
Submitted:
17 April 2024
Posted:
18 April 2024
You are already at the latest version
Abstract
High resolution information on nucleotide arrangement in the genome of any or-ganism is critical to understand its genetic makeup, function, and evolutionary his-tory. For years, DNA sequencing has provided crucial breakthroughs in the field of genomics and molecular biology. With the advent of technology, Next Generation Sequencing has now transformed the whole genome sequencing prospects, thus unveiling the novel and interesting features of an organism genome that can behold. Next generation sequencing platforms offer realistic approach to identify and anal-yses whole genome with high throughput, accuracy, scalability in a very short time span. Information obtained can be further used in improving our existing healthcare systems, designing novel and personalized therapeutic modules, making the envi-ronment sustainable and proactively preparing ourselves for future pandemics and outbreaks. This article discusses the various NGS technologies and approaches, their advantages, drawbacks, and futuristic usage in different fields. Though still in nascent stages and marred with flaws, with ongoing advancement, NGS will indeed have promising impacts in the future.
Keywords:
Next generation sequencing
; High throughput sequencing
; Illumina Sequencing
; Oxford nanopore sequencing
; Single Cell transcriptomics
; Precision Medicine
1. Introduction
An individual’s DNA is the blueprint of that individual’s traits. Although the human genome project was completed in 2003[1] multiple genomic projects to determine the genetic makeup of other organisms are still ongoing[1]. To completely identify the order of nucleotide arrangement in any organism, nucleotide sequencing holds the key[2]. Determining the complete genetic makeup of any organism will help us to identify single-nucleotide polymorphisms (SNPs)/genetic variations which can lead to the development of various diseases including cardiovascular, neurological, infectious, cancers, and autoimmune disorders[3].
Mutations within both coding and non-coding regions can result in abnormal protein production, which can cause multiple pathophysiology [4]. Throughout history, mutations are also a driving force of evolution. These mutations also result in the emergence of pathogenic species/strains [5] and should be studied extensively for the betterment of the human race and environment [6]. Comparing the genetic makeup of a healthy individual with a diseased one aids in the identification of various ailments, including genetic disorders [7], can act as a diagnostic marker to determine the prognosis of any disease [8], and can distinguish between numerous pathogenic microorganisms [9]. Knowledge of the nucleotide arrangement of any given individual also allows for effective and enhanced personalized medical care [10]. Earlier Sanger sequencing was the most popular method to decode any given DNA sequence [11]. This, along with the Maxam-Gilbert method[11] formed the basis of first generation sequencing [11]. However, these traditional methods of DNA sequencing were complicated and cumbersome when it comes to the whole genome sequencing (WGS) of any given organism [12]. Furthermore, low throughput, higher requirement of starting samples, time taken to analyze the results, and its inability to detect variability in the genetic pattern in parallel samples makes Sanger sequencing an expensive and unreliable methodology for sequencing studies[13]. With the rise in pandemics, rare genetic disorders, climate change, and environmental pollution, it has now become necessary to invest in novel methods of nucleic acid sequencing for better accuracy and affordability. Because of the inability of traditional sequencing methods to effectively handle large-scale genome projects with scalability, accuracy and low throughput and turnaround being an issue, it is critical to develop new sequencing technologies for advancing genomic research and applications. High throughput sequencing (HTS) with the capabilities of massively parallel sequencing, speed, and accuracy has revolutionized the total genome sequencing project of any organism [14]. Also, because its application can be further extended to clinical and translational aspects, agriculture, and environmental health, HTS thus is now becoming a benchmark in the new era of genomics and molecular biology research [15].
High throughput sequencing includes next-generation sequencing methods, with a high demand of low-cost sequencing [16]. It ensures high throughput with massively parallel sequencing by running millions of sequences simultaneously. High throughput sequencing provides a low-cost, effective DNA sequencing and its modification. Many such techniques have been developed, which have made a tremendous change in the field of genome sequencing. The ultimate goal of genome sequencing is to ensure preventive and personalized medicine in curing diseases[17], to identify and differentiate pathogenic species/strains or superbugs, in the diagnosis of communicable and non-communicable diseases, and to create aviable environment and future [18]. This review details the advent of the high-throughput sequencing, how its functions, limitations and its future application in multiple fields has the ability to transform our present into a better future, with the promise of superior healthcare, healthy lifestyle, and sustainable environment.
2. High Throughput Sequencing
High Throughput Sequencing (HTS) can be further divided into three classes of second, third, and fourth generation sequencing (Figure 1). Each of these classes contains different ways to prepare the DNA sample for sequencing studies and distinctly sequence them which are discussed below.
2.1. Second Generation Sequencing
Second generation sequencing methods allow us to sequence the amplified fragmented DNA population. The various second-generation sequencing methods are as follows:
2.1.1. Illumina Sequencing
For quick, precise sequencing, both sample amplification and sequencing by synthesis (SBS) steps are incorporated by Illumina’s high throughput sequencing technology. This involves the identification of nucleotides while the amplification of fragmented DNA is ongoing simultaneously. Illumina sequencing mainly involves four steps: sample preparation, cluster generation, sequencing, and data analysis [19,20]. Sample preparation involves fragmentation of the purified DNA using restriction enzymes. The generated DNA fragments are further ligated with adaptors of known nucleotide sequences and amplified subsequently. Two different types of adaptors are used: PCR adapters and index adapters. PCR adapters (common to all the fragments) are required to attach DNA fragments to the flow cell to facilitate amplification. In contrast, index adapters enable sample multiplexing and further recognition of amplified products during data analysis [21]. Small nucleotide sequences, termed as unique indices or barcodes, are integrated within the index adapters during library building. These indices precisely mark individual samples, allowing several samples to be sequenced in a single run. (SASI-Seq). Once the libraries are ready, the samples are denatured and then subjected to flow cells to initiate sequencing. A flow cell is a specialized chip type with oligonucleotide sequences complementary to PCR adaptors to facilitate base pairing with fragmented sample DNA (Figure 2) [19,20]. The first step in the flow cell is DNA amplification. By polymerase chain reaction, the complementary sequence to the single-stranded DNA is synthesized. After that, synthesized dsDNA is denatured, and the unbound single-stranded DNA is washed off from the flow cell. Now, the adaptors on the free end of the synthesized single strand get hybridized with its complementary nucleotides present on the flow cell by bending, thus forming a bridge-like structure. Hence, this process is termed bridge building. This bridged DNA is amplified again through PCR, known as bridge amplification. This whole process is repeated multiple times to generate clusters of amplified DNAs (Figure 2). Once cluster generation is complete, reverse strands are cleaved and washed off while 3’ ends of sense strands are blocked to prevent any unwanted binding [16,19,20]. Sequencing is commenced by the hybridization of the sequencing primer to the amplified DNA clusters, and the addition of distinctly fluorescent labelled nucleotides according to complementary bases sequentially present in the DNA sample (Figure 2). Distinct fluorescent signal is produced based on the base-pairing of complementary nucleotides (Figure 2)[22]. The emitted light intensity at each cluster is recorded, and then, based on base calling algorithms, the sequence of each cluster is determined. Since this process simultaneously involves the amplification and sequencing of samples, hence it is termed as sequencing by synthesis. This process produces millions of reads presenting all the fragments[20,23]. Unique indices help to demultiplex the whole sequencing run by sorting out the different fastq files for different libraries. Reads with identical base call stretches are grouped. Contiguous sequences are produced by pairing forward and reverse reads. For variant identification, these contiguous sequences are aligned back to the reference genome[20,23]. Compared to conventional sequencing techniques like Sanger sequencing, this technology has many advantages. Multiple strands can be sequenced simultaneously, and the demultiplexed data can be obtained quickly because of the automated nature of the process [20,23].
2.1.2. Roche 454 Sequencing
The principle of Roche 454 sequencing involves the amplification of DNA fragments captured on microbeads using emulsion followed by pyrosequencing that involves detecting released pyrophosphate and light emission upon nucleotide incorporation. The basic steps involved include sample preparation, loading of DNA samples on beads, emulsion PCR, and sequencing during synthesis respectively [16,24,25].Sample DNA is first fragmented into shorter double-stranded fragments of ~400 to 600 base pairs. These fragments are then made blunt ended using enzymes, followed by the addition of adaptors A and B. These adaptors are used as priming sites for both amplification and sequencing (Figure 3). Additionally, adaptor B is biotinylated, which is used for the immobilization of DNA fragments on streptavidin-coated beads (Figure 3). After the ligation of adaptors, double-stranded DNA is denatured using sodium hydroxide [24,25]. Biotinylated single stranded DNA fragments are mixed with streptavidin coated beads for immobilization. This whole reaction takes place in a medium containing a mixture of oil and buffer with PCR reagents. Once the DNA fragments get bound to beads; the whole mixture is vigorously shaken to form emulsions/droplets encapsulating the DNA bound beads [26]. Captured DNA beads are then subjected to PicoTiter Plate which has 1.6 million wells (Figure 3) [26]. Each well is preloaded with enzyme beads containing sequencing enzyme and ATP sulfurylase. Amplification and sequencing are initiated after the addition of primers complementary to the adaptors (Figure 3). Once primer extension begins, each pyrophosphate (PPi) released during the addition of nucleotide is detected with a series of enzymatic reactions (Figure 3). A, T, G, C nucleotides are introduced in sequential manner, one nucleotide at a time (Figure 3). Once a nucleotide is base paired pyrophosphate (PPi) is released as a byproduct (Figure 3). This PPi is then converted to ATP-by-ATP sulfurylase enzyme (Figure 3) which is used by luciferase enzyme to generate light further detected by CCD detector. Strength of the signal is directly proportional to the nucleotides incorporated [27]. Roche-454 is particularly beneficial for accurately sequencing fragments of about 300-800bp. However, low output, issues with homopolymers, and expensive reagents are some of the notable drawbacks of Roche-454 sequencing [28].
2.1.3. SOLiD DNA Sequencing
SOLiD (sequencing by oligonucleotide ligation and detection) DNA sequencing was developed by George M. Church at Harvard University [29]. Like Illumina and Roche 545 sequencing it also involves fragmentation of DNA and adaptors attachment to the fragmented DNA [29]. Fragmentation of genomic DNA is done via nebulization, sonication, or digestion. In nebulization, compressed nitrogen gas is used to force DNA through a small hole, creating mechanically sheared fragments. In sonication, ultrasonic waves are used to shear DNA by resonance vibration and digestion involves the usage of restriction enzymes to cleave DNA. Two different types of adaptors are used for 5’ and 3’ end of fragmented DNA. These adaptors contain specific sequence required for attachment of DNA to the glass slide and initiating the sequencing reaction [29,30,31]. Once the DNA is attached amplification is initiated through emulsion PCR by using the primers complimentary to the adaptor region (Figure 4). This process creates numerous copies of each DNA fragment, resulting in a large number of identical templates for sequencing [25,30,31]. The sequencing reaction is initiated after the amplification with the addition of fluorescently labelled 8-mer sequencing primer. It is a short stretch of DNA fragment in which 1st and 2nd nucleotides position are occupied by known bases. The 3rd, 4th, and 5th bases are degenerate while the 6th to 8th bases are inosine bases in which the 8th base is bound to a specific fluorescent dye (Figure 4) [32]. If the first two nucleotides of 8-mer complement the template DNA, it gets hybridized to the template DNA. DNA ligase then ligate this 8-mer oligonucleotide to the primer (Figure 4). Following ligation, silver ions are used to cleave the phosphorothioate linkage between the 5th and 6th nucleotide resulting in release of fluorescent dye. This cleavage allows the fluorescence to be measured (Figure 4). [32]. This process is repeated till the whole ssDNA gets complementarily hybridized with 8-mer oligonucleotides. The fluorescent signals obtained are converted into digital data, and various algorithms are then used to interpret and assemble the sequence information [30,31,33]. This method is very advantageous to achieve accurate sequencing as a particular position is read multiple times with different probes, however, one of the major drawbacks is the long turnover rates [34].
2.1.4. Ion Torrent Sequencing
Ion Torrent sequencing technique involves detection of the hydrogen ions that are released during DNA synthesis. The targeted nucleic acid, which needs to be sequenced, is fragmented, and adaptors are attached to the ends (Figure 5) [35]. These adaptors allow the DNA fragments to bind to the microwells of Ion Torrent chips, which are specialized semiconductor device (Figure 5) [36]. Once bound, DNA polymerase is introduced to initiate amplification using nucleotides added sequentially to the microwells of the ion Torrent chip (Figure 5). If a complementary nucleotide is incorporated into the opposite strand, there will be a change in pH due to the release of H+ ions (Figure 5). This chemical shift will be translated into digital signals through the semi-conductor chip, which is recorded and further used for base calling (Figure 6). ([37,38] . Ion Torrent sequencing has benefits because of its affordability, scalability, and quick turnaround time and is better suited for short fragments [39].
2.2. Third Generation Sequencing
Third generation sequencing uses a completely different approach where the targeted nucleic acid can be sequenced from longer samples at a single molecule level, thus giving rise to long sequencing read outputs in relatively lesser time[40]. The various third-generation sequencing techniques are as follows:
2.2.1. PacBio Sequencing
PacBio sequencing created by Pacific Biosciences is often referred to as single-molecule real-time (SMRT) sequencing. The key principle of PacBio sequencing involves observing the real-time incorporation of nucleotides by a DNA polymerase enzyme as it synthesizes complementary DNA strand from a template strand. This process utilizes zero-mode waveguides (ZMWs), which are tiny wells where the sequencing reactions take place. Each ZMW contains a single DNA polymerase molecule and a DNA template strand [41,42,43].Direct genomic DNA or substantially long fragments of DNA samples can be sequenced accurately through PacBio sequencing [44]. Adapters are attached to the DNA sample for the circularization of the target DNA. These circular DNAs are then attached to the bottom of the individual ZMWs, followed by introduction of nucleotides tagged with different fluorescent markers into ZMW to initiate sequencing process (Figure 6) [44]. Fluorescent signal is emitted as DNA polymerase add nucleotides complementary to the template (Figure 6). Distinct fluorescent signals are generated based on the nucleotides added and this raw data is collected, and processed to generate DNA sequence (Figure 6) [45]. PacBio sequencing is done in real time with no pauses, resulting in longer sequencing reads in lesser time [45]. Although PacBio sequencing offers several benefits, it also has significant drawbacks, like higher read error rates when compared to short-read sequencing techniques like Illumina. To counter this PacBio uses variety of error correction algorithms and data merging form multiple sequencing runs [33,41,42,43].
2.2.2. Helicos Sequencing
Helicos sequencing, also known as Heliscope Single Molecule Sequencing (SMS) was developed by Helicos Biosciences [46]. Helicos sequencing involves direct sequencing of the cellular nucleic acids at single nucleotide resolution [47,48,49]. Sample preparation, immobilization, and sequencing-by-synthesis are the primary steps followed by fluorescence imaging and data analysis in this technique. The target DNA sample is sheared into smaller fragments. The fragmented nucleic acid molecules undergo ‘polyadenylation’ 3’ end by using terminal transferase enzyme (Figure 7) [46]. This polyA tailing is required for the hybridization of the DNA fragments to the polyT sequence attached to the flow cell (Figure 7) [50]. Following immobilization, DNA polymerase enzyme and distinctly fluorescent labelled nucleotides are added to initiate sequencing in sequential manner. Complementary nucleotides will be added to the extending template followed by the washing off of any unbound bases (Figure 7). After every round of template extension, the flow cell is scanned for the fluorescent signal generated by cleavage of the fluorescent dye from the incorporated nucleotide before next round of extension can begin. This continues until the whole DNA fragment is sequenced [46,50]. Despite many advantages, Helicos sequencing faced challenges in throughput, cost-effectiveness, and error rates compared to other NGS technologies. As a result, it did not gain widespread adoption in the NGS market and was eventually discontinued as a commercial offering [46,47,48,49].
2.2.3. Nanopore-Based Sequencing
Nanopore based sequencing technology was first commercialized by Oxford Nanopore Technologies (ONT) and can be used to directly sequence genomic DNA or RNA [51]. The sequencer generally measures the changes in electrical signal (based on each individual nucleotides molecular weight) as DNA/RNA passes through protein nanopores which make this technology extremely convenient for long read sequencing without the need of amplification. ONT does not normally include the fragmentation of the samples. However, it has been shown that the fragmented sample, when attached to adaptors, can enhance the accuracy of the nanopore sequencing [52]. Fragments are passed through a protein nanopore, which measures electrical conductivity distinguishably based on the molecular weights of nucleotides (Figure 8). These signatures are recorded, and the sequence is deduced using specialized software (Figure 8). The resulting sequence data can be further examined and put together to rebuild the organism’s whole genome or transcriptome [53,54,55,56].
Nanopore-based sequencing techniques are appealing among scientists because of the advantages they offer, including vast parallel sequencing, long reads, high data volume and quality, and associated lower costs [57]. These techniques use nanometer-sized pores, either biological or synthetic/solid-state, to accomplish their goals. α-Hemolysine, MspA and bacteriophage phi29 are the three most extensively studied biological/protein nanopores, which are embedded into lipid bilayer, liposomes and polymer film (Figure 9) and are extensively used in ONT [57]. Si3N4 and SiO2 are, however synthetic or solid-state nanopores which are thermally, mechanically and chemically more stable than biological nanopores [58].
Nanopore-based sequencer does not require dNTPs, polymerase or ligase enzyme. Also, sequencing data can be obtained with less amount of samples [59]. Sequencing of plant genome becomes easier using nanopore-based sequencers, which was earlier difficult with conventional sequencing techniques because of the large genome size [59]. These techniques have the capability to impact time-critical tasks like identifying pathogens by offering speedy and accurate identification in real-time [60]. Although these sequencing technologies offer multiple advantages, they also involve some limitations, such as high error rates, single base recognition, DNA velocity, ultra-precise detection, etc. Scientists are actively making an effort to resolve these obstacles and significantly raise the accuracy, speed, and usefulness of nanopore-based DNA sequencing technology [61].
2.3. Fourth Generation Sequencing
Fourth-generation sequencing technologies offer another exciting horizon in the field of genomics and molecular biology, with the ability to connect spatial and sequence information in ways that will significant influence research and diagnoses in a variety of biomedical fields [61]. Massive Parallel Spatially Resolved Sequencing [62] and Single Cell In Situ Transcriptomics are two revolutionary fourth-generation sequencing technologies [63]. The key benefit of these technologies are that they enable direct sequencing of genetic material inside morphologically maintained cells and tissues, providing valuable spatial information in addition to sequence information [64]. Beyond the present third-generation sequencing platforms, fourth-generation sequencing is a speculative advancement that offers substantial advances in speed, precision, and usefulness [64].
3. Applications
In the past few years, next generation sequencing has changed the paradigm of genome sequencing. It has become a critical tool in medical research, diagnostic, therapeutics, environmental, and evolutionary studies [65]. It has been used worldwide by researchers to address a wide variety of biological problems. NGS was first employed to determine single nucleotide variations (SNVs), insertion and deletions called indels, and structural variations in DNA sequence [65]. However, with the advancement in technology, NGS has come a long way and now being widely used in different fields.
In many medical complexities, gene therapies are opening up new avenues for treatment. Severe Combined Immune Deficiency (SCID) was the very first hereditary disorder for which gene therapy was found to be effective [66]. As SCID is caused by mutation in genes, understanding these mutations and their subsequent pathology through NGS leads to the development of effective gene therapy for SCID [67,68].
Whole genome sequencing (WGS) is one of the comprehensive ways to examine the entire genome of an individual organism. Monitoring outbreaks of infectious diseases, recognizing genetic diseases, and describing mutations that lead to the development of cancer have all been made possible by genetic information obtained via whole genome sequencing [69]. Advancing Real-Time Infection Control Network (ARTIC), an international collaboration involving various organizations like Wellcome Sanger Institute and various other researchers around the world, had developed a whole genome sequencing (WGS) approach for SARS-CoV-2 by using Oxford Nanopore sequencing technology [70]. Immunoinformatic-based vaccination research against SARS-CoV-2 was sparked by viral genome sequencing, which resulted in the development of COVID-19 vaccines such as mRNA, viral vectors and protein subunit vaccines [71].
Similarly, the 1000 Bull Genome Project was initiated to analyse the bovine genomes various cattle breeds worldwide for enhancing the productivity and controlling animal disease etc. Through HTS, this project has not only identified various genetic variants to improve milk productions and enhanced disease resistance capacity by selective breeding but also mutations which could be lethal to the livestock [72]. Such is the success of this project that the investigators are planning to further extend the same analysis on other farm animals to enhance the livelihood of farmers [73].
The Centre for Disease Control and Prevention (CDC) in the United States incorporates pathogen genomics into all of its infectious disease programs [74]. To better understand pathogen genetics, pathogen genomics entails DNA/RNA sequencing of pathogens. This is critical in understanding the pathogen evolution, detecting epidemics, comprehending patterns of transmission, and designing targeted therapies and vaccines. This represents a major move towards a more accurate and successful approach in public health management and disease prevention techniques, which requires high throughput sequencing techniques [75].
The introduction of next-generation sequencing techniques has rendered metagenomic sequencing a feasible method for use in clinical evaluations [76]. Metagenomic high throughput sequencing (mHTS), detects and identifies various pathogenic or non-pathogenic micro-organisms of animal and plant origin [77]. Within the leaf samples of grapevine plants, high-throughput sequencing has identified seven viruses and two viriods [78]. Identification of grapevine pathogen, their interactions with host plant leads to the development of innovative control strategies and improved practices over time [79]. This will further increase the fruit yield and will help in selective breeding of disease resistant crops. The ability of cold-resistant bacteria to breakdown petroleum products in the Arctic [80] has been demonstrated using gene sequencing, which offers significant new details about their ability to adapt and the hydrocarbon degradation capabilities of these bacteria [81]. This discovery is extremely critical in addressing major environmental problems arising from petroleum spillage, which is the foremost concern for marine flora and fauna [82].
The Cancer Genome Atlas (TCGA), the publicly financed initiative and Genome India Project (GIP) make use of next-generation sequencing techniques and microarray based high-throughput methods to study genetic abnormalities in various types of cancer [83]. TCGA has discovered many cancer driver genes that led to the development of tumours and the advancement of cancer [84]. A groundbreaking program in cancer genomics has molecularly described more than 20,000 primary cancer cases and comparable normal specimens from 33 different cancer types [85].Genome India Project (GIP) also seeks to generate personalized medicine by employing patient’s genome to predict and control illnesses. For instance, differences in individual genomes of South Asians and Africans could account for the fact that heart diseases are more prominent in south Asians while African populations are mainly affected from strokes [86].
High-throughput accuracy with little turnover time of NGS had made this technology suitable for the advancement of precision medicines. Precision medicine involves the knowledge of individual genomic details to tailor treatment according to the individual needs. High-throughput sequencing creates datasets that will help in the development of drugs and patient stratification for better treatment [87]. NGS has made oncological care more precise and personalized by using genetic data to customize treatment plans based on the unique features of each patient’s cancer. NGS is expected to become more and more important in the era of precision medicine as technology develops and sequencing prices come down [88]. Trastuzumab, bevacizumab, cetuximab, and panitumumab are a few of the well-known precision medications developed with the help of NGS for metastatic colorectal cancer treatment [89].
NGS has been of tremendous help in understanding our environment. The bacterium Deinococcus radiodurans was the very first free-living organism to undergo whole genome sequencing [90]. How this bacterium is highly resistant to DNA damage from a variety of ionizing rays, ultraviolet rays, oxidizing elements, and electrophilic mutagens was revealed through whole genome sequencing [91]. It was revealed that this bacterium harbours two copies of DNA repair enzymes, which enables them to survive the hostile environment enriched with ionizing radiation [92]. This finding has enabled us to consider using Deinococcus radiodurans in the treatment of wastewater contaminated with radioactive uranium thus solving the major environmental concern generated from nuclear waste [92].
Infection in healthcare is continuously rising. Overuse of antibiotics and their release into our environment results with rise in many multi-drug resistance strains also known as “Superbugs”. These new pathogens not only pose formidable health challenges against humans but can also cause life-threatening conditions in animals and livestock. Whole genome sequencing now has revealed genetic variations that can transform a bacterium into a superbug. Examples of such superbugs are S. aureus [93], V. cholerae, C. difficile, K. pneumoniae, P. aeruginosa, P. mirabilis etc. [94,95]. NGS is now being extensively used to identify other superbugs so that proactive measures can be taken to stop their future outbreak [94].
Instead of identifying variations in a limited number of genes, whole exome sequencing (WES) enables the detection of multiple variations in the protein-coding region of any gene. Whole Exome Sequencing (WES) through NGS is a promising method for identifying potential mutations that can cause diseases because most known disease-causing mutations occur in exons. Rare modifications within the KRT82 gene have been identified via exome sequencing in cases of the autoimmune disease Alopecia Areata [96]. Whole exome sequencing was also used to identify variations in the SLC34A1 in Chinese Han Kids suffering from rare condition known as urolithiasis, a condition causing renal dysfunction[97]. This approach has led to the early detection of this disease among various cohorts, which can ultimately aid us in the development of timely interventions [98]. Next-generation sequencing techniques also help to investigate the structure of different RNAs which provides insights into the structure-function relationship of any RNA in various disorders [99,100]. NGS coupled with mutational profiling has been used to determine RNA structure of T. brucei telomerase RNA causing trypanosomiasis [101], SARS-CoV2 genomic RNA [102,103], Dengue virus [104], Chikungunya virus [105], Plasmodium falciparum parasite [106], rotavirus [107], and various long non-coding RNA and miRNA giving rise to various disorders like miR-675 [108], MALAT1 [109] and HOTAIR [110].
Cell identity, functioning, and behaviour are significantly influenced by various epigenetic regulators. Epigenetic regulators have the ability to regulate any cellular process and control gene expression without altering the gene sequence of any organisms [111]. Nucleotide modifications are one of the epigenetic signatures which are ubiquitously present genome wide [112]. They not only dictate the cellular process temporally but can also mask the immunogenic effect of host towards the invading pathogen thus making the latter stealthy [112]. Identification of these epigenetic signatures will be pivotal in determining the underlying processes which drives the virulence and pathogenicity, and disease development [113]. Targeted insertion of promoters (TIP-Seq)[114], Serial analysis of gene expression (ChIP-SAGE)[115], Paired-end ditag sequencing (ChIP-PET)[116], and Next-generation sequencing (ChIP-Seq)[117] are the best high-throughput sequencing tools to identify various epigenetic signatures present on DNA [118]. Transposase-Accessible Chromatin (ATAC-seq) coupled with high-throughput sequencing technique is a popular way to identify accessible chromatin regions for gene expression [119]. Recently, oxford nanopore sequencing has been used to identify modifications present on RNA [120]. These modifications act as epigenetic regulator to control gene expression in many organisms including but not limited to humans [121], bacteria [122], and viruses [122]. In fact, viral RNA modifications mimics the host RNA, thus masking them from the host immune response [123].
Single-cell sequencing technique is the most advanced method for deciphering the variability and complexity of DNA and RNA transcripts inside a specific cell [124]. This is employed to acquire information about gene expression or mutations spatially and temporally at the level of a single cell. In disease-like conditions, such as cancer, the DNA of each cancerous cell can be sequenced to reveal information about mutations or genetic variations altering normal physiology of each cell. So, transcriptome characterization at single-cell resolution in millions of individual cells has been rendered possible by these techniques [125].
NGS is also employed for Cell-free DNA (cfDNA) sequencing that involves analysing free, or non-cellular DNA from biological samples such as plasma, urine, and cerebrospinal fluid (CSF) in the laboratory [126]. The main goal of this technique is to search for genomic variants/biomarkers linked to various genetic medical conditions in various biological fluids. With the help of high-throughput sequencing, Cell-free DNA (cfDNA) sequencing has been used to diagnose tumours, neurological and cardiovascular disorder etc [127]. To provide efficient, targeted therapies, recognizing particular genetic modifications, determining prognosis, and tracking treatment efficacy over time, cfDNA sequencing is becoming a crucial tool [128].
4. Conclusions
With the tremendous increase in viral pandemics, multiple genomic disorders, environmental pollution and the emergence of drug resistant pathogenic strains, it has now become a challenging task to understand the nature and decode the structure of nucleic acids. Gene sequencing has become the best possible approach to tackle these problems and overcome any issues associated with them. Genome sequencing continues to emerge and are being extensively used in the biomedical research and transforming clinical medicine. NGS technologies offer an impressive range of applications to elucidate RNA’s role in biologics and genetic information locked inside the long strands of DNA to read the sequence content. This review details various NGS techniques which will help in knowledge consolidation and dissemination and enables researchers to make informed decisions in the field of molecular biology. There are billions of species present in the ecosystem, and millions of genes and metagenomes are waiting to be sequenced; all these comprehensive studies provide a base for the future of genomics.
5. Future Perspectives
Through the analysis of unique genetic variations, high throughput sequencing techniques can further assist in the development of precision medicine and personalized vaccines. HTS or NGS can be further used in forensics to build a more precise phenotypic profile of people. It will also help scientists to comprehend the role of microbial communities over environmental alterations like pollution, global warming, destruction of habitats, and emergence of superbugs. Agricultural sectors can also employ HTS technologies to examine soil microbes and plant genomes to develop precision techniques that can further increase agricultural yields by addressing drought, high salinity, nutritional values, and productivity. Although the data analysis complexity as well as expense of sequencing technologies and related studies, might be inaccessible to masses, and healthcare systems making it one of the major bottlenecks, however with advancements in technologies, high throughput sequencing techniques will continue to shape current research and development efforts in a variety of scientific, clinical and environmental fields.
Author Contributions
Writing, Original draft preparation- D.B.D., Writing- S.S.N., Review- S.Y.M., Review and Editing- S.A.S., Review and Editing- N.S., Conceptualization, Supervision Writing, Review and Editing- A.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work received no external funding.
Institutional Review Board Statement
Not Applicable
Informed Consent Statement
Not Applicable
Data Availability Statement
Not Applicable
Acknowledgements
The authors would like to acknowledge Department of Biotechnology & Department of Pharamceutics, NIPER Raebareli, for constant support during the preparation of this manuscript. Dr. Abhishek Dey acknowledges the Department of Biotechnology, Govt. of India for the Ramalingaswami Re-entry fellowship (BT/RLF/Re-entry/02/2021).
Conflicts of Interest
The authors declared no conflict of interest.
This article bears NIPER-R communication number 585.
References
- Aganezov, S.; et al. A complete reference genome improves analysis of human genetic variation. Science 2022, 376, eabl3533. [Google Scholar] [CrossRef]
- Shendure, J.; et al. DNA sequencing at 40: past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.R.; Keats, B.J.; Sherman, S.L. Population genetics, in Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics; Elsevier, 2019; pp. 359–373. [Google Scholar]
- Scacheri, C.A.; Scacheri, P.C. Mutations in the non-coding genome. Current opinion in pediatrics 2015, 27, 659. [Google Scholar] [CrossRef]
- Loewe, L.; Hill, W.G. The population genetics of mutations: good, bad and indifferent; The Royal Society, 2010; pp. 1153–1167. [Google Scholar]
- Hershberg, R. Mutation—the engine of evolution: studying mutation and its role in the evolution of bacteria. Cold Spring Harbor perspectives in biology 2015, 7, a018077. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; et al. The genetic basis of disease. Essays in biochemistry 2018, 62, 643–723. [Google Scholar] [CrossRef]
- Bossuyt, P.M.; et al. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative. 2003.
- Ehrlich, G.D.; Hiller, N.L.; Hu, F.Z. What makes pathogens pathogenic. Genome biology 2008, 9, 1–7. [Google Scholar] [CrossRef]
- Goetz, L.H.; Schork, N.J. Personalized medicine: motivation, challenges, and progress. Fertility and sterility 2018, 109, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef]
- Mooney, S.D. Progress towards the integration of pharmacogenomics in practice. Human genetics 2015, 134, 459–465. [Google Scholar] [CrossRef]
- Perrier, L.; et al. Cost of genome analysis: the sanger sequencing method. Value in Health 2015, 18, A353. [Google Scholar] [CrossRef]
- Normand, R.; Yanai, I. An introduction to high-throughput sequencing experiments: design and bioinformatics analysis. Deep Sequencing Data Analysis 2013, 1–26. [Google Scholar]
- Genomics, C. Sanger sequencing: Introduction, principle, and protocol. 2020, CD Genomics Blog. Recuperado el.
- Del Vecchio, F.; et al. Next-generation sequencing: recent applications to the analysis of colorectal cancer. J Transl Med 2017, 15, 246. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; et al. Structural annotation of equine protein-coding genes determined by mRNA sequencing. Anim Genet 2010, 41 (Suppl 2), 121–130. [Google Scholar] [CrossRef] [PubMed]
- Donkor, E.S. Sequencing of bacterial genomes: principles and insights into pathogenesis and development of antibiotics. Genes 2013, 4, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 2010, 2010, pdb-prot5448. [Google Scholar] [CrossRef] [PubMed]
- Bronner, I.F. Improved Protocols for Illumina Sequencing. Current Protocols in Human Genetics. Current Protocols in Human Genetics 2014, 79. [Google Scholar]
- Glenn, T.C.; et al. Adapterama I: universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). PeerJ 2019, 7, e7755. [Google Scholar]
- Sinha, R.; et al. Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. BioRxiv 2017, 125724. [Google Scholar]
- Yang, X.; et al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC bioinformatics 2013, 14, 1–4. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr Protoc Mol Biol 2018, 122, e59. [Google Scholar] [CrossRef]
- Liu, L.; et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Klein, H.-U.; et al. R453Plus1Toolbox: an R/Bioconductor package for analyzing Roche 454 Sequencing data. Bioinformatics 2011, 27, 1162–1163. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; et al. ART: a next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef]
- Luo, C.; et al. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One 2012, 7, e30087. [Google Scholar]
- Hedges, D.J.; et al. Comparison of three targeted enrichment strategies on the SOLiD sequencing platform. PloS one 2011, 6, e18595. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Verma, V.K. Next-Generation Sequencing and Its Application: Empowering in Public Health Beyond Reality, in Microbial Technology for the Welfare of Society. 2019. p. 313-341.
- Ari, Ş.; Arikan, M. Next-Generation Sequencing: Advantages, Disadvantages, and Future, in Plant Omics: Trends and Applications. 2016. p. 109-135.
- Shendure, J.; et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef]
- Garrido-Cardenas, J.A.; et al. DNA Sequencing Sensors: An Overview. Sensors (Basel) 2017, 17. [Google Scholar] [CrossRef]
- Pickrell, W.O.; Rees, M.I.; Chung, S.-K. Next generation sequencing methodologies-an overview. Advances in protein chemistry and structural biology 2012, 89, 1–26. [Google Scholar] [PubMed]
- Malapelle, U.; et al. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. Journal of clinical pathology 2015, 68, 64–68. [Google Scholar] [CrossRef]
- Merriman, B.; R&, *!!! REPLACE !!!*; D Team, I.T.; Rothberg, J.M. T.; Rothberg, J.M. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef]
- Lahens, N.F.; et al. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genomics 2017, 18, 602. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; et al. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J Clin Pathol 2015, 68, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Davies, K. It’s “Watson Meets Moore” as Ion Torrent Introduces Semiconductor Sequencing. Bio-IT World 2010. [Google Scholar]
- Bleidorn, C. Third generation sequencing: technology and its potential impact on evolutionary biodiversity research. Systematics and biodiversity 2016, 14, 1–8. [Google Scholar] [CrossRef]
- Zhang, W.; Jia, B.; Wei, C. PaSS: a sequencing simulator for PacBio sequencing. BMC Bioinformatics 2019, 20, 352. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genomics Proteomics Bioinformatics 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Wang, Y.; et al. Direct Pacbio sequencing methods and applications for different types of DNA sequences. bioRxiv 2023. [Google Scholar] [CrossRef]
- Coupland, P.; et al. Direct sequencing of small genomes on the Pacific Biosciences RS without library preparation. Biotechniques 2012, 53, 365–372. [Google Scholar] [CrossRef]
- Milos, P.M. Helicos single molecule sequencing: unique capabilities and importance for molecular diagnostics. Genome Biology 2010, 11 (Suppl 1), I14. [Google Scholar] [CrossRef]
- Thompson, J.F.; Raz, T.; Milos, P.M. Helicos Single-Molecule Sequencing for Accurate Tag-Based RNA Quantitation. Tag-Based Next Generation Sequencing 2011, 353–365. [Google Scholar]
- Thompson, J.F.; Steinmann, K.E. Single molecule sequencing with a HeliScope genetic analysis system. Curr Protoc Mol Biol 2010, 92, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Milos, P.M. The properties and applications of single-molecule DNA sequencing. Genome biology 2011, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K. Single-molecule DNA sequencing technologies for future genomics research. Trends in biotechnology 2008, 26, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Nanopore, O. Oxford Nanopore announcement sets sequencing sector abuzz. Nature biotechnology 2012, 30, 295. [Google Scholar]
- Gunter, H.M.; et al. Library adaptors with integrated reference controls improve the accuracy and reliability of nanopore sequencing. Nature Communications 2022, 13, 6437. [Google Scholar] [CrossRef]
- Dumschott, K.; et al. Oxford Nanopore sequencing: new opportunities for plant genomics? J Exp Bot 2020, 71, 5313–5322. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly . Genomics Proteomics Bioinformatics 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol 2019, 20, 129. [Google Scholar] [CrossRef]
- Jain, M.; et al. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 2016, 17, 239. [Google Scholar]
- Kumar, A.; Sharma, V.K.; Kumar, P. Nanopore sequencing: The fourthgeneration sequencing. J Entom Zool Stud 2019, 7, 1400–1403. [Google Scholar]
- Feng, Y.; et al. Nanopore-based fourth-generation DNA sequencing technology. Genomics, proteomics & bioinformatics 2015, 13, 4–16. [Google Scholar]
- Gaur, R.; et al. Sequencing Technologies: Introduction and Applications. Int. J. Hum. Genet 2019, 19, 123–133. [Google Scholar]
- Lin, Y.; et al. Application of nanopore adaptive sequencing in pathogen detection of a patient with Chlamydia psittaci infection. Frontiers in Cellular and Infection Microbiology 2023, 13, 1064317. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; et al. Fourth generation of next-generation sequencing technologies: Promise and consequences. Human mutation 2016, 37, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Strell, C.; et al. Placing RNA in context and space–methods for spatially resolved transcriptomics. The FEBS journal 2019, 286, 1468–1481. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science 2021, 371, eaax2656. [Google Scholar] [CrossRef]
- Mignardi, M.; Nilsson, M. Fourth-generation sequencing in the cell and the clinic. Genome medicine 2014, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ratan, A.; et al. Identification of indels in next-generation sequencing data. BMC bioinformatics 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Fischer, A.; Hacein-Bey-Abina, S. Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med 2020, 217. [Google Scholar] [CrossRef]
- Strand, J.; et al. Second-tier next generation sequencing integrated in nationwide newborn screening provides rapid molecular diagnostics of severe combined immunodeficiency. Frontiers in immunology 2020, 11, 545364. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Hacein-Bey-Abina, S. Gene therapy for severe combined immunodeficiencies and beyond. Journal of Experimental Medicine 2019, 217, e20190607. [Google Scholar] [CrossRef] [PubMed]
- Forde, B.M.; et al. Clinical Implementation of Routine Whole-genome Sequencing for Hospital Infection Control of Multi-drug Resistant Pathogens. Clin Infect Dis 2023, 76, e1277–e1284. [Google Scholar] [CrossRef]
- Márquez, S.; et al. Genome sequencing of the first SARS-CoV-2 reported from patients with COVID-19 in Ecuador. MedRxiv 2020. [Google Scholar]
- Quer, J.; et al. Next-generation sequencing for confronting virus pandemics. Viruses 2022, 14, 600. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Daetwyler, H.D. 1000 bull genomes project to map simple and complex genetic traits in cattle: applications and outcomes. Annual review of animal biosciences 2019, 7, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Weldenegodguad, M.; et al. Whole-genome sequencing of three native cattle breeds originating from the northernmost cattle farming regions. Frontiers in genetics 2019, 9, 728. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.L.; et al. Pathogen genomics in public health. New England Journal of Medicine 2019, 381, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Sintchenko, V.; Holmes, E.C. The role of pathogen genomics in assessing disease transmission. Bmj 2015, 350. [Google Scholar] [CrossRef]
- Lopez-Labrador, F.X.; et al. Recommendations for the introduction of metagenomic high-throughput sequencing in clinical virology, part I: Wet lab procedure. J Clin Virol 2021, 134, 104691. [Google Scholar] [CrossRef]
- Bik, H.M.; et al. Metagenetic community analysis of microbial eukaryotes illuminates biogeographic patterns in deep-sea and shallow water sediments. Molecular Ecology 2012, 21, 1048–1059. [Google Scholar] [CrossRef]
- Diaz-Lara, A.; et al. High-Throughput Sequencing of Grapevine in Mexico Reveals a High Incidence of Viruses including a New Member of the Genus Enamovirus. Viruses 2023, 15, 1561. [Google Scholar] [CrossRef]
- Massart, S.; et al. Virus Detection by High-Throughput Sequencing of Small RNAs: Large-Scale Performance Testing of Sequence Analysis Strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.M.; et al. Crude Oil Degradation in Temperatures Below the Freezing Point by Bacteria from Hydrocarbon-Contaminated Arctic Soils and the Genome Analysis of Sphingomonas sp. AR_OL41. Microorganisms 2023, 12, 79. [Google Scholar] [CrossRef]
- Waldrop, M.P.; et al. Permafrost microbial communities and functional genes are structured by latitudinal and soil geochemical gradients. The ISME Journal 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, M.; et al. Bioremediation by oil degrading marine bacteria: An overview of supplements and pathways in key processes. Chemosphere 2022, 303, 134956. [Google Scholar] [CrossRef]
- Goel, N.; Khandnor, P. TCGA: A multi-genomics material repository for cancer research. Materials Today: Proceedings 2020, 28, 1492–1495. [Google Scholar]
- Bailey, M.H.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary Oncology/Współczesna Onkologia 2015, 2015, 68–77. [Google Scholar] [CrossRef]
- Sengupta, D.; et al. Population stratification and underrepresentation of Indian subcontinent genetic diversity in the 1000 genomes project dataset. Genome biology and evolution 2016, 8, 3460–3470. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nature reviews Drug discovery 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Pereira, M.A.; et al. Application of Next-Generation Sequencing in the Era of Precision Medicine, in Applications of RNA-Seq and Omics Strategies - From Microorganisms to Human Health. 2017.
- Naithani, N.; et al. Precision medicine: Uses and challenges. Med J Armed Forces India 2021, 77, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Matrosova, V.Y.; et al. High-quality genome sequence of the radioresistant bacterium Deinococcus ficus KS 0460. Stand Genomic Sci 2017, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; et al. Genome of the extremely radiation-resistant bacterium Deinococcus radiodurans viewed from the perspective of comparative genomics. Microbiol Mol Biol Rev 2001, 65, 44–79. [Google Scholar] [CrossRef]
- Liu, F.; Li, N.; Zhang, Y. The radioresistant and survival mechanisms of Deinococcus radiodurans. Radiation Medicine and Protection 2023, 4, 70–79. [Google Scholar] [CrossRef]
- Lindsay, J.A.; Holden, M.T. Staphylococcus aureus: superbug, super genome? Trends in microbiology 2004, 12, 378–385. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiology and molecular biology reviews 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Shelenkov, A. Whole-Genome Sequencing of Pathogenic Bacteria-New Insights into Antibiotic Resistance Spreading. Microorganisms 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Erjavec, S.O.; et al. Whole exome sequencing in Alopecia Areata identifies rare variants in KRT82. Nat Commun 2022, 13, 800. [Google Scholar] [CrossRef]
- Wang, Z.; et al. Use of whole-exome sequencing to identify novel monogenic gene mutations and genotype–phenotype correlations in Chinese Han children with urolithiasis. Frontiers in Genetics 2023, 14, 1128884. [Google Scholar] [CrossRef]
- Rajagopal, A.; et al. Exome sequencing identifies a novel homozygous mutation in the phosphate transporter SLC34A1 in hypophosphatemia and nephrocalcinosis. The Journal of Clinical Endocrinology & Metabolism 2014, 99, E2451–E2456. [Google Scholar]
- Smola, M.J.; et al. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nature protocols 2015, 10, 1643–1669. [Google Scholar] [CrossRef]
- Spitale, R.C.; et al. RNA SHAPE analysis in living cells. Nature chemical biology 2013, 9, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; et al. In vivo architecture of the telomerase RNA catalytic core in Trypanosoma brucei. Nucleic Acids Research 2021, 49, 12445–12466. [Google Scholar] [CrossRef] [PubMed]
- Schlick, T.; et al. To knot or not to knot: multiple conformations of the SARS-CoV-2 frameshifting RNA element. Journal of the American Chemical Society 2021, 143, 11404–11422. [Google Scholar] [CrossRef]
- Dey, A.; et al. Abolished frameshifting for predicted structure-stabilizing SARS-CoV-2 mutants: Implications to alternative conformations and their statistical structural analyses. bioRxiv 2024. [Google Scholar]
- Boerneke, M.A.; et al. Structure-first identification of RNA elements that regulate dengue virus genome architecture and replication. Proceedings of the National Academy of Sciences 2023, 120, e2217053120. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.A. Using SHAPE-MaP to model RNA secondary structure and identify 3′ UTR variation in chikungunya virus. Journal of virology 2020, 94, e00701-20. [Google Scholar] [CrossRef]
- Alvarez, D.R.; et al. The RNA structurome in the asexual blood stages of malaria pathogen plasmodium falciparum. RNA biology 2021, 18, 2480–2497. [Google Scholar] [CrossRef]
- Coria, A.; et al. Rotavirus RNA chaperone mediates global transcriptome-wide increase in RNA backbone flexibility. Nucleic Acids Research 2022, 50, 10078–10092. [Google Scholar] [CrossRef]
- Dey, A. , Structural Modifications and Novel Protein-Binding Sites in Pre-miR-675—Explaining Its Regulatory Mechanism in Carcinogenesis. Non-coding RNA 2023, 9, 45. [Google Scholar] [CrossRef]
- Monroy-Eklund, A.; et al. Structural analysis of MALAT1 long noncoding RNA in cells and in evolution. RNA 2023, 29, 691–704. [Google Scholar] [CrossRef]
- He, S.; Liu, S.; Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC evolutionary biology 2011, 11, 1–14. [Google Scholar] [CrossRef]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philosophical Transactions of the Royal Society B: Biological Sciences 2009, 364, 3403–3418. [Google Scholar] [CrossRef]
- Wu, D.; et al. Epigenetic mechanisms of immune remodeling in sepsis: targeting histone modification. Cell death & disease 2023, 14, 112. [Google Scholar]
- Kasuga, T.; Gijzen, M. Epigenetics and the evolution of virulence. Trends in microbiology 2013, 21, 575–582. [Google Scholar] [CrossRef]
- Bartlett, D.A.; et al. High-throughput single-cell epigenomic profiling by targeted insertion of promoters (TIP-seq). Journal of Cell Biology 2021, 220, e202103078. [Google Scholar] [CrossRef]
- Tuteja, R.; Tuteja, N. Serial analysis of gene expression: applications in human studies. BioMed Research International 2004, 2004, 113–120. [Google Scholar] [CrossRef]
- Li, G.; et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome biology 2010, 11, 1–13. [Google Scholar] [CrossRef]
- Steinhauser, S.; et al. A comprehensive comparison of tools for differential ChIP-seq analysis. Briefings in bioinformatics 2016, bbv110. [Google Scholar] [CrossRef]
- Halabian, R.; et al. Laboratory methods to decipher epigenetic signatures: a comparative review. Cellular & Molecular Biology Letters 2021, 26, 1–30. [Google Scholar]
- Grandi, F.C.; et al. Chromatin accessibility profiling by ATAC-seq. Nature protocols 2022, 17, 1518–1552. [Google Scholar] [CrossRef]
- Stephenson, W.; et al. Direct detection of RNA modifications and structure using single-molecule nanopore sequencing. Cell genomics 2022, 2. [Google Scholar] [CrossRef]
- Leger, A.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nature communications 2021, 12, 7198. [Google Scholar] [CrossRef]
- Fleming, A.M.; et al. Direct nanopore sequencing for the 17 RNA modification types in 36 locations in the E. coli ribosome enables monitoring of stress-dependent changes. ACS Chemical Biology 2023, 18, 2211–2223. [Google Scholar] [CrossRef]
- Prezza, G.; et al. Improved bacterial RNA-seq by Cas9-based depletion of ribosomal RNA reads. Rna 2020, 26, 1069–1078. [Google Scholar] [CrossRef]
- Eberwine, J.; et al. The promise of single-cell sequencing. Nat Methods 2014, 11, 25–27. [Google Scholar] [CrossRef]
- Shen, X.; et al. Recent advances in high-throughput single-cell transcriptomics and spatial transcriptomics. Lab Chip 2022, 22, 4774–4791. [Google Scholar] [CrossRef]
- Alborelli, I.; et al. Cell-free DNA analysis in healthy individuals by next-generation sequencing: a proof of concept and technical validation study. Cell death & disease 2019, 10, 534. [Google Scholar]
- Esposito Abate, R.; et al. Next generation sequencing-based profiling of cell free DNA in patients with advanced non-small cell lung cancer: advantages and pitfalls. Cancers 2020, 12, 3804. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.J.; Jardin, F. , cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals (Basel) 2021, 14. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Various high throughput sequencing methods. High throughput sequencing mainly divides into second, third and fourth generation. Illumina, Roche 454 and SOLiD sequencing comes under second generation sequencing methods. Third generation sequencing methods include PacBio, Helicos sequencing and Nanopore-based sequencing techniques. Massive Parallel Spatially Resolved Sequencing and Single Cell In Situ Transcriptomics come under the fourth generation high throughput sequencing technique.
Figure 1.
Various high throughput sequencing methods. High throughput sequencing mainly divides into second, third and fourth generation. Illumina, Roche 454 and SOLiD sequencing comes under second generation sequencing methods. Third generation sequencing methods include PacBio, Helicos sequencing and Nanopore-based sequencing techniques. Massive Parallel Spatially Resolved Sequencing and Single Cell In Situ Transcriptomics come under the fourth generation high throughput sequencing technique.
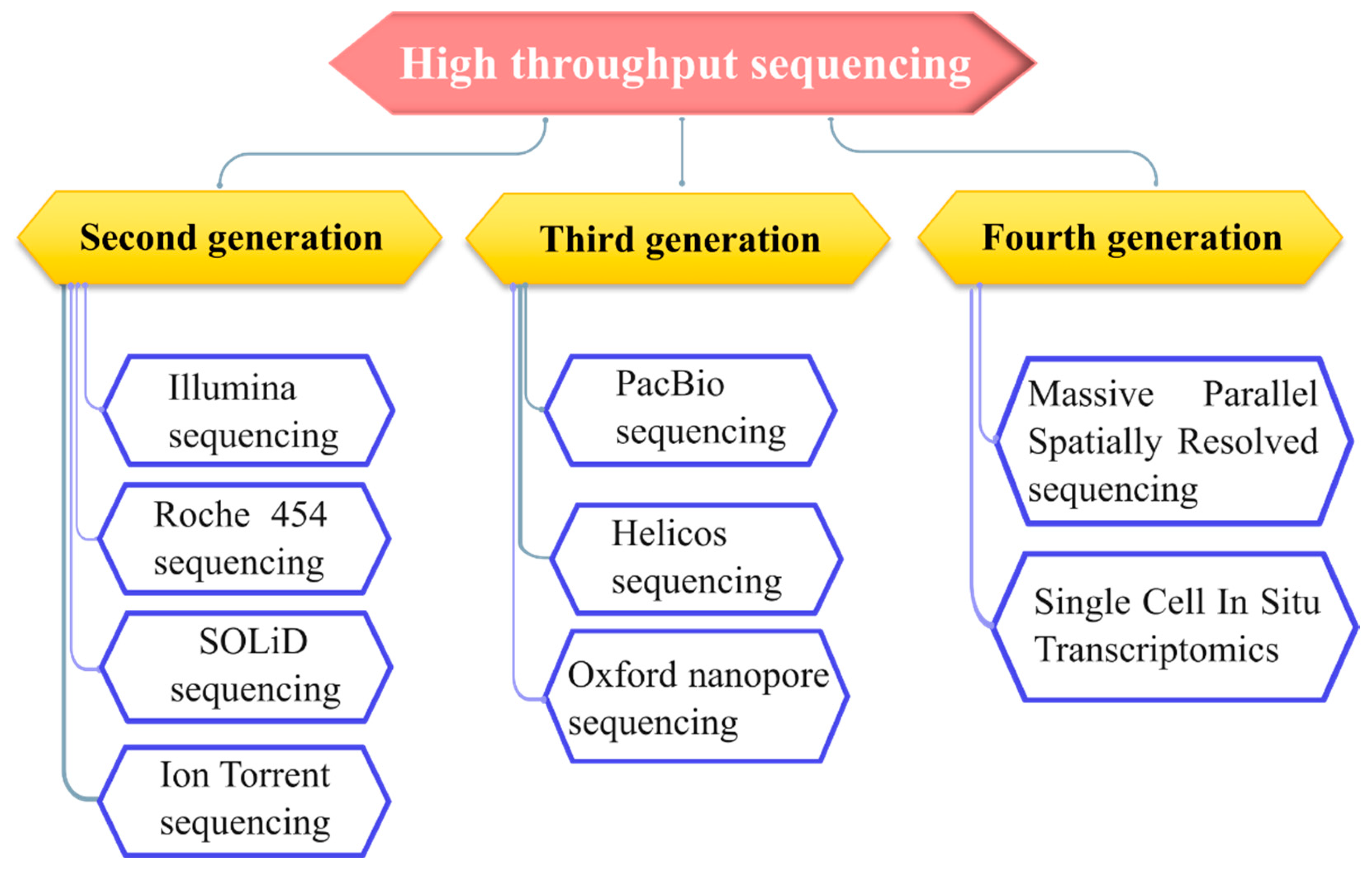
Figure 2.
Steps involved in Illumina sequencing. Library generation involves the fragmentation of genomic DNA and attachment of adaptors to both ends of. Bridge building and bridge amplification is done on the flow cells for cluster generation. Sequencing is initiated by the addition of fluorescently labeled nucleotides. Resultant reads were demultiplexed and are compared back to reference sequence.
Figure 2.
Steps involved in Illumina sequencing. Library generation involves the fragmentation of genomic DNA and attachment of adaptors to both ends of. Bridge building and bridge amplification is done on the flow cells for cluster generation. Sequencing is initiated by the addition of fluorescently labeled nucleotides. Resultant reads were demultiplexed and are compared back to reference sequence.
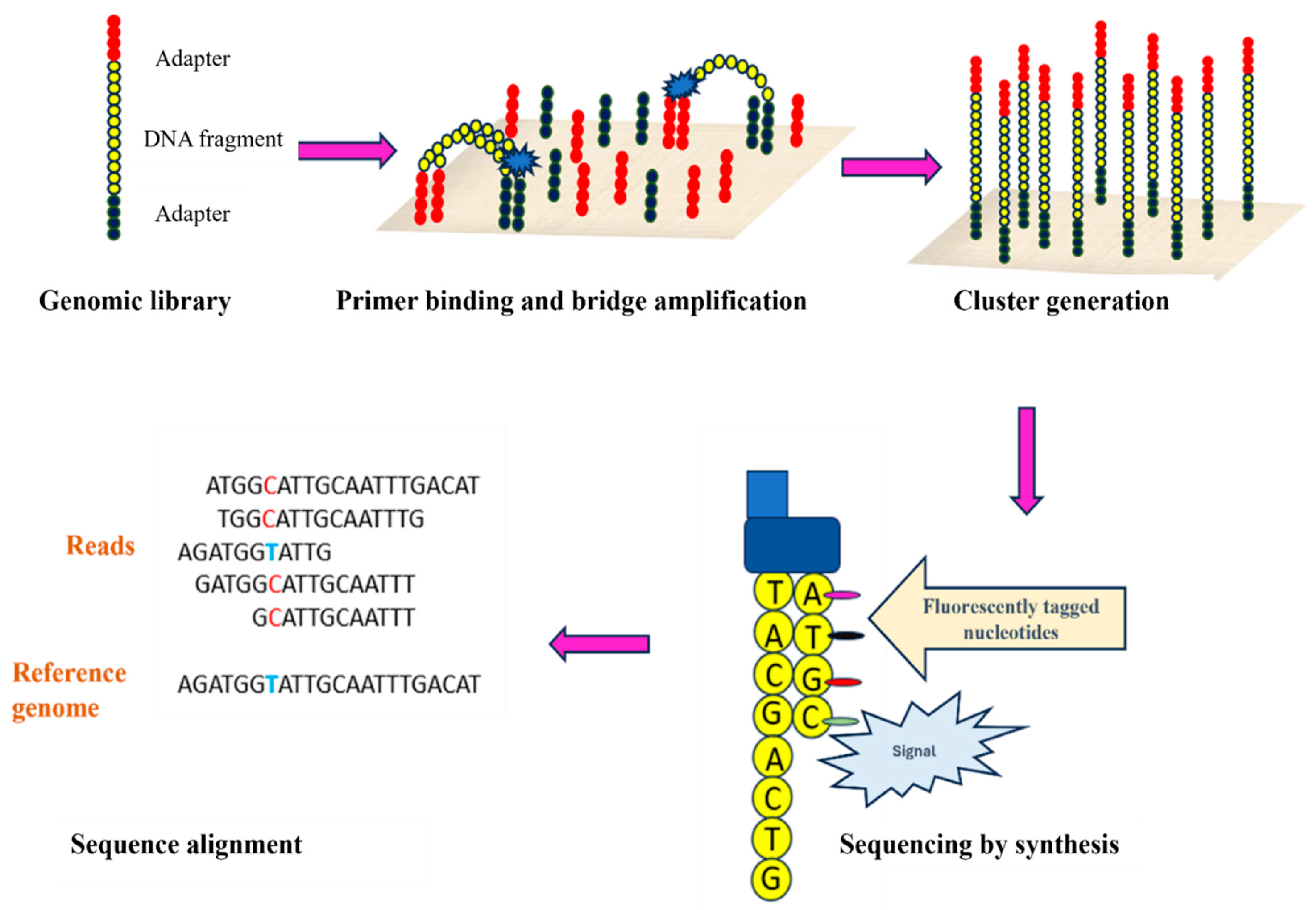
Figure 3.
Steps involved in Roche sequencing. Different adapters are attached to the DNA fragment where one adapter B is biotinylated. This library is further immobilized onto a streptavidin-coated bead. Immobilized libraries are subjected to emulsion PCR to produce multiple DNA copies followed by addition into PicoTitre plate for sequencing .
Figure 3.
Steps involved in Roche sequencing. Different adapters are attached to the DNA fragment where one adapter B is biotinylated. This library is further immobilized onto a streptavidin-coated bead. Immobilized libraries are subjected to emulsion PCR to produce multiple DNA copies followed by addition into PicoTitre plate for sequencing .
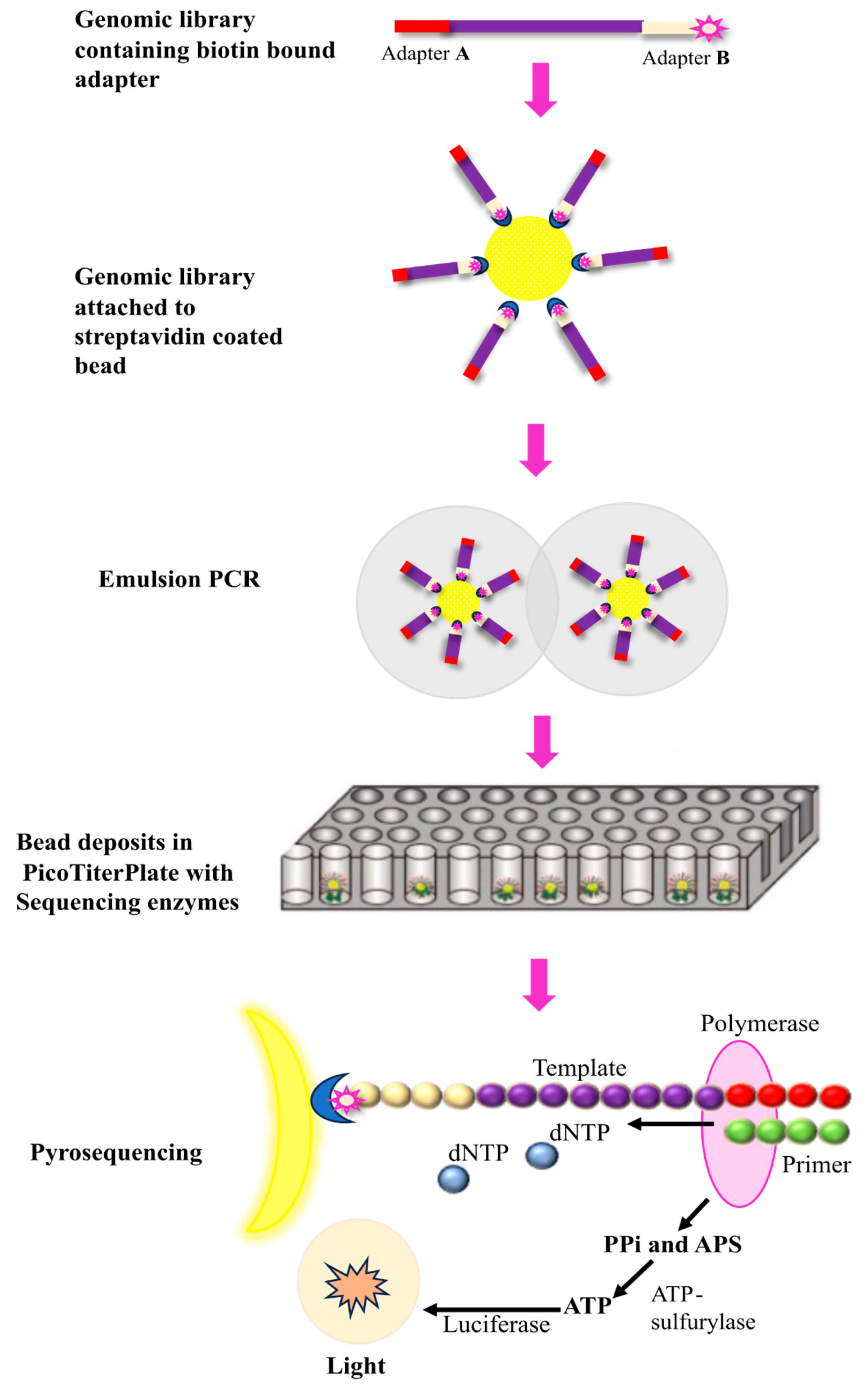
Figure 4.
Steps involved in SOLiD sequencing. Step.1 Multiple libraries are produced via emulsion PCR and immobilized and amplified onto a glass slide. Step.2 involved addition of fluorescently labeled 8-mer nucleotide single-stranded DNA containing phosphorothioate linkage between 5th and 6th nucleotide to initiate sequencing. Step.3 When the first two nucleotides form complementary base pairing with the template strand, phosphorothioate linkage is cleaved thus generating fluorescent emission.
Figure 4.
Steps involved in SOLiD sequencing. Step.1 Multiple libraries are produced via emulsion PCR and immobilized and amplified onto a glass slide. Step.2 involved addition of fluorescently labeled 8-mer nucleotide single-stranded DNA containing phosphorothioate linkage between 5th and 6th nucleotide to initiate sequencing. Step.3 When the first two nucleotides form complementary base pairing with the template strand, phosphorothioate linkage is cleaved thus generating fluorescent emission.
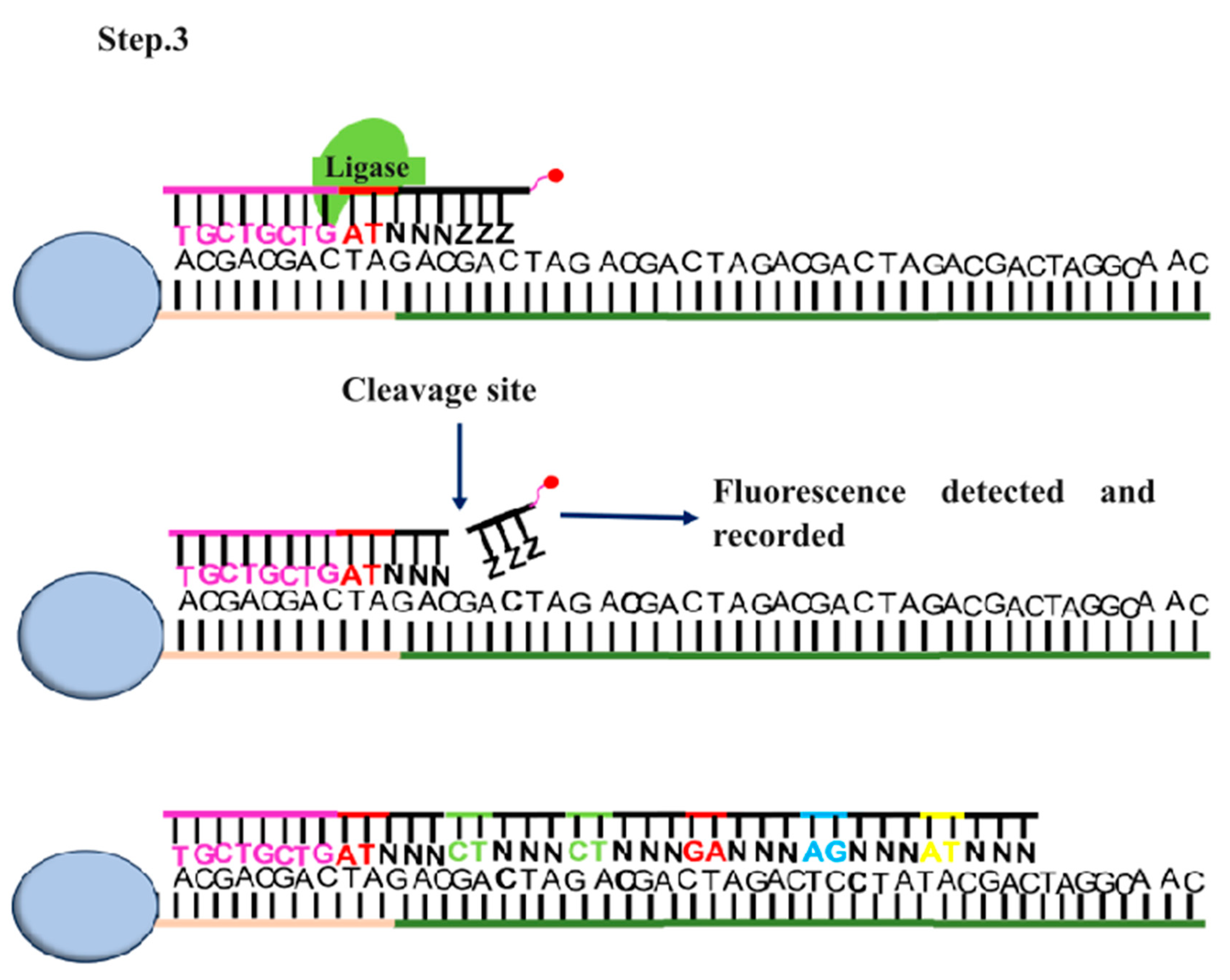
Figure 5.
Steps involved in Ion Torrent sequencing. 1. DNA in which nucleotide sequence is to be recognized is obtained. 2. Libraries are prepared by attaching adapters to both ends of DNA fragments. 3. Prepared genomic libraries are allowed to bind with bead. 4. Libraries loaded beads are then added to ion sensitive chip to initiate sequencing. 5. During nucleotide incorporation, release of H+ ions cause pH fluctuations. 6. Based on pH signals, specialized software produces a sequence of base calls.
Figure 5.
Steps involved in Ion Torrent sequencing. 1. DNA in which nucleotide sequence is to be recognized is obtained. 2. Libraries are prepared by attaching adapters to both ends of DNA fragments. 3. Prepared genomic libraries are allowed to bind with bead. 4. Libraries loaded beads are then added to ion sensitive chip to initiate sequencing. 5. During nucleotide incorporation, release of H+ ions cause pH fluctuations. 6. Based on pH signals, specialized software produces a sequence of base calls.

Figure 6.
Steps involved in PacBio Sequencing. DNA fragments are attached to particular adapters from both ends and form circular DNA. In a zero-mode waveguide (ZMW) chamber, the real-time incorporation of nucleotides is done by DNA polymerase. Different fluorescent signals produce raw data which is further processed to obtain a sequence of sample.
Figure 6.
Steps involved in PacBio Sequencing. DNA fragments are attached to particular adapters from both ends and form circular DNA. In a zero-mode waveguide (ZMW) chamber, the real-time incorporation of nucleotides is done by DNA polymerase. Different fluorescent signals produce raw data which is further processed to obtain a sequence of sample.
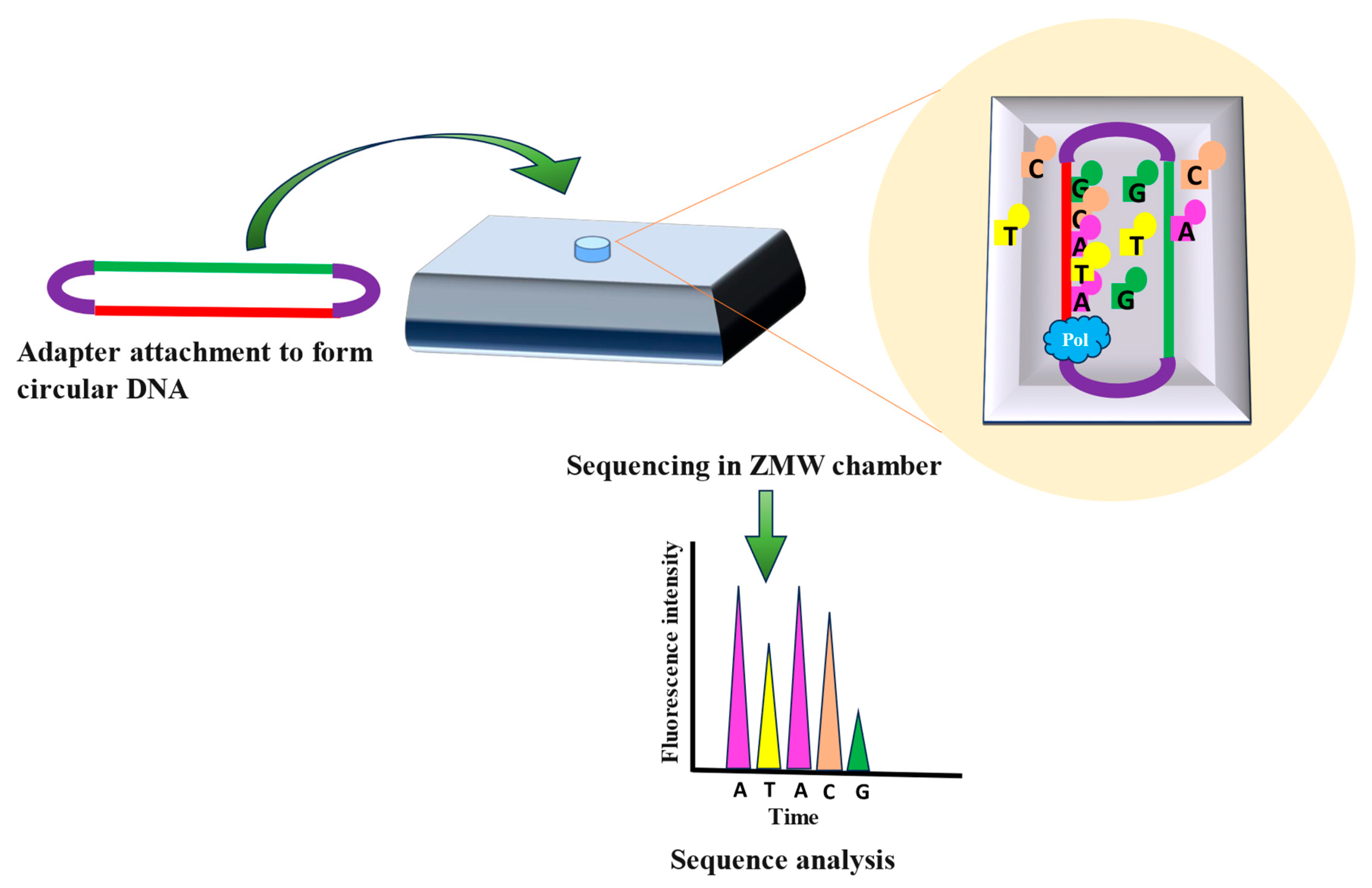
Figure 7.
Steps involved in Helicos sequencing. 1. Fragment generation of sample DNA. 2. Fragmented DNA is polyadenylated. 3. The template strand is allowed to attach to the flow cell with the help of Poly-T tail which is already attached to the flow cell. 4. Sequencing starts with the addition of fluorescent-labeled nucleotides. 5. Imaging data allows for the reconstruction of DNA sequences for sample template.
Figure 7.
Steps involved in Helicos sequencing. 1. Fragment generation of sample DNA. 2. Fragmented DNA is polyadenylated. 3. The template strand is allowed to attach to the flow cell with the help of Poly-T tail which is already attached to the flow cell. 4. Sequencing starts with the addition of fluorescent-labeled nucleotides. 5. Imaging data allows for the reconstruction of DNA sequences for sample template.
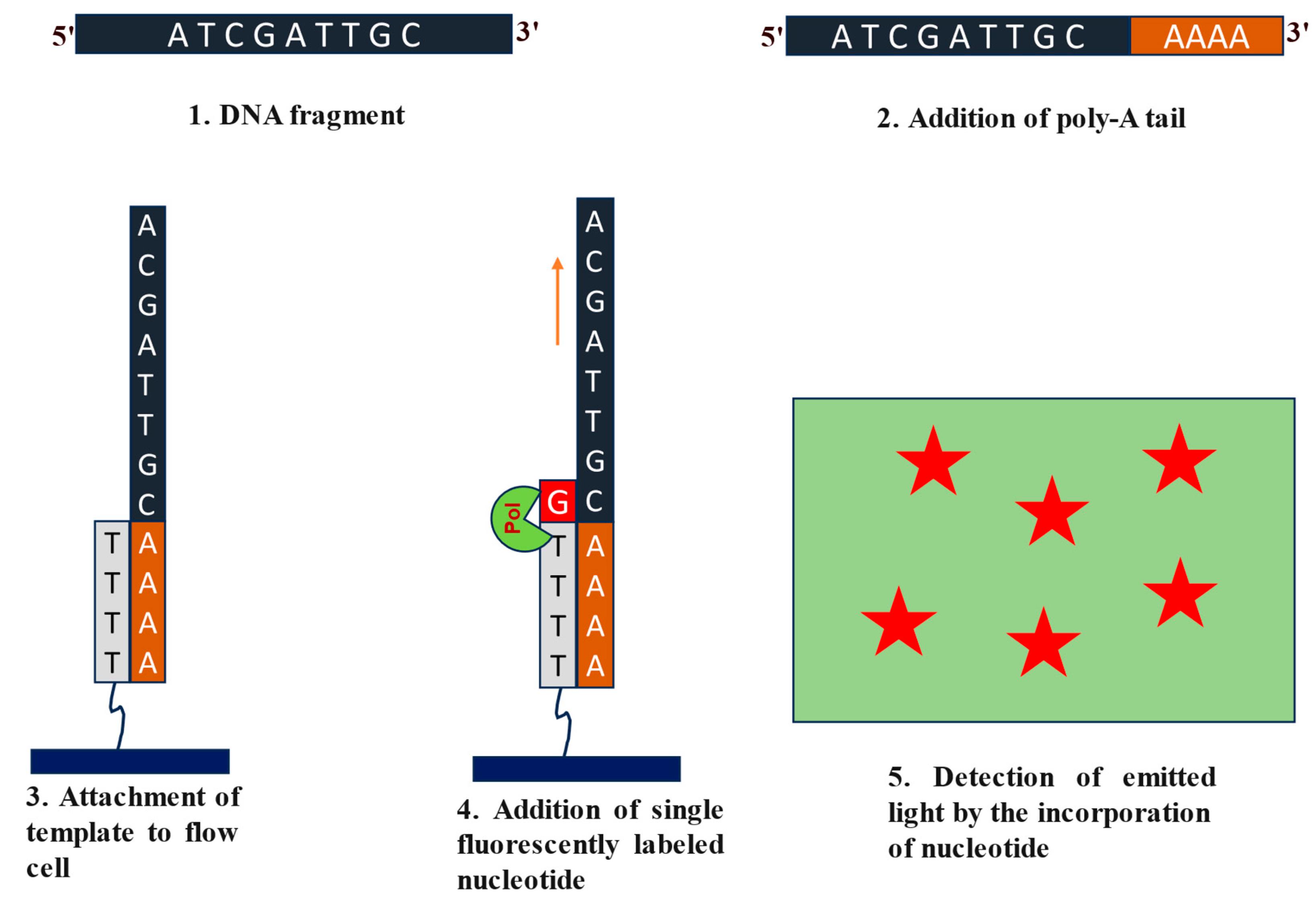
Figure 8.
Oxford nanopore sequencing technique. Genomic libraries prepared from targeted DNA are put into a nanopore sequencer. Ionic current flowing through the nanopore gets disrupted when genomics library is passed through it. Disturbances in electrical signals are recorded and base calling data is produced by specialized software.
Figure 8.
Oxford nanopore sequencing technique. Genomic libraries prepared from targeted DNA are put into a nanopore sequencer. Ionic current flowing through the nanopore gets disrupted when genomics library is passed through it. Disturbances in electrical signals are recorded and base calling data is produced by specialized software.
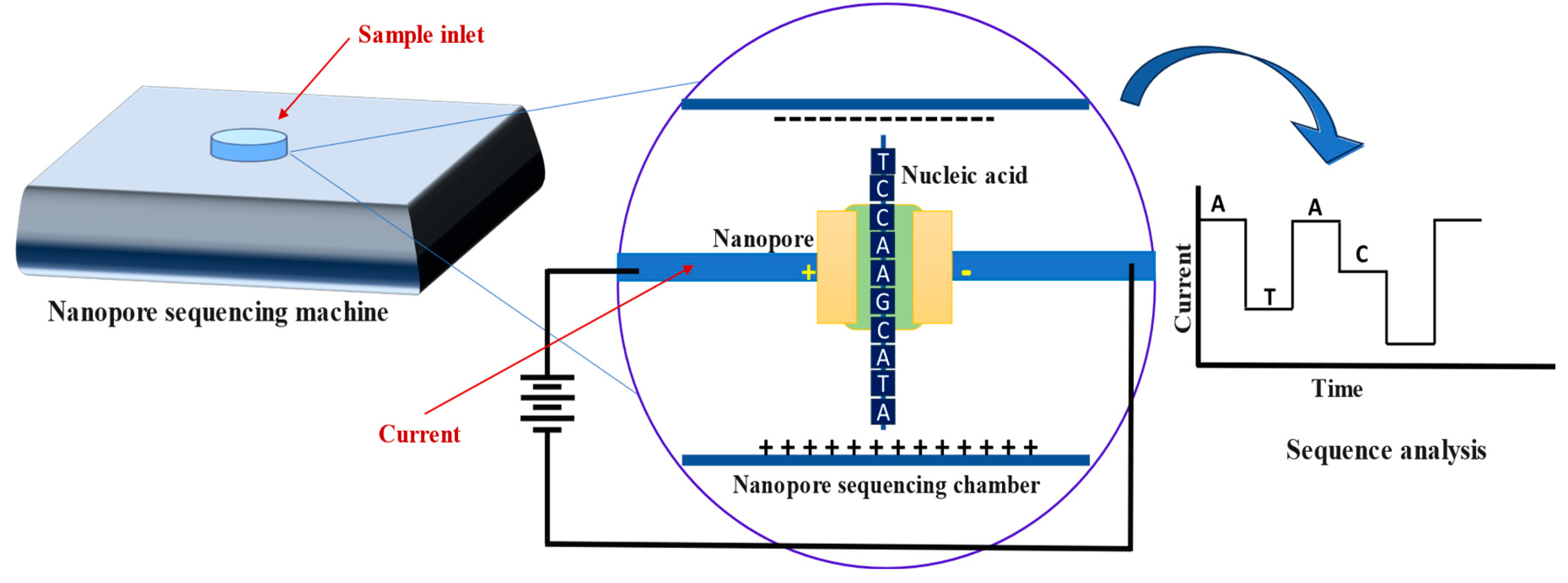
Figure 9.
Top and side views of different nanopores. a) Heptameric α-hemolysin toxin from Staphylococcus aureus b) Octameric MspA porin from mycobacterium smegmatis c) Dodecamer connector channel from bacteriophage phi29 packaging motor.
Figure 9.
Top and side views of different nanopores. a) Heptameric α-hemolysin toxin from Staphylococcus aureus b) Octameric MspA porin from mycobacterium smegmatis c) Dodecamer connector channel from bacteriophage phi29 packaging motor.
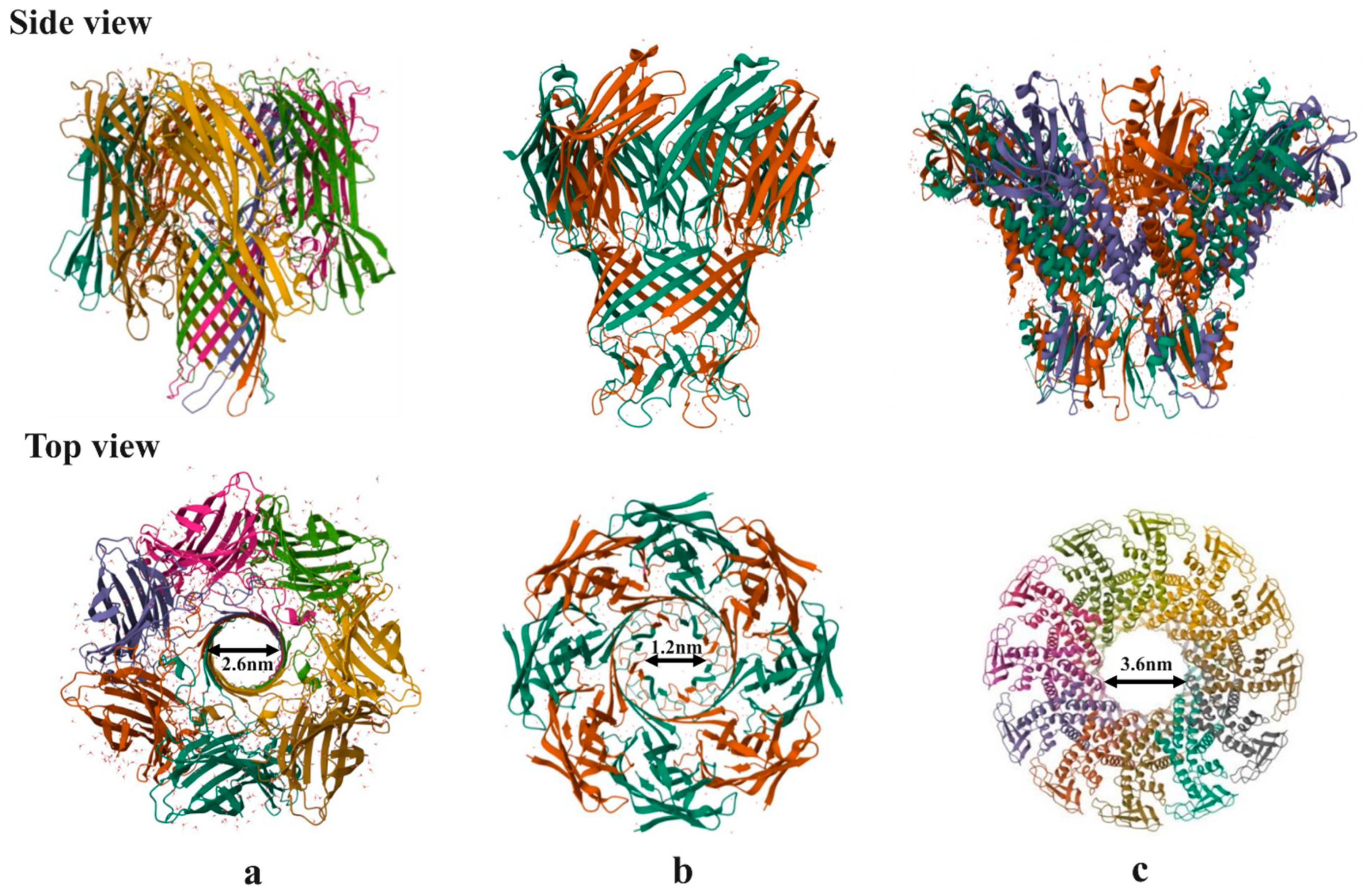
Figure 10.
Applications of high throughput sequencing. In future high throughput sequencing has multiple applications in various fields such as in environment risk management, diagnostics or biomarkers, precision therapeutics, forensics, virus screening, evolutionary genomics etc.
Figure 10.
Applications of high throughput sequencing. In future high throughput sequencing has multiple applications in various fields such as in environment risk management, diagnostics or biomarkers, precision therapeutics, forensics, virus screening, evolutionary genomics etc.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



