
Submitted:
18 April 2024
Posted:
18 April 2024
You are already at the latest version
Abstract
Bicyclic peptides have attracted the interest of pharmaceutical companies because of their remarkable properties putting them on a new path in medicine. Their conformational rigidity that improves proteolytic stability, leads to rapid penetration into tissues via any possible route of administration and elimination, occurring by renal metabolism, is of great importance for people with a history of liver diseases. In addition, each ring can function independently, making them extremely versatile molecules for further optimization. In this paper we compared the potentiometric and spectroscopic properties studied by UV-Vis, MCD, and EPR, of four synthetic analogues of the bi-cyclic peptide c(PKKHP-c(CFWKTC)-PKKH). In particular we correlated the structural and spectral properties of complexes with coordinating abilities toward Cu(II) ions of MCL (Ac-PKKHPc(CFWKTC)PKKH-NH2) that contains the unbinding cycle and N- and C-terminal linear parts, with two histidine residues, one per part; two monocyclic ligands containing one histidine residue, both in the N-terminal position i.e., MCL2 (Ac-PKKHPc(CFWKTC)PKKS-NH2) and in the C-terminal position i.e., MCL3 (Ac-PKKSPc(CFWKTC)PKKH-NH2), respectively, and the linear structure LNL (Ac-PKKHPSFWKTSPKKH-NH2).
Keywords:
bi-cyclopeptide
; histidine
; metal complex
; copper ions
; stability constant
; spectroscopic studies
1. Introduction
Cyclic peptides include in their structure or are themselves rings of polypeptide chains, which can be synthesized by selective side-chain to side-chain or head to tail cyclization strategies, respectively. Homodetic cyclopeptides are based on lactam analogues, compared to heterodetic ones based for example on the classical side-chain to side-chain disulfide bridges but also on other proteolytically stable non-native bonds such as triazolyl bridges [1].
The cyclization affects the stiffening of the structure [2] and allows the mimicry of the secondary structure of some protein fragments [3]. Moreover, the cyclic structure promotes the anchoring of metal ions by amino acid side chains [4]. Studies have proven that cyclic peptides show specificity in forming bonds with certain metal ions. Experiments with native cyclopeptides like oxytocin and vasopressin have proven that despite their lack of coupling to metal ions in nature, they show high ability and efficiency to form bonds with particular metal ions [5,6,7,8].
There are many cyclic peptides with antibacterial, anticancer, immunosuppressive, or toxic properties [9,10]. They are increasingly used in medicine, showing better biological activity than linear peptides [11]. But also natural bicyclic peptides are known to be produced by different organisms. Romidepsin, actinomycin D, moroidin and celogentin C are some examples of natural bicyclic peptides displaying different activities, such as inhibition of tubulin polymerization, prevention of the actin filaments polarization, inhibition of RNA polymerase II, among others [12,13,14]. Interestingly the concept of Bicycles® [15] attracted more and more pharmaceutical companies as synthetic bicyclic peptides rotationally limited by symmetrical small molecule scaffolds that constrain the molecule into rigid two-loop structures. Their versatile peptide scaffold allows iterative and fast optimization processes to increase affinity and selectivity to difficult-to-drug targets. Bicycles are made stable through optimization and their low molecular weight delivers favorable pharmacokinetics, enabling deep tissue penetration, high solubility and flexibility in the areas of manufacturing, route of administration and formulation. The routes of administration of drugs containing bicyclic peptides feature a wide range of choices. Their additional advantage is rapid penetration into tissues. Furthermore, elimination occurs by renal metabolism, which can be of great importance in the use of these compounds in patients with a history of liver disease. The advantage of bicycle peptides is their size and increased conformational rigidity, which improves proteolytic stability in the blood. In addition, each ring can function independently of the other. Moreover, studies point to their use as platforms due to their biocompatibility, similarity, and chemical diversity with previously known native proteins [16,17].
The presence of a second cycle results in an even greater increase in biological stability and ability to bind to the receptor, as each ring in the bicyclic structure can function independently, which makes these peptides bifunctional [18]. Thus, bicycle peptides offer high expectations for the development of new drug carriers due to their similarity with proteins [19].
Previous studies on coordination properties of the bicyclic peptide with one non-binding cycle and a second cycle with two histidine residues as anchoring groups have shown that the bicyclic structure strongly influences the efficacy of the Cu(II) coordination [20], which may be related to the H-bond formation [21].
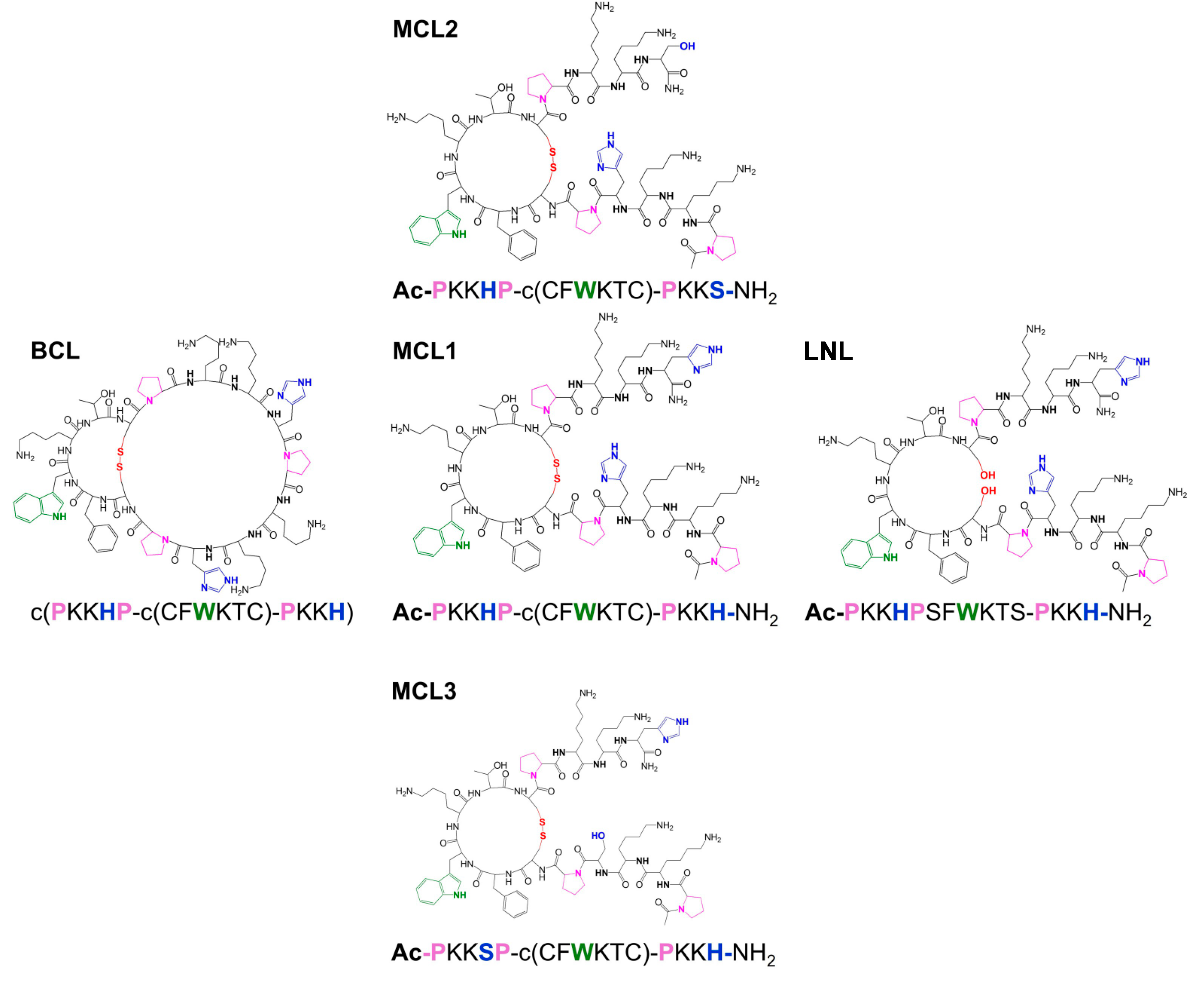
The investigations of the binding abilities of other bicyclopeptides with the c(PKKHP-c(CFWKTC)-PKKH) (BCL) sequence have shown that, above pH 7.4, the peptide forms copper complexes with the untypical transition at 483 nm in the UV-Vis spectra [22]. Owing to this fact we propose herein the results of the studies of four different monocyclic analogues of the original bicyclopeptide BCL. The first, MCL1 (sequence: Ac-PKKHPc(CFWKTC)PKKH-NH2, Bachem AG, Bubendorf, Switzerland), is composed of the unbinding cycle and two linear parts, N- and C-terminal, with two histidine residues, one per part. The next two monocyclic ligands contain one histidine residue in the N-terminal (MCL2: Ac-PKKHPc(CFWKTC)PKKS-NH2) or C-terminal positions (MCL3: Ac-PKKSPc(CFWKTC)PKKH-NH2), respectively, and the last ligand has a linear structure (LNL: Ac-PKKHPSFWKTSPKKH-NH2).
Scheme 1.
Structures of the synthetic peptides BCL, MCL1, MCL2, MCL3, and LNL.
2. Results and Discussion
The analyses of the acid-base properties of MCL1 allowed to determine all protonation constants related to the five side chains of Lys residues and two of the imidazole rings of both His residues (Table1). The values of all protonation constants are comparable to those previously obtained for BCL [22].
The MCL1 and LNL peptides have in their structure the same type and number of deprotonating groups as BCL. Due to this fact, the stoichiometry of the formed complexes can be compared. Both peptides have two separated His residues and based on the fact that imidazole nitrogen is the anchoring group for Cu(II) ions, three systems with nL:nCu(II) = 2:1 (S1), nL:nCu(II) = 1:1 (S2) and nL:nCu(II) = 1:2 (S3) of MCL1 and LNL were investigated with potentiometry. Both MCL1 and LNL form mono- and dinuclear complexes as BCL (Table 1, Figure 1).
MCL1, as well as LNL form the CuH5L species as first complex. The appearance of this complex in both systems is related to the dissociation of two protons from the ligand molecules. In the case of BCL, the CuH5L is characterized by the {2 NIm} binding mode [22]. The value of the calculated stability constants depends on the protonation state of the ligand. It is therefore more advantageous to compare the values of the constants independently of the protonation of the ligand, calculated as logβ* = logβCuHnL – logβHmL. The value of the corrected stability constants, logβ*MCL1 = 4.52 and logβ*LNL = 4.08, are significantly lower than logβ*BCL = 5.68, which supports the involvement of only one imidazole donor in copper binding [25]. In contrast to BCL, which forms the final mononuclear species CuH3L with the {NIm, 2Namide} coordination manner, MCL1 and L form the CuH-3L species with the {NIm, 3Namide} binding mode.
MCL1, in both linear parts, has His residues and due to this fact, it is difficult to define which linear part (N- or C-terminal) favors copper coordination. Because of this, its binding abilities were compared to the analogues having one His residue in the structure (MCL2 in the N-terminal part and MCL3 in the C-terminal part). MCL2 and MCL3 form the CuH4L complex as the first. Its formation, as it was found is related to the dissociation of two protons. The value of the corrected stability constant (logβ*MCL2 = 5.27 and logβ*MCL3 = 6.16) is significantly higher than the stability of MCL1. This result strongly supports different binding mode: one imidazole and one amide moiety.
Both MCL2 and MCL3 form the final species with the {Nim, 3Namide} binding mode that was observed in the MCL1 system. The stability of this species formed by MCL1 is almost the same as the stability of MCL3 (Table 2), which strongly suggests that the linear C-terminal part of the MCL1 sequence is favored.
The di-nuclear complexes existing in the studied system are the second group. Similarly to the BCL systems, the formation of these complexes is promoted with the increase of the Cu(II) concentration concerning the ligand concentration.
As it was mentioned above, in the UV-Vis spectra of all BCL systems [22] is present the untypical transition at 483 nm. Figure 2 shows the UV-Vis spectra obtained for the MCL1-Cu(II) in the systems with the 2:1 (S1), 1:1 (S2), and 1:2 (S3) molar ratios and 1:1 (S2) and 1:2 (S3) systems of the LNL.
The absorption spectra obtained for both systems of the linear peptide LNL, are typical of copper complexes of the peptide and are in good agreement with the potentiometric results. Analyzing the absorption spectra obtained for the MCL1 peptide, we observed that the spectra of the system S3 are similar to those obtained for the LNL. The difference can be observed for the two first systems (S1 and S2) with transition below 500 nm (what was observed for the BCL [22]).
The transition located below 500 nm appears above pH 8. In these conditions in both systems, two complexes dominate the mononuclear CuH3L and the di-nuclear Cu2HL. The concentration of the CuH3L decreases with the change of the molar ratio MCL1:Cu(II) = 2:1 > 1:1 (in the system with the molar ratio MCL1:Cu(II) = 1:2 its concentration = 0) and it can be assumed that the presence of the transition below 500 nm is determined by the mononuclear species.
In the unbinding cycle of the BCL as well as of the MCL1 peptide, the Trp moiety is present. Based on the fact that Trp gives the characteristic transition in the MCD spectra (Figure 3), all MCL1-Cu(II) systems were investigated with MCD.
In all systems with metal ions are present two transitions: one positive at 650 nm and one negative at 500 nm, which are related to the d-d transitions. Below 400 nm are present the transitions characteristic for Trp, CTNim → Cu(II), and CTNim → Cu(II). The pH-dependent changes in the MCD spectra well correlate with the potentiometric results.
The detailed analysis of the Trp region of the Cu(II) complexes concerning the free ligand may give information on the possible interaction of Cu(II) with the Trp side chain. Based on the potentiometric results, the MCD spectra were analyzed at pH 8 and 11 for S1 and S3 molar ratios.
At pH 8 in the first system (S1) exist two main complexes: the dominant CuH3L and Cu2HL whilst in the system with the nL:nCu(II) = 1:2 (S3) the dominant is the Cu2HL. In the CuH3L species, the metal ion is coordinated by three N-donors: one imidazole and two amides. The formation of the Cu2HL complexes is related to the loss of six protons from the ligand molecule. Based on the fact that in these conditions all Lys residues are protonated, there are two main possibilities for binding of both Cu(II) ions: {1NIm, 1Namide}/{1NIm, 3Namide} or {1NIm, 2Namide}/{1NIm, 2Namide}. At pH 8.5 the corrected MCD spectra (complex spectrum – ligand spectrum) of both systems are almost the same. This result strongly supports the same binding mode of copper ions in the mono- as well as in the dinuclear complexes (Figure 3). A similar situation is observed at pH 11 where the final complexes are formed. In the first system dominates the CuH-3L species whilst in the second dominates the Cu2H-5L complex. The corrected spectra are the same what confirms the same {1NIm and 3Namide} donors in the coordination sphere of the Cu(II) ions. Moreover, the region characteristic for the Trp residue is not affected, which strongly supports that the bound Cu(II) ions do not affect the Trp residue, and the presence of this additional transition below 500 nm is not related to an interaction of the side chain of Trp with metal ions.
The next step of the presented studies was the analysis of the EPR spectra for the BCL-Cu(II) and MCL1-Cu(II) systems.
The investigated BCL-Cu(II) systems were systems with all three molar ratios at pH 6.45, 8.4, and 11. The comparison of the EPR spectra obtained for all BCL systems at pH = 6.45 is shown in Figure 4. At this pH in all systems, the CuH5L species is dominating but other complexes are also present. Due to this fact, it is difficult to obtain the EPR parameters for this species. However in the system with 1:2 molar ratio, EPR parameters characteristic for the Cu(II)aq species can be identified, in good agreement with the potentiometric results. The EPR spectra from pH 8.4 (Figure 4) of all BCL systems are very similar with AII = 169 G and gII = 2.234 parameters and support the presence of three N-donors in the coordination sphere of the Cu(II) ion. The EPR parameters: AII = 192 G and gII=2.187 found at pH 11, confirm the existence of the 4N-type of the complex.
In relation to our previous results [22], the untypical transition below 500 nm is present in all systems of MCL1. Due to this fact two MCL1-Cu(II) systems i.e., the nL:nCu(II) = 1:1 and nL:nCu(II) = 1:2, were investigated and compared to BCL in the same conditions. The comparison of the spectra of the investigated systems around pH = 6.45 as well as pH = 11 are very similar and support the same number of N-donors and similar environment of the metal ion. These results suggest that the presence of the transition below 500 nm is not related to the interaction of the additional group with the metal ion.
3. Materials and Methods
3.1. Synthesis of the Peptides LNL, MCL2, and MCL3
Materials for Peptide Synthesis
Fmoc-L-Pro-OH; Fmoc-L-Lys(Boc)-OH; Fmoc-L-Ser(tBu)-OH; Fmoc-L-Cys(Trt)-OH; Fmoc-L-Phe-OH; Fmoc-L-Trp(Boc)-OH; Fmoc-L-Thr(tBu)-OH; Fmoc-L-His(Trt)-OH, CH3CN, TFA, EtOAc, DMSO, DIC, Oxyma Pure, DMF, Piperidine, Acetic anhydride, DCM, Et2O were purchased from Sigma-Aldrich (Milan, Italy).
3.2. Peptide sequences
LNL: Ac-PKKHPSFWKTSPKKH-NH2
MCL2: Ac-PKKHPc(CFWKTC)PKKS-NH2
MCL3: Ac-PKKSPc(CFWKTC)PKKH-NH2
The peptides LNL, MCL2, and MCL3 were synthesized using fully automated induction-assisted solid-phase peptide synthesis (Gyros Protein Technologies, PurePrep® Chorus, Tucson AZ, USA). The Fmoc/tBu induction-assisted SPPS protocol, performed on a 50 μmol scale, consisted of the following steps:
- Swelling in EtOAc/DMSO (4:1 v/v, 3 mL) for 30 minutes at room temperature.
- Fmoc removal for 1 minute at 90°C using 20% piperidine in DMF (2 mL).
- Washings with EtOAc/DMSO (4:1 v/v, 3 x 2 mL).
- Coupling: Fmoc-L-aa-OH, DIC, Oxyma Pure (5:5:5), 2 minutes at 90°C in DMF.
- Washings with EtOAc/DMSO (4:1 v/v, 2 mL).
Peptide elongation involved repeating the following general protocol for each amino acid adequately orthogonally protected, including Fmoc-L-His(Trt)-OH, Fmoc-L-Lys(Boc)-OH, Fmoc-L-Pro-OH, Fmoc-L-Cys(Trt)-OH, Fmoc-L-Thr(tBu)-OH, Fmoc-L-Trp(Boc)-OH, Fmoc-L-Phe-OH, and Fmoc-L-Ser(tBu)-OH.
Both deprotection and coupling reactions occurred in a glass vessel under mechanical mixing and nitrogen bubbling. Following the last cycle of Fmoc removal, the N-terminal position was acetylated using a 10% v/v solution of acetic anhydride in DMF (2 mL).
After washing with DMF and DCM (3 x 3 mL/g resin), the resin was dried under vacuum. Peptide cleavage from the resin, along with deprotection of acid-labile amino acid side-chains, was achieved by treating the peptide-resin with a cocktail of TFA/TIS/DODT/H2O (10 mL, 94:2.5:2.5:1) for 2 hours at room temperature under mechanical shaking. The resin was then filtered and rinsed with fresh TFA. The cleavage mixture was precipitated by adding ice-cold Et2O (35 mL). The resulting crude peptide precursor was washed with ice-cold Et2O (35 mL) and dried under vacuum.
The crude peptides with free cysteine residues MCL2 and MCL3 were solubilized in a mixture of water and CH3CN (1:1) and stirred for approximately 15 minutes to facilitate the formation of intramolecular disulfide bonds. After complete dissolution, additional water was added to the reaction mixture to achieve a final concentration of 5.3 mM. Initially, the pH that was measured 2.5, was adjusted to 9.5 by adding NH4OH (7.5%). H2O2 (0.8 eq) was then added, and the mixture was mechanically stirred for 2 hours at 350 rpm, at room temperature. Finally, the reaction was quenched by adding TFA to readjust the pH to 2.5. The reaction mixture was subsequently lyophilized without further evaporation.
The crude peptides were purified via reverse-phase flash chromatography, monitored by a UV detector (Teledyne ISCO CombiFlash NextGen 300+, Lincoln NE, USA) using a SNAP Ultra C18 column (30 g), with a Column Volume (CV) of 45 mL; flow rate set at 25 mL/min; eluents composed of 0.1% TFA in H2O (A) and 0.1% TFA in CH3CN (B); gradient: 3 CV A, followed by 10 CV from 0% to 50% B, and ending with 3 CV of B.
Subsequent lyophilization yielded the desired peptides as a white powder, with HPLC purity of 98.5% for LNL, 98.5% for MCL2, and 99.1% for MCL3.
The eluted fractions were subjected to analysis by RP-UHPLC-MS using a Thermo Scientific Ultimate 3000 equipped with a diode array detector and a Thermo Scientific-MSQ PLUS, and employing a C18 Waters Acquity CSH™ column (130Å, 1.7 µm, 2.1 × 100 mm). Operating conditions were maintained at a temperature of 318.15 °K, with a flow rate of 0.5 mL/min, eluents consisting of 0.1% TFA in H2O (A) and 0.1% TFA in CH3CN (B), and monitoring at λ 215 nm. The gradient ranged from 25% to 60% B in A over 5 minutes.
LNL: Rt: 3.22 min, [M+2H]2+ calcd. 938.12; [M+2H]2+ found 938.42.
MCL2: Rt: 3.57 min, [M+2H]2+ calcd. 928.14; [M+2H]2+ found 928.38.
MCL3: Rt: 3.58 min, [M+2H]2+ calcd. 928.14; [M+2H]2+ found 928.35.
RP-UHPLC traces and ESI-MS spectra of LNL, MCL2, and MCL3 are reported in the Supplementary materials.
3.3. Potentiometric Measurements
Potentiometric measurements were performed at 25 °C using a Metrohm semi-micro combination electrode and a pH-meter system calibrated with HCl at a hydrogen ion concentration. The pH-metric titrations were carried out in an ionic strength I=0.1 mol L-1 KCl/HCl solution with a sample volume of 1.5 mL at a ligand concentration of 6x10-4 mol L-1. A 2 mL micrometer syringe was used to introduce KOH. It included 0.1 mol L-1 of KOH. The pH range of 2.5 to 11.5 served as the measurement range. The titration curves, HYPERQUAD [23], and SUPERQUAD [24] software were used to calculate the stability constants and stoichiometry of the complexes.
3.4. Spectroscopic Measurements
Using a Varian Cary 50 Bio spectrophotometer (Varian Inc., USA), visible spectra in the 300–800 nm range were captured at 25 °C. For all systems under investigation, magnetic circular dichroism (MCD) spectra were captured at 25 °C using a Jasco J-1500 spectrometer (Jasco, Japan). The MCD spectra were captured using a Permanent Magnet PM-491 accessory and were collected in a magnetic field of +1.6 T in the N/S field direction. All spectroscopic measurements were collected in the pH range of 2.5 to 11. The reactions were carried out in a 0.3 mol L1 KCl solution with a ligand concentration of 6x10-4 mol L-1. 2.0 mL was used for each sample. With the aid of a Mettler Toledo pH meter and modest additions of concentrated KOH and HCl solutions, the pH values were determined. The electron paramagnetic resonance (EPR) spectra were recorded on Bruker ELEXSYS E500 CW-EPR, X-Band spectrometer, equipped with ER 036TM NMR Teslameter and E 41 FC frequency counter. A microwave power of 10 mW, a modulation frequency of 100 kHz, a modulation amplitude of 10 G, a time constant of 82 ms, and a conversion time of 164 ms were adopted. The samples were sealed in quartz tubes and placed inside a standard EPR quartz tube for measurements.
4. Conclusions
We report herein the study of four analogues of the bicyclic peptide with the sequence c(PKKHP-c(CFWKTC)-PKKH) (BCL) and the role of the structural aspects on the coordination abilities of the bi-cyclopeptide with the unbinding cycle -c(CFWKTC)-, are presented. The analysis of the potentiometric results have shown that the bi-cyclic structure promotes the involvement of the side chain imidazole donors in the Cu(II) binding. Based on the results obtained for the mono-cyclic analogues containing two His residues (MCL1: Ac-PKKHPc(CFWKTC)PKKH-NH2) and one His residues (MCL2: Ac-PKKHPc(CFWKTC)PKKS-NH2 and MCL3: Ac-PKKSPc(CFWKTC)PKKH-NH2), respectively, it can be assumed that the coordination of the histidine moiety as anchoring group is promoted by its location in the peptide sequence further from the nonbinding cycle that strongly influences the involvement of the amide donors in the Cu(II) coordination. The spectroscopic studies, based on MCD and EPR, support the hypothesis that neither the direct interaction of tryptophan side chain with the Cu(II) ion nor other groups affect the appearance of an unusual band below 500 nm in the UV-Vis spectrum.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: RP-UHPLC trace of LNL; Figure S2: ESI-MS spectrum of LNL; Figure S3: RP-UHPLC trace of MCL2; Figure S4: ESI-MS spectrum of MCL2; Figure S5: RP-UHPLC trace of MCL3; Figure S6: ESI-MS spectrum of MCL3.
Author Contributions
Conceptualization - J.B. and A.M.P.; methodology - J.B. and A.M.P.; solution investigation and data curation of MCL1 - J.B. and A.M.; solution investigation of MCL2, MCL3, and LNL - J.B. and A.Le.; data curation of MCL2, MCL3, and LNL - J.B., A.Li., P.S., and F.D.; peptide synthesis and characterization - A.M.P. and L.P.; EPR investigation - A.Li., A.Le., and J.B.; writing the original draft - J.B., A.Li., P.S., F.D.; writing and editing the final version of the manuscript - J.B., A.M.P., L.P., A.Li., P.S., F.D., A.M., and A.Le.; supervision - J.B. and A.M.P.; project administration - J.B. and A.M.P.; funding acquisition - J.B. and A.M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Wroclaw Medical University, Poland, grant number SUBZ.DO80.22.024. L.P. is the recipient of the PhD fellowship PON Research and Innovation 2014–2020 (D.M 1061/2021) XXXVII Cycle in Chemical Sciences: “Greening peptide chemistry, a necessary step to the future”, funded by MUR/EU-FSE. The project was funded in part by the TTPep project in the context of 5th seed funding call of EUniWell.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- D’Ercole, A. , Sabatino G., Pacini L., Impresari E., Capecchi I., Papini A. M., Rovero P. On-resin microwave-assisted copper-catalyzed azide-alkyne cycloaddition of H1-relaxin B single chain ‘stapled’analogues. Peptide Sci. 2020, 112, e24159. [Google Scholar] [CrossRef]
- Bockus, A.T. , McEwen C. M., Lokey R.S. Form and function in cyclic peptide natural products: a pharmacokinetic perspective, Curr. Top. Med. Chem., 2013, 13, 821–836. [Google Scholar]
- Testa, C. , Papini A.M., Chorev M., Rovero P. Copper-catalyzed azide-alkyne cycloaddition (CuAAC)-mediated macrocyclization of peptides: impact on conformation and biological activity. Curr. Top. Med. Chem. 2018, 18, 591–610. [Google Scholar] [CrossRef]
- Brzeski, J. , Wyrzykowski D., Chylewska A., Makowski M., Papini A. M., Makowska J. Metal-Ion Interactions with Dodecapeptide Fragments of Human Cationic Antimicrobial Protein LL-37 [hCAP(134–170)]. J. Phys. Chem. B 2022, 126, 36–6911. [Google Scholar] [CrossRef]
- (a) Bal W., Kozlowski H., Lammek B., Rolka K., Pettit L.D. Potentiometric and spectroscopic studies of the Cu (II) complexes of Ala-Arg8-vasopressin and oxytocin: two vasopressin-like peptides. J. Inorg. Biochem. 1992, 45 , 193-202. (b) Uber D., Wyrzykowski D., Tiberi C., Sabatino G., Żmudzińska W., Chmurzyński L, Papini A.M., Makowska, J. Conformation-dependent affinity of Cu (II) ions peptide complexes derived from the human Pin1 protein: ITC and DSC study. J. Therm. Anal. Calorim. 2017, 127, 1431-1443. (c) Gellini C., Sabatino G., Papini A. M., Muniz-Miranda, M, SERS study of a tetrapeptide based on histidine and glycine residues, adsorbed on copper/silver colloidal nanoparticles. J. Raman Spectrosc. 2014, 45(6), 418-423. (d) Żamojć K., Wyrzykowski D., Sabatino G., Papini A. M., Wieczorek R., Chmurzyński L., Makowska J. Key role of histidine residues orientation in affinity binding of model pentapeptides with Ni2+ ions: A theoretical supported experimental study. J. Mol. Liq. 2021, 341, 117414. [CrossRef]
- Lammek, B. , Rekowski P., Kupryszewski G., Melin P., Ragnarsson U. Synthesis of arginine-vasopressins, modified in positions 1 and 2, as antagonists of the vasopressor response to the parent hormone. J. Med. Chem. 1988, 31, 603–606. [Google Scholar] [CrossRef]
- Liu, D. , Seuthe A. B., Ehrler O.T., Zhang X., Wyttenbach T., Hsu J.F., Bowers M.T. Oxytocin-receptor binding: why divalent metals are essential, J. Am. Chem. Soc. 2005, 127, 2024–2025. [Google Scholar]
- Pearlmutter, F. , Soloff M. S. Characterization of the metal ion requirement for oxytocin-receptor interaction in rat mammary gland membranes. J. Biol. Chem. 1973, 254, 3899. [Google Scholar]
- Sobell H., M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 5328–5331. [Google Scholar] [CrossRef]
- Karim M. R., U. , Zhou Y. In, T., Harunari E., Oku N., Igarashi N., Y., Nyuzenamides A and B: Bicyclic Peptides with Antifungal and Cytotoxic Activity from a Marine-Derived Streptomyces sp. Org. Lett. 2021, 23, 2109–2113. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K. , Katoh T., Shimodaira H., Oda A., Takahashi O., Ishioka C., Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors. Cancer Sci. 2012, 103, 1994–2001. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.C. , Fernandez A.M., Lassota P., Ireland C.M., Barrows L.R. Inhibition of tubulin polymerization by vitilevuamide, a bicyclic marine peptide, at a site distinct from colchicine, the vinca alkaloids, and dolastatin 10. Biochem. Pharmacol. 2002, 63, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F. , Xiang L., Zhou Q., Carralot J.P., Prunotto M., Niederfellner G., Pastan I. Actinomycin D enhances killing of cancer cells by immunotoxin RG7787 through activation of the extrinsic pathway of apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 10666–10671. [Google Scholar] [CrossRef] [PubMed]
- Ji, X. , Nielsen A.L., Heinis C.H. Cyclic Peptides for Drug Development. Angew. Chem. Int. Ed. 2024, 63, e202308251–1. [Google Scholar] [CrossRef] [PubMed]
- Creative Peptides https://www.pepdd.com/services/bicyclic-peptides.html.
- Ibraheem D., Elaissari A., Fessi H. Administration strategies for proteins and peptides. Int. J. Pharm. 2014, 477(1-2), 578–589.
- Quartararo, J.S. , Eshelman M. R., Peraro L., Yu H., Baleja J. D., Lin Y. S., Kritzer J. A. A bicyclic peptide scaffold promotes phosphotyrosine mimicry and cellular uptake. Bioorg. Med. Chem. 2014, 22, 6387–6391. [Google Scholar] [CrossRef]
- Saijo, K. , Katoh T. , Shimodaira H., Oda A., Takahashi O., Ishioka C., C. Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors. Cancer Sci. 2012, 103, 1994–2001. [Google Scholar] [PubMed]
- Marciniak, A. , Witak W., Pieniężna A., Brasun J. The binding ability of a bicyclic somatostatin analogue towards Cu(II) ions. Chem. 2020; 17, e200030. [Google Scholar]
- Marciniak, A. , Witak W., Sabatino G., Papini A.M., Brasuń J. Detailed Insight into the Interaction of Bicyclic Somatostatin Analogue with Cu(II) Ions. Int. J. Mol. Sci. 2020, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A. , Pacini L., Papini A.M., Brasuń J. Bicyclopeptides: a new class of ligands for Cu(II) ions. Dalton Trans. 2022, 51, 13368–13375. [Google Scholar] [CrossRef]
- Gans, P. , Sabatini A., Vacca A. SUPERQUAD: an improved general program for computation of formation constants from potentiometric data. J. Chem. Soc. Dalt. Trans. 1985, 1195–1200.
- Gans, P. , Sabatini A., Vacca A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Kotynia, A. , Pap J.S, Brasuń J. The binding abilities of homodetic cyclic His-peptides toward copper ions. Inorg. Chim. Acta 2018, 472, 3–11. [Google Scholar] [CrossRef]
Figure 1.
The coordination profile for BCL, MCL1, and LNL.
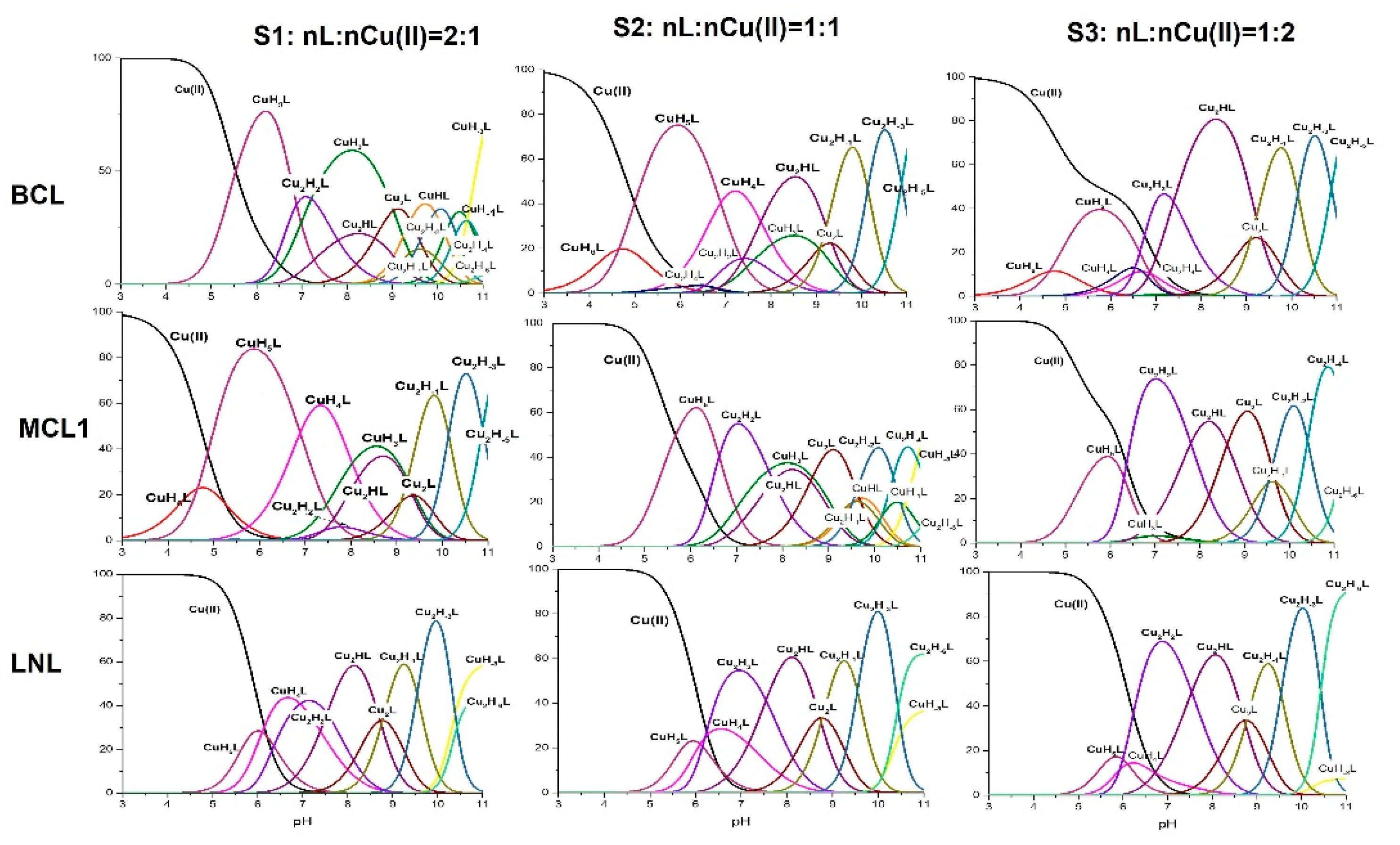
Figure 2.
The pH-dependent absorption, UV-Vis spectra of the LNL peptide: S2 and S3 systems and of the MCL1 peptide: S1, S2, S3 systems.
Figure 2.
The pH-dependent absorption, UV-Vis spectra of the LNL peptide: S2 and S3 systems and of the MCL1 peptide: S1, S2, S3 systems.
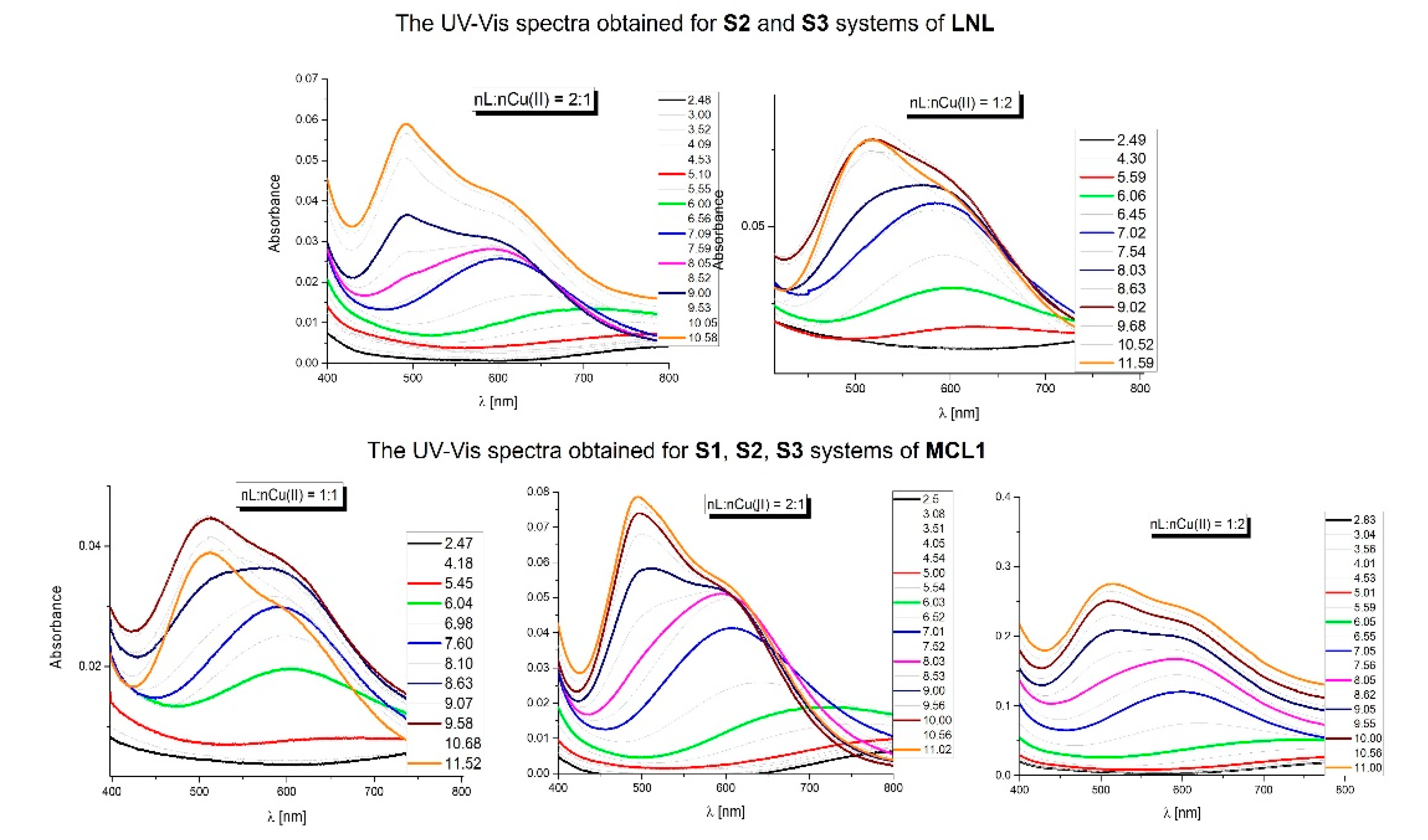
Figure 3.
The pH-dependent MCD spectra of the MCL1 peptide in all systems, of the free MCL1 peptide and the corrected MCD spectra obtained at pH 8 and 11.
Figure 3.
The pH-dependent MCD spectra of the MCL1 peptide in all systems, of the free MCL1 peptide and the corrected MCD spectra obtained at pH 8 and 11.
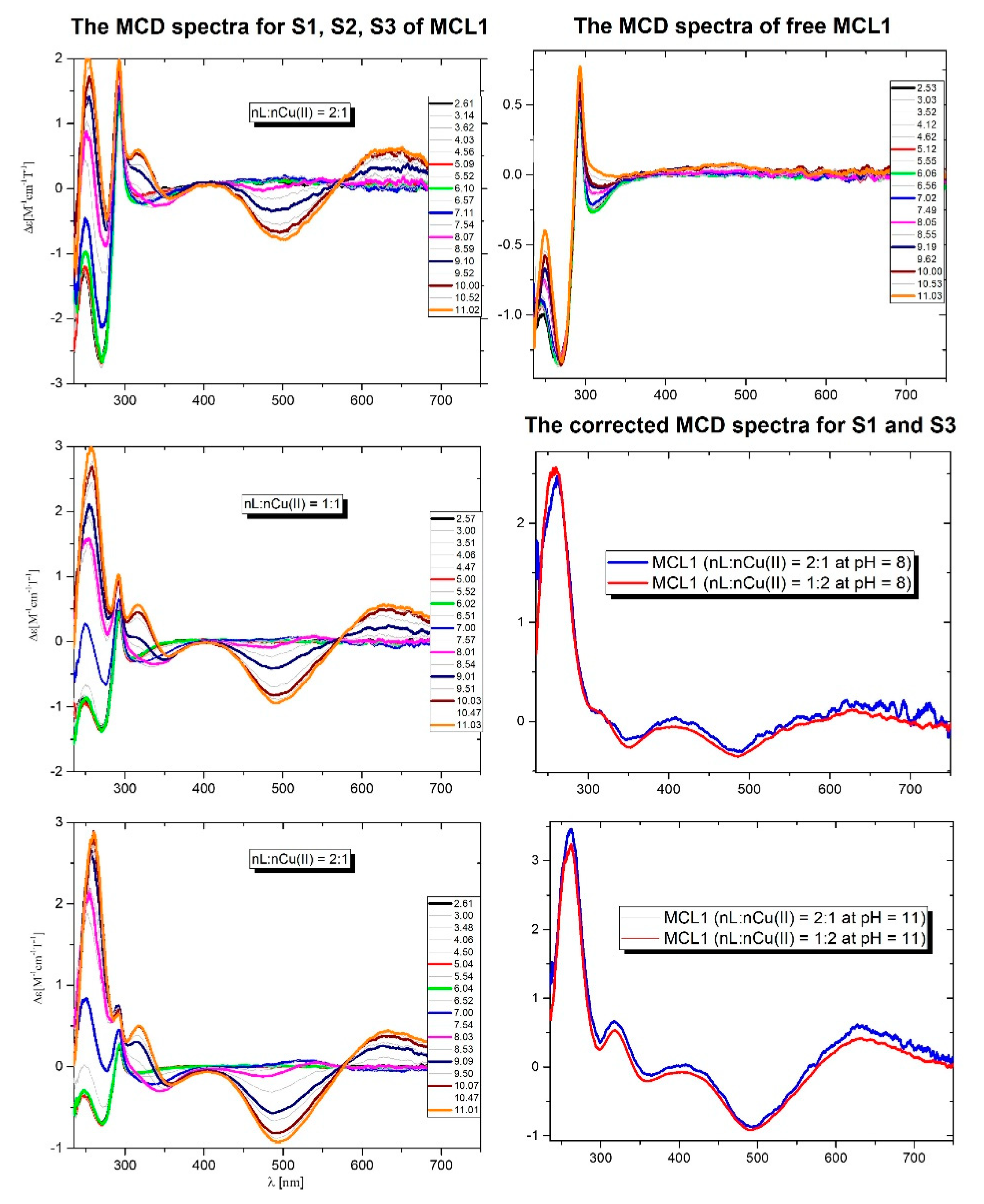
Figure 4.
The pH-dependent EPR spectra of the BCL peptide in all systems obtained at selected pH and comparison of the EPR spectra of the BCL and MCL1 peptides in the S1 and S3 systems obtained at pH 6.5 and 11.
Figure 4.
The pH-dependent EPR spectra of the BCL peptide in all systems obtained at selected pH and comparison of the EPR spectra of the BCL and MCL1 peptides in the S1 and S3 systems obtained at pH 6.5 and 11.
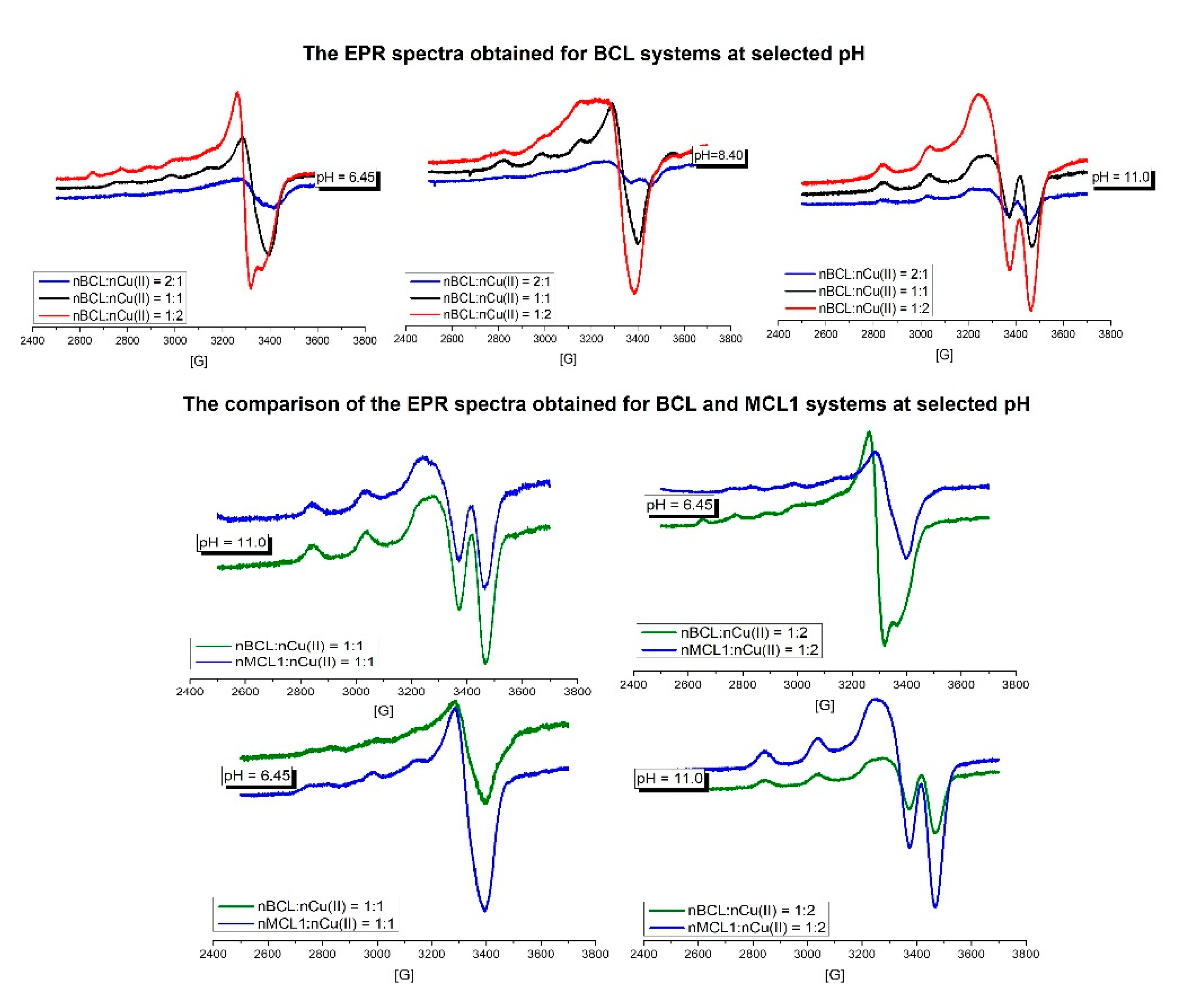

Table 1.
The protonation and stability constants of Cu(II)-complexes of BCL [22], MCL1, and LNL peptides.
Table 1.
The protonation and stability constants of Cu(II)-complexes of BCL [22], MCL1, and LNL peptides.
| Species | BCL [22] | MCL1 | LNL | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Logβ | logK | Logβ | logK | logβ | logK | ||||||||
| AH | 10.78 | 10.78 | 10.93 +/- 0.10 | 10.93 | 10.22 +/- 0.15 | 10.22 | |||||||
| AH2 | 21.23 | 10.45 | 21.26 +/- 0.06 | 10.33 | 20.31 +/- 0.09 | 10.09 | |||||||
| AH3 | 31.47 | 10.24 | 31.42 +/- 0.09 | 10.16 | 30.49 +/- 0.08 | 10.18 | |||||||
| AH4 | 41.33 | 9.86 | 41.07 +/- 0.05 | 9.65 | 39.71 +/- 0.04 | 9.22 | |||||||
| AH5 | 50.59 | 9.26 | 50.07 +/- 0.06 | 9.00 | 49.02 +/- 0.05 | 9.31 | |||||||
| AH6 | 57.11 | 6.52 | 56.74 +/- 0.07 | 6.67 | 55.72 +/- 0.05 | 6.70 | |||||||
| AH7 | 62.78 | 5.67 | 62.17 +/- 0.07 | 5.43 | 61.32 +/- 0.05 | 5.60 | |||||||
| Cu (II) complexes | |||||||||||||
| BCL [22] | MCL1 | LNL | |||||||||||
| Logβ | logK | logβ | logK | logβ | logK | ||||||||
| 60.91 | 4.64 | - | - | - | - | ||||||||
| 56.27 | 6.89 | 54.59 +/- 0.02 | 13.97 | 53.10 +/- 0.06 | 6.09 | ||||||||
| 49.38 | 8.03 | - | - | 47.01 +/- 0.05 | 65.6 | ||||||||
| 41.35 | - | 40.62 +/- 0.06 | 18.47 | - | - | ||||||||
| - | - | 22.15 +/- 0.09 | 20.32 | - | - | ||||||||
| - | - | 1.83 +/- 0.09 | 21.31 | - | - | ||||||||
| - | - | - 19.48 +/- 0.05 | - | - 18.59 +/- 0.06 | - | ||||||||
| 52.59 | 13.06 | - | - | - | - | ||||||||
| 39.53 | 7.29 | 38.48 +/- 0.03 | 7.80 | 38.03 +/- 0.04 | 7.54 | ||||||||
| 32.14 | 9.35 | 30.68 +/- 0.04 | 8.61 | 30.49 +/- 0.05 | 8.72 | ||||||||
| 22.79 | 9.09 | 22.07 +/- 0.04 | 9.70 | 21.77 +/- 0.07 | 8.77 | ||||||||
| 13.70 | 20.27 | 12.37 +/- 0.07 | 9.52 | 13.00 +/- 0.05 | 19.08 | ||||||||
| - | - | 2.85 +/- 0.04 | 20.81 | - | - | ||||||||
| - 6.57 | 21.76 | - | - | - 6.08 +/- 0.05 | 31.26 | ||||||||
| - | - | -17.96 +/- 0.04 | 22.60 | - | - | ||||||||
| - 28.33 | - | - | - | - | - | ||||||||
| - | - | -40.56 +/- 0.05 | - | - 37.34 +/- 0.05 | - | ||||||||
Table 2.
The protonation and stability constants of Cu(II)-complexes of MCL2 and MCL3 peptides.
| Species | MCL2 | MCL3 | ||
|---|---|---|---|---|
| Logβ | logK | Logβ | logK | |
| AH | 10.32 +/- 0.13 | 10.32 | 10.63 +/- 0.13 | 10.63 |
| AH2 | 20.99 +/- 0.02 | 10.67 | 20.62 +/- 0.12 | 9.99 |
| AH3 | 30.71 +/- 0.08 | 9.72 | 30.79 +/- 0.11 | 10.17 |
| AH4 | 40.58 +/- 0.03 | 9.87 | 40.53 +/- 0.05 | 9.74 |
| AH5 | 49.40 +/- 0.04 | 8.82 | 49.38 +/- 0.07 | 8.85 |
| AH6 | 55.13 +/- 0.04 | 5.73 | 55.13 +/- 0.07 | 5.75 |
| Cu (II) complexes | ||||
| MCL2 | MCL3 | |||
| Logβ | logK | Logβ | logK | |
| 45.85 +/- 0.14 | 6.25 | 46.69 +/- 0.04 | 7.04 | |
| 39.60 +/- 0.04 | 8.76 | 39.65 +/- 0.05 | 9.39 | |
| 30.84 +/- 0.07 | 9.30 | 30.26 +/- 0.12 | 8.82 | |
| 21.54 +/- 0.06 | 20.20 | 21.44 +/- 0.06 | 19.84 | |
| 1.34 +/- 0.06 | 22.08 | 1.60 +/- 0.07 | 21.36 | |
| - 20.74 +/- 0.08 | - 19.76 +/- 0.08 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



