Submitted:
17 April 2024
Posted:
17 April 2024
You are already at the latest version
Abstract
The p53 tumor suppressor protein shows antiviral activity. The replication of viruses is also antagonized by interferons (IFNs) – cytokines, which control immunity genes by the STAT transcription factors. The best studied interferons belong to type I (e.g. IFNα) and type II (IFNγ) groups. IFNα and IFNγ both induce activating phosphorylation of STAT1 on Tyr701. Previously, we noticed that p53 activates SOCS1, which is a negative regulator of STAT1 phosphorylation. Thus, we hypothesized that p53 by activating SOCS1 expression reduces phosphorylation of STAT1 and attenuates activation of genes stimulated either by IFNα or IFNγ. To test this hypothesis, we exposed p53-proficient and p53-deficient cells to strong p53 activators, and either to IFNα or IFNγ. Subsequently, we estimated the phosphorylation status of STAT1 and the expression of genes regulated by either of these cytokines. Strong activation of p53 reduced the phosphorylation of STAT1 on Tyr701, however surprisingly it did not lead to reduced expression of most tested interferon-stimulated genes. On contrary, p53 and IFNγ synergized in the activation of CASP1, IFIT1 and IFIT3 genes. We conclude that the interactions between p53 and interferon-activated pathways are more complicated than initially expected, but the apparent cooperation between p53 and IFNγ deserves further studies.
Keywords:
p53
; interferon
; STAT1
; NK-92
; caspase-1
; innate immunity
1. Introduction
The p53 protein coded by TP53 is best-known for inhibition of the cell cycle and activation of apoptosis [1]. However, p53 protein acts not only as the tumor suppressor, but also shows antiviral activity, e.g. by stimulating the expression of innate and adaptive immunity genes. For this reason, p53 is a frequent target of inactivation by proteins coded by viruses [2]. Notably, p53 was discovered as a protein bound and inactivated by large T antigen of the SV40 virus [3]. Furthermore, p53 is a transcription regulator, which directly activates at least several hundred of genes [4]. There is also a large group of genes repressed by p53, however, this repression is executed indirectly, by activating the gene for p21 protein, which inhibits cyclin-dependent kinases, what in turn promotes the formation of repressive complexes on genes coding for cell cycle genes [5]. Interestingly, p53 activates the expression of many genes, which are also stimulated by interferons [6].
Interferons are cytokines, which regulate both innate and adaptive immunity and play a major role in the defense against viruses and bacteria. Interferons are strong activators of gene expression. Their activity is mediated by transcription factors from the STAT family. There are three types of interferons, each signals through different receptors. The best-studied are interferons α (IFNα) and β (IFNβ) (type I) and interferon γ (IFNγ, type II). IFNα can be secreted by all cells, whereas IFNγ is secreted only by T cells and natural killer cells. The activity of interferons-α and interferon-γ critically depends on STAT1 transcription factor. Binding of type I interferons to their cognate receptor induces its dimerization, which in turn activates two kinases JAK1 and TYK2. Activated kinases phosphorylate interferon receptor on target tyrosine residues, what creates a docking site for STAT1 and STAT2 transcription regulators, which become phosphorylated on a critical tyrosine residues – Tyr701 on STAT1 and Tyr690 on STAT2. Phosphorylated STAT1 and STAT2 bind to the IRF9 protein and as a trimer known as interferon-stimulated gene factor 3 (ISGF3) bind to the DNA sequence known as interferon-stimulated response element (ISRE) and activate a set of interferon-stimulated genes (ISGs). IFNγ binds to its own receptor and induces the activating phosphorylation of STAT1 on Tyr701. Phosphorylated STAT1 forms a homodimer known as γ-activated factor (GAF) and activates its own set of genes. The general term interferon-stimulated genes is slightly misleading because type I and type II interferons activate different but overlapping sets of genes [7].
The signaling pathways activated by interferon-α or interferon-γ have negative regulators. One of them is the SOCS1 protein, which prevents STAT1 phosphorylation. The SOCS1 gene is activated by interferon-γ and it is an important element of the negative feedback loop in this signaling pathway [8,9]. Previously, we found that the SOCS1 gene is positively regulated by p53. We tested the regulation of SOCS1 by p53 because we noticed that strong activation of p53 was associated with the reduced phosphorylation of STAT1 on Tyr701 initiated by IFNα. The logical explanation of this observation was that p53 activates the negative regulator of phosphorylation of STAT1 on Tyr701. We found that in p53-deficient cells the activation of SOCS1 is attenuated, what indicates that p53 at least indirectly activates the expression of SOCS1 [10]. Thus, based on this observation, we hypothesized that strong activation of p53 can attenuate the expression of genes regulated by phosphorylated STAT1, e.g., the genes activated by either interferon α or γ. We started this study by testing this hypothesis.
2. Results
2.1. p53 Can Either Activate or Repress SOCS1 Expression Depending on the Cell Line
In our earlier paper, we demonstrated that the negative regulator of STAT1 activity – the SOCS1 protein is coded by a p53-activated gene [10]. Strong activation of p53 either by camptothecin or by the mixture of actinomycin D and nutlin-3a (A+N) led to upregulation of SOCS1 mRNA and protein. This was associated with attenuated phosphorylation of STAT1 in cells exposed to IFNα1 and A+N [10]. In the first step of the current project, we tested if various activators of p53 are able to upregulate SOCS1 in other cell lines. U-2 OS cells, with wild-type p53, are frequently employed in transcriptomic studies to search for p53-regulated genes [4]. As a positive control, alongside with U-2 OS cells, we exposed the A549 cell line to actinomycin D, nutlin-3a, combination of both substances (A+N) or a camptothecin, as demonstrated on Figure 1A. Expectedly, in A549 cells SOCS1 expression was upregulated by camptothecin and by A+N. However, in U-2 OS cells, the results were surprising. First, we noticed strong, constitutive expression of SOCS1. Second, we noticed that following exposure to actinomycin D, nutlin-3a or A+N the expression of SOCS1 was significantly attenuated. This suggested that activation of p53 in this cell line leads to repression of highly expressed SOCS1. In another cell line with wild-type p53 – NCI-H292, in control conditions the expression of SOCS1 was undetectable but was strongly upregulated following exposure either to camptothecin or A+N (Figure 1B). In contrast to this, in p53-null cells - NCI-H1299, the expression of SOCS1 was easily detectable in cells growing in control conditions but was not upregulated following any treatment conditions. Thus, cells growing in standard conditions differ in the expression level of SOCS1. Moreover, SOCS1 may respond differently to activation of p53, in some cells it is upregulated (A549, NCI-H292) and in some cells it is downregulated (U-2 OS).
To test the hypothesis that p53 is responsible for SOCS1 repression in U-2 OS cells, we prepared by CRISPR/Cas9 technology the clones with p53 knockout. Two control clones and four knockout clones were exposed to A+N or to nutlin-3a (Figure 1C). In the control clones, both treatment modalities led to a strong repression of SOCS1 as in the parental cell line. However, in all p53 knockout clones, the nutlin-3a (but not A+N) lost the ability to repress SOCS1. Thus, p53 can either upregulate or downregulate the expression of SOCS1 depending on cellular context. Furthermore, the ability of p53 to downregulate SOCS1 strictly depends on the mode of p53 activation. Only p53 activated by nutlin-3a but not by camptothecin or A+N combination is able to downregulate the expression of SOCS1 in U-2 OS cells. The plausible explanation will be presented in the Discussion.
2.2. Treatment with A+N Attenuates Phosphorylation of STAT1 Initiated by IFNα1
In order to compare how the activation of p53 by A+N modulates the response of cells to interferon-α1, we pre-exposed A549 or U-2 OS cells to A+N for 24h (control cells were mock-treated) and subsequently IFNα1 was added either to cells treated with A+N or to cells growing in control conditions as shown on Figure 2A. In both cell lines, the exposure to IFNα1 resulted in phosphorylation of STAT1 on Tyr701 (Y701), what is consistent with known molecular consequences of IFNα1 activity. However, in A549 cells the exposure to A+N strongly attenuated the phosphorylation of STAT1, which was accompanied by upregulation of SOCS1, whereas in U-2 OS cells the phosphorylation of STAT1 was not altered significantly even though the expression of SOCS1 significantly dropped. Therefore, in case of U-2 OS cells there is no relationship between expression of SOCS1 and the phosphorylation status of STAT1. Hence, further experiments were performed using A549 cells.
Next, we compared how strong or weak activation of p53, by A+N or nutlin-3a respectively, modulates phosphorylation of STAT1. A549 cells were pre-exposed either to A+N or nutlin-3a and after 24h the cells were additionally treated with IFNα1 as shown on Figure 2B. The level of activation of p53 is visualized by its phosphorylation on Ser37 [11]. Pre-exposure to A+N attenuated phosphorylation of STAT1, whereas pre-exposure to nutlin-3a had weak effect. Consistent with previous findings, SOCS1 was upregulated only in cells exposed to A+N. In this experiment, we additionally examined the expression of MX1 protein, which is encoded by the archetypal IFNα-activated gene [12]. Surprisingly, the phosphorylation level of STAT1 was not associated with expression of MX1, which was almost identical in every case, when the cells were exposed to IFNα1 (Figure 2B). This is very odd, because phosphorylation of STAT1 is considered as measure of its activation, hence we expected lower expression of MX1 in cells pre-exposed to A+N and treated with IFNα1. Thus, even though pre-treatment with A+N activates p53 and leads to increased expression of SOCS1 and this in turn is linked to diminished phosphorylation of STAT1 on Tyr701, this does not impact on the expression of the gene (MX1) activated by the complex containing STAT1. This indicates that our understanding of the mechanisms of activation of interferon-regulated genes are far from satisfactory.
To further explore the relationship between the phosphorylation status of STAT1 and the expression of MX1 we performed similar experiment (pretreatment with A+N with the subsequent treatment with IFNα1), however this time we exposed cells to various concentrations of the cytokine. The results are presented on Figure 3A. Pre-exposure to A+N reduced the phosphorylation of STAT1 triggered by IFNα1. It was visible in concentration of IFNα1 ranging from 0.05-1.0 ng/ml; however, it did not modulate the expression of MX1. Comparison of the amount of phospho-STAT1 between various experimental conditions shows that in spite of similar phospho-STAT1 level the expression of MX1 is clearly different (compare the lanes 5 versus 6, 7 versus 9 and 7 versus 8). This experiment clearly confirms the lack of correlation between the amount of phosphorylated STAT1 and the expression of MX1. However, it shows that lower concentration of the cytokine is associated with lower expression of MX1. Thus, even though the activation of p53 is associated with reduced phosphorylation of STAT1, it does not affect the expression of the gene regulated by phospho-STAT1.
2.3. p53 Attenuates the Phosphorylation of STAT1 Initiated by IFNα1
The exposure of cells to A+N attenuates the phosphorylation of STAT1. Does A+N act in this regard through activation of p53? To answer this question we tested how the p53 status modulates the phosphorylation of STAT1. We employed a mixture of clones of A549 cells with attenuated expression of p53. The p53-deficient cells were prepared as described previously [13]. IFNα1 acting alone induced strong phosphorylation of STAT1, which was not modulated by p53 status (Figure 3B, lanes 7, 8). Pre-exposure to A+N significantly attenuated phosphorylation of STAT1 induced by IFNα1 but only in cells with wild-type p53 (Figure 3B, lanes 5, 7). In p53-deficient cells, pre-exposure to A+N only slightly reduced the phosphorylation of STAT1 (Figure 3B, lanes 6, 8). As in previous experiments, the phosphorylation of STAT1 was not correlated with the expression of MX1 (compare lanes 5 and 6). Another gene activated by IFNα1 – IRF7 [14] was not affected either. Thus, the activation of p53 by A+N reduces the phosphorylation of STAT1 but it has no measurable influence on the expression of the STAT1 target genes, MX1 or IRF7.
2.4. Treatment with A+N Reduces Phosphorylation of STAT1 but Does Not Reduce the Expression of Most Tested Interferon-Stimulated Genes
The lack of association between the phosphorylation status of STAT1 and the expression of STAT1 targets – the MX1 and IRF7 is surprising. In order to find out how other genes activated by IFNα1 are modulated by A+N and by p53 status, we first treated p53 proficient and p53-deficient cells as presented on Figure 3B and then we isolated RNA and performed gene expression analysis by semi-quantitative RT-PCR (Figure 4). We found that for p53-proficient cells, the pre-treatment with A+N did not reduce the expression of any gene with the exception of IFI6. Furthermore, in case of IFI6 the knockdown of p53 prevented the reduction of expression by A+N. Thus, the expression of IFI6 was roughly correlated with the phosphorylation status of STAT1. The p53 status significantly modulates the expression of IFI44 in cells exposed to A+N and IFNα1. In p53-deficient cells this gene is more strongly activated. Thus, p53 negatively regulated expression of IFI44 in cells exposed to IFNα1. In case of other genes, the treatment with A+N did not reduce and in some cases it even augments their expression, when combined with IFNα1. An extreme example is the IFI27 gene, which is not activated by A+N, it is strongly activated by IFNα (approximately 250-fold), but when A+N and the cytokines are combined the expression of this gene is augmented even further (to approximately 800-fold) without measurable influence from p53. Thus, it appears that A+N and IFNα cooperate in activation of IFI27 in p53-independent manner. Similar cooperation between A+N and IFNα1 in gene activation (although not as spectacular as in the case if IFI27) was observed for OAS1, IFIT3, DDX60, IFIT2. We noticed that p53 regulates the expression of two genes in cells exposed only to A+N – IFIT3 and OAS1. Their expression is attenuated in p53-deficient cells exposed to A+N. Thus, these two genes appear as p53-activated genes. However, when the cells are additionally exposed to IFN-a, the influence of p53 on the expression of OAS1 is no longer measurable.
Interestingly, we found that in all treatment conditions the expression of IFITM3 gene is higher in p53-deficient cells (Figure 4). This is consistent with the observations of Wang et al. [15], who demonstrated that p53 negatively regulates the expression of this gene. The OAS3 gene is activated only by IFNα1 and neither exposure to A+N nor p53 status modulate its expression. In this regard it is similar to MX1 gene.
To summarize the RT-PCR analysis, we can conclude that various interferon-stimulated genes (ISGs) respond in different fashion to A+N, IFNα1, the co-treatment (A+N+IFNα1) and the status of p53. A+N appears to collaborate with IFNα in activation of some genes, but it happens in p53-independent a manner (e.g. IFI27, DDX60). P53 promotes the expression of some interferon-stimulated genes (OAS1, IFIT3). On the other hand, some of the examined ISGs, e.g., IFITM3, IFI44, are negatively regulated by p53. Thus, in our experimental conditions the relationships between the signaling systems governed by p53 and interferon-α1 are not simple, even though p53 clearly participates in reducing the phosphorylation of STAT1 and reduces expression of some interferon-stimulated genes. Moreover, our treatment modality (A+N) clearly collaborates with IFNα1 in the activation of some genes but the p53 status has no impact on this collaboration. Thus, A+N may be important regulator of innate immunity by the mechanism, which is unknown, but is clearly independent of p53.
2.5. p53 and IFNγ Synergize in the Activation of a Subset of Interferon-Stimulated Genes
The STAT1 protein is also activated by phosphorylation in response to other interferon – interferon-γ. Thus, SOCS1 activated by p53 may also influence the signaling pathway of this cytokine. Hence, we decided to examine how the activation of p53 by A+N impacts on the outcome of IFNγ treatment. First, we performed the time-course experiment exposing A549 cells to A+N, IFNγ or A+N and IFNγ for 6, 12, 24 and 48 hours. By Western blotting, we examined the activation status of p53 (total protein expression and its phosphorylation on Ser37), the phosphorylation of STAT1 (Tyr701) and the expression of genes known to be activated by this cytokine (IRF1, SOCS1, CASP1, IFIT1 and IFIT3). This Western blot shows several interesting points (Figure 5). The activation of the p53 protein rose steadily during whole treatment time with A+N, while the phosphorylation of STAT1 reached its peak after 6 h from the start of IFN-γ treatment and then gradually declined. A similar pattern was observed for the expression of SOCS1 and IRF1 in cells exposed to the interferon. The expression of CASP1 and IFIT1 increased steadily in cells exposed to A+N or in cells exposed to IFN-γ. When the two treatment modalities were combined, the expression of CASP1 and IFIT1 was further enhanced, reaching its peak after 48 h. Interestingly, at 24 h time point phosphorylation of STAT1 and the level of IRF1 were lower in cells co-exposed to A+N and interferon than in cells treated with the cytokine alone. This suggests that at some time point, the activation of p53 can reduce the phosphorylation of STAT1. On the other hand, it appears that p53 and IFN-γ cooperate in the activation of CASP1 and IFIT1.
To find out if p53 participates in the regulation of STAT1 phosphorylation and the expression of IRF1 and CASP1, we perform the treatment of p53 proficient and deficient cells as shown on Figure 6. This experiment confirms our previous observations that SOCS1, IRF1 and CASP1 are regulated by p53 because in p53-deficient cells A+N is unable to upregulate their expression. However, if the cells are additionally exposed to IFNγ, the effect of p53 on the expression of IRF1 and SOCS1 is lost. This is in stark contrast to the expression of CASP1. A+N and IFNγ clearly synergize in activation of its expression. Moreover, this effect strictly depends on p53 because in p53-deficient cells the effect of A+N is completely lost. This experiment also confirms that the exposure to A+N reduces the phosphorylation of STAT1 and the expression of IRF1 in cells exposed to IFNγ. In p53-deficient cells the STAT1 phosphorylation is stronger than in p53-proficient cells, what clearly suggests that p53 contributes to reduction of STAT1 activation triggered by A+N.
To find out how A+N and IFNγ cooperate in the regulation of other genes in p53 proficient and p53-deficient cells we performed RT-PCR analysis on RNA samples isolated from cells exposed as shown on Figure 6. The results are presented on Figure 7. We selected genes considered as activated by IFNγ. Alike in the case of IFNα1, we did not see a single pattern of cooperation between A+N and IFNγ. In p53-proficient cells, the expression of only two genes matched the phosphorylation status of STAT1 (lower in A+N+IFN-γ than in IFN-γ) – WARS1 and TAP2. The expression of TAP2 is additionally influenced by p53 status. IFI16 and SOCS1 show a very similar expression pattern in these treatment conditions – they are weakly upregulated by IFNy and strongly upregulated by A+N, but only in p53-proficient cells what is consisted with observations that they are p53-regulated genes [10,16]. The expression of IRF9 and IFIH1 is governed principally by IFNγ and A+N does not reduce its expression. Expression of IRF1 is governed by p53 in cells exposed to A+N, and this combination together with IFNγ do not collaborate in its upregulation. In this context, the expression pattern of CASP1 stands out. First, there is clear synergy between A+N and IFNγ in activation of this gene. Its upregulation by any treatment is very strong (up to 300-fold). The p53 is strictly required for its upregulation by A+N with or without IFNγ. Therefore, RT-PCR confirms the conclusion from Western blotting data that p53 and IFN-γ strongly synergize in the activation of CASP1 gene.
Because p53 is progressively activated during the treatment with A+N and the phosphorylation of STAT1 and the expression of some genes activated by IFNγ is the strongest after 6 h of cytokine treatment, our next experiment had a different setup. First, we exposed the cells for 24 h to A+N and then we additionally exposed them for 6 h to IFNγ as demonstrated on Figure 8. The Western blot shows that in p53-proficient cells the treatment with A+N slightly reduce the phosphorylation of STAT1. SOCS1 is activated in a p53-dependent manner only in cells exposed to A+N. The activation of this gene is much stronger after 6 h of exposure to IFNγ than after 30 h of exposure to A+N. Thus, in these experimental conditions IFNγ is a much stronger activator of SOCS1 than p53. A similar conclusion can be drawn from Figure 5. Consistent with previous experiment, this blot shows a very strong synergy between p53 and IFN-γ in the activation of CASP1. Next, we performed RT-PCR on RNA samples isolated from cells treated as shown on Figure 8. The results are presented on Figure 9. In these experimental conditions IRF1 is more potently activated by the cytokine than by p53 and these two molecules do not cooperate in activation of this gene. On the other hand, the IFI16 gene is more potently activated by p53 than by IFNγ and these two molecules appear to cooperate additively in its activation. A+N and IFNγ also cooperate in activation of ICAM1, but p53 does not seem to participate in this process. And finally, we observed a very strong and synergistic cooperation between p53 and IFNγ in the activation of CASP1, IFIT1 and IFIT3, the synergy being the strongest in case of the CASP1 gene. Thus, even though the treatment with A+N reduces the phosphorylation of STAT1 initiated by IFNγ, A+N does not reduce the expression of most examined genes activated by this cytokine, on the contrary, it cooperates with this cytokine in activation of a subset of genes, especially the ones upregulated late during exposure to IFNγ.
2.6. The Synergy between A+N and IFN-γ in Activation of CASP1 Is Cell-Type Specific
Next we tested if the synergy between A+N and IFN-γ in activation of CASP1 gene occurs in other cell types. We employed another cell line with wild-type p53 derived from lung cancer (NCI-H292). The cells were exposed to A+N, IFN-α, IFN-γ or the combination of these factors as shown on Figure 10A. We noticed that treatment with A+N attenuated phosphorylation of STAT1 and that both interferons strongly induced the expression of CASP1 and IRF1, which were not modified by A+N. This experiment confirms that in NCI-H292 cells, A+N treatment does not modulate the expression of the tested interferon-stimulated genes (CASP1, IRF1) (Figure 10A). To find out whether the synergy between p53 and IFNγ in upregulation of CASP1 occurs in non-cancerous cells, we exposed normal human fibroblasts (GM07492) as shown on Figure 10B. We examined the activation of p53, phosphorylation of STAT1 and the expression of both CASP1 and IRF1. Expectedly, exposure to the cytokine induced phosphorylation of STAT1 and strong expression of IRF1, however, exposure to A+N did not reduce the phosphorylation of STAT1. CASP1 was slightly upregulated by IFN-γ or by A+N, however, when the two treatments were combined the upregulation of CASP1 was very strong. Thus, the normal cells can respond by activation of CASP1 when they are simultaneously exposed to IFNγ and a factor, which strongly activates p53.
The negative effect of A+N on the phosphorylation status of STAT1 was observed in at least two cell types A549 and NCI-H292. Next, we tested the impact of p53 on STAT1 phosphorylation in another model of p53-deficient cells. We prepared p53-deficient NCI-H460 cells and we exposed them to A+N and IFN-γ as shown on Figure 10C. The exposure to A+N reduced phosphorylation of STAT1, however, the p53 status did not modify this effect. Thus, A+N can regulate the innate immunity through modulation of STAT1 phosphorylation; however, it can happen both in p53-dependent and independent fashion. In NCI-H460 cells, A+N does not appear to collaborate with IFNy in the activation of CASP1 gene. In this experiment, A549 cells served as a positive control for the collaboration between A+N and IFNy.
To further test the hypothesis, that A+N can reduce the phosphorylation of STAT1 triggered by IFNγ without the participation of p53, we employed the p53-null cell line, NCI-H1299. The cells were exposed as shown on Figure 11A. In addition to A+N co-treatment, we exposed cells separately to each of these compounds. In these p53-null cells, A+N co-treatment reduced the phosphorylation of STAT1 triggered by IFNγ. Unexpectedly, actinomycin D and nutlin-3a acting solo demonstrated a very similar effect. In spite of reduced level of phospho-STAT1, the expression of genes stimulated by IFNγ (IRF1, IFIH1, IFIT1, IFIT3) was not changed (Figure 11A). To find out whether actinomycin D and nutlin-3a collaborate in reducing STAT1 phosphorylation in A549 cells, we treated them as shown on Figure 11B. In these cells, both compounds clearly synergized in reducing STAT1 phosphorylation. Interestingly, both compounds independently collaborated with IFN-y in the activation of IFIT1.
Several of our experiments suggested that A+N combination can exert some effect on the expression of interferon-stimulated genes in p53-independent fashion. This is surprising because nutlin-3a was designed to specifically activate p53 by preventing interaction with its negative regulator, the MDM2 protein. In our earlier study [13], we demonstrated that A+N combination can activated some interferon-stimulated genes (IFIT1, IFIT3, IFIH1) in clones of p53-null A549 cells prepared using CRISPR/Cas9 technology and in p53-null NCI-H1299 cells. We decided to find out if this effect stems from the concerted action of both compounds or whether only one of them is responsible. Hence, we exposed NCI-H1299 cells for 48h as shown on Figure 11C. These data demonstrated that both actinomycin D and nutlin-3a synergize in activation of these genes. The effect is best visible in the case of IFIT3 gene. Thus, these two compounds can synergize in p53-independent fashion in the activation of some interferon-stimulated genes. It is a very important finding because it indicates that the antagonist of the p53-MDM2 interaction can modify the physiology of cells even in the absence of wild-type p53.
3.7. Actinomycin D and Nutlin-3a Can Sensitize both p53 Proficient and p53-Deficient Cells to the Killing Effect of NK-92 Cells
Our current and previous analyses demonstrated that co-treatment of p53-proficient and p53-deficient cells with A+N can upregulate the expression of many innate immunity genes [10]. Thus, these two drugs may, in principle, modulate the response of cancer cells to the effector cells of innate immunity. One of such effectors are natural killer (NK) cells. There is a commercially available cell line, NK-92, derived from lymphoma, which has the properties of NK cells. These cells are also used for experimental anti-cancer in vivo therapies in humans [17]. We employed these cells to find out if pre-treatment with A+N sensitizes cancer cells to the killing effect of natural killer cells. First, we pretreated A549 cells with actinomycin D and nutlin-3a for 48h (control cells were mock-treated) and subsequently the control and treated cells were incubated either with empty medium or with NK-92 cells at two ratios (NK-92:A549) 1:1 or 5:1 (Fig.12A). With increasing ratios, the number of control A549 cells decreased. The pretreatment of cells with A+N sensitized them to the killing effect of NK-92 cells. The low viability of cells pre-exposed to A+N and co-incubated with NK-92 cells did not result from the cytostatic effect of A+N, because we normalized viability of cells pre-exposed to A+N and incubated without NK-92 cells to 100%. Thus, incubation of mock-treated A549 cells with NK-92 cells (1:1 ratio) reduced the viability of cancer cells from 100% to approximately 70%, whereas the incubation of pre-treated A549 cells with NK-92% reduced the viability of cancer cells from 100% to approximately 20%. Because the NK-92 cells used at 1:1 ratio killed the substantial fraction of cancer cells, we used this ratio in the following experiments.
Next, A549 cells were preexposed to either actinomycin D, to nutlin-3a, or to both compounds (A + N) and then they were co-incubated with NK-92 cells. The co-treatment resulted in stronger sensitization that the treatment with each compound separately (Figure 12B). Subsequently, we wanted to find out if the sensitizing effect can be visible in other cell line – NCI-H460 cells. The result was very similar to what we observed in A549 cells (Figure 12C). Interestingly, the sensitizing effect of A+N was also present in the p53-null cell line NCI-H1299 (Figure 12D). These cells were slightly more sensitive to NK-92 cells even when growing in control conditions. However, when these cells were exposed separately to actinomycin D or nutlin-3a the sensitizing effect of mono-treatment was not observed (Figure 12E). However, it emerged when the two compounds were combined together. To find out, if p53 contributes to the sensitizing effect of A+N, we performed this experiment on p53-deficient and p53-proficient A549 cells. We observed that more p53-deficient cells survived the co-incubation with NK-92 cells (Figure 12F). Although the difference did not reach the statistical significance, our observations strongly suggest that the combined exposure to A+N makes various cancer cells better targets for natural killer cells and that both p53-dependent and independent mechanisms contribute to this effect. To observe long term outcome of the pre-treatment with A+N and co-incubation with NK-92 cells, we employed a different experimental setting. The adherent cancer cells (A549 control or exposed to A+N) were co-incubated with NK-92 cells (growing in suspension) on 6-well plates for 24 h. Subsequently, the medium was removed and the adherent cells were allowed to recover and grow in fresh medium for 5 to 7 days (three biological replicates were performed). We allowed cells in the control well (exposed neither to A+N nor to NK-92 cells) to grow to a very dense monolayer to better visualize the scarcity of cancer cells surviving the exposure to A+N and NK-92 at 5:1 ratio (Figure 12G). Note that at these conditions even the cells exposed only to A+N were able to regrow, forming an almost complete monolayer. This experiment shows that hardly any cancer cells exposed to A+N survive the co-incubation with NK-92 cells, what indicates that some innate immunity systems boosted by A+N efficiently sensitize cancer cells to the effectors of innate immunity.
3. Discussion
The interactions between signaling pathways governed by interferons and by p53 were presented in our recent review [18] and in reviews prepared by others [2,19]. However, it must be stressed that the name interferons comprises distinct protein families, which are synthesized by various cells in reaction to diverse stimuli and elicit differing effects in target cells. Thus, speaking about interferon one must be explicit about the type of the cytokine. In this work we selected interferons belonging to two types – type I interferon-α1 and type II interferon-γ. Previously, we demonstrated that important negative regulator of cytokine signaling (including type I and type II interferons) – the SOCS1 gene, is activated in p53-dependent fashion in A549 lung cancer cell line [10]. We started the current study with testing whether SOCS1 is induced by various p53 activators in other cancer cell lines. We found that this protein is upregulated by strong p53 activators in A549 and NCI-H292 cells (lung cancers), which express a low level of SOCS1 during normal growth conditions. Expectedly, in p53-null lung cell line NCI-H1299, neither p53 activator stimulated expression of SOCS1. Surprisingly, in U-2 OS cell line (osteosarcoma), which expresses high amount of SOCS1 in normal growth conditions, the expression of this protein was repressed by actinomycin D, nutlin-3a and both substances acting together (A+N). However, in p53 knockout clones, nutlin-3a but not A+N, was not able to repress SOCS1. Thus, in this cell line, the expression of SOCS1 is repressed by p53 activated by nutlin-3a but not by p53 activated by A+N. In our opinion, this is a very interesting observation, which should be extended in further studies. Why is p53 able to suppress SOCS1 when activated by nutlin-3a and is not able to do so when activated by A+N? This is another observation, which supports the idea that p53 activated by various stress factors has different biological properties. Moreover, our observation suggests that the direction of the change of a gene expression by p53 protein may depend on the steady state expression level of the gene. In cells with low level, the activated p53 may stimulate expression, whereas in cells with high level, the activated p53 may repress it. Our observations are consistent with the published transcriptomic data, which demonstrated the upregulation of SOCS1 by p53 in 12 reports and its downregulation by p53 in 9 reports [4]. Our data also suggest, that cancer cells differ in the steady-state level of SOCS1 what is consistent with data published by others [20]. Interestingly, depending on the context, SOCS1 can behave either as an oncogene or as a tumor suppressor [21]. It may be hypothesized that cancer cells with high expression of SOCS1 are more resistant to the anticancer properties of interferon-y. Thus, the level of this cytokine in tumor microenvironment might determine the selection pressure for expression of SOCS1.
Next we found that p53 activated by A+N attenuates the phosphorylation of STAT1 on Tyr701 induced by interferon-α1. Thus, p53 definitely has the potential to attenuate the expression of genes activated by this cytokine. However, surprisingly, attenuated phosphorylation of STAT1 did not translate into attenuated expression of two genes stimulated by IFNα1 – MX1 and IRF7. To study the expression of more interferon-stimulated genes, we performed RT-PCR analysis. We selected 9 genes activated by IFNα. We observed very variable pattern of gene regulation. Activation of three genes (IFI6, IFI44, IFITM3) by IFNα1 was attenuated by p53. In case of other genes, A+N and IFNα1 collaborated in their activation. In case of IFI27 the synergy between A+N and IFN α1 was very strong, however it was not influenced by p53 status. IFI27 protein may be involved in regulation of cell sensitivity to pro-apoptotic stimuli [22]. This experiment leads to two conclusions. First, p53 is able to attenuate the expression of only a subset of genes activated by IFNα1, what is consistent with the ability of p53 to attenuate phosphorylation of STAT1. Second, A+N treatment can collaborate with this cytokine in upregulation of a subset of genes, but p53 does not participate in it. This points toward the existence of a p53-independent mechanism able to activate innate immunity genes, which is triggered by actinomycin D and nutlin-3a. The biggest riddle of the experiments with IFNα1 is why there is no universal correlation between the phosphorylation of STAT1 and the expression level of most tested genes activated by this cytokine. With dose-response treatment there is clear correlation – both phosphorylation of STAT1 and expression of MX1 increase with the increasing concentration of interferon-α1. However, despite clearly different phosphorylation status of STAT1 between p53-proficient and deficient cells, the expression level of MX1 is virtually identical. The obvious explanation is that the relationship between phosphorylation status of STAT1 and the activity of transcription complex, which it forms together with STAT2 and IRF9 is more complicated than expected.
The ability of p53 to attenuate the expression of a subset of genes stimulated by IFNα was observed by Wang et al. [15]. They noticed this in case of genes for interferon-induced transmembrane proteins – IFITM1, IFITM2 and IFITM3. We also observed that IFITM3 is repressed by p53 (Figure 4). Wang et al. [15] observed that activation of p53 leading to repression of interferon-induced transmembrane proteins facilitates the infectivity if influenza A virus. On the other hand Muñoz-Fontela et al. [23] observed that infected mouse and human cells with functional p53 displayed markedly decreased viral replication early after infection. This inhibition of viral replication was mediated by a p53-dependent enhancement of IFN signaling, specifically the induction of some interferon-stimulated genes. These apparent discrepancies pose a serious biological question. Why does p53, which stimulates the expression of STING1 gene [10], which is the stimulator of interferon genes (hence the name), attenuate the expression of some genes induced by interferon-α? We believe that this system is very complicated because it is the result of long-lasting arms race between viruses and cells, and a measure “devised” by one system (e.g. a virus) is offset by a countermeasure “devised” by the other system (e.g. a cell). What we observe is an evolutionary snapshot of the current state of the battle. It is also possible that inhibition of STAT1 phosphorylation by p53 is an element of the negative-feedback loop of this signaling system. Our experiment (Figure 4) also shows that p53 and interferon-α activate common genes, e.g., IFIT3 and OAS1. We conclude it from the observation that expression of both genes is attenuated in p53-deficient cells exposed to A+N. Thus, p53 can attenuate the expression of some genes activated by IFN-a1 (e.g. IFITM3), it can activate some IFN-α-target genes in the absence of this cytokine (OAS1), however p53 does not participate in regulation of some genes, which are activated by concerted action of IFN-a1 and A+N (e.g. IFI27). This clearly points to conclusion that each interferon-stimulated gene in addition to being stimulated by these cytokines, has its own program of regulation when interferon is combined with other stress factors.
Having examined how p53 impacts on the activation of genes regulated by IFNα1, we studied how p53 regulates the expression of genes activated by IFNγ. The time course experiment involving co-treatment of cells with A+N and IFNy revealed that after 24 h of incubation, activation of p53 was associated with attenuated phosphorylation of STAT1 (Figure 5). Moreover, we found that p53 contributes to the repression of STAT1 phosphorylation in cells exposed to IFNγ (Fig.6). Reduced phosphorylation of STAT1 was reflected in reduced expression of IRF1, which is a gene activated by IFNy early during exposure (reviewed by Castro et al., [24]). Interestingly, this correlation was not visible in case of other early gene – SOCS1 (Figure 6). Thus, in case of IFNγ, we have close correlation between phosphorylation status of STAT1 and the expression of at least one major IFNγ-target gene (IRF1). However, when the expression of other interferon targets was examined, the picture became more complicated. Some genes are activated by IFNγ early during exposure and some genes are activated late (Figure 5). This was observed by others [25]. According to our data, IRF1 and SOCS1 belong to early genes and CASP1, IFIT1 and IFIT3 belong to the late genes (Figure 5). Others demonstrated that IFNγ induces expression of IRF1, which in turn activates CASP1 gene [26]. We noticed that co-treatment with IFNγ and A+N synergistically induces the expression of three late genes CASP1, IFIT1 and IFIT3 (Figure 5, 7 and 9). This synergy is lost in p53-deficient cells. The strongest synergy was observed in case of activation of CASP1 gene. This is original and very important observation because it indicates that these two signaling systems strongly promote cellular functions executed by this caspase. The best studied function of CASP1 is the induction of pyroptosis – regulated form of death, which releases pro-inflammatory factors [27]. What is the molecular mechanism of synergy between IFNγ and p53 in activation of CASP1 remains a puzzle. The late genes activated by IFNγ are not induced by the dimer of phosphorylated STAT1, but by the product of the early gene - IRF1, which is a transcription factor [24]. Interestingly IRF1 is also activated by p53, at least in A549 cells (Figure 7, 9 and [13]). Thus, at least in principle, IRF1 upregulated by IFNγ and by p53 may converge on CASP1 promoter and stimulate its activity. But it is very likely that other factors also play a role. Definitely p53 and IFNγ can act in concert to additively or synergistically stimulate the expression of some genes despite p53-dependent attenuation of STAT1 phosphorylation. It was best visible, when we employed a different treatment strategy – first treatment with A+N for 24 h (to upregulate the p53 target genes) and then exposure to IFNγ for 6 h. Following this treatment pattern we observed by RT-PCR strong cooperation of p53 and IFNγ in upregulation of CASP1, IFIT1 and IFIT3 and probably of ICAM1 – another target of IFN-y (Figure 9). In case of IFI16 gene, positively regulated by both p53 [16] and IFNγ [28], these two proteins act additively in its activation. Thus, different genes regulated by both IFNγ and p53 show variable response to simultaneous activation of both signaling systems. Our experiment also support the notion that interferon-γ and p53 have many common target genes. Moreover, they activate genes coding for proteins participating in the same biological process. This results in common biological outcomes of p53 activation and exposure to IFNγ, e.g., improvement of antigen presentation [29,30], sensitization of cells to apoptosis induced by FAS ligand [31,32] and inhibition of the cell cycle [5,33]. Our observations suggest that IFNγ and p53 may sensitize at least some cancer cells or normal human fibroblasts to CASP1-dependent pyroptosis. In addition to CASP1, the aforementioned IFI16 is another component of pyroptotic infalmmasome [34] activated by both p53 and IFNγ.
We also made another important observation. Nutlin-3a designed to specifically activate p53 by antagonizing its negative regulator – MDM2 protein, can exert p53 independent effect on cells when combined with actinomycin D or even acting alone. It can attenuate phosphorylation of STAT1 in p53-null NCI-H1299 cells and it synergizes with actinomycin D in activation of some interferon-stimulated genes in NCI-H1299 cells, most notably IFIT3. It would be interesting to find out by transcriptomic methods what other genes are activated in p53-null cells by common action of actinomycin D and nutlin-3a. The co-treatment with these two compounds also sensitizes NCH-H1299 cells to the killing effect of natural killer cells NK-92 (Figure 12). The mechanism of this biological effect is not known, however we can speculate that A+N strongly activates some of the innate immunity genes, which make cancer cells better targets for natural killer cells. However, p53 can also contribute to the killing of cancer cells by natural killer cells (Figure 12). The sensitizing mechanism has been partially unveiled by published observations. In cancer cells p53 activates the expression of genes, which encode the surface proteins acting as activators of natural killer cells. These proteins are ULBP1 and ULBP2 [35,36]. Interestingly, our recently published transcriptomic data are consistent with these observations demonstrating that co-treatment with A+N strongly upregulates the expression of ULBP2 gene in A549 and in NCI-H460 cells [6]. Thus, actinomycin D and nutlin-3a can sensitize cancer cells to the killing effect of natural killer cells and this effect is governed by both p53-dependent and p53-independent mechanisms and while we start to understand the former, the latter are completely unknown. The drug-induced sensitization of cancer cells to the killing by natural killer cells was already observed by others and generally involves modulation of protein expression on the surface of cancer cells [37]. However, our drug combination (A+N) was not previously tested and the mechanism of sensitization is only starting to be deciphered. The intriguing finding is that A+N can promote the innate immunity in p53-independent fashion.
4. Materials and Methods
4.1. Cell Culture and Treatment
A549, U-2 OS and NCI-H292 cell lines from ATCC (Manassas, VA, USA) were cultured in low-glucose DMEM supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA). NCI-H460 (ATCC) were cultured in RPMI-1640 with 4.5 g/l glucose, supplemented with 2 mM glutamine and 1 mM sodium pyruvate. NCI-H1299 cells were cultured in RPMI-1640 with 1g/l glucose supplemented with 10% FBS. NK-92 cells (from ATCC) were cultured on RPMI-1640 medium supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 20 ng/ml interleukin-2 (Miltenyi Biotech, Bergisch Gladbach, Germany), 12.5% FBS, 12.5% horse serum (Biowest, Nuaillé, France). GM07492 normal human fibroblasts (Coriell Cell Repositories, Camden, NJ) were cultured in low glucose DMEM supplemented with 15% FBS. All media were supplemented with penicillin/streptomycin solution. Cells were incubated at 37oC, 5% CO2 with saturating humidity.
Stock solutions of chemicals were prepared in DMSO (Dimethyl Sulfoxide): actinomycin D (10 µM; Sigma-Aldrich, St. Louis, MO, USA), camptothecin (10 mM; Calbiochem-Merck, Darmstadt, Germany), and nutlin-3a (10 mM; Selleck Chemicals LLC, Houston, TX, USA). The stock solutions were diluted in culture medium to the following concentrations: 5 nM actinomycin D, 5 µM nutlin-3a, and 5 µM camptothecin. Control cells were mock-treated with a medium containing DMSO. Interferons α1 and γ were purchased from Cell Signalling Technology (Danvers, MA, USA). The stock solutions were prepared in sterile water at 75 μg/ml (IFNα1) or 100 μg/ml (IFNγ). The final concentrations are given in Results.
4.2. Generation of p53-Deficient Cells
4.3. Western Blotting
The preparation of the whole-cell lysates using IP buffer, supplemented with protease and phosphatase inhibitors were described previously [10]. Aliquots of lysates (35–50 µg) were separated by SDS-PAGE on 8% or 13% gels and electro-transferred onto PVDF membranes. Before incubation with primary antibody, the membranes were incubated for 1 h at room temperature in blocking solution (5% skim milk in PBS with 0.1% Tween-20). The anti-phospho-Ser37-p53, anti-phospho-Tyr701-STAT1 (D4A7), anti-STAT1, anti-MX1, anti-IRF1 and anti-IFIT1 (D2X9Z) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Anti-p53 (DO-1), and loading control anti-HSC70 (B-6) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-SOCS1 antibody was (clone 4H1) was from EMD Millipore (Temecula, CA, USA). Anti-CASP1 (ab179515) and anti-IFIT3 (ab95989) antibodies were from Abcam (Cambridge, UK). Anti-GAPDH loading control antibody was from Merck (Sigma) (Darmstadt, Germany). Anti-IRF7 antibody was from Proteintech (Rosemont, IL). All incubations with primary antibodies were performed overnight at 4oC in blocking solution. HRP-conjugated secondary antibodies (anti-mouse, anti-rabbit) were detected by chemiluminescence (SuperSignalWest Pico or SuperSignal West Femto Chemiluminescent substrate (Thermo Fisher Scientific, Waltham, MA, USA).
4.4. Gene Expression Analysis by Semi-Quantitative Real-Time PCR
After the treatment, cells were harvested by trypsinization, washed with PBS, frozen on dry ice and stored at -80oC. Total RNA samples were isolated using the RNeasy mini kit (Qiagen, Hilden, Germany). The cDNA was synthesised with MuLV reverse transcriptase and random hexamers (Applied Biosystems, Foster City, CA, USA). Gene expression was measured using Real-Time 2x PCR Master Mix SYBR (A&A Biotechnology, Gdynia, Poland). The sequence of primers used for RT-PCR are given in supplementary Table S1. Amplification was performed on a CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). In each RT-PCR run, cDNA samples were amplified in triplicate. A relative quantitation of mRNA was carried out using the ΔΔCT method with ACTB or GAPDH as a reference. Mean and standard deviation were calculated from three biological replicates.
4.5. Killing of Cancer Cells by NK-92 Cells
Cancer cells growing in respective culture media on 6-cm culture plates were either mock-treated or exposed for 48 h to the drugs or their combinations at concentrations given above. Subsequently, the cells were trypsynized, washed with PBS, centrifuged and the cell pellets were suspended in the medium for NK-92 cells. Subsequently, cancer cell (control or treated) were seeded into the wells of 96-well plates together with NK-92 cells at rations given in the results. We seeded 3 600 cancer cells in one well in 100 µl of medium for NK-92 cells. For the control, cancer cells were seeded without addition of NK-92 cells. For each experimental condition we seeded cells on 3 wells. The co-incubation lasted for 24 h. After this time, we removed the medium and replaced it with the relevant medium for cancer cells. The surviving cells were allowed to recover for 72 h. After this time, the metabolic activity of cells was measured using MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay, cat no. G3582, Promega, Madison, WI). The metabolic activity of cells, which were not incubated with NK-92 cells was set as 100%. To macroscopically visualize adherent A549 cells surviving the co-treatment with NK-92 cells (growing in suspension) we employed the following protocol. A549 cells were treated with A+N or mock-treated for 48 h in DMEM. Subsequently, the cells were trypsynized, counted and 100 000 cells were seeded onto wells of 6-well plate. The NK-92 cells were counted and they were seeded onto the wells of 6-well plate for co-incubation with A549 cells. We used two NK-92:A549 ratios – 1:1 and 5:1. In control wells, A549 were incubated only with RPMI medium. The cells were incubated with 2 ml culture medium (RPMI) for NK-92 cells per well. After 24-h co-incubation, the RPMI medium was removed and the attached cells were allowed to recover and grow in fresh DMEM for 5-7 days. Subsequently, the medium was removed, the cells were rinsed with PBS and fixed by incubation with -20oC methanol for 10 min. Fixed and dried cells were stained with 0.01% crystal violet for 2 min. Finally, the cells were washed in distilled water, dried, and the plates were scanned.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: The sequence of RT-PCR primers.
Author Contributions
Conceptualization, M.R., A.B. and B.Ł-S; methodology, M.R., A.B., B. Ł.-S., A.G.-K., M.K.; validation, A.G.-K., and M.K.; formal analysis, B. Ł.-S., A.B.; investigation, A.B., B.Ł-S., A.G.-K., M.K.; writing— M.R.; writing—review and editing, B.Ł-S., A.B.; visualization, M.R., A.B., B.Ł.-S.; supervision, M.R.; project administration, M.R.; funding acquisition, M.R. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was funded by National Science Center (NCN), Poland, grant number 2019/35/O/NZ5/02600 to MR.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
We created no new data, which are not published.
Acknowledgments
We thank Patrycja Jakubowska for technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Baker SJ, Vogelstein B. p53: a tumor suppressor hiding in plain sight. J Mol Cell Biol. 2019 Jul 19;11(7):536-538. PMID: 31276589; PMCID: PMC6736432. [CrossRef]
- Aloni-Grinstein R, Charni-Natan M, Solomon H, Rotter V. p53 and the Viral Connection: Back into the Future ‡. Cancers (Basel). 2018 Jun 4;10(6):178. PMID: 29866997; PMCID: PMC6024945. [CrossRef]
- Levine AJ. The many faces of p53: something for everyone. J Mol Cell Biol. 2019 Jul 19;11(7):524-530. PMID: 30925588; PMCID: PMC6736316. [CrossRef]
- Fischer M, Schwarz R, Riege K, DeCaprio JA, Hoffmann S. TargetGeneReg 2.0: a comprehensive web-atlas for p53, p63, and cell cycle-dependent gene regulation. NAR Cancer. 2022 Mar 23;4(1):zcac009. PMID: 35350773; PMCID: PMC8946727. [CrossRef]
- Uxa S, Bernhart SH, Mages CFS, Fischer M, Kohler R, Hoffmann S, Stadler PF, Engeland K, Müller GA. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019 Sep 26;47(17):9087-9103. PMID: 31400114; PMCID: PMC6753476. [CrossRef]
- Łasut-Szyszka B, Gdowicz-Kłosok A, Małachowska B, Krześniak M, Będzińska A, Gawin M, Pietrowska M, Rusin M. Transcriptomic and proteomic study of cancer cell lines exposed to actinomycin D and nutlin-3a reveals numerous, novel candidates for p53-regulated genes. Chem Biol Interact. 2024 Apr 1;392:110946. Epub 2024 Mar 7. PMID: 38460933. [CrossRef]
- Philips RL, Wang Y, Cheon H, Kanno Y, Gadina M, Sartorelli V, Horvath CM, Darnell JE Jr, Stark GR, O’Shea JJ. The JAK-STAT pathway at 30: Much learned, much more to do. Cell. 2022 Oct 13;185(21):3857-3876. PMID: 36240739; PMCID: PMC9815833. [CrossRef]
- Sakamoto H, Kinjyo I, Yoshimura A. The janus kinase inhibitor, Jab/SOCS-1, is an interferon-gamma inducible gene and determines the sensitivity to interferons. Leuk Lymphoma. 2000 Jun;38(1-2):49-58. PMID: 10811447. [CrossRef]
- Sharma J, Larkin J 3rd. Therapeutic Implication of SOCS1 Modulation in the Treatment of Autoimmunity and Cancer. Front Pharmacol. 2019 Apr 11;10:324. PMID: 31105556; PMCID: PMC6499178. [CrossRef]
- Krześniak M, Zajkowicz A, Gdowicz-Kłosok A, Głowala-Kosińska M, Łasut-Szyszka B, Rusin M. Synergistic activation of p53 by actinomycin D and nutlin-3a is associated with the upregulation of crucial regulators and effectors of innate immunity. Cell Signal. 2020 May;69:109552. Epub 2020 Feb 4. PMID: 32032660; PMCID: PMC7126238. [CrossRef]
- Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, Fornace AJ Jr. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999 Dec 1;18(23):6845-54. PMID: 10581258; PMCID: PMC1171747. [CrossRef]
- Horisberger MA, Gunst MC. Interferon-induced proteins: identification of Mx proteins in various mammalian species. Virology. 1991 Jan;180(1):185-90. PMID: 1984648. [CrossRef]
- Łasut-Szyszka B, Małachowska B, Gdowicz-Kłosok A, Krześniak M, Głowala-Kosińska M, Zajkowicz A, Rusin M. Transcriptome Analysis of Cells Exposed to Actinomycin D and Nutlin-3a Reveals New Candidate p53-Target Genes and Indicates That CHIR-98014 Is an Important Inhibitor of p53 Activity. Int J Mol Sci. 2021 Oct 14;22(20):11072. PMID: 34681730; PMCID: PMC8538697. [CrossRef]
- Ma W, Huang G, Wang Z, Wang L, Gao Q. IRF7: role and regulation in immunity and autoimmunity. Front Immunol. 2023 Aug 10;14:1236923. PMID: 37638030; PMCID: PMC10449649. [CrossRef]
- Wang B, Lam TH, Soh MK, Ye Z, Chen J, Ren EC. Influenza A Virus Facilitates Its Infectivity by Activating p53 to Inhibit the Expression of Interferon-Induced Transmembrane Proteins. Front Immunol. 2018 May 31;9:1193. PMID: 29904383; PMCID: PMC5990591. [CrossRef]
- Song LL, Alimirah F, Panchanathan R, Xin H, Choubey D. Expression of an IFN-inducible cellular senescence gene, IFI16, is up-regulated by p53. Mol Cancer Res. 2008 Nov;6(11):1732-41. Epub 2008 Oct 30. PMID: 18974396. [CrossRef]
- Klingemann H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy. 2023 May;25(5):451-457. Epub 2023 Jan 6. PMID: 36610812. [CrossRef]
- Łasut-Szyszka B, Rusin M. The Wheel of p53 Helps to Drive the Immune System. Int J Mol Sci. 2023 Apr 21;24(8):7645. PMID: 37108808; PMCID: PMC10143509. [CrossRef]
- Carlsen L, Zhang S, Tian X, De La Cruz A, George A, Arnoff TE, El-Deiry WS. The role of p53 in anti-tumor immunity and response to immunotherapy. Front Mol Biosci. 2023 Aug 1;10:1148389. PMID: 37602328; PMCID: PMC10434531. [CrossRef]
- Zhang J, Li H, Yu JP, Wang SE, Ren XB. Role of SOCS1 in tumor progression and therapeutic application. Int J Cancer. 2012 May 1;130(9):1971-80. Epub 2012 Jan 11. PMID: 22025331. [CrossRef]
- Beaurivage C, Champagne A, Tobelaim WS, Pomerleau V, Menendez A, Saucier C. SOCS1 in cancer: An oncogene and a tumor suppressor. Cytokine. 2016 Jun;82:87-94. Epub 2016 Jan 19. PMID: 26811119. [CrossRef]
- Cheriyath V, Leaman DW, Borden EC. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J Interferon Cytokine Res. 2011 Jan;31(1):173-81. Epub 2010 Oct 12. PMID: 20939681; PMCID: PMC6468951. [CrossRef]
- Muñoz-Fontela C, Macip S, Martínez-Sobrido L, Brown L, Ashour J, García-Sastre A, Lee SW, Aaronson SA. Transcriptional role of p53 in interferon-mediated antiviral immunity. J Exp Med. 2008 Aug 4;205(8):1929-38. Epub 2008 Jul 28. PMID: 18663127; PMCID: PMC2525597. [CrossRef]
- Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol. 2018 May 4;9:847. PMID: 29780381; PMCID: PMC5945880. [CrossRef]
- Sekrecka A, Kluzek K, Sekrecki M, Boroujeni ME, Hassani S, Yamauchi S, Sada K, Wesoly J, Bluyssen HAR. Time-dependent recruitment of GAF, ISGF3 and IRF1 complexes shapes IFNα and IFNγ-activated transcriptional responses and explains mechanistic and functional overlap. Cell Mol Life Sci. 2023 Jun 22;80(7):187. PMID: 37347298; PMCID: PMC10287828. [CrossRef]
- Kim EJ, Lee JM, Namkoong SE, Um SJ, Park JS. Interferon regulatory factor-1 mediates interferon-gamma-induced apoptosis in ovarian carcinoma cells. J Cell Biochem. 2002;85(2):369-80. PMID: 11948692. [CrossRef]
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007 Sep;14(9):1590-604. Epub 2007 Jun 29. PMID: 17599095; PMCID: PMC3345951. [CrossRef]
- Trapani JA, Browne KA, Dawson MJ, Ramsay RG, Eddy RL, Show TB, White PC, Dupont B. A novel gene constitutively expressed in human lymphoid cells is inducible with interferon-gamma in myeloid cells. Immunogenetics. 1992;36(6):369-76. PMID: 1526658. [CrossRef]
- Martini M, Testi MG, Pasetto M, Picchio MC, Innamorati G, Mazzocco M, Ugel S, Cingarlini S, Bronte V, Zanovello P, Krampera M, Mosna F, Cestari T, Riviera AP, Brutti N, Barbieri O, Matera L, Tridente G, Colombatti M, Sartoris S. IFN-gamma-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine. 2010 Apr 30;28(20):3548-57. Epub 2010 Mar 19. PMID: 20304037. [CrossRef]
- Braun MW, Iwakuma T. Regulation of cytotoxic T-cell responses by p53 in cancer. Transl Cancer Res. 2016 Dec;5(6):692-697. PMID: 28944167; PMCID: PMC5607642. [CrossRef]
- Müller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest. 1997 Feb 1;99(3):403-13. PMID: 9022073; PMCID: PMC507813. [CrossRef]
- Selleck WA, Canfield SE, Hassen WA, Meseck M, Kuzmin AI, Eisensmith RC, Chen SH, Hall SJ. IFN-gamma sensitization of prostate cancer cells to Fas-mediated death: a gene therapy approach. Mol Ther. 2003 Feb;7(2):185-92. PMID: 12597906. [CrossRef]
- Harvat BL, Seth P, Jetten AM. The role of p27Kip1 in gamma interferon-mediated growth arrest of mammary epithelial cells and related defects in mammary carcinoma cells. Oncogene. 1997 May 1;14(17):2111-22. PMID: 9160891. [CrossRef]
- Fan Z, Chen R, Yin W, Xie X, Wang S, Hao C. Effects of AIM2 and IFI16 on Infectious Diseases and Inflammation. Viral Immunol. 2023 Sep;36(7):438-448. Epub 2023 Aug 16. PMID: 37585649. [CrossRef]
- Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011 Sep 15;71(18):5998-6009. Epub 2011 Jul 15. PMID: 21764762. [CrossRef]
- Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, Szekely L, Kärre K, Carbone E, Selivanova G. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle. 2011 Oct 1;10(19):3346-58. Epub 2011 Oct 1. PMID: 21941086. [CrossRef]
- Cifaldi L, Locatelli F, Marasco E, Moretta L, Pistoia V. Boosting Natural Killer Cell-Based Immunotherapy with Anticancer Drugs: a Perspective. Trends Mol Med. 2017 Dec;23(12):1156-1175. Epub 2017 Nov 10. PMID: 29133133. [CrossRef]
Figure 1.
Expression of SOCS1 in response to the treatment with actinomycin D and nutlin-3a is regulated in cell-specific manner. A. B. Levels of p53, SOCS1 and loading control (HSC70) in indicated cell lines exposed to actinomycin D (ActD), nutlin-3a (Nut), both compounds acting together (A+N) or camptothecin (CPT) for 48 h. Control cells (Con) were mock-treated. C. Levels of p53, SOCS1 and loading control in p53-proficient (Con) and knockout clones (p53-KO) of U-2 OS cell line exposed to A+N, nutlin-3a (Nut) or mock-treated (C) for 48 h. # indicates the clone number.
Figure 1.
Expression of SOCS1 in response to the treatment with actinomycin D and nutlin-3a is regulated in cell-specific manner. A. B. Levels of p53, SOCS1 and loading control (HSC70) in indicated cell lines exposed to actinomycin D (ActD), nutlin-3a (Nut), both compounds acting together (A+N) or camptothecin (CPT) for 48 h. Control cells (Con) were mock-treated. C. Levels of p53, SOCS1 and loading control in p53-proficient (Con) and knockout clones (p53-KO) of U-2 OS cell line exposed to A+N, nutlin-3a (Nut) or mock-treated (C) for 48 h. # indicates the clone number.
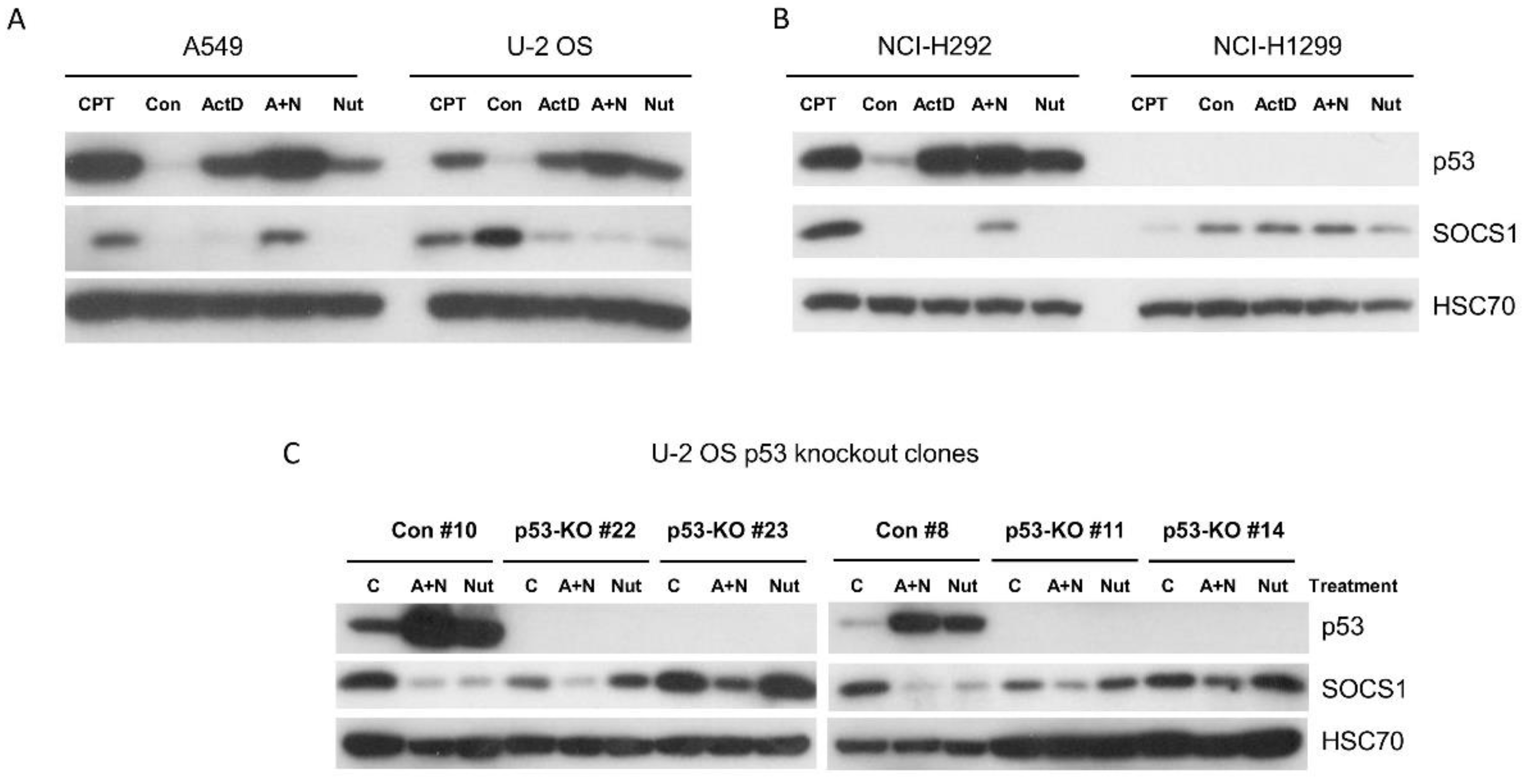
Figure 2.
Exposure of cells to actinomycin D and nutlin-3a (A+N) reduces phosphorylation of STAT1 on Tyr(Y)-701 in cells exposed to interferon-α1 (IFNα1). A. B. The expression of indicated proteins in cell lines pre-exposed to A+N, nutlin-3a (Nut) or mock-treated (Con) for 24 h and subsequently mock-treated (Con), exposed to A+N, Nut, A+N with 1ng/ml IFNα1, Nut with 1ng/ml IFNα1 or alone with 1ng/ml IFNα1 for the next 24 h. In A GAPDH was loading control.
Figure 2.
Exposure of cells to actinomycin D and nutlin-3a (A+N) reduces phosphorylation of STAT1 on Tyr(Y)-701 in cells exposed to interferon-α1 (IFNα1). A. B. The expression of indicated proteins in cell lines pre-exposed to A+N, nutlin-3a (Nut) or mock-treated (Con) for 24 h and subsequently mock-treated (Con), exposed to A+N, Nut, A+N with 1ng/ml IFNα1, Nut with 1ng/ml IFNα1 or alone with 1ng/ml IFNα1 for the next 24 h. In A GAPDH was loading control.
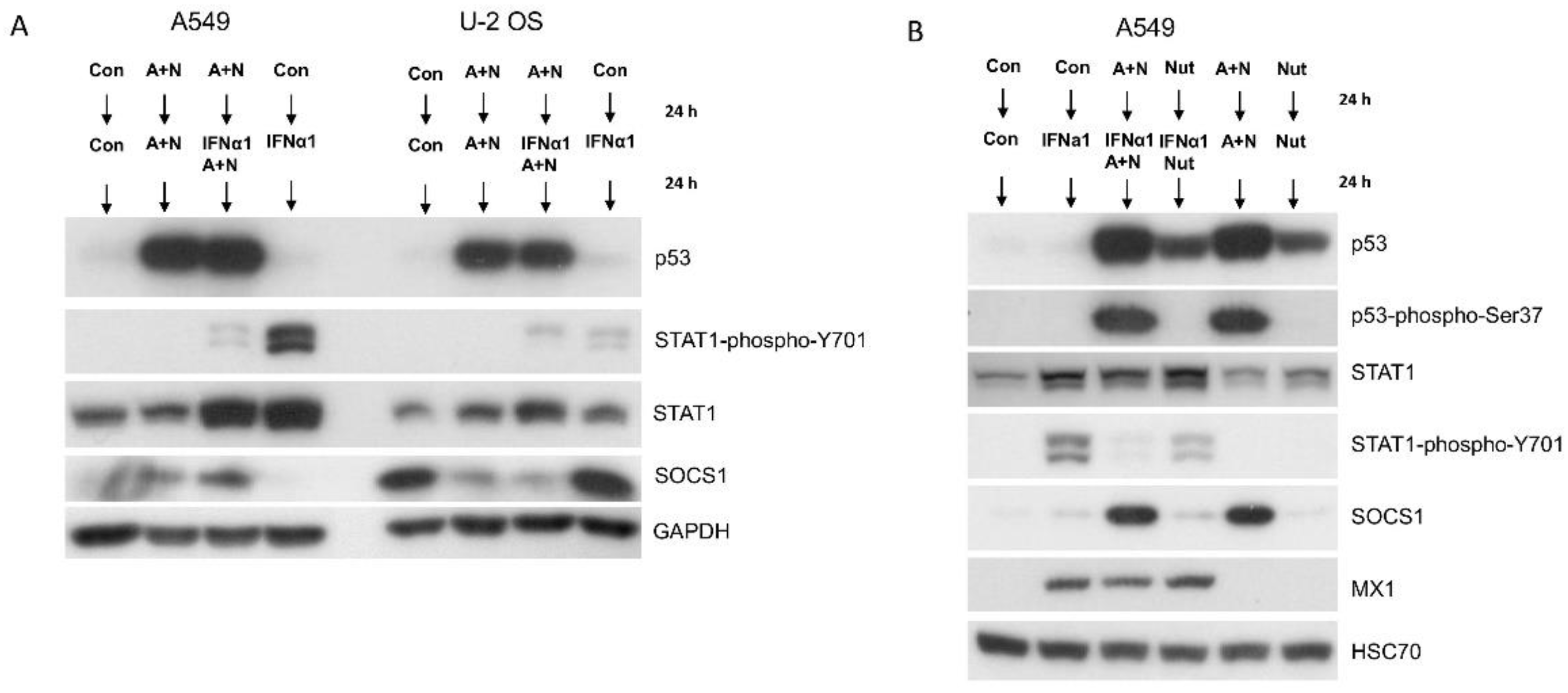
Figure 3.
The modulation of STAT1 phosphorylation by A+N does not translate into modulated expression of genes activated by IFNα1. A. A549 cells were exposed using the procedure presented in Figure 2A, various concentrations of IFNα1 were used. Subsequently the expression of indicated proteins was determined by Western blotting. The expression of phospho-STAT1 was visualized by long and sort (se) exposures. B. A549 cells (p53 proficient and p53-deficient) were exposed as indicated and the expression of relevant proteins or their modified forms was determined by Western blotting. IFNα1 was used at 1ng/ml concentration. The lower panel shows the results of the independent experiment with IFNα1 0.5 ng/ml concentration.
Figure 3.
The modulation of STAT1 phosphorylation by A+N does not translate into modulated expression of genes activated by IFNα1. A. A549 cells were exposed using the procedure presented in Figure 2A, various concentrations of IFNα1 were used. Subsequently the expression of indicated proteins was determined by Western blotting. The expression of phospho-STAT1 was visualized by long and sort (se) exposures. B. A549 cells (p53 proficient and p53-deficient) were exposed as indicated and the expression of relevant proteins or their modified forms was determined by Western blotting. IFNα1 was used at 1ng/ml concentration. The lower panel shows the results of the independent experiment with IFNα1 0.5 ng/ml concentration.
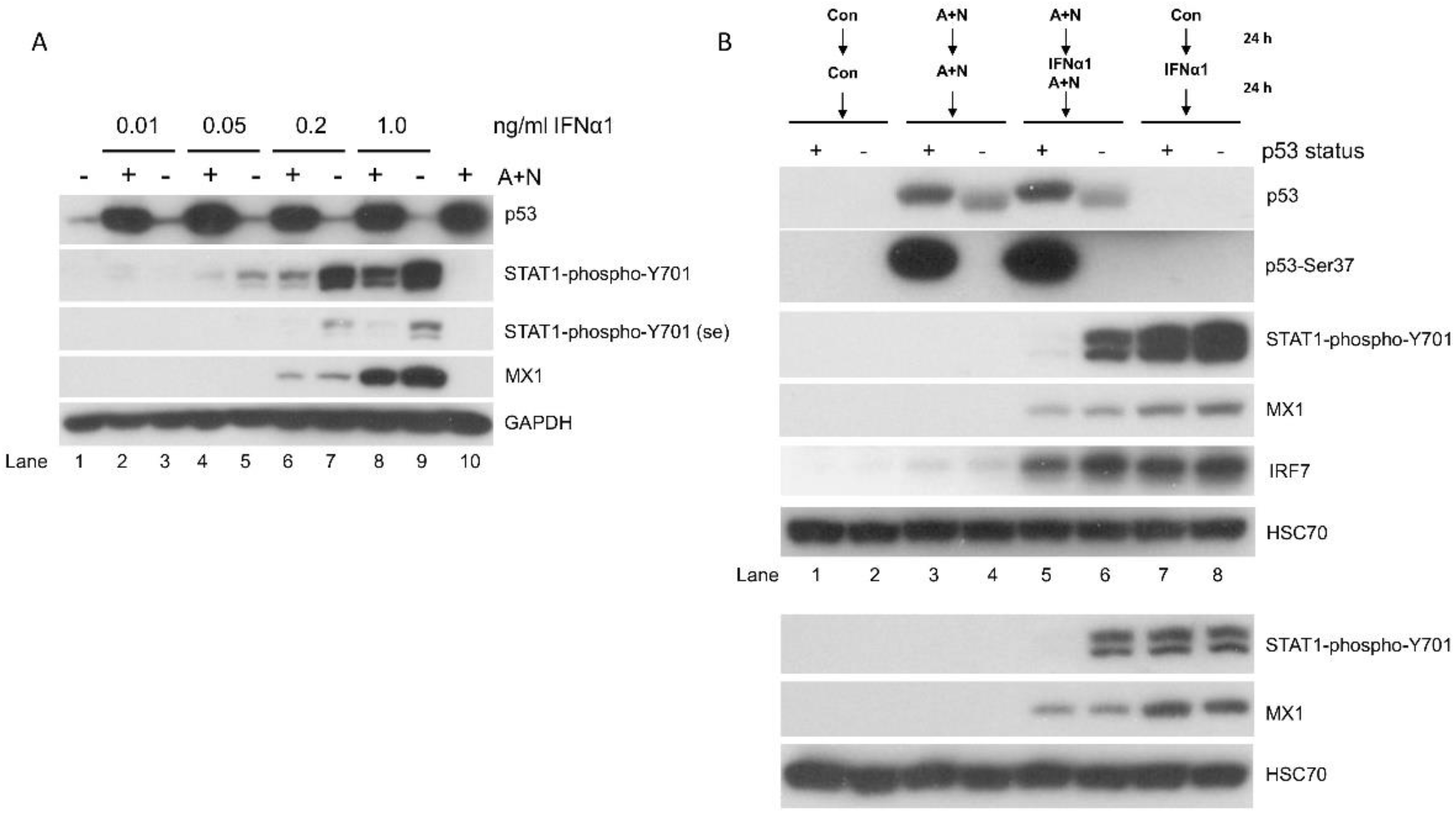
Figure 4.
p53 modulates the expression of only a subset of genes activated by IFNα1. The p53-proficient (red bars) and p53-deficient (blue bars) A549 cells were exposed to A+N, A+N with IFNα1 and to IFNα1 using the two-step procedure presented on Figure 2A. IFNα1 concentration was 0.5 ng/ml. Subsequently, the expression of indicated genes was determined by RT-PCR. Three biological replicates were performed. ACTB gene was used as a reference gene. The influence of p53 status was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=10%). Additionally, the statistical significance was calculated for p53-proficient cells treated with A+N vs. A+N+IFNα and A+N+IFNα vs. IFNα. For this purpose calculations were made using the unpaired t test with Welch’s correction for each group separately (* p < 0,05, ** p<0,01, *** p<0,001). All tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
Figure 4.
p53 modulates the expression of only a subset of genes activated by IFNα1. The p53-proficient (red bars) and p53-deficient (blue bars) A549 cells were exposed to A+N, A+N with IFNα1 and to IFNα1 using the two-step procedure presented on Figure 2A. IFNα1 concentration was 0.5 ng/ml. Subsequently, the expression of indicated genes was determined by RT-PCR. Three biological replicates were performed. ACTB gene was used as a reference gene. The influence of p53 status was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=10%). Additionally, the statistical significance was calculated for p53-proficient cells treated with A+N vs. A+N+IFNα and A+N+IFNα vs. IFNα. For this purpose calculations were made using the unpaired t test with Welch’s correction for each group separately (* p < 0,05, ** p<0,01, *** p<0,001). All tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
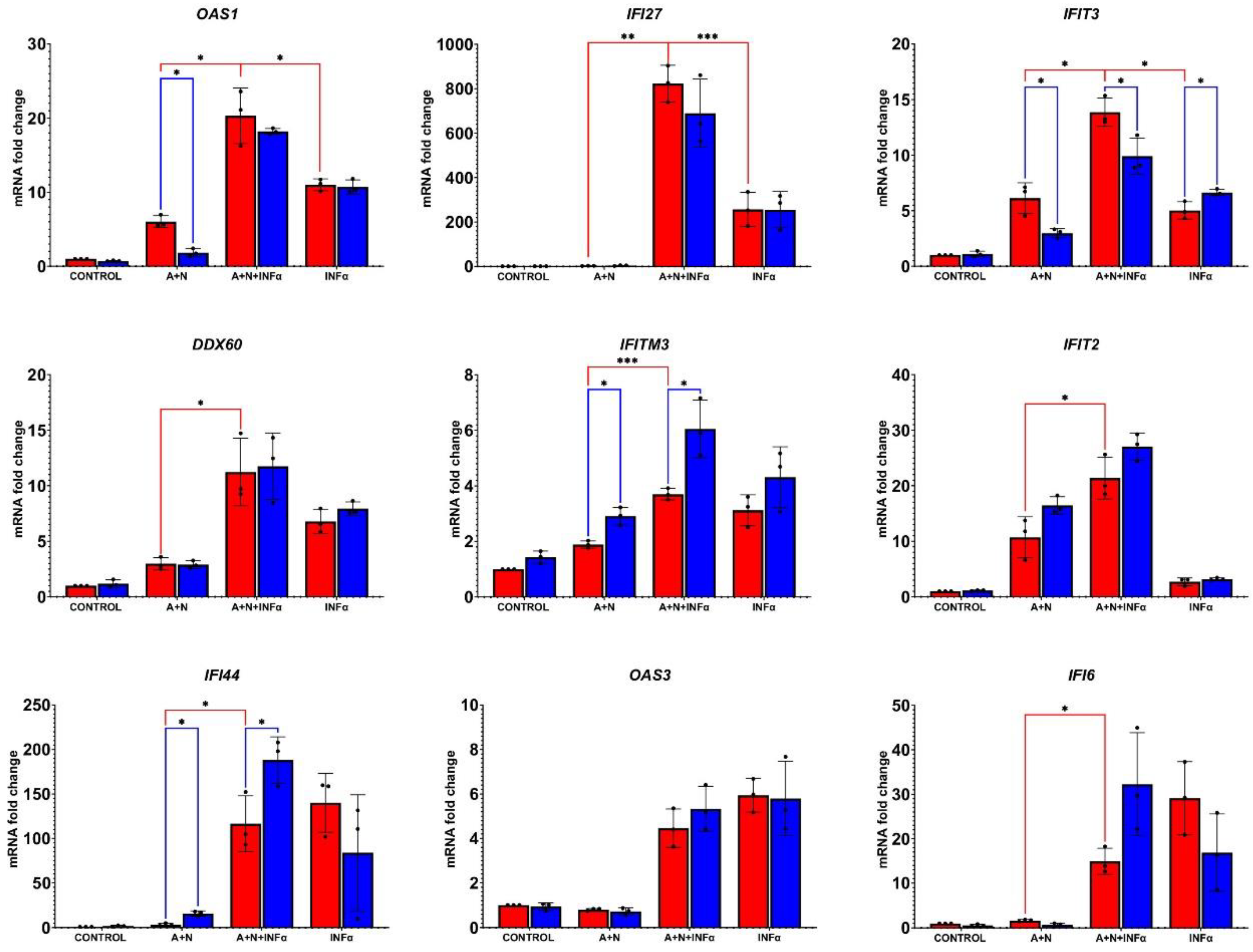
Figure 5.
A+N and interferon-γ (IFNγ) synergize in activation of a subset of innate immunity genes. A549 cells were exposed to A+N, A+N with IFNγ or to IFNγ alone for increasing number of hours. IFNγ was used at 1 ng/ml. Subsequently, the expression of selected proteins was determined using Western blotting.
Figure 5.
A+N and interferon-γ (IFNγ) synergize in activation of a subset of innate immunity genes. A549 cells were exposed to A+N, A+N with IFNγ or to IFNγ alone for increasing number of hours. IFNγ was used at 1 ng/ml. Subsequently, the expression of selected proteins was determined using Western blotting.
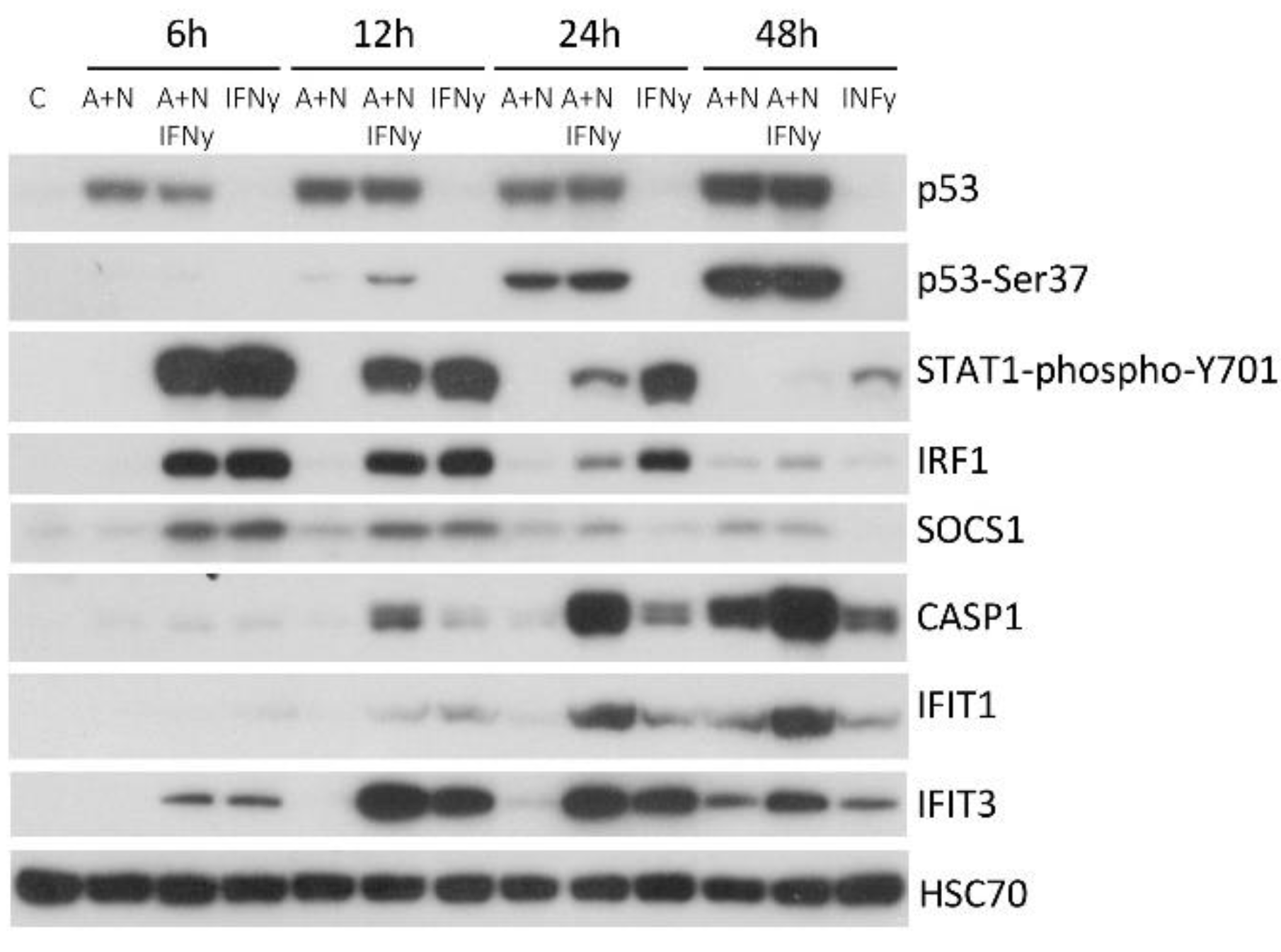
Figure 6.
p53 has some effect on reducing phosphorylation of STAT1 induced by IFNγ. P53-proficient and p53-deficient A549 cells were exposed as indicated for 24 h. IFNγ was used at 1ng/ml concentration. The expression of indicated proteins was determined by Western blotting. The lower panel shows the results of an independent experiment.
Figure 6.
p53 has some effect on reducing phosphorylation of STAT1 induced by IFNγ. P53-proficient and p53-deficient A549 cells were exposed as indicated for 24 h. IFNγ was used at 1ng/ml concentration. The expression of indicated proteins was determined by Western blotting. The lower panel shows the results of an independent experiment.
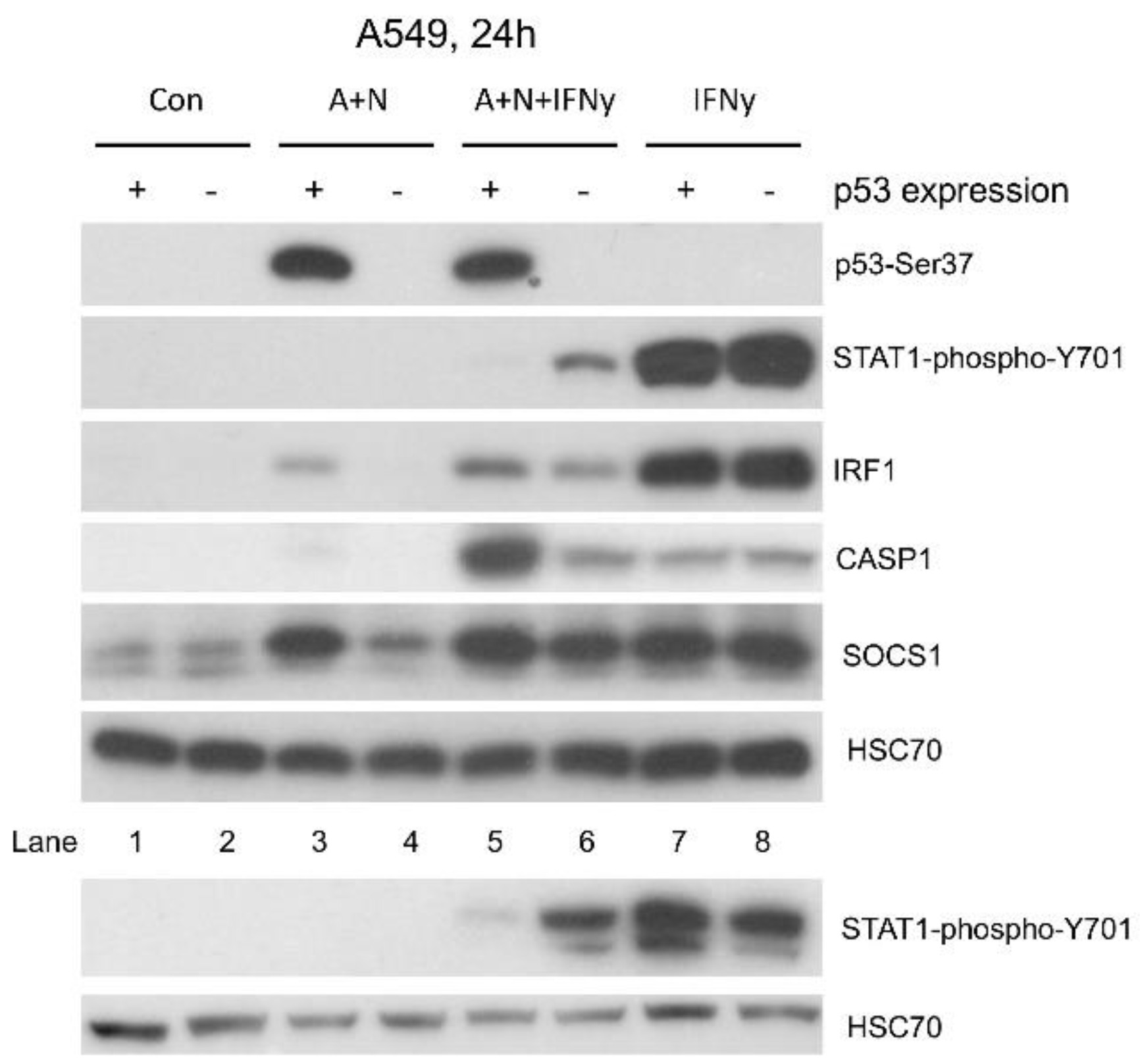
Figure 7.
Activated p53 and IFNγ synergize in stimulation of CASP1 gene coding for pro-pyroptotic caspase-1. The p53-proficient (red bars) and p53-deficient (blue bars) A549 cells were exposed to A+N, A+N with IFNγ and to IFNγ alone for 24 h. Control cells were mock-treated. IFNγ concentration was 1 ng/ml. Subsequently, the expression of indicated genes was determined by RT-PCR. Three biological replicates were performed. ACTB was used as a reference gene. The influence of p53 status was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=10%). Additionally, statistical significance was calculated for p53 proficient cells treated with A+N vs. A+N+IFNγ and A+N+IFNγ vs. IFNγ. For this purpose calculations were made using the unpaired t test with Welch’s correction or Mann Whitney test for each group separately (* p < 0,05, ** p<0,01, *** p<0,001). All tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
Figure 7.
Activated p53 and IFNγ synergize in stimulation of CASP1 gene coding for pro-pyroptotic caspase-1. The p53-proficient (red bars) and p53-deficient (blue bars) A549 cells were exposed to A+N, A+N with IFNγ and to IFNγ alone for 24 h. Control cells were mock-treated. IFNγ concentration was 1 ng/ml. Subsequently, the expression of indicated genes was determined by RT-PCR. Three biological replicates were performed. ACTB was used as a reference gene. The influence of p53 status was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=10%). Additionally, statistical significance was calculated for p53 proficient cells treated with A+N vs. A+N+IFNγ and A+N+IFNγ vs. IFNγ. For this purpose calculations were made using the unpaired t test with Welch’s correction or Mann Whitney test for each group separately (* p < 0,05, ** p<0,01, *** p<0,001). All tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
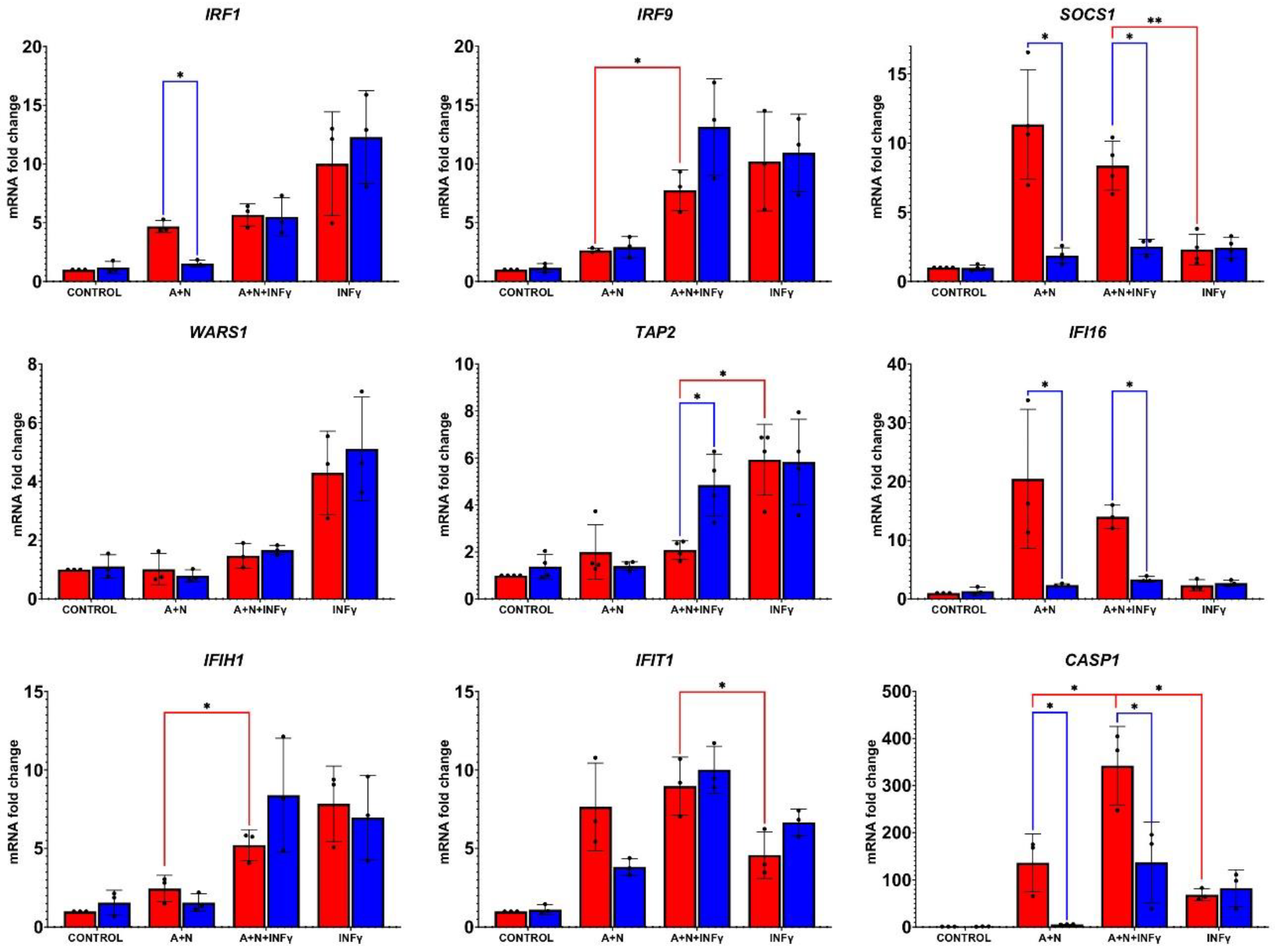
Figure 8.
The synergy between activated p53 and IFNγ results in strong upregulation of caspase-1 (CASP1) protein. The expression of indicated proteins in A549 cell line (p53-proficient and p53-deficient) pre-exposed to A+N, or mock-treated (Con) for 24 h and subsequently mock-treated (Con), exposed to A+N, A+N with 1 ng/ml IFNγ, or alone with 1ng/ml IFNγ for the next 6 h.
Figure 8.
The synergy between activated p53 and IFNγ results in strong upregulation of caspase-1 (CASP1) protein. The expression of indicated proteins in A549 cell line (p53-proficient and p53-deficient) pre-exposed to A+N, or mock-treated (Con) for 24 h and subsequently mock-treated (Con), exposed to A+N, A+N with 1 ng/ml IFNγ, or alone with 1ng/ml IFNγ for the next 6 h.
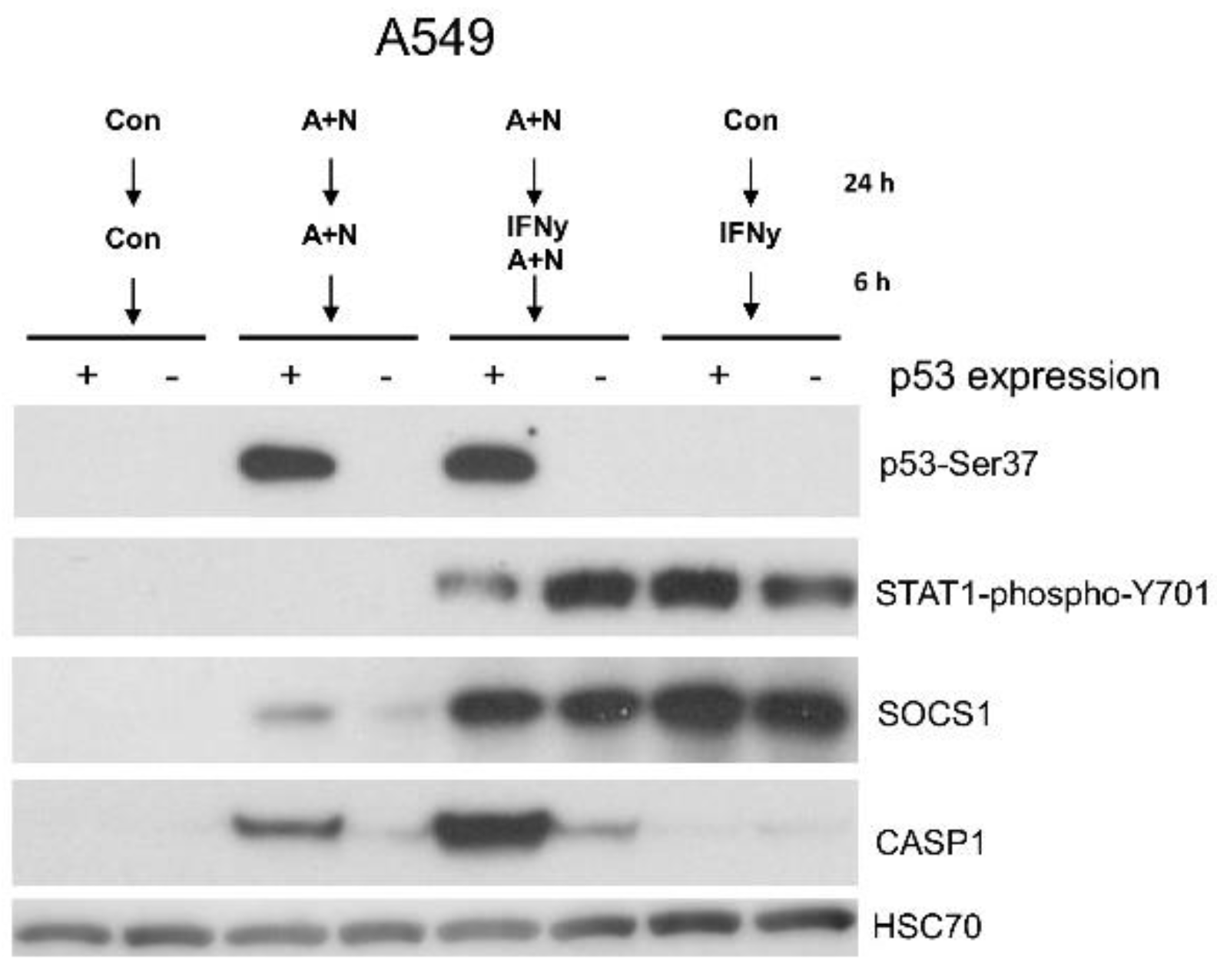
Figure 9.
The activated p53 and IFNγ synergize in stimulation of a subset of innate immunity genes. The p53-proficient (red bars) and p53-deficient (blue bars) A549 cells were exposed to A+N, A+N with IFNγ and to IFNγ alone as described in Figure 8 (treatment mode 24 h + 6 h). Control cells were mock-treated. IFNγ concentration was 1 ng/ml. Subsequently, the expression of indicated genes was determined by RT-PCR. Three biological replicates were performed. GAPDH was used as a reference gene. The influence of p53 status was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=10%). Additionally, statistical significance was calculated for p53-proficient cells treated with A+N vs. A+N+IFNγ and A+N+IFNγ vs. IFNγ. For this purpose calculations were made using the unpaired t test with Welch’s for each group separately (* p < 0,05, ** p<0,01, *** p<0,001). All tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
Figure 9.
The activated p53 and IFNγ synergize in stimulation of a subset of innate immunity genes. The p53-proficient (red bars) and p53-deficient (blue bars) A549 cells were exposed to A+N, A+N with IFNγ and to IFNγ alone as described in Figure 8 (treatment mode 24 h + 6 h). Control cells were mock-treated. IFNγ concentration was 1 ng/ml. Subsequently, the expression of indicated genes was determined by RT-PCR. Three biological replicates were performed. GAPDH was used as a reference gene. The influence of p53 status was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=10%). Additionally, statistical significance was calculated for p53-proficient cells treated with A+N vs. A+N+IFNγ and A+N+IFNγ vs. IFNγ. For this purpose calculations were made using the unpaired t test with Welch’s for each group separately (* p < 0,05, ** p<0,01, *** p<0,001). All tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
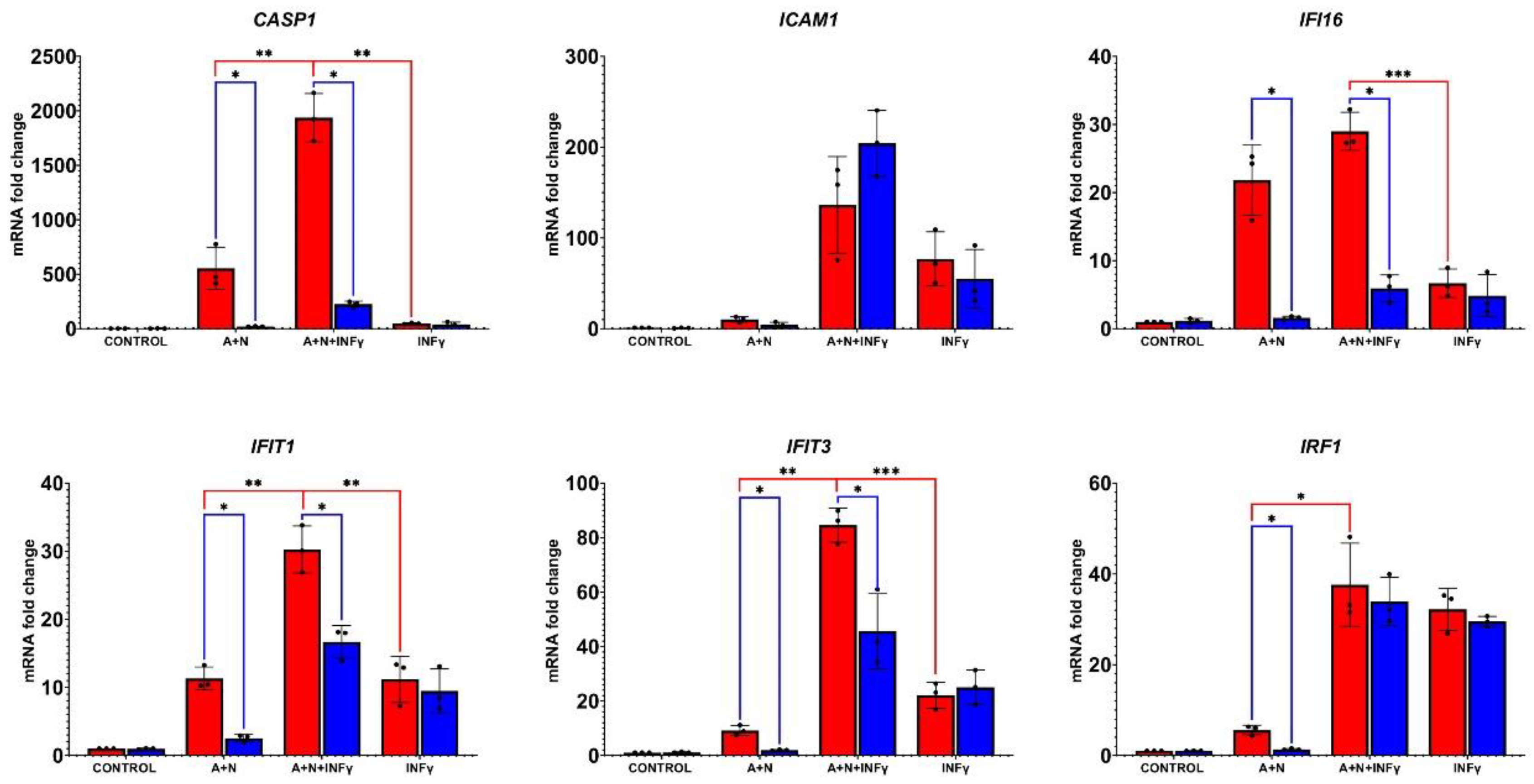
Figure 10.
The collaboration between p53 and IFNγ in activation of CASP1 is cell-specific. A. The Western blot of lysates from NCI-H292 lung cancer cell line exposed to A+N, IFNα1 (1 ng/ml), IFNγ (1 ng/ml) or the indicated combinations for 24 h. B. The Western blot of lysates from GM07492 normal human fibroblasts exposed to A+N, IFNγ (1 ng/ml) or the their combination for 24 h. C. The p53-proficient and p53-deficient NCI-H460 cells were exposed to A+N, IFNγ (1 ng/ml) or their combination for 24 h. Simultaneously, A549 cells were exposed in similar fashion as a positive control. The expression of indicated proteins was examined by Western blotting.
Figure 10.
The collaboration between p53 and IFNγ in activation of CASP1 is cell-specific. A. The Western blot of lysates from NCI-H292 lung cancer cell line exposed to A+N, IFNα1 (1 ng/ml), IFNγ (1 ng/ml) or the indicated combinations for 24 h. B. The Western blot of lysates from GM07492 normal human fibroblasts exposed to A+N, IFNγ (1 ng/ml) or the their combination for 24 h. C. The p53-proficient and p53-deficient NCI-H460 cells were exposed to A+N, IFNγ (1 ng/ml) or their combination for 24 h. Simultaneously, A549 cells were exposed in similar fashion as a positive control. The expression of indicated proteins was examined by Western blotting.

Figure 11.
Actinomycin D and nutlin-3a collaborate in regulation of innate immunity in p53-independent fashion. A. B. The indicated cell lines were exposed as shown for 24 h to actinomycin D (ActD), nutlin-3a (Nut), both compounds (A+N) with or without IFNγ (1 ng/ml). The protein expression was detected by Western blotting. C. Protein expression in p53-null NCI-H1299 cells exposed for 48 h to indicated compounds and their combination. CPT – camptothecin at 5 µM concentration.
Figure 11.
Actinomycin D and nutlin-3a collaborate in regulation of innate immunity in p53-independent fashion. A. B. The indicated cell lines were exposed as shown for 24 h to actinomycin D (ActD), nutlin-3a (Nut), both compounds (A+N) with or without IFNγ (1 ng/ml). The protein expression was detected by Western blotting. C. Protein expression in p53-null NCI-H1299 cells exposed for 48 h to indicated compounds and their combination. CPT – camptothecin at 5 µM concentration.
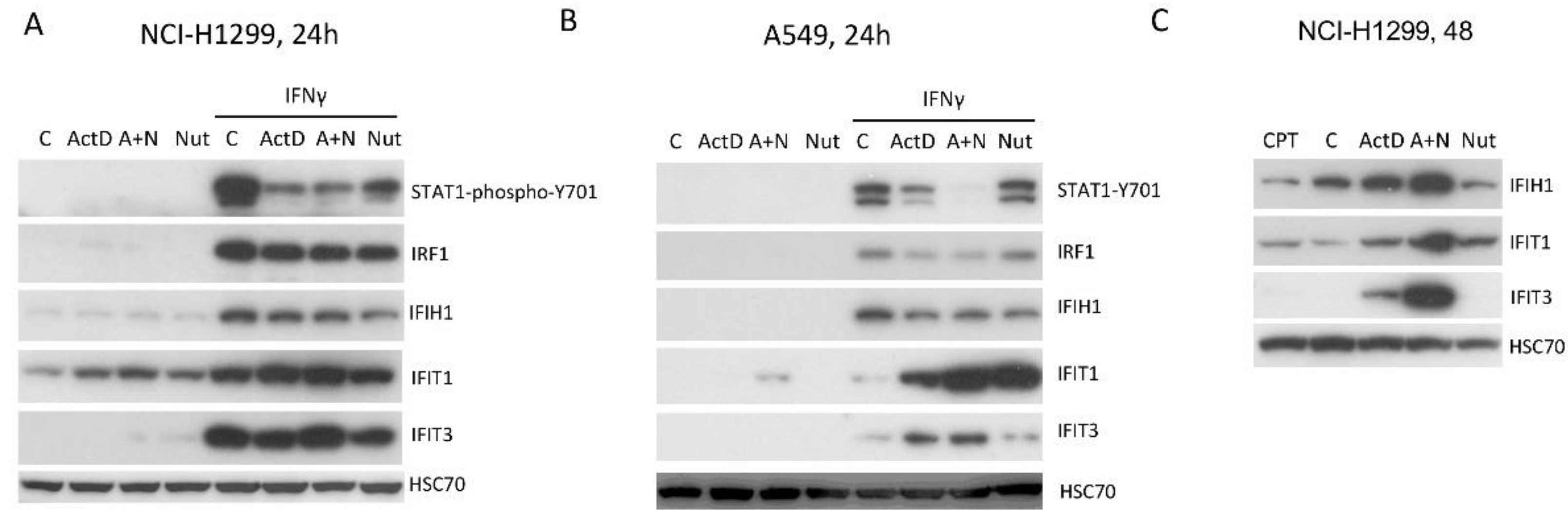
Figure 12.
Actinomycin D and nutlin-3a co-treatment sensitizes cancer cells to the killing by NK-92 natural killer cells. A. The results of MTS assay of A549 cells, which were either mock-treated (DMSO) or exposed to A+N for 48 h. After the treatment, the cells were trypsinized, counted and incubated either with empty medium or with NK-92 cells at two ratios (NK-92:A549 – 1:1 or 5:1) for 24 h. After change of culture medium the A549 cells were allowed to recover for 72 h before their viability was measured by MTS assay. To visualize only the effect of NK-92 cells, the viability of A549 cells, which were incubated without NK-92 cells was set to 100%. Four biological replicates were performed. The influence of A+N was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=5%). B. The results of MTS assay of A549 cells performed as in A. Four biological replicates were performed. Before the co-incubation with NK-92 cells, A549 cells were exposed for 48 h with actinomycin D (ActD), nutlin-3a (Nut) or to both compounds (A+N). Also here, the viability of cells exposed to drugs and not co-incubated with NK-92 cells was set to 100%. The impact of substances were calculated using Brown-Forsythe and Welch ANOVA test. The statistical significance shown in the graph calculated by Dunnett’s T3 multiple comparisons test (* p < 0,05, ** p<0,01, *** p<0,001). C. D. The results of MTS assay on indicated cell lines pre-exposed to A+N or mock-treated (DMSO) for 48 h and co-incubated with NK-92 cell line (1:1 ratio) for 24 h. Other conditions were as described in A. E. The results of MTS assay performed on NCI-H1299 cells as described in B. The influence of A+N w calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=5%). F. The results of MTS assay performed on p53-proficient (CRISPR-Con) and p53-deficient (CRISPR-p53) A549 cells. The setup of experiment was as described above – 48 h exposure to A+N or mock-treatment (DMSO) with subsequent 24 h incubation either with medium or with NK-92 cells (1:1 ratio) with subsequent 72 h recovery. Three biological replicates were performed. The influence of A+N w calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=5%). G. A549 cells were treated with A+N (48 h) or mock-treated as controls (Con), seeded onto 6-well plate in medium or with NK-92 cells at ratio 1:1 or 5:1 (NK-92:A549). After 24-h co-incubation the medium was removed and the attached A549 cells were allowed to recover for 5-7 days. Subsequently the cells were fixed and stained with crystal violet. Three biological replicates are shown. All statistical tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
Figure 12.
Actinomycin D and nutlin-3a co-treatment sensitizes cancer cells to the killing by NK-92 natural killer cells. A. The results of MTS assay of A549 cells, which were either mock-treated (DMSO) or exposed to A+N for 48 h. After the treatment, the cells were trypsinized, counted and incubated either with empty medium or with NK-92 cells at two ratios (NK-92:A549 – 1:1 or 5:1) for 24 h. After change of culture medium the A549 cells were allowed to recover for 72 h before their viability was measured by MTS assay. To visualize only the effect of NK-92 cells, the viability of A549 cells, which were incubated without NK-92 cells was set to 100%. Four biological replicates were performed. The influence of A+N was calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=5%). B. The results of MTS assay of A549 cells performed as in A. Four biological replicates were performed. Before the co-incubation with NK-92 cells, A549 cells were exposed for 48 h with actinomycin D (ActD), nutlin-3a (Nut) or to both compounds (A+N). Also here, the viability of cells exposed to drugs and not co-incubated with NK-92 cells was set to 100%. The impact of substances were calculated using Brown-Forsythe and Welch ANOVA test. The statistical significance shown in the graph calculated by Dunnett’s T3 multiple comparisons test (* p < 0,05, ** p<0,01, *** p<0,001). C. D. The results of MTS assay on indicated cell lines pre-exposed to A+N or mock-treated (DMSO) for 48 h and co-incubated with NK-92 cell line (1:1 ratio) for 24 h. Other conditions were as described in A. E. The results of MTS assay performed on NCI-H1299 cells as described in B. The influence of A+N w calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=5%). F. The results of MTS assay performed on p53-proficient (CRISPR-Con) and p53-deficient (CRISPR-p53) A549 cells. The setup of experiment was as described above – 48 h exposure to A+N or mock-treatment (DMSO) with subsequent 24 h incubation either with medium or with NK-92 cells (1:1 ratio) with subsequent 72 h recovery. Three biological replicates were performed. The influence of A+N w calculated using unpaired t test with Welch’s correction taking into account the False Discovery Rate (FDR=5%). G. A549 cells were treated with A+N (48 h) or mock-treated as controls (Con), seeded onto 6-well plate in medium or with NK-92 cells at ratio 1:1 or 5:1 (NK-92:A549). After 24-h co-incubation the medium was removed and the attached A549 cells were allowed to recover for 5-7 days. Subsequently the cells were fixed and stained with crystal violet. Three biological replicates are shown. All statistical tests were performed using GraphPad Prism version 10.2.2 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
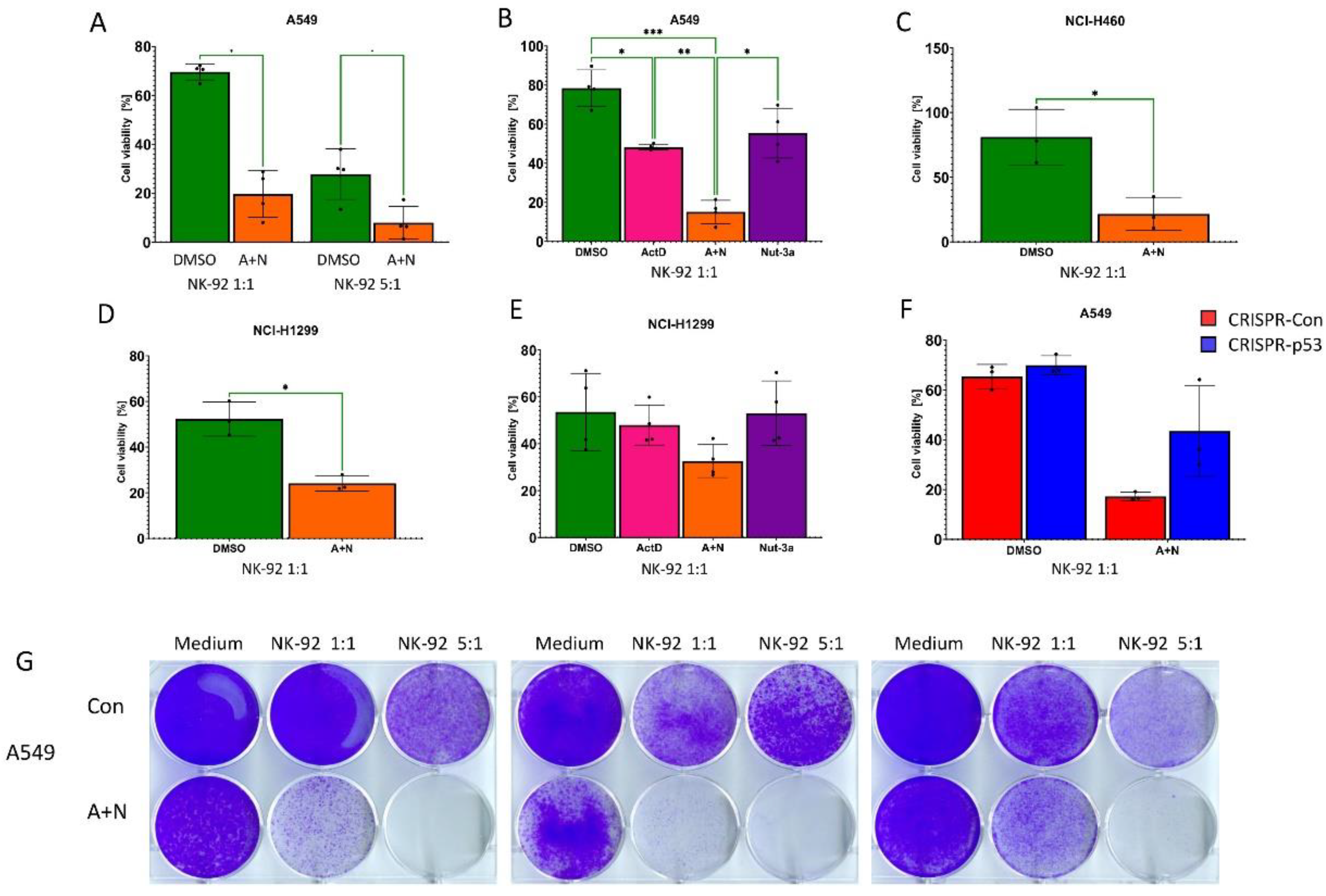
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



