
Submitted:
18 April 2024
Posted:
23 April 2024
You are already at the latest version
Abstract
p-Toluenesulfonic acid is commercially available, non-toxic, cheap organic compound with robust stability. p-Toluenesulfonic acid is solid, non-corrosive and a strong organic acid that can be handled easily. Due to this p-toluenesulfonic acid could be the better replacement of other conventional acids like nitric acid, sulfuric acid, perchloric acid and AlCl3 in organic transformations. Moreover, p-toluenesulfonic acid could be used synergistically with other mode of catalysis like transition metal catalysis and photocatalysis. In recent years, the use of this solid organic acid in organic and polymer synthesis has grown tremendously. In this review, we present an overview of p-toluenesulfonic acid promoted various organic reactions with particular emphasis to the more recently reported methods. In some cases, we also detailed the understanding of the reactions by providing the reaction mechanism. Despite these advances, there are still many opportunities for the development of new reactions using PTSA. Industry and academia have become more conscious to adopt sustainable chemical processes using safer reagents and solvents and the use of PTSA in this context is highly significant. This review will inspire researchers for further reaction development employing p-toluenesulfonic acid.
Keywords:
p-toluenesulfonic acid
; tosic acid
; catalytic
; organic reactions
; multicomponent reactions
1. Introduction
p-Toluenesulfonic acid has attracted much attention over the years as a catalyst for various organic transformations due to its superior stability, air and water compatibility, non-toxicity, ease of operation, ready availability, inexpensiveness, and strong organic acidic nature. p-Toluenesulfonic acid is an organic compound with molecular formula CH3C6H4SO3H, and it is abbreviated as PTSA, Tosh or p-TSA. It is a white solid that is soluble in water, alcohols, and other polar organic solvents. Generally, TsOH refers to the monohydrate, TsOH. H2O. p-Toluenesulfonic acid is a strong organic acid, approaximately a million times stronger than benzoic acid. It is one of the few strong acids that is solid and can be conveniently weighed. Unlike other strong mineral acids (especially nitric acid, sulfuric acid, and perchloric acid), TsOH is non-oxidizing. PTSA is an environmentally benign, non-volatile, recyclable solid acid catalyst. Solid acid catalysts have been widely used in different industrial purposes. Still a significant number of acid catalyzed reactions are carried out using conventional acids, such as H2SO4 and AlCl3 and the processes are typically associated with problems of high toxicity, corrosion, and use of large amounts of catalyst. So, the replacement of liquid acids with environmentally friendly solid acids is highly desirable.
Herein we wish to demonstrate the application of this versatile and powerful acid in plethora of organic reactions leading to the formation of bioactive molecules and useful building blocks. To the best of our knowledge, so far only one review article is available in the literature, [1] date back to 2011. We believe that this is high time to summarize the application of p-toluenesulfonic acid in organic transformations. The review is divided into four section: 1. application of PTSA as a catalyst; 2. application of PTSA as a reagent; 3. application of PTSA in combination with metal salts and other reagents; 4. application of PTSA in ionic liquids
2. Advances of p-Toluenesulfonic Acid Promoted Reactions in Organic and Polymer Chemistry
2.1. Application of p-Toluenesulfonic Acid as a Catalyst
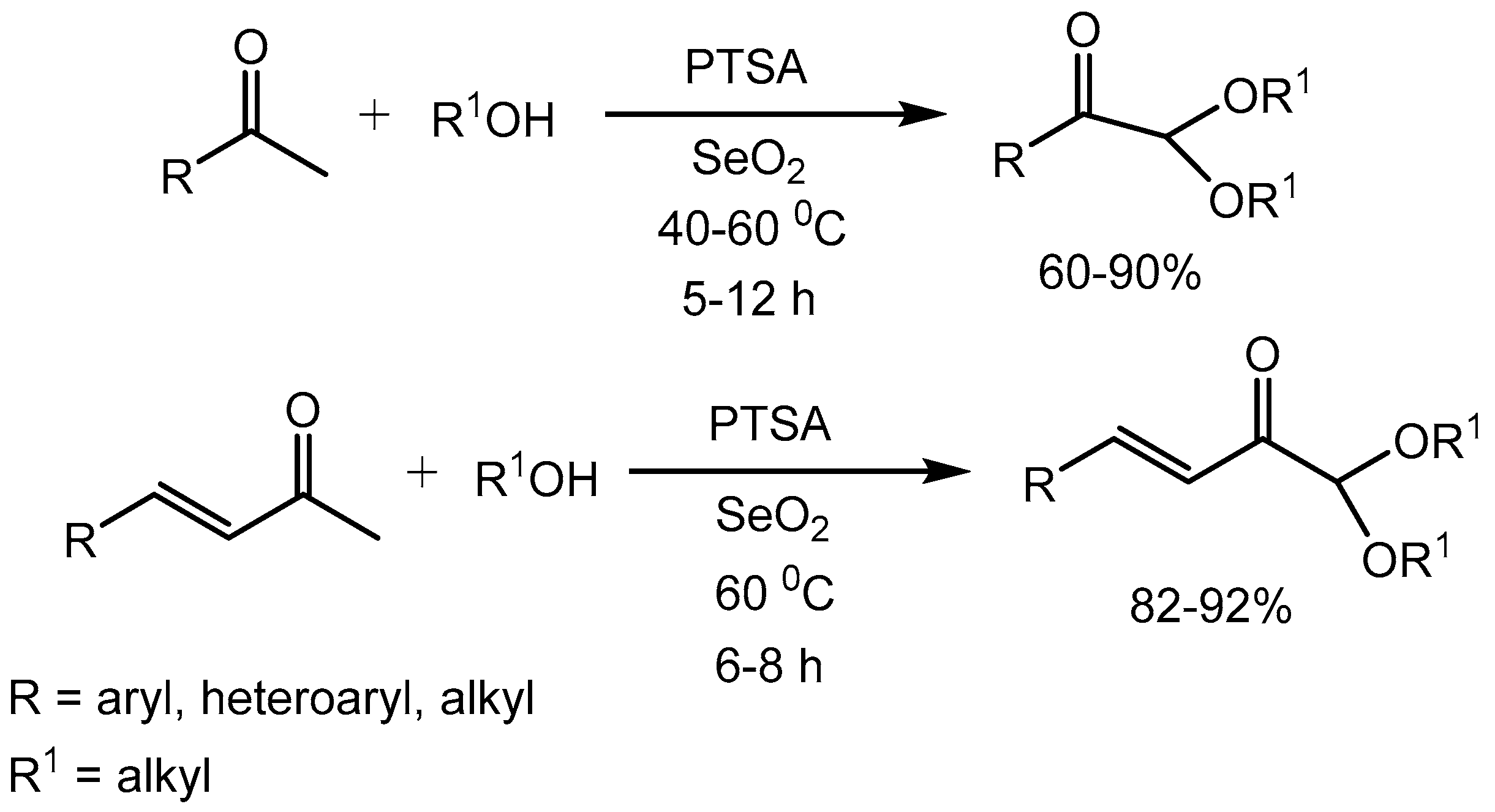
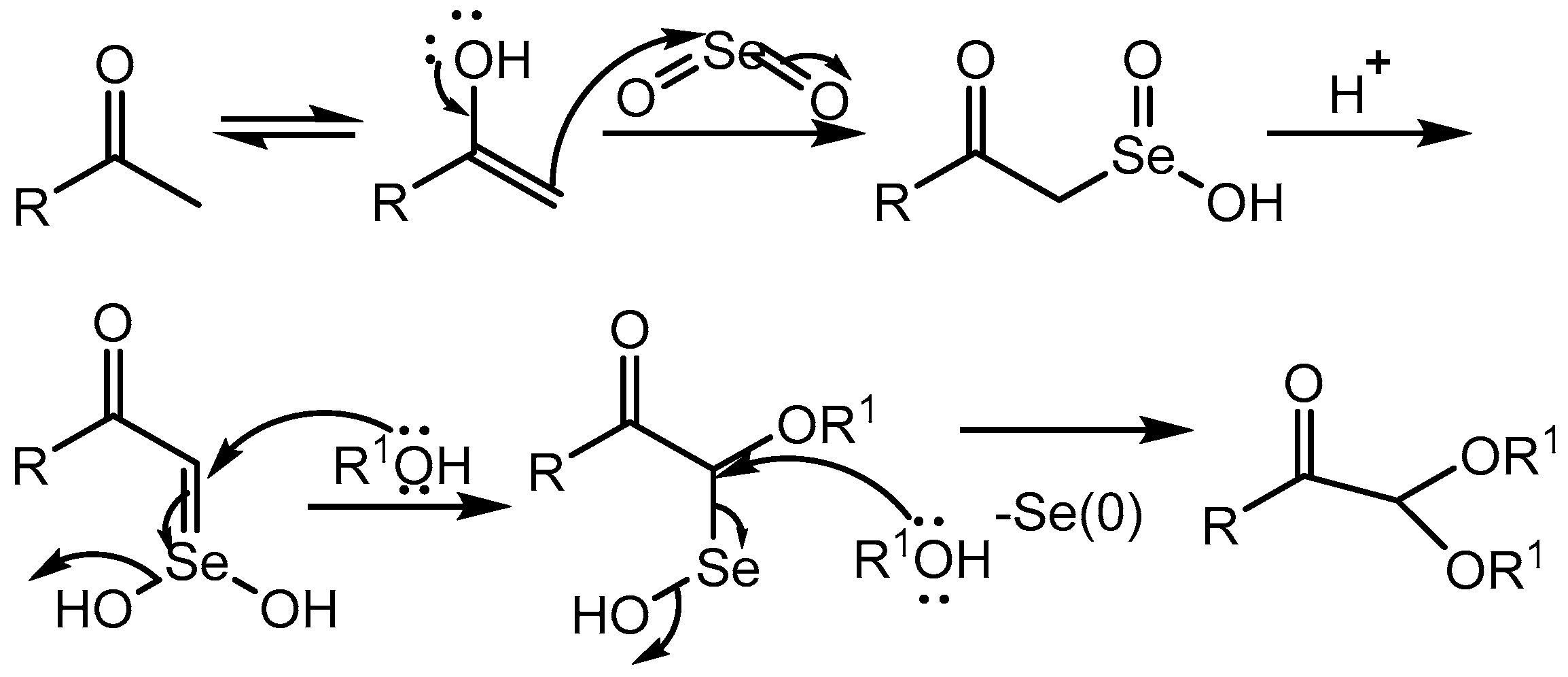
Myrboh group [2] reported p-toluenesufonic acid (PTSA) catalysed reaction of aryl methyl ketones and alkyl methyl ketones with a variety of aliphatic alcohols in the presence of selenium dioxide leading to the generation of α-ketoacetals in 60-90% yields (Scheme 1). These compounds are considered as important building blocks in organic synthesis and many biologically active compounds are synthesized using α-ketoacetals as key intermediates. The methodology was further extended to the substituted benzylidine acetones to produce the corresponding acetals in 82-92% yields (Scheme 1).
The mechanism of the reaction is proposed (Scheme 2) based on their earlier works [3] on selenium dioxide. The precipitation of elemental selenium occurred in the reaction.
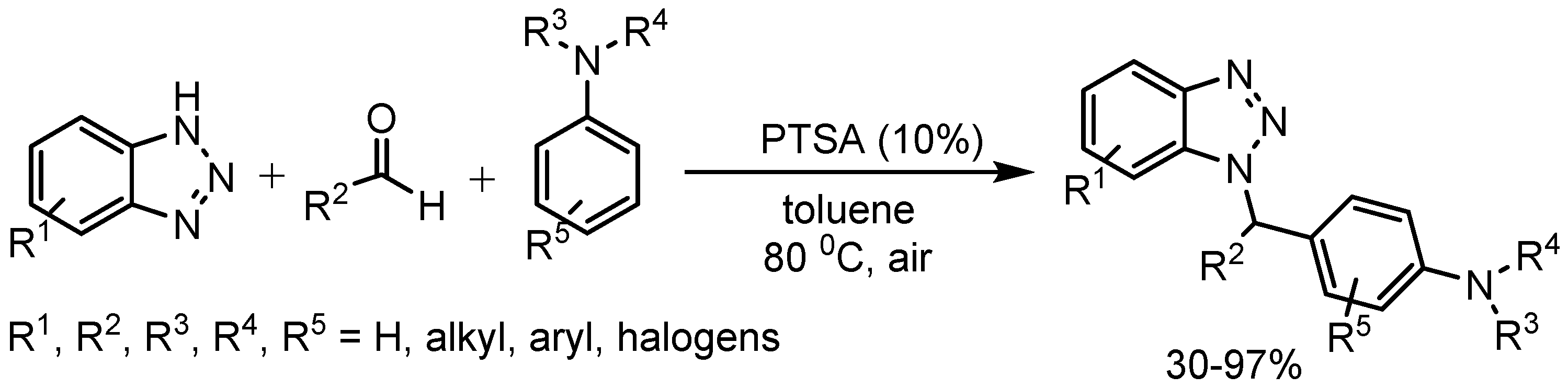
Xiuling Cui et al. [4] described an efficient synthesis of N-alkyl benzotriazoles in 30-97% yields (Scheme 3) via a condensation reaction involving benzotriazoles, aldehydes and tertiary anilines using catalytic amount of p-toluenesulfonic acid (PTSA). This methodology showed high atom economy, excellent regioselectivity and good functional group tolerance. Mechanistic investigations indicated that, presumably the reaction moved through an aza quinone methide intermediate.
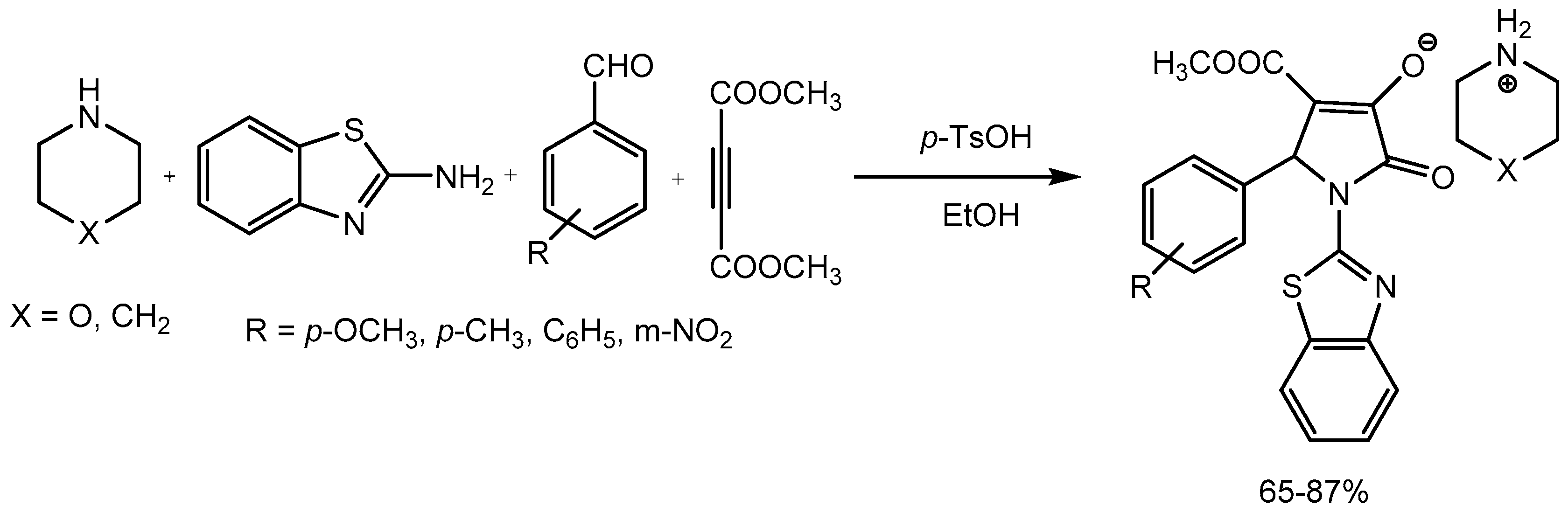
Yan group [5]reported a four-component reaction involving 2-aminobenzothiazole, aromatic aldehydes, acetylenedicarboxylate and piperidine or pyrrolidine in presence of catalytic amount of p-toluenesulfonic acid in ethanol resulted the formation of functionalized morpholinium or piperidinium 2-pyrrolidinon-3-olates in 65-87% yields (Scheme 4). This protocol could potentially be used for other complex heterocycles synthesis useful in medicinal chemistry.
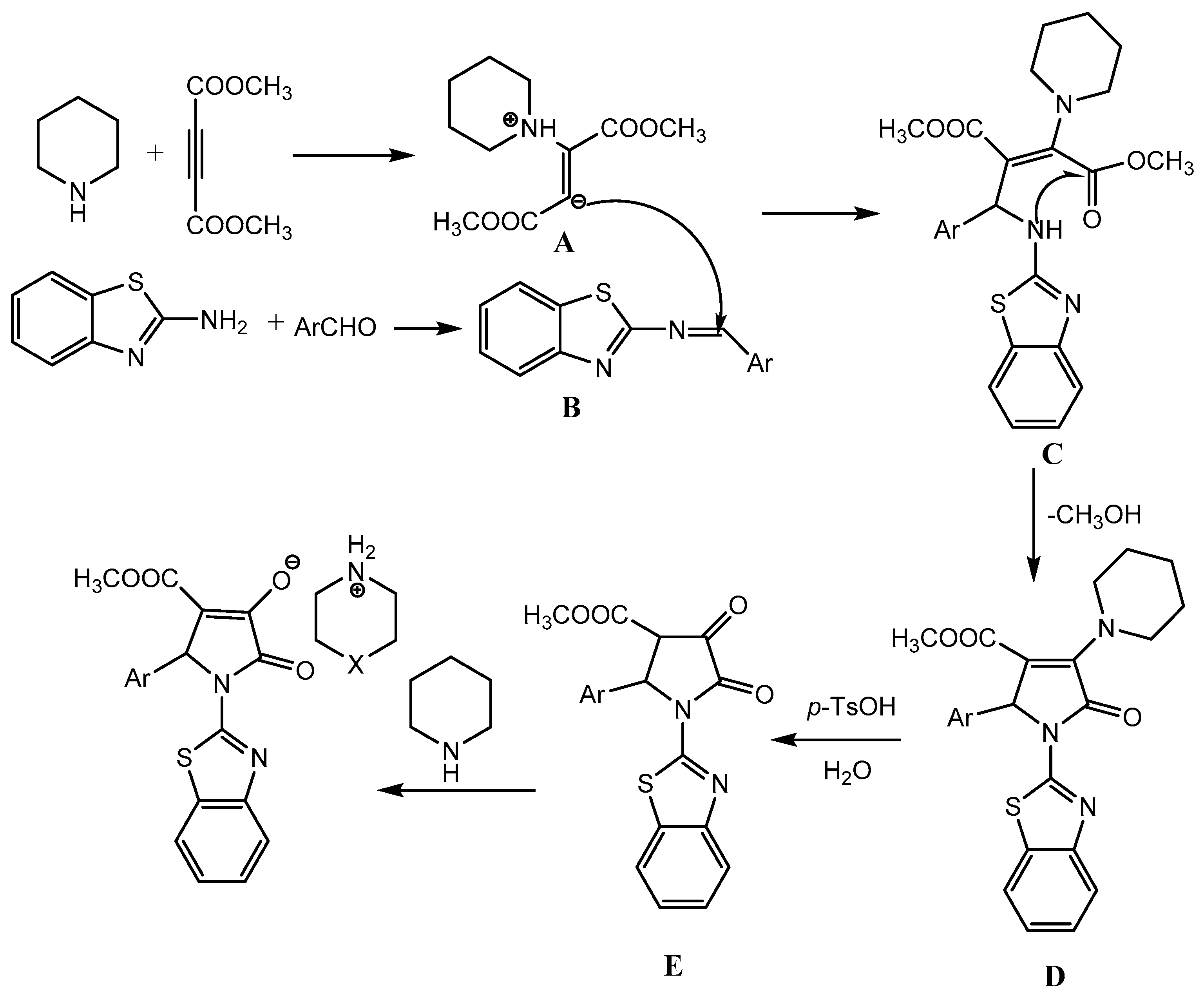
In the proposed mechanistic pathway, piperidine reacts with acetylenedicarboxylate to produce 1,3-dipolar intermediate A, which then react with aldimine B to generate intermediate C. Intramolecular cyclization of C to give intermediate D, which in presence of of p-toluenesulfonic acid form the product (Scheme 5).
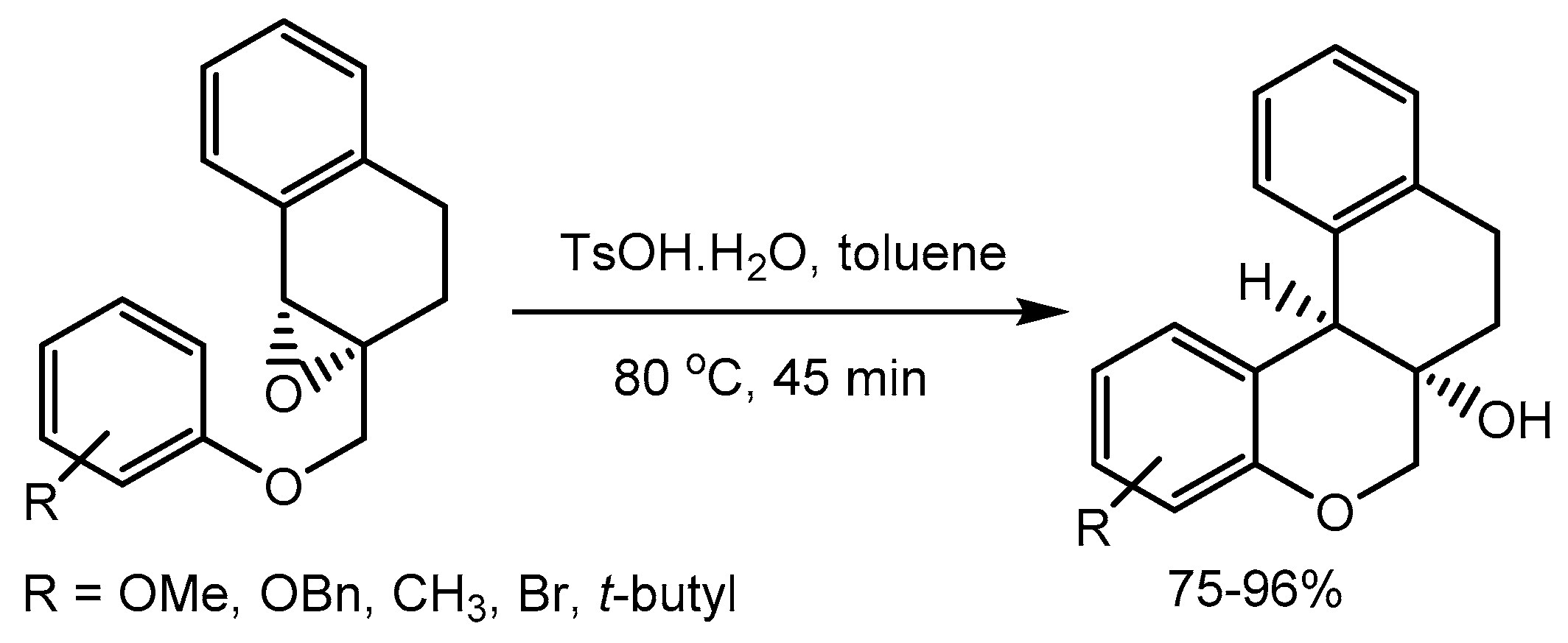
Gogoi et al. [6] described diastereoselective synthesis of chroman-fused tetralins as B-ring-modified analogues of (±)-brazilin in 75-96% yields. The key step of this transformation is p-toluenesulfonic acid catalysed intramolecular Friedel–Crafts epoxy–arene cyclization of 1-tetralone-derived glycidyl ethers (Scheme 6). This work can be considered as a useful addition in the diversity-oriented synthesis of natural-product like molecules.
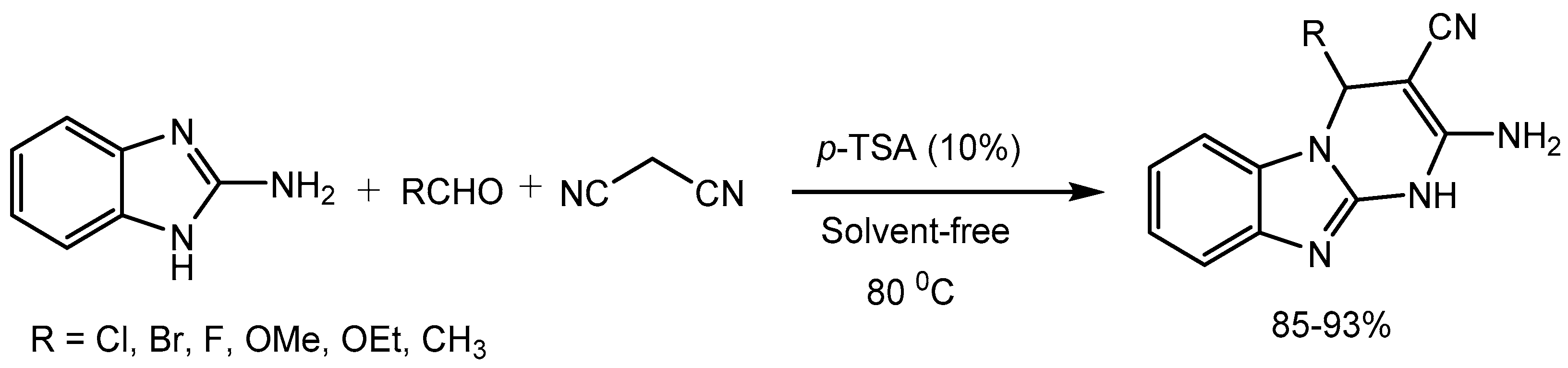
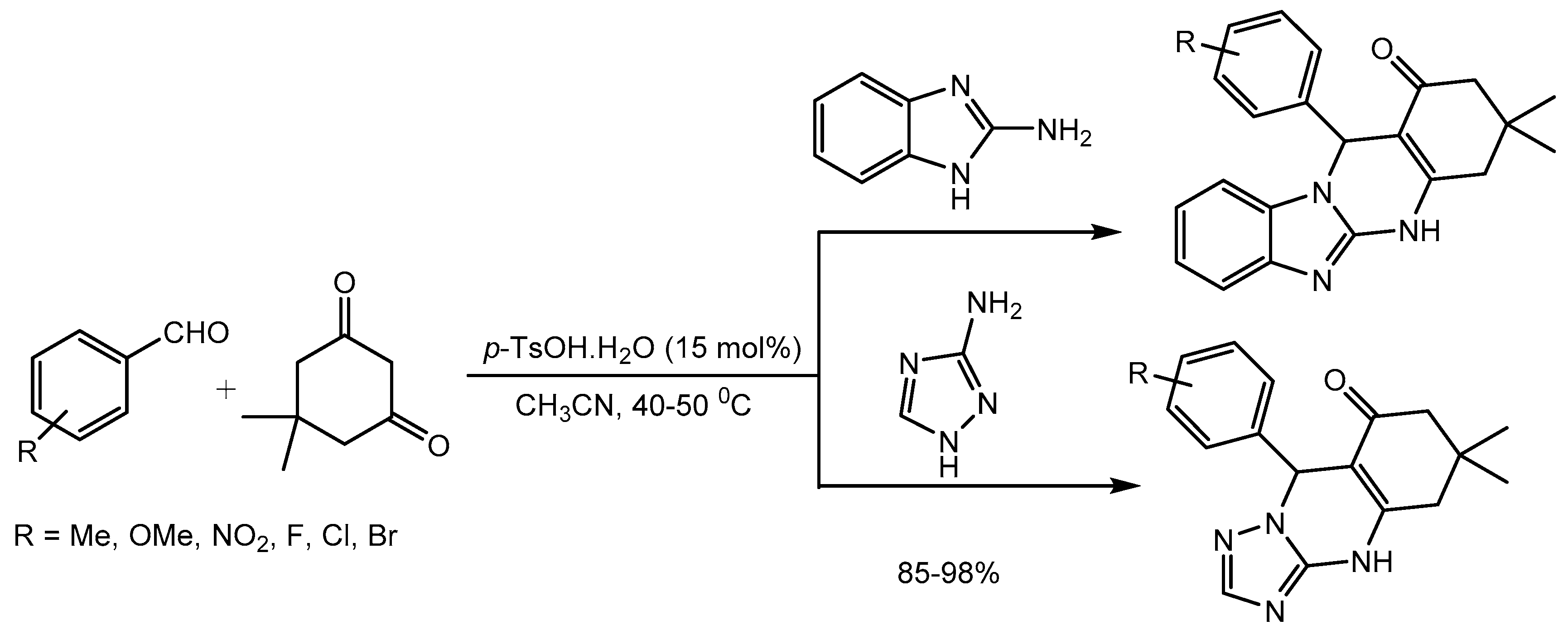
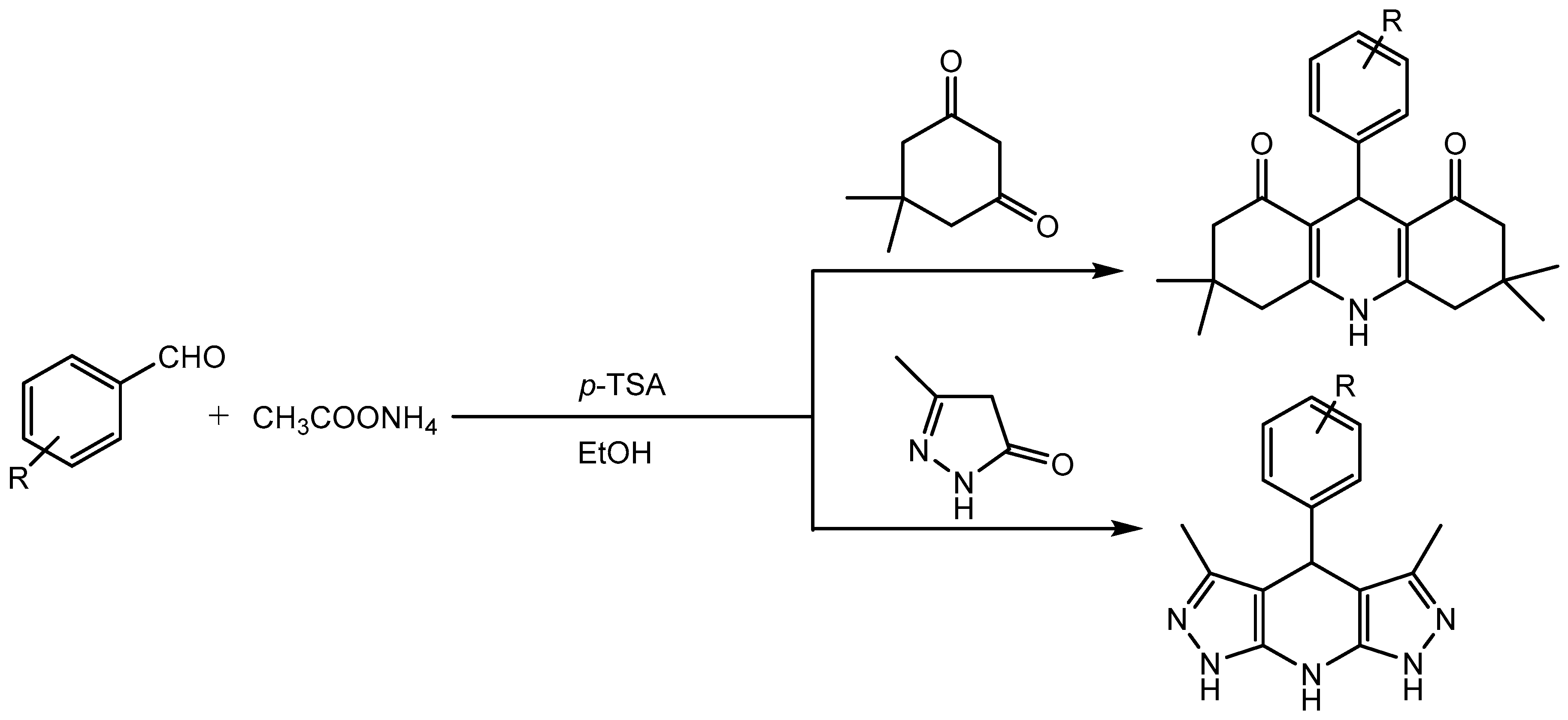
Jeong research group [7] developed a simple, efficient, and green procedure for one-pot synthesis of 2-amino-4-substituted-1,4- dihydrobenzolo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitriles in 85-93% yields via multi-component condensation of 2-aminobenzimidazole, aldehydes and malononitrile in the presence of a catalytic amount of p-toluenesulfonic acid (Scheme 7). Multicomponent reactions (MCRs) have emerged as one of the most powerful tools in organic synthesis. The idea of involving three or more reagents in a one-pot transformation and generating the products with complexity and diversity while incorporating most of the atoms from the reagents in the final structure. MCRs are considered green and sustainable approach as it increases the atom and step economy, decreases the time and labor required in linear syntheses and reduces the waste. Pyrimidinobenzimidazoles motifs have broad range of biologically activities and are present in many pharmaceuticals and agrochemicals. [8]
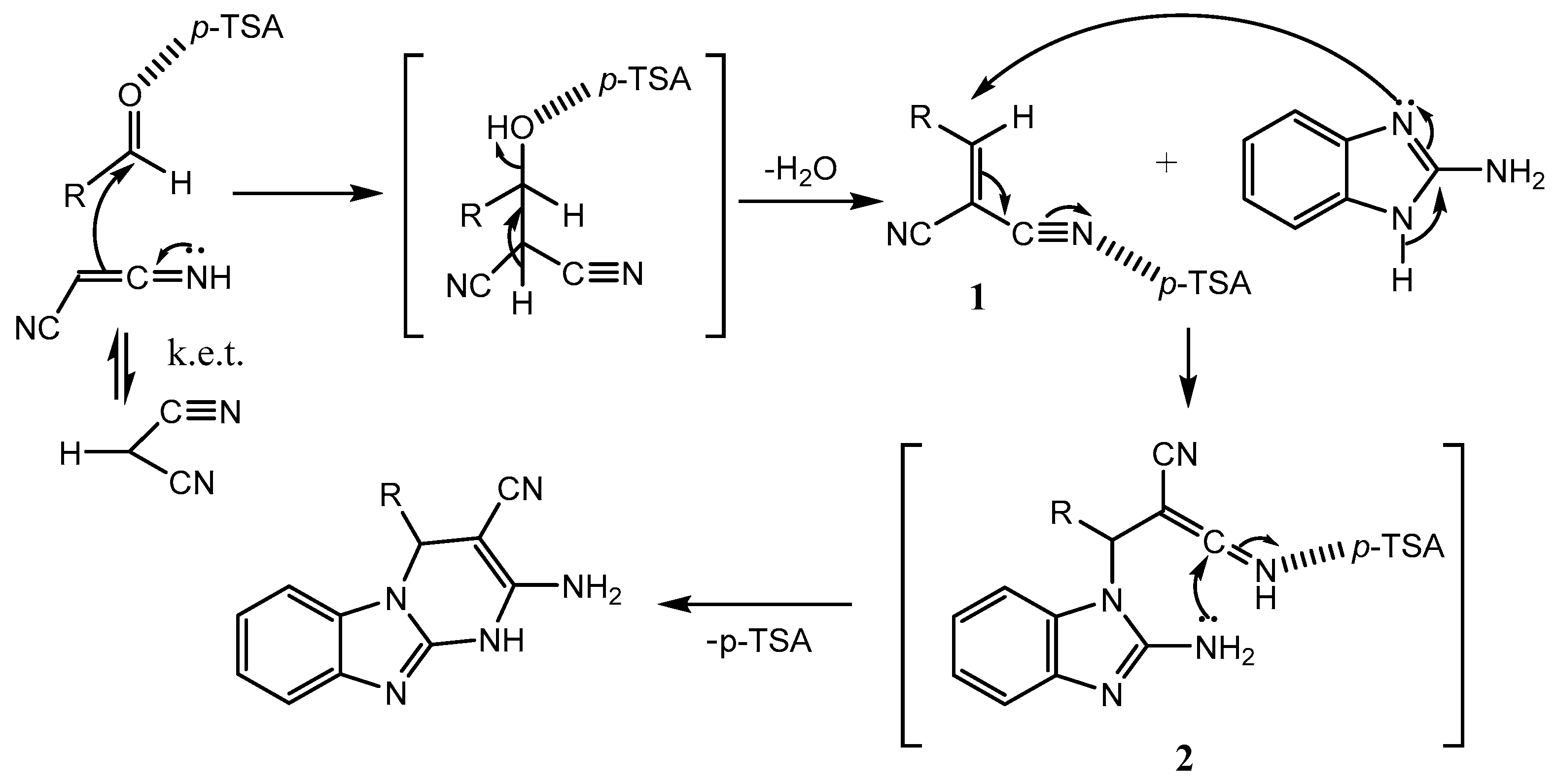
An interesting mechanism of the reaction was proposed to showcase the catalytic activity of PTSA (Scheme 8). Firstly, a Knoevenagel condensation between malononitrile and aldehyde occurs to form the intermediate 1 in the presence of p-TSA, which immediately reacts with 2-aminobenzimidazole via Michael addition to generate the intermediate 2. Finally, intramolecular cyclisation of intermediate 2 produces the product.
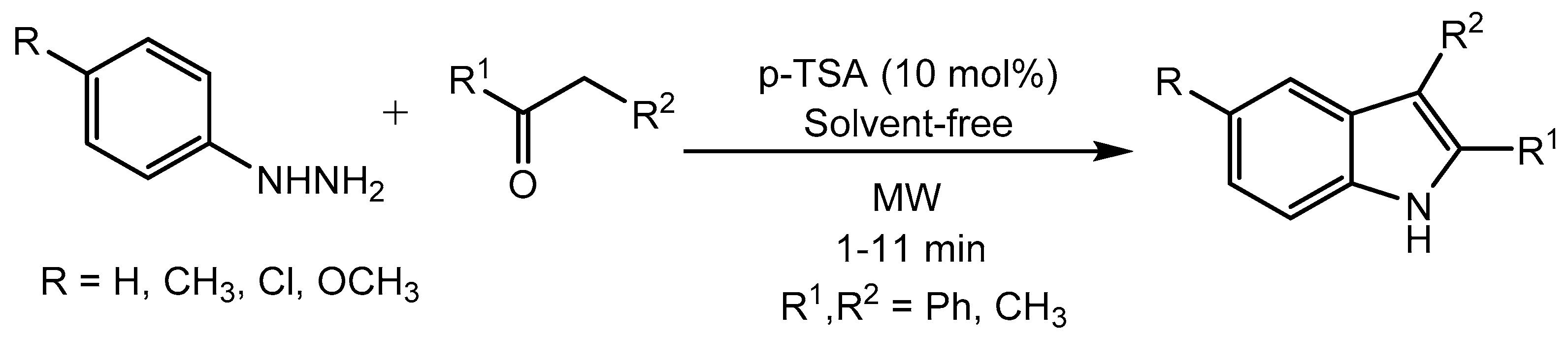
Creencia et al. [9] reported p-toluenesulfonic acid (p-TSA) catalyzed microwave (MW) assisted one-pot synthesis of indole derivatives under green and solvent-free condition. The indole derivatives were prepared from the reaction between phenylhydrazines and enolizable ketones via Fischer-indole process (Scheme 13). Indole moiety is found in different classes of pharmaceutically active molecules.
Scheme 9.
P-TSA catalysed indole synthesis under microwave irradiation.
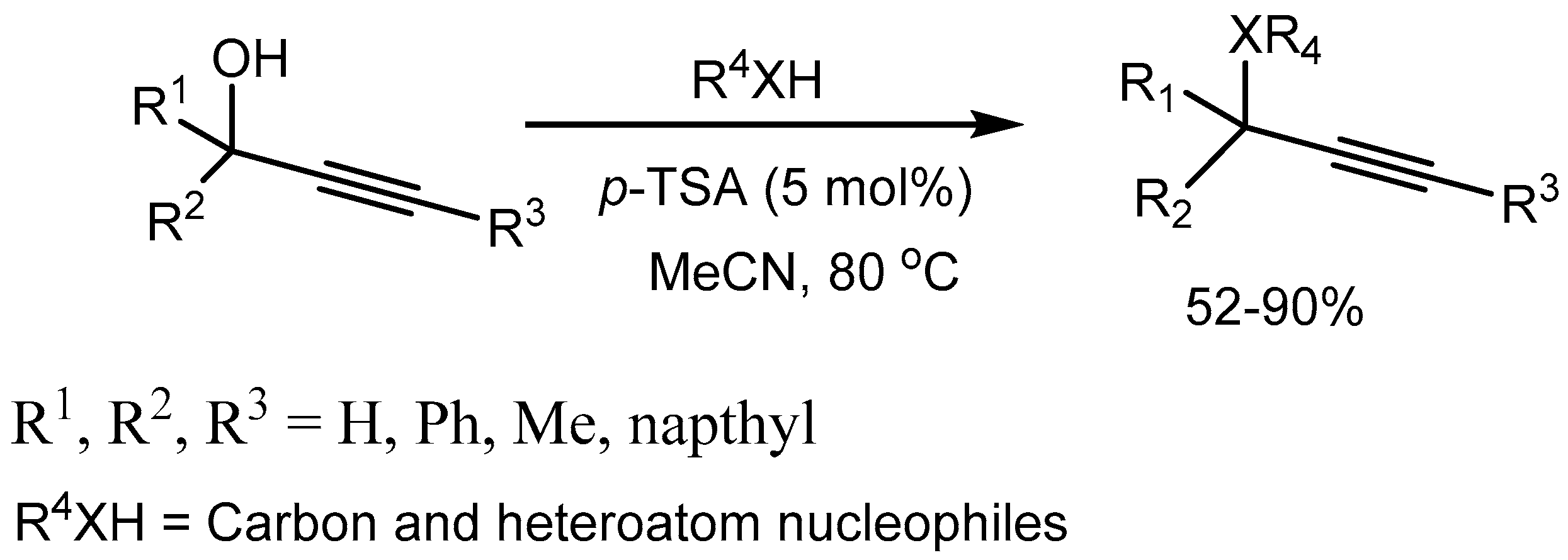
Sanz etal [10] reported p-toluenesulfonic acid catalysed direct nucleophilic substitutions of the hydroxy groups of propargylic alcohols with a large variety of carbon- and heteroatom-centered nucleophiles to generate the products in 52-90% yields under mild conditions (Scheme 10).
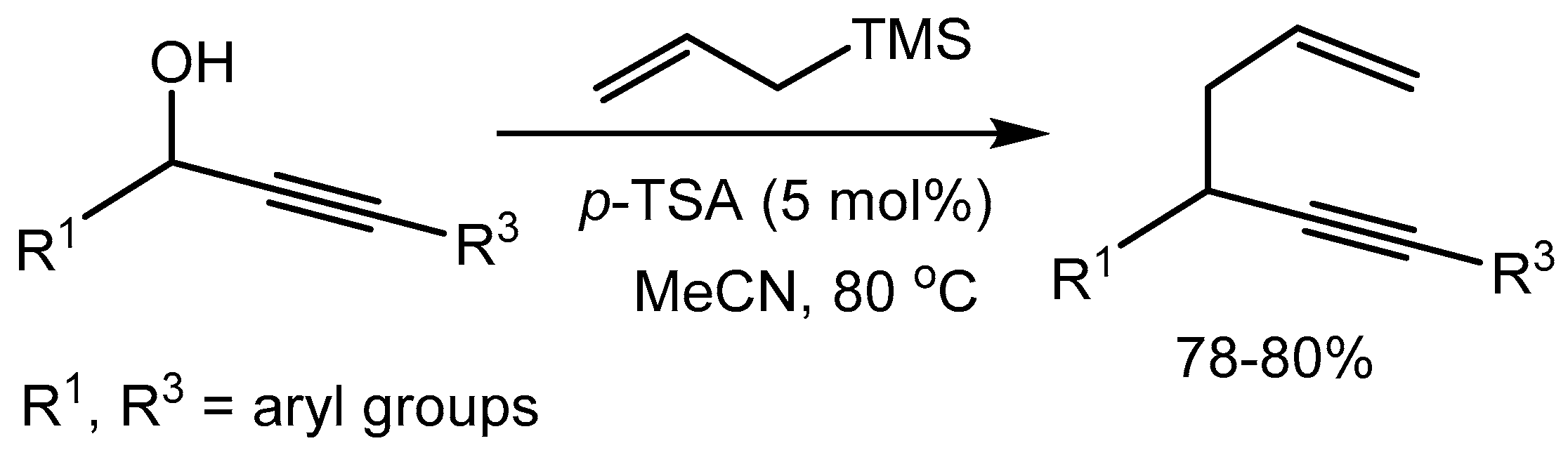
The group nicely applied this methodology for direct propargylic substitution of hydroxy groups with carbon-centered nucleophile, like allyltrimethylsilane to synthesize the corresponding 1,5-enynes in 78-80% yields (Scheme 11). This methodology provides an attractive alternative for the construction of sp3 –sp3 carbon–carbon bonds without using any metallic catalyst.
Jia group [11] described a green and economical process of PTSA catalysed cyclotrimerization of aryl methyl ketones for sustainable production of 1,3,5-triarylbenzenes in 61-91% yields (Scheme 12).
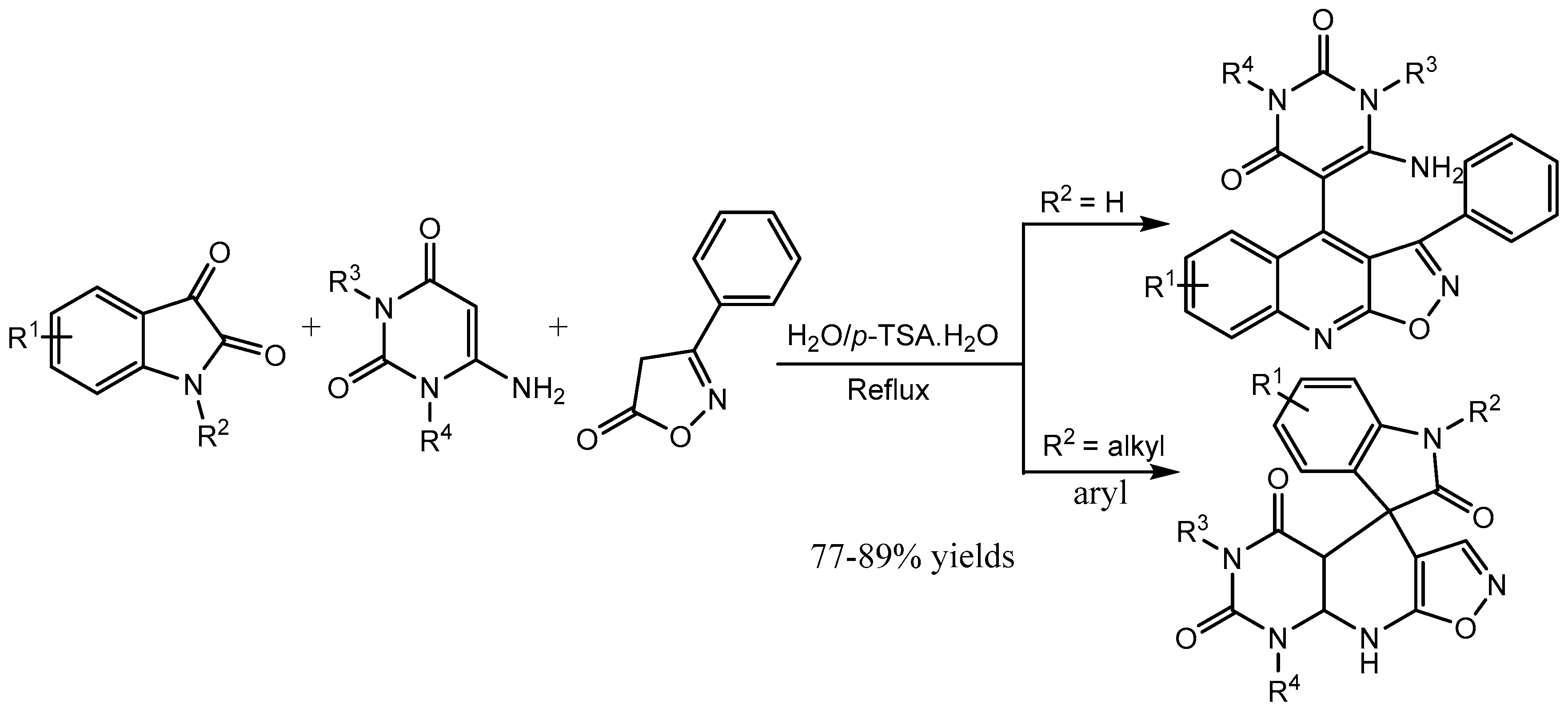
Perumal and co-workers [12] reported a green and efficient one-pot method for the regioselective synthesis of novel 6-amino-5-(3-phenyl-isoxazolo[5,4-b]quinolin-4-yl)pyrimidine-2,4(1H,3H)-diones and 3-methyl-1-phenyl-4-(3-phenylisoxazolo-[5,4-b]quinolin-4-yl)-1H-pyrazol-5-amines via the cleavage of the isatin C–N bond followed by a ring expansion reaction using environmentally benign p-toluenesulfonic acid as a catalyst (Scheme 13). An exciting feature of this reaction is that the formation of product depends on the nature of the group attached to the isatin ring nitrogen atom. This method is attractive because of short reaction time, an excellent yield (77-89%), practical simplicity and high regioselectivity.
Scheme 13.
Synthesis of isoxazoloquinolines and spiroxindoles.
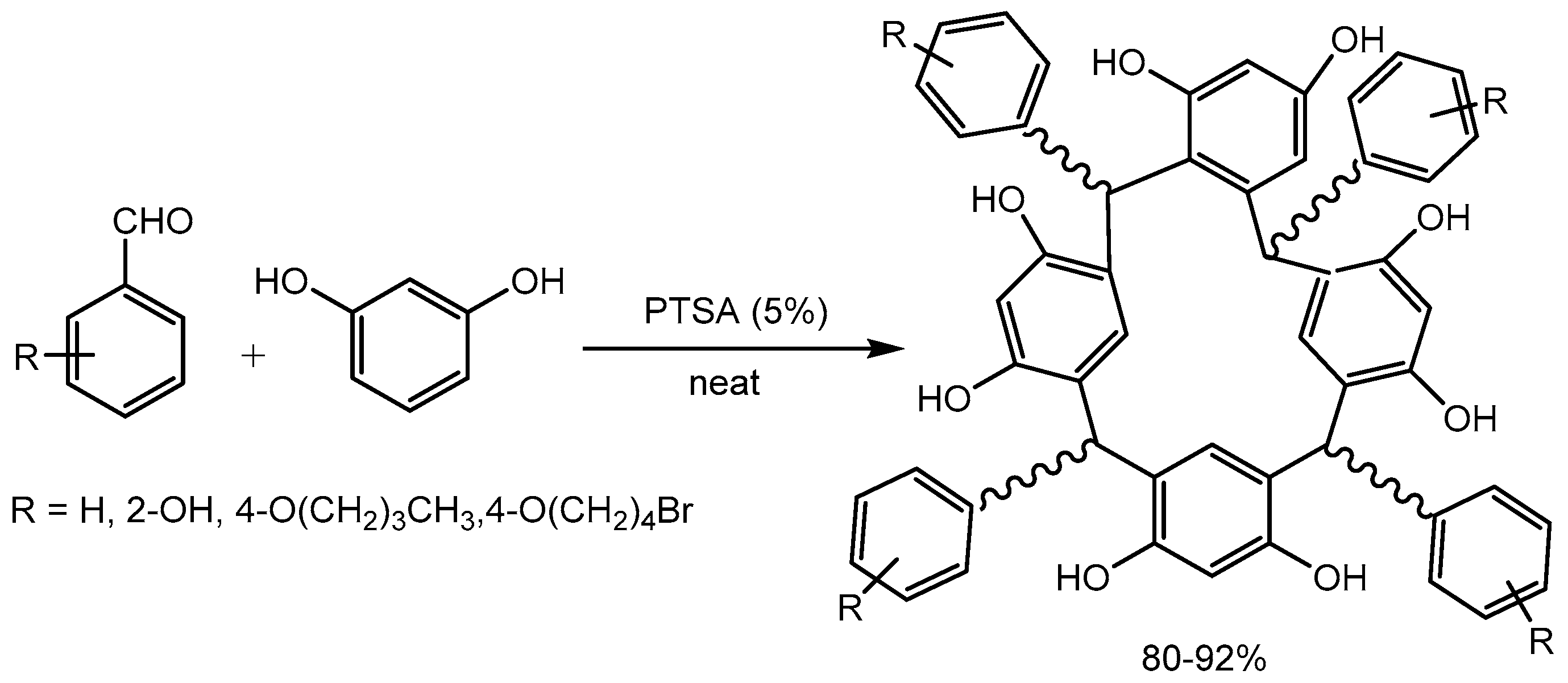
Scott research group [13] developed a green and efficient approach for the synthesis of calix[4]resorcinarenes by the direct reaction of aldehyde and resorcinol in the presence of catalytic amount of p-toluenesulfonic acid (Scheme 14). Calix[4]resorcinarenes were prepared in high yield (80-92%) and purity at ambient temperature under solvent-free conditions. Calix[4]resorcinarenes have wide range of application such as supramolecular tectons, DNA recognition, as components in liquid crystals, metal ion extraction agents, selective membranes, gas adsorption, nanotubes, and catalysis.
Atwood and coworkers [14] reported a solvent free protocol for the single step direct synthesis of pyrogallol[4]arene from isovaleraldehyde and pyrogallol in the presence of catalytic amount of p-toluenesulfonic acid (Scheme 15). The spontaneous self-assembly of these supramolecular building blocks produce nano-sized molecular capsules in the absence of solvent.
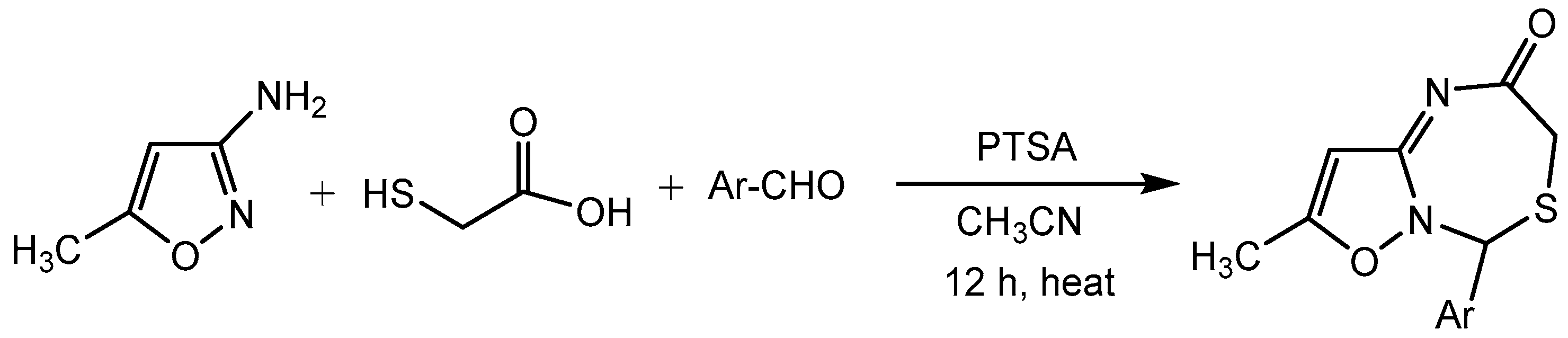
Rajanarendar group [15] disclosed a green approach for the efficient synthesis of novel isoxazolo[2,3-c][1,3,5]thiadiazepin-2-ones by one-pot three-component Domino reaction using p-toluene sulfonic acid (PTSA) as a catalyst (Scheme 16). Based on the versatile bioactivities of thiadiazepines and isoxazoles, it is promising that the design of thiadiazepine implanted with isoxazole framework might result in the discovery of new drug candidates.
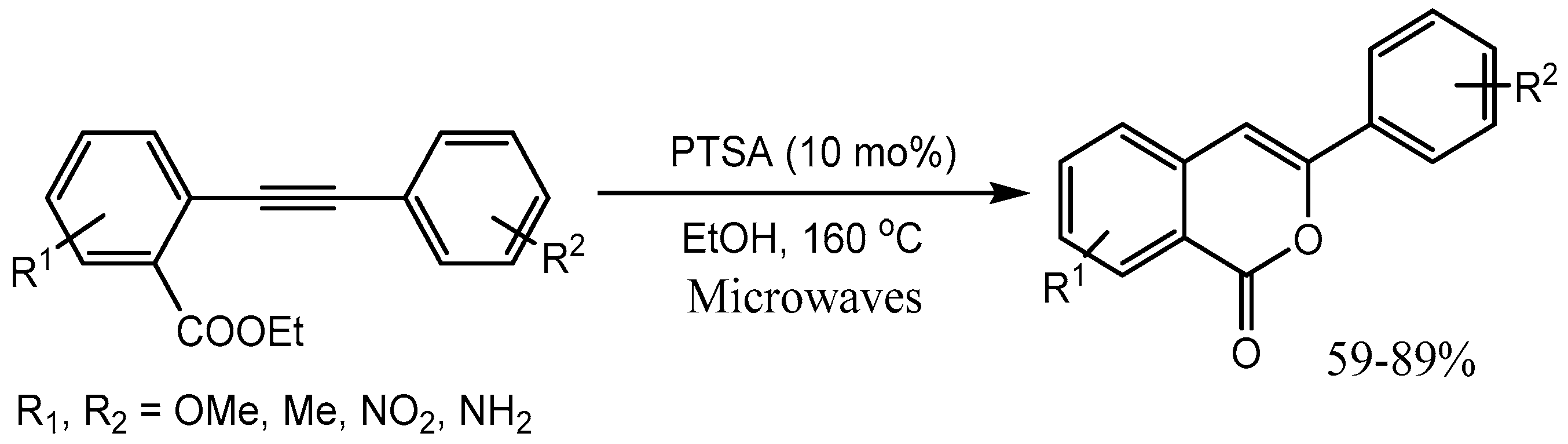
Bras et al. [16] reported p-toluene sulfonic acid (PTSA) catalysed annulation of various functionalized diarylalkynes for the synthesis of variety of 3-aryl-substituted isocoumarins in 59-89% yields. This metal-free process operated under microwave irradiation (Scheme 17). Isocoumarins are prevalent motifs in many natural products that exhibit a broad range of biological activities.
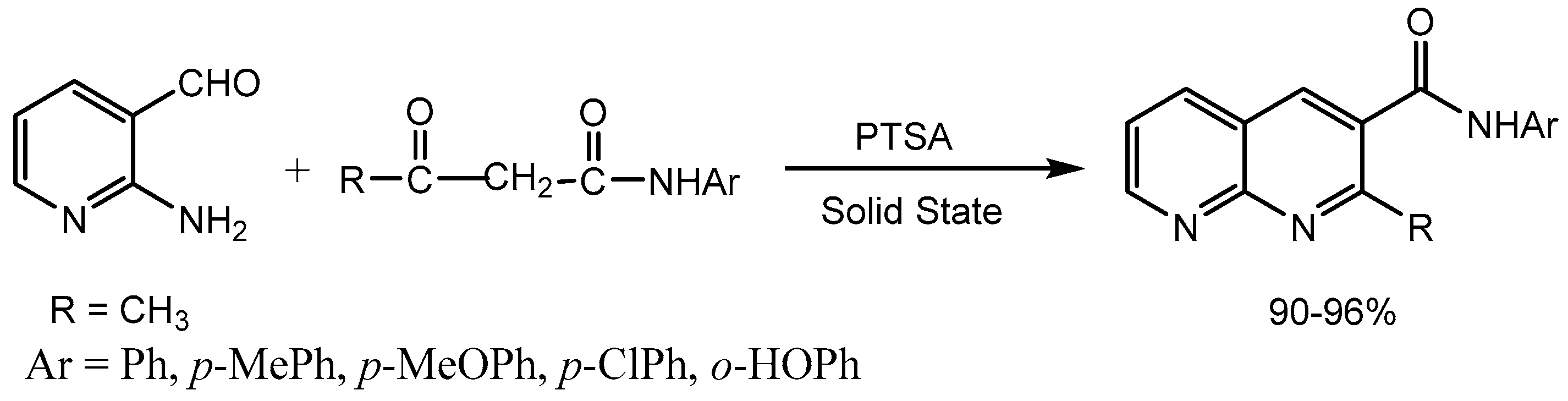
Mogilaiah group [17] developed p-toluene sulfonic acid (PTSA) catalysed Friedlander condensation of various active methylene compounds with 2-aminonicotinaldehyde in the solid state to produce corresponding 1,8-naphthyridines at room temperature in excellent yields (90-96%) with high purity (Scheme 18).
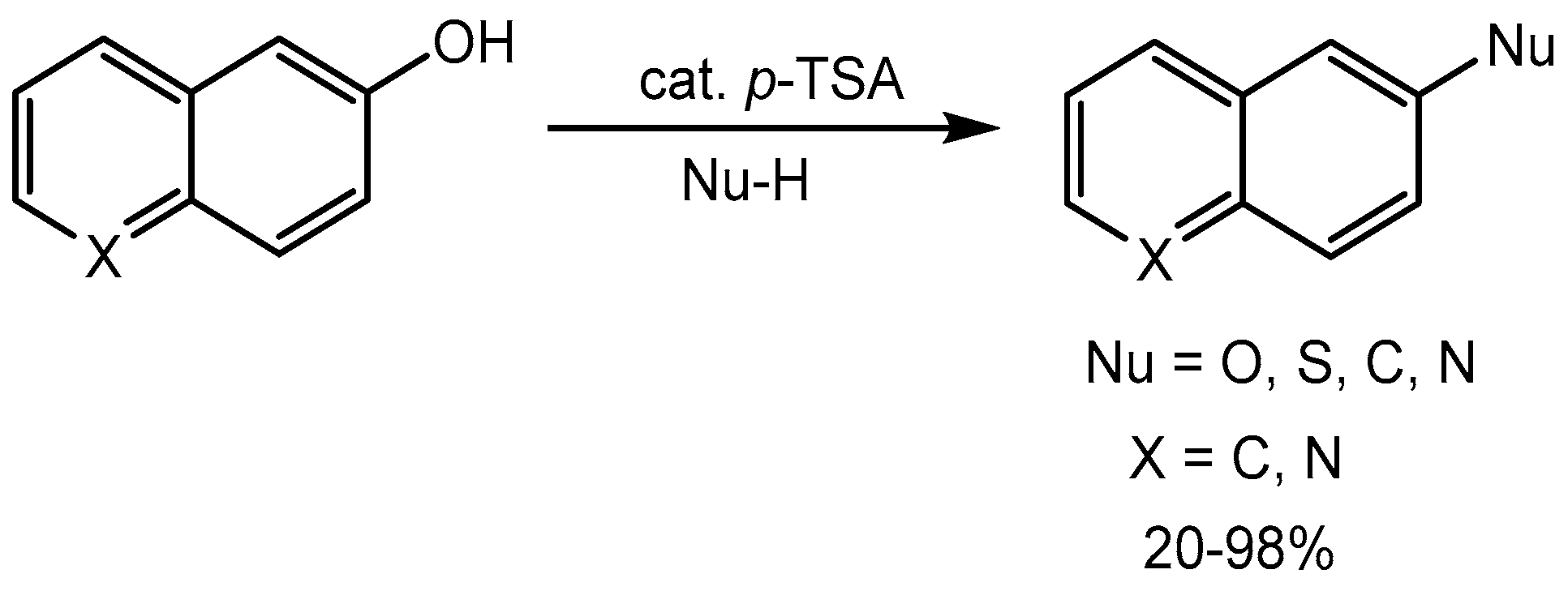
Biswas group [18] reported p-toluene sulfonic acid (P-TSA) catalysed nucleophilic substitution of hydroxyl groups of naphthol and tautomerizable phenol derivatives by O-, S-, N-, and C-centered nucleophiles under solvent-free reaction conditions to produce the products in 20-98% yields (Scheme 19).
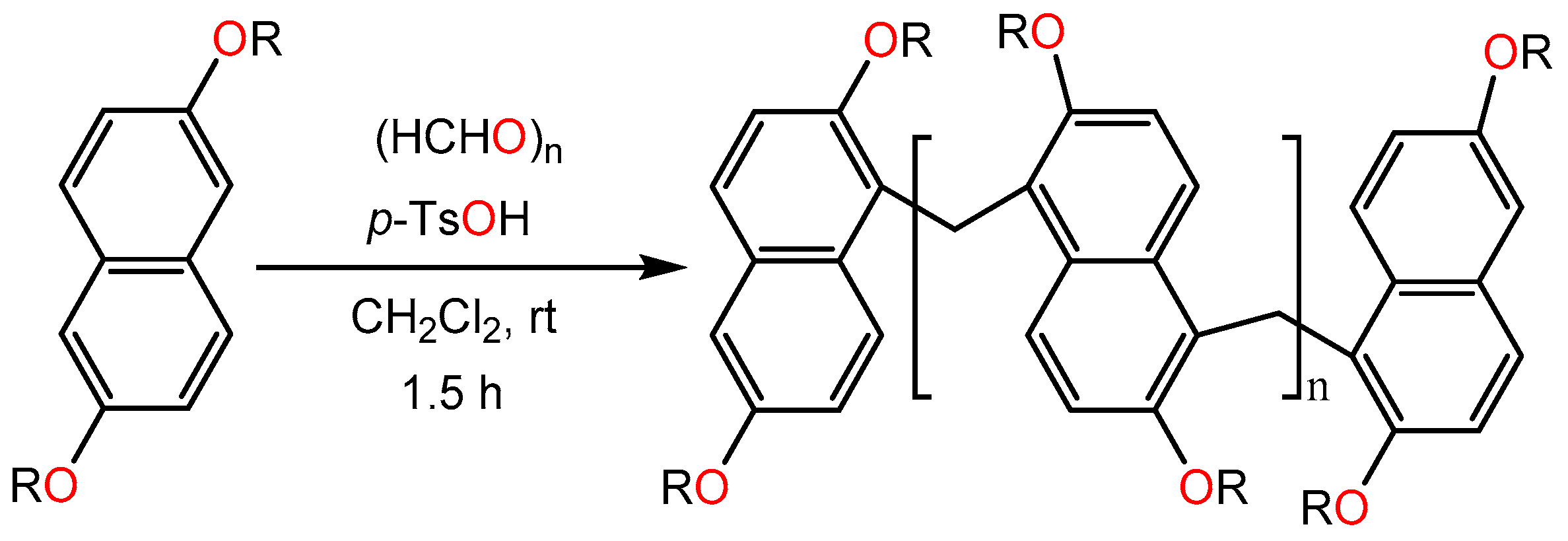
Pan et al. [19] described a regioselective route for the synthesis of methylene-bridged naphthalene oligomers from 2,6- dialkoxyl naphthanene and paraformaldehyde using catalytic amount of p-toluene sulfonic acid (P-TsOH) (Scheme 20). Methylene-bridged aromatic macrocycles are considered as versatile supramolecular hosts in host−guest chemistry.
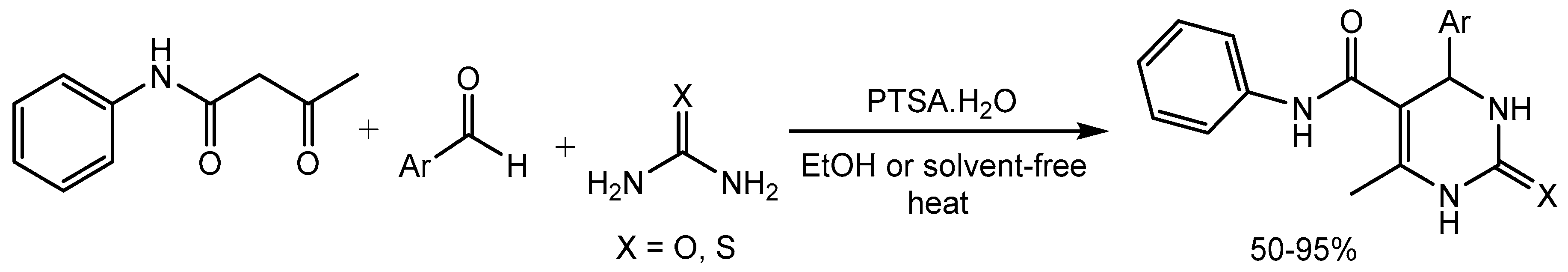
Gündoğan group [20] disclosed a fast and efficient method for the synthesis of a variety of 1,2,3,4-tetrahydro 2-pyrimidinone/thione derivatives (Scheme 21) bearing a phenylcarbamoyl group at C-5 position via one-pot three-component Biginelli condensation reaction using acetoacetanilide, aromatic aldehydes and urea/thiourea in the presence of a catalytic amount of p-toluenesulfonic acid monohydrate (PTSA·H2O) in 50-95% yields. Since pyrimidines have different biological properties, the synthesized compounds have potential applications in medicinal chemistry.
Mousavi and Maghsoodlou [21] reported a one-pot three-component reaction of dimedone, aldehydes and 2-aminobenzimidazole or 3-amino-1,2,4-triazole for the synthesis of biologically important benzimidazoquinazolinone and triazoloquinazolinone derivatives under mild conditions using p-toluenesulfonic acid monohydrate as catalyst (Scheme 22). This metal-free environmentally friendly protocol has high yields (85-98%), shorter reaction times and no column chromatographic separation. Multicomponent reactions (MCRs), leading to interesting heterocyclic scaffolds, are particularly useful for combinatorial chemistry and medicinal chemistry.
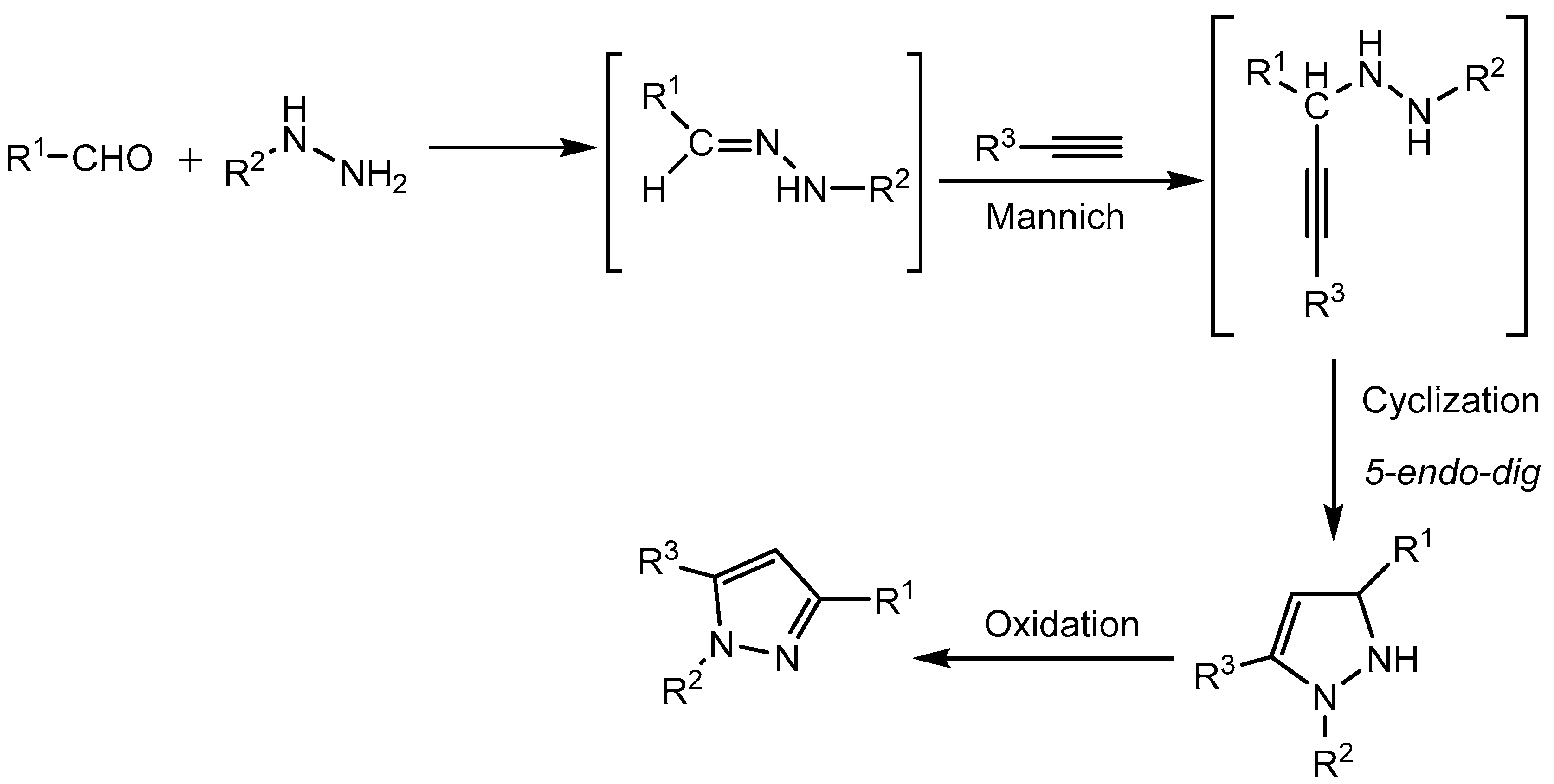
Wang and co-workers [22] developed a convenient one pot three component reaction of aldehydes, hydrazines and alkynes for the synthesis of 1,3,5- trisubstituted pyrazoles derivatives in 45-88% yields using p-toluenesulfonic acid monohydrate (PTSA) as catalyst (Scheme 23).
PTSA acts as a multifunctional catalyst and promotes three transformations in tandem, Mannich-type reaction, 5-end-dig cyclization and oxidation in a single reaction vessel to produce pyrazole derivatives (Scheme 24).
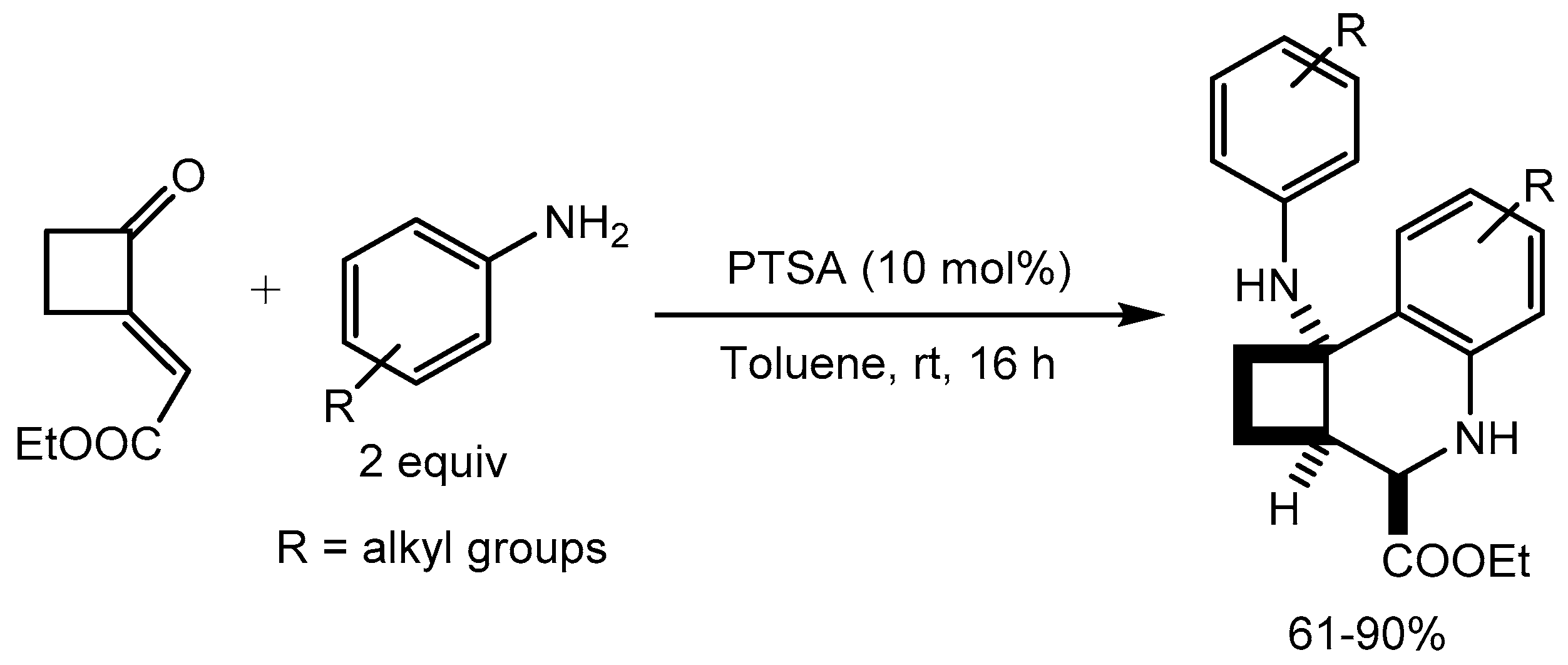
Marras etal [23] disclosed p-toluenesulfonic acid monohydrate (PTSA) catalyzed novel tandem aza-Michael addition-aza-Friedel–Crafts cyclization process, for the synthesis of highly functionalized cyclobuta-fused tetrahydroquinoline carboxylic esters in 61-90% yields from anilines and 2-alkylenecyclobutanones at room temperature. The cyclization is highly stereoselective resulting in the formation of three contiguous stereocenters (Scheme 25).
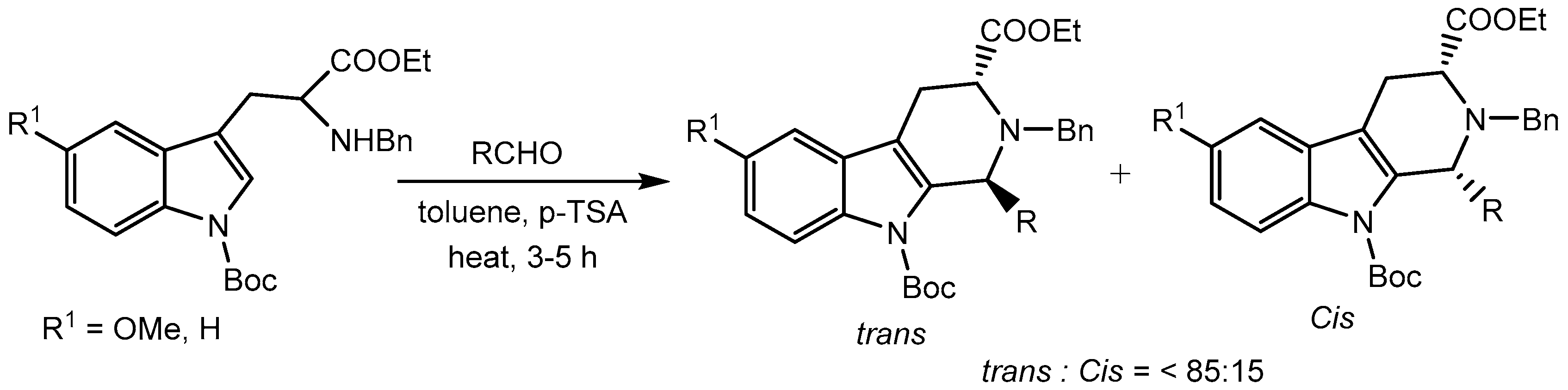
Hennkens et al [24] reported p-toluenesulfonic acid (p-TSA) catalysed Pictet-Spengler (P-S) reaction between Nα-Boc protected tryptophans and a series of aldehydes for the synthesis of biologically important tetrahydro-β-carbolines (THBCs) with the desired trans diastereoselectivity (Scheme 26).
Elwahy group [25] described a p-toluenesulfonic acid (p-TSA) catalysed facile Multicomponent reaction (MCR) of dimedone or pyrazolone with bis-aldehydes and ammonium acetate in ethanol under conventional heating as well as under microwave irradiation for the synthesis of bis(hexahydroacridine-1,8-diones) and bis(tetrahydrodipyrazolo[3,4-b:4’,3’-e]pyridines) derivatives (Scheme 27). This protocol is attractive because of operational simplicity, mild and green conditions, good yields, and easy purification of the products. Acridine derivatives and pyrazolopyridines attracted considerable attentions due to their wide range of biological activities.
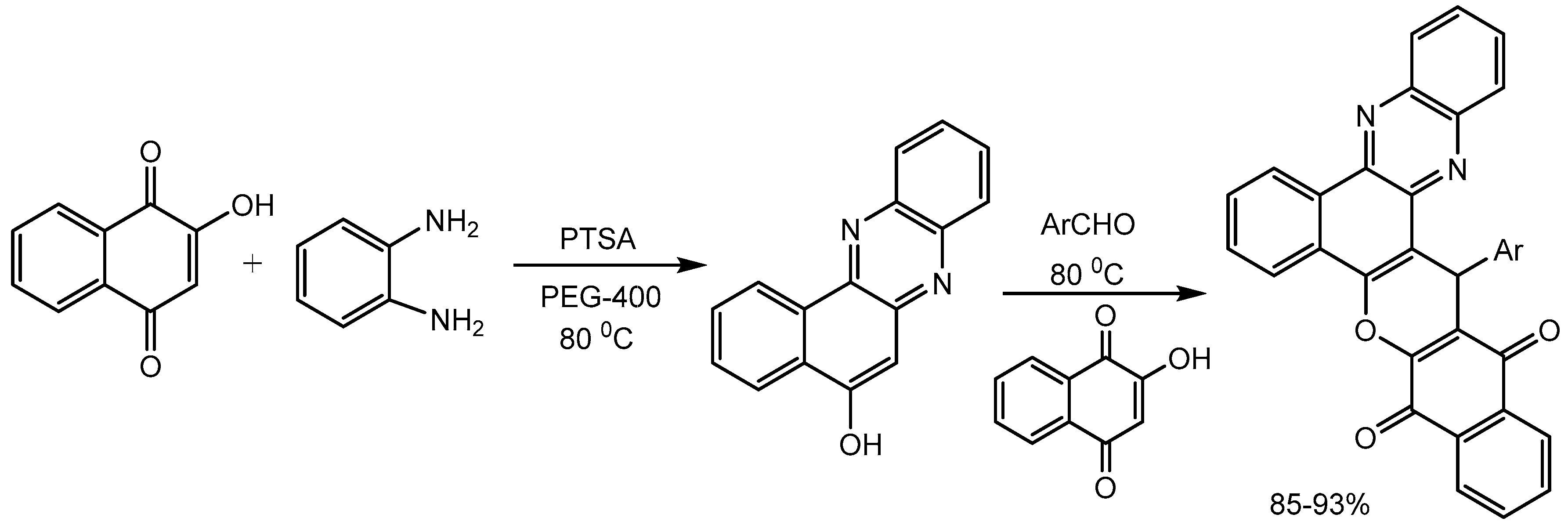
Mohebat et al. [26] developed an efficient p-toluenesulfonic acid catalysed one-pot four-component domino reactions of 2-hydroxynaphthalene-1,4- dione, o-phenylenediamine, aromatic aldehydes using polyethylene glycol as solvent for the synthesis of 11H-benzo[a]benzo[6,7]chromeno[2,3-c]phenazine-11,16(17H)-dione derivatives in 85-93% yields (Scheme 28). This domino protocol produces biologically important heterocycles with the formation of C–C, C=C, C–N, C=N, C–O bonds in a single operation.
Khosropour and Khodaei [27] reported an efficient and straightforward approach for the synthesis of 14-alkyl or aryl-14-H-dibenzo[a,j]xanthenes in 80-96% yields via p-toluenesulfonic acid (p-TSA) catalysed one-pot condensation of β-naphthol with alkyl or aryl aldehydes in solution and solvent-free Conditions (Scheme 29).
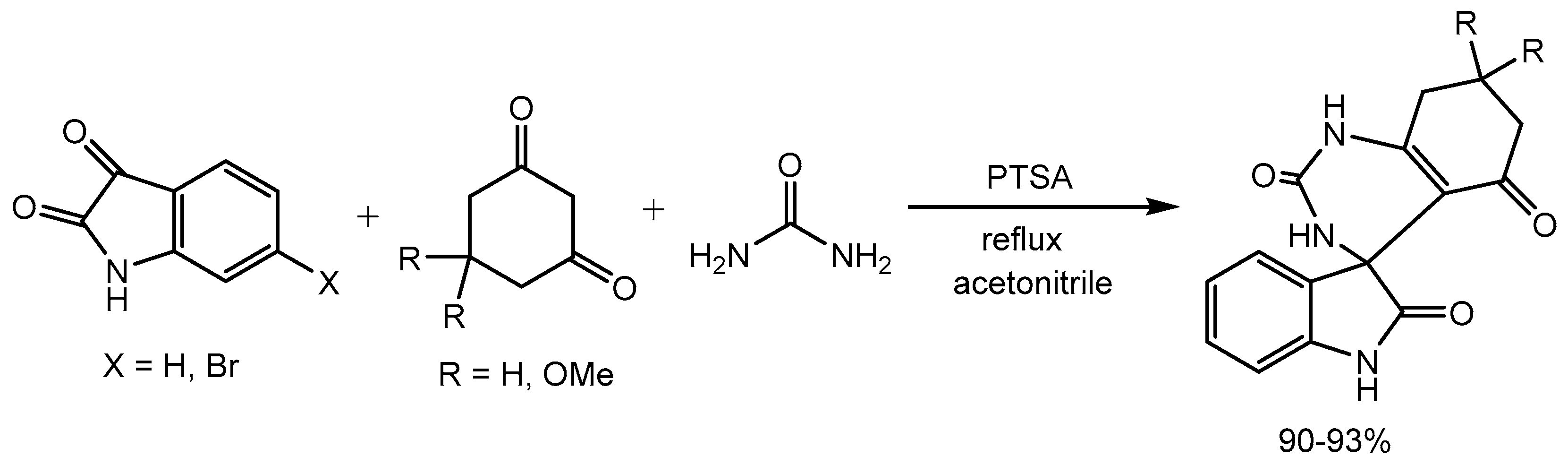
Baghernejad group [28] elaborated p-toluenesulfonic acid (PTSA) catalysed one-pot synthesis of spirooxindoles in 90-93% yields via three-component reaction of urea, isatin, and 1,3-dicarbonyl compounds in acetonitrile (Scheme 30). Spirooxindole skeleton are present in many natural products and biologically active compounds.
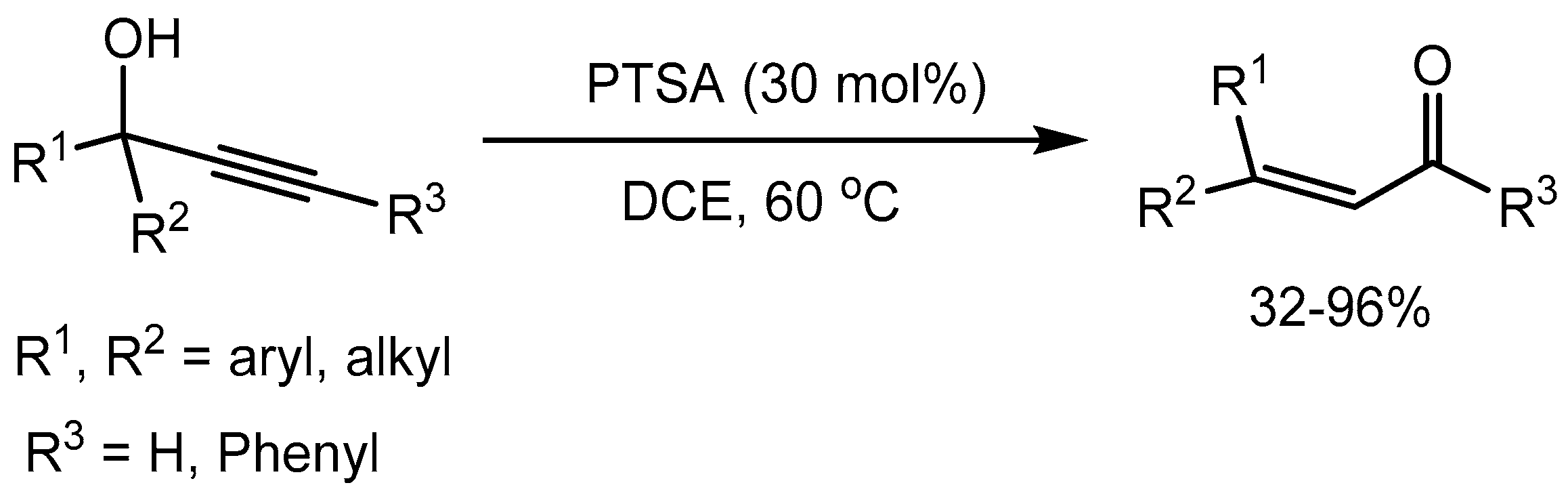
Park etal [29] described an efficient and simple protocol of p-toluenesulfonic acid (PTSA) catalysed Meyer–Schuster rearrangement of propargyl alcohols into α,β-unsaturated carbonyl compounds in 32-96% yields in 1,2-dichloroethane (Scheme 31).
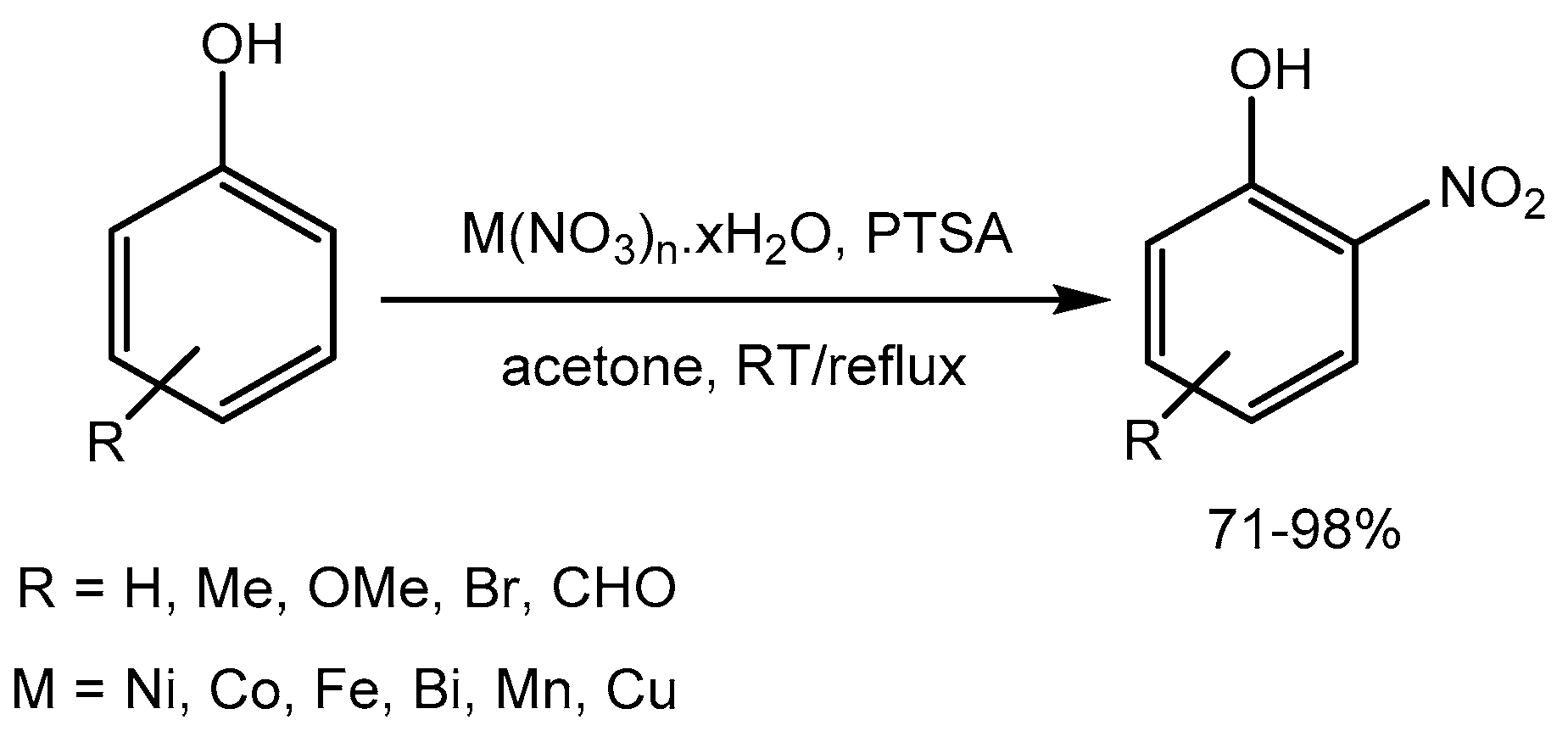
V. Anuradha et al. [30] disclosed a p-toluenesulfonic acid (PTSA) catalysed highly regiospecific mononitration of phenols using a variety of metal nitrates to obtain o-nitrophenols in 71-98% yields. Nitro phenols are intermediates for fine chemicals, pharmaceuticals, and agrochemicals (Scheme 32).
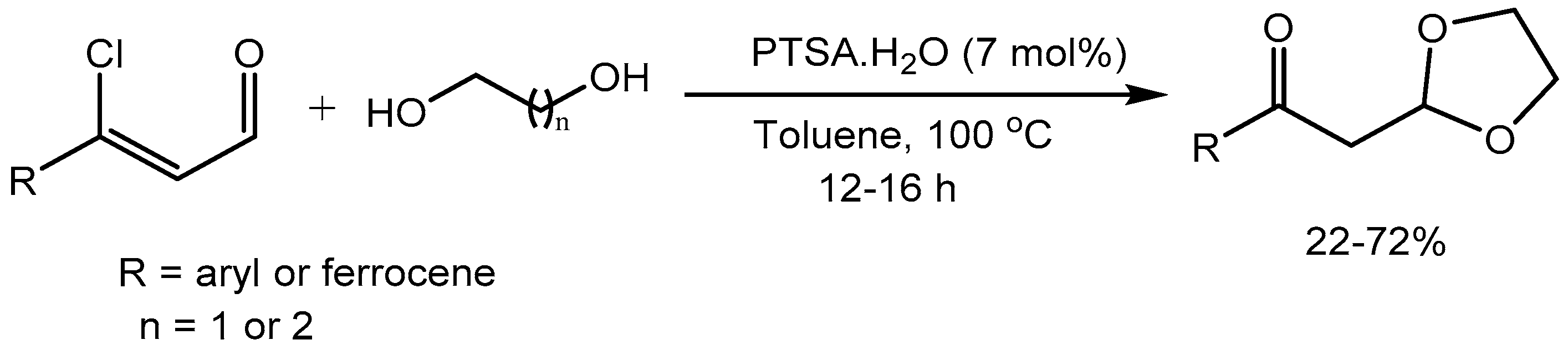
Joshi research group [31] developed a metal-free, p-toluenesulfonic acid (PTSA) catalyzed facile synthesis of β-ketoacetal from β-chlorocinnamaldehyde and dihydroxy alcohols in 22-72% yields (Scheme 33). PTSA catalysed selective protection of the aldehydic group to form the β-chloroacetal and the subsequent dechlorination by H2O resulted the formation of β-ketoacetal. In this transformation multitasking nature of PTSA is observed.
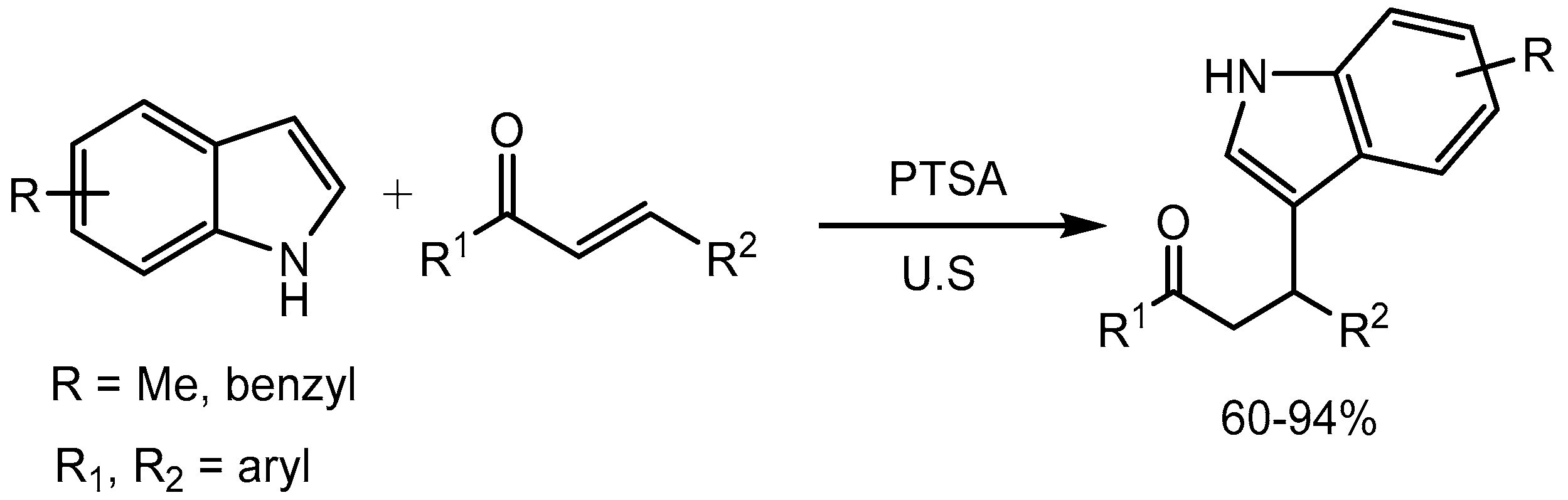
Ji and Wang [32] reported p-toluenesulfonic acid (PTSA) catalyzed Michael addition of indole to α,β-unsaturated ketones for the expeditious synthesis of β-indolylketones in 60-94% yields under ultrasonic irradiation (Scheme 34). β-indolylketones are considered important building blocks for the synthesis of natural products and other biologically active compounds.
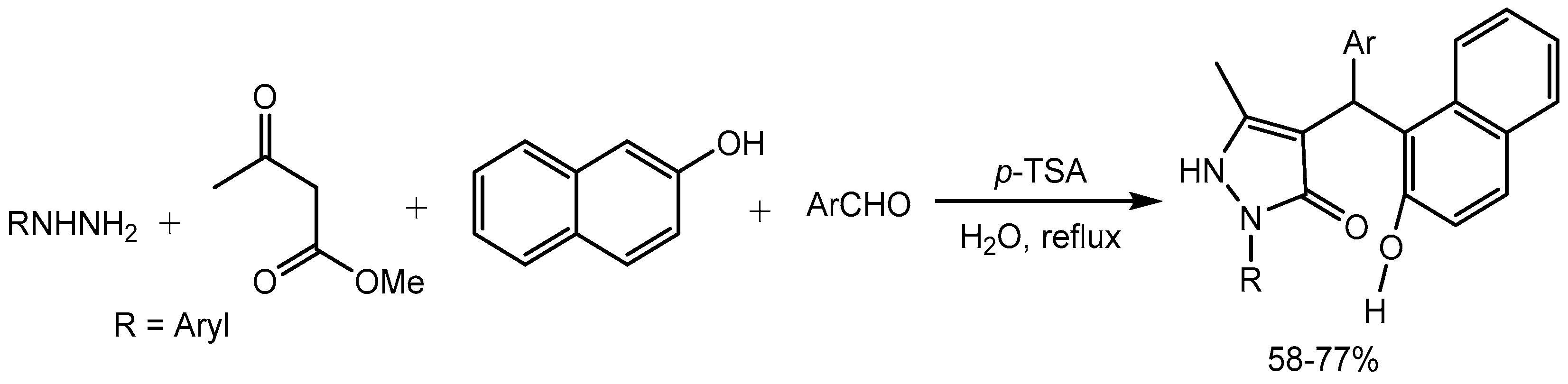
Perumal research group [33] reported expedient synthesis of a series of 2-aryl-5-methyl-2,3-dihydro-1H-3-pyrazolones via one-pot, four-component sequential reactions of phenylhydrazine, methyl acetoacetate, aromatic aldehydes and β-naphthol in the presence of p-toluenesulfonic acid in water. The products were obtained in 58-77% yields and the compounds were screened for in-vitro antimycobacterial activity against Mycobacterium tuberculosis H37Rv (MTB). One of the compound 4-[(2,4-dichlorophenyl)(2- hydroxy-1-naphthyl)methyl]-2-(4-fluorophenyl)-5-methyl-2,3-dihydro-1H-3-pyrazolone displays the maximum potency in the series with a minimum inhibitory concentration (MIC) of 1.6 μM against MTB, being 2.94 and 4.75 times more active than antibiotic ciprofloxacin and ethambutol respectively (Scheme 35).
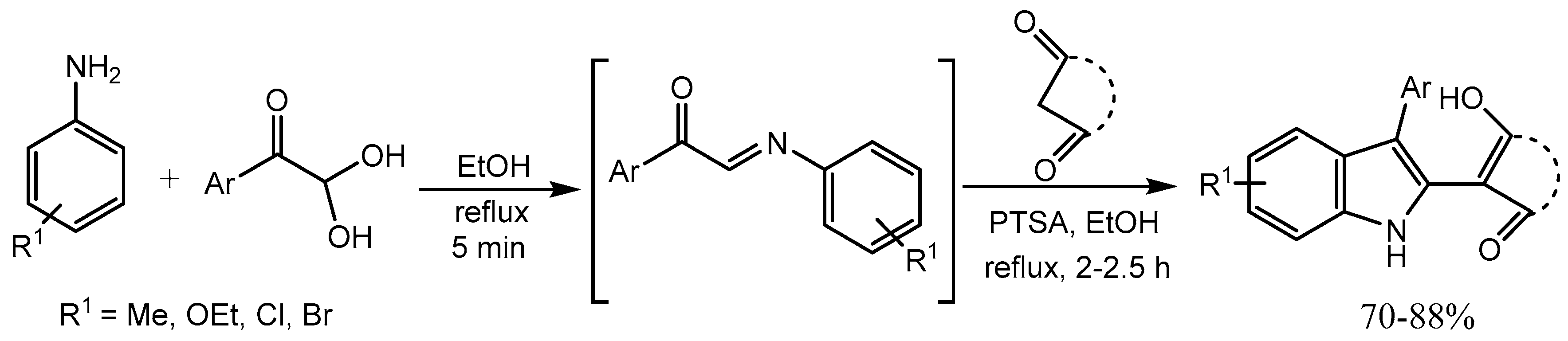
Naidu et al. [34] developed a simple and efficient one pot three components protocol for the synthesis of highly functionalized indoles in 70-88% yield in an easy workup procedure. In presence of PTSA, the reaction occurred via reductive alkylation of α-keto imines, followed by a cyclization process (Scheme 36). Indole moiety is present in diverse biologically active natural products and pharmaceuticals.
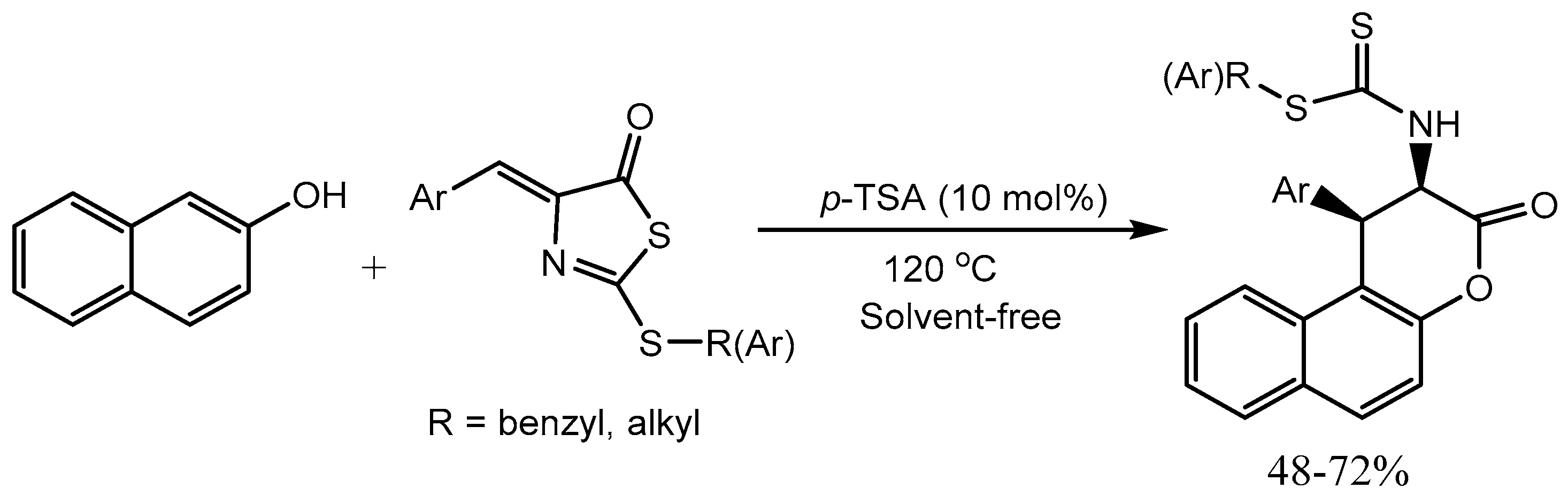
Halimehjani and Khoshdoun [35] disclosed an efficient PTSA catalysed synthesis of functionalized 1,2-dihydrobenzo[f]chromen-3-ones and 3,4- dihydrochromen-2-ones via tandem esterification/ intramolecular 1,4-addition-type Friedel−Crafts alkylation reaction of phenols or naphthols with Olefinic Thioazlactones. The reaction proceeds with high yields (48-72%) and diastereoselectivity (Scheme 37). Due to the widespread occurrence and broad range of biological activities chromenes, chromanes and their benzo-fused derivatives are highly attractive synthetic targets.
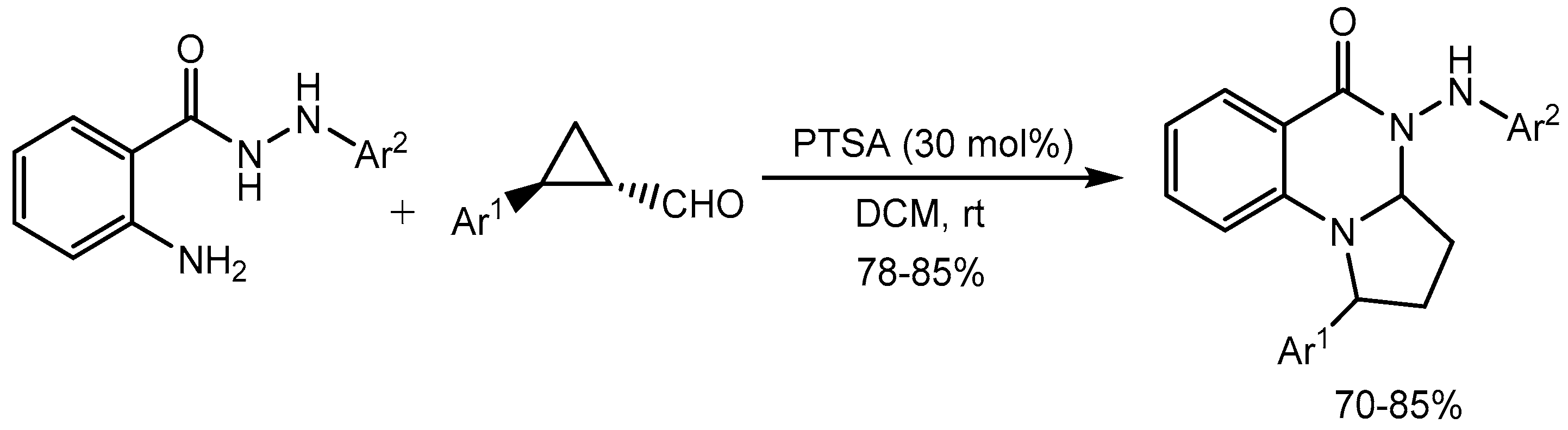
Singh et al. [36] described p-toluene sulfonic acid (PTSA) catalysed regioselective annulation of cyclopropane aldehydes with N′-aryl anthranil hydrazides for the synthesis of tetrahydropyrrolo[1,2-a]quinazolin-5(1H)ones (Scheme 38). The reaction involves domino imine formation followed by intramolecular cyclization and nucleophilic ring opening of the cyclopropyl ring to form the desired products in good to excellent yield (70-85%) with complete regioselectivity. This methodology provides a simple and step-efficient synthesis of pyrroloquinazolinone derivatives which is an important core of a variety of natural products and biologically active molecules.
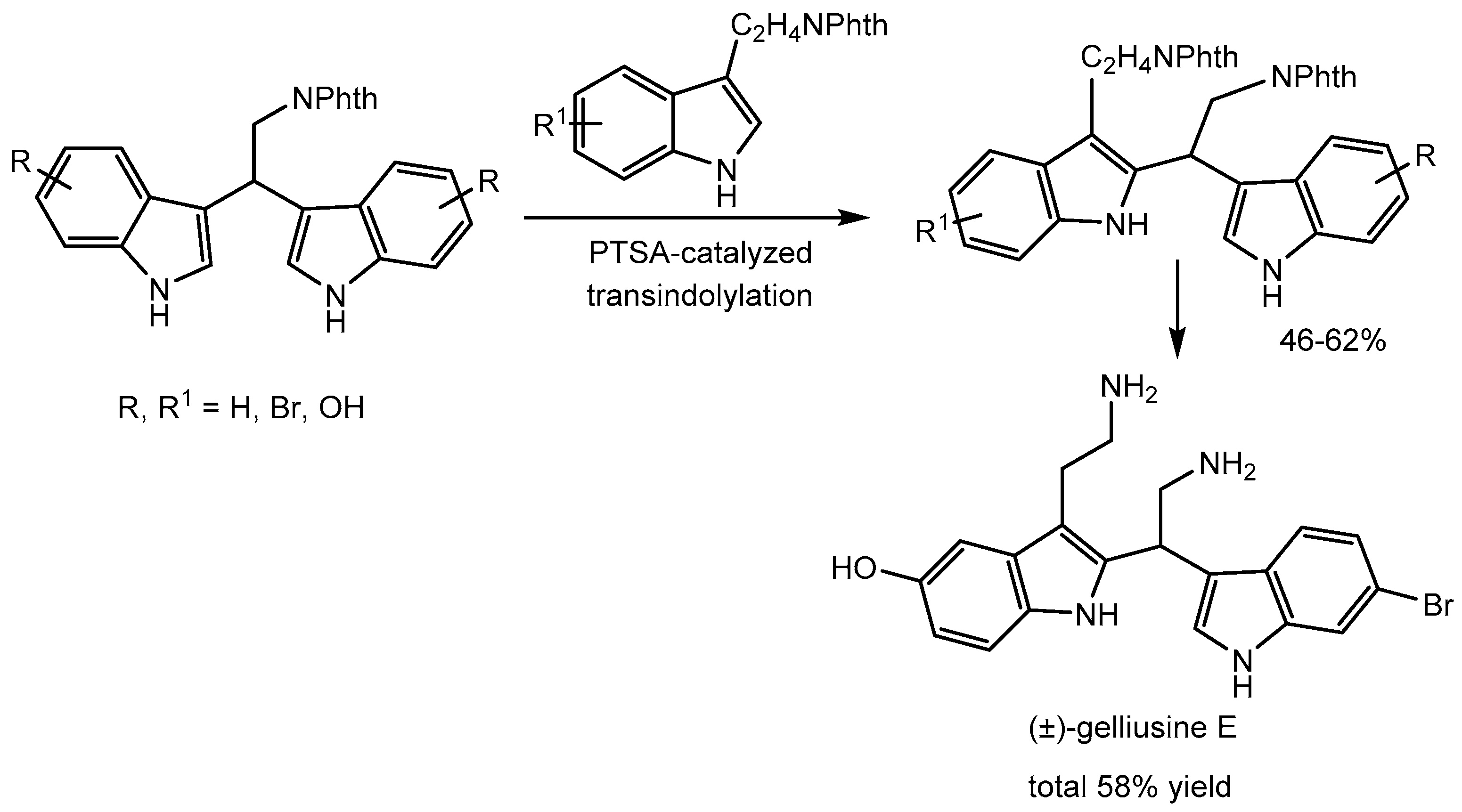
Jaratjaroonphong and co-workers [37] reported a novel strategy of PTSA-catalyzed transindolylation of the readily synthesized 3,3′-bis(indolyl)ethylamines (3,3′-BIEAs) with tryptamine derivatives in 46-62% yields. The group has nicely applied this strategy for the first and short total synthesis of the marine sponge 2,3′-bis(indolyl)ethylamine (2,3′-BIEA) alkaloid (±)-gelliusine E in 58% yield. This modular approach allows the rapid synthesis of other members of the 2,3′-BIEA family. Additionally, cytotoxicity of these alkaloids in breast cancer cells was investigated (Scheme 39).
Meng etal [38] disclosed the method for the synthesis of novel bis-furan diepoxide (OmbFdE) via a mild and scalable route, and applied in epoxy resins in an eco-friendly manner, with the resulting materials showing good thermal stability. Difuranic dialdehyde, the precursor the difuranic diepoxide monomer (OmbFdE) was synthesized from HMF in the presence of catalytic p-toluenesulfonic acid. Further reduction and etherification yielded the bis-furan diepoxide product on a large scale (Scheme 40).
Secci group [39] described a general strategy for the synthesis of arylthio cyclopropyl carbaldehydes and ketones via p-toluene sulfonic acid (PTSA) catalyzed arylthiol addition followed by ring contraction reaction. The methodology produced a variety of cyclopropyl carbaldehydes in 78-96% yields (Scheme 41).
Baire and Tharra [40] reported p-toluene sulfonic acid (PTSA) catalysed nucleophilation of propargylic alcohols via Meyer–Schuster rearrangement (M–S) for the synthesis of α-arylenone esters in 47-83% yields (Scheme 42). This protocol involves cis-enoate assisted reverse polarization of the M–S intermediate allenyl cation. To showcase the application of this method, the α-arylenone esters was successfully converted to the biologically important molecules such as pyrazoles and 4,5-seco-abietane.
Enaminone chemistry has attracted enormous attention in recent years because enaminones are a versatile intermediate used for the synthesis of various heterocyclic compounds and the enaminone framework is also found in many bioactive molecules. Yao group [41] developed a one-pot strategy for the synthesis of α-bromo enaminones in 29-94% yields via reactions of 3-bromopropenals with anilines in the presence of catalytic amount of p-toluenesulfonic acid monohydrate (TsOH·H2O) without using an external brominating agent (Scheme 43).
Ren and co-workers [42] described a convenient approach for the synthesis of 3-difluoroalkyl phthalides from phthalaldehydic acids and difluoroenoxysilanes using catalytic amount of p-toluenesulfonic acid monohydrate (PTSA). The reaction provided a variety of 3- difluoroalkyl phthalides and cyclic difluoroalkyl ethers up to 99% yield (Scheme 44).
Ma and his group [43] disclosed p-toluenesulfonic acid monohydrate (TsOH·H2O) catalysed unprecedented cascade reaction involving C(sp2)–H addition to carbonyl and the C(sp2)–CN/C(sp2)–H coupling of 2-(2-oxo-2-arylethyl)benzonitriles with indoles. The metal-free cascade approach is operationally simple and represent a new route for the synthesis of benzo[a]carbazole derivatives with a broad substrate scope and 38-97% yields (Scheme 45).
Cankařová and co-workers [44] reported a p-toluenesulfonic acid (p-TSA) catalysed four-component condensation of an isocyanide, amine, aldehyde, and amide leading to the generation of diversely substituted mesoionic 1H-imidazol-3-ium-4-olates in one step in 10-50% yields. The mesoionic compounds formed are structurally related to sydnones and münchnones and direct precursors for anionic N-heterocyclic carbenes. Mild reaction conditions, atom economy and time efficiency make this protocol attractive (Scheme 46).
Guan group [45] described a p-toluenesulfonic acid (p-TsOH) promoted 1,3-dipolar cycloaddition of nitroolefins and sodium azide for the rapid synthesis of valuable 4-aryl-NH-1,2,3-triazoles in high yields (66-97%). 1,2,3-Triazoles have been widely applied in medicinal chemistry, agrochemistry, and materials chemistry (Scheme 47).
Wang group [46] developed an efficient methodology for the synthesis of substituted 2, 2-aminobenzophenone derivatives and aromatic β-ketosulfones. The reactions were carried out using p-toluenesulfonic acid monohydrate under microwave irradiation. The synthetic route is atom-economical and a series of pharmaceutically active 3-arylsulfonylquinolines were prepared in 72-94% yields (Scheme 48).
Singh and co-workers [47] reported the p-toluenesulfonic acid catalyzed divergent synthetic routes toward 3-aryl coumarins and indenes via annulations of ketene dithioacetals. The process is transition-metal and oxidant free and these important moieties were obtained in more than 90% yield under mild conditions (Scheme 49).
Huang group [48] developed an efficient method for regioselective acyl migration of benzoyl ester catalyzed by p-toluenesulfonic acid monohydrate (TsOH.H2O) for accessing wide range of orthogonally protected monosaccharides, which are useful intermediates for the synthesis of natural oligosaccharides (Scheme 50).
Khan and co-workers [49] disclosed metal- and solvent-free environmentally benign synthesis of 2,4-diarylquinolines via a one-pot three component reaction of arylamine, aryl aldehyde, and aryl acetylene using 30mol% p-toluenesulfonic acid monohydrate (p-TSA·H2O). The substrate scope of the reaction is wide and the products were obtained in 60-96% yields (Scheme 51).
Khan group [50] also developed a metal- and solvent-free highly efficient and straightforward synthetic route for the synthesis of 2-benzyl-3-arylquinoline derivatives from aryl amines and styrene oxides in the presence of 20 mol% p-toluenesulfonic acids (Scheme 52). The reaction also can produce 2,3-dialkylquinoline derivative using aliphatic epoxide. The metal- and solvent-free reaction conditions, operational simplicity, broad substrate scope, good to excellent yields (60-89%) of the products, and the formation of one C-N and two C-C bonds in a single step make this protocol attractive.
Wang and co-workers [51] reported a metal-free environmentally friendly approach for the synthesis of biologically important cyclohepta[b]indole in 38-80% and furo[3,4-b]carbazole frameworks in 52-85% yields respectively via a three-component reaction of indoles, tertiary propargylic alcohols, and activated alkynes using catalytic amount of p-toluenesulfonic acid (p-TsOH.H2O) (Scheme 53). A gram-scale reaction and further synthetic transformations of the products were also performed to demonstrate the practicality of the methodology.
Baskaran group [52] developed a facile domino strategy involving a sequence of semipinacol rearrangement, iterative aldol condensation, and iso-Nazarov cyclization reactions for the synthesis of biologically active cyclopent-2-enones in 55-83% yields via p-toluene sulfonic acid (p-TSOH) promoted reactions of aryloxirane and aryl aldehyde (Scheme 54).
Taylor and co-workers [53] reported para-toluenesulfonic acid (p-TSA) catalyzed desilylative heterocyclisation of tert-butyldimethylsilyl (TBS) protected γ-hydroxy-α,β-unsaturated ketones for the synthesis of substituted furans in 18-89% yields. The reaction proceeds at room temperature under mild conditions (Scheme 55).
Xinxin etal [54] described an approach of p-toluenesulfonic acid catalyzed intramolecular amination of allylic alcohols for efficient access to biologically important multisubstituted indolizines. This metal-free process enabled the divergent synthesis of these important frameworks in high yields (Scheme 56).
Anada group [55] reported p-toluenesulfonic acid (p-TsOH.H2O) catalyzed transition metal-free intramolecular 7-endo hydroarylation reaction of 1,5-diaryl-1-pentynes using 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as a solvent in 53-97% yields (Scheme 57). To demonstrate the potential of this methodology, KGP-18, an analogue of the anticancer natural product combretastatin A4 was synthesized.
Maiti and co-workers [56] described an unprecedented p-toluenesulfonic acid (p-TsOH) catalyzed [5+1] and [4+1] annulation of cyclic anhydrides or o-formylbenzoates with o-alkynylanilines for the construction of valuable fused-N-heterocycles, isoindolo/pyrido/pyrrolo-quinolinediones and isoindolo-indolones (Scheme 58). The process is metal-free, operationally simple, highly regioselective, atom economical, high yielding. All these features make this protocol sustainable.
Tandon group [57] developed a p-toluenesulfonic acid (p-TsOH) catalyzed regioselective direct carboxamidation reaction of 2-indolylmethanols with readily available isocyanoesters/isocyanides. The reaction produced benzylic regioselective amides in 67–86% yield under mild conditions (Scheme 59). This methodology provides alternative access to traditional metal-free carboxamidation approach via C–C and C–O bond formation with high atom economy.
Zhan etal [58] reported a [3 + 2] cycloaddition reaction of various 3-vinylindoles and (indol-2-yl)diphenylmethanols catalyzed by p-TsOH to synthesize functionalized cyclopenta[b]indoles in good yields (58-88%) and with high diastereoselectivity (Scheme 60).
Makino group [59] reported the first total synthesis of cadinane sesquiterpenoid alanense A via an intramolecular dehydrative Friedel–Crafts alkylation of 2,5-diaryl-2-pentanol. The combination of p-toluenesulfonic acid (p-TsOH.H2O) as a catalyst and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as a solvent provides 1,1-disubstituted tetrahydronaphthalene in 97% yield (Scheme 61).
2.2. Application of p-Toluenesulfonic Acid as a Reagent
Bhowmik research group [60] developed a new class of bisquinoline diamines photoactive amorphous molecular materials with high glass transition temperatures and high thermal stabilities. The intermediate bisquinoline dinitro compounds was synthesized by p-toluenesulfonic acid (PTSA) mediated Friedländer condensation reaction in 50-97% yields. Finally the dinitro compounds was reduced to bisquinoline diamines in 55-80% yields (Scheme 62).
Bhowmik et al. [61] elaborated a nice application of tosic acid for the synthesis of 2,4,6-tris(4-substituted phenyl)pyrylium tosylates from para-substituted benzaldehydes and para-substituted acetophenones in 17-21% yields in one pot. Further these pyrylium tosylate salts were converted to the corresponding triflimide salts in 81-93% yields. Study of the optical and thermal properties of these salts indicated that some of these pyrylium salts emitted strong blue and green light and showed excellent thermal stability (Scheme 63).
The group [62] also showed the application of pyrylium tosylates to prepare a new series of poly(pyridinium salt)s with tosylate counterion that exhibited both lyotropic liquid-crystalline and light-emitting properties (Scheme 64).
Bhowmik research group [63] disclosed a safe and inexpensive method for the synthesis of a series of four substituted 2,4,6-triphenylpyrylium tosylate salts in 11-28% yields using p-toluenesulfonic acid monohydrate and studied their optical spectroscopy and laser potential (Scheme 65).
Zhu group [64] disclosed p-toluenesulfonic acid promoted annulation of 2-alkynylanilines with activated ketones for efficient synthesis of 4-alkyl-2,3-disubstituted quinolines in 66-99% yields (Scheme 66). The substituents at the other end of the triple bond of 2-alkynylanilines makes this method valuable to access diversified 4-alkylquinolines, which are difficult to obtain by classical methods like Friedländer reaction.
Yan and co-workers [65] reported an unprecedented PTSA-promoted sequential furan annulation and aromatization in one pot for the construction of various highly substituted benzo[b]furan derivatives in 45-99% yields. Benzo[b]furans are privileged structures found in numerous natural products and biologically active compounds and this motif is also used in organic semiconductor and organic light-emitting devices (Scheme 67).
Li and co-workers [66] described an efficient and environmentally friendly approach for cyclotrimerisation of alkynes leading to the synthesis of 1,3,5-trisubstituted benzenes in 78-95% yields using p-toluenesulfonic acid monohydrate (p-TsOH·H2O) under solvent-free conditions. This method provides good to excellent yields and overcomes the shortcomings of previous methods for the synthesis of substituted benzenes (Scheme 68).
Babu et al. [67] disclosed a high-speed deprotection of Boc group from Boc amino acids and protected peptide esters using p-toluenesulfonic acid. The reaction was carried out in toluene under microwave irradiation and found to be complete in 30 s with 8-96% yields. The strategy can be extended for the concomitant removal of the Boc group and the formation of C-benzyl amino acid esters in single step (Scheme 69).
2.3. Application of p-Toluenesulfonic Acid in Combination with Metal Salts and Other Reagents
Augustine and co-workers [69] discovered that PTSA-ZnCl2 is an efficient and mild catalyst for the synthesis of 3,5-disubstituted-1,2,4-oxadiazoles from amidoximes and organic nitriles in 77-95% yields. The 1,2,4-oxadiazole moiety has been proved to be a stable ester or amide bioisostere and is found in several drug molecules [70] (Scheme 71).
Ghorai and co-workers [71] developed an interesting PTSA promoted efficient cyclization for the synthesis of biologically important 3,4-dihydropyrazino[1,2-a]indoles in 57-70% yields via an unprecedented Pictet-Spengler-detosylation cascade. This method would find tremendous utility of accessing medicinally important polycyclic indole-fused heterocycles (Scheme 72).
Zhou and co-workers [72] described a p-toluenesulfonic acid (p-TsOH.H2O) promotrd Fe-catalyzed coupling reaction between oxime ester and benzothiazole involving C–C bond cleavage of oxime ester via a single-electron transfer process. The coupling worked efficiently in water under mild conditions to produce alkyl nitrile substituted benzothiazole derivatives in 37-92% yields (Scheme 73).
Taniguchi [73] reported a ZnI2-catalyzed addition of ammonium thiocyanate to olefins in the presence of p-toluenesulfonic acid (p-TsOH.H2O) and tetrabutylammonium iodide. The reaction proceeds by a Markovnikov-type hydroisothiocyanation of alkenes followed by a radical isomerization, and gives the corresponding isothiocyanates selectively in good yields (17-88%) (Scheme 74).
Ghorai and co-workers [74] developed a mild and efficient method for the one-pot stereospecific synthesis of highly functionalized imidazolidines and oxazolidines via SN2-type ring-opening of the aziridines and epoxides with amines and subsequent intramolecular cyclization with aldehydes catalysed by p-toluenesulfonic acid (PTSA). The methodology tolerates wide range of functional groups and provide the desired products in high yields (up to 92%) with excellent stereoselectivities (de, ee > 99%) (Scheme 75). Interestingly, imidazolidines were formed as the cis-isomers, under the reaction conditions whereas oxazolidines were produced as trans-isomers exclusively. Imidazolidine and oxazolidine moieties are prevalent in bioactive natural products and pharmaceuticals.
Wu et al. [75] disclosed the rapid synthesis of Luotonin A derivatives with up to 97% yield by merging visible-light photoredox and acid catalysis using eosin Y as the photocatalyst and p-toluenesulfonic acid monohydrate (TSOH.H2O) as the co-catalyst. The reaction proceeds through Povarov cycloaddition followed by visible-light mediated dehydrogenation. Luotonin A is a cytotoxic alkaloid reported to inhibit topoisomerase I and the compound has anticancer activity (Scheme 76).
Substituted xanthene moiety exhibits a wide range of activity in biological and material sciences.
Deore and De [76] described a visible light-mediated approach for Cross-Dehydrogenative Coupling (CDC) of Xanthene with β-keto moieties using MoS2 Quantum Dot (QD) as a photoredox catalyst. This methodology is attractive as it does not require pre-functionalized starting material and the reaction conditions are mild and water is used as solvent , substrate scope is broad, yields of the products are good (42-94%) and recyclability of catalyst up to six cycles without loss of yield and selectivity (Scheme 77).
Shingare group [77] developed a highly efficient and sustainable protocol for the synthesis of dihydropyrano[2,3-c]pyrazolesin in 82-94% yields in water using polystyrene-supported p-toluenesulfonic acid as reusable catalyst (Scheme 78). Dihydropyrano[2,3-c]pyrazoles exhibit diverse biological activities. [78]
Ganga etal [79] disclosed synthesis of perfumery chemical-jasminaldehyde under solvent-free condition using p-toluene sulfonic acid (PTSA)-MCM-41 as a green, efficient, and reusable heterogeneous catalyst via cross-aldol condensation of active methylene bearing aliphatic aldehydes with aromatic aldehydes under solvent and metal-free condition. The PTSA-MCM-41 catalyst displayed high efficiency and selectivity in cross-aldol condensation reaction and was reusable (5 cycles) with no apparent loss in activity (Scheme 79).
Jeong research group [80] described polystyrene supported p-toluenesulfonic acid (PS-PTSA) promoted one-pot multicomponent reaction of aldehydes, nitroalkane, amine and active methylene compound for the synthesis of diversified pyrrole derivatives via cross-coupling-cyclization–oxidation under microwave irradiation. This solvent-free and metal-free environmentally benign protocol provides remarkable advantages such as good to excellent yields (78-93%), shorter reaction time and easy work-up procedure (Scheme 80).
Substituted quinoline motifs are present in numerous biologically active natural products and pharmaceuticals. Hence synthesis of these compounds is of much importance. Niggemann and Stopka [81] reported a metal free carboamination of unactivated alkynes for the synthesis of highly substituted quinolones in 48-93% yields in the presence of PTSA and HNTf2. Mechanistic studies indicated that the reaction proceeds via a highly reactive vinyl cation in a C–C bond formation – Schmidt reaction sequence. These findings indicate that PTSA could be used in combination with other Brønsted acid catalyst in a synergistic mode (Scheme 81).
Tang et al. [82] developed a p-toluenesulfonic acid catalyzed fluorination of α-branched ketones for the construction of fluorinated quaternary carbon centers in 23-94% yields. This method is environmentally benign, operationally simple and has a broad substrate scope. The structural motif bearing a quaternary C–F center is widely present in pharmaceuticals, agrochemicals, and functional materials (Scheme 82).
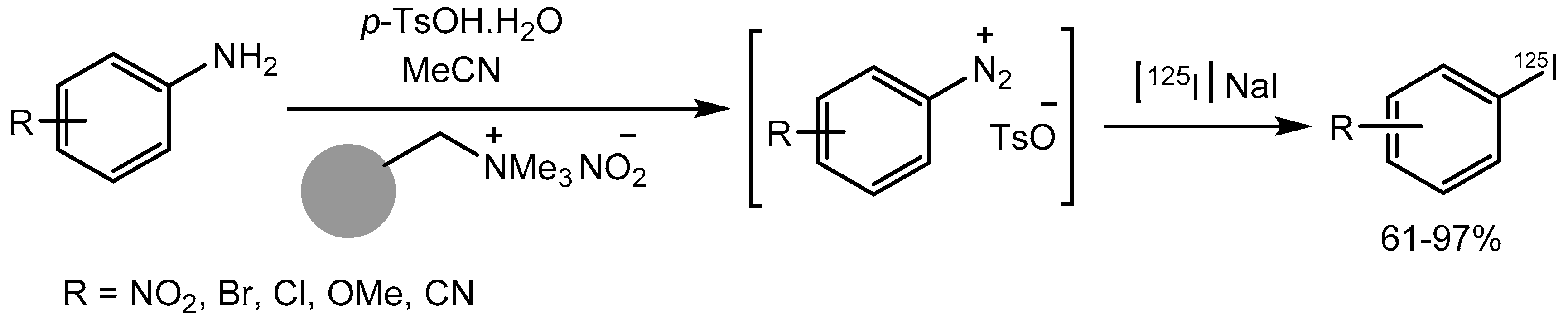
The radionuclide labelled molecules and nuclear imaging techniques has become important for the study of dynamic biochemical processes at the cellular levels useful in drug discovery process. In this context, Sloan group [83] reported a mild and efficient one-pot tandem protocol for incorporation of radioactive iodine into aryl amines via stable diazonium salts synthesized using p-toluenesulfonic acid monohydrate. The method is operationally simple and tolerate broad range of functional groups, allowing late-stage, rapid access to a variety of 125I-labelled aryl compounds in 61-97% yields and SPECT radiotracers (Scheme 58).
Scheme 83.
One-pot radioiodination of anilines using p-TsOH.H2O.
Liu et al. [84] reported the Diels–Alder reaction of [60]fullerene (C60) with ferrocenes bearing electron-withdrawing groups to afford single isomers of [2 + 4] cycloadducts of C60 in 22-59% yields (Scheme 84). Mechanistic studies indicate that in the presence of oxidant K2S2O8 and p-toluenesulfonic acid cyclopentadienes are in situ generated from electron-deficient ferrocenes which then undergoes [2 + 4] cycloadditions with dienophiles.
Sutherland and co-workers [85] also showed that multibond forming tandem reactions of anilines could be possible via stable aryl diazonium salts generated in situ using a polymer-supported nitrite reagent and p-toluenesulfonic acid. This stable aryl diazonium tosylate salts effectively converted to Heck coupled products in 46-87% yields via one-pot tandem process. This one-pot tandem process was extended for the direct synthesis of 3,4-dihydroquinolin-2-ones using 2-nitroanilines as substrates in 57-79% yields (Scheme 85). The synthetic utility of this method was demonstrated by the synthesis of quinolinone-based sodium ion channel modulator.
Chakrabarty group [86] described the efficient preparation of 2-Substituted and 1,2-disubstited benzimidazoles in 26-87% and 35-84% yields respectively from o-phenylenediamines and various aryl aldehydes using p-toluenesulphonic acid (5 mol%)-on-silica gel as a cheap and environmentally benign catalyst (Scheme 86). Benzimidazoles are useful compounds having wide range of biological activities.
Sutherland group [87] also reported a mild and efficient one-pot synthesis of N-substituted 1,2,3-benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)- dioxides in 51-91% yields (Scheme 87). The method involves the diazotisation of 2-aminobenzamides and 2-aminobenzenesulfonamides using a polymer-supported nitrite reagent and p-tosic acid to form stable aryl diazonium tosylate salts, followed by intramolecular cyclisation to produce the desired products. The reaction was compatible with a broad range of aryl functional groups and amide/sulfonamide-substituents and application of this protocol was demonstrated with the preparation of an α-amino acid containing 1,2,3-benzotriazin-4(3H)-one. Benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)-dioxides are privileged structures present in a wide range of pharmaceutically relevant compounds.
Saejong et al. [88] developed an efficient dual organocatalytic strategy for the synthesis of coumarin derivatives. A combination of p-toluenesulfonic acid monohydrate (PTSA.H2O) and piperidine effectively catalyzed the cyclization between salicylaldehydes and alkynoic esters to afford various coumarin derivatives in 9-80% yields and high regioselectivity. Mechanistic studies indicated that the conjugate addition between piperidine and alkynoic esters played a crucial role in the reaction (Scheme 88).
Jadhav and co-workers [89] developed a metal-free approach the synthesis of 2,5-furandicarboxylic acid (FDCA) from 5-hydroxymethylfurfural (HMF), fructose and glucose using a p-toluene sulfonic acid (p-TSA)-derived heterogeneous solid acid catalyst (p-TSA–POM). The synthesis proceeds via the conversion of HMF, fructose and glucose to 2,5-diformylfuran (DFF) using the p-TSA–POM catalyst and subsequent oxidation of DFF to FDCA using oxone (Scheme 89).
Olofsson group [90] reported one-pot facile synthesis of neutral and electron-rich [hydroxy(tosyloxy)iodo]arenes, (HTIBs), also known as Koser’s Reagent from iodine or aryl iodides using p-toluenesulfonic acid monohydrate (TsOH.H2O) in 23-95% yields. The reaction conditions are mild and avoid the use of expensive iodine(III) precursors and generate variety of HTIBs which are useful reagents for wide range of synthetic transformations (Scheme 90).
Wang et al. [91] described a metal-free efficient phosphorylation process via a Sandmeyer-type reaction from aryl amines employing p-toluenesulfonic acid monohydrate (TsOH.H2O). The reaction proceeds smoothly at room temperature and tolerates a wide range of functional groups. The phosphorylation products were obtained in 25-99% yields. This method is thus considered to be valuable for the formation of aromatic carbon−phosphorus bonds (Scheme 91).
Huang etal [92] disclosed a new and metal-free method for the synthesis of diverse 2,4-disubstituted quinolines in 22-67% yields via the reactions of anilines, α-keto acids and alkyl lactates in the presence of p-toluene sulfonic acid (p-TSA) and tert-butyl peroxybenzoate (TBPB) (Scheme 92). This method resulted the construction of new C=C double, C–C single and C=N double bonds without producing any organic mass-based side product. The anti-inflammatory activity of the quinolines has also been investigated.
2.4. Application of p-Toluenesulfonic Acid in Ionic Liquids
Ionic liquids are generally organic salts which comprise of organic cation and suitable inorganic or organic anion. In recent years, ionic liquids have emerged as one of the most promising green solvents for organic reactions, due to their high thermal stability, non-volatility, non-flammability and recyclability. Because of negligible vapour pressure, ionic liquids are considered an environmentally benign alternative to traditional volatile and flammable organic solvents. Ionic liquids are found to be an efficient reaction media for different catalytic avenues, like transition metal-based catalysts, Lewis acid catalysts, and enzyme catalysts. p-Toluenesulfonic acid (pTSA) have also been used to catalyze reactions in ionic liquids which would be discussed in this section. Due to their high polarity, reactions in ionic liquids have different kinetic and thermodynamic behavior than classical solvents, which often leads to improved efficiency of the reactions.
Ponzinibbio group [94] described the synthesis of α-2-deoxyglycosides in ionic liquid ([bmim]BF4) by stereoselective glycosylation of endo-glycals with various o-nucleophiles in the presence of catalytic amount of p-Toluenesulfonic acid (pTSA) in 71–95 % yield (Scheme 94). High yields of the products and reusable reaction medium (pTSA/[bmim][BF4] ionic liquid) with consistent activity makes this protocol green and sustainable.
Jiang and co-workers [95] developed p-toluenesulfonic acid (p-TSA) functionalized imidazole ionic liquids encapsulated into bismuth SBA-16 as high-efficiency catalysts for Friedel–Crafts acylation reaction of anisole with acetic anhydride in 69% yield (Scheme 95).
Nguyen group [96] disclosed “PTSA/1-alkyl-3-methylimidazolium ionic liquids” as reusable couple for efficient halogenation of fatty diols (Scheme 96). The kinetics of halogenation of fatty dialcohols proved the A2 (or SN2c) mechanism in which monohalogenation was four-fold faster than dihalogenation.
Zhao etal [97] described the synthesis of functional binuclear ionic liquids based on bis-(3-methyl-l-imidazole)butylidene double p-toluene sulfonic acid salt (Im-PTSA) and bis-(1-pyridine)butylidene double p-toluene sulfonic acid salt (Py-PTSA) and studied the catalytic activity of the binuclear ionic liquids for the esterification of succinic acid with ethanol (Scheme 97). The results show that under the optimized conditions, diethyl succinate was obtained in 93.6% and the selectivity was near up to 100%. Im-PTSA was recycled and reused at least 8 times without significant decrease in activity after drying under vacuum.
Khurana group [98] developed p-toluenesulfonic acid (pTSA) catalyzed multi-component reaction of β-naphthol, aromatic aldehydes, and cyclic 1,3-dicarbonyl compounds in ionic liquid([bmim]BF4) for the one pot synthesis of a series of 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-ones (Scheme 98). High yields of the products (80-92%) and reusable reaction medium makes this protocol efficient and environmentally benign.
A plausible mechanism for the pTSA catalyzed formation of 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one derivatives is proposed in Scheme 99.
3. Conclusions
In this review, versatile applications of p-toluenesulfonic acid in various organic reactions for synthesizing useful building blocks, heterocyclic compounds, polymers and supra molecules have been depicted. Several new synthetic approaches have been discussed for functional group transformations, photoactive materials synthesis, multicomponent reactions (MCRs) for bioactive molecule synthesis, liquid-crystalline and light-emitting materials synthesis, cyclization reaction, substitution and condensation reaction, Michael type addition reaction, tandem reaction, rearrangement reaction, transition metal catalysed reaction, privileged heterocycles and radio-labelled compound synthesis, photochemical reaction, cycloaddition reaction and Cross-Dehydrogenative Coupling (CDC). This review also demonstrates the potential of polystyrene, zeolite and silica supported p-toluenesulfonic acid in different organic reactions. We have also described the use of PTSA in environmentally benign ionic liquids for different organic transformations. PTSA promoted synthesis has produced structures with interesting properties. The summary of the synthesis and useful features will be helpful to researchers working in the fields of chemical, biological and materials sciences. We present several reactions here exemplifying the multiple roles of PTSA. Since the discovery, great progress has been made regarding the use of PTSA in synthetic organic transformations. In conclusion, the advances in synthetic methodology using PTSA and a better understanding of the reaction mechanisms have been very significant. Therefore, we expect that this review will encourage more organic chemists to continue investing their creative strength in the discovery of new reactions using this environmentally benign solid organic acid with novel mechanisms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Pradip Bhowmik; Software, Pradip Bhowmik; Supervision, Pradip Bhowmik; Writing – original draft, Biplab Banerjee and Haesook Han; Writing – review & editing, Biplab Banerjee, Haesook Han and Pradip Bhowmik.
Funding
This work is in part supported by the NSF under Grant No. 0447416 (NSF EPSCoR RING-TRUE III), NSF-Small Business Innovation Research (SBIR) Award (Grant OII-0610753), NSF-STTR Phase I IIP-0740289, and NASA GRC Contract No. NNX10CD25P. Financial support is also provided by University Grants Commission, New Delhi through Grant No.F.4−5/2018(FRP-Start-up-grant)(Cycle-IV)(BSR) and by SERB-DST, New Delhi through Grant No. CRG/2020/003773. .
Acknowledgments
P.K.B. and H.H. thank the University of Nevada Las Vegas for the support of this research. B.B thank the Vice Chancellor of Central University of Punjab, Bathinda, for providing necessary facilities for this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baghernejad, B. Application of p-toluenesulfonic Acid (PTSA) in Organic Synthesis. Current Organic Chemistry 2011, 15, 3091–3097. [Google Scholar] [CrossRef]
- Shangpliang, O.R.; Wanniang, K.; Kshiar, B.; Marpna, I.D.; Lipon, T.M.; Mizar, P.; Myrboh, B. PTSA-catalyzed reaction of alkyl/aryl methyl ketones with aliphatic alcohols in the presence of selenium dioxide: A protocol for the generation of an α-ketoacetals library. ACS omega 2019, 4, 6035–6043. [Google Scholar] [CrossRef] [PubMed]
- Kharkongor, I.; Myrboh, B. One-pot synthesis of α-ketoacetals from aryl methyl ketones in the presence of selenous acid catalyzed by boron trifluoride etherate. Tetrahedron letters 2015, 56, 4359–4362. [Google Scholar] [CrossRef]
- Wang, X.; Jia, C.; Feng, Y.; Wang, L.; Cui, X. One-Pot Synthesis of N-Alkyl Benzotriazoles via a Brønsted Acid-Catalyzed Three-Component Reaction. Adv. Synth. Catal. 2018, 360, 374–378. [Google Scholar] [CrossRef]
- Gao, H.; Sun, J.; Yan, C.-G. Four-component reaction of cyclic amines, 2-aminobenzothiazole, aromatic aldehydes and acetylenedicarboxylate. Beilstein J. Org. Chem. 2013, 9, 2934–2939. [Google Scholar] [CrossRef] [PubMed]
- Gogoi, D.; Devi, R.; Pahari, P.; Sarma, B.; Das, S.K. cis-Diastereoselective synthesis of chroman-fused tetralins as B-ring-modified analogues of brazilin. Beilstein J. Org. Chem. 2016, 12, 2816–2822. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Oh, J.; Jeong, Y.T. p-Toluenesulfonic acid-catalyzed one-pot synthesis of 2-amino-4-substituted-1,4-dihydrobenzo [4, 5] imidazolo [1, 2-a] pyrimidine-3-carbonitriles under neat conditions. C. R. Chimie 2014, 17, 484–489. [Google Scholar] [CrossRef]
- Nofal, Z.; Fahmy, H.; Mohamed, H. Synthesis, antimicrobial and molluscicidal activities of new benzimidazole derivatives. Arch. Pharm. Res. 2002, 25, 28–38. [Google Scholar] [CrossRef]
- Creencia, E. C.; Tsukamoto, M.; Horaguchi, T. , One-pot-one-step, microwave-assisted Fischer indole synthesis. Journal of Heterocyclic Chemistry 2011, 48, 1095–1102. [Google Scholar] [CrossRef]
- Sanz, R.; Martínez, A.; Álvarez-Gutiérrez, J.M.; Rodríguez, F. Metal-Free Catalytic Nucleophilic Substitution of Propargylic Alcohols. Eur. J. Org. Chem. 2006, 1383–1386. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.; Li, C.; Yin, K.; Ye, D.; Jia, X. PTSA-catalyzed green synthesis of 1, 3, 5-triarylbenzene under solvent-free conditions. Green Chem. 2010, 12, 1370–1372. [Google Scholar] [CrossRef]
- Poomathi, N.; Mayakrishnan, S.; Muralidharan, D.; Srinivasan, R.; Perumal, P.T. Reaction of isatins with 6-amino uracils and isoxazoles: isatin ring-opening vs. annulations and regioselective synthesis of isoxazole fused quinoline scaffolds in water. Green Chem. 2015, 17, 3362–3372. [Google Scholar] [CrossRef]
- Roberts, B.A.; Cave, G.W.; Raston, C.L.; Scott, J. L. Solvent-free synthesis of calix [4] resorcinarenes. Green Chem. 2001, 3, 280–284. [Google Scholar] [CrossRef]
- Antesberger, J.; Cave, G. W. V.; Ferrarelli, M. C.; Heaven, M. W.; Raston, C. L.; Atwood, J. L. Solvent-free, direct synthesis of supramolecular nano-capsules. Chem. Commun. 2005, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Rajanarendar, E.; Venkateshwarlu, P.; Krishna, S.R.; Reddy, K.G.; Thirupathaiah, K. One-Pot Three Component Domino Reaction for the Synthesis of Novel Isoxazolo [2, 3-c][1, 3, 5] Thiadiazepin-2-Ones Catalyzed by PTSA—A Green Chemistry Approach. Green and Sustainable Chemistry 2015, 5, 107–114. [Google Scholar] [CrossRef]
- Le Bras, G.; Hamze, A.; Messaoudi, S.; Provot, O.; Le Calvez, P.-B.; Brion, J.-D.; Alami, M. Synthesis of isocoumarin via PTSA-catalyzed annulation of diarylalkynes. Synthesis 2008, 1607–1611. [Google Scholar]
- Mogilaiah, K.; Sudhakar, G. R. PTSA-catalyzed Friedlander condensation in the solid state. Ind. J. Chem. 2003, 42B, 1170–1171. [Google Scholar]
- Mishra, A.K.; Biswas, S. Brønsted acid catalyzed functionalization of aromatic alcohols through nucleophilic substitution of hydroxyl group. J. Org. Chem. 2016, 81, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.-J.; Ye, G.; Jia, F.; He, Z.; Ke, H.; Yao, H.; Fan, Z.; Jiang, W. Regioselective synthesis of methylene-bridged naphthalene oligomers and their host–guest chemistry. J. Org. Chem. 2017, 82, 9570–9575. [Google Scholar] [CrossRef]
- Gülten, Ş.; Gezer, U.; Gündoğan, E.A. Fast and Efficient One-Pot Three-Component Synthesis of Some 1, 2, 3, 4-Tetrahydro-6-methyl-N-phenyl-5-pyrimidinecarboxamide Derivatives via Biginelli Condensation Reaction. Letters in Organic Chemistry 2020, 17, 366–371. [Google Scholar] [CrossRef]
- Mousavi, M.R.; Maghsoodlou, M.T. Catalytic systems containing p-toluenesulfonic acid monohydrate catalyzed the synthesis of triazoloquinazolinone and benzimidazoquinazolinone derivatives. Monatsh Chem 2014, 145, 1967–1973. [Google Scholar] [CrossRef]
- Liu, P.; Pan, Y.-M.; Xu, Y.-L.; Wang, H.-S. PTSA-catalyzed Mannich-type–cyclization–oxidation tandem reactions: one-pot synthesis of 1, 3, 5-substituted pyrazoles from aldehydes, hydrazines and alkynes. Org. Biomol. Chem. 2012, 10, 4696–4698. [Google Scholar] [CrossRef] [PubMed]
- Marras, V.; Caboni, P.; Secci, F.; Guillot, R.; Aitken, D.J.; Frongia, A. A Brønsted acid catalyzed tandem reaction for the diastereoselective synthesis of cyclobuta-fused tetrahydroquinoline carboxylic esters. Org. Biomol. Chem. 2021, 19, 8912–8916. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Tanaka, H. Syntheses of 1, 2, 3, 4-tetrahydroisoquinolines from N-sulfonyl-phenethylamines and aldehydes. Chem. Pharm. Bull. 1977, 25, 1732–1739. [Google Scholar] [CrossRef]
- Salama, S.K.; Darweesh, A.F.; Abdelhamid, I.A.; Elwahy, A.H. p-TSA Catalyzed One-Pot Synthesis of Some Novel Bis (Hexahydroacridine-1, 8-Diones) and Bis (Tetrahydrodipyrazolo [3, 4-b: 4′, 3′-e] Pyridines) Derivatives. Polycyclic Aromatic Compounds 2021, 41, 1392–1405. [Google Scholar] [CrossRef]
- Mohebat, R.; Yazdani Elah Abadi, A.; Maghsoodlou, M.-T.; Mohammadi, M. PTSA-catalyzed four-component domino reactions for the one-pot synthesis of functionalized 11H-benzo [a] benzo [6, 7] chromeno [2, 3-c] phenazine-11, 16 (17H)-diones in PEG. Res Chem Intermed 2016, 42, 5915–5926. [Google Scholar] [CrossRef]
- Khosropour, A.R.; Khodaei, M.M.; Moghannian, H. A facile, simple and convenient method for the synthesis of 14-alkyl or aryl-14-H-dibenzo [a, j] xanthenes catalyzed by pTSA in solution and solvent-free conditions. Synlett 2005, 0955–0958. [Google Scholar] [CrossRef]
- Khorshidi, M.; Heravi, M.M.; Beheshtia, Y.S.; Baghernejad, B. Novel One-Pot Synthesis of New Oxindole Derivatives Catalyzed by PTSA. Synth. Commun. 2011, 41, 2899–2904. [Google Scholar] [CrossRef]
- Park, J.; Yun, J.; Kim, J.; Jang, D.-J.; Park, C.H.; Lee, K. Brønsted Acid–Catalyzed Meyer–Schuster rearrangement for the synthesis of α, β-unsaturated carbonyl compounds. Synth. Commun. 2014, 44, 1924–1929. [Google Scholar] [CrossRef]
- Anuradha, V.; Srinivas, P.; Aparna, P.; Rao, J.M. p-Toluenesulfonic acid catalyzed regiospecific nitration of phenols with metal nitrates. Tetrahedron letters 2006, 47, 4933–4935. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Ali, M.; Sharma, K.N.; Joshi, R.K. Metal-free, PTSA catalyzed facile synthesis of β-ketoacetal from β-chlorocinnamaldehyde. Tetrahedron Letters 2018, 59, 3188–3193. [Google Scholar] [CrossRef]
- Ji, S.-J.; Wang, S.-Y. An expeditious synthesis of β-indolylketones catalyzed by p-toluenesulfonic acid (PTSA) using ultrasonic irradiation. Ultrasonics sonochemistry 2005, 12, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, P.; Perumal, S.; Yogeeswari, P.; Sriram, D. A facile four-component sequential protocol in the expedient synthesis of novel 2-aryl-5-methyl-2, 3-dihydro-1H-3-pyrazolones in water and their antitubercular evaluation. Eur. J. Med. Chem. 2011, 46, 4530–4536. [Google Scholar] [CrossRef] [PubMed]
- Naidu, P.S.; Kolita, S.; Sharma, M.; Bhuyan, P.J. Reductive alkylation of α-keto imines catalyzed by PTSA/FeCl3: Synthesis of indoles and 2, 3′-biindoles. J. Org. Chem. 2015, 80, 6381–6390. [Google Scholar] [CrossRef] [PubMed]
- Ziyaei Halimehjani, A.; Khoshdoun, M. Tandem esterification/1, 4-addition-type Friedel–Crafts alkylation reactions of phenols/naphthols with olefinic thioazlactones: access to functionalized 1, 2-dihydrobenzo [f] chromen-3-ones and 3, 4-dihydrochromen-2-ones. J. Org. Chem. 2016, 81, 5699–5704. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, N.; Banerjee, P. Regioselective Brønsted Acid-Catalyzed Annulation of Cyclopropane Aldehydes with N′-Aryl Anthranil Hydrazides: Domino Construction of Tetrahydropyrrolo [1, 2-a] quinazolin-5 (1 H) ones. J. Org. Chem. 2020, 85, 3393–3406. [Google Scholar] [CrossRef] [PubMed]
- Chantana, C.; Sirion, U.; Iawsipo, P.; Jaratjaroonphong, J. Short total synthesis of (±)-gelliusine E and 2, 3′-bis (indolyl) ethylamines via PTSA-catalyzed transindolylation. J. Org. Chem. 2021, 86, 13360–13370. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Zeng, Y.; Zhu, G.; Zhang, J.; Chen, P.; Cheng, Y.; Fang, Z.; Guo, K. Sustainable bio-based furan epoxy resin with flame retardancy. Polymer Chemistry 2019, 10, 2370–2375. [Google Scholar] [CrossRef]
- Porcu, S.; Luridiana, A.; Martis, A.; Frongia, A.; Sarais, G.; Aitken, D.J.; Boddaert, T.; Guillot, R.; Secci, F. Acid-catalyzed synthesis of functionalized arylthio cyclopropane carbaldehydes and ketones. Chem. Commun. 2018, 54, 13547–13550. [Google Scholar] [CrossRef]
- Tharra, P.; Baire, B. The Z-enoate assisted, Meyer–Schuster rearrangement cascade: unconventional synthesis of α-arylenone esters. Chem. Commun. 2016, 52, 12147–12150. [Google Scholar] [CrossRef]
- Suresh, S.; Bhimrao Patil, P.; Yu, P.H.; Fang, C.C.; Weng, Y.Z. ; Kavala,V.; Yao, C.F. A Study of the Reactions of 3-Bromopropenals with Anilines for the Synthesis of α-Bromo Enaminones. Adv. Synth. Catal. 2021; 363, 4915–4925. [Google Scholar]
- Liu, S.; Li, Y.; Wang, F.; Ma, C.; Yang, G.; Yang, J.; Ren, J. p-Toluenesulfonic acid-catalyzed reaction of phthalaldehydic acids with difluoroenoxysilanes: Access to 3-difluoroalkyl phthalides. Synthesis 2022, 54, 161–170. [Google Scholar]
- Tang, L.; Jiang, S.; Huang, X.; Song, Z.; Wang, J.-b.; Ma, M.; Chen, B.; Ma, Y. Cascade of C(sp2)–H Addition to Carbonyl and C(sp2)–CN/C(sp2)–H Coupling Enabled by Brønsted Acid: Construction of Benzo [a] carbazole Frameworks. Org. Lett. 2022, 24, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Cankařová, N.; Nemec, I.; Krchňák, V. p-TSA-Mediated Four-Component Reaction: One-Step Access to Mesoionic 1H-Imidazol-3-ium-4-olates, Direct NHC Precursors. Adv. Synth. Catal. 2022, 364, 2996–3003. [Google Scholar] [CrossRef]
- Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. p-Toluenesulfonic acid mediated 1, 3-dipolar cycloaddition of nitroolefins with NaN3 for synthesis of 4-aryl-NH-1, 2, 3-triazoles. Org. Lett. 2014, 16, 5728–5731. [Google Scholar] [CrossRef]
- Chan, C.-K.; Lai, C.-Y.; Lo, W.-C.; Cheng, Y.-T.; Chang, M.-Y.; Wang, C.-C. p-TsOH-mediated synthesis of substituted 2, 4-diaryl-3-sulfonylquinolines from functionalized 2-aminobenzophenones and aromatic β-ketosulfones under microwave irradiation. Org. Biomol. Chem. 2020, 18, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Singh, S. P.; Sharma, P.; Singh, A. , Brønsted acid catalyzed annulations of ketene dithioacetals: synthesis of 3-aryl coumarins and indenes. Org. Biomol. Chem. 2022, 20, 8907–8911. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-Y.; Liu, A.-L.; Fan, H.-J. S.; Wang, L.; Xu, Z.-N.; Ding, X.-G.; Huang, B.-S. , TsOH-catalyzed acyl migration reaction of the Bz-group: innovative assembly of various building blocks for the synthesis of saccharides. Org. Biomol. Chem. 2023, 21, 1537–1548. [Google Scholar] [CrossRef]
- Faraz, S.; Yashmin, S.; Marathe, M. D.; Khan, A. T. , Environmentally benign synthesis of 2, 4-diarylquinolines under metal-& solvent-free conditions. Tetrahedron Letters 2023, 120, 154433. [Google Scholar]
- Faraz, S.; Kumar, M.; Khan, A. T.; Ponneganti, S.; Radhakrishnanand, P. , Metal-and solvent-free synthesis of 2-benzyl-3-arylquinoline using a pseudo-three-component reaction. Tetrahedron Letters 2023, 115, 154283. [Google Scholar] [CrossRef]
- Yin, H.; Wu, Y.; Jiang, Y.; Wang, M.; Wang, S. , Synthesis of Cyclohepta [b] indoles and Furo [3, 4-b] carbazoles from Indoles, Tertiary Propargylic Alcohols, and Activated Alkynes. Org. Lett. 2023, 25, 3078–3082. [Google Scholar] [CrossRef]
- Reddy, N. R.; Gouse, S.; Selvaraju, S.; Baskaran, S. , Domino Semipinacol/Iterative Aldol/Iso-Nazarov Cyclization to Triaryl-cyclopentenone: Enantioselective Synthesis of Combretastatin A-4 Analogues. Org. Lett. 2022, 24, 4240–4245. [Google Scholar] [CrossRef]
- Babcock, E. G.; Rahman, M. S.; Taylor, J. E. , Brønsted acid-catalysed desilylative heterocyclisation to form substituted furans. Org. Biomol. Chem. 2023, 21, 163–168. [Google Scholar] [CrossRef]
- Lv, X.; Gao, P.; Zhao, X.; Jiang, Z. , Metal-Free Construction of Multisubstituted Indolizines via Intramolecular Amination of Allylic Alcohols. J. Org. Chem. 2023, 88, 9459–9468. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Sueki, S.; Anada, M. , Brønsted Acid-Catalyzed Intramolecular 7-endo Hydroarylation Reaction of 1, 5-Diaryl-1-pentynes. Adv. Synth. Catal. 2023, 365, 1471–1476. [Google Scholar] [CrossRef]
- Das, A.; Ajarul, S.; Debnath, S.; Hota, P.; Maiti, D. K. , Bro̷nsted Acid-Catalyzed [5+ 1] and [4+ 1] Annulation of Cyclic Anhydrides with o-Alkynylanilines to Construct Fused-N-Heterocycles. J. Org. Chem. 2023, 88, 15073–15084. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, T. Y.; Bisht, S.; Chorol, S.; Bhujbal, S. M.; Bharatam, P. V.; Tandon, V. , Bronsted Acid-Catalyzed Regioselective Carboxamidation of 2-Indolylmethanols with Isonitriles. J. Org. Chem. 2023, 88, 10412–10425. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.-C.; Sun, J.; Sun, Q.; Han, Y.; Yan, C.-G. , Acid-Modulated Construction of Cyclopenta [b] indole and Cyclohepta [b] indole via Unprecedented C3/C2 Carbocation Rearrangement. J. Org. Chem. 2023, 88, 5440–5456. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Fukuda, R.; Sueki, S.; Anada, M. , Total Synthesis of Alanense A through an Intramolecular Friedel–Crafts Alkylation. J. Org. Chem. 2024, 89, 2050–2054. [Google Scholar] [CrossRef]
- Bhowmik, P.K.; Nedeltchev, A.K.; Han, H.; Jo, T.S.; Koh, J.J.; Senthilkumar, L. Umadevi, P. Photoactive amorphous molecular Materials based on bisquinoline diamines and their synthesis by Friedländer condensation reaction. J. Photochem. Photobiol. A: Chem. 2014; 283, 45–55. [Google Scholar]
- Bhowmik, P.K.; Lee, C.I.; Koh, J.J.; Han, H.; Jubair, A.; Kartazaev, V.; Gayen, S.K. Synthesis, optical, and thermal properties of 2,4,6-tris(4-substituted phenyl)pyrylium tosylates and triflimides. J. Mol. Struc. 2020, 1202, 127325. [Google Scholar] [CrossRef]
- Bhowmik, P.K.; Burchett, R.A.; Han, H.; Cebe, J.J. Synthesis and characterization of poly(pyridinium salt)s with organic counterion exhibiting both lyotropic liquid-crystalline and light-emitting properties. Macromolecules 2001, 34, 7579–7581. [Google Scholar] [CrossRef]
- Koh, J.J.; Lee, C.I.; Ciulei, M.A.; Han, H.; Bhowmik, P. K.; Kartazaev, V.; Gayen, S.K. Synthesis, optical spectroscopy and laser potential of pyrylium tosylates. J. Mol. Struc. 2018, 1171, 458–465. [Google Scholar] [CrossRef]
- Peng, C.; Wang, Y.; Liu, L.; Wang, H.; Zhao, J.; Zhu, Q. p-Toluenesulfonic Acid Promoted Annulation of 2-Alkynylanilines with Activated Ketones: Efficient Synthesis of 4-Alkyl-2, 3-Disubstituted Quinolines. Eur. J. Org. Chem. 2010, 818–822. [Google Scholar] [CrossRef]
- Ao, J.; Liu, Y.; Jia, S.; Xue, L.; Li, D.; Tan, Y.; Qin, W.; Yan, H. Acid-promoted furan annulation and aromatization: An access to benzo [b] furan derivatives. Tetrahedron 2018, 74, 433–440. [Google Scholar] [CrossRef]
- Gao, Q.; Bao, F.-p.; Feng, X.-j.; Pan, Y.-m.; Wang, H.-s.; Li, D.-p. An efficient approach to the cyclotrimerisation of alkynes: solvent-free synthesis of 1, 3, 5-trisubstituted benzenes using p-toluenesulfonic acid monohydrate. Arkivoc 2013, 3, 49–60. [Google Scholar] [CrossRef]
- Babu, V.V.S.; Patil, B.S.; Vasanthakumar, G.R. MW-Enhanced High-Speed Deprotection of Boc Group Using p-TsOH and Concommitant Formation of N-Me-Amino Acid Benzyl Ester p-TsOH Salts. Synth. Commun. 2005, 35, 1795–1802. [Google Scholar] [CrossRef]
- Tian, S.; Wang, C.; Xia, J.; Wan, J.P.; Liu, Y. Transition Metal-Free, Free-Radical Sulfenylation of the α-C (sp3)− H Bond in Arylacetamides and Its Application Toward 2-Thiomethyl Benzoxazoles Synthesis. Adv. Synth. Catal. 2021, 363, 4627–4631. [Google Scholar] [CrossRef]
- Augustine, J.K.; Akabote, V.; Hegde, S.G.; Alagarsamy, P. PTSA− ZnCl2: An efficient catalyst for the synthesis of 1, 2, 4-oxadiazoles from amidoximes and organic nitriles. J. Org. Chem. 2009, 74, 5640–5643. [Google Scholar] [CrossRef]
- Zhang, H.-Z.; Kasibhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Ollis-Mason, K.; Qiu, L.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery and structure− activity relationship of 3-aryl-5-aryl-1, 2, 4-oxadiazoles as a new series of apoptosis inducers and potential anticancer agents. J. Med. Chem. 2005, 48, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Wani, I.A.; Das, S.; Mondal, S.; Ghorai, M.K. Stereoselective construction of pyrazinoindoles and oxazinoindoles via ring-opening/pictet-spengler reaction of aziridines and epoxides with 3-methylindoles and carbonyls. J. Org. Chem. 2018, 83, 14553–14567. [Google Scholar] [CrossRef]
- Zhou, W.; Long, Y.; Xiang, H.; Xu, B.; Zhou, X. Iron-catalyzed C–C bond cleavage of oximes for direct coupling of benzothiazole in water. J. Org. Chem. 2023, 88, 4875–4879. [Google Scholar] [CrossRef]
- Taniguchi, N. Zinc-Catalyzed Markovnikov-Type Hydroisothiocyanation of Alkenes with Ammonium Thiocyanate. Synlett 2023, 34, 73–76. [Google Scholar] [CrossRef]
- Tarannum, S.; Sk, S.; Das, S.; Wani, I. A.; Ghorai, M. K. Stereoselective syntheses of highly functionalized imidazolidines and oxazolidines via ring-opening cyclization of activated aziridines and epoxides with amines and aldehydes. J. Org. Chem. 2019, 85, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Dong, W.; Fan, S.; Yuan, Y.; Liang, C.; Chen, A.; Yin, Z.; Zhang, Z. Rapid Synthesis of Luotonin A Derivatives via Synergistic Visible-Light Photoredox and Acid Catalysis. J. Org. Chem. 2022, 87, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Deore, J.P.; De, M. Photoredox C(sp3)−C(sp3) Cross-Dehydrogenative Coupling of Xanthene with β-keto Moiety using MoS2 Quantum Dot (QD) Catalyst. Adv. Synth. Catal. 2022, 364, 3049–3058. [Google Scholar] [CrossRef]
- Chaudhari, M.A.; Gujar, J.B.; Kawade, D.S.; Jogdand, N.R.; Shingare, M.S. A highly efficient and sustainable synthesis of dihydropyrano [2, 3-c] pyrazoles using polystyrene-supported p-toluenesulfonic acid as reusable catalyst. Cogent Chemistry 2015, 1, 1063830. [Google Scholar] [CrossRef]
- Abdelrazek, F.M.; Metz, P.; Metwally, N.H.; El-Mahrouky, S.F. Synthesis and molluscicidal activity of new cinnoline and pyrano [2, 3-c] pyrazole derivatives. Archiv der Pharmazie: An International Journal Pharmaceutical and Medicinal Chemistry, 2006; 339, 456–460. [Google Scholar]
- Ganga, V.S.R.; Abdi, S.H.; Kureshy, R.I.; Noor-ul, H.K.; Bajaj, H.C. p-Toluene sulfonic acid (PTSA)-MCM-41 as a green, efficient and reusable heterogeneous catalyst for the synthesis of jasminaldehyde under solvent-free condition. Journal of Molecular Catalysis A: Chemical, 2016; 420, 262–271. [Google Scholar]
- Shinde, V.V.; Lee, S.D.; Jeong, Y.S.; Jeong, Y.T. p-Toluenesulfonic acid doped polystyrene (PS-PTSA): solvent-free microwave assisted cross-coupling-cyclization–oxidation to build one-pot diversely functionalized pyrrole from aldehyde, amine, active methylene, and nitroalkane. Tetrahedron Letters 2015, 56, 859–865. [Google Scholar] [CrossRef]
- Stopka, T.; Niggemann, M. Metal free carboamination of internal alkynes–an easy access to polysubstituted quinolines. Chem. Commun. 2016, 52, 5761–5764. [Google Scholar] [CrossRef]
- Tang, S.-Z.; Bian, H.-L.; Zhan, Z.-S.; Chen, M.-E.; Lv, J.-W.; Xie, S.; Zhang, F.-M. p-Toluenesulfonic acid catalysed fluorination of α-branched ketones for the construction of fluorinated quaternary carbon centres. Chem. Commun. 2018, 54, 12377–12380. [Google Scholar] [CrossRef]
- Sloan, N. L.; Luthra, S. K.; McRobbie, G.; Pimlott, S. L.; Sutherland, A. A one-pot radioiodination of aryl amines via stable diazonium salts: preparation of 125 I-imaging agents. Chem. Commun. 2017, 53, 11008–11011. [Google Scholar] [CrossRef]
- Liu, Z.; Yin, Z.-C.; Lu, W.-Q.; Zhou, D.-B.; Wang, G.-W. Unexpected Diels–Alder reaction of [60] fullerene with electron-deficient ferrocenes as cyclopentadiene surrogates. Chem. Commun. 2021, 57, 13389–13392. [Google Scholar] [CrossRef]
- Faggyas, R.k.J.; Grace, M.; Williams, L.; Sutherland, A. Multibond forming tandem reactions of anilines via stable aryl diazonium salts: one-pot synthesis of 3, 4-dihydroquinolin-2-ones. J. Org. Chem. 2018, 83, 12595–12608. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, M.; Mukherjee, R.; Karmakar, S.; Harigaya, Y. Tosic acid-on-silica gel: a cheap and eco-friendly catalyst for a convenient one-pot synthesis of substituted benzimidazoles. Monatsh Chem 2007, 138, 1279–1282. [Google Scholar] [CrossRef]
- McGrory, R.; Faggyas, R. J.; Sutherland, A. One-pot synthesis of N-substituted benzannulated triazoles via stable arene diazonium salts. Org. Biomol. Chem. 2021, 19, 6127–6140. [Google Scholar] [CrossRef] [PubMed]
- Saejong, P.; Somprasong, S.; Rujirasereesakul, C.; Luanphaisarnnont, T. Direct Synthesis of Coumarin Derivatives from Alkynoic Esters via Dual Organocatalysis. Synlett 2022, 33, 1399–1404. [Google Scholar]
- Tamboli, A.T.B.; Kirdant, S.P.; Jadhav, V.H. Metal-free approach towards efficient synthesis of FDCA using a p-toluene sulfonic acid (p-TSA)-derived heterogeneous solid acid catalyst and oxone over two steps from HMF, fructose and glucose. New J. Chem. 2022, 46, 10272–10279. [Google Scholar] [CrossRef]
- Merritt, E.A.; Carneiro, V.M.; Silva Jr, L.F.; Olofsson, B. Facile synthesis of Koser’s reagent and derivatives from iodine or aryl iodides. J. Org. Chem. 2010, 75, 7416–7419. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, D.; Mo, F.; Zhang, Y.; Wang, J. Metal-free aromatic carbon–phosphorus bond formation via a sandmeyer-type reaction. J. Org. Chem. 2016, 81, 11603–11611. [Google Scholar] [CrossRef]
- Huang, L.; Yang, L.; Wan, J.-P.; Zhou, L.; Liu, Y.; Hao, G. Metal-free three-component assemblies of anilines, α-keto acids and alkyl lactates for quinoline synthesis and their anti-inflammatory activity. Org. Biomol. Chem. 2022, 20, 4385–4390. [Google Scholar] [CrossRef]
- Kanaujiya, V. K.; Tiwari, V.; Baranwal, S.; Srivastava, V.; Kandasamy, J. Denitrosation of Aryl-N-nitrosamines by a Transnitrosation Strategy Using Ethanethiol and p-Toluenesulfonic Acid under Mild Reaction Conditions. Synlett 2023, 34, 970–974. [Google Scholar]
- Dı´az, G.; Ponzinibbio, A.; Bravo, R. D. pTSA/[bmim][BF4] Ionic Liquid: A Powerful Recyclable Catalytic System for the Synthesis of α-2-Deoxyglycosides. Top Catal, 2012, 55, 644–648. [Google Scholar] [CrossRef]
- Gao, G.; Zhao, Q.; Yang, C.; Jiang, T. p-Toluenesulfonic acid functionalized imidazole ionic liquids encapsulated into bismuth SBA-16 as high-efficiency catalysts for Friedel–Crafts acylation reaction. Dalton Trans. 2021, 50, 5871–5882. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.-P.; Kirilov, P.; Matondo, H.; Baboulène, M. The reusable couple “PTSA/1-alkyl-3-methylimidazolium ionic liquids”: excellent reagents–catalysts for halogenation of fatty diols. Journal of Molecular Catalysis A: Chemical 2004, 218, 41–45. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, M.; Ge, J.; Zhang, J.; Ren, P. Synthesis of binuclear ionic liquids and their catalytic activity for esterification. Chin. J. Org. Chem. 2012, 32, 2382. [Google Scholar] [CrossRef]
- Khurana, J. M.; Magoo, D. pTSA-catalyzed one-pot synthesis of 12-aryl-8, 9, 10, 12-tetrahydrobenzo [a] xanthen-11-ones in ionic liquid and neat conditions. Tetrahedron Letters 2009, 50, 4777–4780. [Google Scholar] [CrossRef]
Scheme 1.
PTSA catalyzed synthesis of α-ketoacetals using selenium dioxide.
Scheme 2.
Plausible mechanism for α-ketoacetal formation.
Scheme 3.
PTSA catalyzed synthesis of N-alkyl benzotriazoles.
Scheme 4.
Synthesis of functionalized pyrrolidinones.
Scheme 5.
Proposed mechanism.
Scheme 6.
Synthesis of chroman-fused tetralins.
Scheme 7.
PTSA catalysed synthesis of 2-amino-4-substituted-1,4- dihydrobenzolo[4,5]imidazolo[1,2-a]pyrimidine-3-carbonitriles.
Scheme 8.
Plausible mechanism for the catalytic activity of p-TSA.
Scheme 10.
P-TSA catalysed propargylic substitution of alkynols with nucleophiles.
Scheme 11.
Propargylic substitution of alkynols with allyltrimethylsilane.
Scheme 12.
Cyclotrimerization of aryl methyl ketones.
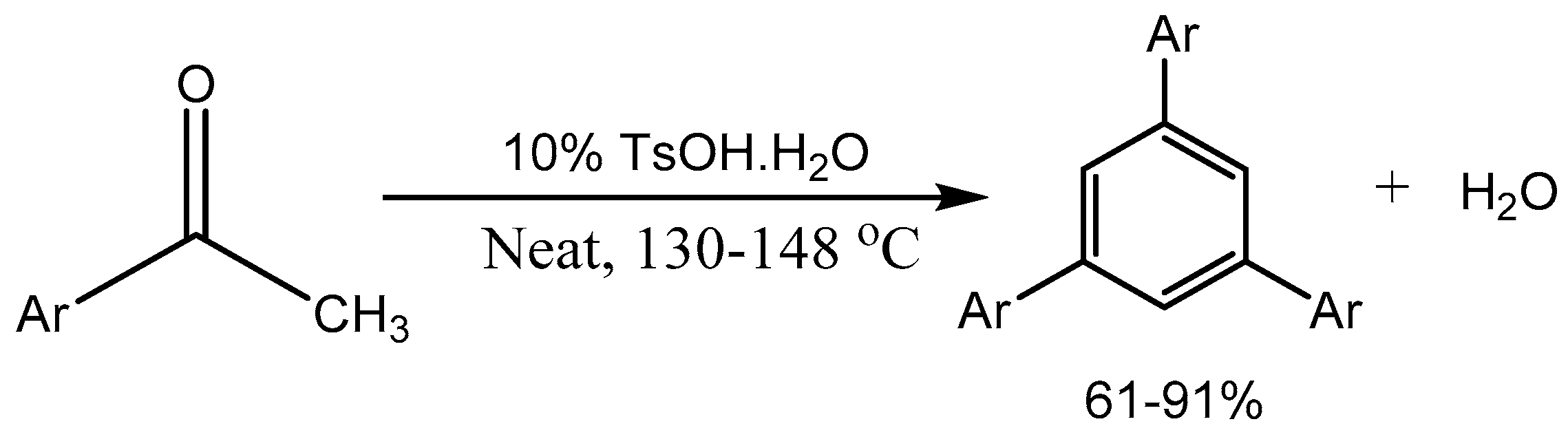
Scheme 14.
Synthesis of calix[4]resorcinarenes.
Scheme 14.
Synthesis of calix[4]resorcinarenes.
Scheme 15.
Synthesis of pyrogallol[4]arene.
Scheme 15.
Synthesis of pyrogallol[4]arene.

Scheme 17.
PTSA catalysed one pot synthesis of 3-aryl-isocoumarins.
Scheme 18.
PTSA catalysed synthesis of 1,8-naphthyridines.
Scheme 19.
P-TSA catalysed nucleophilic substitution of naphthyl C−OH bonds.
Scheme 20.
P-TsOH catalysed synthesis of methylene-bridged naphthalene oligomers.
Scheme 21.
PTSA·H2O catalysed synthesis of 1,2,3,4-tetrahydro 2-pyrimidinone/thione derivatives.
Scheme 22.
P-TsOH·H2O catalysed synthesis of benzimidazoquinazolinone and triazoloquinazolinone derivatives.
Scheme 22.
P-TsOH·H2O catalysed synthesis of benzimidazoquinazolinone and triazoloquinazolinone derivatives.
Scheme 23.
PTSA catalysed synthesis of 1,3,5- trisubstituted pyrazoles.
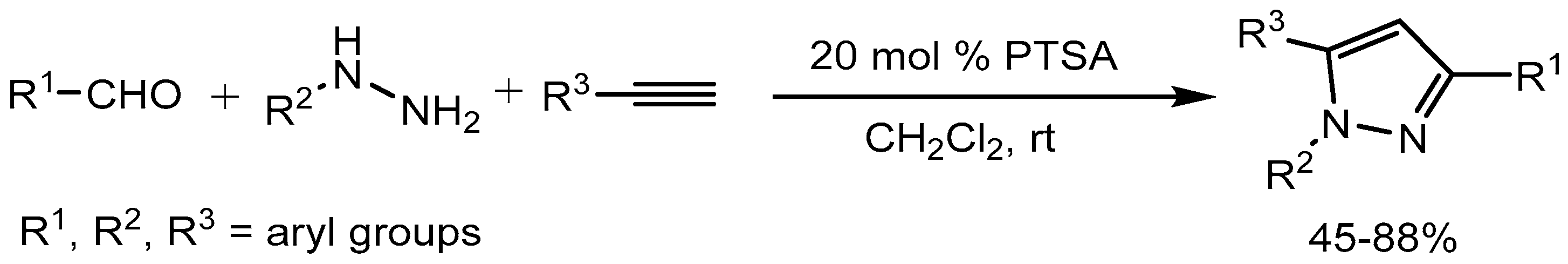
Scheme 24.
PTSA catalysed tandem process involving Mannich-type–cyclization–oxidation.
Scheme 25.
PTSA catalysed synthesis of cyclobuta-fused tetrahydroquinoline carboxylic esters.
Scheme 26.
PTSA catalysed synthesis of tetrahydro-β-carbolines (THBCs).
Scheme 27.
PTSA catalysed synthesis of hexahydroacridines and tetrahydrodipyrazolopyridines via three component reaction.
Scheme 27.
PTSA catalysed synthesis of hexahydroacridines and tetrahydrodipyrazolopyridines via three component reaction.
Scheme 28.
Synthesis of 11H-benzo[a]benzo[6,7]chromeno[2,3-c]phenazine-11,16(17H)-dione derivatives in the presence of catalytic PTSA.
Scheme 29.
Synthesis of 14-alkyl- or aryl-14-H-dibenzo[a,j]xanthenes using catalytic amount of p-TSA.
Scheme 29.
Synthesis of 14-alkyl- or aryl-14-H-dibenzo[a,j]xanthenes using catalytic amount of p-TSA.
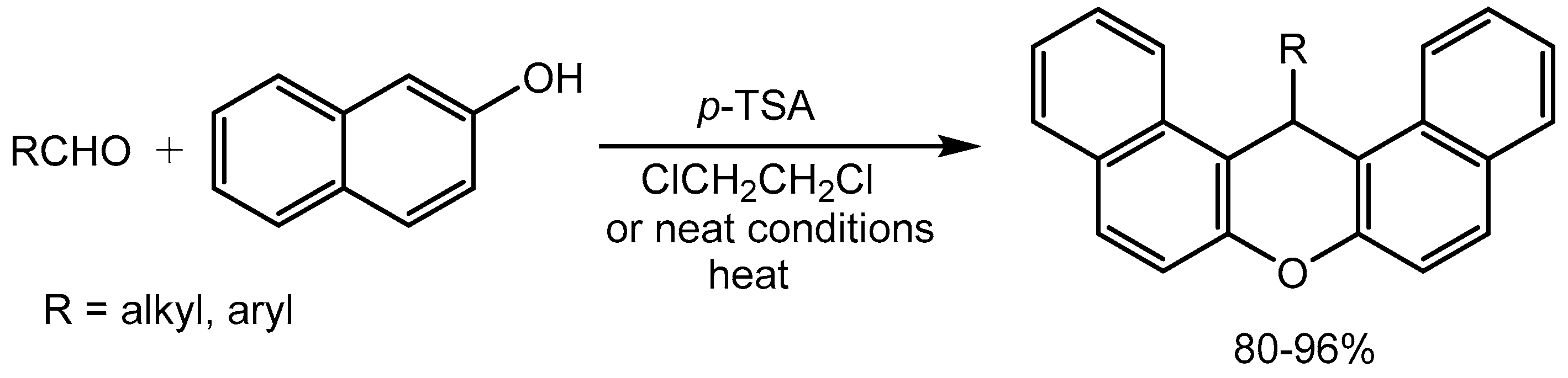
Scheme 30.
Synthesis of spirooxindoles catalyzed by PTSA.
Scheme 31.
PTSA–catalyzed Meyer–Schuster rearrangement.
Scheme 32.
PTSA–catalysed nitration of phenols.
Scheme 33.
Synthesis of β-ketoacetal using catalytic PTSA.
Scheme 34.
PTSA–catalysed synthesis of β-indolylketones under ultrasonic irradiation.
Scheme 35.
Synthesis of 2-aryl-5-methyl-2,3-dihydro-1H-3-pyrazolones in the presence of p-TSA.
Scheme 36.
Synthesis of highly functionalized indoles using PTSA.
Scheme 37.
P-TSA catalysed Synthesis of 1,2-Dihydrobenzo[f]chromen-3-one.
Scheme 38.
PTSA catalysed synthesis of pyrroloquinazolinone derivatives.
Scheme 39.
PTSA catalysed transindolylation for the synthesis of (±)-gelliusine E.
Scheme 40.
Synthesis of bis-furan diepoxide (OmbFdE) in the presence of catalytic PTSA.
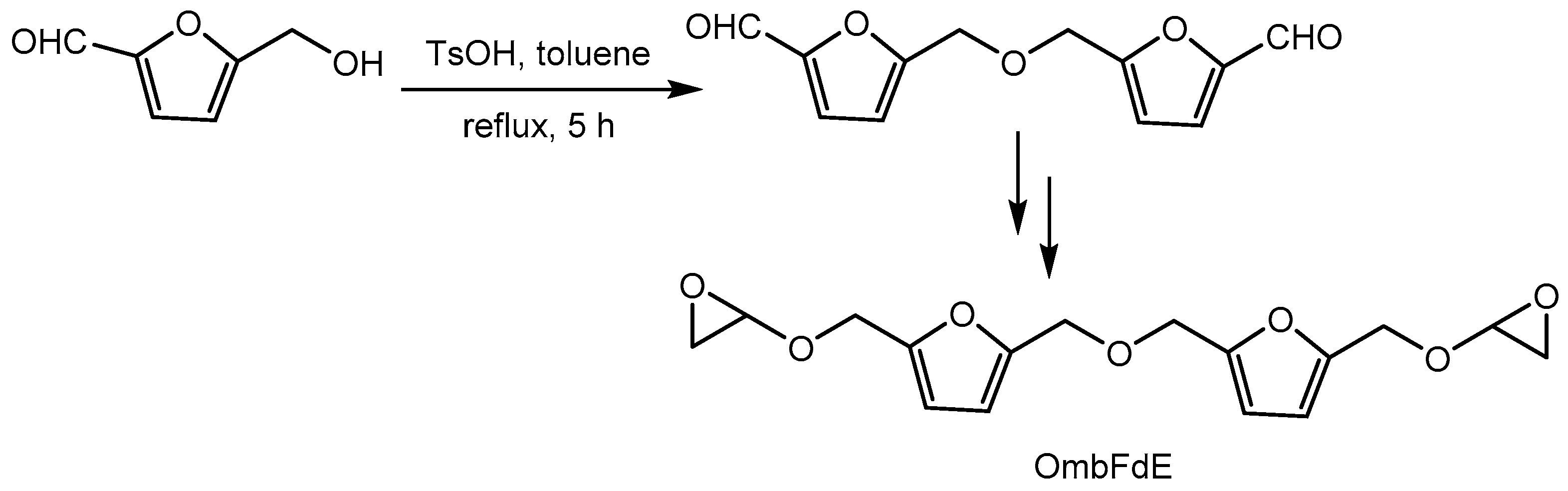
Scheme 41.
PTSA catalysed synthesis of arylthio cyclopropyl carbaldehydes.
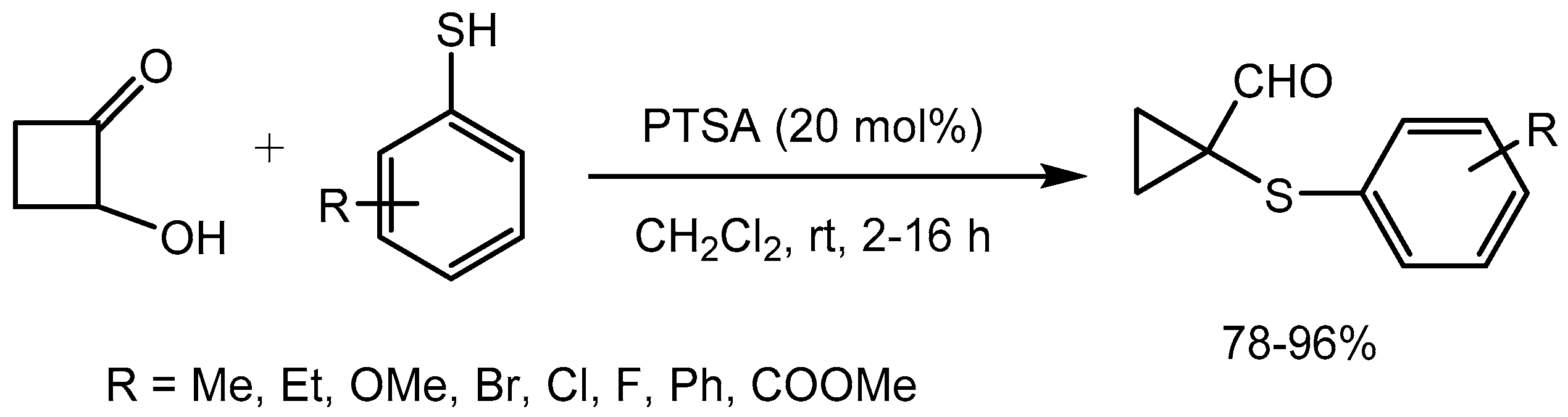
Scheme 42.
Z-enoate assisted, nucleophilic intercepted M–S rearrangement.
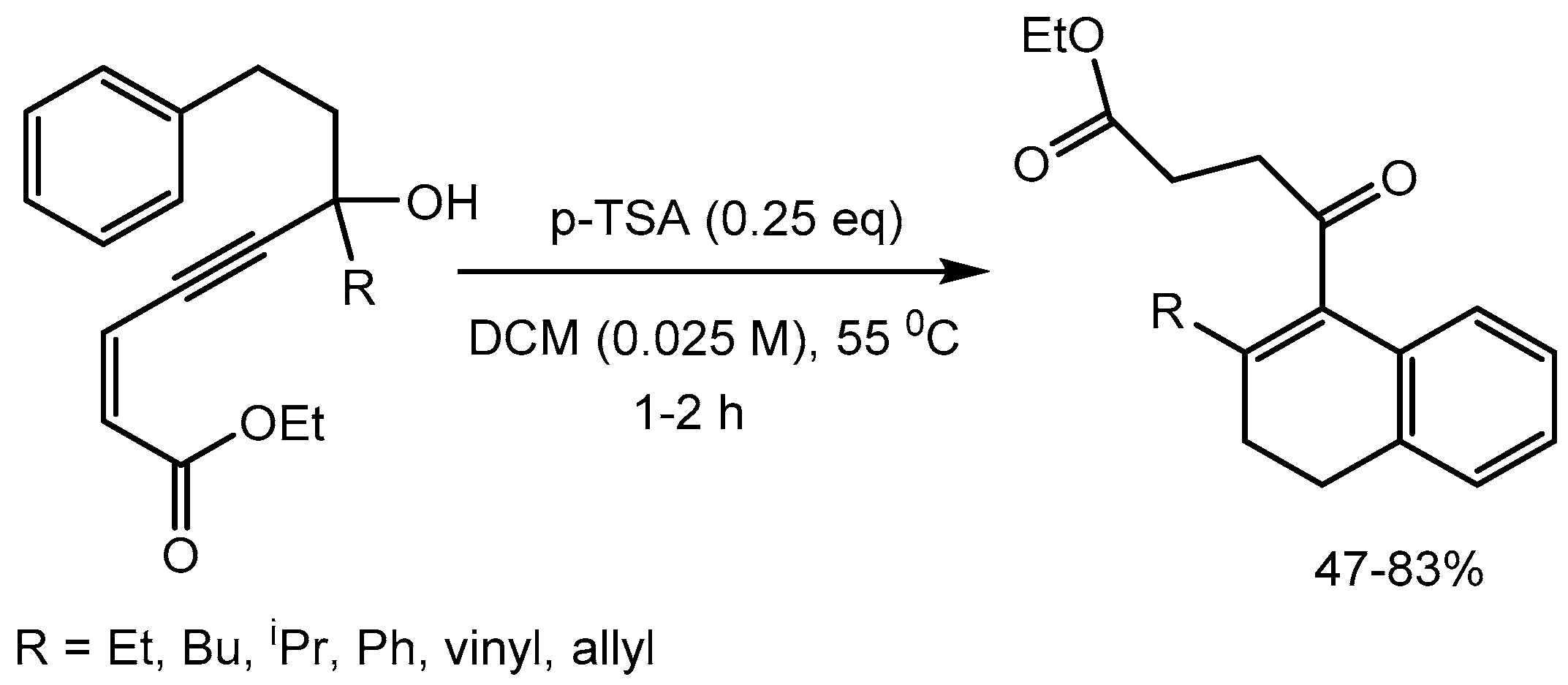
Scheme 43.
One-pot synthesis of α-bromo enaminones using catalytic p-toluenesulfonic acid monohydrate.
Scheme 43.
One-pot synthesis of α-bromo enaminones using catalytic p-toluenesulfonic acid monohydrate.
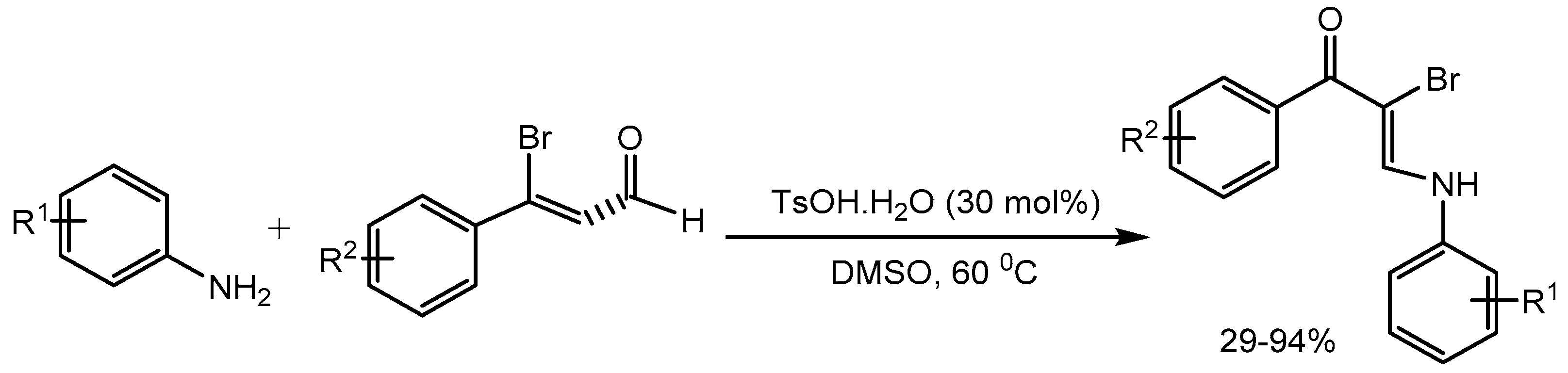
Scheme 44.
Synthesis of 3-difluoroalkyl phthalides using catalytic PTSA.
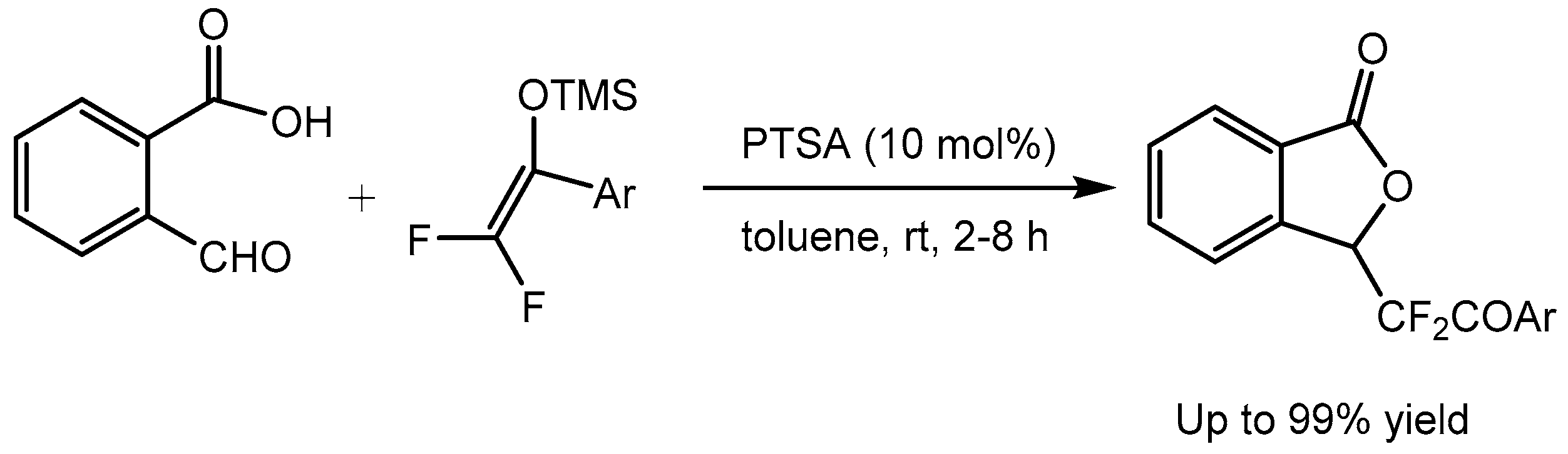
Scheme 45.
TsOH·H2O catalysed cascade reaction of 2-(2-oxo-2-arylethyl)benzonitriles with indoles.
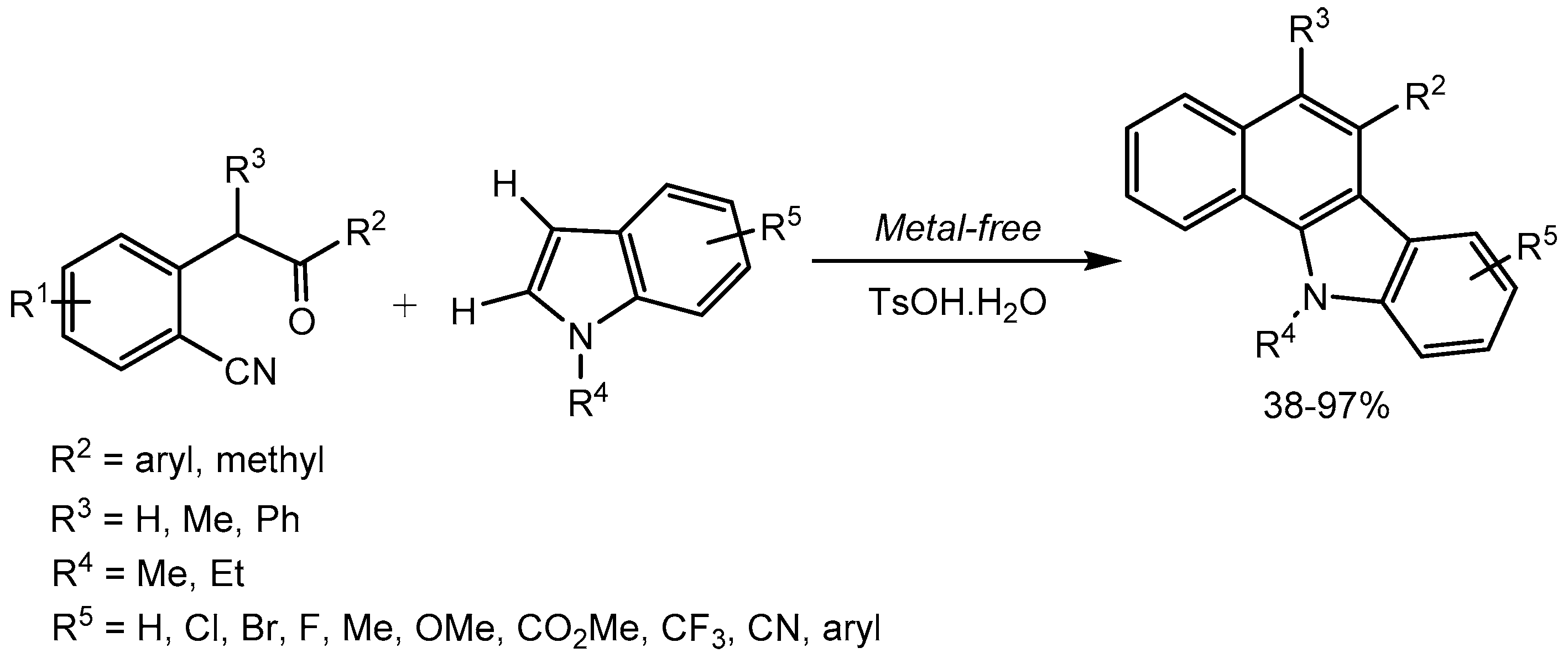
Scheme 46.
p-TSA catalysed four-component condensation of an isocyanide, amine, aldehyde, and amide.
Scheme 46.
p-TSA catalysed four-component condensation of an isocyanide, amine, aldehyde, and amide.
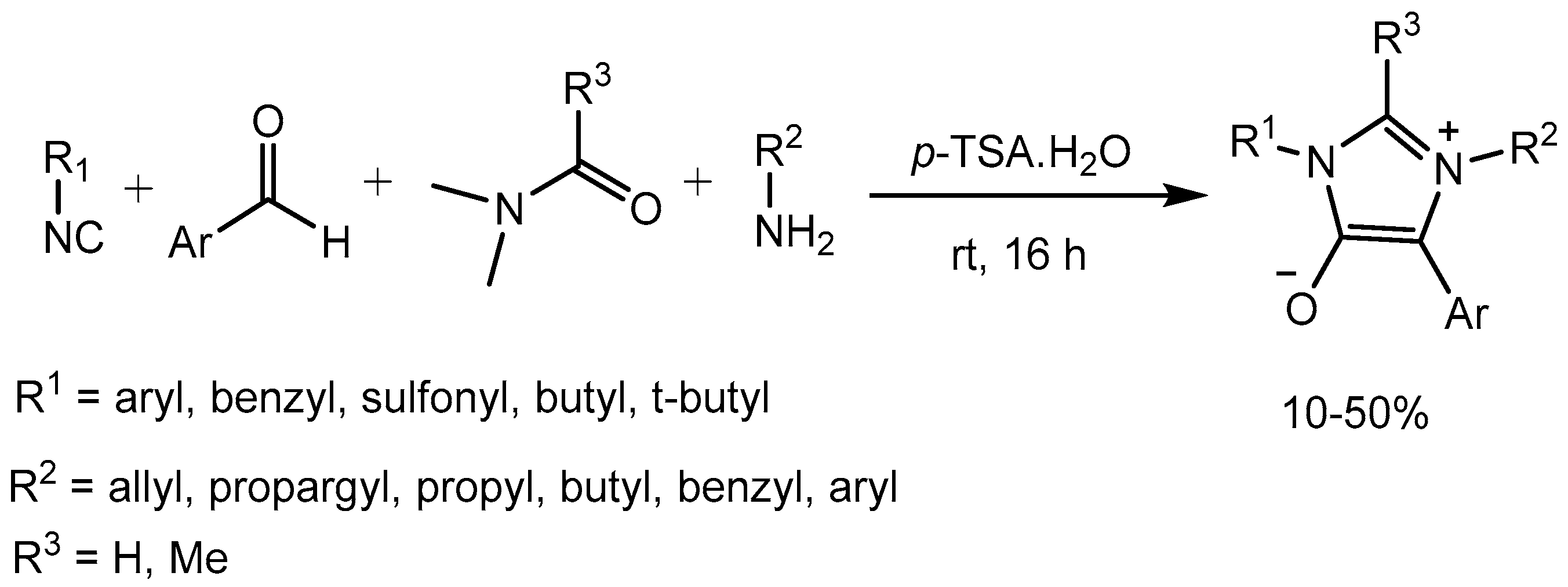
Scheme 47.
p-TsOH promoted 1,3-dipolar cycloaddition of nitroolefins and sodium azide.
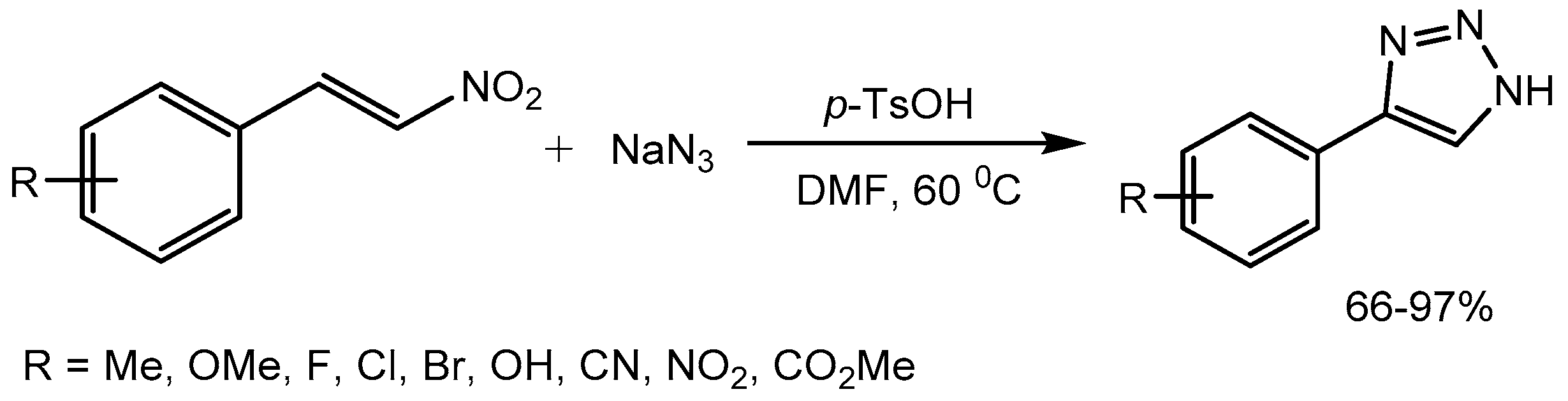
Scheme 48.
Synthesis of substituted 2,4-diaryl-3-sulfonylquinolines using p-TsOH.H2O..
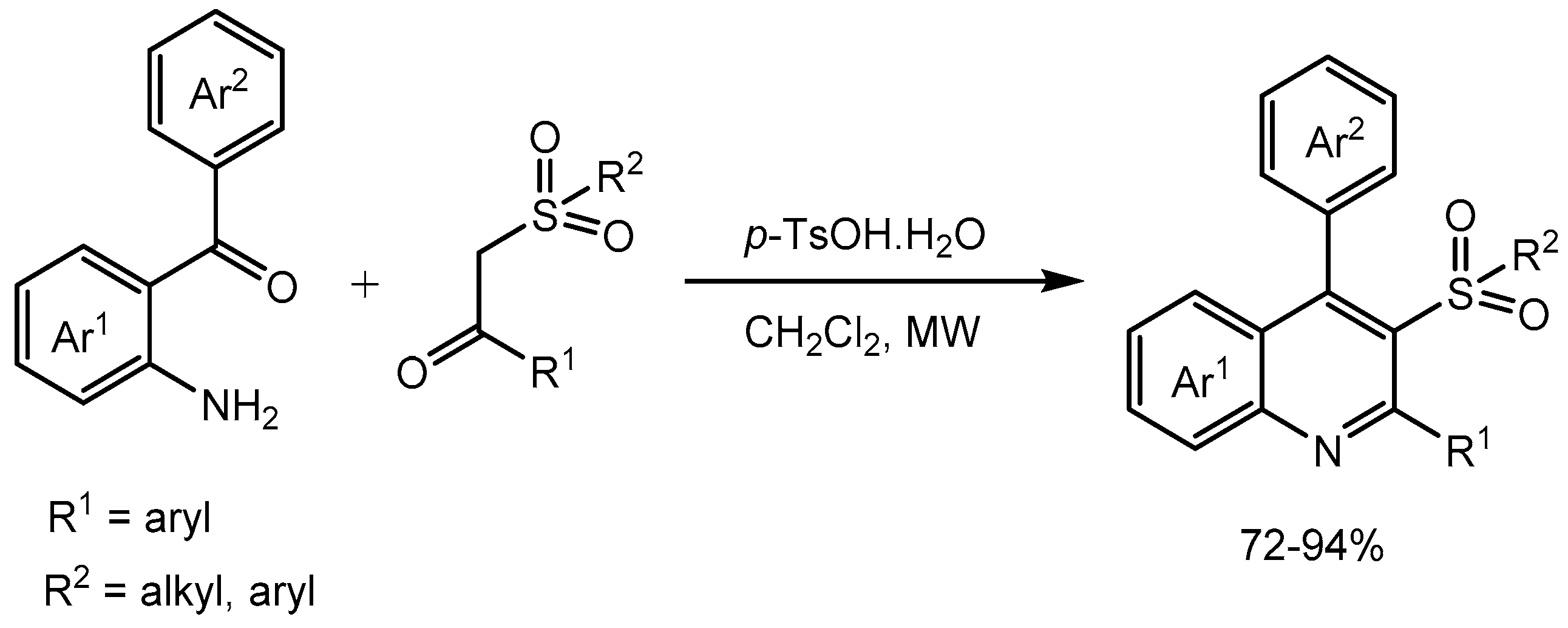
Scheme 49.
PTSA catalyzed synthesis of 3-aryl coumarins and indenes.
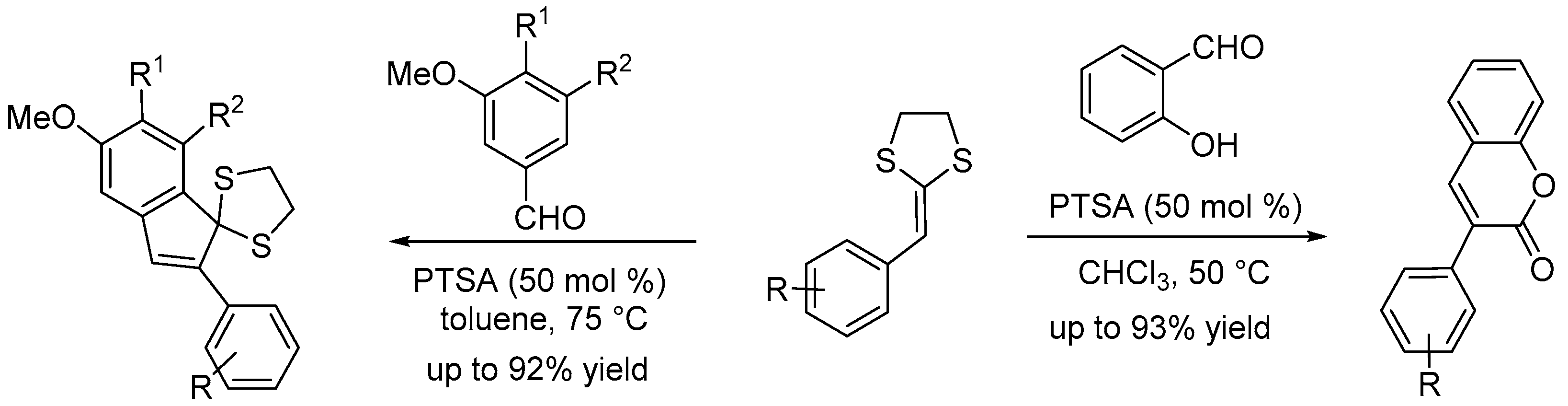
Scheme 50.
TsOH.H2O catalyzed synthesis of diverse orthogonally protected monosaccharides.
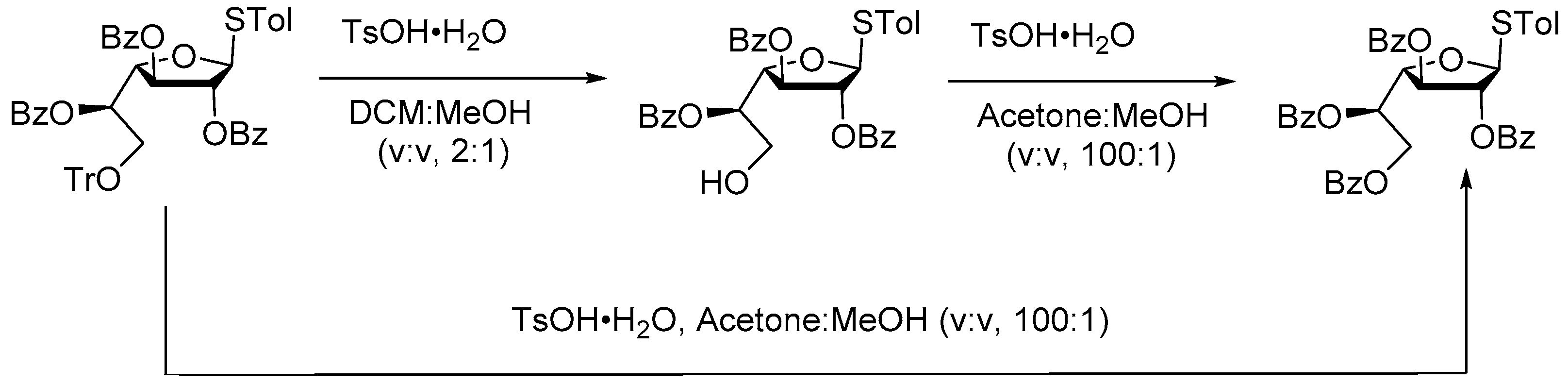
Scheme 51.
p-TSA·H2O catalyzed synthesis of 2,4-diarylquinolines.
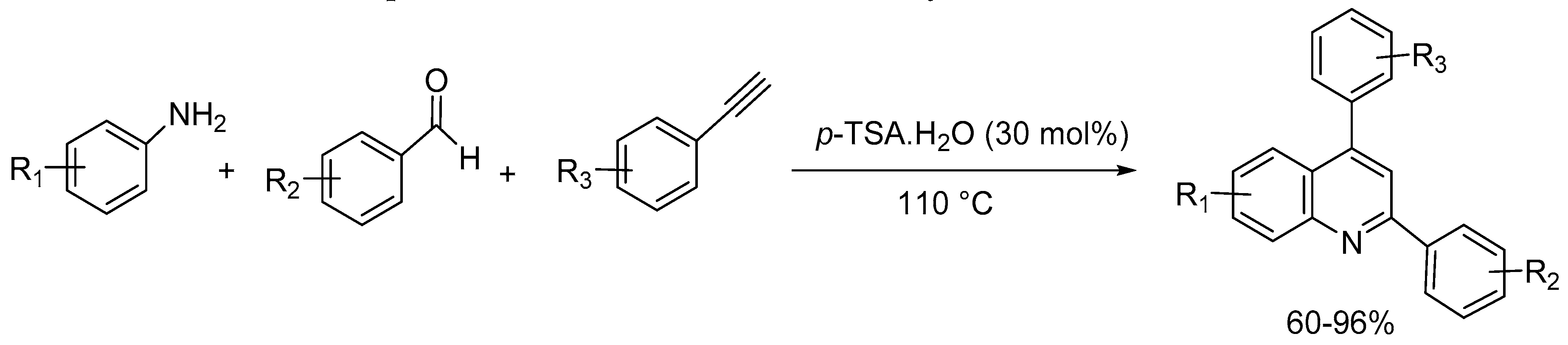
Scheme 52.
p-TSA·H2O catalyzed synthesis of 2-benzyl-3-arylquinoline derivatives.
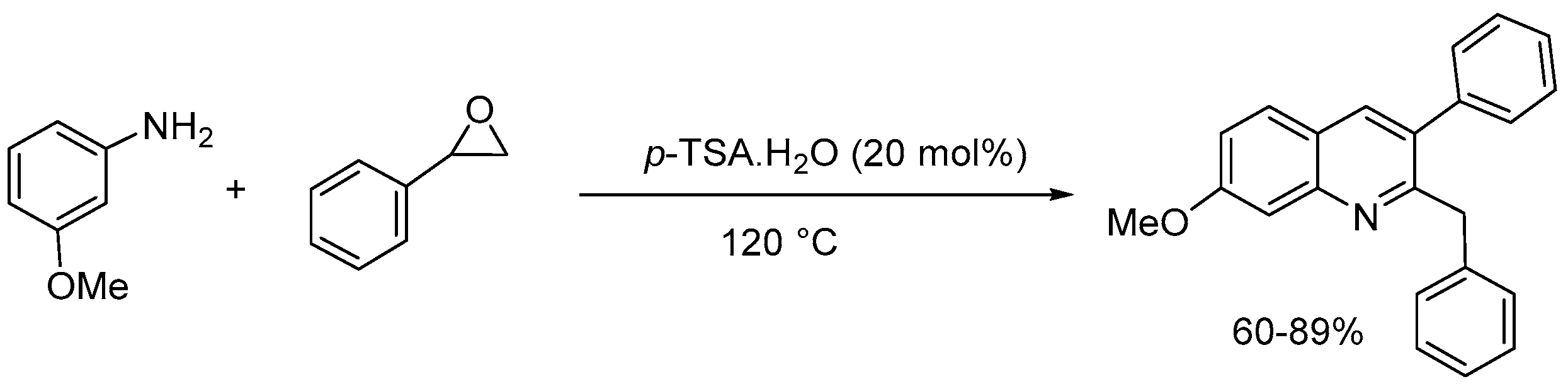
Scheme 53.
Synthesis of cyclohepta[b]indoles and furo[3,4-b]carbazoles using p-TsOH.H2O.
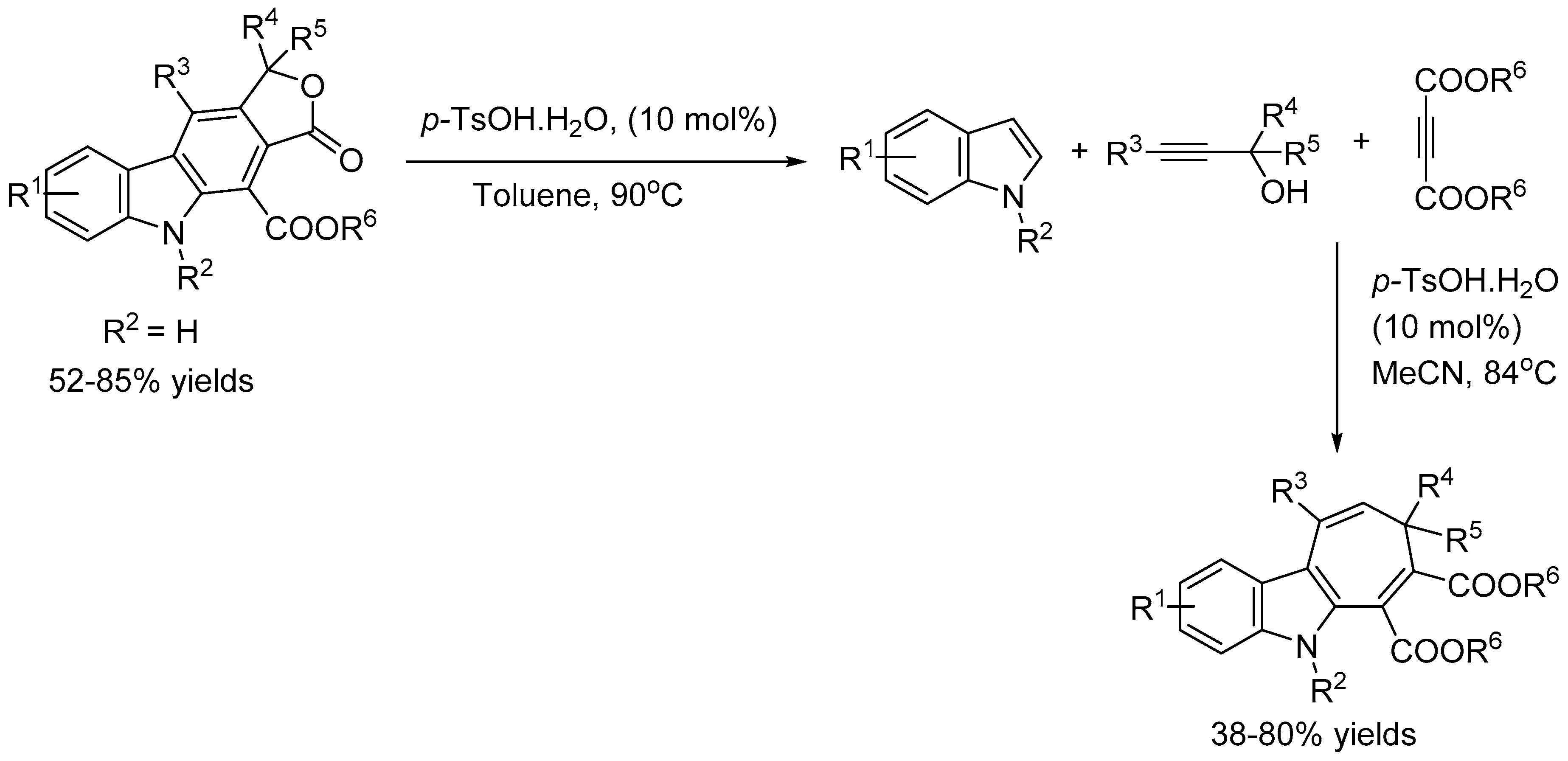
Scheme 54.
P-TSOH promoted synthesis of biologically important cyclopent-2-enones.
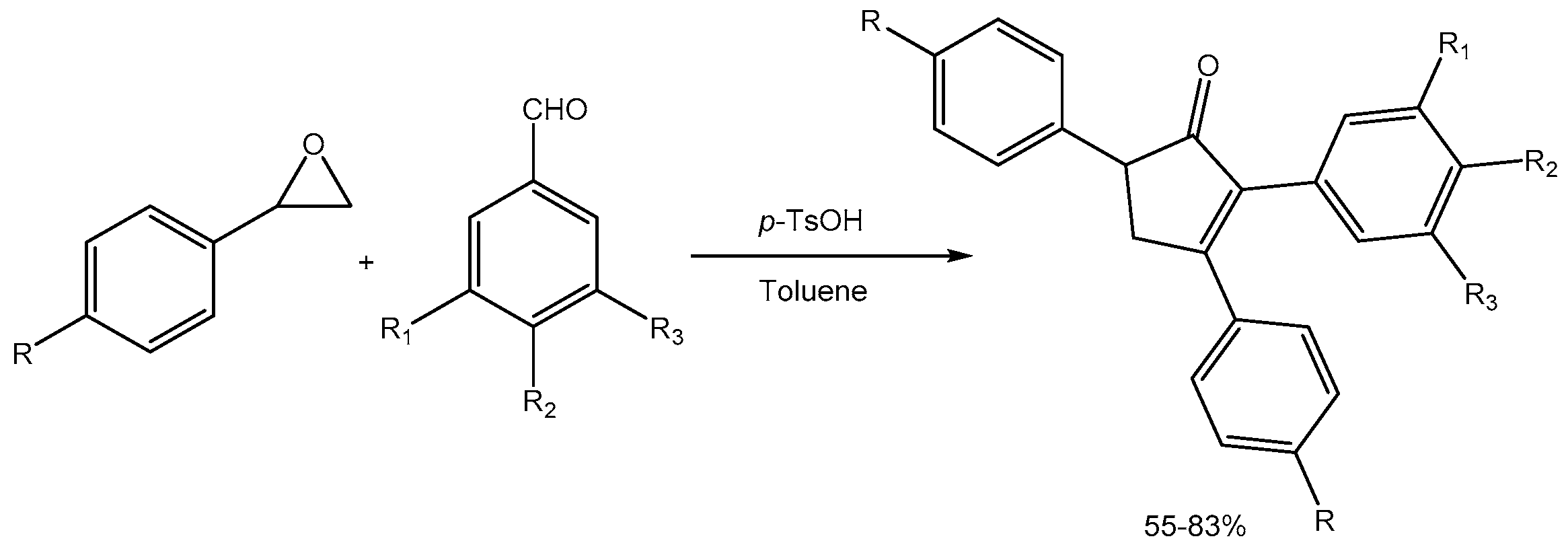
Scheme 55.
Synthesis of substituted furans catalyzed by p-TSA.
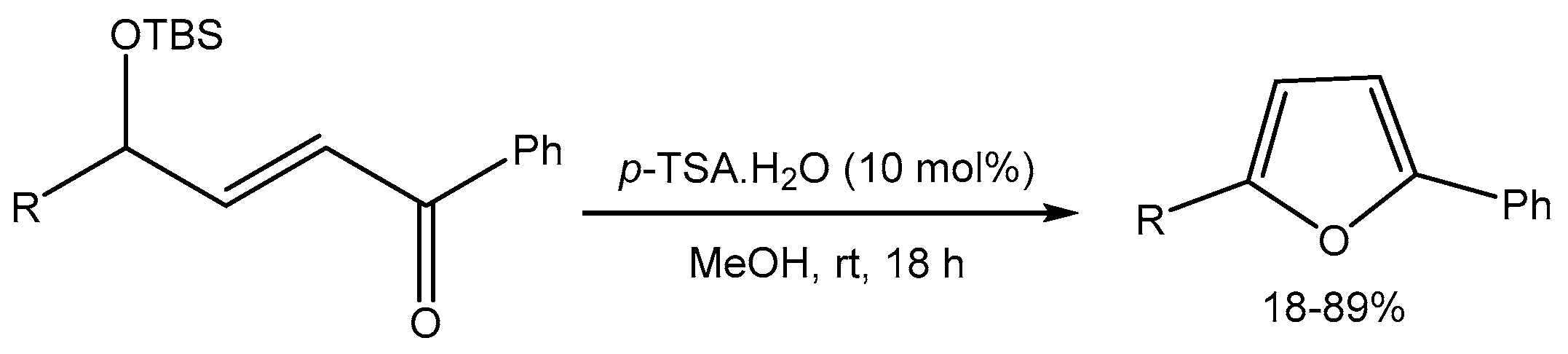
Scheme 56.
P-TSOH catalyzed synthesis of biologically important multisubstituted indolizines.
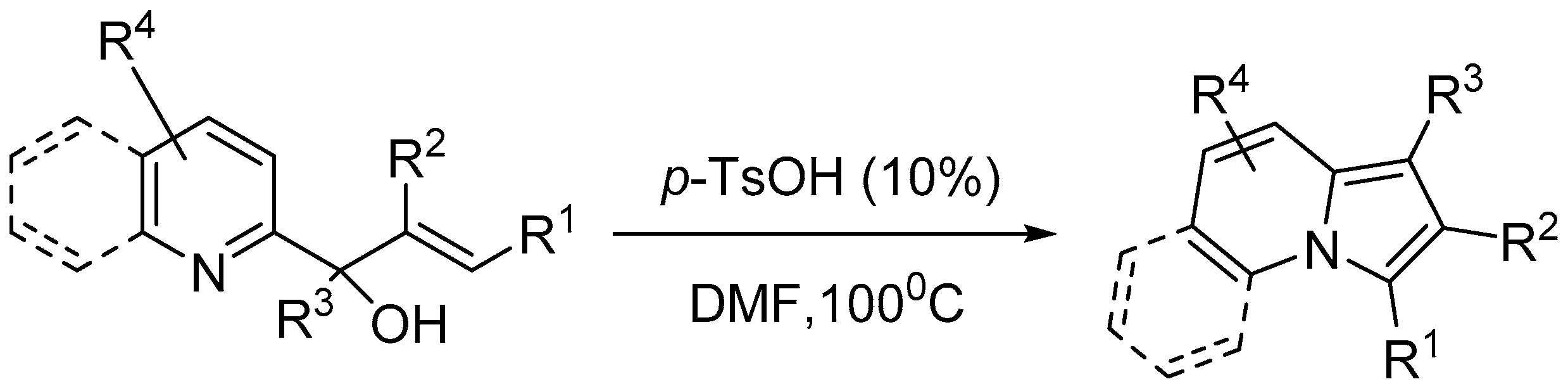
Scheme 57.
p-TsOH.H2O catalyzed intramolecular 7-endo hydroarylation reaction.
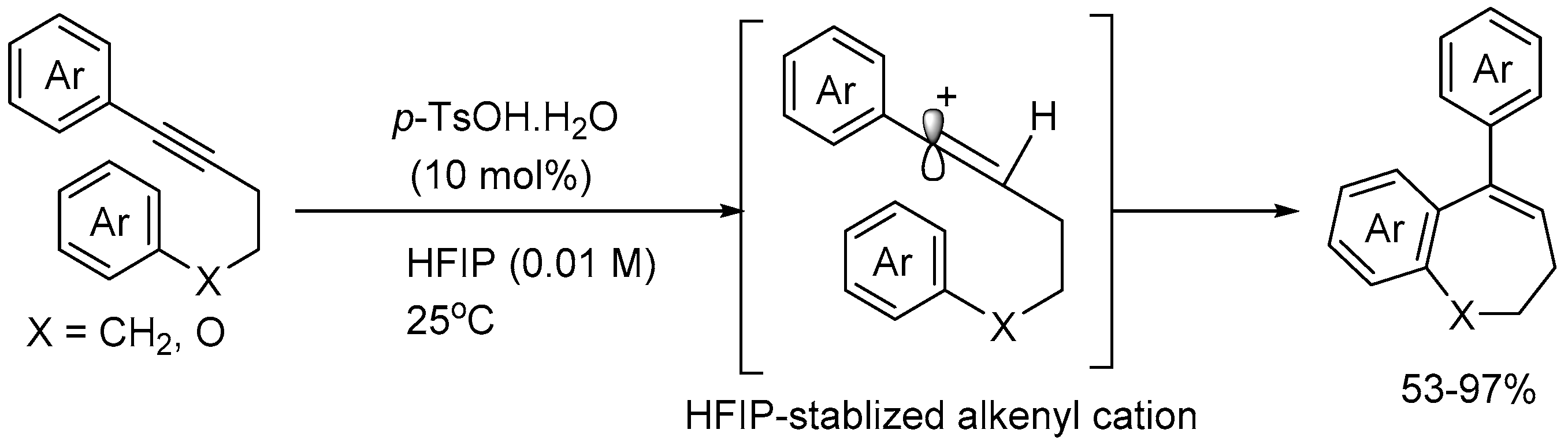
Scheme 58.
p-TsOH catalyzed synthesis of valuable fused-N-heterocycles.
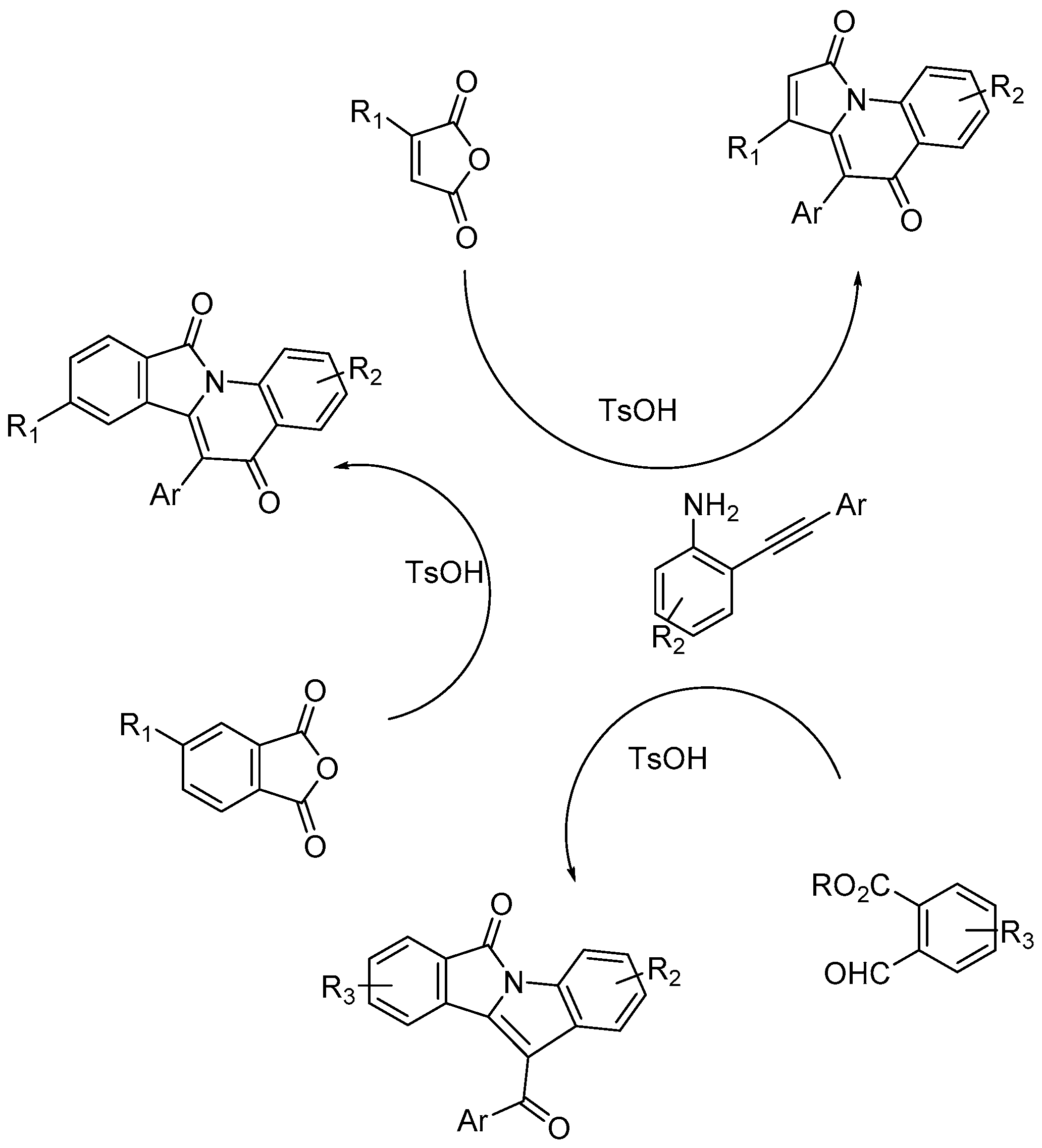
Scheme 59.
Regioselective direct carboxamidation reaction of 2-indolylmethanols catalyzed by p-TsOH.
Scheme 59.
Regioselective direct carboxamidation reaction of 2-indolylmethanols catalyzed by p-TsOH.
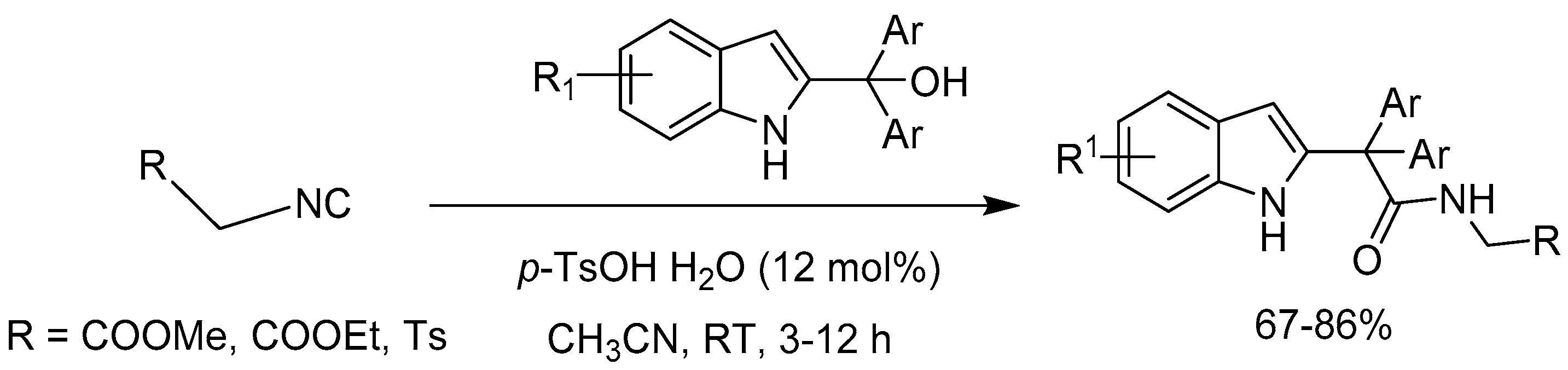
Scheme 60.
Synthesis of cyclopenta[b]indoles catalyzed by p-TsOH.H2O.
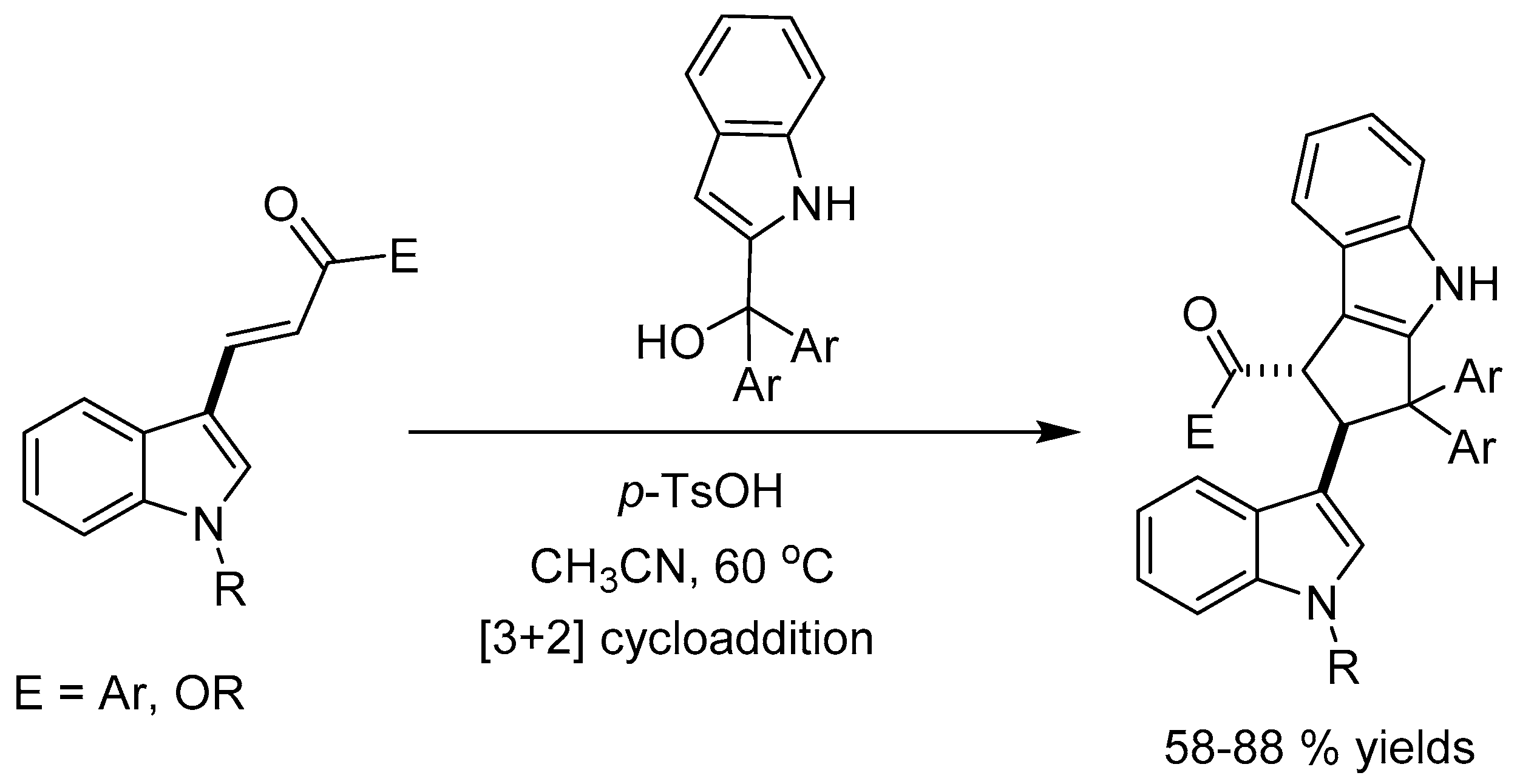
Scheme 61.
Total synthesis of sesquiterpenoid alanense A via p-TsOH.H2O catalyzed Friedel–Crafts alkylation reaction.
Scheme 61.
Total synthesis of sesquiterpenoid alanense A via p-TsOH.H2O catalyzed Friedel–Crafts alkylation reaction.
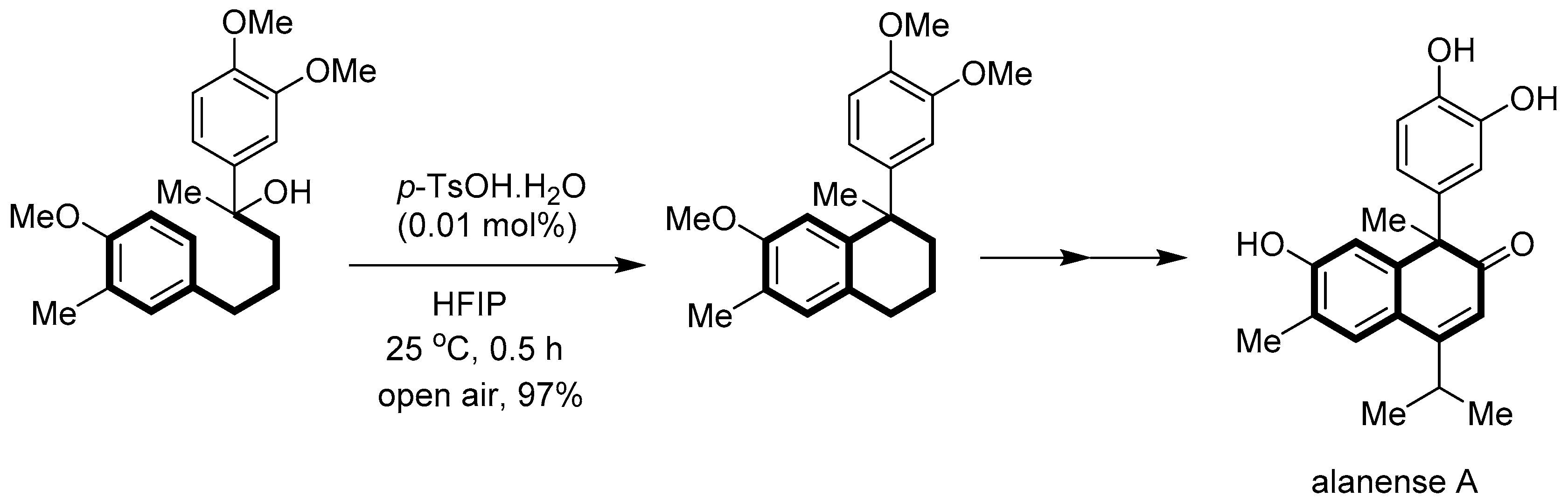
Scheme 62.
Synthesis of bisquinoline diamines.
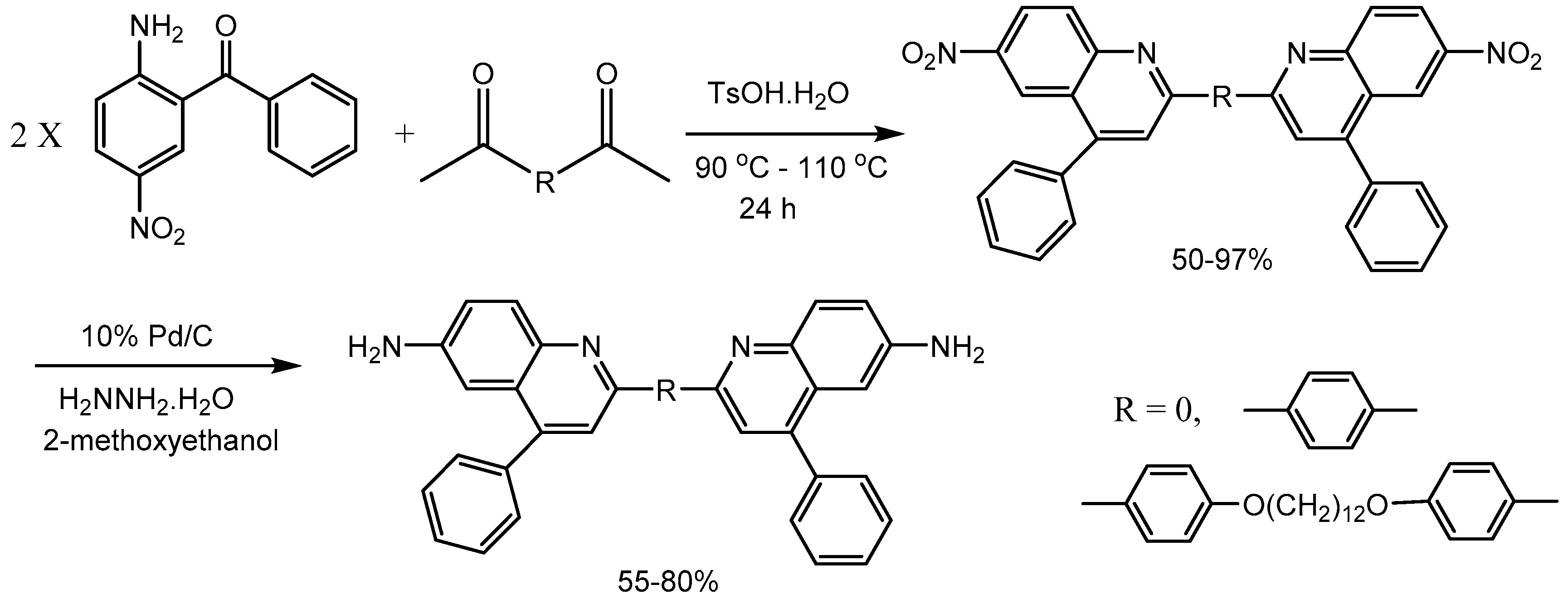
Scheme 63.
Synthesis of pyrylium tosylates and triflimides.
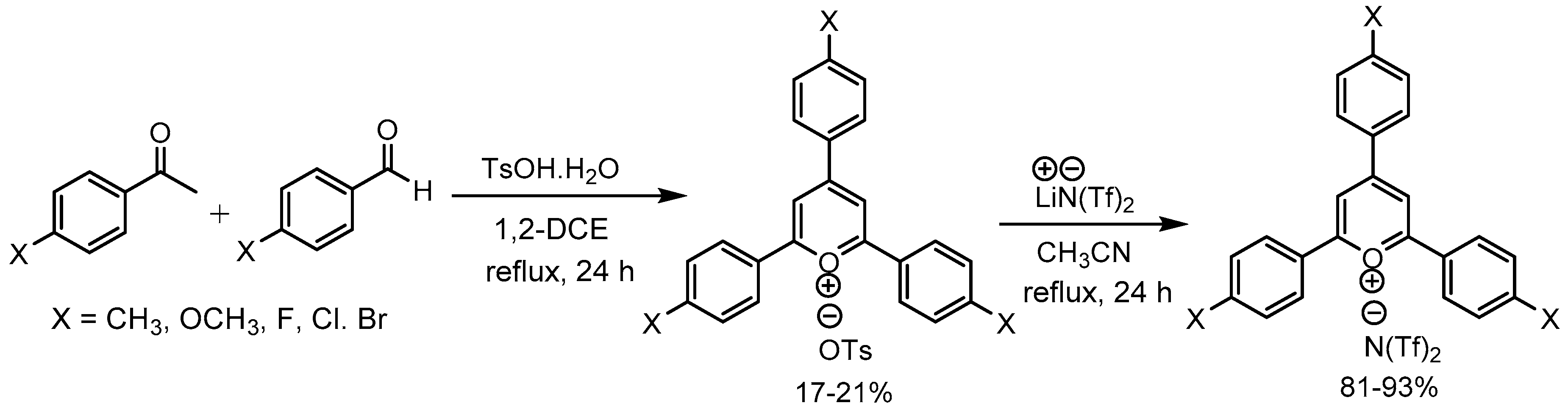
Scheme 64.
Synthesis of poly(pyridinium salt)s.
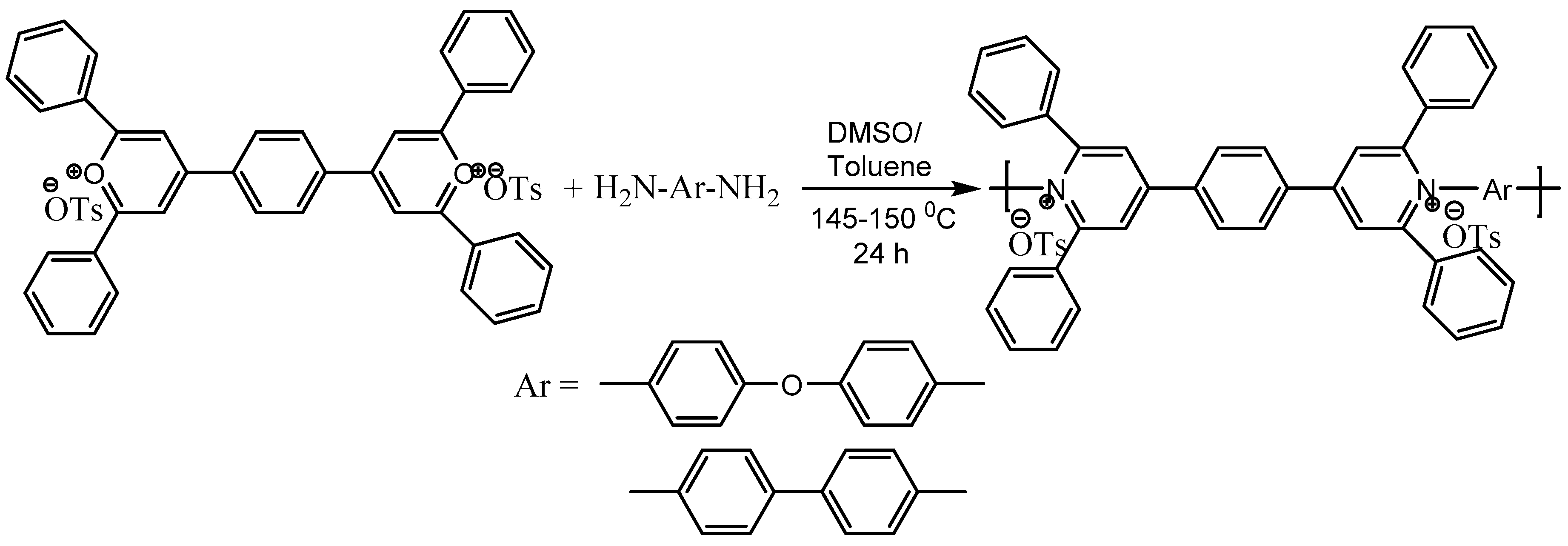
Scheme 65.
Synthesis of four substituted 2,4,6-triphenylpyrylium tosylate salts.
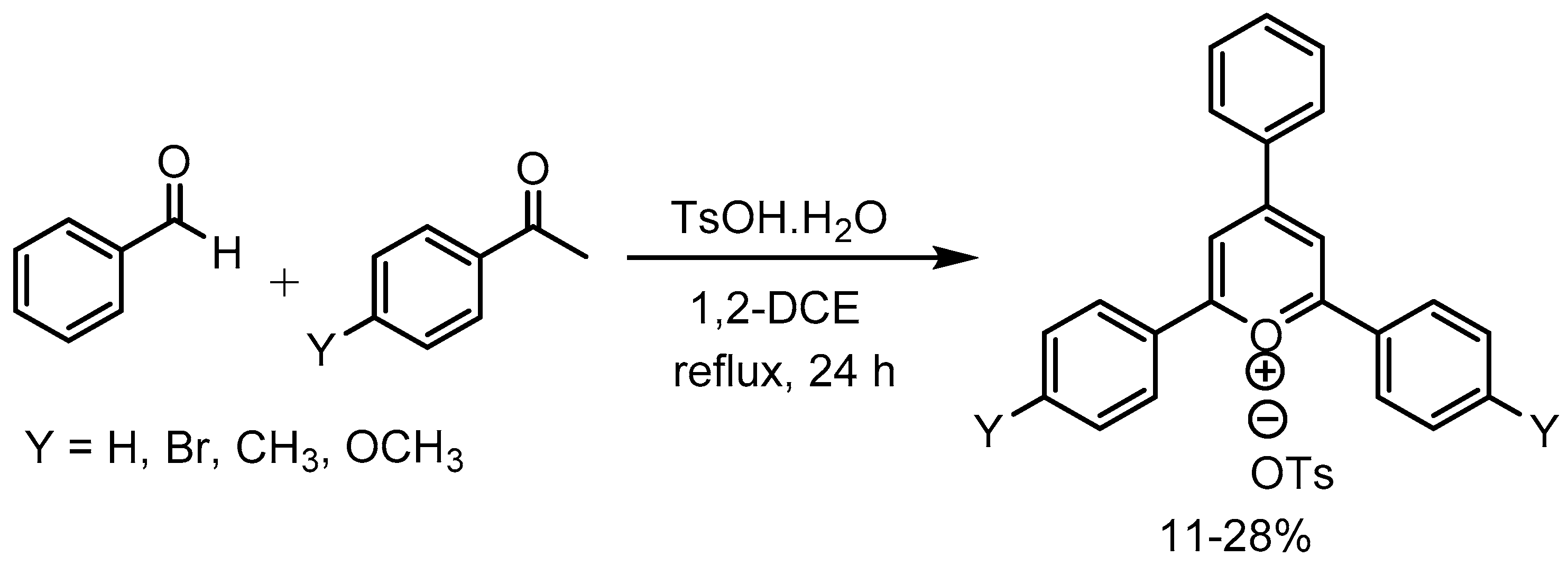
Scheme 66.
p-Toluenesulfonic acid promoted annulation of 2-alkynylanilines with activated ketones.
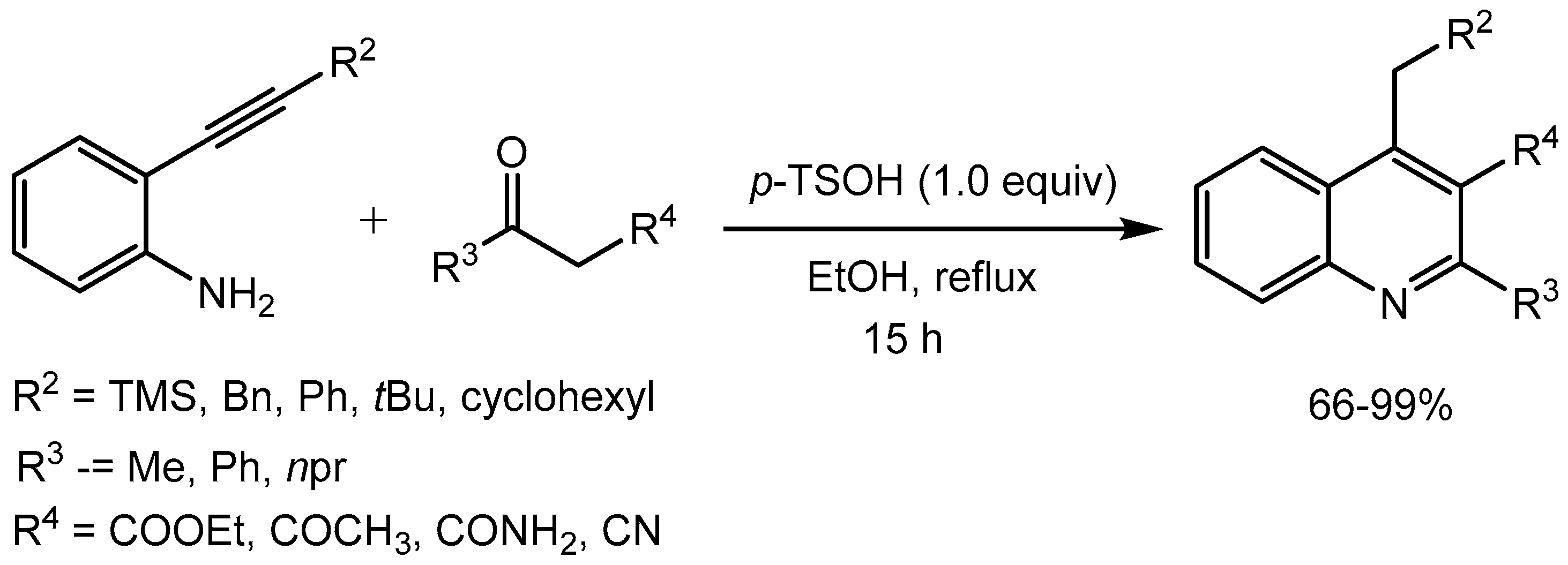
Scheme 67.
PTSA–catalyzed one pot construction of benzo[b]furan derivatives.
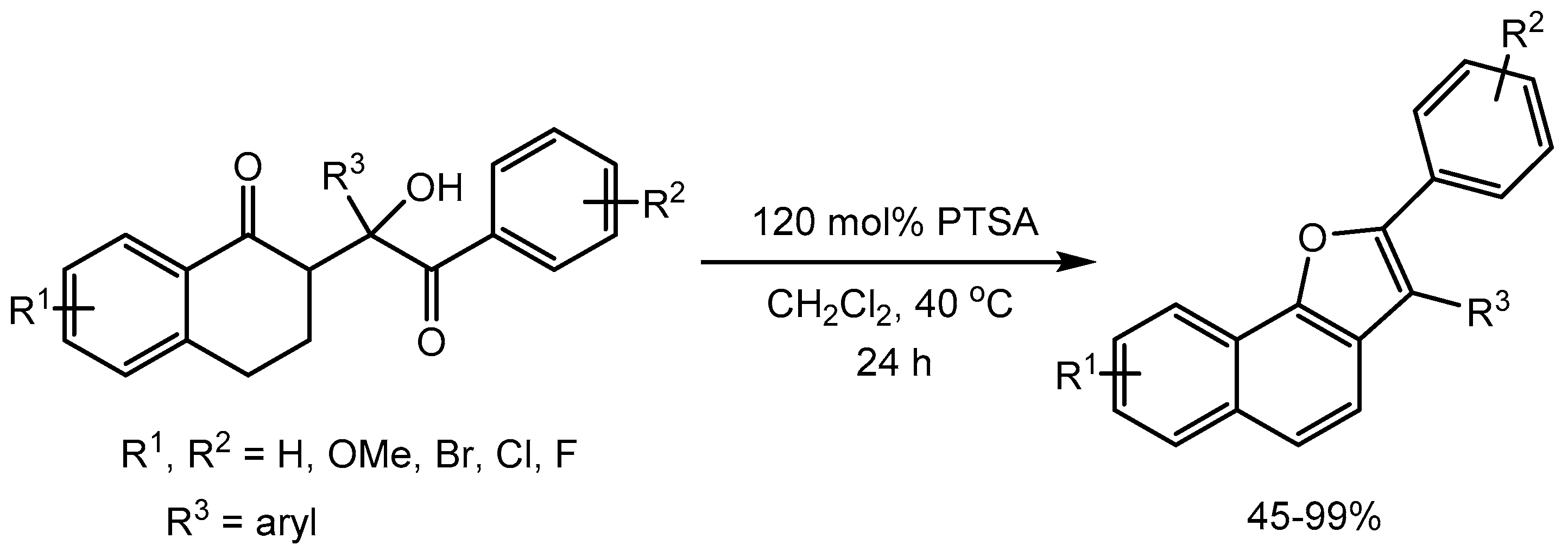
Scheme 68.
Solvent-free synthesis of substituted benzenes using p-TsOH·H2O.
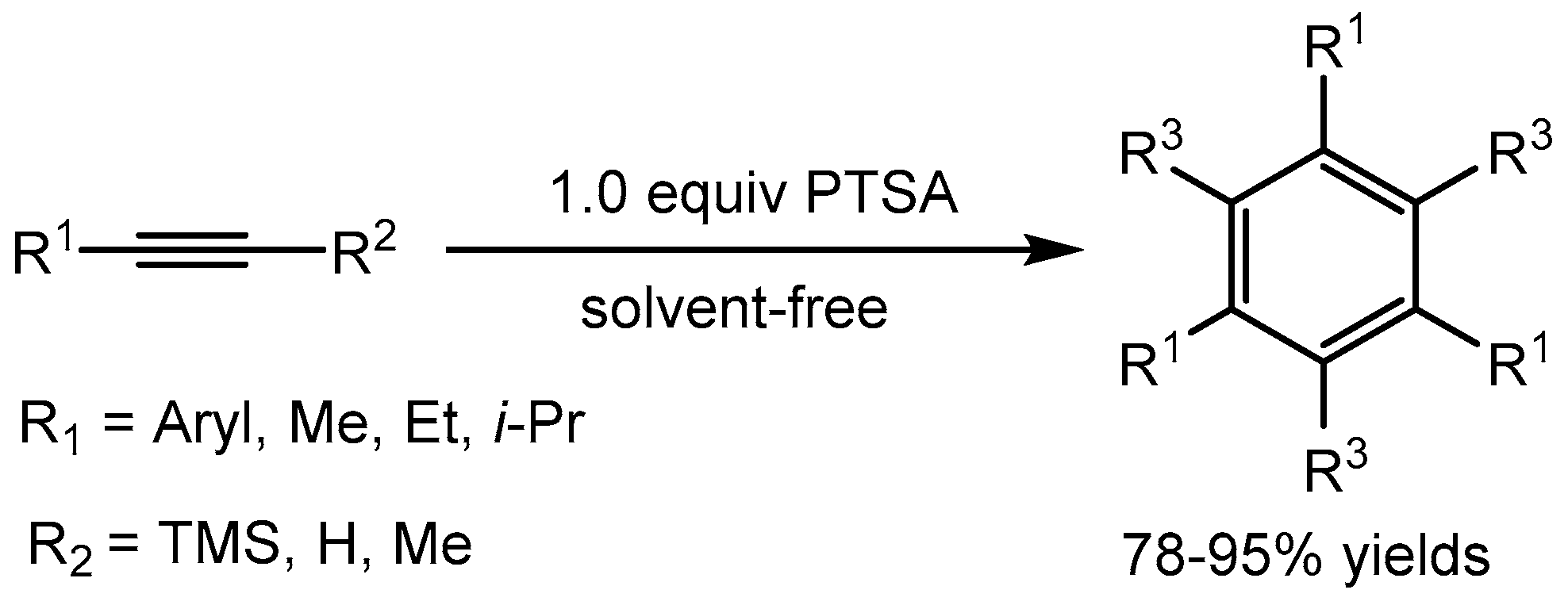
Scheme 69.
Deprotection of Boc group and formation of N-Me amino acid benzylester TsOH salt under MW irradiation.
Scheme 69.
Deprotection of Boc group and formation of N-Me amino acid benzylester TsOH salt under MW irradiation.
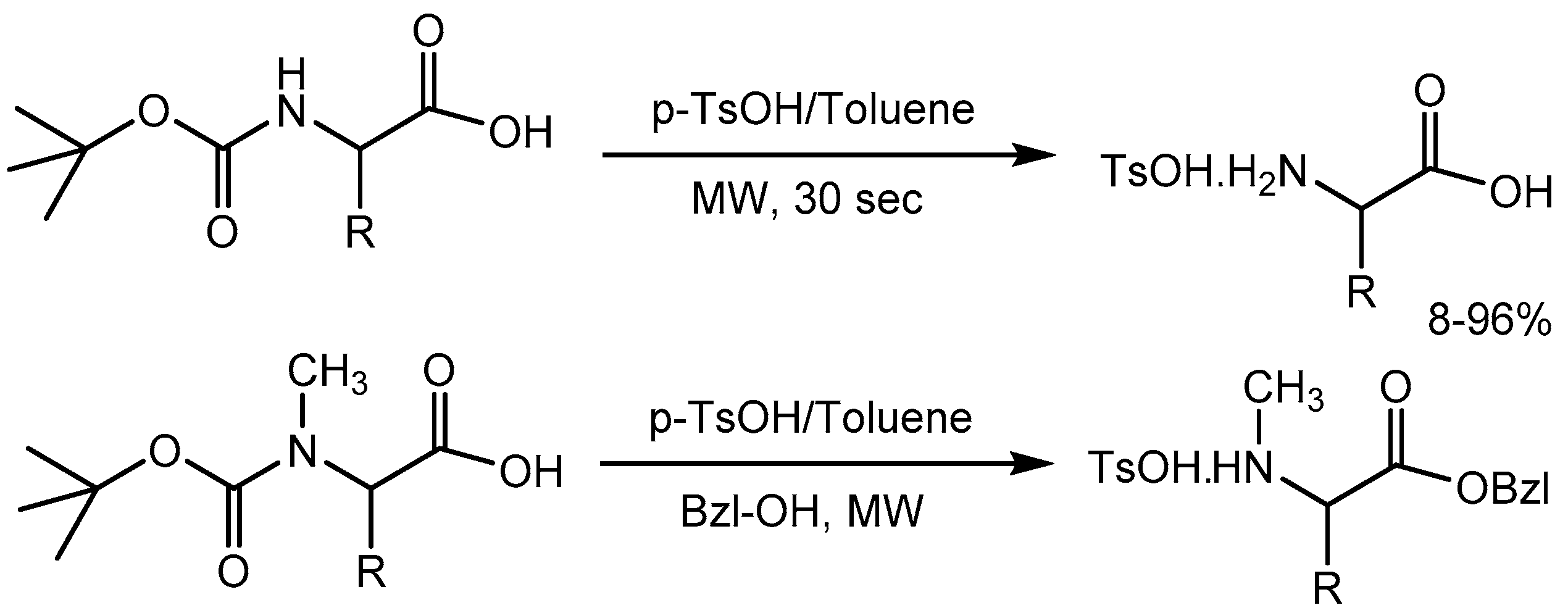
Scheme 70.
p-TSA promoted Synthesis of 2-thiomethyl benzoxazoles.
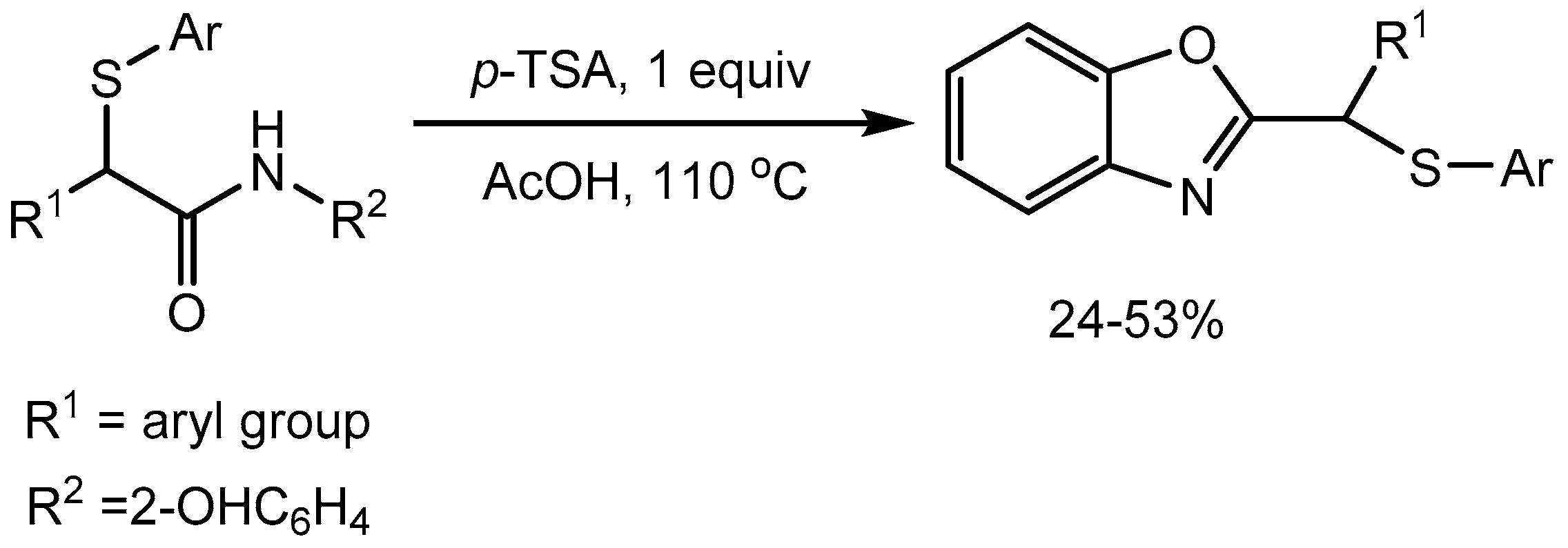
Scheme 71.
PTSA-ZnCl2 catalysed synthesis of 3,5-disubstituted-1,2,4-oxadiazoles.
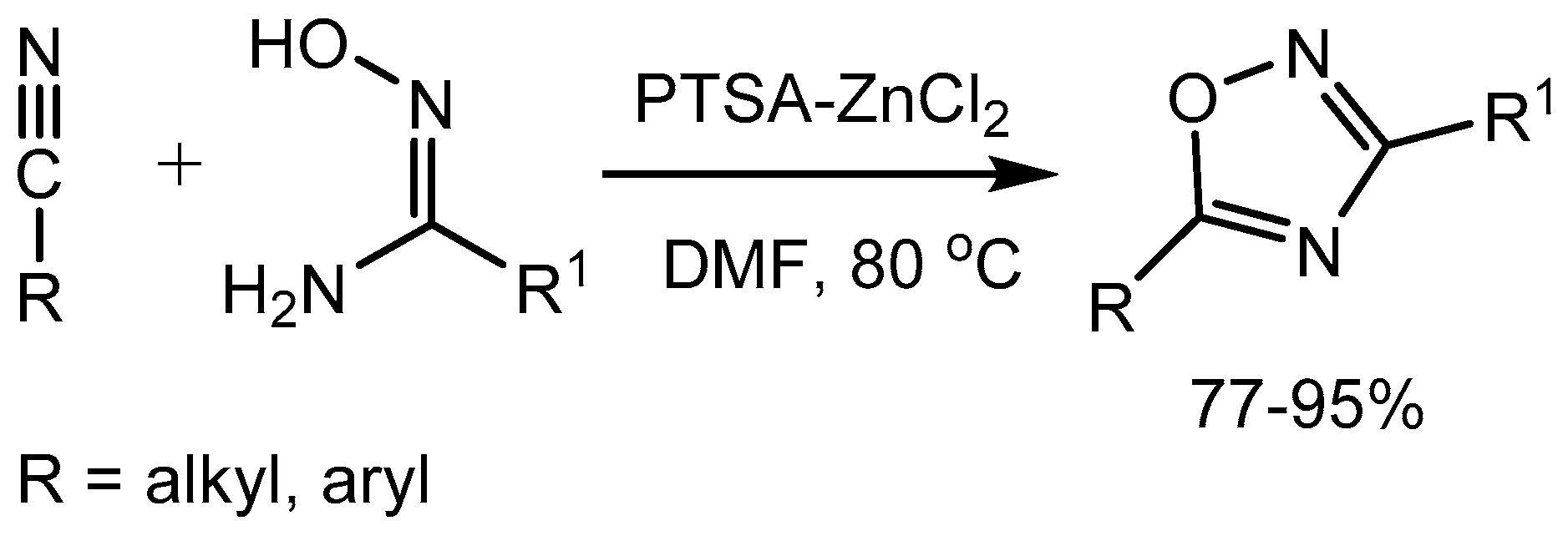
Scheme 72.
PTSA promoted synthesis of 3,4-dihydropyrazino[1,2-a]indoles.
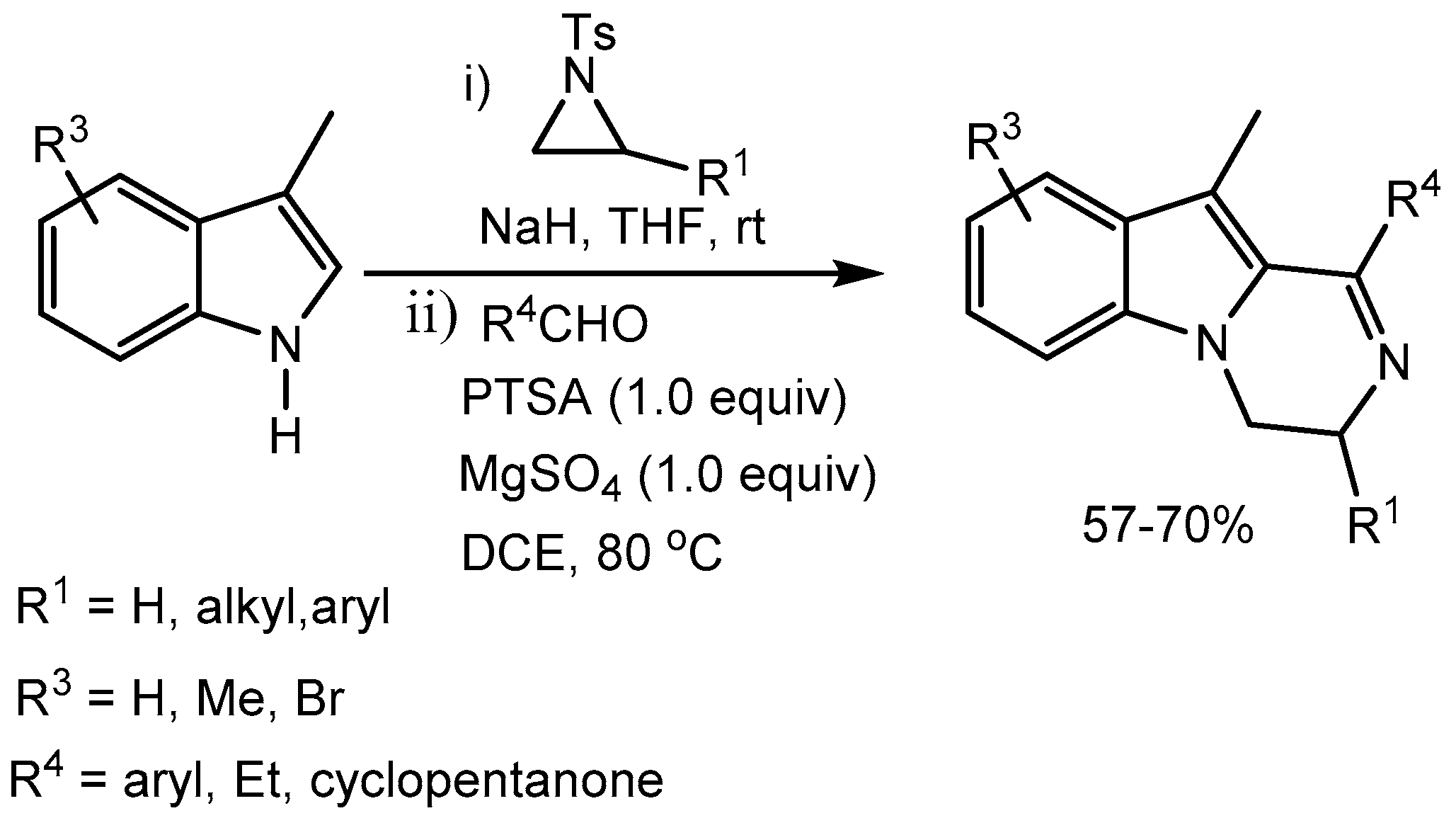
Scheme 73.
p-TsOH.H2O promoted Fe-catalyzed synthesis of alkyl nitrile substituted benzothiazole derivatives.
Scheme 73.
p-TsOH.H2O promoted Fe-catalyzed synthesis of alkyl nitrile substituted benzothiazole derivatives.
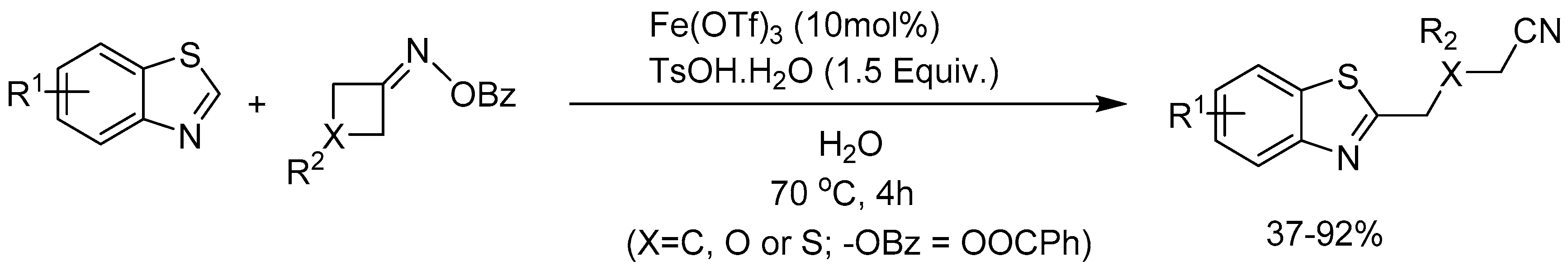
Scheme 74.
Hydroisothiocyanation of alkenes in the presence of p-TsOH.H2O.
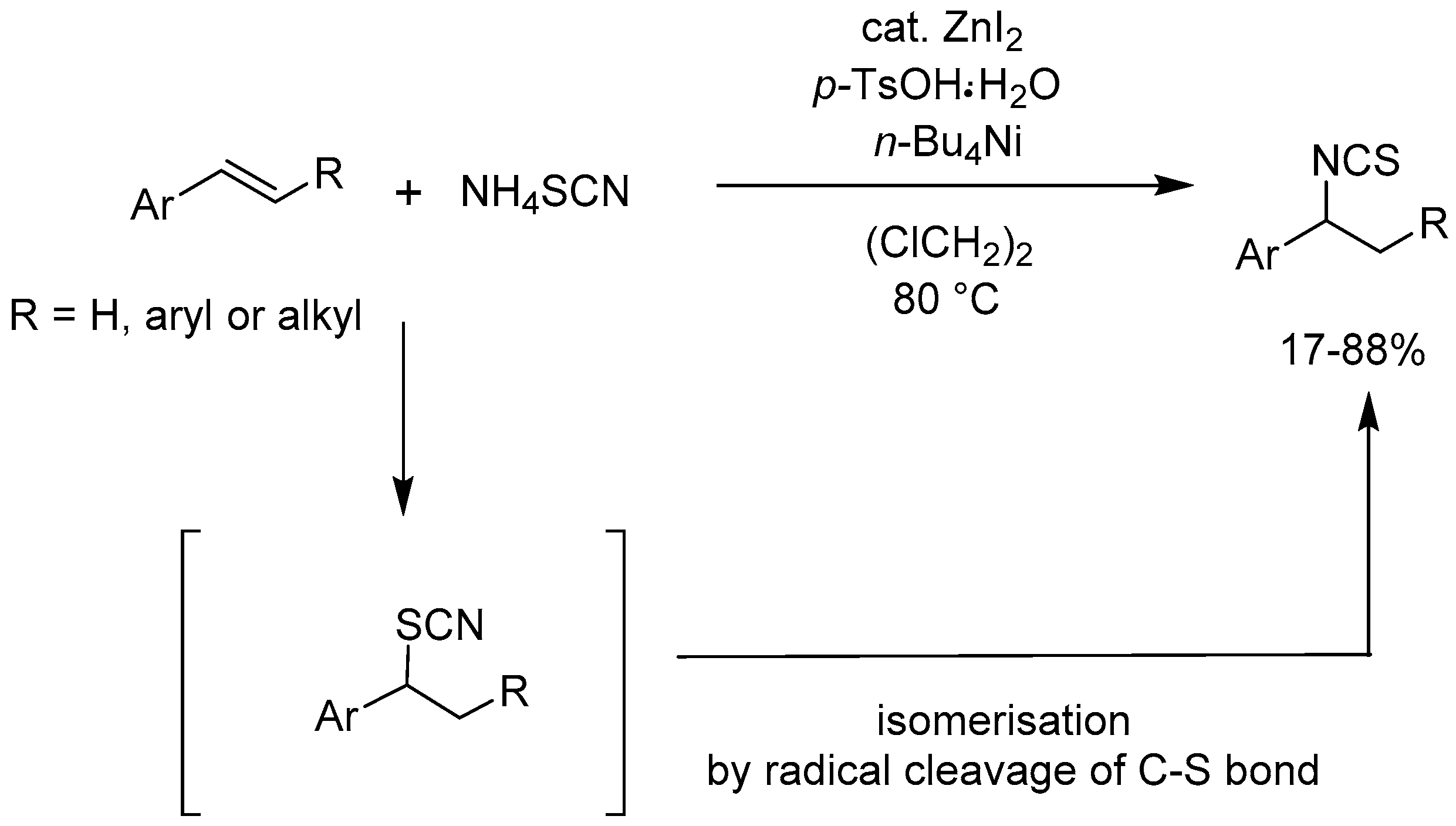
Scheme 75.
One-pot synthesis of imidazolidines and oxazolidines using catalytic PTSA.
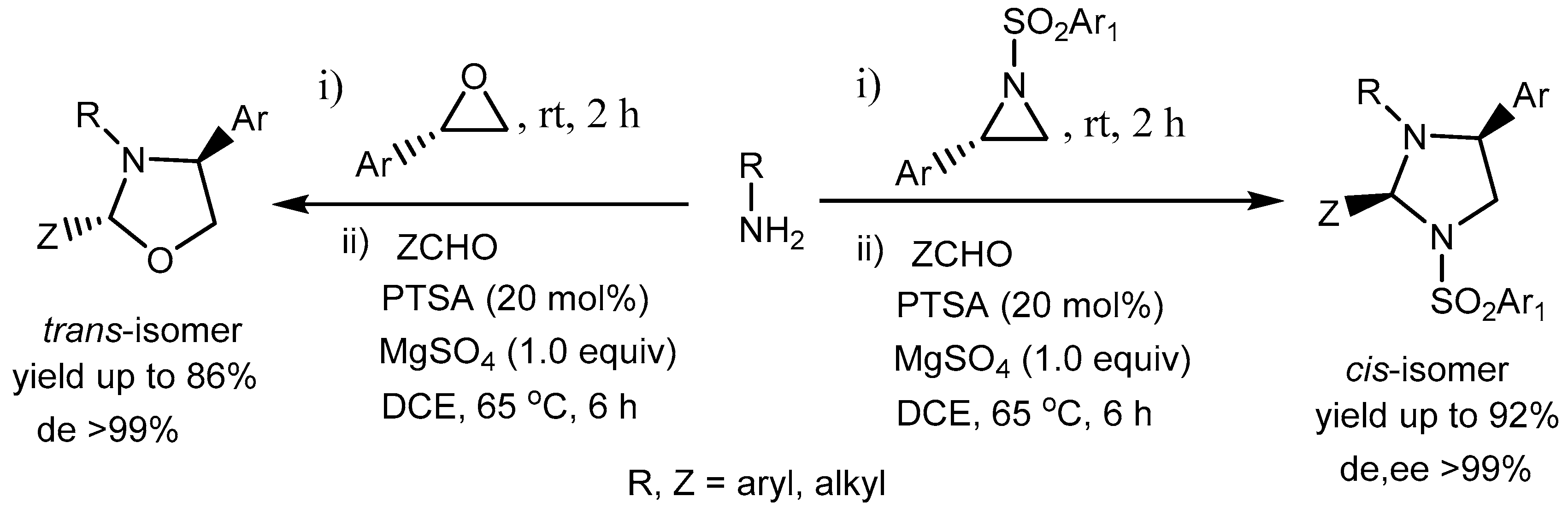
Scheme 76.
Synthesis of Luotonin A derivatives via synergestic visible-light photoredox and acid catalysis.
Scheme 76.
Synthesis of Luotonin A derivatives via synergestic visible-light photoredox and acid catalysis.
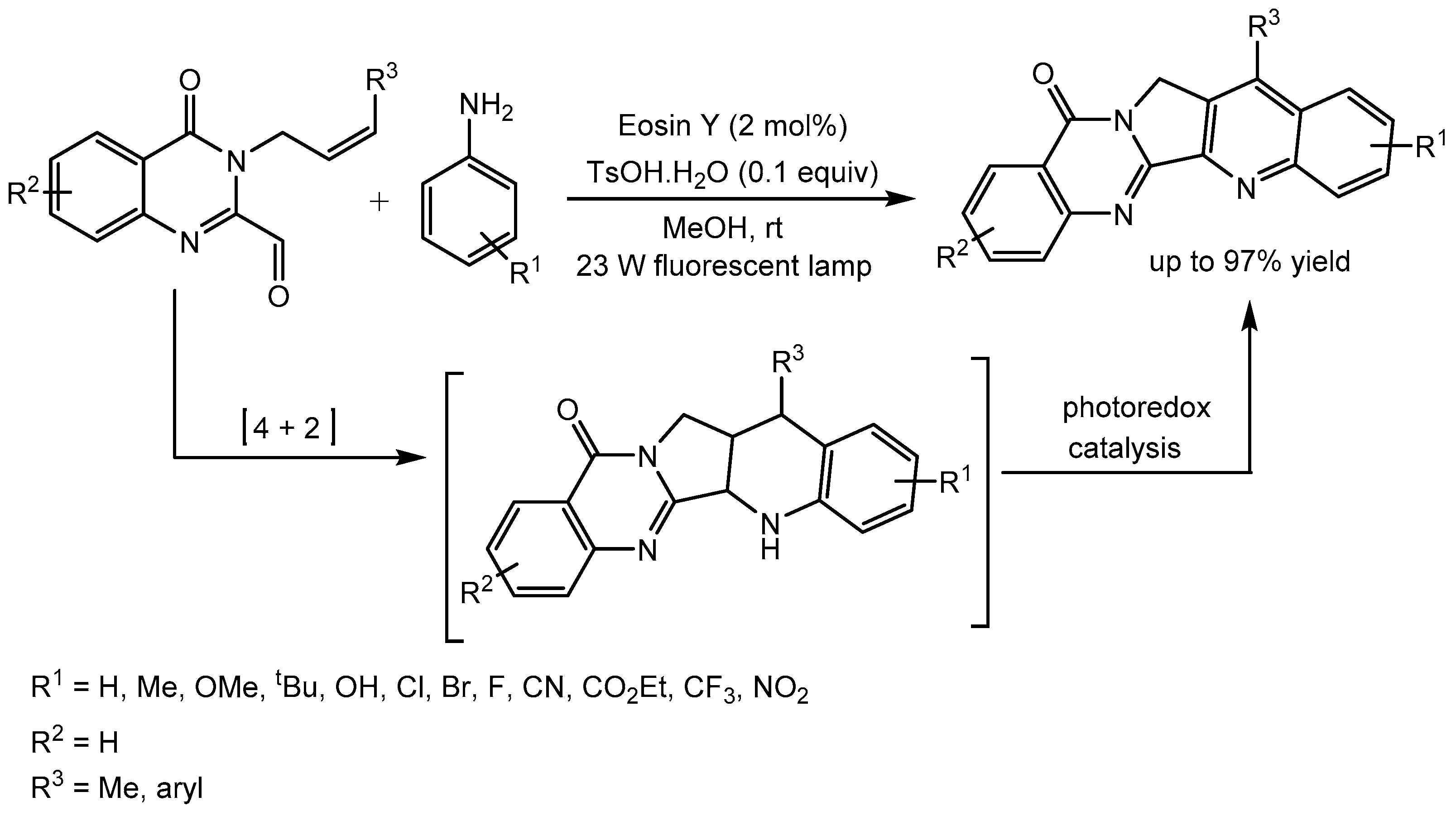
Scheme 77.
PTSA promoted Cross-Dehydrogenative Coupling (CDC) of Xanthene with β-keto moieties.
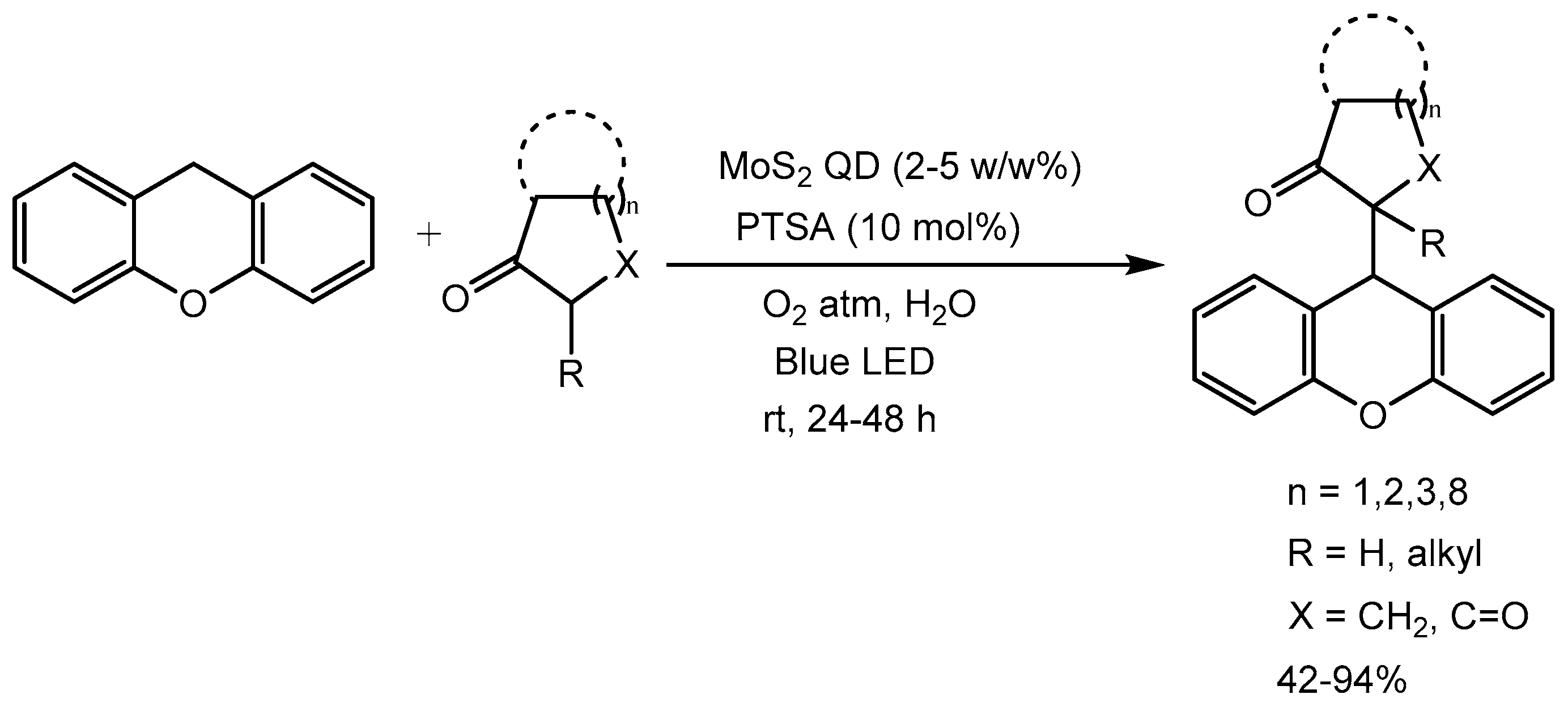
Scheme 78.
Polystyrene-supported P-TSA catalysed dihydropyrano[2,3-c]pyrazoles synthesis.
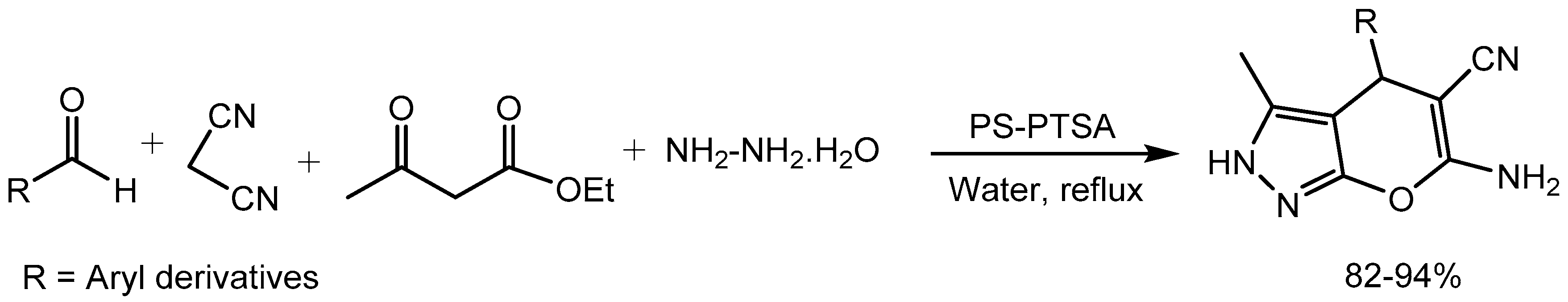
Scheme 79.
PTSA catalysed synthesis of Jasminaldehyde.
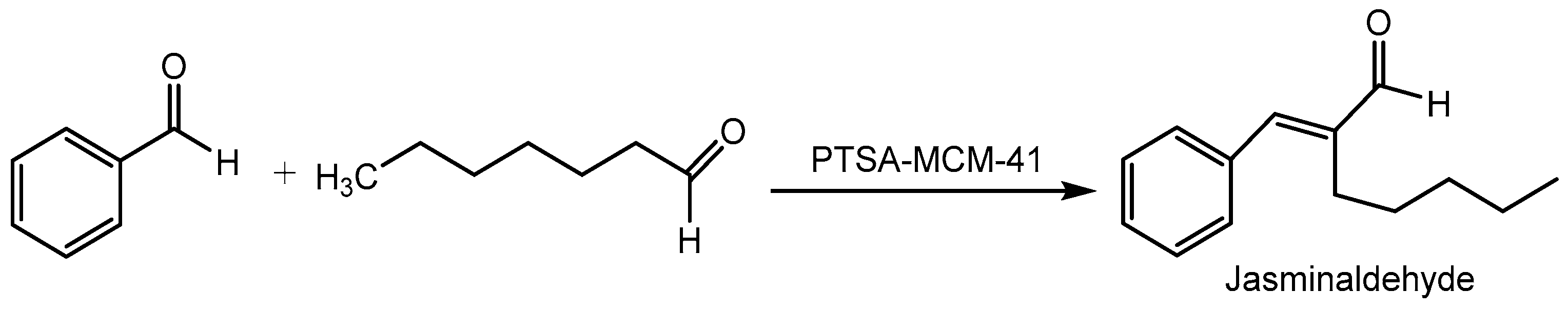
Scheme 80.
Synthesis of tetrasubstituted pyrrole derivative catalyzed by PS-PTSA.
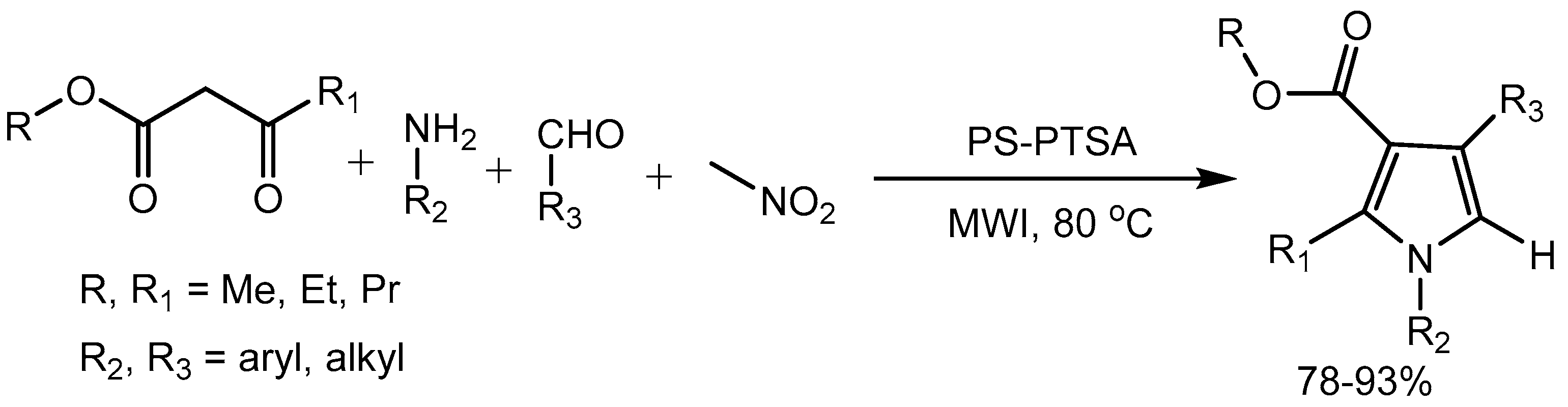
Scheme 81.
Synthesis of quinolines via synergistic catalysis using PTSA and HNTf2.
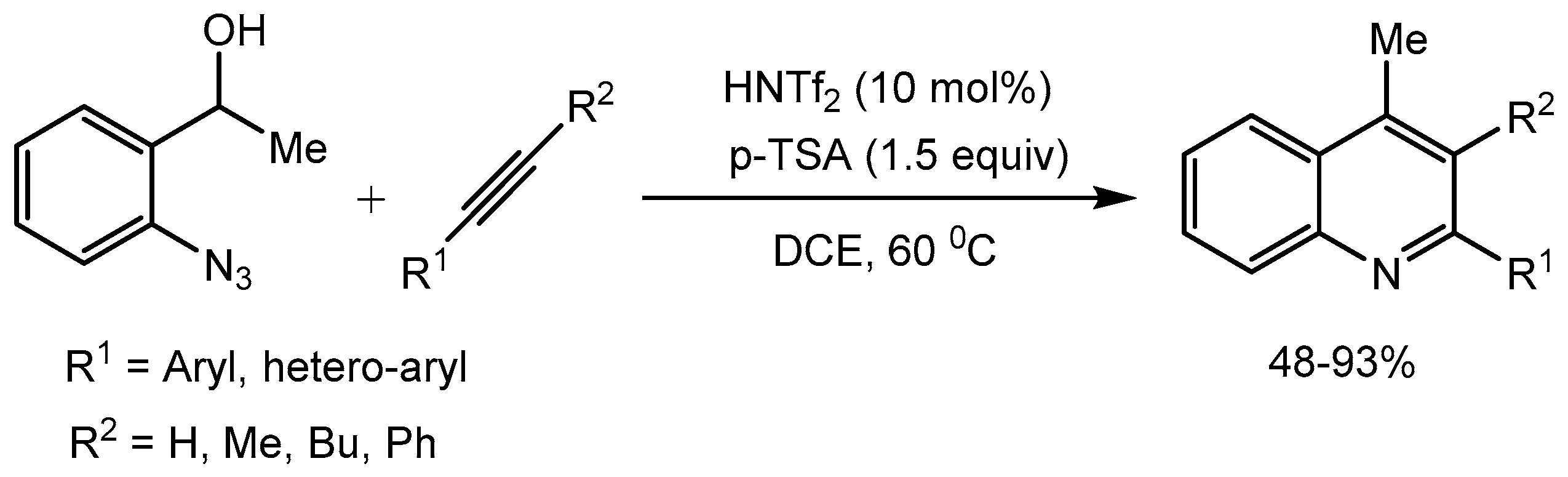
Scheme 82.
PTSA catalysed construction of the quaternary C–F bond from ketone derivatives.
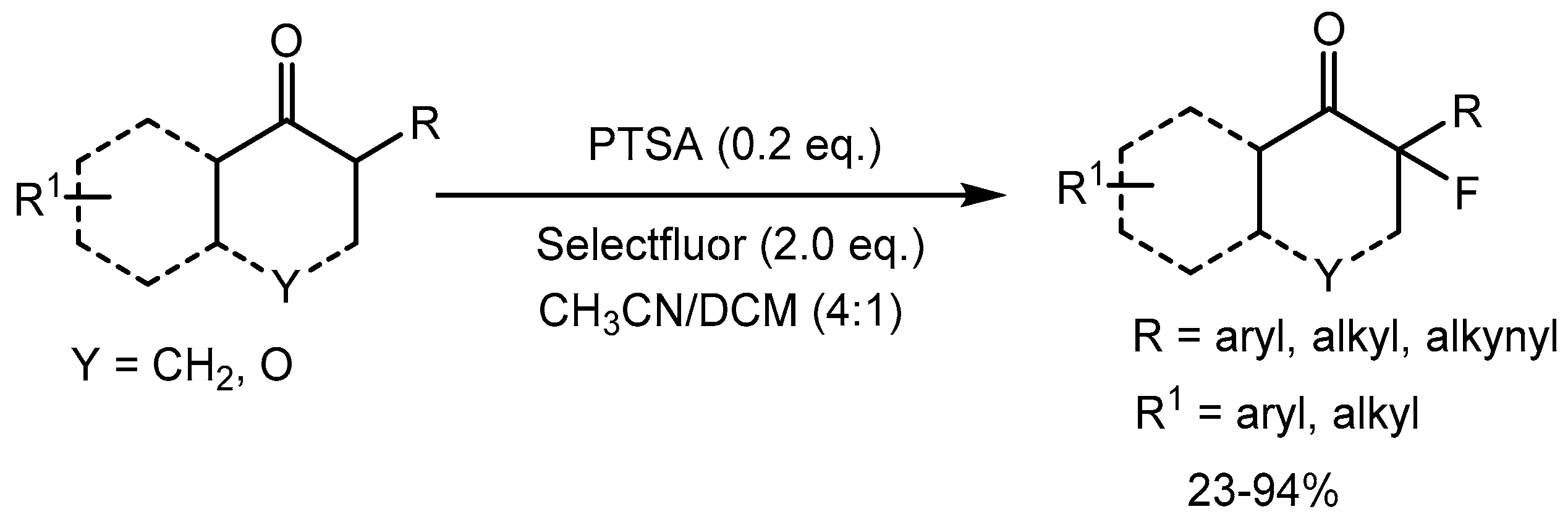
Scheme 84.
PTSA promoted reaction of [60]fullerene with electron-deficient ferrocenes.
Scheme 84.
PTSA promoted reaction of [60]fullerene with electron-deficient ferrocenes.
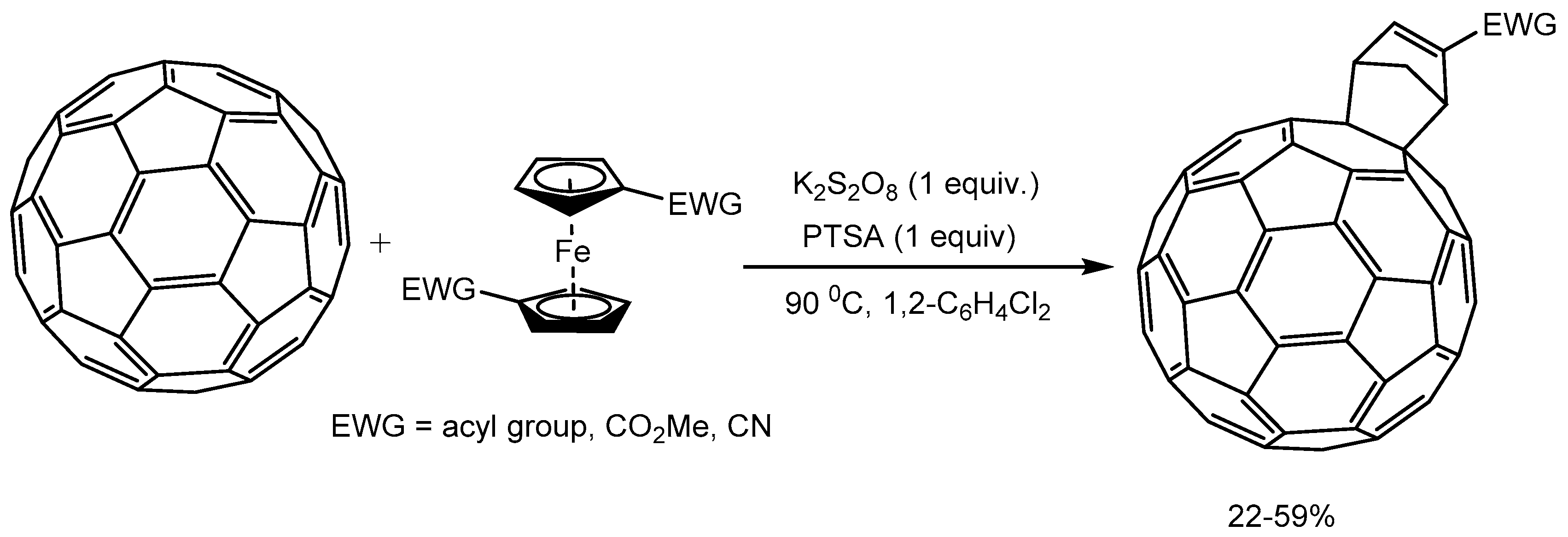
Scheme 85.
PTSA promoted one-pot tandem processes using stable aryl diazonium tosylate salts.
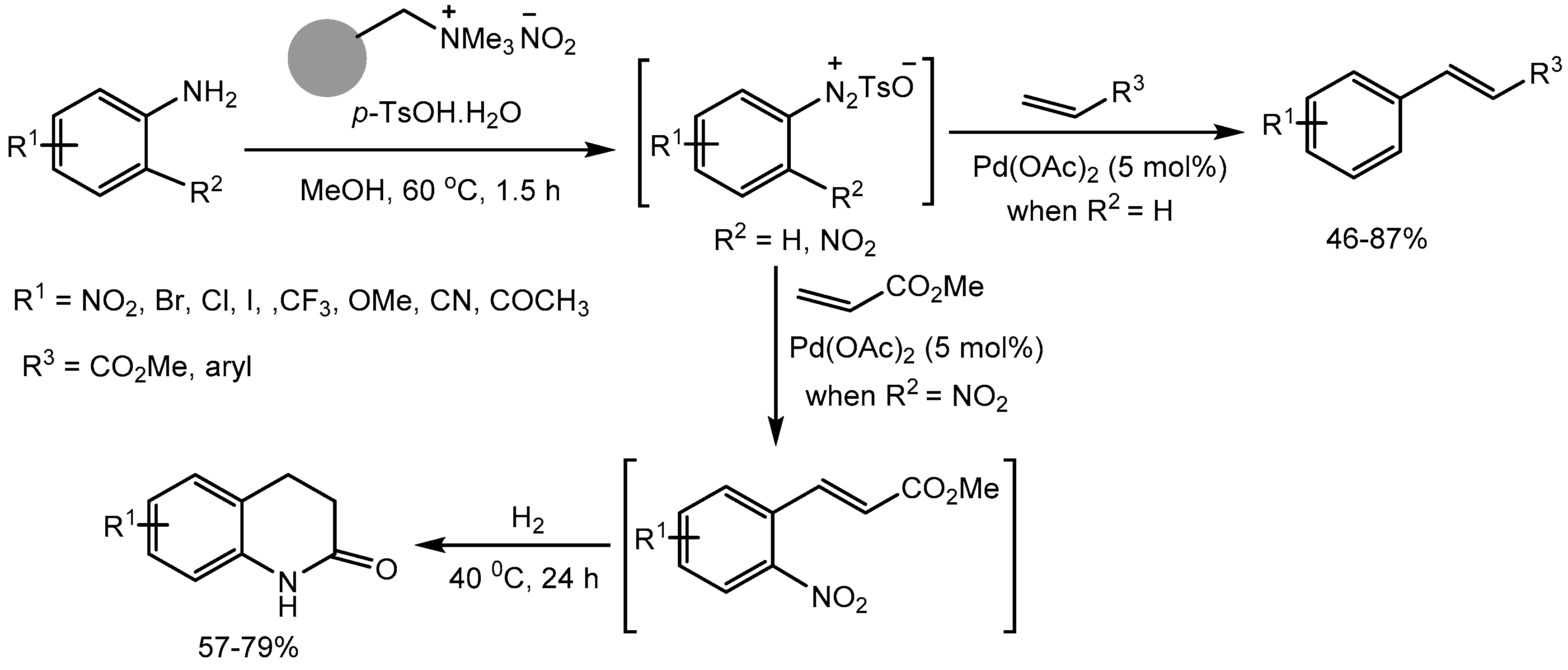
Scheme 86.
Synthesis of substited benzimidazoles using tosic acid-on-silica gel.
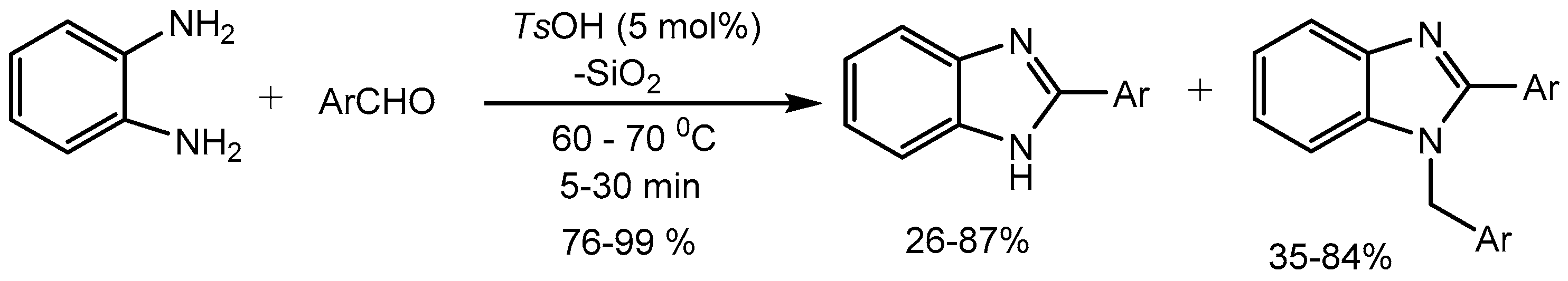
Scheme 87.
Synthesis of N-substituted 1,2,3-benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)- dioxides using p-tosic acid.
Scheme 87.
Synthesis of N-substituted 1,2,3-benzotriazin-4(3H)-ones and benzothiatriazine-1,1(2H)- dioxides using p-tosic acid.
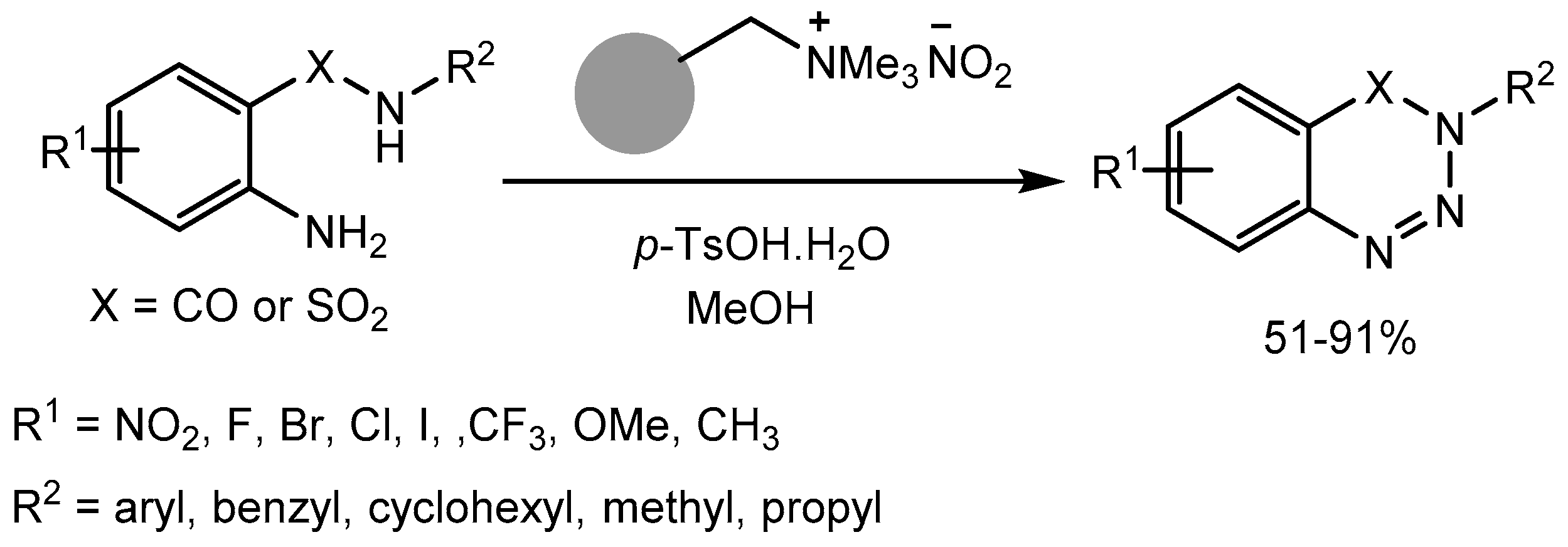
Scheme 88.
Dual organocatalytic synthesis of coumarin derivatives.

Scheme 89.
Synthesis of 2,5-furandicarboxylic acid (FDCA) using the p-TSA–POM catalyst and oxone.
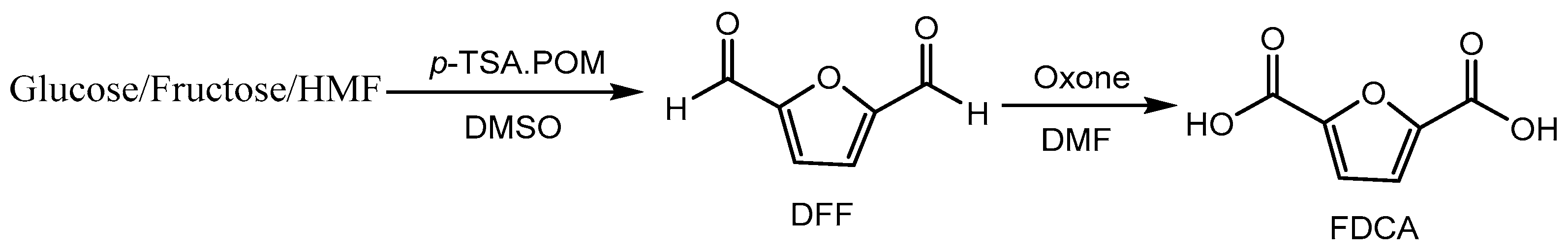
Scheme 90.
Synthesis of Substituted HTIBs using TsOH·H2O.
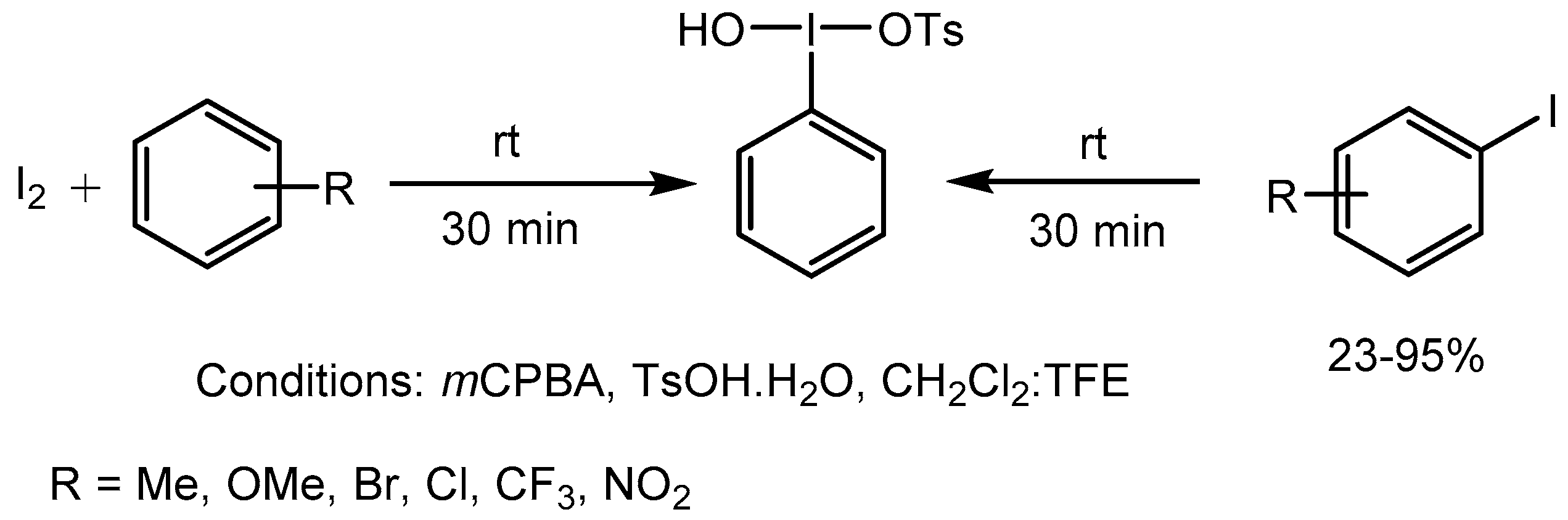
Scheme 91.
TsOH.H2O promoted metal-free carbon−phosphorus bond formation. .
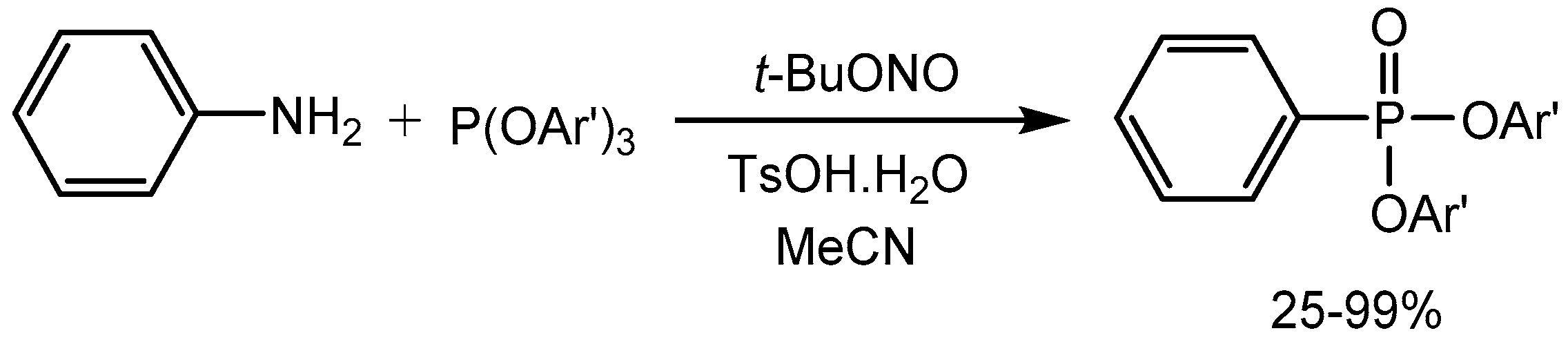
Scheme 92.
P-TSA catalyzed synthesis of 2,4-disubstituted quinolines.
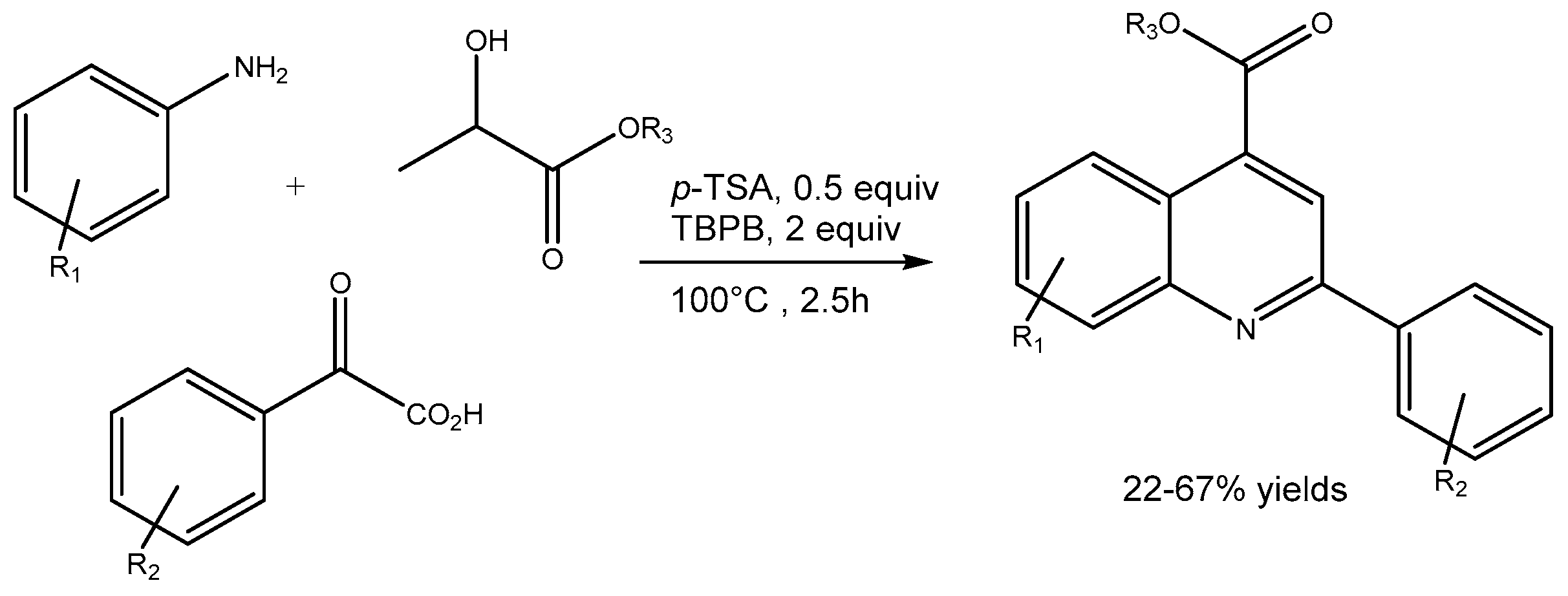
Scheme 93.
P-TSA catalyzed synthesis of amines via denitrosation of aryl-N-nitrosamines.
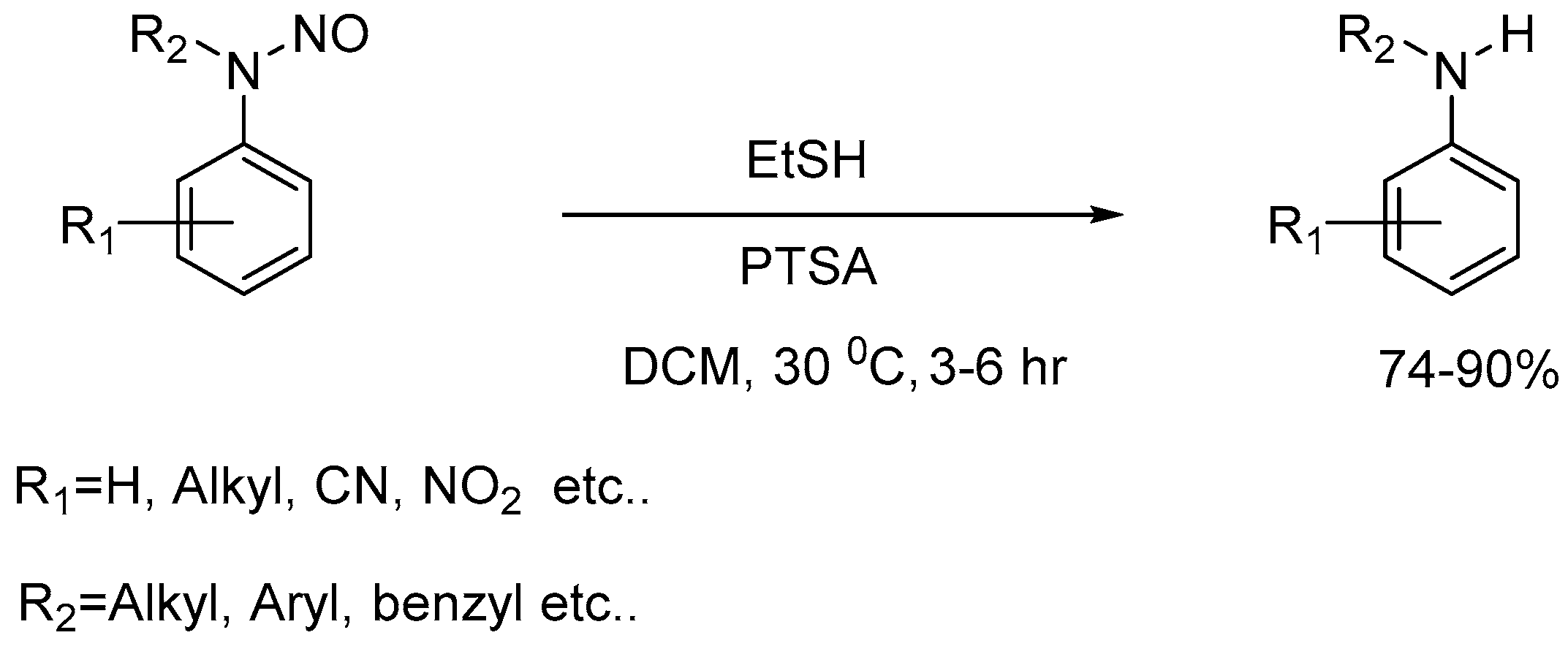
Scheme 94.
pTSA catalyzed synthesis of α-2-deoxyglycosides in ionic liquid.
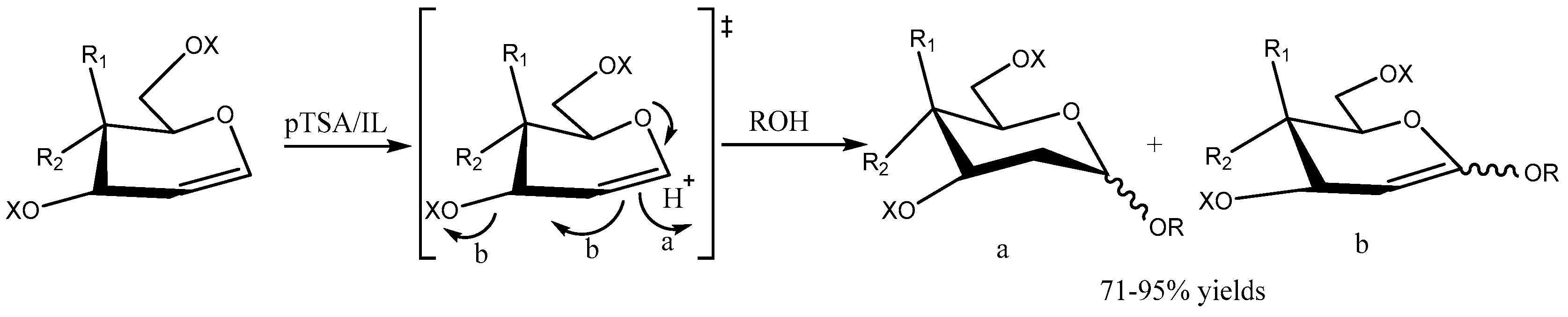
Scheme 95.
Friedel–Crafts acylation reaction catalyzed by p-TSA functionalized imidazole ionic liquids encapsulated into bismuth SBA-16.
Scheme 95.
Friedel–Crafts acylation reaction catalyzed by p-TSA functionalized imidazole ionic liquids encapsulated into bismuth SBA-16.
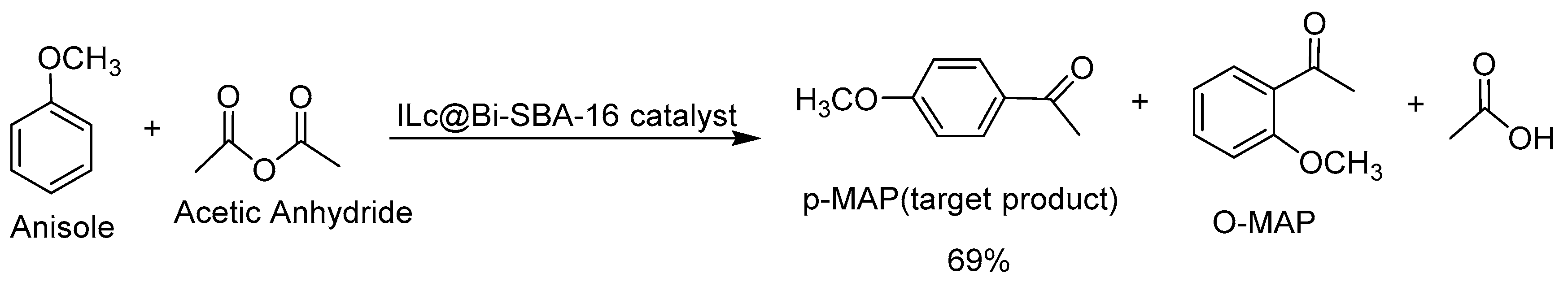
Scheme 96.
Halogenation of fatty diols by “PTSA/1-alkyl-3-methylimidazolium ionic liquids”.

Scheme 97.
Esterification of acid using bis-(3-methyl-l-imidazole)butylidene double p-toluene sulfonic acid salt (Im-PTSA).
Scheme 97.
Esterification of acid using bis-(3-methyl-l-imidazole)butylidene double p-toluene sulfonic acid salt (Im-PTSA).
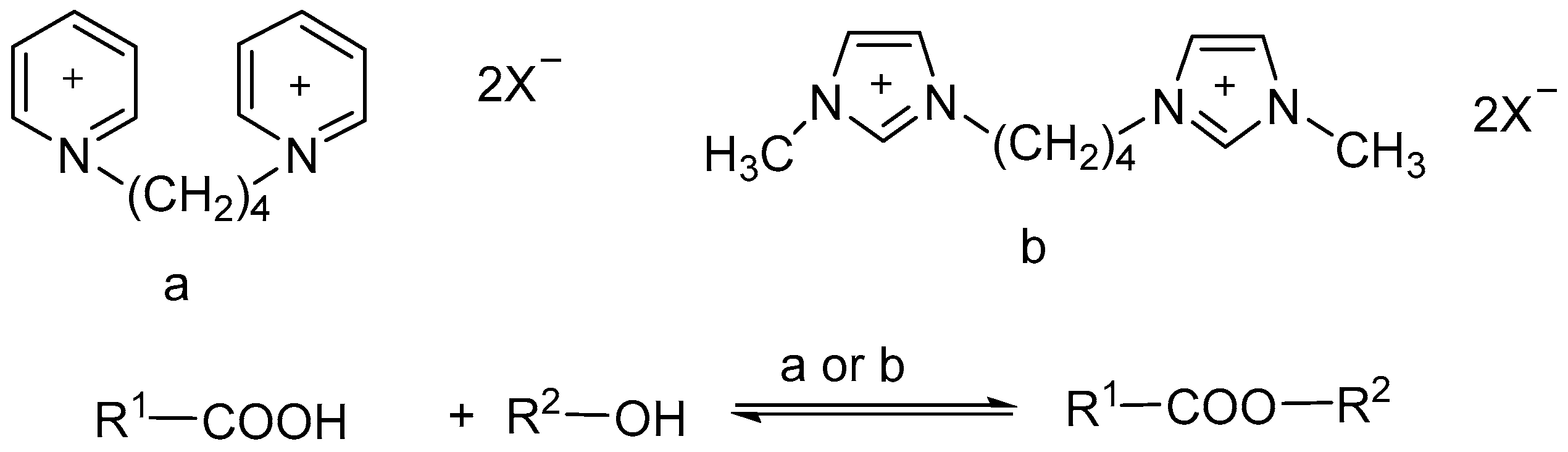
Scheme 98.
pTSA catalyzed multi-component synthesis of 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-ones in ionic liquid.
Scheme 98.
pTSA catalyzed multi-component synthesis of 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-ones in ionic liquid.
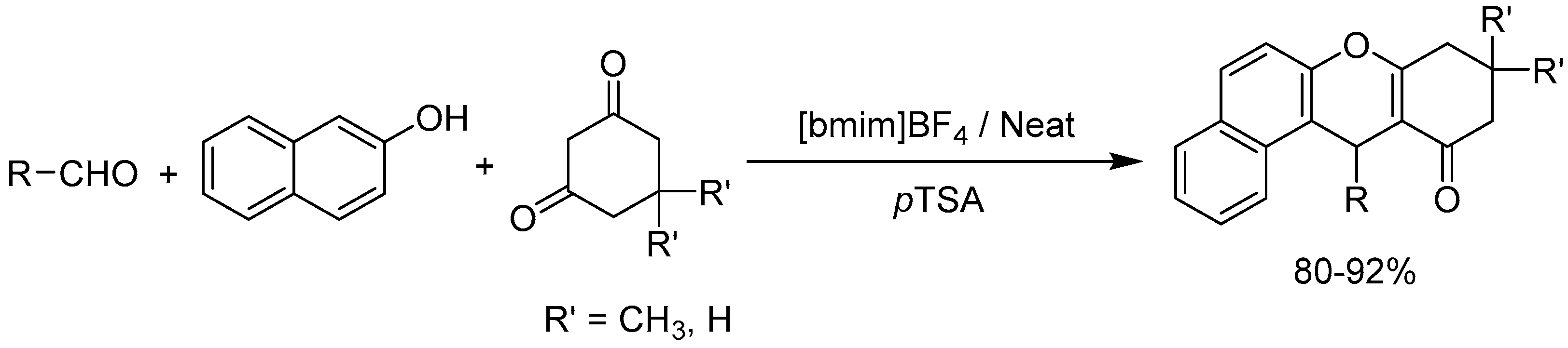
Scheme 99.
Plausible mechanism of 12-aryl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-ones formation.
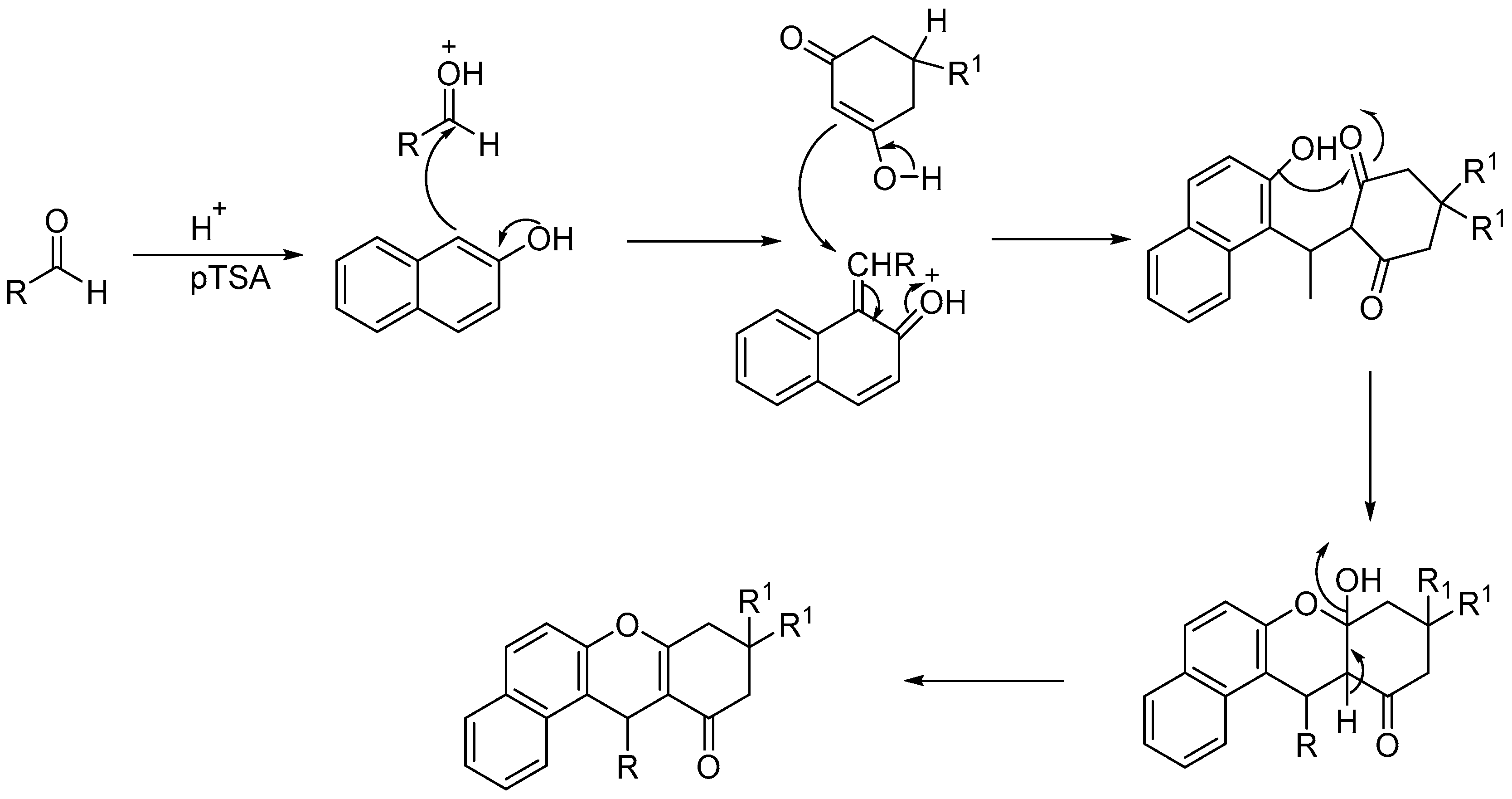
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



