Submitted:
18 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
The many limitations of implementing anticancer strategies under the term “precision oncology” have been extensively discussed. While some authors propose promising future directions, others are less optimistic and use phrases such as illusion, hype and false hypotheses. The reality is revealed by practicing clinicians and cancer patients in various online publications, one of which has stated that “in the quest for the next cancer cure, few researchers bother to look back at the graveyard of failed medicines to figure out what went wrong.” The message is clear: novel therapeutic strategies with catchy names (e.g., synthetic “lethality”) have not fulfilled their promises despite decades of extensive research and clinical trials. The main purpose of this review is to discuss key challenges in solid tumor therapy that surprisingly continue to be overlooked by the nomenclature committee on cell death and numerous other authors. These challenges include: the impact of chemotherapy-induced genome chaos (e.g., multinucleation) on resistance and relapse, oncogenic function of caspase 3, cancer cell anastasis (recovery from late stages of apoptosis), and pitfalls of ubiquitously used preclinical chemosensitivity assays (e.g., cell “viability” and tumor growth delay studies in live animals) that score such pro-survival responses as “lethal” events. Studies outline herein underscore the need for new direction in the management of solid tumors.
Keywords:
solid tumor therapy
; intratumor heterogeneity
; polyploid giant cancer cells
; senescence
; anastasis
; oncogenic caspases
; Phoenix Rising
; treacherous apoptosis
; preclinical assays
; precision oncology
1. Introduction
1.1. Expert Opinions on Solid Tumor Therapy
In 2016 Vinay Prasad published a Perspective article in Nature on the illusion of precision oncology in which he concluded that “precision oncology is inspirational. What doctor or patient would not want to harness genetics to tailor a therapy to an individual? But travelling back in a time machine is also inspirational. Who would not want to wind back the clock to remove their cancer before it spreads? In both cases, however, as of 2016, the proposal is neither feasible, cost-effective nor assured of future success. Yet in only one of these cases does the rhetoric so far outpace the reality that we risk fooling even ourselves” [1].
In 2017 Kaiser Health News article was published online by Liz Szabo which contains input from oncologists and families of cancer patients regarding how “cancer treatment hype gives false hope to many patients” [2]. On the topic of being continuously bombarded with the news and scientific publications on how oncology is making significant progress and inroads in terms of winning the war against cancer, Dr. Otis Brawley, previous (2007-2018) chief medical officer at the American Cancer Society, stated that “We’re starting to believe our own (hogwash)...The consequences are real — and they can be deadly. Patients and their families have bought into treatments that either don’t work, cost a fortune or cause life-threatening side effects.”
Since then numerous articles have been published in scientific journals (e.g., [3,4,5,6,7,8,9]) or posted online (e.g., [10,11,12,13,14]) that appear to support Prasad’s predictions. In 2023, for example, John Horgan [14] posted a blog in which he addressed “Big business, big hype (of cancer industry)…Little net progress besides anti-smoking efforts…The false comfort of survival rates…Corruption in the cancer industry…”
1.2. Objectives
The purpose this review is to highlight several well established therapy-induced responses that underlie resistance and relapse, but surprisingly continue to be widely overlooked. The graphic abstract regarding apoptosis is presented in Figure 1. The take home messages are listed in Appendix A, which include an urgent need for a paradigm change to “stay away” form apoptosis-promoting strategies.
Specifically, this review is in four parts: (i) a brief discussion on the consequences of information-generating approaches (“omics”) to cancer therapy (sections 2); (ii) the contribution of genome chaos (e.g., polyploidy/multinucleation) to chemotherapy resistance (sections 3); the “dark” side of apoptosis in solid tumor therapy (section 4); and (iii) exploiting the apoptotic threshold for managing solid tumors (section 5).
2. Danger of Information-Generating Approaches to Cancer Biology: The Current State of Confusion
2.1. Off-Target Effects of Drugs Designed for Targeted Therapies
Over the past three decades, many cancer researchers and investigators in the pharmaceutical industry have expressed heightened excitement regarding the application of rapid screens and validation studies to identify drugs that are capable of targeting specific proteins. These so-called small molecule inhibitors have been developed to be used for novel anticancer strategies under the term precision oncology with catchy names (e.g., targeted therapy, personalized medicine, synthetic “lethality”). Unfortunately, the majority (~97%) of such anticancer drugs that undergo clinical trials fail to advance to receive FDA approval [10,11,12,13,14,15,16]. For those few drugs that are approved, the results often turn out to be disappointing once they are administered in non-trial settings [13,14,15]. This is partly because of how trials are conducted, involving subjects who are ”handpicked and generally in reasonable physical shape,” the practicing oncologist Azra Raza asserts in her book entitled “The First Cell: And the Costs of Pursuing Cancer to the Last” [15].
In 2019, Lin et al. [16] reported studies that were designed to investigate target specificity of a set of cancer drugs that were in various stages of clinical trials. All drugs that were tested by these authors were shown to have off-target effects. In other words, many compounds that were identified/developed for “targeted therapies” and had entered clinical trials were not functioning for the purpose that they were developed. This means that patients who participate in these clinical studies “risk their lives on treatments that end up in the dustbin" [12].
Since 2019 various groups have reported similar off-target effects for a number of other small molecule inhibitors. These include poly (ADP-ribose) polymerase (PARP) inhibitors, which have been developed and evaluated in clinical trails for targeted therapies [19]. Unlike most of the drugs tested by Lin et al. [16], the PARP inhibitors olaparib, rucaparib, niraparib and talazoparib have received FDA approval for various malignancies.
A summery of the results reported by Lin et al. [16] together with their clinical implications have been discussed in a blog by Julia Belluz [12]. The blog contains input from the Nobel Prize Laureate William Kailen who stated that “I hope this paper will help people see the need to raise to bar in terms of how we choose and validate cancer drug targets.” The blog also contains the following statement by Belluz: “In the quest for the next cancer cure, few researchers bother to look back at the graveyard of failed medicines to figure out what went wrong” [12]. (Our group has been in the same boat, as stated previously [20,21].)
This is one of the main goals of the proceeding discussions. Namely, to try to shed some light on what went wrong in the “graveyard” of precision oncology.
2.2. Precision Oncology for Treating Patients with Solid Tumors: Ever-Increasing Information and More (Empty) Promises
A decade ago, Robert Weinberg published a Leading Edge Essay article entitled “Coming Full Circle—From Endless Complexity to Simplicity and Back Again” in which he discussed the consequences of a merely reductionist approach to cancer research. The reductionistic paradigm refers to the hypothesis that biological systems can be fruitfully investigated at the lowest possible level [22]. In this essay, Weinberg noted that, since the 1980s, the need for generating large data sets led to the “omics” era of cancer research, which included studying genomes, transcriptomes, proteomes, epigenomes, kinomes, methylomes, glycomes, and matrisomes. Each one of these “omics” branches led to accumulating staggering amounts of information. Unfortunately, however, we can’t really assimilate and interpret most of the accumulated data [22]. More recently, William Kailen also discussed the danger of relying on large amounts of data sets, often crammed into a single manuscript, without undertaking in-depth biological assessments to validate overly broad claims in terms of potential clinical relevance [11,23]. Kaelin used the analogy of “building with straw” for such information-generating publications and stated that the “real advances are built with bricks, not straw” [23]. Kaelin also cautioned the scientific community in terms of drawing “wishy-washy” conclusions from ubiquitous use of “down” assays (see below) to clinical relevance. (Key points raised by Weinberg, Kailen and other leading oncologists in the context of solid tumor therapy have been recently reviewed [24].)
The commonly used “down” assays include immunoblotting (to assess decreased global expression of genes/proteins under study), decreased cell proliferation, and decreased tumor growth in live animals [11,23]. A major shortcoming of such assays is that they overlook the complexity and heterogeneity that exists within a tumor (intratumor heterogeneity) ([24,25]; also see Figure 2). Another shortcoming pertains to experimental design in most apoptosis and cell “viability” studies (see section 5 below).
2.3. Anticancer Strategies Targeting the p53 “Firework”
In 2017 Kastenhuber and Lowe [26] discussed the difficulties associated with targeting the p53 network in the context of cancer therapy and concluded that, despite over three decades of intensive research and numerous discoveries, “a clear appreciation of how and in what contexts p53 exerts its diverse effects remains unclear.” Since then, many other authors have discussed the increasing challenges encountered in p53/p21-based cancer therapeutic strategies (too many to cite!). These challenges in part pertain to proteoforms of p53 (and its family members, p63 and p73) [26,27,28,29,30], which can be generated by posttranslational modifications, alternative splicing, mutations, and a combination of these events [27,30]. Uveresky [27] has appropriately used the term “p53 firework” (rather than network) to illustrate the mind-numbing complexity of p53 proteoforms.
The firework extends to p53 downstream events, given its well known function as a transcription regulator. A meta analysis reported in 2017 identified over 3,500 genes that are directly upregulated by p53 [31]. In addition, p53 interacts with a large number of proteins [32] and contributes to the stability of the epigenetic state [33,34].
An illustration of p53 interacting proteins is available online (cited in [32]). Figure 3 contains this version of “p53 firework” together with key functions of the p53 target p21WAF1 (p21). Like p53, p21 regulates gene expression both directly and indirectly by modulating the epigenetic landscape. (Multifunctional nature of p21 beyond its canonical role in regulating cell cycle progression has been reviewed [35].)
Huge efforts have been devoted to predict the biological outputs of the molecule(s) under study merely based on such perplexing signaling networks. As an example, Markowska et al. [36] have recently presented a “firework” of overlapping signaling pathways that regulate chromatin remodeling, DNA damage response and cell cycle. The authors used a uniquely comprehensive workflow and identified several pairs of genes, related to these processes, that might display synthetic “lethal” interactions. (This phenomenon refers to genetic interactions between two genes, where inactivation of each individual gene is compatible with a viable phenotype, whereas co-inactivation of both genes is anticipated to result in lethality [36].)
The danger of relying on such information generating approach for predicting the potential cell fate outcomes is that key discoveries of fundamental clinical relevance might be overlooked. For example, in most reviews on different strategies of precision oncology (e.g., engaging apoptosis [37,38,39,40,41,42], targeting the p53 pathway [43,44,45,46,47], synthetic “lethality” [48,49,50,51,52,53,54]) there is no mention of “outliers” within a tumor that are known to underlie resistance and relapse. These outliers include cancer cells with various manifestations of genome chaos and apoptotic cancer cells that survive and generate aggressive variants (see sections 3 and 4).
With respect to synthetic “lethality” targeting the DNA damage response, several challenges in implementing this strategy were discussed by Lord and associates in a landmark review that was published in 2017 [53]. Since then the challenges have mounted, which led Groelly et al. [49] to caution that such an approach might not work for complex diseases such as cancer.
Please note: The current review is focused on solid tumors and thus the conclusions drawn may or may not pertain to other malignancies. In addition, quotation marks are used for the term “lethality” throughout this review given that the in vitro colony formation and other preclinical assays that are widely used to assess “lethality” are not designed to distinguish dead cancer cells and dying cancer cells that can recover and contribute to disease relapse.
3. Impact of Genome Chaos on Cancer Cell Resistance To Therapeutic Agents
One of the reasons why the metaphoric “war on cancer” has not been won is the ability of cancer cells to evolve and undergo phenotypic changes (plasticity). Ironically, these adaptation processes are greatly enhanced by our anticancer treatment strategies. In other words, the various cytotoxic treatment options that are designed to destroy cancer cells (e.g., via regulated cell death) can paradoxically trigger the creation of more aggressive “monsters" [55,56,57,58,59]. This rather bizarre process of acquired therapy resistance of cancer cells is characterized by rapid and extensive genome restructuring episodes (i.e., through non-mutational/non-genetic events) and has been termed “genome chaos” by Heng and coworkers ([56,57]).
3.1. Therapy-Induced Dormancy via polyploidy/Multinucleation
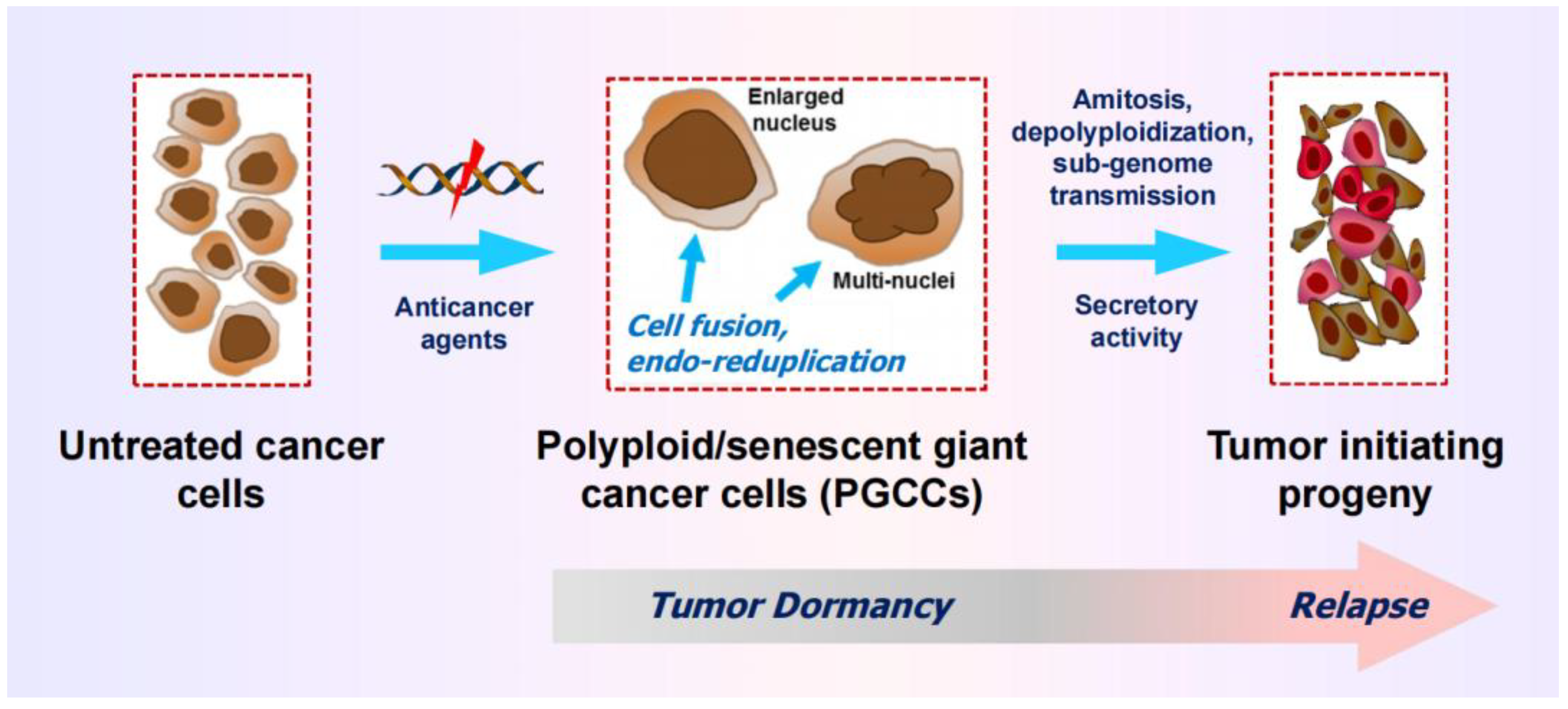
A subset of cancer cells with chaotic genome are readily distinguishable from the bulk of cancer cells by virtue of their enormous size and massive nuclear contents (i.e., highly enlarged nucleus, multiple nuclei, and/or multiple micronuclei). These so-called polyploid giant cancer cells (PGCCs) can enter a state of transient dormancy (active sleep) after they are formed, but remain viable, secrete growth promoting factors, and exhibit the ability to undergo depolyploidization as well as “neosis” (nuclear budding and bursting), ultimately giving rise to stem-cell like progeny that repopulate the tumor (reviewed in [25]; also see Figure 4).
The mechanisms of formation and fate of PGCCs as well as their prevalence and prognostic value across different cancer types are well established and extensively discussed, albeit by only a handful of authors who reply on time lapse microscopy and other single cell assays (e.g., [59,60,61,62,63,64,65,66,67]).
3.2. The Creation of PGCCs Complicates the Interpretation of Chemosensitivity Data
PGCCs are not considered by most authors who have discussed the challenges and promises of precision oncology, synthetic “lethality,” artificial intelligence, and p53/p21-based therapies (too many to cite!). Possible reasons for this serious oversight has been pointed out [24,25], which includes misinformation perpetuated by the misguided use of preclinical chemosensitivity studies (e.g., the colony formation assay, multiwell plate “viability” assays, and tumor growth studies in live animals).
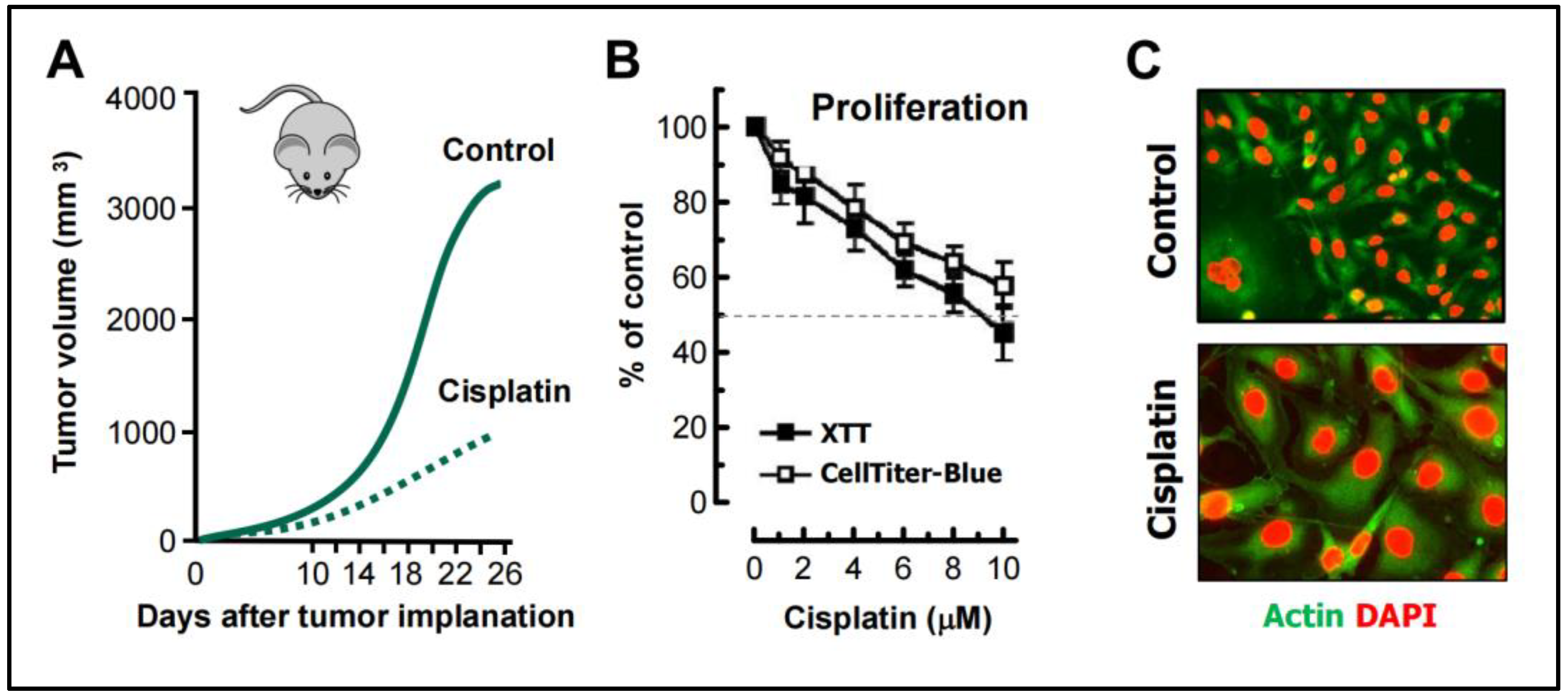
The in vivo and in vitro cisplatin-triggered responses presented below illustrates the consequences of relying on such assays. The tumor growth data shown in Figure 5 are reproduced from a study reported by Weng et al. [68], and the cell-based data are reproduced from our published work [69]. The following observations should be noted:
- Weng et al [68] studied the mechanism of cisplatin resistance in B16-F10 (mouse melanoma) cells grown in C57BL (immune proficient) mice. As expected, cisplatin treatment resulted in significant reduction (by ~75%) of tumor volume compared to the control group (Figure 5A). Tumors in cisplatin-treated mice were highly enriched with PGCCs which were shown to be more malignant than the parental cancer cells. A similar observation was reported by Puig et al [70] with a rat colon carcinoma isografts established in immunocompetent rats. In the latter study, tumor repopulation resulting from PGCCs was observed late times (>40 days) after cisplatin treatment (for details, see [25]).
- Figure 5B shows cisplatin sensitivity of MDA-MB-231 breast carcinoma cells evaluated by the 96-well plate XTT and CellTiter-Blue assays. The IC50 (half-maximum inhibitory concentration) vales were ~10 µM.
- Further studies involving single cell assays demonstrated that cisplatin sensitivity reflected proliferation arrest via the creation of PGCCs (predominantly mononucleated giants; Figure 5C), and that virtually all emerging PGCCs remained adherent to the culture dish and remained viable and metabolically active [69].
Data interpretation: The data obtained by such “down” assays (Figure 5A,B) are typically misinterpreted to represent cancer cell death. However, based on single cell evaluations (Figure 5C and [68,69,70]), as we stated previously ([20,25]), these chemosensitivity assays measure the conversion of dangerous (proliferating) cancer cells to potentially even more dangerous (proliferation arrested) “monsters” (e.g., PGCCs) that are known to repopulate the tumor.
3.3. Common Features of PGCCs and Senescent Cancer Cells
Therapy-induced dormancy in p53 wild-type cancer cells predominantely reflects p21-mediated senescence. Landmark studies reported from the laboratory of Igor Robinson over two decades ago demonstrated that p21 orchestrates this response by transcriptionally upregulating senescence genes and down regulating gene involved in mitosis (reviewd in [71]).
It was originally assumed by us [72] and many other groups that therapy-induced senescence might be a permanent cell fate (i.e., reflecting “reproductive” death [73]). Now we know that: (i) senescence can be reversible, particularly in the absence of p16INK4A (p16) function [71,74]; (ii) engaging regulated cell death via apoptosis in senescent cancer cells (e.g., MCF7 breast carcinoma cells that lack p16 function) can accelerate the reversal of senescence rather than leading to lethality [75]; (iii) senescent cancer cells that emerge following chemotherapy exposure gain survival advantage by frequently engulfing neighboring (senescent or non-senescent) tumor cells [76]; and (iv) senescence can be accompanied by polyploidy/multinucleation and the acquisition of several stem-cell like characteristics [70,77,78].
In the context of cancer therapy, giant cancer cells with a highly enlarged nucleus (e.g., Figure 1C), multiple nuclei, multiple multinuclei, with or without senescence features, together with senescence cells with modest (if any) nuclear abnormalities, exhibit the following common properties: they enter a state of dormancy (and thus might be scored “dead” in various chemosensitivity assays), remain viable, secrete growth promoting factors, and can give rise to rapidly proliferating progeny with stem cell-like properties (reviewed in, e.g., [25]).
4. Rethinking Pro-Apoptotic Strategies to Combat Solid Tumors
Apoptosis is the most extensively studied form of regulated cell death which has been traditionally considered to be irreversible once a particular biochemical point-of-no-return is activated. For educational purposes, Taylor et al. [79] used the analogy of demolishing a building to illustrate the complex process of apoptosis. Demolition of a building needs to be carried out in a safe, precise and orderly manner to avoid damage to surrounding buildings, and also the resulting debris need to be removed to make room to build a new structure. Previously it was assumed that cell demolition via apoptosis follows a similar scenario [79]. In the past 15 years, however, numerous groups have demonstrated that apoptosis is a reversible process, and that triggering cancer cell apoptosis can fuel tumorigenesis.
The processes of cell survival after engaging apoptosis and other modes of regulated cell death are well established and extensively reviewed (e.g., [80,81,82,83,84,85,86,87,88,89,90,91,92,93]). Some key discoveries are outlined below to illustrate the basis for the main purpose of the current review: a paradigm shift in oncology to focus on apoptosis-suppressing (rather than promoting) strategies
4.1. Dark side of Apoptosis in Solid Tumor Therapy
- Caspase 3, which has canonically served as a marker of programmed cell death, is known to promote survival through processes that are called “Phoenix Rising” [82,88,89] or “Failed Apoptosis” [80,83,86]. These processes are initiated by DNA double-strand breaks (e.g., arising in the course of regulated cell death), resulting in activation of stress response signaling pathways (e.g., orchestrated by ATM) and caspase 3-mediated secretion of prostaglandine E2 and other prosurvival factors. (This calls for revisiting thousands of articles that have relied on caspase 3 activation as a marker of cell “lethality.”)
- The term “anastasis” refers to a homeostatic process that enables mammalian cells to recover after engaging regulated cell death [81,84,85,87,94,95]]. Like prosurvival function of caspase 3, anastasis poses an obvious challenge in cancer therapy. Thus, cancer cells triggered to undergo apoptosis (e.g., in response to chemotherapeutic drugs) can recover from late stages of apoptosis (even after formation of apoptotic bodies) [91,94] and give rise to aggressive variants with increased aneuploidy [94,95]. (This calls for revisiting thousands of articles that use the terms “apoptosis” and “lethality” interchangeably!)
- Studies with adenocarcinoma tumor samples have revealed the presence of densely populated apoptotic cells within an individual tumor [96]. Such “apoptotic islands” contribute to intratumor heterogeneity and are associated with cancer cell survival [96]. This process of cancer cell survival - called “Treacherous Apoptosis” by Dhanasekaran [90] - cannot be recapitulated by the conventional preclinical “down” assays listed in Figure 1.
- In a recent report, Park et al [97] demonstrated that apoptotic cancer cells can promote metastasis through a process called nuclear expulsion. This involves the release of chromatin and associated proteins (nuclear expulsion products; NEPs) by apoptotic cells, which bind to neighboring cancer cells and leads to their metastatic outgrowth in an ERK-dependent manner. NEPs were detected in cancer cell lines as well as tumor biopsies from patients with different types of solid tumors. The authors concluded that apoptosis-induced nuclear expulsion might be a generalized phenotype in cancer that underlies metastatic spread. The relationship (if any) between this process and treacherous apoptosis remains to be determined.
- Clinical studies reported since the 1990s have revealed that solid tumors with high apoptotic index tend to have a poor prognosis. The dark side of apoptosis based on clinical outcomes was well documented over a decade ago [98,99,100], and has been fairly recently reviewed for colon cancer [101,102] and breast cancer [103,104,105].
4.2. Cancer Stem Cell Survival after Engaging Apoptosis
Since anastasis is a homeostasis cell recovery process, it is likely to occur all cell types including stem cells. In 2016, Jinesh and coworkers reported an anastasis-like phenomenon of survival in cancer stem cells after engaging apoptosis [106]. One of the typical morphological changes of apoptotic cells is the formation of membrane blebs, which are initially small but their size gradually increases during the progression of apoptosis, eventually leading to formation of apoptotic bodies (large extracellular vesicles), that are subsequently phagocytosed [107]. Apoptotic cancer stem cells were shown to avoid death (phagocytosis) by fusing their blebs to form blebbishield. This intriguing phenomenon (blebbishield emergency program) will undoubtedly attract more attention and its relation to anastasis determined.
4.3. Questions
Collectively, these observations raise two fundamental questions. First, do TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining, Annexin V staining and other apoptosis assays measure cancer cell death in preclinical studies? Second, why are novel anticancer strategies centered on promoting cancer cell apoptosis rather than avoiding/suppressing this “treacherous” response? Perhaps because many authors still consider apoptosis to be a permanent cell fate (e.g., [37,38,39,40,41,42,43,44,45,46]).
5. Exploiting the Apoptotic Threshold for Managing Solid Tumors
5.1. Apoptosis-Promoting Preclinical Chemosensitivity Studies Often Generate Clinically Irrelevant Information
Figure 6 presents a summary of cisplatin-induced responses in human solid tumor-derived cell lines that were reported over a decade ago (reviewed in [108]). The following observations should be noted:
- Cisplatin concentrations between 5 and 10 µM used in cell-based studies are determined to be comparable to concentrations that may be achieved in tumor/tissues of treated patients and tumor-bearing laboratory animals [70]. Higher concentrations result in severe side effects in animal studies [70]. Thus, ~10 µM cisplatin is denoted as the maximum tolerated dose (MTD) in Figure 6.
- The basis for this discrepancy is known [109]. Relatively low concentrations of cisplatin induce sufficient amounts of DNA lesions that inhibit cell proliferation, whereas very high drug concentrations are needed to damage the cytoplasmic compartments to engage apoptosis.
Figure 6.
Apoptotic threshold in human HCT116 colon carcinoma cells treated with the chemotherapeutic drug cisplatin. The apoptotic trend shown by the sold red curve is based on caspase 3 activation reported by Berndtsson et al. [109] in 2006. Since then, other groups have used different assays and confirmed that very high concentrations of cisplatin is required to engage apoptosis in a significant proportion of cells within HCT116 cultures. For example, 60 µM cisplatin induced ~20% apoptosis [111], and ~100 µM cisplatin induced ~30% [112] and ~60% [113] apoptosis in this cell line. In our own work [110], we observed higher levels of apoptosis compared to other groups but, consistent with other groups, we also observed marginal levels of apoptosis at the MTD (~10 µM). These various apoptotic response reported from different laboratories are marked with the two red dashed lines. MTD, maximum tolerated dose.
Figure 6.
Apoptotic threshold in human HCT116 colon carcinoma cells treated with the chemotherapeutic drug cisplatin. The apoptotic trend shown by the sold red curve is based on caspase 3 activation reported by Berndtsson et al. [109] in 2006. Since then, other groups have used different assays and confirmed that very high concentrations of cisplatin is required to engage apoptosis in a significant proportion of cells within HCT116 cultures. For example, 60 µM cisplatin induced ~20% apoptosis [111], and ~100 µM cisplatin induced ~30% [112] and ~60% [113] apoptosis in this cell line. In our own work [110], we observed higher levels of apoptosis compared to other groups but, consistent with other groups, we also observed marginal levels of apoptosis at the MTD (~10 µM). These various apoptotic response reported from different laboratories are marked with the two red dashed lines. MTD, maximum tolerated dose.
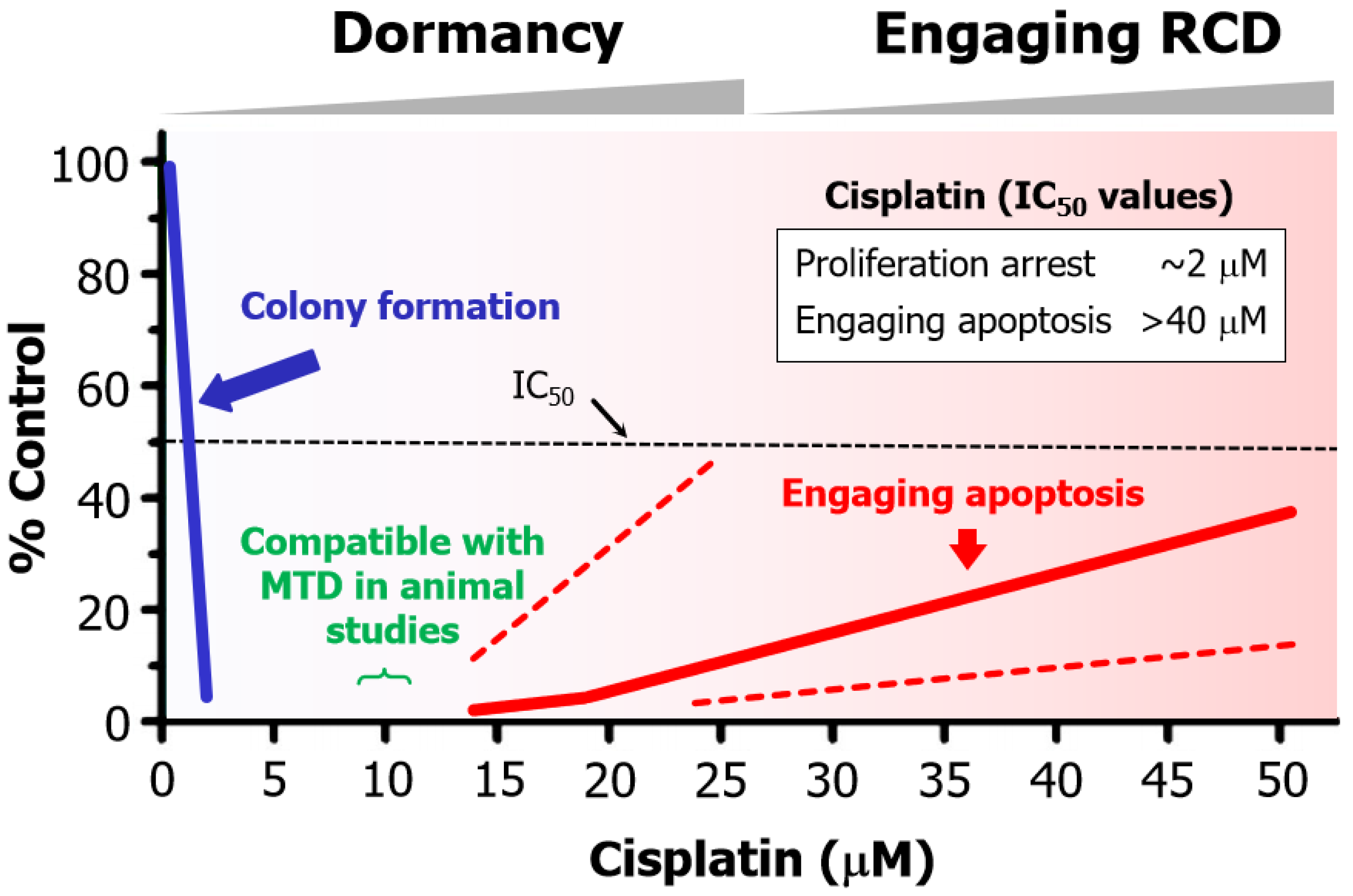
In short, cell “viability” (e.g., 96-well plate colorimetric) and apoptosis studies that call for continuous treatments with very high concentrations of chemotherapeutic drugs (e.g., >10 µM cisplatin) to achieve the IC50 values have probably generated clinically irrelevant information. The danger of relying on such assays for chemosensitivity assessment has been discussed by us [20,24,84] and others [85,114,115]
5.2. Chemotherapy-Induced Cancer Cell Dormancy: lesser Evil Than Apoptosis?
The difference in drug concentrations that are required to induce proliferation arrest versus apoptosis is striking. We have presented the trend for cisplatin (Figure 6) and oxaliplatin [116], but similar differences are reported for virtually all anticancer agents.These observations led us to suggest that, in the absence of a “magic bullet” for treating cancer, perhaps cancer cell dormancy might be a more favorable clinical outcome than apoptosis simply because relatively low/moderate doses of therapeutic agents that are required to trigger dormancy “will undoubtedly cause less unwanted side effects, such as compromising patient’s immune system, as compared to high doses required to induce apoptosis” [117].
6. Conclusions
6.1. Preclinical “Down” Assays have Caused More Harm Than Benefit
For nearly half a century, most cancer researchers and oncologists have relied on the ubiquitous use of “down” assays (e.g., decreased proliferation, decreased tumor growth, etc.) in concert with the omics technologies to draw conclusions of clinical relevance. Despite generating huge amounts of bewildering information, modern therapeutic strategies under the term precision oncology have not fulfilled their promises for treating patients with solid tumors.
Such information-generating strategies, which includes targeting p53, are largely centered on the hypothesis that engaging apoptosis in cancer cells will eventually lead to their demise. This is surprising because clinical studies published since the 1990s have revealed that this rather simplistic hypothesis is not tenable. Consistent with these clinical observations, in the past decade numerous authors have demonstrated that cancer cells not only secrete pro-survival factors, which is mediated by caspase 3, but also exhibit the ability to recover from late stages of apoptosis (even after formation of apoptotic bodies) via the homeostatic process of anastasis.
A paradigm shift is urgently needed in terms of how potential anticancer drugs are identified and validated (i.e., by single cell techniques and not the conventional “down” assays) as well as the rationale for this endeavor (i.e., suppressing, rather than promoting regulated cell death). Exploiting the apoptotic threshold (Figure 6) and the antiapoptotic property of wild-type p53 signaling [25,118] might prove to be instrumental in this regard.
This paradigm shift needs to encompass cancer cells with chaotic genome in general, and PGCCs in particular. To this end, it is curious that these tumor repopulating giants still continue to be ignored by the majority of precision oncology community members. “Are misleading/inappropriate preclinical assays to be blamed” [24]? If so, then it is reasonable to conclude that widely used preclinical “dawn” assays have derailed cancer research for decades.
6.2. Conflicting Reports on Cancer Cell Fate After Engaging Apoptosis
Comprehensive review articles have been published by two members of the nomenclature committee on cell death (NCCD), Douglas Green and associates [92] and Boris Zhivotovsky and associates [85], as well as other authors (e.g., [93]), in which they have extensively discussed the mechanisms of cancer cell survival after engaging regulated cell death. These articles have concluded that “…the goal of most cancer therapies (to induce regulated cell death in cancer cells) directly brings about the cellular changes that we seek to avoid” [92], “…the identification of the anastasis phenomenon and the accumulated data on it indicate the importance of developing a new direction in the study of tumor resistance” [85], and “…while we know that cells respond differently to caspase activation, we still don’t fully understand where this heterogeneity comes from…what is the point of no return in apoptotic commitment?” [93].
On the other hand, a number of other comprehensive reviews on apoptosis in cancer [37,38,39,40,41,42] and related topics (e.g., p53-based therapies [43,44,45,46,47], synthetic “lethality” [48,49,50,51,52,53,54]) have been recently published in high impact factor journals that completely ignore the dark side of apoptosis in cancer therapy. These include a recent Nature review article published by the NCCD which states (under the FACTS) that “In mammalian organisms, executioner caspases are activated after cells are already committed to die” [37]. What is the reason that the NCCD members, which includes over 300 world-class experts, have provided an impressive account of apoptotic pathways in relation to cancer (and other diseases) in a literal “dictionary-style” article, but have left out fundamental responses listed in Figure 1 (anastasis, Phoenix Rising, etc) that are known to underlie cancer resistance and disease recurrence? “I wouldn’t pretend to know.” (This quote is from Weinberg's 2014 Leading Edge Essay [22] when he was discussing how this data-generating dilemma of cancer research will play out.)
6.3. Intratumor Heterogeneity: How Complex Does it Get?
Therapy-resistance responses of cancer cells are not limited to those discussed in the previous sections. The immune system, for example, plays an important role in therapy outcome [24], which underscores the limitations of preclinical anticancer studies that reply on immune compromised animals (e.g., nude mice). Other therapy-resistance responses that contribute to intratumor heterogeneity have been discussed. These include: (i) atavistic reprogramming, which refers to the activation of ancestral resistance genes [24], some of which might contribute to cancer cell escape from immune editing; (ii) cell fusion [20], which is known to promote the creation of PGCCs and facilitate their migration []; (iii) cancer cell dormancy reflecting drug-tolerant persister cells [20]; and (iv) the ability of p21 to promote cancer cell survival following clinically relevant chemotherapy exposure through a process called the “p21 Goldilocks Zone for Proliferation” [119].
6.4. Where Will All This Lead?
Just thinking about all these complex processes makes ones head spin, but unfortunately this is the established reality. We will be fooling ourselves if we continue to “reduce” this mind-numbing complexity to highly simplistic hypotheses, such as the notion that cancer cells treated with therapeutic agents either repair their genome and survive, or die via apoptosis and other means.
Time will tell whether there will be a significant improvement in solid tumor therapy, or whether precision oncology will remain an illusion as predicted by Prasad few years ago [1]. Until then, hopefully taking heed of advice of numerous pioneers in cancer research, including those noted herein will prevent the undertaking of unnecessary clinical trials to test flawed strategies based on faulty hypotheses.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
Appendix A
Figure A1.
take home messages (FACTS) of the studies discussed in the current review.

References
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [CrossRef]
- Szabo, L. Cancer treatment hype gives false hope to many patients. Kaiser Health News (2017)https://www.usatoday.com/story/news/2017/04/27/cancer-treatment-hype-gives-false-hope-many-patients/100972794/ (accessed on 15 April 2024).
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11.
- Joyner, M.J.; Paneth, N. Promises, promises, and precision medicine. J. Clin. Invest. 2019, 129, 946–948.
- Marine, J.-C.; Dawson, S.-J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [CrossRef]
- Pich, O.; Bailey, C.; Watkins, T.B.K.; Zaccaria, S.; Jamal-Hanjani, M.; Swanton, C. The translational challenges of precision oncology. Cancer Cell 2022, 40, 458–478. [CrossRef]
- Heng, J.; Heng, H.H. Genome chaos, information creation, and cancer emergence: Searching for new frameworks on the 50th anniversary of the “war on cancer”. Genes 2022, 13, 101.
- Lohse, S. Mapping uncertainty in precision medicine: A systematic scoping review. J. Eval. Clin. Pract. 2023, 29, 554–564. [CrossRef]
- Fojo, T. Journeys to failure that litter the path to developing new cancer therapeutics. JAMA Netw. Open 2023, 6, e2324949. [CrossRef]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 2024, 14, 49–65. [CrossRef]
- Kailen, W.G. Preclinical cancer target validation: How not to be wrong. NIH Wednesday Afternoon Lectures (WELS) series, January 24, 2018 [https://videocast.nih.gov/watch=27066] (accessed on 15 April 2024).
- Belluz, J. Most cancer drugs fail in testing. This might be a big reason why. Science _VOX blog (2019) https://www.vox.com/2019/9/16/20864066/cancer-studies-fail (accessed on 15 April 2024).
- Horgan, J. The cancer industry: hype vs. reality. Cancer medicine generates enormous revenues but marginal benefits for patients (2020) https://www.scientificamerican.com/blog/cross-check/the-cancer-industry-hype-vs-reality/ (accessed 15 April 2024).
- Horgan, J. The cancer industry: Hype versus reality (2023) https://johnhorgan.org/cross-check/the-cancer-industry-hype-versus-reality (accessed on 15 April 2024).
- Azra, R. The First Cell: And the Human Costs of Pursuing Cancer to the Last. New York, Basic Books, 2019.
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta. Pharm. Sin. B 2022, 12, 3049−3062.
- Sadri, A. Is target-based drug discovery efficient? Discovery and “off-target” mechanisms of all drugs. J. Med. Chem. 2023, 66, 12651−12677. [CrossRef]
- Bruin, M.A.C.; Sonke, G.S.; Beijnen, J.H.; Huitema, A.D.R. Pharmacokinetics and harmacodynamics of PARP inhibitors in oncology. Clin, Pharmacokinet. 2022, 61, 1649–1675.
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [CrossRef]
- Mirzayans, R. When therapy-induced cancer cell apoptosis fuels tumor relapse. Onco 2024, 4, 37–45. [CrossRef]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [CrossRef]
- Kailen, W.G. Publish Houses of Brick, not Mansions of Straw. Nature 2017, 5454, 387.
- Mirzayans, R.; Murray, D. What are the reasons for continuing failures in cancer therapy? Are misleading/inappropriate preclinical assays to be blamed? Might some modern therapies cause more harm than benefit? Int. J. Mol. Sci. 2022, 23, 13217.
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and treatment resistance of solid tumors with a focus on polyploid/senescent giant cancer cells (PGCCs). Int. J. Mol. Sci. 2023, 24, 11534. [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [CrossRef]
- Uversky, V.N. p53 proteoforms and intrinsic disorder: An illustration of the protein structure-function continuum concept. Int. J. Mol. Sci. 2016, 17, 1874. [CrossRef]
- Ranjan, A.; Iwakuma, T. Emerging non-canonical functions and regulation of p53. Int. J. Mol. Sci. 2018, 19, 1015. [CrossRef]
- Horvat, A.; Tadijan, A.; Vlaši´c, I.; Slade, N. p53/p73 Protein network in colorectal cancer and other human malignancies. Cancers 2021, 13, 2885.
- Montero-Calle, A.; Garranzo-Asensio, M.; Torrente-Rodríguez, R.M.; Ruiz-Valdepeñas Montiel, V.; Poves, C.; Dziaková, J.; Sanz, R.; Díaz del Arco, C.; Pingarrón, J.M.; Fernández-Aceñero, M.J.; et al. p53 and p63 Proteoforms Derived from Alternative Splicing Possess Differential Seroreactivity in Colorectal Cancer with Distinct Diagnostic Ability from the Canonical Proteins. Cancers 2023, 15, 2102. [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [CrossRef]
- Su, Y.; Sai, Y.; Zhou, L.; Liu, Z.; Du, P.; Wu, J.; Zhang, J. Current insights into the regulation of programmed cell death by TP53 mutation in cancer. Front Oncol. 2022, 12, 1023427.
- Levine, A.J.; Berger, S.L.The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [CrossRef]
- Sandoval, J.E.; Reich, N.O. p53 and TDG are dominant in regulating the activity of the human de novo DNA methyltransferase DNMT3A on nucleosomes. J. Biol. Chem. 2021, 296, 100058. [CrossRef]
- Murray, D.; Mirzayans, R.; McBride, W.H. Defenses against pro-oxidant forces—Maintenance of cellular and genomic integrity and longevity. Radiat. Res. 2018, 190, 331–349. [CrossRef]
- Markowska, M.; Budzinska, M.A.; Coenen-Stass, A.; Kang, S.; Kizling, E.; Kolmus, K.; Koras, K.; Staub, E.; Szczurek, E. Synthetic lethality prediction in DNA damage repair, chromatin remodeling and the cell cycle using multi-omics data from cell lines and patients. Sci Rep. 2023, 13, 7049. [CrossRef]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis. P.; Alnemri. E.S.; Altucci, L.; et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097−1154. [CrossRef]
- Biswas, U.; Roy, R.; Ghosh, S.; Chakrabarti, G. The interplay between autophagy and apoptosis: its implication in lung cancer and therapeutics. Cancer Lett. 2024, 585, 21666. [CrossRef]
- Kim, R.; Kin, T.; Beck, W.T. Impact of complex apoptotic signaling pathways on cancer cell sensitivity to therapy. Cancers 2024, 16, 984. [CrossRef]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell death. Cell 2024, 187, 235−256.
- Kayagaki, N.; Webster, J.D.; Newton, K. Control of cell death in health and disease. Annu. Rev. Pathol. 2024, 19, 157−180. [CrossRef]
- Kulbay, M.; Paimboeuf, A.; Ozdemir, D.; Bernier, J. Review of cancer cell resistance mechanisms to apoptosis and actual targeted therapies. J. Cell. Biochem. 2022; 123, 1736–1761. [CrossRef]
- Tuval, A.; Strandgren, C.; Heldin, A.; Palomar-Siles, M.; Wiman, K.G. Pharmacological reactivation of p53 in the era of precision anticancer medicine. Nat. Rev. Clin. Oncol. 2024, 21, 106–120. [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [CrossRef]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death and Dis. 2022, 13, 974. [CrossRef]
- Levine, A.J. Targeting the p53 protein for cancer therapies: The translational impact of p53 research. Cancer Res. 2022, 82, 362–364. [CrossRef]
- Brown, D.W.; Beatty, P.H.; Lewis, J.D. Molecular targeting of the most functionally complex gene in precision oncology: p53. Cancers 2022, 14, 5176. [CrossRef]
- Murai, J.; Pommier, Y. BRCAness, homologous recombination deficiencies, and synthetic lethality. Cancer Res. 2023, 83, 1173–1174.
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94. [CrossRef]
- Cong, K.; Cantor, S.B. Exploiting replication gaps for cancer therapy. Mol. Cell 2022, 82, 2363–2369. [CrossRef]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [CrossRef]
- Lord. C.J.; Ashworth, A. PARP Inhibitors: The first synthetic lethal targeted therapy. Science 2017, 355, 1152–1158.
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613-623.
- Heng, J.; Heng, H.H. Genome chaos, information creation, and cancer emergence: searching for new frameworks on the 50th anniversary of the “war on cancer”. Genes 2022, 13, 101.
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press: Cambridge, MA, USA, 2019; p. 556.
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and genome chaos: Changing the system inheritance. Genes 2019, 10, 366. [CrossRef]
- Shapiro, J.A. How chaotic is genome chaos? Cancers 2021, 13, 1358.
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [CrossRef]
- 60Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [CrossRef]
- Coward, J.; Harding, A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 2014, 4, 123. [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid giant cancer cells (PGCCs): The evil roots of cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [CrossRef]
- Dudkowska, M.; Staniak, K.; Bojko, A.; Sikora, E. Chapter Five - The role of autophagy in escaping therapy-induced polyploidy/senescence Author links open overlay panel. Adv. Cancer Res. 2021, 150, 209–247.
- Zhang, J.; Qiao, Q.; Xu, H.; Zhou, R.;Liu, X. Human cell polyploidization: The good and the evil. Semin. Cancer Biol. 2022, 81, 54–63. [CrossRef]
- Liu, J.; Niu, N.; Li, X.; Zhang, X.; Sood, A.K. The life cycle of polyploid giant cancer cells and dormancy in cancer: Opportunities for novel therapeutic interventions. Semin. Cancer Biol. 2022, 81, 132–144. [CrossRef]
- Trabzonlu, L.; Pienta, K.J.; Trock, B.J.; De Marzo, A.M.; Amend. S.R. Presence of cells in the polyaneuploid cancer cell (PACC) state predicts the risk of recurrence in prostate cancer. Prostate 2023, 83, 277–285. [CrossRef]
- Weng, C.H.; Wu, C.S.; Wu, J.c.; Kung, M. L; Wu, M.H.; Tai, M.H. Cisplatin-induced giant cells formation is involved in chemoresistance of melanoma cells. Int. J. Mol. Sci. 2020, 21, 7892. [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Impact of chemotherapeutic drugs on cancer cell proliferation, morphology and metabolic activity. J. Cancer Biol. Res. 2018, 6, 1118.
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [CrossRef]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715.
- Barley, R.D.C.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fifibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543.
- Brown, J.M.; Wouters, B.G. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999, 59, 1391–1399.
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joe,l S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–83.
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [CrossRef]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; Jackson, J.G. hemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844.
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. The. 2018, 19, 63–75. [CrossRef]
- Czarnecka-Herok, J.; Sliwinska, M.A.; Herok, M.; Targonska, A.; Strzeszewska-Potyrala, A.; Bojko, A.; Wolny, A.; Mosieniak, G.; Sikora, E. Therapy-induced senescent/polyploid cancer cells undergo atypical divisions associated with altered expression of meiosis, spermatogenesis and EMT genes. Int. J. Mol. Sci. 2022, 23, 8288. [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [CrossRef]
- Ichim, G.; Tait, S.W.G. A fate worse than death: apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [CrossRef]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442.
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.-Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [CrossRef]
- Berthenet, K.; Castillo Ferrer, C.; Fanfone, D.; Popgeorgiev, N.; Neves, D.; Bertolino, P.; Gibert, B.; Hernandez-Vargas, H.; Ichim, G. Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep. 2020, 31, 107731. [CrossRef]
- Mirzayans, R.; Murray, D. Do TUNEL and other apoptosis assays detect cell death in preclinical studies? Int. J. Mol. Sci. 2020, 21, 9090.
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return journey from cell death. Cancers 2021, 13, 3671. [CrossRef]
- Castillo Ferrer, C.; Berthenet, K.; Ichim, G. Apoptosis—Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463.
- Mohammed, R.N.; Khosravi, M.; Rahman, H.S.; Adili, A.; Kamali, N.; Soloshenkov, P.P.; Thangavelu, L.; Saeedi, H.; Shomali, N.; Tamjidifar, et. al. Anastasis: cell recovery mechanisms and potential role in cancer. Cell Commun. Signal. 2022, 20, 81.
- Corsi, F.; Capradossi, F.; Pelliccia, A.; Briganti, S.; Bruni, E.; Traversa, E.; Torino, F.; Reichle, A.; Ghibelli, L. Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (CRAC): A simple in vitro model of Phoenix Rising in prostate cancer. Int. J. Mol. Sci. 2022, 23, 1152. [CrossRef]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [CrossRef]
- Dhanasekaran, R. Treacherous apoptosis—Cancer cells sacrifice themselves at the altar of heterogeneity. Hepatolog. 2022, 76, 549–550. [CrossRef]
- Jinesh, G.G.; Brohl, A.S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 2022, 7, 296. [CrossRef]
- Kalkavan, H.; Rühl, S.; Shaw, J.J.P.; Green, D.R. Non-lethal outcomes of engaging regulated cell death pathways in cancer. Nat. Cancer 2023, 4, 795–806. [CrossRef]
- Nano, M.; Montell, D.J. Apoptotic signaling: Beyond cell death. Semin, Cell Dev. Biol. 2024, 156, 22–34. [CrossRef]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [CrossRef]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43.
- Khatib, S.A.; Ma, L.; Dang, H.; Forgues, M.; Chung, J.-Y.; Ylaya, K.; Hewitt, S.M.; Chaisaingmongkol, J.; Rucchirawat, M.; Wang, X.W. Single-cell biology uncovers apoptotic cell death and its spatial organization as a potential modifier of tumor diversity in HCC. Hepatology 2022, 76, 599–611. [CrossRef]
- Park, W.Y.; Gray, J.M.; Holewinski, R.J.;Andresson, T.; So, J.S.; Carmona-Rivera, C.; Hollander, M.C.; Yang, H.H.; Lee, M.; Kaplan, M.J.; et al. Apoptosis-induced nuclear expulsion in tumor cells drives S100a4-mediated metastatic outgrowth through the RAGE pathway. Nat. Cancer 2023, 4, 419–435. [CrossRef]
- Shekhar, M.P.V. The dark side of apoptosis. In Molecular Mechanisms of Tumor Cell Resistance to Chemotherapy, Resistance to Targeted Anti-Cancer Therapeutics 1; Bonavida, B., Ed.; Springer: New York, NY, USA, 2013; pp. 245–258.
- Wang, R.A.; Li, Q.L.; Li, Z.S.; Zheng, P.J.; Zhang, H.Z.; Huang, X.F.; Chi, S.M.; Yang, A.G.; Cui, R. Apoptosis drives cancer cells proliferate and metastasize. J. Cell. Mol. Med. 2013, 17, 205–211. [CrossRef]
- Wang, R.A.; Li, Z.S.; Yan, Q.G.; Bian, X.W.; Ding, Y.Q.; Du, X.; Sun, B.C.; Zhang, X.H. Resistance to apoptosis should not be taken as a hallmark of cancer. Chin. J. Cancer 2014, 33, 47–50. [CrossRef]
- Huang, Q.; Li, S.; Cheng, I.; Deng, M.; He, X.; Wang, Z.; Yang, C.H.; Zhao, X.Y.; Huang, J. High expression of anti-apoptotic protein Bcl-2 is a good prognostic factor in colorectal cancer: Result of a metaanalysis.World J. Gastroenterol. 2017, 23, 5018–5033. [CrossRef]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of Caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [CrossRef]
- Pu, X.; Storr, S.J.; Zhang, Y.; Rakha, E.A.; Green, A.R.; Ellis, I.O.; Martin, S.G. Caspase-3 and caspase-8 expression in breast cancer: caspase-3 is associated with survival. Apoptosis 2017, 22, 357–368. [CrossRef]
- Lindner, A.U.; Lucantoni, F.; Varešlija, D.; Resler, A.; Murphy, B.M.; Gallagher, W.M.; Hill, a.D.K.; Young, L.S.; Prehn, J.H.M. Low cleaved caspase-7 levels indicate unfavourable outcome across all breast cancers. Mol. Med. 2018, 96, 1025–1037. [CrossRef]
- Yang, X.; Zhong, D.N.; Qin, H.; Wu, P.R.; Wei, K.L.; Chen, G.; He, R.Q.; Zhong, J.C. Caspase-3 over-expression is associated with poor overall survival and clinicopathological parameters in breast cancer: a meta-analysis of 3091 cases. Oncotarget 2018, 9, 8629–8641. [CrossRef]
- Jinesh, G. G.; Kamat, A. M. Endocytosis and serpentine filopodia drive blebbishield-mediated resurrection of apoptotic cancer stem cells. Cell Death Disco. 2016, 2, 15069. [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [CrossRef]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The growing complexity of cancer cell response to DNA-damaging agents: Caspase 3 mediates cell death or survival? Int. J. Mol. Sci. 2016, 17, 708.
- Berndtsson, M.; Hägg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int. J. Cancer 2007, 120, 175–180. [CrossRef]
- 1Murray, D.; Mirzayans, R. Cellular responses to platinum-based anticancer drugs and UVC: Role of p53 and implications for cancer therapy. Int. J. Mol. Sci. 2020, 21, 5766. [CrossRef]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.Q.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, A.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [CrossRef]
- Barkinge, J.L.; Gudi, R.; Sarah, H.; Chu, F.; Borthakur, A.; Prabhakar, B.S.; Prasad, K.V.S. The p53 induced Siva-1 plays a signifi cant role in cisplatin-induced apoptosis. J. Carcinog. 2009, 8, 2.
- Zhang, H.; Sun, J.; Ma, R.; Zhao, S. Role of episamarcandin in promoting the apoptosis of human colon cancer HCT116 cells through the PI3K-Akt signaling pathway. Evid. Based Complement. Alternat. Med. 2021, 2021, 9663738. [CrossRef]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [CrossRef]
- Nicoletto, R.E.; Ofner, C.M. Cytotoxic mechanisms of doxorubicin at clinically relevant concentrations in breast cancer cells. Cancer Chemother. Pharmacol. 2022, 89, 285–311. [CrossRef]
- Gourdier, I.; Crabbe, L.; Andreau, K.; Pau, B.; Kroemer, G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene 2004, 23, 7449–7457. [CrossRef]
- Mirzayans, R. When Therapy-Induced Cancer Cell Apoptosis Fuels Tumor Relapse. Onco. 2024, 4, 37–45. [CrossRef]
- Jänicke, R.U.; Sohn, D.; Schulze-Osthoff, K. The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death Differ. 2008, 15, 959–976. [CrossRef]
- Hsu, C.H.; Altschuler, S.J.; Wu, L.F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 2019, 178, 361–373. [CrossRef]
Figure 1.
Dark side of apoptosis in treating solid tumor malignancies based on preclinical studies (left) and the outcome of clinical studies (right), which pose the indicated question.
Figure 1.
Dark side of apoptosis in treating solid tumor malignancies based on preclinical studies (left) and the outcome of clinical studies (right), which pose the indicated question.
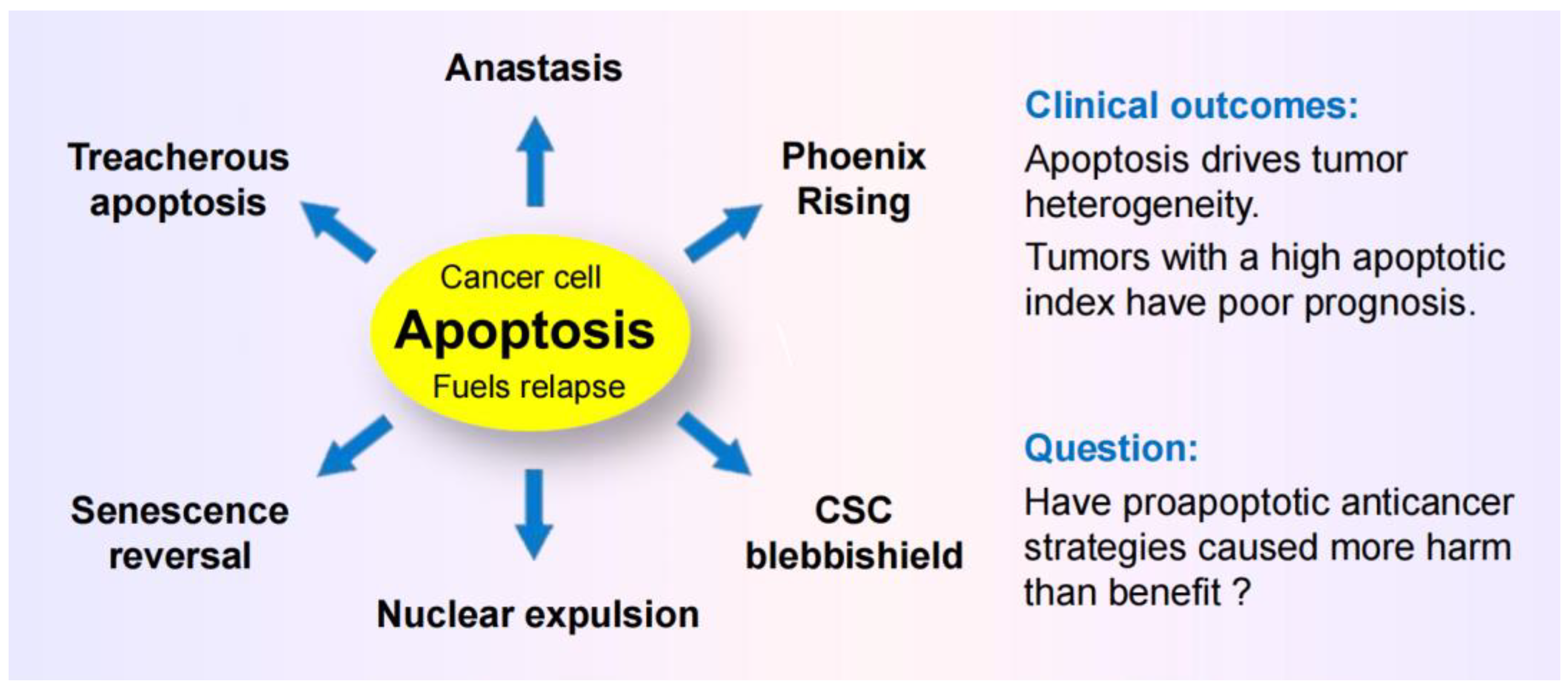
Figure 2.
Preclinical “down” assays that are widely used to identify novel anticancer drugs and treatment strategies (left) measure averaged responses of a large number of cells. Such assays are not designed to recapitulate the complexity and heterogeneity that exists within a tumor in terms of therapy resistance (right).
Figure 2.
Preclinical “down” assays that are widely used to identify novel anticancer drugs and treatment strategies (left) measure averaged responses of a large number of cells. Such assays are not designed to recapitulate the complexity and heterogeneity that exists within a tumor in terms of therapy resistance (right).
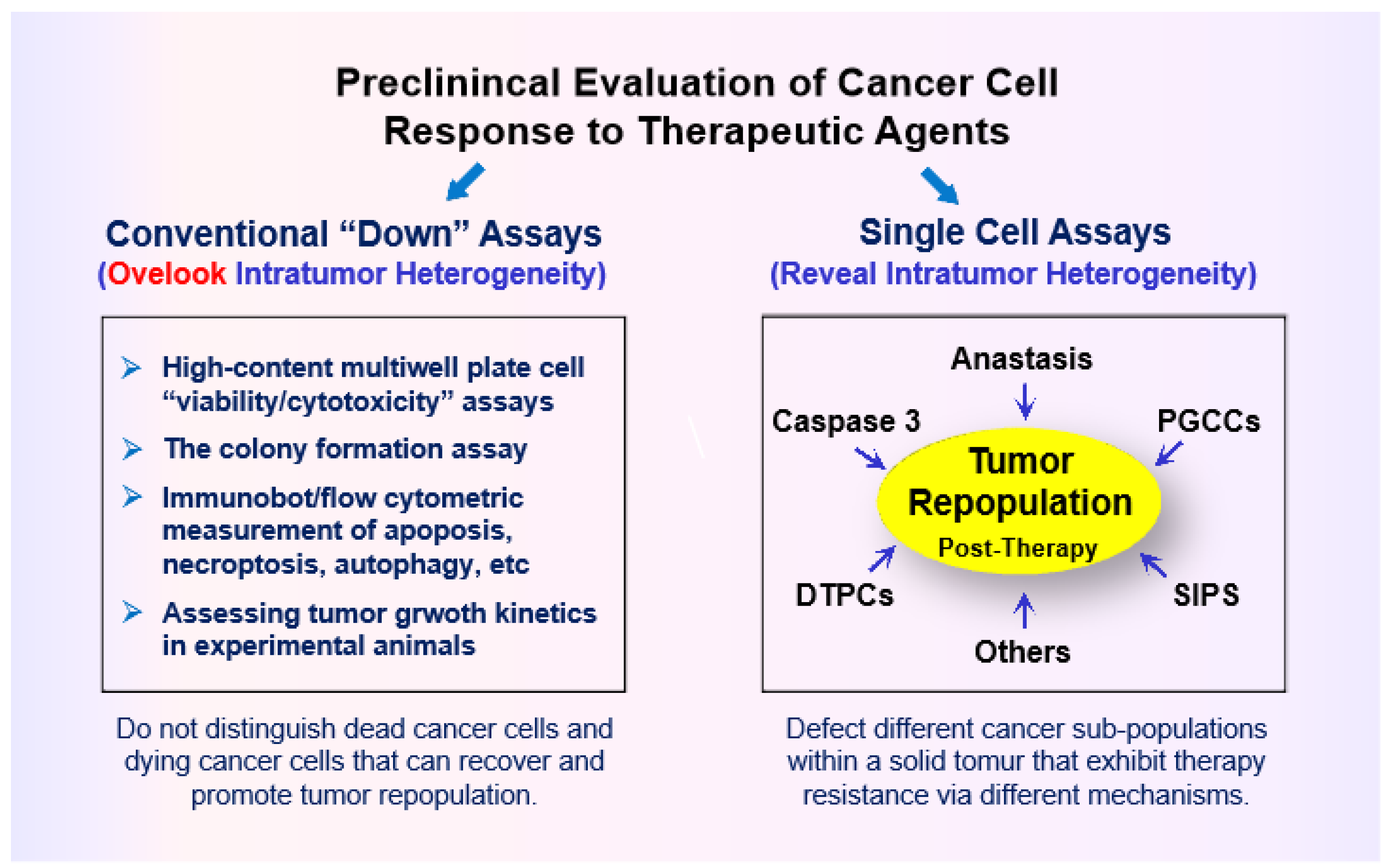
Figure 3.
(A) Some p53 interacting proteins that create the p53 “firework” (free online available information presented in [32]). (B) Multiple functions of p21. CDK, cyclin-dependent kinase; ASK, apoptosis signal-regulating kinase; DNMT1. DNA methyltransferase 1.
Figure 3.
(A) Some p53 interacting proteins that create the p53 “firework” (free online available information presented in [32]). (B) Multiple functions of p21. CDK, cyclin-dependent kinase; ASK, apoptosis signal-regulating kinase; DNMT1. DNA methyltransferase 1.
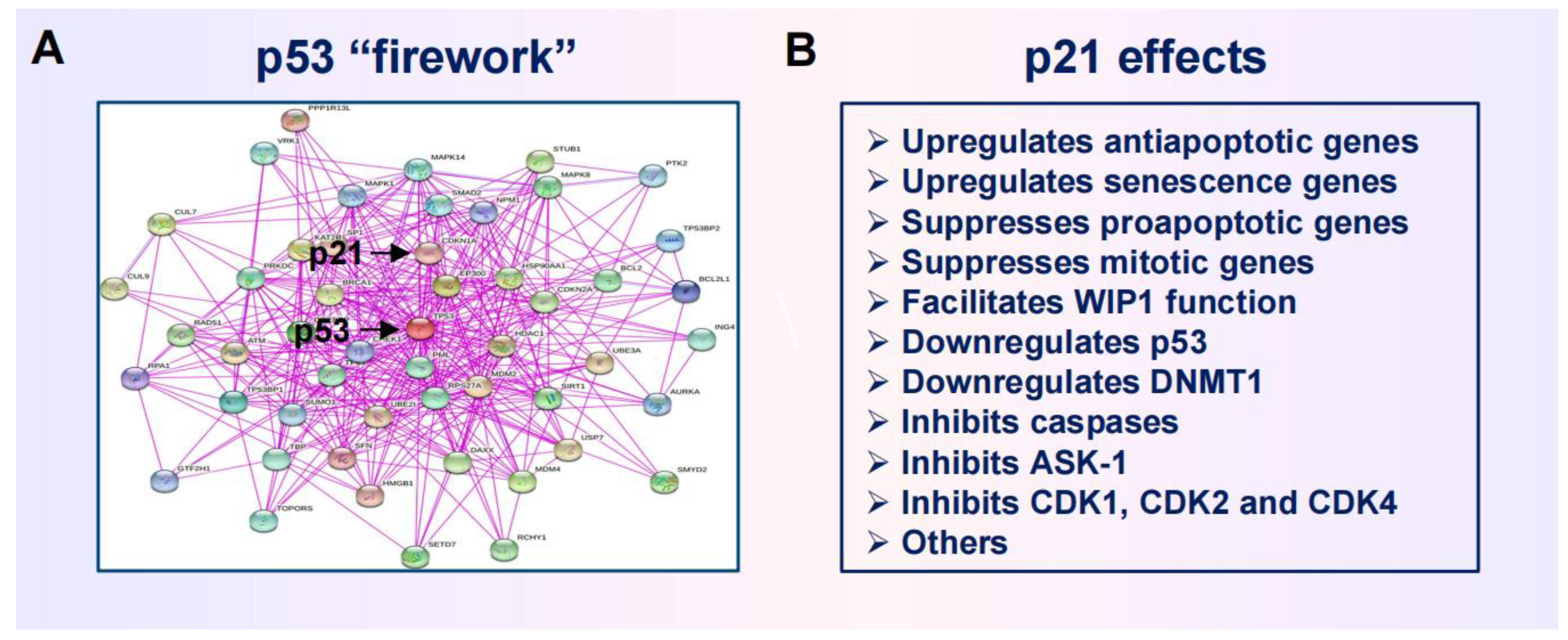
Figure 3.
Cartoon illustrating the development and fate of polyploid giant cancer cells (PGCCs), containing a highly enlarged nucleus or mutiple nuclei. PGCCS often enter a state of dormancy after they are formed, but remain viable and secrete growth promoting factors. PGCCs can also give rise to therapy resistant and tumor repopulating progeny through amitotic cell division, depolyploidization, and sub-genome transmission of nuclear material into surrounding cells. For further details, see [25].
Figure 3.
Cartoon illustrating the development and fate of polyploid giant cancer cells (PGCCs), containing a highly enlarged nucleus or mutiple nuclei. PGCCS often enter a state of dormancy after they are formed, but remain viable and secrete growth promoting factors. PGCCs can also give rise to therapy resistant and tumor repopulating progeny through amitotic cell division, depolyploidization, and sub-genome transmission of nuclear material into surrounding cells. For further details, see [25].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



