver qPCR analysis for indicated gene expression, normalized by ribosomal protein 36b4 (n = 11-15). (D) Liver sections stained with hematoxylin and eosin (H&E) or that underwent immunohistochemistry (IHC) with antibody detecting ki67 or p62, with scale indicated in panel. (E) Steatosis and inflammation in liver determined using H&E-stained sections, and % of ki67 positive cells and % area of p62 determined using IHC (n = 8-17). Data in A-C and E are mean ± standard error of the mean, with individual data points shown. The p-value was determined by multiple unpaired t-tests, with adjustment for multiple comparisons.
Figure 1.
HFFC diet promotes weight gain and MASH. Mice were fed control diet or diet enriched with high fat, fructose, and 1% cholesterol (HFFC) for 24 weeks. A-B) % change in body weight relative and % liver to body weight ratio (A) and levels of triglyceride and cholesterol in liver (B), comparing diets (n = 11-17). C) Liver qPCR analysis for indicated gene expression, normalized by ribosomal protein 36b4 (n = 11-15). (D) Liver sections stained with hematoxylin and eosin (H&E) or that underwent immunohistochemistry (IHC) with antibody detecting ki67 or p62, with scale indicated in panel. (E) Steatosis and inflammation in liver determined using H&E-stained sections, and % of ki67 positive cells and % area of p62 determined using IHC (n = 8-17). Data in A-C and E are mean ± standard error of the mean, with individual data points shown. The p-value was determined by multiple unpaired t-tests, with adjustment for multiple comparisons.
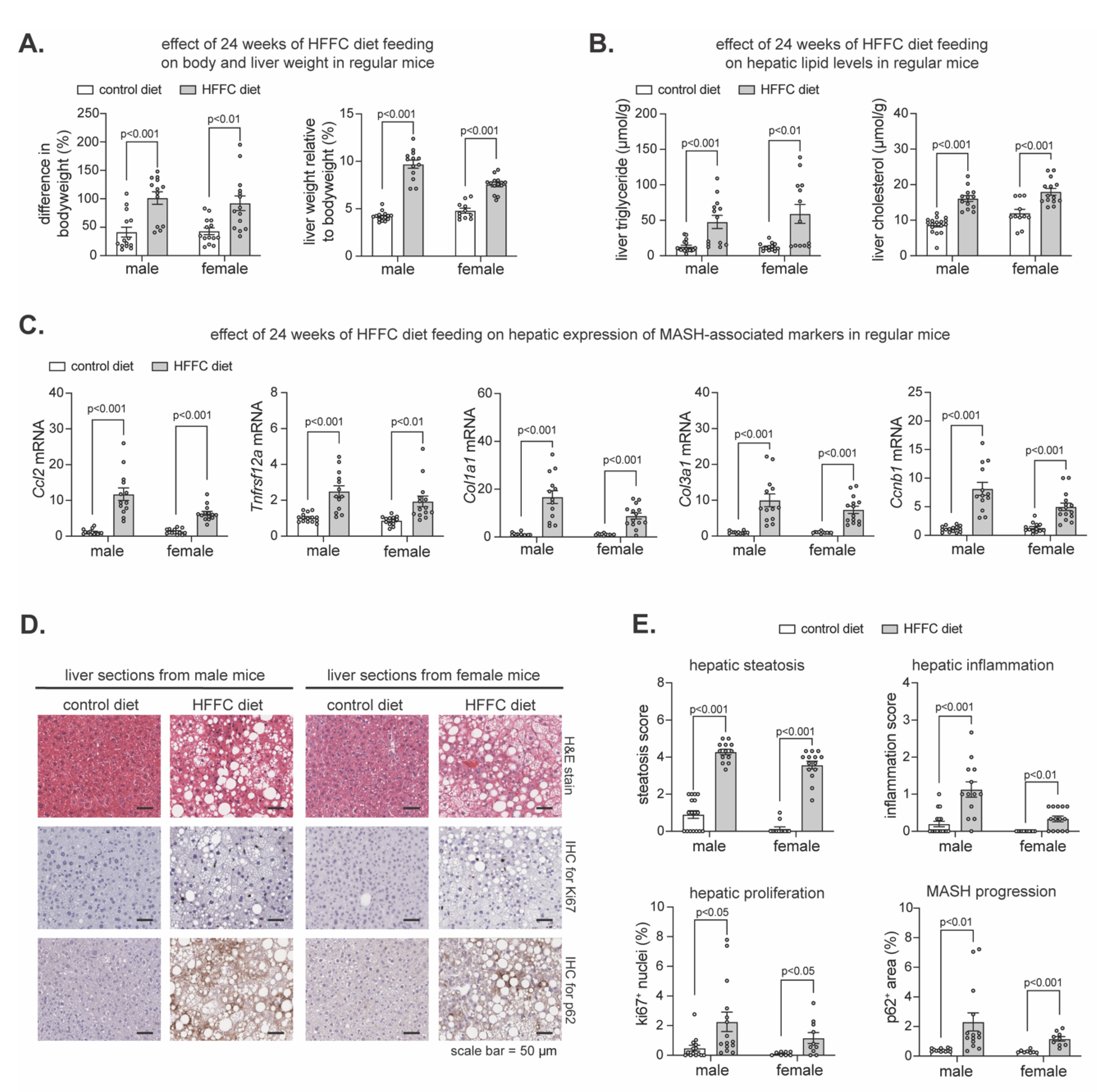
Figure 2.
Effect of hepatocyte deficiency for Nrf1, Nrf2, or both in mice chronically fed HFFC diet. Mice were fed HFFC diet for 24 weeks. In (A–C), mice were infected with indicated virus on week 22. In (D), mice were infected on week 16. (A) Liver sections stained with hematoxylin and eosin, with scale indicated in panel, and steatosis and inflammation in liver (n = 5-14). (B) Levels of triglyceride and cholesterol in liver (n = 6-14). (C) Liver qPCR analysis for indicated gene expression, normalized by ribosomal protein 36b4 (n = 5-14). (D) Representative liver sections that underwent immunohistochemistry (IHC) with antibody detecting ki67 or p62, with scale indicated in panel, and % of ki67 positive cells and % area of p62 (n = 6-23). Data are mean ± standard error of the mean, with individual data points shown (males = circles; females = triangle). In (A–C), the p-value was determined by two-way analysis of variance, with Sidak post-test. In D), the p-value was determined by one-way analysis of variance, with Dunnett post-test.
Figure 2.
Effect of hepatocyte deficiency for Nrf1, Nrf2, or both in mice chronically fed HFFC diet. Mice were fed HFFC diet for 24 weeks. In (A–C), mice were infected with indicated virus on week 22. In (D), mice were infected on week 16. (A) Liver sections stained with hematoxylin and eosin, with scale indicated in panel, and steatosis and inflammation in liver (n = 5-14). (B) Levels of triglyceride and cholesterol in liver (n = 6-14). (C) Liver qPCR analysis for indicated gene expression, normalized by ribosomal protein 36b4 (n = 5-14). (D) Representative liver sections that underwent immunohistochemistry (IHC) with antibody detecting ki67 or p62, with scale indicated in panel, and % of ki67 positive cells and % area of p62 (n = 6-23). Data are mean ± standard error of the mean, with individual data points shown (males = circles; females = triangle). In (A–C), the p-value was determined by two-way analysis of variance, with Sidak post-test. In D), the p-value was determined by one-way analysis of variance, with Dunnett post-test.
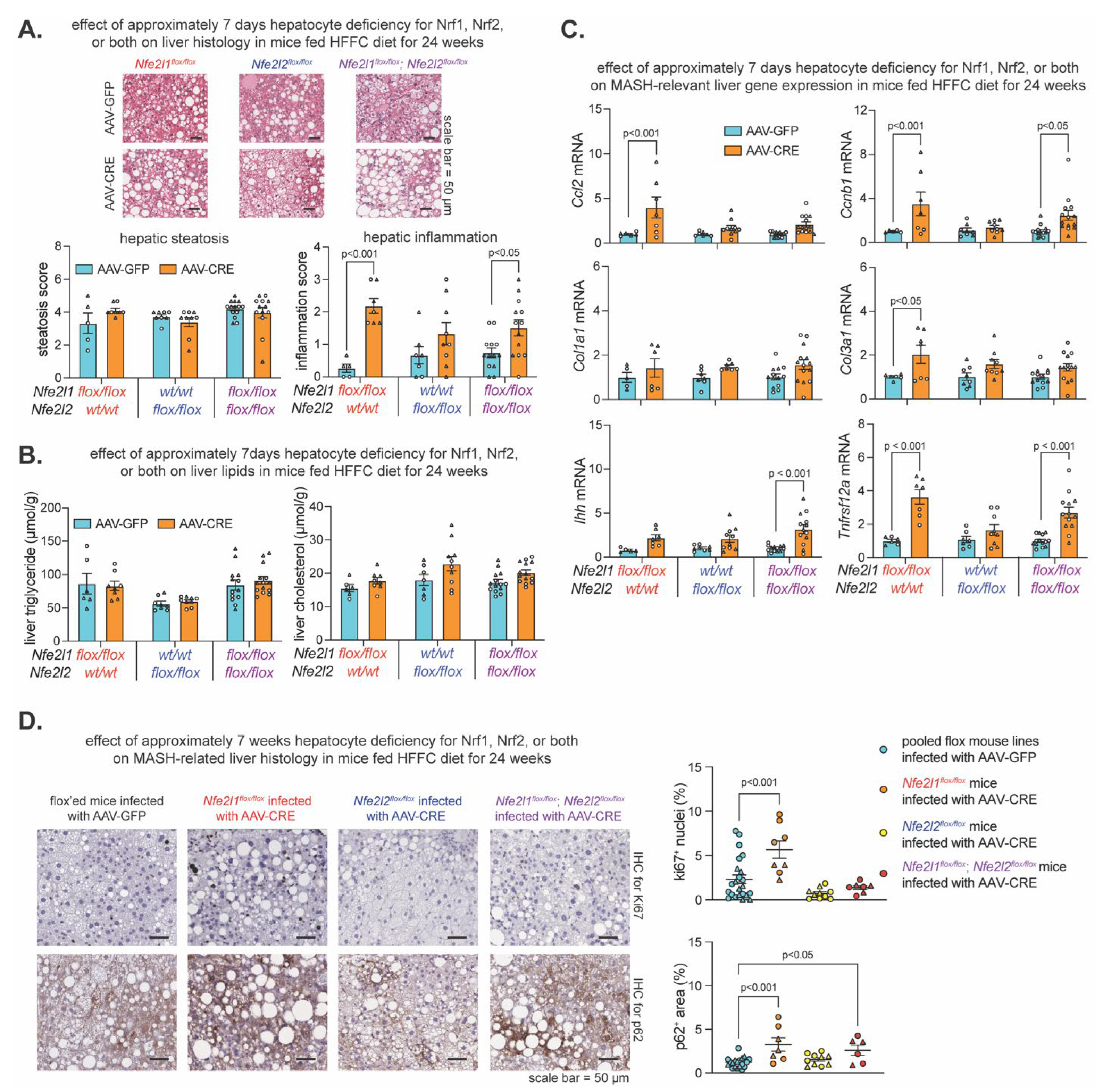
Figure 3.
Effect of hepatocyte deficiency for Nrf1, Nrf2, or both on liver tumor development. (A) Study design showing that mice were fed HFFC diet with 2% cholesterol for 52 weeks and infected on week 16, 32, and 48 with indicated virus. Liver tumor analysis was done at the endpoint. B-C) % incidence of liver tumors (B) and the number of tumors per liver (C) in males (n = 5-7) and females (n = 5-6). (D) Volume of largest liver tumor in males (n = 5-7) and females (n = 5-6). Data are mean ± standard error of the mean, with individual data points shown. The p-value was determined by two-way analysis of variance, with Sidak post-test.
Figure 3.
Effect of hepatocyte deficiency for Nrf1, Nrf2, or both on liver tumor development. (A) Study design showing that mice were fed HFFC diet with 2% cholesterol for 52 weeks and infected on week 16, 32, and 48 with indicated virus. Liver tumor analysis was done at the endpoint. B-C) % incidence of liver tumors (B) and the number of tumors per liver (C) in males (n = 5-7) and females (n = 5-6). (D) Volume of largest liver tumor in males (n = 5-7) and females (n = 5-6). Data are mean ± standard error of the mean, with individual data points shown. The p-value was determined by two-way analysis of variance, with Sidak post-test.
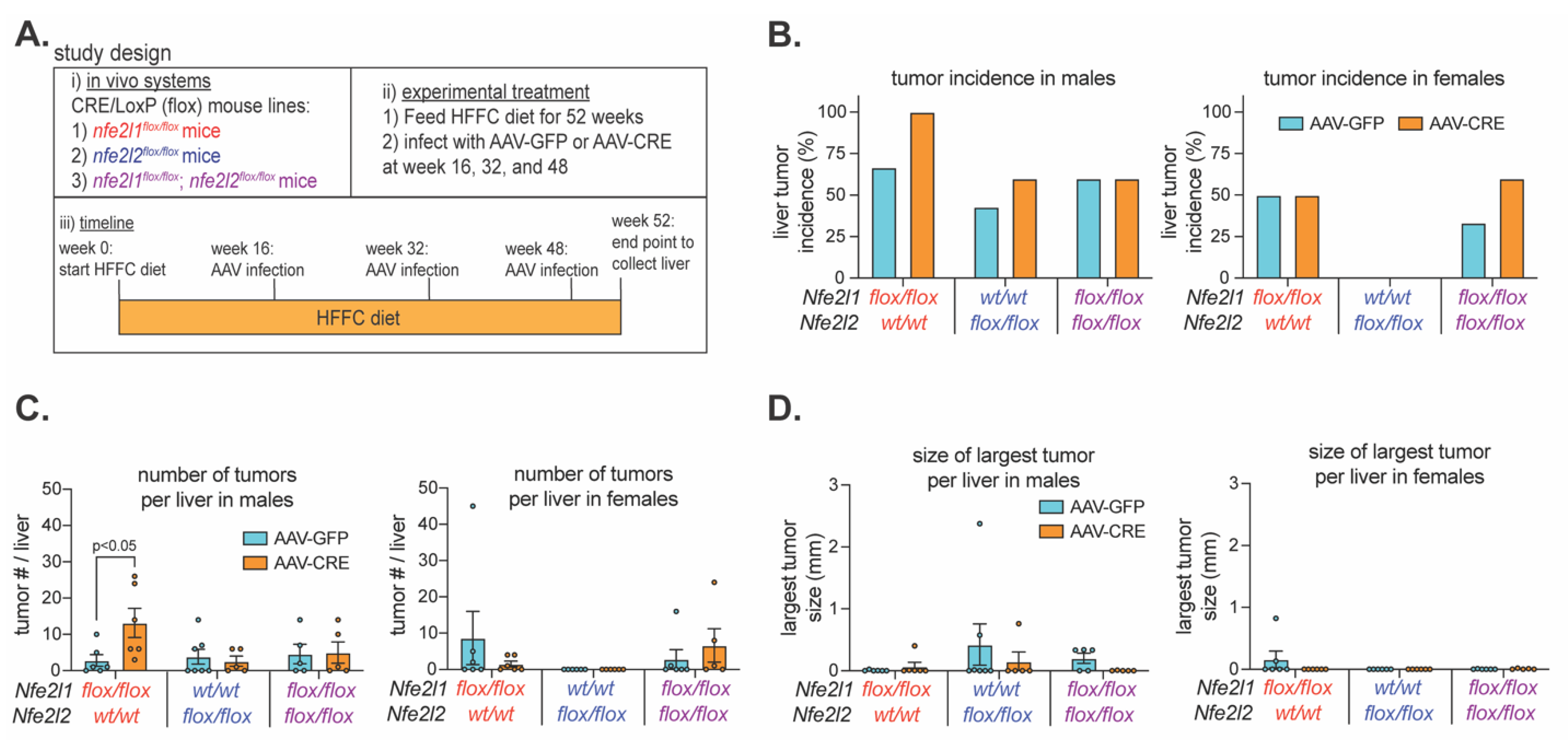
Figure 4.
Combined effect of Nrf2-inducing drug bardoxolone and Nrf1 overexpression on MASH-linked fibrosis. (A) Study design showing C57bl/6J mice that were fed HFFC diet with 2% cholesterol for 24 weeks. Mice were injected with carbon tetrachloride once per week from week 0-15 to induce liver fibrosis. On week 16-24, mice were treated as indicated with modulators of Nrf1 and Nrf2 activity. Liver analysis was done at the endpoint. (B) Liver sections stained with hematoxylin and eosin, with scale indicated in panel, and corresponding score for steatosis and inflammation in liver (n = 12-15). (C) Liver sections stained with Masson Trichrome, with scale indicated in panel, and corresponding score for liver fibrosis (n = 10-14). (D) Liver qPCR analysis for indicated gene expression, normalized by ribosomal protein 36b4 (n = 11-15). Data are mean ± standard error of the mean, with individual data points shown (males = circles; females = triangle). The p-value was determined by one-way analysis of variance, with Dunnett post-test.
Figure 4.
Combined effect of Nrf2-inducing drug bardoxolone and Nrf1 overexpression on MASH-linked fibrosis. (A) Study design showing C57bl/6J mice that were fed HFFC diet with 2% cholesterol for 24 weeks. Mice were injected with carbon tetrachloride once per week from week 0-15 to induce liver fibrosis. On week 16-24, mice were treated as indicated with modulators of Nrf1 and Nrf2 activity. Liver analysis was done at the endpoint. (B) Liver sections stained with hematoxylin and eosin, with scale indicated in panel, and corresponding score for steatosis and inflammation in liver (n = 12-15). (C) Liver sections stained with Masson Trichrome, with scale indicated in panel, and corresponding score for liver fibrosis (n = 10-14). (D) Liver qPCR analysis for indicated gene expression, normalized by ribosomal protein 36b4 (n = 11-15). Data are mean ± standard error of the mean, with individual data points shown (males = circles; females = triangle). The p-value was determined by one-way analysis of variance, with Dunnett post-test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



