
Submitted:
17 April 2024
Posted:
19 April 2024
You are already at the latest version
Abstract
Experimental evidence suggests that chronic intermittent hypoxia (CIH), a major hallmark of obstructive sleep apnea (OSA), boosts carotid body (CB) responsiveness, thereby causing in-creased sympathetic activity, arterial and pulmonary hypertension, and cardiovascular disease. An enhanced circulatory chemoreflex, oxidative stress, and NO signaling appear to play im-portant roles in these responses to CIH in rodents. Since the guinea pig has a hypofunctional CB (i.e., it is a natural CB knockout), in this study we used it as a model to investigate the CB de-pendence of the effects of CIH on pulmonary vascular responses, including those mediated by NO, by comparing them with those previously described in the rat. We have analyzed pulmonary artery pressure (PAP), the hypoxic pulmonary vasoconstriction (HPV) response, endothelial function both in vivo and in vitro, and vascular remodeling (intima-media thickness, collagen fiber content, and vessel lumen area). We demonstrate that 30 days of exposure of guinea pigs to CIH (FiO2, 5% for 40 s, 30 cycles/h) induces pulmonary artery remodeling but does not alter en-dothelial function or the contractile response to phenylephrine in these arteries. In contrast, CIH exposure increased the systemic arterial pressure, and enhanced the contractile response to phenylephrine while decreasing endothelium-dependent vasorelaxation to carbachol in the aorta, without causing its remodeling. We conclude that since all of these effects are independent of CB sensitization, there must be other oxygen sensors, beyond the CB, with the capacity to alter the autonomic control of the heart and vascular function and structure in CIH.
Keywords:
Systemic and pulmonary hypertension
; intermittent hypoxia
; hypoxic pulmonary vasoconstriction
; carotid body
; autonomic nervous system
; obstructive sleep apnea
; endothelial function
; vessel remodeling
; guinea pig
1. Introduction
Obstructive sleep apnea (OSA), characterized by the repeated occurrence of apneas and hypopneas during sleep, is one of the most common chronic diseases, affecting up to nearly one billion people worldwide [1]. This breathing disorder is strongly associated with cardiovascular disease, as sleep apnea is widely recognized as a major independent risk factor for hypertension, atherogenesis, coronary heart disease, cardiac arrhythmias, heart failure, and stroke [2,3].
Among the pathophysiological mechanisms of OSA, intermittent hypoxia (IH) caused by repetitive hypoxia-reoxygenation cycles is thought to be the key intermediary mechanism leading to cardiovascular morbidity and mortality [4,5,6]. However, the mechanisms underlying the association between OSA and cardiovascular disease are not fully understood. In this regard, several animal models have been developed to study the effects of chronic intermittent hypoxia (CIH) on the cardiovascular system. Numerous studies in rodents support the hypothesis that CIH is responsible for increased arterial blood pressure, structural vascular remodeling, altered vascular reactivity, and progression of atherosclerosis observed in OSA.
There is considerable evidence that, when exposed to CIH, the carotid body (CB), the major peripheral oxygen sensor, becomes pathologically hyperactive, leading to chronic activation of chemoreflexes which increase afferent traffic to the brainstem, respiratory plasticity, and sympathetic outflow responsible for the increased blood pressure [7,8]. CB denervation or ablation blocks CIH-induced changes in ventilation and systemic hypertension, demonstrating that chemoreflexes are essential not only for the development of respiratory plasticity but also for the cardiovascular events. In addition, CIH promotes oxidative stress and inflammation in a CB-dependent and CB-independent manner [9], which may contribute to the onset of vascular dysfunction and the development of sustained hypertension in CIH-exposed animals and in patients with OSA [10,11,12].
It has also been suggested that OSA may contribute to the development of pulmonary hypertension (PH) and right ventricular failure through pulmonary vasoconstriction secondary to hypoxia. However, other studies suggest that CIH is not sufficient to cause sustained PH, as occurs with sustained alveolar hypoxia, for example in patients with chronic obstructive pulmonary disease (COPD) and in animal models with chronic sustained hypoxia (CSH). A recent study by our group in rats [13] showed that CIH, like CSH, produces an elevation in pulmonary arterial pressure (PAP), although smaller than that caused by CSH, in addition to different effects on ventricular hypertrophy and the contractile properties of pulmonary arteries (PA). The development of left rather than right ventricular hypertrophy may be secondary to the development of systemic hypertension in CIH rats. Regarding contractile properties, our results showed that CIH exposure slightly increased phenylephrine-induced contraction but had no effect on hypoxic pulmonary vasoconstriction (HPV) or endothelium-dependent relaxation in rat PA [13]. It has also been reported that mice exposed to CIH had lungs with significantly more muscularized small pulmonary vessels (positive staining for myosin) compared to normoxic mice [14].
Because we are interested in evaluating the involvement of the CB in the cardiovascular and pulmonary effects of CIH, we recently developed a model of CIH in the guinea pig, an animal with a hypoxia-insensitive CB that can be considered a natural CB knockout. In a previous work [15], we demonstrated that CIH in the guinea pig induced sympathetic hyperactivity with increased circulating catecholamines that promoted cardiovascular adaptations such as increased heart rate and mean arterial systemic pressure, without significant changes in the hypoxic ventilatory response, i.e., effects that were apparently not mediated by CB chemoreflex activation.
Here, we hypothesize that prolonged exposure to CIH in the guinea pig may alter pulmonary hemodynamics and cause PH, as in rats [13], if these effects are CB-independent. To date, no study has investigated pulmonary vascular function using the same experimental model of CIH in the guinea pig. Therefore, the present study was designed to characterize the functional consequences of chronic exposure to intermittent hypoxia on PAP, the HPV response, endothelial function in both in vivo and in vitro (arterial rings) vessels, vascular remodeling by proliferating PASMCs (intima-media thickness; IMT) and vessel lumen area), or expression of endothelium-derived pulmonary vasodilators in the guinea pig.
2. Results
2.1. Animal’s General Status, Body Weight, and Hematocrit
All the animals completed the study in good condition. The body weight, arterial gasometry and hematocrit of both CIH and C groups after the 30-day CIH period are shown in Table 1. Body weight was significantly lower in the CIH than in the C group (p<0.01). CIH caused a significant decrease in weight gain from the first week of exposure. As a result, at the end of the study (day 30), body weight in the experimental group was significantly lower than in the control group. Hematocrit was not significantly different in the hypoxic with respect to the normoxic group although EPO increased in CIH animals (p < 0.05). These effects of CIH were the same as those observed by [15].
2.2. Pulmonary Hemodynamics and Right Ventricle Hypertrophy
To assess the impact of CIH exposure on systemic and pulmonary hemodynamics, direct measurements of arterial pressure in the carotid and main PA were obtained by catheterization in guinea pigs ventilated with 21% O2, except during the measurements shown in Figure 1F. When compared with the control group, CIH exposure increased the mean systemic arterial blood pressure (MABP) from 37 ± 2 mmHg to 45 ± 3 mmHg (p < 0.05; Figure 1A) as both systolic and diastolic pressures were increased in CIH-exposed guinea pigs, although the pulse pressure (PP) did not change significantly (Fig 1B). However, a significantly increased heart rate (25%) was seen in CIH guinea pigs compared to the control group (245 ± 5 vs 195 ± 3 beats/m; ****p < 0.0001; Figure 1C).
To determine the impact of blood pressure alterations on ventricular cardiac mass, the Fulton index was calculated (Figure 1D). CIH-exposed guinea pigs did not show RV hypertrophy, as the RV/LV+S ratio was not different from the C group.
Finally, baseline normoxic PA pressure was found not to be significantly different in CIH compared to controls (10.8 ± 0.8 vs 10.1 ± 0.4 mmHg; Figure 1E). PAP similarly increased to ~15 mmHg during acute hypoxic challenge (10% O2 during 3 min) in both C and CIH groups reflecting a completely functional HPV in this experimental model (Fig 1E). In summary, in the guinea pig, CIH increases MABP and HR, but it does not change the Fulton index, PAP or HPV.
2.3. Vascular Contractility and Endothelial Function
The mechanical properties and reactivity of pulmonary, aorta and carotid arteries were assessed. Table 2 shows the vascular responses to 80 mM KCl in the aorta, carotid and PA. The maximal contraction induced by KCl in aorta was lower in animals exposed to CIH (but not significantly) whereas in the pulmonary and carotid arteries it was similar to that in the C group.
Figure 2 shows the vascular contractile responses to cumulative doses of phenylephrine (PE; 0.1 -30 µM) in the aorta (panel A), carotid (panel B) and pulmonary artery (panel C), and the effect of the NOS inhibitor, L-NAME. PE induced contraction was higher at all concentrations in the aorta of CIH compared to C guinea pigs in the presence or absence of NO (L-NAME pretreatment). In contrast, the response to PE was virtually identical at all doses in carotid arteries from C and CIH animals in both the presence and absence of L-NAME. The low contractile values reached in both systemic arteries (~20% KPSS) are striking.
In PA, the PE-induced contraction was relatively larger than in the systemic arteries (>50% KPSS response) but was lower at all concentrations in the CIH than in the C group. Inhibition of eNOS similarly enhanced contraction in both groups (dashed line), ruling out a higher production of NO in CIH guinea pigs as the cause of lower contractibility than in C guinea pigs.
Figure 2 shows that L-NAME almost tripled the 30 µM PE contraction in the carotid artery (panel E) of both CIH than C guinea pigs. In contrast, the response to PE was not significantly different in the presence or absence of L-NAME in aorta (panel D) and PA (panel F) from both groups. These data suggest that a1-receptor activation evokes NO release which greatly limits its ability to vasoconstrict the carotid artery.
Figure 3 shows the changes in arterial tension caused by cumulative concentrations of carbachol (10nM–10µM) on top of a maximal PE contraction. This protocol was used to test endothelial functionality, because carbachol releases NO from the endothelial layer of the artery. In the aorta (panel A), endothelium dependent vasorelaxation was significantly blunted at each concentration in CIH compared to controls (p < 0.05), indicating the presence of endothelial dysfunction in this artery. Concomitant treatment with L-NAME almost completely abolished the endothelium-dependent vasodilator response in the CIH group and strongly depressed it in the C group. In contrast, no differences were observed in endothelium-dependent vasorelaxation in carotid (panel B) and PA (panel C) in the CIH group compared to the C group (50% and 75% reduction, respectively). The effect of carbachol was completely reversed by L-NAME in the pulmonary artery whereas L-NAME induced reversal was only partial (~50%) in the carotid artery. In PA from both groups the response to carbachol with or without L-NAME was very similar, indicating a lack of endothelial dysfunction in CIH.
Figure 3 (D, E, F) shows that the effect of L-NAME on 10 µM carbachol relaxation was greater in the aorta (85% vs 60% reversal; panel D) but similar in PA (panel F) of the CIH group. In the carotid artery (panel E), the reversal of the L-NAME effect was only partial (~50%) but was similar in the two groups.
2.4. Morphometric and Histological Assessments of Vascular Remodeling
To evaluate the morphology of the aorta, carotid artery and PA from C and CIH guinea pigs, Masson's trichrome staining was performed on 5-µm thick arterial sections from a total of 6 C and 6 CIH guinea pigs. Figure 4 shows sections of aorta, carotid artery, and PA from Control (top) and CIH (bottom) guinea pigs taken with the 4X or 10X objective with a magnified area taken with the 20X objective. This staining allows the visualization of the muscle layer (pink) and the collagen (blue green) present in the arteries to analyze the morphology of the vessels.
Figure 5 shows the morphometric and histological analysis for each artery in both conditions. The luminal area of the aorta (Figure 5A), carotid artery (Figure 5B), and intralobar PA (Figure 5C) was not modified by exposure to CIH in guinea pigs. Although intima media thickness (IMT) was not altered in the aorta (Figure 5D) and carotid artery (Figure 5E), it was significantly increased in the PA (63.7 ± 4.0 µm vs 52.9 ± 2.5 µm in C; p<0.05), as shown in Figure 5E. A significant increase in the % collagen occurs in the carotid artery as shown in Figure 5H (13.1 ± 1.5% vs 9.7 ± 0.9% in C; p<0.05) and in the PA as shown in Figure 5I (16.0 ± 1.8 vs 12.0 ± 1.1 in C; p<0.5), suggesting an induction of collagen synthesis during CIH exposure. No significant changes were observed in the aorta (Figure 5G).
2.5. NO Bioavailability: Plasma Nitrites and Nitrates and L-Arginine and Its Metabolites
Circulating vasoactive factors were studied to determine the status of the substances most likely to affect the endothelial function in guinea pig vessels and the effect of CIH on them. First, the bioavailability of NO was assessed by quantifying nitrites and nitrates, both metabolites of NO, and L-arginine, a natural precursor of NO, and its metabolites, ADMA and SDMA. As shown in Figure 6A, plasma levels of nitrites and nitrates were doubled in animals exposed to CIH (69 ± 14 µM vs 37 ± 4 µM in C; p<0.05). L-Arg (125.9 ± 9.1 vs 111.4 ± 6.7) and SDMA (4.5 ± 0.4 vs 3.2 ± 0.2) increased in the CIH group, whereas ADMA and L-homoarginine were similar in both groups.
2.6. Plasma Endothelin-1 (ET-1), Angotensin II (ANG II), Atrial Natriuretic Peptide (ANP), Vascular Endothelial Growth Factor (VEGF) and Catecholamines
Table 3 shows that there was no effect of CIH on plasma levels of ET-1 or ANG II. ANP and VEGF were also not altered by CIH exposure. In contrast, significant differences were observed in plasma catecholamines. Norepinephrine (NE) was significantly higher in the CIH guinea pigs than in the controls (p<0.05). Plasma epinephrine (E) was also significantly higher in CIH guinea pigs (p<0.05).
3.7. Aconitase: Fumarase Activity Ratio and Nuclear Factor Kappa B (NF-kB)
Neither the ratio of aconitase to fumarase (0.33 ± 0.05 vs 0.30 ± 0.04 in C group), an index of mitochondrial oxidative stress, nor NF-kB (1.02 ± 0.09 vs 1.01 ± 0.06 mU/mL in C group), an index of inflammation, changed in the liver from guinea pigs exposed to CIH.
3. Discussion
This study represents the first investigation of pulmonary vascular function in guinea pigs exposed to chronic intermittent hypoxia. Intermittent hypoxia is considered the main feature of OSA associated with respiratory events that could affect sympathetic tone and arterial pressure [7,16], vascular structural remodeling and function [17,18,19], and atherosclerosis progression [20,21].
In the 1990s, Fletcher and others proposed that CIH-induced CB hyperactivity may be the driver of many of the effects that lead to hypertension and cardiovascular events in animals exposed to CIH [7,10,22,23,24] and in patients with OSA [8,25]. CB denervation or ablation blunts CIH induced sympathetic nervous system hyperactivity and systemic hypertension [11,26]. However, it has been questioned whether an intact CB is essential to produce these effects [27]. There is evidence that neurons of the rostral ventrolateral medulla (RVL), which controls spinal vasomotor neurons, are excited by systemic hypoxia in CB-denervated rats, whereas RVL respiratory neurons are inhibited, demonstrating that the CB per se is not obligatory for hypoxic recruitment of sympathetic neurons [28]. Hypoxia can excite putative C1 presympathetic neurons though a P2Y1 receptor-dependent mechanism [29]. There is also evidence for direct effects of CIH on respiratory neurons in the medulla oblongata and altered respiratory autonomic coupling with implications for cardiovascular control [30].
In this context, we developed a model of CIH in the guinea pig as an experimental paradigm of OSA (FiO2 5%, 8 h/day, 30 days) with the unique feature that this hypoxia-adapted rodent has a hypofunctional CB. The guinea pig CB lacks O2-sensitive K+ channels and therefore exposure to hypoxia does not induce glomus cell depolarization and a neurosecretory response [31], which is essential for chemoafferent signaling from the CB. In summary, the guinea pig can be considered a natural CB knockout species.
As expected, CIH exposure did not potentiate CB responses in the guinea pig OSA model, in contrast to the robust responses induced in rats. However, CIH was associated with sympathetic hyperactivity [15], and it promoted cardiovascular adjustments by increasing heart rate and mean arterial blood pressure without causing ventricular hypertrophy. Thus, CIH did not sensitize the CB chemoreceptor response to hypoxia in guinea pigs but promoted CB-independent cardiovascular adjustments [15]. The aim of the present study was to extend this preliminary cardiovascular study by characterizing the functional consequences of CIH exposure on the structure and responsiveness of the guinea pig vasculature. Therefore, aorta, carotid, and pulmonary artery vasoreactivity was assessed by wire myography, and vessel remodeling by tissue staining; MABP and PAP were also recorded in vivo. To our knowledge, this is the first study to evaluate the consequences of CIH exposure on the function of guinea pig arteries from the systemic and pulmonary circulations. Our results show that guinea pigs developed hypertension and PA remodeling after four weeks of exposure to CIH. In contrast, CIH did not alter PAP, HPV, or Fulton index during this period, nor did it induce endothelial dysfunction in PA.
These results agree with other studies in rats suggesting that CIH can cause a moderate (10–16 mmHg) increase in MABP [7,23,32]. In contrast, other authors observed no change in MABP after 14 days [19] or even 70 days of CIH [33] in rats. Lucking et al. [27] also found that arterial blood pressure was unaffected by exposure to CIH in guinea pigs, although a β-adrenoceptor-dependent tachycardia was evident compared with controls. A direct comparison between our data and those of other studies in rats and guinea pigs is difficult because of differences in the species and CIH protocols used, such as the severity of hypoxia, the number and duration of hypoxic cycles, the methods used to measure MABP, and the lack of studies in guinea pigs. However, it is striking that the positive results were similar in both species, demonstrating that CB involvement is not strictly necessary to induce either an increase in MABP or tachycardia, although they do not rule out the possibility that it may potentiate them.
Changes in the levels of several mediators have been suggested to contribute to OSA-associated cardiovascular morbidity. OSA patients exhibit a decreased response to NO and an increased activation of the sympathetic, angiotensin II and endothelin systems [34,35,36]. We therefore investigated the effect of CIH on the vasorelaxation induced by carbachol, a cholinergic agonist which promotes endothelial NO release, and also on the contraction induced by phenylephrine, an adrenergic agonist that mimics sympathetic stimulation, in our model of CIH guinea pig. Furthermore, we measured plasma endothelin-1, plasma NE and E and angiotensin II levels.
Interestingly, CIH produced a different pattern of functional and structural effects in each of the three arteries we investigated. CIH had no effect on the responses to high K+, PE or carbachol in the carotid artery. The carbachol response was greatly depressed by L-NAME in these arteries, as it was in the aorta and PA, indicating that it was mainly mediated by NO release in all three vessels. It is notable that NO release also appeared to be strongly suppressing the PE contraction, as evidenced by the marked increase in the PE contraction in the presence of L-NAME. The enhancement of vasoconstriction by eNOS blockade is thought to be associated with the presence of myoendothelial gap junctions, which provide a pathway by which the increase in smooth muscle cell [Ca2+]i evoked by vasoconstrictors can also raise [Ca2+]i in the endothelium, causing a stimulation of eNOS which curbs the contraction [37]. The observation that L-NAME similarly enhanced the PE contraction in carotid arteries from the control and CIH groups suggests that this mechanism of NO release was also not altered by exposure to intermittent hypoxia.
CIH also had no effect on the contractions evoked by high K+ or hypoxia, or on carbachol-induced vasorelaxation, in PA. However, it decreased the amplitude of the PE contraction by ~30%. In line with the lack of effect of CIH on endothelium-dependent vasorelaxation, the depression of the PE response persisted in the presence of L-NAME, again suggesting that CIH does not affect endothelial NO production in these arteries. We have previously shown that exposure of rats to an identical CIH protocol similarly did not alter either HPV or carbachol-induced vasorelaxation in PA, although the response to PE was modestly increased [13]. It is possible that the increased pulmonary artery collagen content induced by CIH could explain the decreased responsiveness of these arteries to PE in guinea pig [38]. Interestingly, CIH also caused a small but significant increase in PAP in rats, whereas in the present study it had no effect on PAP in guinea pigs. This suggests that a fully functional CB may be required for CIH to induce pulmonary hypertension.
In the aorta, as in the other two arteries, CIH had no significant effect on the high K+ contraction. However, the constriction elicited by PE was increased and carbachol-induced vasorelaxation was attenuated. Interestingly, L-NAME had little effect on the amplitude of the PE contraction in the aorta, consistent with the lack of myoendothelial gap junctions reported in this artery [39]. These results suggest that CIH is likely to enhance aortic vascular tone in vivo, especially in the face of the increased sympathetic drive which occurs under these conditions.
These data are not consistent with previous findings in rats showing that exposure to CIH (FiO2 5%, 8 h/day for 35 days) did not alter the endothelium-dependent relaxation elicited by acetylcholine in the aorta [40]. Furthermore, whereas they found that aorta from control and CIH rats had similar contractile responses to KCl, norepinephrine, angiotensin II, and endothelin-1, the contractile response to endothelin-1 was weakly but significantly increased in carotid artery from CIH rats in the same study [40]. A heterogeneous distribution in the receptor density of vasoactive agents in different vascular beds, such as endothelin-1 [41], has been proposed to account for the changes in the response to different vasoactive agents [40]. In the same line, a heterogeneous distribution of adrenoceptors in arteries has also been reported and could explain the different effect on aorta and carotid artery that we found in the guinea pig after CIH exposure [42].
Intermittent hypoxia also had different effects on the structure of each artery. Despite altering contractile function in the aorta, CIH did not affect IMT thickness or the collagen content in this artery. In pulmonary artery, CIH increased the accumulation of collagen fiber and also the intima-media thickness, whereas in the carotid artery only the collagen fiber content was enhanced.
Patients with OSA have been shown to have increased carotid artery IMT [43]. IMT remodeling is considered an early predisposing event in atherosclerosis and plaque formation and is associated with increased cardiovascular risk [44]. Our results showed that CIH promotes vascular remodeling with increased IMT in the pulmonary artery but not in the aorta and carotid artery of guinea pigs. Vascular remodeling is the result of interactions between multiple biochemical mechanisms such as oxidative stress, tissue inflammation, endothelial dysfunction, and sympathetic overactivation and blood pressure overload [45,46]. The increased carotid artery IMT in patients with OSA appeared to be associated with a rise in serum inflammatory markers. In guinea pigs, although CIH caused sympathetic overactivation, it did not evoke generalized endothelial dysfunction, and did not increase liver NF-kB or the aconitase to fumarase ratio, indicative of mitochondrial oxidative stress and inflammation, suggesting that it may cause less oxidative stress and inflammation than it does in rats [47]. Although we cannot rule out the possibility that CIH was eliciting oxidative stress and/or inflammation in other regions of the body, these results suggest that these effects of CIH could generally be less pronounced in the guinea pig than in rats. This may account for the lack of an increase in carotid artery IMT which we observed.
Apparently, however, pulmonary artery remodeling in this species is more sensitive to CIH, since both collagen fiber content and intima-media thickness were increased. IMT, elastin fiber disruption, and interlaminar collagen accumulation induce arterial stiffness, thereby contributing to vascular resistance and arterial blood pressure elevation [5,48]. Remodeling of the adventitia, media, and intima in the pulmonary arterial bed has also been associated with increased pulmonary pressure. Several studies correlate intermittent hypoxia and pulmonary hypertension in mouse and rat models [14,49,50], including those from our laboratory [13]. In contrast, other studies suggest that intermittent apnea-related hypoxia is not sufficient to cause sustained PH [51,52]. We observed that 4 weeks of CIH did not cause PH in guinea pigs, possibly because the extent of remodeling which developed was insufficient to affect baseline pulmonary vascular resistance. However, we cannot exclude the possibility that a prolonged exposure to CIH may result in greater structural and functional damage to the pulmonary circulation, including an increase in PAP.
It is interesting to note that elevated levels of NO were found in CIH guinea pigs (Figure 6) when plasma NO metabolites (nitrites and nitrates) were measured as an index of NO bioavailability [53]. NO oxidation products nitrite and nitrate are recognized as sources of recycled NO. The activation of the nitrate-nitrite-NO pathway is an alternative source of NO that may have an anti-hypertensive effect [54]. This conversion may be particularly effective under conditions of hypoxia and acidosis, such as occur in skeletal muscle during exercise. It is noteworthy that the increase in plasma NO metabolites in guinea pig was similar to that we observed using the same CIH protocol in the rat [13], suggesting that this effect occurs independently of the CB and does not require a rise in plasma angiotensin II or endothelin.
Conversely, no changes were found in plasma levels of L-arginine or its metabolite asymmetric dimethylarginine (ADMA), a recognized cardiovascular risk factor [55]. In contrast, plasma levels of symmetric dimethylarginine (SDMA), an isomer of ADMA, were elevated (≈66%) in CIH-exposed guinea pigs with systemic arterial hypertension but not pulmonary hypertension. ADMA and SDMA result from post-translational methylation of L-arginine residues in proteins by protein arginine methyltransferases (PMRTs). Therefore, their levels increase after proteolysis of methylated proteins [56]. ADMA acts as an endogenous competitive inhibitor of NOS [56,57], and although SDMA does not directly inhibit NOS, it competes with L-arginine for cellular uptake [56]. Elevated levels of ADMA, but not SDMA, have been implicated in endothelial dysfunction in many pathological conditions [58,59,60]. Further studies are needed to investigate a possible correlation between SDMA levels and CIH exposure effects.
One of the hallmark effects of CIH in rodents [61] is a rise in the plasma endothelin concentration. There is evidence that this, as well as an increased expression of ETA receptors, contributes to systemic vascular constriction and a rise in MABP in rats, and to the hypersensitivity of the CB evoked by CIH [41,62]. Endothelin has also been implicated in CIH-induced vascular remodeling in mice [18,63]. In humans, however, although OSA has been shown to increase plasma endothelin levels [64] it is unclear whether this is a direct effect or is due to co-morbidities associated with this condition [65]. Also, a role for endothelin in chronic OSA-associated hypertension appears not to have been confirmed, and endothelin has been shown not to contribute to the hemodynamic effects of acute intermittent hypoxia in humans [66]. We observed that CIH had no effect on plasma ET-1, suggesting that a degree of vascular remodeling of carotid and pulmonary arteries can occur independently of an increase in ET-1. However, in view of the finding by Gras et al [18] that the increase in the aortic IMT thickness in mice was prevented by the ETA/B receptor antagonist bosentan, it is possible that the lack of effect of CIH on the IMT in the aorta and carotid arteries may have been due to the absence of a rise in ET-1 in the guinea pig.
CIH in animal models has also been shown to increase plasma angiotensin II levels [26]. Increased circulating angiotensin II levels contribute to the rise in MABP induced by CIH [67], in part by acting on CNS blood pressure-regulating centers to increase sympathetic outflow [6,69]. Angiotensin II has also been shown to promote the CB hypersensitivity [70], oxidative stress [71,72] and pulmonary vascular remodeling [73] associated with CIH/OSA. In guinea pigs, however, we found that CIH did not affect plasma levels of angiotensin II, suggesting that the rise in MABP it evokes in this species does not depend on an increase in its circulating concentration. In addition, it appears that the increase in IMT thickness and collagen content we observed did not require an increase in circulating angiotensin II levels, although we cannot exclude the possibility that local activation of the renin-angiotensin-aldosterone system within the lung could have occurred and contributed to pulmonary artery remodeling.
Finally, the findings on plasma levels of catecholamines deserve a few remarks. In CIH guinea pigs, there was a large increase in plasma levels of CA, both NE and E. Since sympathetic nerve endings are the origin of plasma NE, whereas E is secreted from the adrenal medulla (AM), plasma levels of NE and E represent an index of general sympathetic tone [74,75]. Interestingly, these results, together with an increase in MABP (10 mmHg) and HR (25% increase), point to an activation of the sympathetic system after CIH in the guinea pig. Given that CIH-induced hypertension and increased sympathetic activity may be mediated by CB overactivity, as it is prevented by chronic bilateral sectioning of the carotid sinus nerve [10], these responses represent a paradoxical result in an animal lacking CB activity and carotid sinus nerve afferent activity [31,76] and a blunted ventilatory response to the full range of hypoxia. These respiratory responses are typical of mammals and humans adapted to high altitude [77]. Furthermore, the absence of hypoventilation during a Dejours test [15] indicates that, unlike in rats and other species, the CB does not contribute to normoxic ventilation in guinea pigs. Conversely, the ventilatory response to hypercapnia is similar before and after CIH treatment and is comparable to that observed in the rat [31], demonstrating that central chemoreceptors respond normally to hypercapnic stimuli in guinea pigs, as reported by Schwenke et al. [76]. Taken together, as noted above, with the sympathetic activation following CIH exposure in guinea pigs, our results demonstrate that other oxygen sensors integrated into the respiratory and sympathetic networks of the brainstem could monitor tissue oxygen in the CNS independently of CB activity and elicit respiratory and/or sympathetic excitatory responses during central hypoxia [78].
In conclusion, the present study demonstrates that 30 days of exposure of guinea pigs to CIH (FiO2, 5% for 40 s, 30 cycles/h) induces pulmonary artery remodeling but does not alter endothelial function or the contractile response to phenylephrine in these arteries. On the other hand, this model was associated with increased MABP, and altered responses to phenylephrine and carbachol in the aorta without changes in its wall structure. Since these alterations occurred in the functional absence of a CB influence, we conclude that they are independent of CB sensitization. Thus, it appears that there must be other oxygen sensors with the capacity to alter the autonomic control of the heart in CIH as well as vascular structure and function. Whether this finding can be extended to OSA patients, and those living and adapted to high altitude, remains to be investigated.
4. Materials and Methods
This investigation conformed to the Guide for Care and Use of Laboratory Animals published by the European Union Directive for Protection of Vertebrates Used for Experimental and Other Scientific Ends (2010/63/EU). Protocols were reviewed and approved by the University of Valladolid Institutional Ethical Committee for Animal Care and Use (Project Approval Ethical Code: 4505502)
4.1. Chronic Intermittent Hypoxia Protocol
Experiments were conducted on 24 male 3-month-old Hartley guinea pigs (GP) from Harlam Iberica housed (3/ cage) in an ad hoc room of the vivarium with a light–dark cycle of 12 h at a room temperature of 20–23°C. Animals were provided standard chow and water supplemented with vitamin C (Farma Bayer, Spain; 1 g/l) ad libitum. After one week of adaptation animals were randomly distributed into two groups of 12. One group was maintained in a normoxic atmosphere (control animals, C) and the other group was exposed to an intermittently hypoxic intermittent atmosphere (CIH animals; 21% O2 -80s/5% O2 – 40 s; 8 h/day) as previously described [15]. Briefly, GP were placed in Plexiglas chambers. A timed solenoid valve system was used to distribute nitrogen and air to each chamber. CIH GP chambers (n = 12) were flushed with air–nitrogen for 20 s to achieve hypoxia (5% O2) followed by 80 s of compressed air to restore normoxia (21% O2) before starting the next hypoxia cycle. Cycles for both groups were repeated 8 h/day for 30 days. The level of FiO2 in the chambers was controlled throughout the hypoxia protocol using an O2 analyzer (Oxydig, DRÄGER, Lübeck, Germany). Body weight was measured weekly.
4.2. In Vivo Pulmonary and Systemic Blood Pressure Measurements
For systemic arterial pressure recordings, anesthetized GP (ketamine 100 mg/Kg + diazepam 2 mg/Kg; i.p.) were tracheostomized and pump-ventilated with room air (CL Palmer, UK; 60 cycles/min and positive expiratory pressure of 2 cm H2O) as previously described (15). The right carotid artery was cannulated and connected to a physiological pressure transducer (AD Instruments, Oxford, UK). Animals were allowed to stabilize for 20 min following surgery and then the baseline heart rate (HR) and arterial blood pressure (ABP) was continuously monitored. Signals were recorded and stored (BIOPAC Systems, Inc. MP 150, Goleta, CA; Acknowledge 3.9.1) for subsequent analysis (heart rate, systolic and diastolic pressure, pulse pressure).
In order to simultaneously measure PAP, in open chest animals the main PA was cannulated through the right ventricle (RV) and connected to a pressure transducer (Transpac IV; ICU Medical, Inc., San Clemente, CA 92673, USA). Systolic and diastolic pressure measurements were performed in normoxic and hypoxic conditions (4 min at FIO2 = 0.10). To record the effect of hypoxia on PAP, the inlet of the respirator was connected to a balloon filled with a mixture of 10% O2 /90% N2. The recovery profile was evaluated by pumping in air with an FIO2= 0.21. Arterial oxygen tension (PaO2) was analyzed in blood sampled from the carotid artery, immediately after hemodynamic measurements. At the end of experiments, animals were euthanized by the administration of a lethal dose of sodium pentobarbital.
4.3. Vessel Reactivity and Endothelial Function
Studies of tension development were performed on rings from aortas, pulmonary and carotid arteries. Following euthanasia, the thoracic aorta and the left carotid artery were excised and transferred to dishes filled with Krebs bicarbonate buffer solution (NaCl 118 mM, KCl 4 mM, MgSO4 1 mM, CaCl2 1.8 mM, KH2PO4 1.0 mM, NaHCO3 24 mM, glucose 5 mM) and cleared of periadventitial tissue under a dissection microscope. Lungs were extracted and the PA isolated and cleaned of fat and connective tissue. Arteries were cut into rings 3 mm long and mounted on a conventional wire myograph (DMT) in Krebs solution maintained at 37° C and bubbled with 21% O2, 5% CO2, remainder N2. Care was taken not to touch the inner surface of the blood vessels.
After a period of stabilization, arteries were contracted using 80 mM KCl to determine their viability and contractile capacity (reference contraction). After a second stabilization period, vascular rings were contracted with cumulative concentrations of phenylephrine (PE; 0.1 µM–30 µM). Contractile responses were expressed as a percentage of the contraction induced by 80 mM KCl. Endothelial function was then assessed by testing the relaxant effect of cumulative doses of carbachol (10 nM–0.1 mM) on rings pre-contracted with 30 µM PE. Relaxant responses to carbachol were expressed as a percentage of phenylephrine-induced tone. The failure of carbachol to elicit relaxation of vessels rings previously incubated with PE was taken as proof of endothelial damage. Endothelium-dependent contractile and relaxation function were assessed by testing the L-NAME (0.1 mM) effect on the dose response contractions to PE (0.1 µM– 30µM) and in on the relaxant effects of carbachol, respectively.
4.4. Fulton Index
To measure possible RV hypertrophy, immediately after death the heart was removed and the RV and the left ventricle plus the septum (LV+S) were dissected out and separately weighed separately, and the ratio between RV and LV+S weights (RV/LV+S) was calculated as the Fulton index.
4.5. Morphometric and Histological Assessments
To assess vessel wall remodeling, aorta, carotid and PA rings were collected at the end of the in vivo analysis. They were fixed in 10% paraformaldehyde for 48 hours at room temperature and stored in PBS at 4° C. For processing, they were embedded in paraffin, sectioned at 5 μm and stained with Masson’s trichrome for histological examination. After staining, the sections were visualized with a NIKON Eclipse 90i Fluorescence Microscope and photographed with a NIKON, model DS-Fi3 CMOS camera (5.9 mP). Images were taken in from serial sections of each tissue at x4, x10 and x20 magnification.
Arterial intima-media thickness, lumen area, and collagen (expressed as a percentage of total IMT area) were measured in five tissue sections per animal using Image J/Fiji software. To quantify the percentage of collagen per vessel area, all photographs were first visualized for standardization, using the color threshold. The same threshold was then applied to all the images.
4.6. L-Arginine and Metabolites
For the simultaneous analysis of the several endogenous substances involved in the NO-generating pathway, the plasma levels of L-arginine, asymmetrical dimethyl-L-arginine (ADMA), and symmetrical dimethyl-L-arginine (SDMA) were measured by a two-step HPLC-FD with fluorescence detection. Plasma samples were prepared adding 1 mM stock solution of the internal standard monomethyl-arginine (MMA) prepared in 10 mM HCl and using a pre-conditioned solid extraction cartridge (Oasis MCX cation-exchange SPE columns (30 mg, 1 cc) supplied by Waters as previously described [15].
Briefly, a 0.2 mL sample was mixed with 0.1 mL of internal standard (MMA) and 0.7 mL of PBS. The Oasis MCX SPE columns were preconditioned with 1 mL of ammonia/water/methanol (10/40/50) and 1 mL of water. After sample addition, columns were consecutively washed with 1 mL of 100 mM HCl and 1 mL of methanol. Analytes were eluted with 1 mL of ammonia/water/methanol (10/40/50). All washing and elution steps were performed by vacuum suction. Column eluates were evaporated under a vacuum lyophilizer and frozen at -20°C until injection into the HPLC-FD. Before injection, the amino acids were derivatized with 0.1 mL of o-phthaldialdehyde (OPA) reagent containing 3- mercaptopropionic acid added to the residue. The derivatized amino acids were separated by isocratic reversed-phase chromatography performed on a C18 column (Strata-XL-C 100 µm, 30 mg/mL Phenomenex) using a mobile phase consisting of potassium phosphate buffer (50 mmol/L, pH 6.5), containing 8.7% acetonitrile at a flow rate of 0.3 mL/min. Fluorescence detection was performed at excitation and emission wavelengths of 340 and 455 nm, respectively. Peaks were quantified based on peak area.
4.7. Plasma Chemistry of Vasoactive Agents
A method based on the Griess reaction was used to measure nitrites and nitrates in plasma as described [13,79]. Briefly, plasma samples were incubated with nitrate reductase (Sigma-Aldrich, Dorset UK) for 2 hours at room temperature; then Griess reagent (Sigma-Aldrich, Dorset, UK) was added to the sample. The absorbance was read at 540 nm with a microplate reader 10 min later and interpolated to a standard curve with different concentrations of sodium nitrate (0– 60µM).
Endothelin-1 (ET-1) and angiotensin II (ANG II) levels were determined by ELISA as previously described [47].
Plasma levels of norepinephrine (NE) and epinephrine (E) were measured by high-pressure liquid chromatography (HPLC) as previously described [13].
The activity of aconitase/fumarase was determined as previously described [47]. Briefly, the liver was homogenized on ice in TES buffer (10 mM Tris, pH 7.4, 250 mM sucrose and 1 mM EDTA; 10mL/g of tissue) and the mitochondria were isolated and centrifuged at 4°C [47]. The resulting pellet was resuspended in 0.5 mL of TES buffer containing 0.2% TritonX-100 to lyse the mitochondria.
The enzymatic reaction was measured for 3 minutes in 30 mM sodium isocitrate, 50 mM sodium isocitrate, 50 mM Tris-HCl, pH 7.4, 0.6 mM MnCl2 for the determination of aconitase activity, and 50 mM sodium malate, 50 mM sodium phosphate buffer, pH 7.4 for the fumarase activity. Enzyme activities were measured spectrophotometrically according to the increase in optical density at 240 nm and expressed as the activity ratio.
NF-ĸB measurement was performed in liver nuclear extracts (Active Motif kit, Belgium) using a p65 TransAM transcription factor assay kit (40096; Active Motif, Belgium). NF-kB activation was detected by incubation of the primary anti-NF-kB antibody with the nuclear samples. The HRP-conjugated anti-IgG secondary antibody allows its detection by spectrophotometry by measuring the absorbance of the samples at 450 nm.
4.8. Drugs
Drugs used were: carbachol, phenylephrine, and L-NAME (all from Sigma-Aldrich; Madrid, Spain). Drugs were kept at -20°C and freshly dissolved in distilled water to the appropriate concentration expressed as final molar concentration.
4.9. Data Presentation and Statistics
Data were evaluated using a Graph Pad Prism Software, version 6 (GraphPad Software Inc., San Diego, USA) and presented as mean ± standard error of the mean (S.E.M.).
Mean value comparisons were performed with unpaired Student’s t-test, and by Two-way ANOVA with Sidak’s multi-comparison test, according to the structure of data. Differences were considered statistically significant at a p-value of <0.05.
Author Contributions
The authors have contributed to the study as follows: conceptualization, AR and PIA; methodology, E.O, J. P-L; EV-P and ID.; software, E.O. and J.P-L; validation, E.O., J. P-L. and E. V-P; formal analysis, E.O and J. P-L.; investigation, E.O and J. P-L; data curation, E.O, J.P-L, P.I.A and A.R; writing—original draft preparation, A.R; writing—review and editing, A.R, E. O and P.I.A.; supervision, J.P-L.; project administration, A.R.; funding acquisition, A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was was supported by grant reference BFU2015-70616-R from MINECOFEDER and by Programa Estrategico IBGM, Escalera de Excelencia, grant number CCVC8485, Consejeria de Educacion, Junta de Castilla y León (Spain).
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Universidad de Valladolid (protocol code 4505502) for studies involving animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. The data are pending to upload to the repository of the University of Valladolid (UVaDoc).
Acknowledgments
The authors thank Ana Gordillo and María Llanos Bravo for their help with the CIH experimental model and technical assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip M.S.; Morrell, M.J.; Nuñez, C.M.; Patel, S.R.; Penzel, T.; Pepin, J.L.; Peppard, P.E.; Sinha, S.; Tufik, S.; Valentine, K; Malhotra, A. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med 2019, 7(8), 687–98. [CrossRef]
- Wolk, R.; Shamsuzzaman, A.S.; Somers, V.K. Obesity, Sleep Apnea, and Hypertension. Hypertension 2003, 42, 1067–74. [CrossRef]
- Lavie, L. Oxidative stress in obstructive sleep apnea and intermittent hypoxia – Revisited – The bad ugly and good: Implications to the heart and brain. Sleep Med Rev 2015, 20, 27–45. [CrossRef]
- Lévy, P.; Kohler, M.; McNicholas, W.T.; Barbé, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pepin J.L. Obstructive sleep apnoea syndrome. Nat Rev Dis Primers 2015, 1(1), 15015. [CrossRef]
- Dematteis, M.; Godin-Ribuot, D.; Arnaud, C.; Ribuot, C.; Stanke-Labesque, F.; Pépin J.L.; Levy, P. Cardiovascular Consequences of Sleep-Disordered Breathing: Contribution of Animal Models to Understanding of the Human Disease. ILAR J 2009, 50(3), 262–81. [CrossRef]
- Ryan, S.; Cummins, E.P.; Farre, R.; Gileles-Hillel, A.; Jun, J.C.; Oster, H.; Pepin, J.L.; Ray D.W.; Reutrakul, S.; Sanchez-de-la-Torre, M.; Tamisier, R.; Almendro,s I. Understanding the pathophysiological mechanisms of cardiometabolic complications in obstructive sleep apnoea: towards personalised treatment approaches. Eur Respir J 2020, 56(2), 1902295. [CrossRef]
- Fletcher, E.C.; Lesske, J.; Qian, W.; Miller, C.C.; Unger, T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension 1992, 19(6), 555–61. [CrossRef]
- Prabhakar, N.R. Carotid body chemoreflex: a driver of autonomic abnormalities in sleep apnoea. Exp Physiol 2016, 101(8), 975–85. [CrossRef]
- Semenza, G.L.; Nanduri, X.; Prabhakar, R. Neural regulation of hypoxia-inducible factors and redox state drives the pathogenesis of hypertension in a rodent model of sleep apnea. J Appl Physiol 2015, 119, 1152–6. [CrossRef]
- Lesske, J.; Fletcher, E.C.; Bao, G.; Unger, T. Hypertension caused by chronic intermittent hypoxia – influence of chemoreceptors and sympathetic nervous system. J Hypertens 1997, 15(12), 1593-603. [CrossRef]
- Del Rio, R.; Andrade, D.C.; Lucero, C.; Arias, P.; Iturriaga, R. Carotid Body Ablation Abrogates Hypertension and Autonomic Alterations Induced by Intermittent Hypoxia in Rats. Hypertension 2016, 68(2), 436–45. [CrossRef]
- Dempsey, J.A.; Veasey, S.C.; Morgan, B.J.; O’Donnell, C.P. Pathophysiology of Sleep Apnea. Physiol Rev 2010, 90(1), 47–112. [CrossRef]
- Prieto-Lloret, J.; Olea, E.; Gordillo-Cano, A.; Docio, I.; Obeso, A.; Gomez-Niño, A.; Aaronson, P.I.; Rocher, A. Maladaptive pulmonary vascular responses to chronic sustained and chronic intermittent hypoxia in rat. Antioxidants 2022, 11(1), 54. [CrossRef]
- Fagan, K.A. Selected Contribution: Pulmonary hypertension in mice following intermittent hypoxia. J Appl Physiol 2001, 90(6), 2502–7. [CrossRef]
- Docio, I.; Olea, E.; Prieto-Lloret, J.; Gallego-Martin, T.; Obeso, A.; Gomez-Niño, A.; Rocher, A. Guinea Pig as a Model to Study the Carotid Body Mediated Chronic Intermittent Hypoxia Effects. Front Physiol 2018, 9, 694. [CrossRef]
- Fletcher, E.C.; Lesske, J.; Behm, R.; Miller, C.C.; Stauss, H.; Unger, T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol 1992, 72(5), 1978–84. [CrossRef]
- Arnaud, C.; Beguin, P.C.; Lantuejoul, S.; Pepin, J.L.; Guillermet, C.; Pelli, G.; Burger, F.; Buatois, V.; Ribuot, C.; Baguet, J.P.; Mach, F.; Levy, P.; Dematteis, M. The Inflammatory Preatherosclerotic Remodeling Induced by Intermittent Hypoxia Is Attenuated by RANTES/CCL5 Inhibition. Am J Respir Crit Care Med 2011, 184(6), 724–31. [CrossRef]
- Gras, E.; Belaidi, E.; Briançon-Marjollet, A.; Pépin, J.L.; Arnaud, C.; Godin-Ribuot, D. Endothelin-1 mediates intermittent hypoxia-induced inflammatory vascular remodeling through HIF-1 activation. J Appl Physiol 2016, 120(4), 437–43. [CrossRef]
- Phillips, S.A.; Olson, E.B.; Morgan, B.J.; Lombard, J.H. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol. 2004, 286(1), H388-93. [CrossRef]
- Arnaud, C.; Poulain, L.; Lévy, P.; Dematteis, M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis 2011, 219(2), 425–31. [CrossRef]
- Drager, L.F.; Yao, Q.; Hernandez, K.L.; Shin, M.K.; Bevans-Fonti, S.; Gay, J.; Sussan, T.E.; Jun, J.C.; Myers, A.C.; Olivecrona, .; Schwartz, A.R.; Halberg, N.; Scherer, P.E.; Semenza, G.L.; Powell, D.R.; Polotsky, V.Y. Chronic intermittent hypoxia induces atherosclerosis via activation of adipose angiopoietin-like 4. Am J Respir Crit Care Med. 2013, 188(2), 240-8. [CrossRef]
- Lai, C.J.; Yang, C.C.; Hsu, Y.Y.; Lin, Y.N.; Kuo, T.B. Enhanced sympathetic outflow and decreased baroreflex sensitivity are associated with intermittent hypoxia-induced systemic hypertension in conscious rats. J Appl Physiol 2006, 100(6), 1974–82. [CrossRef]
- Lucking, E.F.; O’Halloran, K.D.; Jones, J.F. Increased cardiac output contributes to the development of chronic intermittent hypoxia-induced hypertension. Exp Physiol 2014, 99(10), 1312–24. [CrossRef]
- Iturriaga, R.; Oyarce, M.P.; Dias, A.C. Role of Carotid Body in Intermittent Hypoxia-Related Hypertension. Curr Hypertens Rep 2017, 19(5), 38. [CrossRef]
- Peppard, P.E.; Young, T.; Palta, M.; Skatrud, J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000, 342(19), 1378-84. [CrossRef]
- Fletcher, E.C. Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol 2001, 90(4), 1600–5. [CrossRef]
- Lucking. E.F.; O’Connor, K.M.; Strain, C.R.; Fouhy, F.; Bastiaanssen, T.F.; Burns, D.P.; Golubeva, A.V.; Stanton, C.; Clarke, G.; Cryan, J.F.; O'Halloran, K.D. Chronic intermittent hypoxia disrupts cardiorespiratory homeostasis and gut microbiota composition in adult male guinea-pigs. EBioMedicine. 2018, 38, 191–205. [CrossRef]
- Sun, M.K.; Reis, D.J. Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am J Physiol. 1994, 266(1), R245-56. [CrossRef]
- Marina, N.; Tang, F.; Figueiredo, M.; Mastitskaya, S.; Kasimov, V.; Mohamed-Ali, V.; Roloff, E.; Teschemacher, A.G.;, Gourine, A.V.; Kasparov, S. Purinergic signalling in the rostral ventro-lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res Cardiol. 2013, 108(1), 317. [CrossRef]
- Machado, B.H.; Zoccal, D.B.; Moraes, D.J.; Neurogenic hypertension and the secrets of respiration. Am J Physiol Regul Integr Comp Physiol. 2017, 312(6), R864–72. [CrossRef]
- Gonzalez-Obeso, E.; Docio, I.; Olea, E.; Cogolludo, A.; Obeso, A.; Rocher, A.; Gomez-Niño, A. Guinea Pig Oxygen-Sensing and Carotid Body Functional Properties. Front Physiol. 2017, 8,285. [CrossRef]
- Greenberg, H.E.; Sica, A.; Batson, D.; Scharf, S.M. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol 1999, 86(1), 298–305. [CrossRef]
- Kraiczi, H.; Magga, J.; Sun, X.Y.; Ruskoaho, H.; Zhao, X.; Hedner, J. Hypoxic pressor response, cardiac size, and natriuretic peptides are modified by long-term intermittent hypoxia. J Appl Physiol 1999, 87(6), 2025–31. [CrossRef]
- Wu, Z.H.; Tang, Y.; Niu, X.; Sun, H.Y. The role of nitric oxide (NO) levels in patients with obstructive sleep apnea-hypopnea syndrome: a meta-analysis. Sleep Breath. 2021, 25(1), 9—16. [CrossRef]
- Moller, D.S.; Lind, P.; Strunge, B.; Pedersen, E.B. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am J Hypertens 2003, 16(4), 274–80. [CrossRef]
- Phillips, B.G.; Narkiewicz, K.; Pesek, C.A; Haynes, W.G.; Dyken, M.E.; Somers, V.K. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J Hypertens 1999, 17(1), 61-6. [CrossRef]
- Lemmey, H.A.; Garland, C.J.; Dora, K.A. Intrinsic regulation of microvascular tone by myoendothelial feedback circuits. Curr Top Membr. 2020, 85, 327–55. [CrossRef]
- Tozzi, C.A.; Poiani, G.J.; Edelman, N.H.; Riley, D.J. Vascular collagen affects reactivity of hypertensive pulmonary arteries of the rat. J Appl Physiol 1989, 66(4), 1730–5. [CrossRef]
- Sandow, S.L.; Gzik, D.J.; Lee, R.M. Arterial internal elastic lamina holes: Relationship to function? J Anat. 2009, 214(2), 258–66. [CrossRef]
- Lefebvre, B.; Godin-Ribuot, D.; Joyeux-Faure, M.; Caron, .;, Bessard, G.; Lévy, P.; Stanke-Labesque, F. Functional assessment of vascular reactivity after chronic intermittent hypoxia in the rat. Respir Physiol Neurobiol 2005, 150(2), 278–86. [CrossRef]
- Allahdadi, K.J.; Walker, B.R.; Kanagy, N.L. Augmented Endothelin Vasoconstriction in Intermittent Hypoxia-Induced Hypertension. Hypertension 2005, 45(4), 705–9. [CrossRef]
- Daly, C.J.; Ross, R.A.; Whyte, J.; Henstridge, C.M.; Irving, A.J.; McGrath, J.C. Fluorescent ligand binding reveals heterogeneous distribution of adrenoceptors and “cannabinoid-like” receptors in small arteries. Br J Pharmacol. 2010, 159(4), 787–96. [CrossRef]
- Minoguchi, K.; Yokoe, T.; Tazaki, T.; Minoguchi, H.; Tanaka, A.; Oda, Okada, S.; Ohta, S.; Naito, H.; Adachi, M. Increased Carotid Intima-Media Thickness and Serum Inflammatory Markers in Obstructive Sleep Apnea. Am J Respir Crit Care Med 2005, 172(5), 625–30. [CrossRef]
- Hodis, H.N.; Mack, W.J.; LaBree, L.; Selzer, R.H; Liu, C.R; Liu, C.H.; Liu, C.H.; Azen, S.P. The role of carotid arterial intima - Media thickness in predicting clinical coronary events. Ann Intern Med. 1998, 128(4), 262–9. [CrossRef]
- Gileles-Hillel, A.; Almendros, I.; Khalyfa, A.; Zhang, S.X.; Wang, Y.; Gozal, D. Early Intermittent Hypoxia Induces Proatherogenic Changes in Aortic Wall Macrophages in a Murine Model of Obstructive Sleep Apnea. Am J Respir Crit Care Med 2014, 190(8), 958–61. [CrossRef]
- Dewan, N.A; Nieto, F.J.; Somers, V.K. Intermittent Hypoxemia and OSA: Implications for Comorbidities. Chest 2015, 147(1), 266–74. [CrossRef]
- Quintero, M.; Gonzalez-Martin, M.C.; Vega-Agapito, V.; Gonzalez, C.; Obeso, A.; Farré, R.; Gonzalez C. The effects of intermittent hypoxia on redox status, NF-κB activation, and plasma lipid levels are dependent on the lowest oxygen saturation. Free Radic Biol Med 2013, 65, 1143–54. [CrossRef]
- Wagenseil, J.E.; Mecham, R.P. Elastin in Large Artery Stiffness and Hypertension. J Cardiovasc Transl Res 2012, 5(3), 264–73. [CrossRef]
- Campen, M.J.; Shimoda, L.A.; O’Donnell, C.P. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol 2005, 99(5), 2028–35. [CrossRef]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.H.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The Role of NADPH Oxidase in Chronic Intermittent Hypoxia-Induced Pulmonary Hypertension in Mice. Am J Respir Cell Mol Biol 2009, 40(5), 601–9. [CrossRef]
- Chaouat, A.; Weitzenblum, E.; Krieger, J.; Oswald, M.; Kessler, R. Pulmonary Hemodynamics in the Obstructive Sleep Apnea Syndrome: Results in 220 Consecutive Patients. Chest 1996, 109(2), 380–6. [CrossRef]
- Yang JZ, Mokhlesi B, Mesarwi OA. Obstructive sleep apnea and pulmonary hypertension: the pendulum swings again. J Clin Sleep Med. 2023 , 19(2), 209-211. [CrossRef]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu. W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int 2005, 67(5), 1739–42. [CrossRef]
- Pinheiro, L.C.; Tanus-Santos, J.E.; Castro, M.M. The potential of stimulating nitric oxide formation in the treatment of hypertension. Expert Opin Ther Targets 2017, 21(5), 543–56. [CrossRef]
- Böger, R.H. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res. 2003, 59(4), 824-33. [CrossRef]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol Res 2009, 60(6), 448–60. [CrossRef]
- Cooke, J.P. Does ADMA Cause Endothelial Dysfunction? Arterioscler Thromb Vasc Biol 2000, 20(9), 2032–7. [CrossRef]
- Surdacki, A.; Nowicki, M.; Sandmann, J.; Tsikas, D.; Boeger, R.H.; Bode-Boeger, S.M.; Kruszelnicka-Kwiatkowska, O.; Kokot, F.; Dubiel, J.S.; Froelich, J.C. Reduced Urinary Excretion of Nitric Oxide Metabolites and Increased Plasma Levels of Asymmetric Dimethylarginine in Men with Essential Hypertension. J Cardiovasc Pharmacol 1999, 33(4), 652-658. [CrossRef]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—An update on patient populations with a wide range of cardiovascular risk. Pharmacol Res 2009, 60(6), 481–7. [CrossRef]
- Böger, R.H.; Hannemann, J. Dual role of the L-arginine–ADMA–NO pathway in systemic hypoxic vasodilation and pulmonary hypoxic vasoconstriction. Pulm Circ 2020, 10(2), 2045894020918850. [CrossRef]
- Kanagy, N.L.; Walker, B.R.; Nelin, L.D. Role of Endothelin in Intermittent Hypoxia-Induced Hypertension. Hypertension 2001, 37(2), 511–5. [CrossRef]
- Rey, S.; Del Rio, R.; Iturriaga, R. Contribution of Endothelin-1 and Endothelin A and B Receptors to the Enhanced Carotid Body Chemosensory Responses Induced by Chronic Intermittent Hypoxia. Adv Exp Med Biol 2008, 605, 228-232. [CrossRef]
- de Frutos, S.; Caldwell, E.; Nitta, C.H.; Kanagy, N.L.; Wang, J.; Wang, W.; Walker, M.K.; Gonzalez Bosc, L.V. NFATc3 contributes to intermittent hypoxia-induced arterial remodeling in mice. Am J Physiol Heart Circ Physiol. 2010, 299(2), H356-63. [CrossRef]
- Gjørup, P.H.; Sadauskiene, L.; Wessels, J.; Nyvad, O.; Strunge, B.; Pedersen, E.B. Abnormally Increased Endothelin-1 in Plasma During the Night in Obstructive Sleep Apnea: Relation to Blood Pressure and Severity of Disease. Am J Hypertens. 2007, 20(1):44–52. [CrossRef]
- Atkeson, A.; Yeh, S.Y.; Malhotra, A.; Jelic, S. Endothelial Function in Obstructive Sleep Apnea. Prog Cardiovasc Dis. 2009, 51(5), 351–62. [CrossRef]
- Limberg, J.K.; Baker, S.E.; Ott, E.P.; Jacob, D.W.; Scruggs, Z.M.; Harper, J.L.; E Manrique-Acevedo, C.M. Endothelin-1 receptor blockade does not alter the sympathetic and hemodynamic response to acute intermittent hypoxia in men. J Appl Physiol 2022, 133(4), 867–75. [CrossRef]
- Marcus, N.J.; Li, Y.L.; Bird, C.E.; Schultz, H.D.; Morgan, B.J. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: Role of the angiotensin II type 1 receptor. Respir Physiol Neurobiol. 2010, 171(1), 36–45. [CrossRef]
- Saxena, A.; Little, J.T.; Nedungadi, T.P.; Cunningham, J.T. Angiotensin II type 1a receptors in subfornical organ contribute towards chronic intermittent hypoxia-associated sustained increase in mean arterial pressure. Am J Physiol Heart Circ Physiol 2015, 308(5), H435–46. [CrossRef]
- Mifflin, S.; Cunningham, J.T.; Toney, G.M.; Mifflin, S. Neurogenic mechanisms underlying the rapid onset of sympathetic responses to intermittent hypoxia. J Appl Physiol 2015, 119, 1441–8. [CrossRef]
- Iturriaga, R.; Andrade, D.C.; Del Rio, R. Enhanced carotid body chemosensory activity and the cardiovascular alterations induced by intermittent hypoxia. Front Physiol 2014, 5, 468. [CrossRef]
- Pialoux, V.; Foster, G.E.; Ahmed, S.B.; Beaudin, A.E.;, Hanly, P.J.; Poulin, M.J. Losartan abolishes oxidative stress induced by intermittent hypoxia in humans. J Physiol 2011, 589(22), 5529–37. [CrossRef]
- Lam, S.Y.; Liu, Y.; Ng, K.M.; Liong, E.C.; Tipoe, G.L.; Leung PS, Fung, L.M. Upregulation of a local renin–angiotensin system in the rat carotid body during chronic intermittent hypoxia. Exp Physiol 2014 , 99(1), 220–31. [CrossRef]
- Morrell, N.W.; Morris, K.G.; Stenmark, K.R. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol 1995, 269(4), H1186–94. [CrossRef]
- Goldstein, D.S.; Eisenhofer, G.; Kopin, I.J. Sources and Significance of Plasma Levels of Catechols and Their Metabolites in Humans. J Pharmacol Exp Ther 2003, 305(3), 800-811. [CrossRef]
- Kjær, M. Adrenal Gland: Fight or Flight Implications for Exercise and Sports. In: The Endocrine System in Sports and Exercise 2005. eds William J., Kraemer W. J., Rogol A. D. p. 194–9. [CrossRef]
- Schwenke, D.O.; Bolter, C.P.; Cragg, P.A. Are the carotid bodies of the guinea-pig functional? Comp Biochem Physiol A Mol Integr Physiol 2007, 146(2), 180–8. [CrossRef]
- Monge, C.; Leon-Velarde, F. Physiological adaptation to high altitude: oxygen transport in mammals and birds. Physiol Rev 1991, 71(4), 1135–72. [CrossRef]
- Zoccal, D.B.; Vieira, B.N.; Mendes, L.R.; Evangelista, A.B.; Leirão, I.P. Hypoxia sensing in the body: An update on the peripheral and central mechanisms. Exp Physiol 2024, 109(4), 461–9. [CrossRef]
- Ricart-Jané, D.; Llobera, M.; López-Tejero, M.D. Anticoagulants and Other Preanalytical Factors Interfere in Plasma Nitrate/Nitrite Quantification by the Griess Method. Nitric Oxide 2002, 6(2), 178–85. [CrossRef]
Figure 1.
1A. Mean systemic arterial blood pressure (mmHg). 1B. Pulse pressure (mmHg). 1C. Heart rate (beats/minute). 1D. Fulton index (right ventricle/left ventricle + septum). 1E. Pulmonary arterial pressure (mmHg) and 1F. Hypoxic pulmonary vasoconstriction (mmHg), in control (C) and CIH guinea pigs in hypoxia (10% O2). Data are expressed as mean ± standard error (SEM) of n=7. Unpaired t-test (Figure 1A, 1B, 1C, 1D and 1F) and Two Way ANOVA (Figure 1E). *p<0.05; **p<0.01; ****p<0.0001.
Figure 1.
1A. Mean systemic arterial blood pressure (mmHg). 1B. Pulse pressure (mmHg). 1C. Heart rate (beats/minute). 1D. Fulton index (right ventricle/left ventricle + septum). 1E. Pulmonary arterial pressure (mmHg) and 1F. Hypoxic pulmonary vasoconstriction (mmHg), in control (C) and CIH guinea pigs in hypoxia (10% O2). Data are expressed as mean ± standard error (SEM) of n=7. Unpaired t-test (Figure 1A, 1B, 1C, 1D and 1F) and Two Way ANOVA (Figure 1E). *p<0.05; **p<0.01; ****p<0.0001.
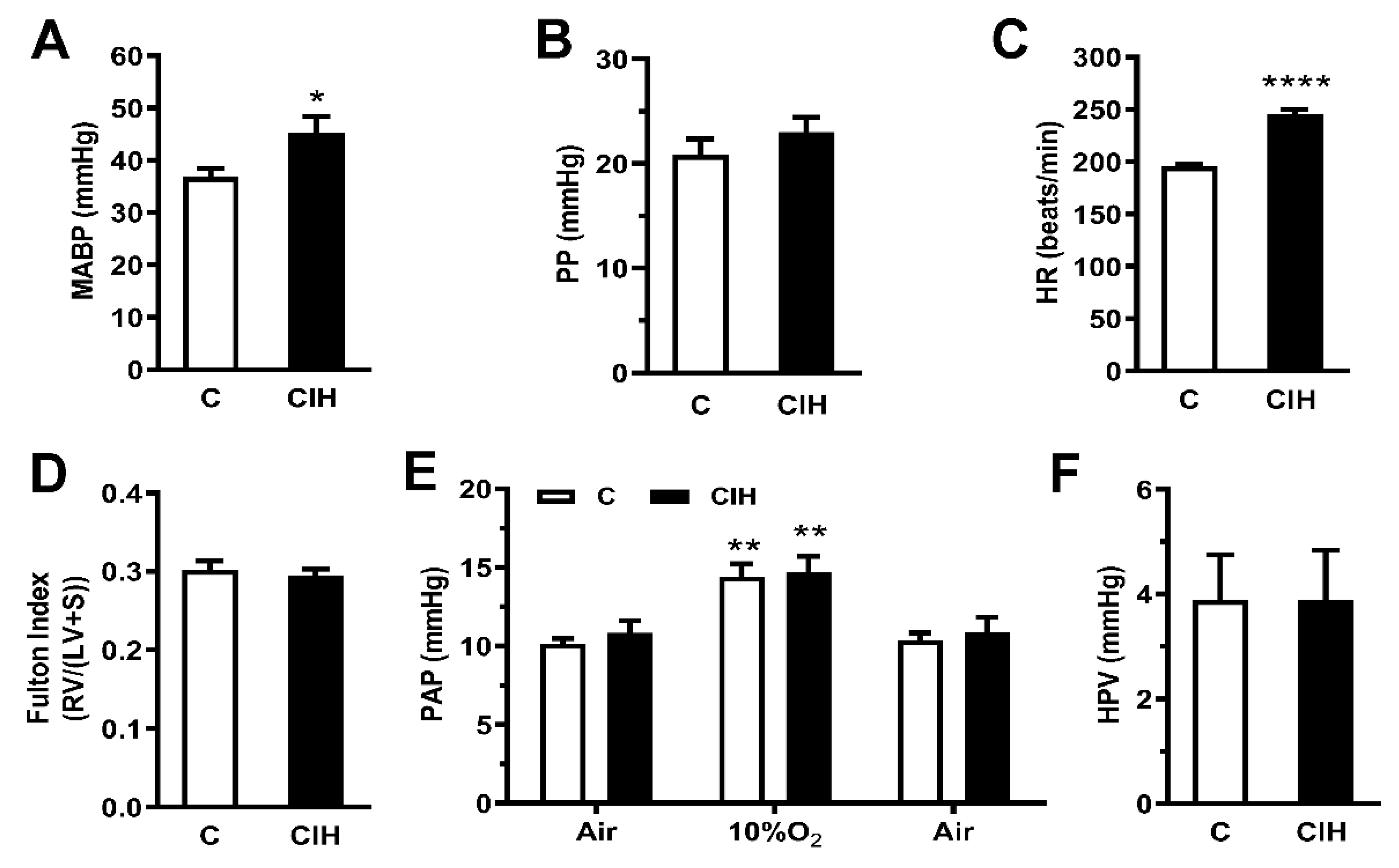
Figure 2.
Vascular contractile responses to PE in aorta (panel A), carotid artery (panel B) and pulmonary artery (panel C) with or without 0.1 mM L-NAME in control and CIH guinea pigs. Data are expressed as mean ± standard error (SEM). Two-way ANOVA Sidak´s multiple comparison test; **p<0.01; ***p<0.001, L-NAME vs its control. Vascular contractile responses to 30 µM PE in aorta (panel D) carotid artery (panel E) and pulmonary artery (panel F) with or without 0.1 mM L-NAME in C and CIH guinea pigs. Data are expressed as mean ± standard error (SEM), n=7-15 for aorta; n=6-19 for carotid artery; n=12-16 for pulmonary artery. Unpaired t-test ***p<0.001 C or CIH vs C + L-NAME or CIH + L-NAME; ###p<0.001 C vs CIH.
Figure 2.
Vascular contractile responses to PE in aorta (panel A), carotid artery (panel B) and pulmonary artery (panel C) with or without 0.1 mM L-NAME in control and CIH guinea pigs. Data are expressed as mean ± standard error (SEM). Two-way ANOVA Sidak´s multiple comparison test; **p<0.01; ***p<0.001, L-NAME vs its control. Vascular contractile responses to 30 µM PE in aorta (panel D) carotid artery (panel E) and pulmonary artery (panel F) with or without 0.1 mM L-NAME in C and CIH guinea pigs. Data are expressed as mean ± standard error (SEM), n=7-15 for aorta; n=6-19 for carotid artery; n=12-16 for pulmonary artery. Unpaired t-test ***p<0.001 C or CIH vs C + L-NAME or CIH + L-NAME; ###p<0.001 C vs CIH.
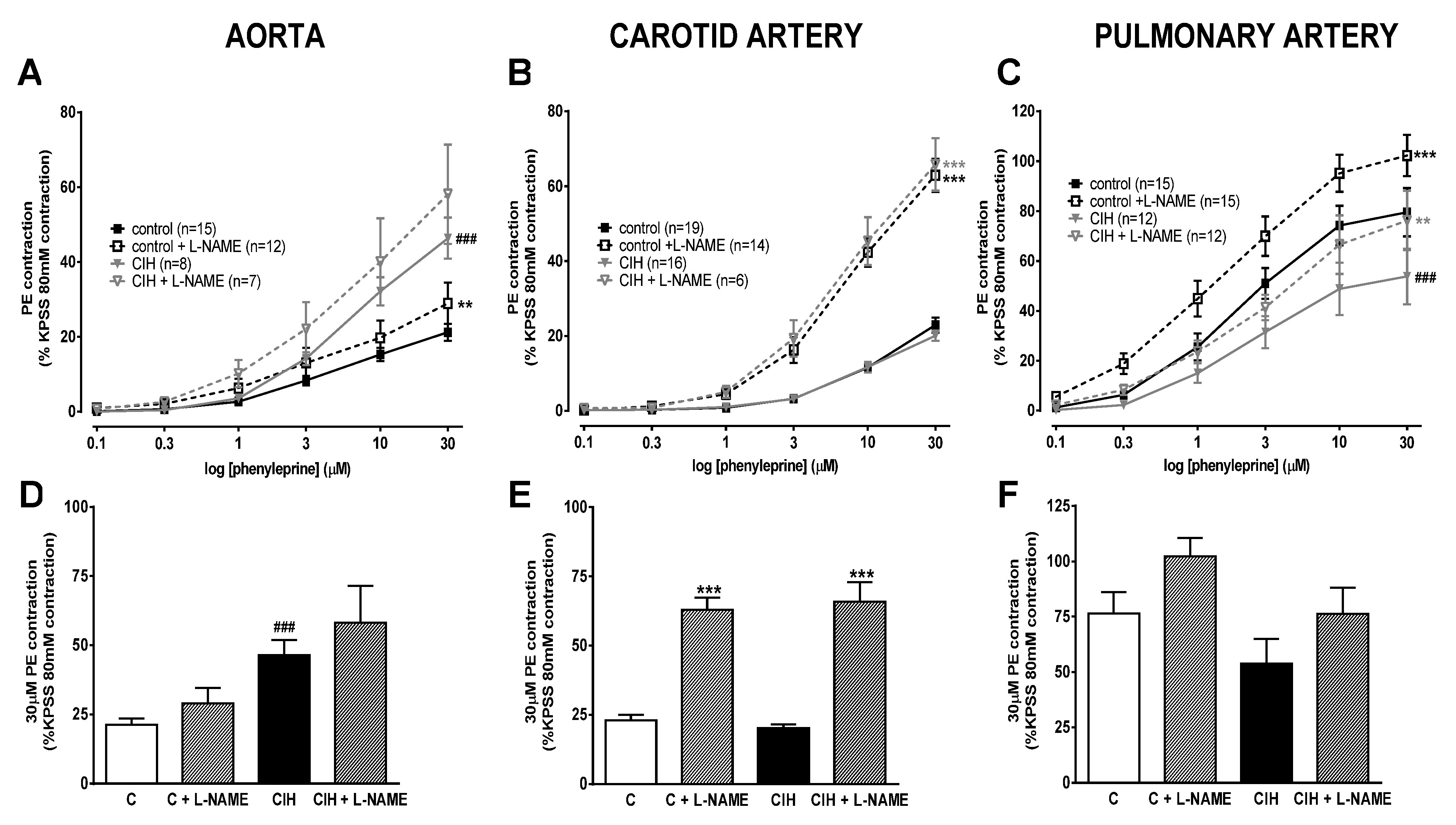
Figure 3.
Vascular relaxation responses to carbachol in aorta (panel A), carotid artery (panel B) and pulmonary artery (panel C), with or without 0.1 mM L-NAME, in C and CIH guinea pigs. Data are expressed as mean ± standard error (SEM). Two-way ANOVA Sidak´s multiple comparison test; ***p<0.001 (C or CIH vs C + L-NAME or CIH + L-NAME); #p<0.05 C vs CIH. Vascular relaxation to 10µM carbachol in aorta (panel D), carotid artery (panel E) and pulmonary artery (panel F) with or without 0.1 mM L-NAME. Data are expressed as mean ± standard error (SEM), n=7-15 for aorta; n=6-19 for carotid artery; n=12-16 for pulmonary artery. Unpaired t-test *p<0.05; **p<0.01; ***p<0.001 (C or CIH vs C + L-NAME or CIH + L-NAME).
Figure 3.
Vascular relaxation responses to carbachol in aorta (panel A), carotid artery (panel B) and pulmonary artery (panel C), with or without 0.1 mM L-NAME, in C and CIH guinea pigs. Data are expressed as mean ± standard error (SEM). Two-way ANOVA Sidak´s multiple comparison test; ***p<0.001 (C or CIH vs C + L-NAME or CIH + L-NAME); #p<0.05 C vs CIH. Vascular relaxation to 10µM carbachol in aorta (panel D), carotid artery (panel E) and pulmonary artery (panel F) with or without 0.1 mM L-NAME. Data are expressed as mean ± standard error (SEM), n=7-15 for aorta; n=6-19 for carotid artery; n=12-16 for pulmonary artery. Unpaired t-test *p<0.05; **p<0.01; ***p<0.001 (C or CIH vs C + L-NAME or CIH + L-NAME).
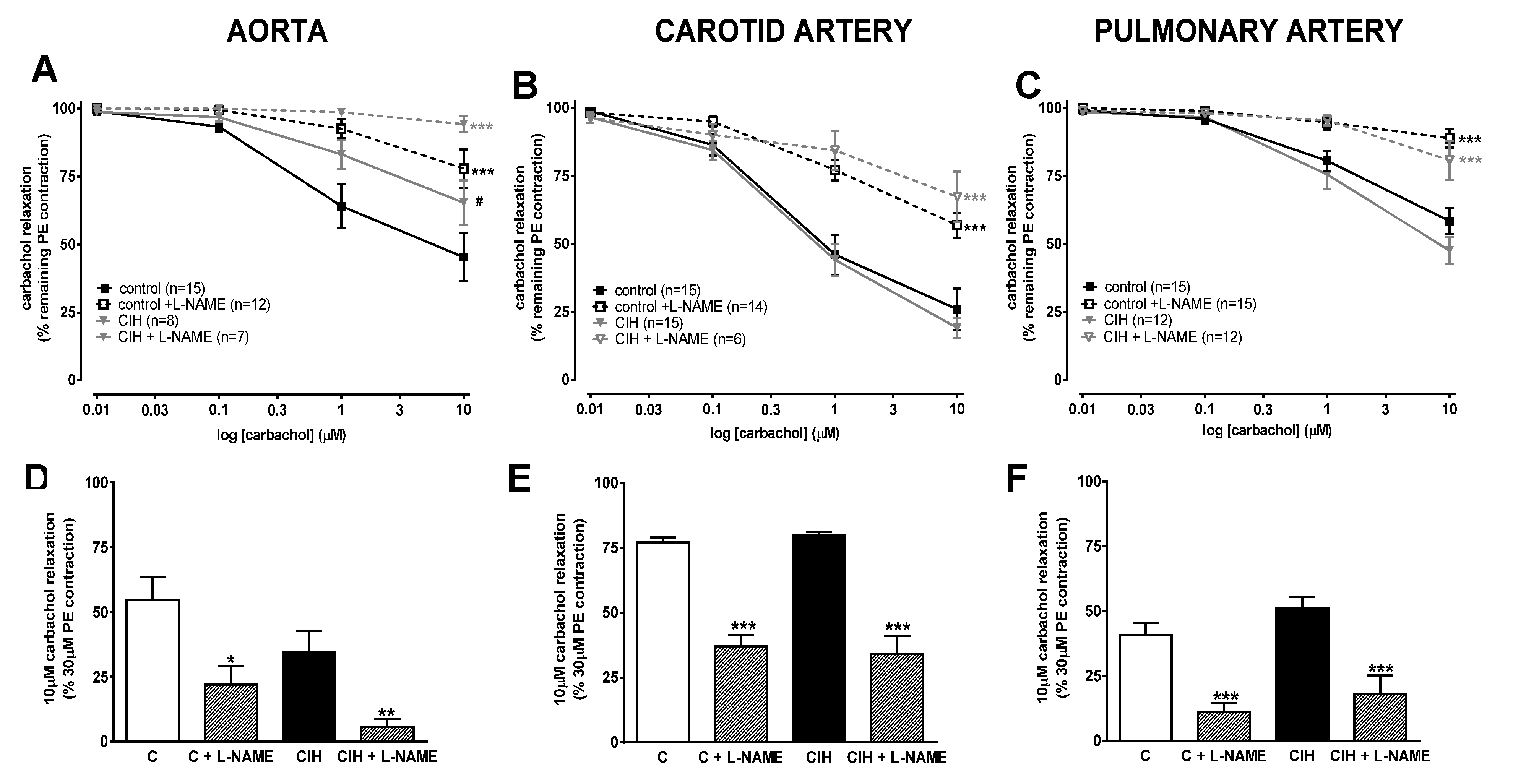
Figure 4.
Representative images of the aorta (left), carotid artery (middle) and PA (right) stained with Masson’s trichrome for C (top) and CIH (button) groups. Scale bars: 60 μm.
Figure 4.
Representative images of the aorta (left), carotid artery (middle) and PA (right) stained with Masson’s trichrome for C (top) and CIH (button) groups. Scale bars: 60 μm.
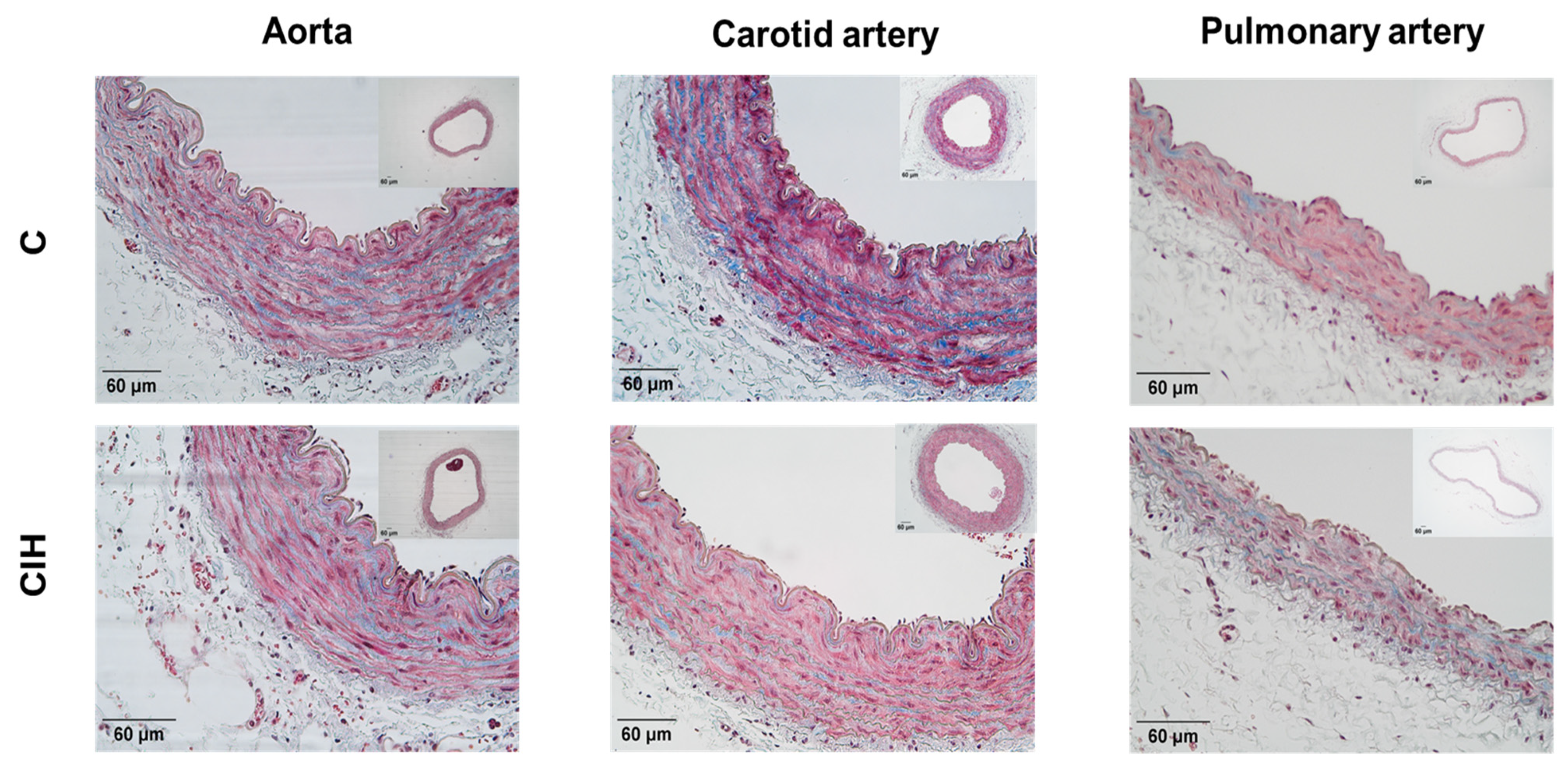
Figure 5.
Morphometric and histologic evaluation in the aorta (left), carotid artery (middle) and PA (right). A, B, C. Lumen area for each artery. D, E, F. Intima-media thickness (IMT) for each artery. G, H, I Collagen deposition expressed as a percentage of the IMT area for each artery. Data are expressed as mean ± standard error (SEM) of quintuplicate measurements from n=4-6 animals. Unpaired t-test *p<0.05.
Figure 5.
Morphometric and histologic evaluation in the aorta (left), carotid artery (middle) and PA (right). A, B, C. Lumen area for each artery. D, E, F. Intima-media thickness (IMT) for each artery. G, H, I Collagen deposition expressed as a percentage of the IMT area for each artery. Data are expressed as mean ± standard error (SEM) of quintuplicate measurements from n=4-6 animals. Unpaired t-test *p<0.05.
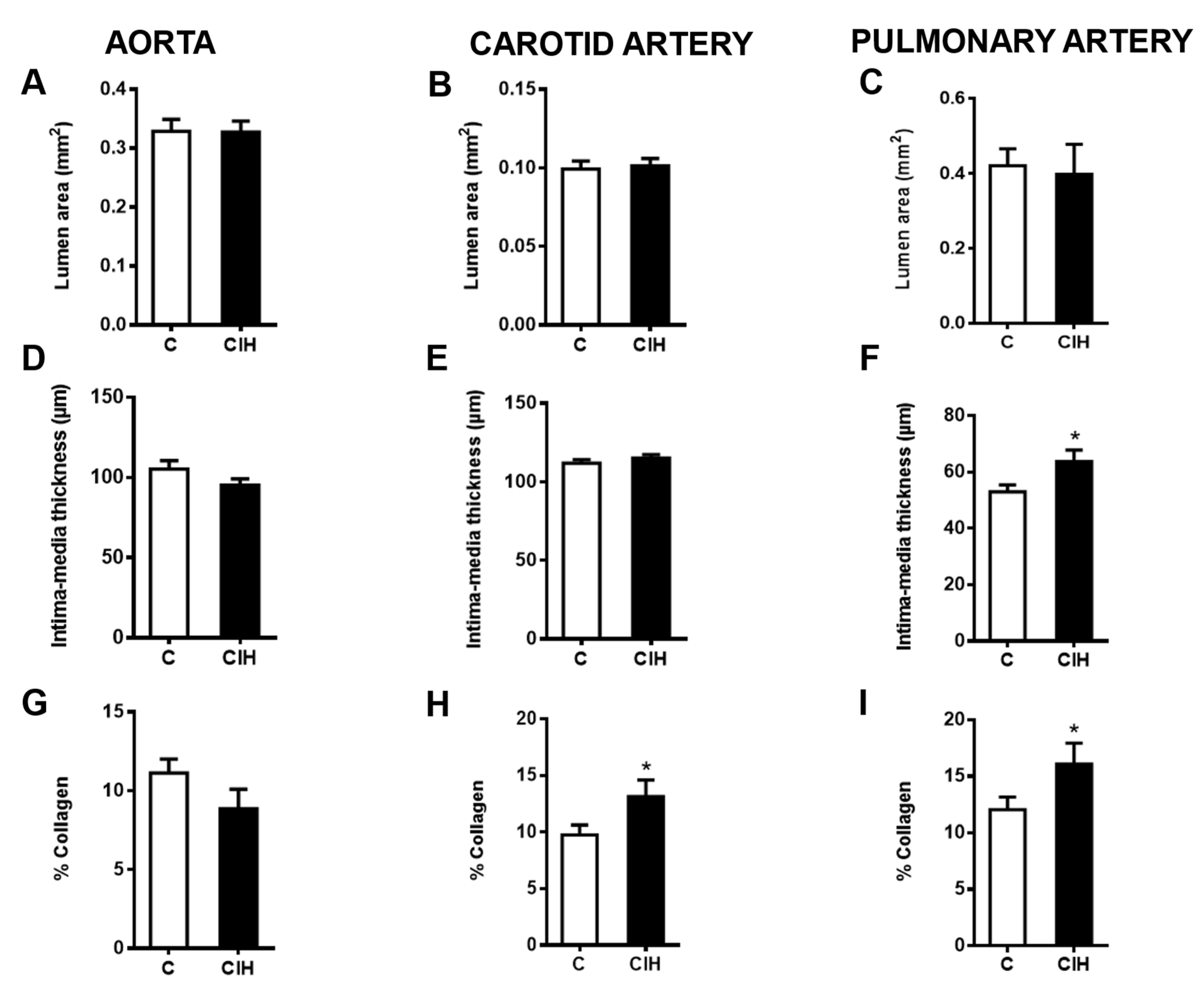
Figure 6.
6A. Plasma levels of L-Arginine (µM) in Control and CIH guinea pigs. 6B. Asymmetric dimethylarginine (ADMA). 6C. L-Arginine/ADMA ratio. 6D. L-Homoarginine (µM). 6E. Symmetric dimethylarginine (SDMA). 6F. Plasma Nitrites (µM) in Control and CIH guinea pigs. Data are expressed as mean ± standard error (SEM) of n=6-10. Unpaired t-test *p<0.05.
Figure 6.
6A. Plasma levels of L-Arginine (µM) in Control and CIH guinea pigs. 6B. Asymmetric dimethylarginine (ADMA). 6C. L-Arginine/ADMA ratio. 6D. L-Homoarginine (µM). 6E. Symmetric dimethylarginine (SDMA). 6F. Plasma Nitrites (µM) in Control and CIH guinea pigs. Data are expressed as mean ± standard error (SEM) of n=6-10. Unpaired t-test *p<0.05.
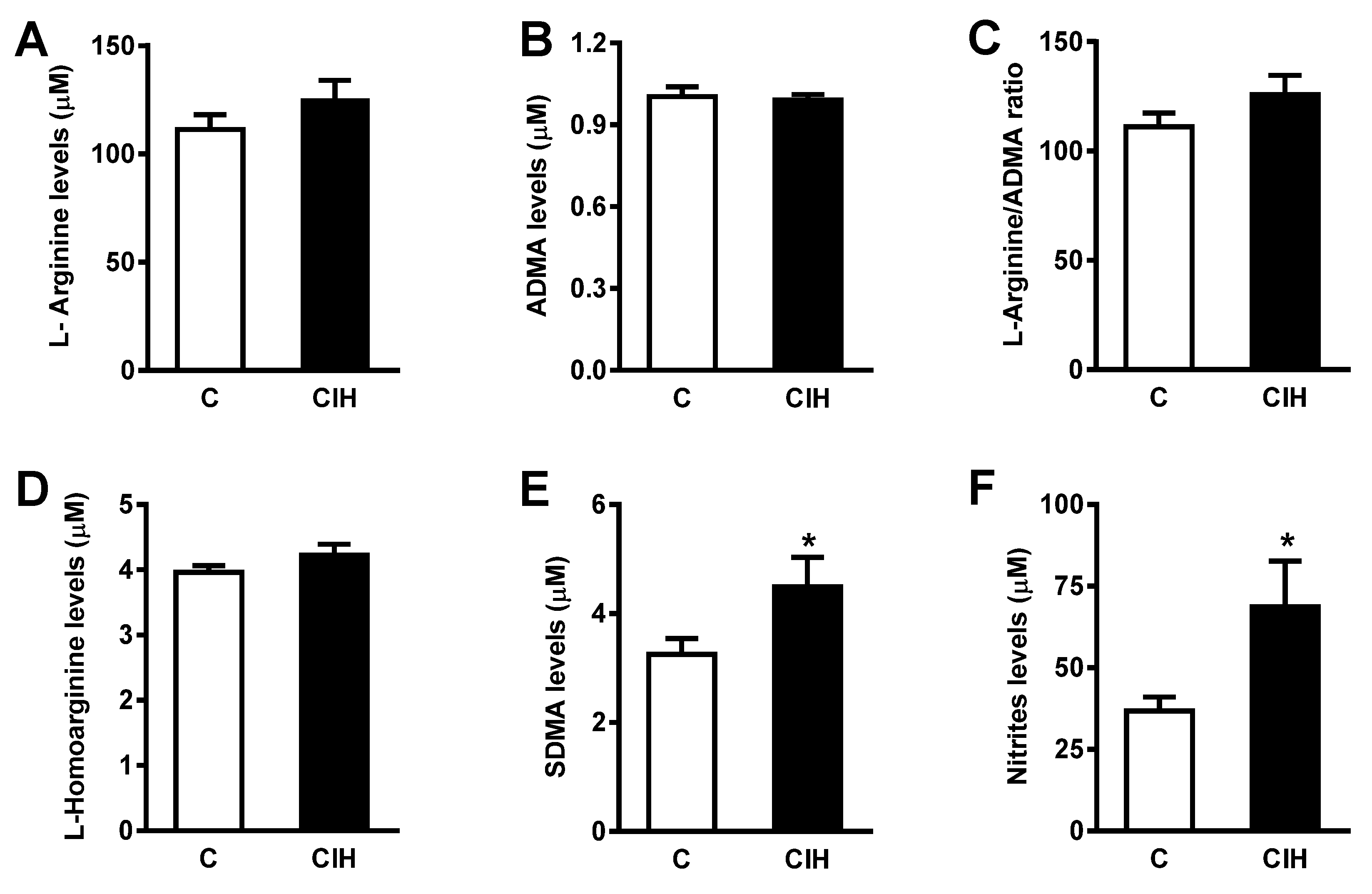

Table 1.
Body weight, erythrocytes, hematocrit, arterial blood PO2 and PCO2 and erythropoietin (EPO) in control (C) and chronic intermittent hypoxia (CIH) guinea pigs. Data are expressed as mean ± standard error (SEM) from n=8 animals. *p<0.05; **p<0.01.
Table 1.
Body weight, erythrocytes, hematocrit, arterial blood PO2 and PCO2 and erythropoietin (EPO) in control (C) and chronic intermittent hypoxia (CIH) guinea pigs. Data are expressed as mean ± standard error (SEM) from n=8 animals. *p<0.05; **p<0.01.
| C | CIH | |
| Body weight (g) | 791 ± 19 | 715 ± 14 ** |
| Hematocrit (%) | 41.5 ± 0.8 | 41.4 ± 0.8 |
| EPO (mU/mL) | 141 ± 17 | 214 ± 52 * |
| pO2 (mm Hg) | 65 ± 6 | 64 ± 6 |
| pCO2 (mm Hg) | 36 ± 2 | 38 ± 3 |
Table 2.
Vascular responses to 80 mM KCl in the aorta, carotid and pulmonary artery in Control (C) and Chronic intermittent hypoxia (CIH) guinea pigs.
Table 2.
Vascular responses to 80 mM KCl in the aorta, carotid and pulmonary artery in Control (C) and Chronic intermittent hypoxia (CIH) guinea pigs.
| AORTA | CAROTID ARTERY | PULMONARY ARTERY | ||||
| C | CIH | C | CIH | C | CIH | |
| Mean (mN) | 9.7 ± 1.4 | 6.5 ± 1.8 | 16.0 ± 1.7 | 16.9 ± 2.4 | 10.0 ± 1.4 | 10.3 ± 0.7 |
| n | 15 | 8 | 19 | 16 | 15 | 12 |
Table 3.
Endothelin-1 (ET-1), Angotensin II (ANG II), Atrial natriuretic peptide (ANP), Vascular endothelial growth factor (VEGF) and Catecholamines, norepinephrine (NE) and epinephrine (E), content in plasma in Control and CIH guinea pigs. Data are expressed as mean ± standard error (SEM) n=7-16. Unpaired t-test *p<0.05.
Table 3.
Endothelin-1 (ET-1), Angotensin II (ANG II), Atrial natriuretic peptide (ANP), Vascular endothelial growth factor (VEGF) and Catecholamines, norepinephrine (NE) and epinephrine (E), content in plasma in Control and CIH guinea pigs. Data are expressed as mean ± standard error (SEM) n=7-16. Unpaired t-test *p<0.05.
| Guinea pig | C | CIH |
|---|---|---|
| ET-1 (pg/mL) | 19.9 ± 1.9 | 21.5 ± 1.8 |
| ANG II (pg/mL) | 355 ± 49 | 412 ± 67 |
| ANP (pg/mL) | 234 ± 15 | 209 ± 6 |
| VEGF (pg/mL) | 2.5 ± 0.2 | 2.6 ± 0.2 |
| NE (pmol/mL) | 7.2 ± 1.7 | 80.7 ± 24.0 * |
| E (pmol/mL) | 11 ± 0.4 | 64 ± 20 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



