
Submitted:
22 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
Pyrazinacenes are linearly fused heteroaromatic rings, with N atoms replacing all apical CH moieties. Component rings may exist in a reduced state having NH groups instead of N, causing cross-conjugation. These compounds have interesting optical and electronic properties, including strong fluorescence in the near infrared region and photocatalytic properties, leading to diverse possible applications in bio-imaging, organic synthesis as well as obvious molecular electronic uses. In this study, we have investigated the behaviour of seven-ring pyrazinacene 2,3,11,12-tetraphenyl-7,16-dihydro-1,4,5,6,7,8,9,12,13,14,15,16,17,18-tetradecaazaheptacene (Ph4H2N14HEPT) with emphasis on protic processes including oxidation, tautomerism, deprotonation and protonation, and the species resulting from those processes. We have used computational methods to optimize the structures of the different species and generate/compare molecular orbital structures. Aromaticity of species generated by the different processes has been assessed using the nucleus independent chemical shifts, and trends in the values have been associated with the different transformations of the pyrazinacene core. The computational data was compared with experimental data obtained from synthetic samples of the molecule tBu8Ph4H2N14HEPT.
Keywords:
pyrazinacene
; acene
; N-heteroacene
; azaacene
; tautomerization
1. Introduction
The term ‘acene’ refers to a series of polycyclic aromatic hydrocarbon compounds composed of linearly fused benzene rings, the archetypal member of which is pentacene [1] (Figure 1) having, as the name suggests, five fused rings. Other members of the series named according to ring multiplicity include tetracene [2], hexacene [3], and heptacene [4]. These compounds are investigated for their organic semiconductor properties although other aspects based on synthetic modifications are available [5,6,7,8]. Longer analogues can behave as 1-dimensional conductors complementary to 2-dimensional semi-metallic graphene [9]. In terms of real-world applications, acenes have appropriate properties for their use in low-cost large screen displays, or as semiconductor ‘inks’ processable by conventional printing techniques [10].
An unfortunate symptom of increasing ring multiplicity in acenes is a diminishing of the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) and an associated reduction in ionization energy, a feature which is detrimental to their stability against oxidation so that longer acenes (pentacene and beyond) require precautions in their applications, or are simply not sufficiently stable [11]. This instability can be accounted for according to Clar’s π–sextet rule since in acenes only every other ring can possess the six localised π electrons necessary to fulfil the rule [12]. The number of rings lacking sextets increases in the longer acenes leading to increasingly poor stability. Recently, there have been several reports of methods for the in-situ generation of extended acenes, which avoid synthetic and stability problems, thus allowing characterization of these fascinating compounds [13,14,15].
Figure 1.
Chemical structures of acenes and pyrazinacenes including examples pentacene and 1,4,5,6,7,8,11,12,13,14-decaazapentacene. Energy-minimized structures of pentacene and decaazapentacene are also shown. At right are shown previously prepared examples of pyrazinacenes: Ph4H2N10PENT (2,3,9,10-tetraphenyl-6,13-dihydro-1,4,5,6,7,8,11,12,13,14-decaazapentacene [16]), Phen-H2N8PENT-CN2 (6,13-Dihydrodipyrido[3,2-a:2',3'-c]-5,6,7,8,11,12,13,14-octaazapentacene-9,10- dicarbonitrile [17]) and, the subject of this work, H2N14HEPT (7,14-dihydro-1,4,5,6,7,8,9,10,13,14,15,16,17,18-tetradecaazaheptacene [18]). Numbering scheme of rings in Ph4H2N14HEPT is also shown (see H2N14HEPT in figure).
Figure 1.
Chemical structures of acenes and pyrazinacenes including examples pentacene and 1,4,5,6,7,8,11,12,13,14-decaazapentacene. Energy-minimized structures of pentacene and decaazapentacene are also shown. At right are shown previously prepared examples of pyrazinacenes: Ph4H2N10PENT (2,3,9,10-tetraphenyl-6,13-dihydro-1,4,5,6,7,8,11,12,13,14-decaazapentacene [16]), Phen-H2N8PENT-CN2 (6,13-Dihydrodipyrido[3,2-a:2',3'-c]-5,6,7,8,11,12,13,14-octaazapentacene-9,10- dicarbonitrile [17]) and, the subject of this work, H2N14HEPT (7,14-dihydro-1,4,5,6,7,8,9,10,13,14,15,16,17,18-tetradecaazaheptacene [18]). Numbering scheme of rings in Ph4H2N14HEPT is also shown (see H2N14HEPT in figure).
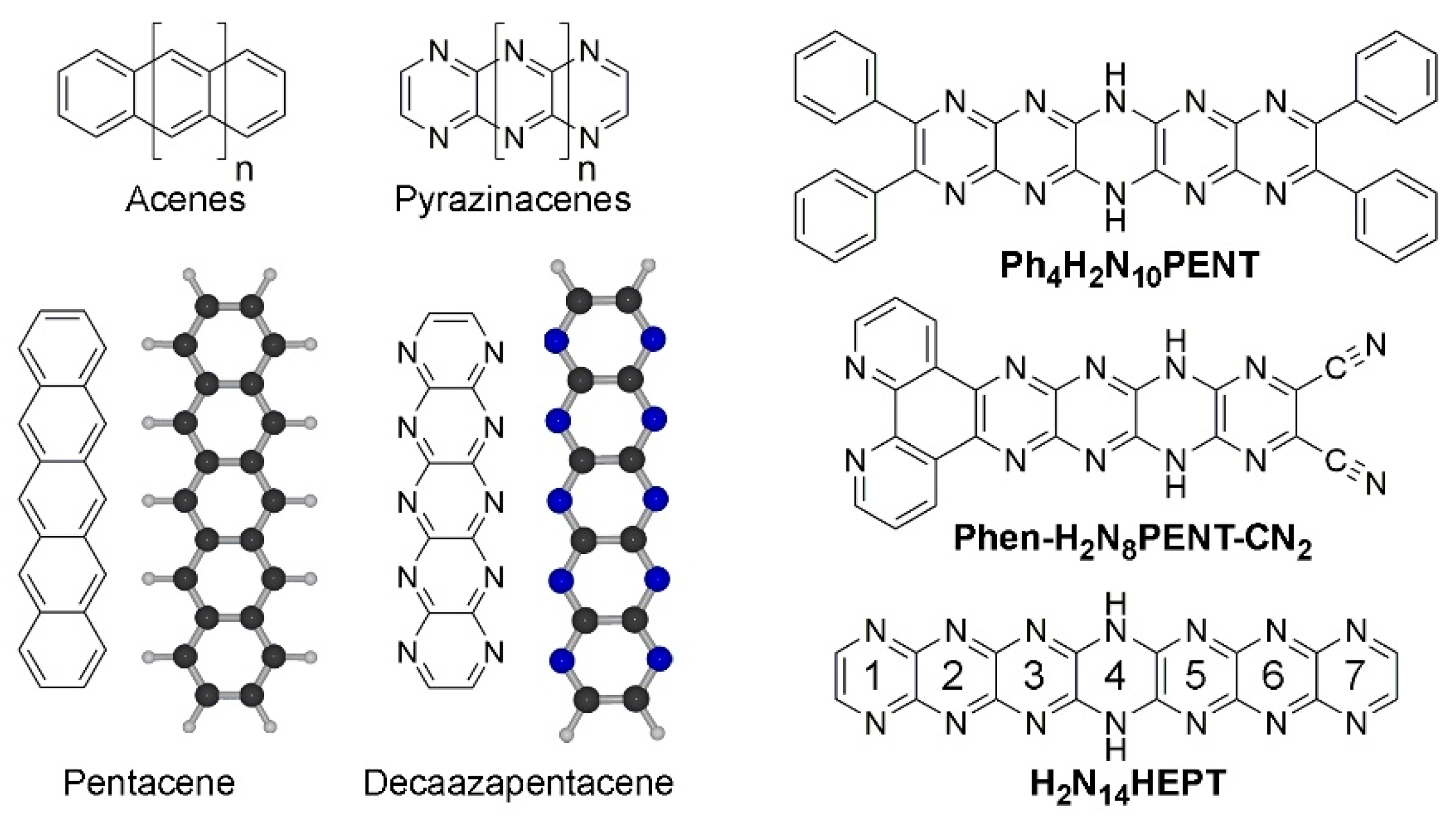
Pyrazinacenes (Figure 1) are N-substituted analogues of acenes which contain linearly fused pyrazine rings, generally with all acene apical atoms being nitrogen atoms [19,20]. The presence of apical nitrogen atoms allows for the presence of reduced (dihydropyrazine) rings (sp3 NH instead of sp2 N) in turn introducing the possibility of multi-stable redox activity or proton delocalization. The compounds can be prepared in a variety of forms including substituted with solubilizing groups (e.g., Ph4H2N10PENT [16], the first synthetically available decaazapentacene, see Figure 1) or with different functionalities (e.g., phenanthrolo-appended Phen-H2N8PENT-CN2 [17] for metal coordination). Pyrazinacenes can be considered as members of the azaacene [21,22] family, and are also a subset of heteroacenes [23,24]. Both classes of compounds are investigated for their potential applications as organic semi-conductors [25,26], for sensing [27], or other properties [28,29]. Acenes and pyrazinacenes are interesting targets for theoretical studies because of predictions that might be made about properties including their electronic structures [30,31,32] or other aspects of the compounds including protic tautomerism [33,34].
In this computational study, the longest of so far synthetically available pyrazinacenes [18], namely 7,16-dihydro-1,4,5,6,7,8,9,10,13,14,15,16,17,18-tetradecaazaheptacene (Ph4H2N14HEPT, Figure 1 and Figure 2), has been considered based on the salient properties of oxidation, protonation/deprotonation, and tautomerism. The compound contains seven linearly fused pyrazine rings with four phenyl substituents at the terminal pyrazine units of the molecule introduced to promote solubility in organic solvents. A single reduced ring is present and is described formally as residing at the centre of the molecule as a dihydropyrazine unit. Although we have previously studied extended reduced pyrazinacenes using computational methods [33,34], this work is of interest from the point-of-view that the chromophore of study has been synthesized in previous work [18] enabling a direct comparison of computational data with some of the experimental results. In this case, there exists the possibility of multiple tautomers with the additional aspects of protonation due to the presence of several basic nitrogen atoms, and deprotonation at the dihydropyrazine unit. We have studied the electronic and optical properties of this chromophore based on these transformations using different computational methods.
2. Results and Discussion
Here, we have focused on several possible transformations of Ph4H2N14HEPT in order of their perceived complexity: i) The simplest possible transformation with the fewest possible products involves 2-electron oxidation with concurrent elimination of protons at 7,14 positions; ii) next, we consider deprotonation where stepwise processes lead respectively to monoanion and dianion species; iii) tautomerization is based on isomerization involving variation of the protons’ locations at the different electronegative atoms, i.e., N atoms, of Ph4H2N14HEPT. For simplicity here, we consider only isomers where these two protons are shifted to the same adjacent ring so that all isomers contain a dihydropyrazine unit as shown in Figure 2. Other isomers where the two protons reside on nitrogen atoms of different pyrazine rings contain quinoidal forms where acene character is lost or interrupted and will be treated elsewhere. iv) Finally, we consider processes involving protonation and, in this case, monoprotonation is the simplest transformation. Protonation-coupled tautomerized structures are also considered. Each of the aforementioned processes has different connotations for the electronic structure and optical properties of the compound and we aimed to predict or assess the effects relative to the known properties of this chromophore [18].
2.1. Experimental vs. Computed Structures
The X-ray crystal structure of tBu8Ph4H2N14HEPT (Figure 2a; here, T0 (Ph4H2N14HEPT) is used to differentiate computed (T0) from experimental (tBu8Ph4H2N14HEPT) structures) was used as a comparison to test the computational functional/basis set. Excellent agreement with only minor bond length deviations of 0.01-0.02 Å were found between the core chromophores of the two structures (see Table 1).
For tBu8H2N14HEPT, its X-ray structure is essentially planar (global torsion angle: 0.7°) while that computed for T0 (of D2 symmetry after optimisation) has torsional distortion (about 23°), as shown in Figure 3. Both X-ray and computed structures confirm the delocalization of the sp3 character of the dihydropyrazine unit leading to planarity of the acene moiety [35]. There is an overall alternation of short and long C-N bond distances in both structures while the interstitial C-C bond lengths reflect their substantial single bond status. The most significant differences between the two structures involve the N-H bond length, which is shorter than expected for tBu8Ph4H2N14HEPT, and the dispositions of the phenyl substituents (see Figure 3). The two protons could not be located during refinement (due to poor crystal quality and lower resolution X-ray data) so were placed using a riding model, which was perturbed during subsequent refinement. Also, crystal packing forces also affect the form of tBu8Ph4H2N14HEPT (especially conformation at the phenyl substituents which bear tBu groups) as well as the presence on one 4-dimethylaminopyridine (4-DMAP) molecule on each side making a direct comparison of the two structures less meaningful. However, the main salient features of the molecule (i.e., almost planar structure, acene-like alternating C-N bond lengths and strong single bond character in interstitial C-C bonds) found in the experimental structure are reproduced in the computed structure.
2.2. Oxidation and Deprotonation
If tautomer T0 is oxidized involving the loss of two protons/electrons, then the compound labelled Ox would be obtained (can also be considered Ph4N14HEPT, labelled Ox here for convenience). This compound has a fully conjugated acene-like structure lacking a reduced dihydropyrazine unit (although the reduced ring in those compounds is also subject to delocalization as has been reported previously [35], and is confirmed here). The HOMO-LUMO gap for Ox of 1.813 eV is much lower than that of T0 (2.499 eV), which is expected for compounds with more highly conjugated electronic structure. Since Ox can be formally considered as T0 that has lost two hydrogen atoms (not protons), the stabilities of its possible singlet and triplet states were assessed. However, it was found that its triplet state is less stable than the singlet state by 14.7 kcal.mol-1, and the resulting SOMO-LUMO gap is only 0.606 eV.
Electronic structures of T0 and Ox are shown in Figure 4. An important feature of the former is the stabilizing distribution of antiaromatic character (introduced by the dihydropyrazine unit, Figure 4a), which yields planar structure despite sp3 character of the reduced N atoms [35]. This promotes acene-like structure in molecular orbitals of T0 [36] as shown in orbitals HOMO-1 up to LUMO+3 (Figure 4b). HOMO-2 and HOMO-3 indicate a propensity for delocalization of N-atom lone pairs indicated by the overlapping orbital lobes of the hexaazaanthracene groups fused to the central dihydropyrazine. This feature is emphasized in Ox (Figure 4c) where HOMO and HOMO-1 (Figure 4d) orbitals exhibit strong lobe overlap between adjacent rings. This aspect of pyrazinacene orbital structure distinguishes them from the CH-acenes although acene character is also retained. The energy order of the orbitals is also quite different: the structures of HOMO-1, HOMO and LUMO orbitals of T0 correspond respectively to HOMO-2, LUMO and LUMO+1 orbitals of Ox. For the latter, two σ orbitals (HOMO-1 and HOMO) lie between π and π* orbitals, which correspond to the HOMO and LUMO orbitals of T0. Thus, the main absorption band of Ox can be assigned to a HOMO-2-to-LUMO transition (π → π*) rather than a HOMO-LUMO electronic transition (σ → π*). Table 2 summarizes this situation where the relevant transition, HOMO-2 to LUMO for compound Ox, is also shown to be red-shifted relative to T0 due to the lower HOMO-LUMO gap of Ox.
Nucleus-Independent Chemical Shifts (NICS) [37] values are used as a measure of local aromaticity across molecules. Artificial protons positioned at the centre of aromatic rings have substantially low chemical shifts, even reaching negative values (-1, -3 ppm…) due to increased shielding although this weakens towards the edges due to deshielding. These values can be used to assess aromatic or non-aromatic character at each 6-membered ring. The NICS(0) index is the value at the centre of the ring while the NICS(1) index is the value 1 Å above the plane of the ring. Increasingly negative NICS indices indicate higher aromaticity with increasingly positive values indicating antiaromaticity. For reference here [38], benzene and pyrazine have NICS(0) values of -8.1 and -5.2 ppm, respectively, with values of NICS(1) around -10.0 ppm for both, due to their essential aromatic character. The two conformers of 1,4-dihydropyrazine (boat and chair) have NICS(0) at +11 and +14 ppm, and NICS(1) +8 and +11 ppm, respectively, due to antiaromaticity.
According to NICS indices for Ox (Figure 4c), pyrazine ring aromaticity increases towards the center of the molecule (similarly to other acenes [39] and N-heteroacenes [40]). In contrast, T0, has the opposite tendency with a peak in aromaticity at the center of the conjugated units (note that T0 can be considered as two hexaazaanthracene units fused through the central dihydropyrazine). In the oxidized compound Ox, NICS values are also more homogeneous because of the increasing similarity in character of the fused pyrazine rings of the H2N14HEPT unit and the lack of a reduced dihydropyrazine ring. The terminal pyrazine rings are however less similar, as has been reported previously [33,34].
The presence of the dihydropyrazine ring introduces the possibility of anionic pyrazinacene frameworks by deprotonation. In this case, one or two protons can be successively removed from the dihydropyrazine ring of T0 [18]. Single deprotonation to the monoanion (MA = [Ph4HN14HEPT]–) is known to occur easily in the presence of weak bases such as potassium carbonate while the formation of the dianion (DA = [Ph4N14HEPT]2–) is known to require a much stronger base such as lithium diethylamide (LDA) [16,18]. The effects of these deprotonation processes have been studied here using computational methods. For successive deprotonations of T0, the HOMO-LUMO gap decreases from 2.50 eV to 2.06 eV for the monoanion MA then to 1.75 eV for the dianion DA, reflecting some decrease in their stability and/or some increase in their aromaticity.
Figure 5 shows the formal chemical structures (Figure 5a,c) and molecular orbital diagrams of MA and DA (Figure 5b,d). The HOMO-LUMO gap of the anions is lower in both cases than that of the neutral tautomer T0. Lone pair character of the molecular orbitals (HOMO-1) lies on the peripheral pyrazine units for MA, and on the three central fused pyrazine units for DA. For MA, its HOMO-2, which is very close in energy to its HOMO-1, also shows contributions at all nitrogen atoms, especially at the central pyrazine unit, while HOMO-4 is highly localised on the central pyrazine ring opposing the remaining proton. Plots of HOMO-1, HOMO-2 and HOMO-4 indicate that contributions from nitrogen lone pairs remain significant after deprotonation with monodeprotonation reducing the symmetry of the electronic structure. The symmetry is restored by the second deprotonation with HOMO-1 (now similar in structure to HOMO of Ox), HOMO-3 and HOMO-4 showing the impact of the presence of multiple nitrogen atoms.
In this case, the pyrazine groups with highest NICS aromaticity are found at the molecular ends remote from the reduced ring with degree of aromaticity increasing regularly moving outwards from the central dihydropyrazine unit. Deprotonation leads to a stepwise evening of NICS values most likely as a result of charge being more highly delocalised over all of the rings in both MA and DA. For this reason, it is not realistic to place localised charges at the central nitrogen atoms of the dihydropyrazine unit (except to denote the chemical structure). The wavelengths of the electronic absorption maxima of the oxidised and dianionic compounds are similar. Initially, this seemed to originate from the similarities in the energies of their HOMO-LUMO gaps and computed structures, which are identical except for the two extra electrons in the dianionic compound. However, on closer observation the structure of Ox is subject to more significant torsional distortion than DA by about 10°, and its bond lengths do not vary in the same way.
For MA and DA, TD-DFT results indicate that absorption maxima are almost exclusively due to HOMO-LUMO (π → π*) transitions, explaining why the energy of the main absorption wavelength is close to the HOMO-LUMO gaps of each compound (see Table 3). Each deprotonation step is associated with a red shift from 550 (T0) to 618 nm (MA) then 618 to 664 nm (DA) with corresponding shifts in fluorescence emission (see Figure 6a). This can be compared directly with the experimentally known behaviour of tBu8Ph4H2N14HEPT during deprotonation using weak and strong bases where respective shifts from 653 to 700 nm then 700 to 741 nm (in THF) have been observed (Figure 6b) [18]. Thus, the TD-DFT results are consistent with the experimental trends. Note that the vibrationally resolved electronic absorption spectra were not computed to confirm the vibronically coupled monotonically diminishing higher energy bands commonly observed in acenes, pyrazinacenes and other chromophores [41]. Finally, regarding Ox, the main electronic absorption earlier assigned to a HOMO-2-to-LUMO transition (π → π*) occurs at 657 nm representing a 107 nm red shift and a substantial stabilizing effect. However, tBu8Ph4H2N14HEPT cannot be obtained in the 2-electron oxidized state so a direct comparison of this value is not available. Despite this, in analogous dihydrooctaazatetracene (H2N8TET) and dihydrodecaazapentacene (H2N10PENT) derivatives, oxidation to their respective N8TET and N10PENT forms is accompanied by 90 nm red shifts in their absorption maxima. It is not clearly understood why tBu8Ph4N14HEPT is not stable although reactivity towards trace water is strongly suspected as the reason for this observation. The strongly basic oxidized state tBu8Ph4N14HEPT might easily abstract a proton from water facilitating the reduction of the resulting protonated state. It is also possible that commonly available oxidants (e.g., PbO2 or high potential quinones such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)) are simply capable of oxidizing the already electron deficient tBu8Ph4H2N14HEPT chromophore. We are currently investigating the role of water in the respective reactions based on the possibility of using extended pyrazinacenes as electro- or photoredox catalysts.
2.3. Tautomerization
Tautomerization is potentially the most significant of the possible properties of H2N14HEPT derivatives because of implications for intramolecular proton transport or hydrogen-bonding guest adaptability [42]. However, it has so far proved the least accessible for study experimentally due to the broadness of the relevant signals in NMR or FTIR spectroscopy, although tautomerization in an N8-tetracene monoanion where two tautomers can coexist, has been reported [34]. Availability of the tautomers of pyrazinacenes has also been established indirectly by N-alkylation of N8-pentacene derivatives [17]. Examples of these two special observable cases of tautomerization are shown in Figure 7. The different possible dihydropyrazine-containing tautomers of Ph4H2N14HEPT (T0, T1, T2, T3) are shown in Figure 2d. Based on experimental observations, tautomerization is favoured only in N8-tetracenes and above because existence of the dihydropyrazine unit at extremities of the pyrazinacenes is strongly energetically disfavoured most likely due to the weaker delocalization of antiaromaticity and the loss of a Clar sextet. Thus, N6-anthraenes exist as a single tautomer while symmetrically substituted N8-tetracenes may exist as two tautomers.
The energies of the different T0–T3 were computed and are compared in Table 4. The tautomers become increasingly unstable as the dihydropyrazine unit shifts away from the centre of the molecule with ΔE and ΔG following a similar pattern. As expected from experiment, T3 is significantly less stable than T1 and T2 accounting for the absence of its N-alkylates in experiments designed to trap the tautomers by derivatization at the nitrogen atoms [17,34]. The HOMO-LUMO gap (see Table 5) is reduced being associated with the increasing extent of the conjugated system (3, 4, 5, then 6 rings), since in general larger conjugated systems exhibit smaller HOMO-LUMO gaps. Related to this are bond lengths in the tautomers (see Supporting Information, Table S1), where C-C bonds at the more highly conjugated side of the compound are lengthened while those at the opposing side (i.e., lower conjugation) become shorter (ca. 0.02 - 0.03 Å). Conversely, the C-N bonds at the more highly conjugated side are shortened significantly (by 0.06 - 0.08 Å). These variations are consistent with the increasing acene character of the increasingly conjugated moiety and low acene character of the opposing side despite the delocalization of the dihydropyrazine unit.
In T0 (see Figure 4a for the NICS values), NICS values indicate a reduction in the antiaromatic character of the dihydropyrazine ring relative to the parent 1,4-dihydropyrazine, while the largest aromaticity is located at a point generally as far from the dihydropyrazine unit as possible without being on the terminal pyrazine group. For regular acenes and azaacenes, aromaticity is generally the greatest at the centre of the molecules. In this case, NICS values indicate that the antiaromatic character of the 1,4-dihydropyrazine ring is essentially delocalized over the molecule and should be associated with the planarization of the molecule. This has been reported previously in the case of dihydro-6,13-diazapentacene [35] although the multiplicity of pyrazine units in T0 led us to confirm this situation. Incidentally, the NICS values of the phenyl groups are similar to those of benzene despite some orbital participation indicated by the frontier molecular orbital structures of T0 (Figure 4b, HOMO, HOMO-1).
For T0–T3 compounds containing 14 N atoms and a single dihydropyrazine unit, the sites at which protons of the reduced ring reside is an important parameter especially in the context of molecular recognition events where proton location would affect intermolecular interactions. Local charge or orbital structure might affect the location of those protons. To investigate this point, natural charges were estimated using natural bond orbital (NBO) analysis revealing that the most highly charged atoms are those of the dihydropyrazine ring, with others being slightly less negative. Notably, terminal pyrazine rings 1 and 7 show a persistent charge of –0.37 which hardly varies except when adjacent to the dihydropyrazine unit.
For electronic absorption spectra (UV-vis), the lowest energy computed wavelength of the absorption maximum is due largely to a HOMO-LUMO electronic transition, and there is a successive 20 – 30 nm red shift in the absorption band for the less stable tautomers. This should be accompanied by a significant Stokes shift (from 35 to 85 nm) of the fluorescence emission band. Calculated UV-vis spectra for T0–T3 are shown in Figure 8. In this case, the red shifts in absorption maxima can be assigned to the increasing extent of the delocalized π electronic system which has a stabilizing effect. It will be difficult experimentally to confirm these values due to the large number of possible tautomers available for Ph4H2N14HEPT (13 possible tautomers in symmetrical derivatives if N-alkylation at rings 1 and 7 is neglected). To obtain pure samples of derivatized tautomers will require extensive chromatographic separation and an informed selection of the derivatizing agent. However, this method can be used to fix electronic structures of the N14 chromophore allowing access to different aromatic and quinoidal forms. We are currently investigating this aspect of the pyrazinacene systems.
2.4. Protonation
Based on the large number of nitrogen atoms in Ph4H2N14HEPT compounds, protonation is the potentially most complicated of its protic processes. For this reason, and because these are currently the least accessible of the real-world isomers, we have studied only selected monoprotonated isomers. There are many possible structures available if a reduced Ph4H2N14HEPT is monoprotonated based on several different possible tautomers (including unsymmetrical ones) and 12 additional possible protonation positions. If the linear symmetry of Ph4H2N14HEPT is taken into account and tautomers are constrained to contain a single neutral dihydropyrazine group, there are 25 possible isomers of [Ph4H3N14HEPT]+. Here we have selected 8 of these using the most stable neutral tautomers as starting points.
The chemical structures of the protonated tautomers studied here are shown in Figure 9. Each protonated tautomer is identified based on the starting tautomer structure and the protonation site. There are three series of isomers: T0-HX, T1-HX and T2-HX where X denotes the protonation site of 4 selected as indicated below T0-H0 in Figure 9. Note that protonation on the dihydropyrazine ring has also been considered (Site 0) although this is highly unlikely given the weak sp3 character of those atoms. Also, T3 has been neglected because of the low stability of that tautomer.
Computed results shown in Table 6 indicate that the stability of the compounds is promoted if protonation is (in order of importance): i) not at the reduced dihydropyrazine ring, ii) not at a terminal pyrazine ring, and iii) separated from the reduced dihydropyrazine by two pyrazine units. Interestingly, the most stable of these compounds (T1-H2) is found in the T1 series rather than in the T0 series as might be expected based on the overall greater stability of T0. Overall, the stabilities suggest that there is a balance between tautomer stability and site of protonation for these isomers, and that protonation might be reasonably used to affect tautomer identity. That is, T0 might rearrange to T1 upon protonation to optimize stability. In solution especially in polar solvents, the situation is likely to be highly complex with protonated tautomers in a fluxional state depending on the acidity and concentration of Ph4H2N14HEPT.
HOMO-LUMO gaps of the protonated compounds are lower than their non-protonated counterparts being the lowest of the compounds discussed here. Essentially, the reduction in gap is inversely connected with the proximity of the protonated ring to the dihydropyrazine unit and is probably related to the relative extent of the non-protonated conjugated system in the different series. In this respect, it should be remembered that the dihydropyrazine ring is effectively delocalized over local non-protonated units. HOMO-LUMO gaps are 0.15–1 eV lower than for the neutral tautomers, and all (except that of T0-H0) are lower than that of compound Ox (1.81 eV) where all pyrazine rings are conjugated. Protonation causes some small variations in molecular geometry (see Supporting Information Table S1 for details) where C-N bonds at the side of the molecule at which protonation occurs are lengthened about the site of the proton, then alternate between shortened and lengthened for each successively remote bond by about 0.02-0.03 Å, up to 0.05 Å. Carbon-carbon bonds shorten somewhat about the site of protonation and lengthen at the remote rings (changes in the range of 0.01-0.02 Å). These changes are likely also connected with variations in local aromaticity. Relative to the parent tautomers, perturbation of aromaticity due to protonation in most of the isomers increases NICS(1) indices for the pyrazine rings on the protonated side by +2 to +5 ppm in the protonated ring and by +1 to +2 ppm in the others (see next section).
A consideration of the charges on the neutral compound T0 might reveal N atom sites preferred for protonation. For T0–T3, charges at the rings are shown in Figure 10. Values of charge indicate that protonation at Site 0 ought to lead to the most stable isomer with Site 2 being least favoured (a charge value around –0.5 is maintained at the dihydropyrazine ring in all of the tautomers). However, considering the total energies of the protonated compounds, protonation appears to be preferred at less negatively charged sites. For T0, protonation can be considered an electrophilic attack and so involves its non-bonding σ molecular orbitals, and the influence of nitrogen lone pairs is important. The MO diagram indicates that HOMO-2 and HOMO-3 have the greatest contribution from the N lone pairs at the 1st, 2nd, 6th and 7th rings. For this reason, isomers protonated at nitrogen atoms of those rings should be the most stable, and this is indeed what is found by calculation, in fact more so for rings 2 and 6.
The isomer T0-H2, the most stable in the T0 family of isomers, was selected to assess the effects of monoprotonation on molecular orbital structure. Figure 11 shows the HOMO and LUMO structures of this isomer. Interestingly, there is almost no evidence for nitrogen lone pair delocalization in T0-H2 with these features only emerging in deeper HOMO-5 and HOMO-6 where phenyl substituents also contribute. In the HOMO, electron density is situated remote from the protonation site while the LUMO is focused about the site. This situation indicates the possibility of electronic push-pull type behaviour or intramolecular electron transfer activity, both of which might be photolytically activated using long wavelength excitation sources.
For T0-H2 and the other stable isomers, the HOMO is quite close in energy (0.36-0.46 eV) to the next occupied orbitals, while the LUMO is separated (by 1 eV or more) from the next vacant molecular orbitals. Hence, electronic excitations must involve HOMO-to-LUMO or HOMO region-to-LUMO transitions. Similar to the neutral compounds, the HOMOs and LUMOs are of π and π* character, respectively. We indeed observe this type of electronic transition (see Table 7) but exclusively HOMO to LUMO (so the wavelengths follow exactly the same evolution as the gaps), similarly to the neutral compounds. However, since the HOMO-LUMO gaps are smaller, the main absorption wavelengths are much larger, in the near infrared (770-930 nm). The absorption wavelengths increase when the proton is further away from the reduced ring and when the reduced ring is closer to the remote rings, with red shifts between 25 and 80 nm from one series to another. However, the main bands are less intense than those of neutral compounds and there are other absorptions in the visible region. The intensities also decrease as the wavelengths increase. Computed absorption spectra are shown in Figure 12 for some monoprotonated tautomers of T0, while some of monoprotonated tautomers of T1 and T2 are available in Supporting Information (Figures S1 and S2).
Based on the calculations, both protonation and deprotonation occur with similar red shifts of the absorption maximum, although this effect is more significant for protonation (for deprotonation, shifts lie in the range 70 and 115 nm, and for protonation, in the range 220 and 360 nm). However, while calculations give a reasonable reproduction of the red shifts incurred by stepwise deprotonation (and also for oxidation of T0), electronic absorption data collected from acidified solutions of T0 are not consistent at first sight with the calculated absorption maxima found here. Figure 12 shows a comparison of calculated (Figure 12a) vs. experimental (Figure 12b) electronic absorption spectra of protonated T0. The experimentally observed electronic absorption maximum for tBu8Ph4H2N14HEPT is actually blue shifted by approximately 100 nm. The reasons for this inconsistency are unclear although solvent or impurities such as water are expected to have a significant effect on the stability of protonated tautomers. In the case of deprotonation and oxidation, electron density can be effectively delocalized intramolecularly minimizing the possible effects of solvation/hydration while the presence of positive charges after protonation might imply interactions with electronegative solvating agents or water for stabilization.
2.5. Trends in Aromaticity Based on NICS
Perturbation of aromaticity involving the processes studied here can be probed computationally using NICS indices [37], the trends can be associated with specific changes in the molecule structures, and visualized. Figure 13 shows variations in NICS(0) (red traces) and NICS(1) (blue traces) indices for T0, Ox, MA and DA. These species are symmetrical about a mirror plane placed perpendicular to the molecules’ long axes at the central ring. Both NICS indices (Figure 13a,b) show symmetric traces based on this. T0, MA and DA formally contain a dihydropyrazine unit whose antiaromaticity results in a strong positive peak in NICS values at the center of the molecule. For MA and DA, deprotonation allows further delocalization of electron density leading to distribution of the positive value across all rings and a significant lowering at the central ring. For Ox, all pyrazine rings have substantial aromatic character and this appears to be focused in intensity at the central pyrazine unit where there are strongly negative values for both NICS indices. Interestingly, terminal pyrazine rings have significantly less negative NICS values again emphasizing the different state of these areas of the molecule. For the tautomers T0-T3, the positive peak in NICS values (see Figure 13c,d) shifts without a substantial change in intensity according to the location of the dihydropyrazine unit, and an increasingly negative peak appears at the central rings (Rings 5, 6) of the widening conjugated region reflecting the locally increasing aromaticity due to the shift of the dihydropyrazine group.
Trends in the NICS indices are summarized for protonated tautomers in Figure 14. Similar to the neutral tautomers, the positive peaks in NICS values coincide with the location of the dihydropyrazine ring and the value magnitude hardly varies. Protonation shifts NICS(0,1) about the location of protonation to less negative values in all cases. In the case of pyridine, NICS values become more negative upon protonation to pyridinium as the electronic structure shifts towards that of benzene with which it is isoelectronic [37]. Less negative NICS for protonated tautomers of T0 might reflect an overall shift of electron density away from the site of protonation as suggested to occur the HOMO structures of T0-H2 (Figure 11b) where several of the occupied molecular orbitals reside largely on the phenyl substituents. Alternatively, increasing NICS values occur for the central rings of the conjugated regions of the molecules perhaps indicating some delocalization of the cationic charge.
3. Methods
Computational Details. Density functional theory (DFT) was used for geometry optimization of Ph4H2N14HEPT [43,44] (and its derivatives) using the Gaussian 16 program package [45], with the B3LYP functional [46,47,48,49] and the all-electron def2-TZVP basis set of the EMSL Basis Set Exchange Library [50,51,52,53,54]. Geometries optimized without any constraint were characterized as being true minima based on a vibrational analysis. Natural charges were computed using the NBO 6.0 program [55]. Electronic absorption (UV-vis) spectra were calculated by using time-dependent DFT (TD-DFT) [56,57], at the B3LYP/def2-TZVP level with only singlet-singlet, i.e., spin-allowed, transitions being computed. UV–vis spectra were simulated based on the TD-DFT transitions and their computed oscillator strengths using the SWizard program [58]. Each transition was associated with a Gaussian function at half-height width (equal to 1500 cm-1). Nucleus-Independent Chemical Shifts (NICS) were estimated based on NMR calculations at B3LYP/def2-TZVP level [37]. The AOMix program was used to calculate the composition of the molecular orbitals [59,60].
4. Conclusions
In summary, N14heptacene and pyrazinacenes in general, provide an opportunity to study molecules capable of undergoing all major protic processes within the same chromophore system. This presents unique opportunities for applications in bio-imaging (for instance, local pH mapping), molecular recognition including systems involving dynamic proton transport, H-bonding catalysis (including photoredox catalysis) and molecular electronics. In this work, we have considered the seven-ring reduced pyrazinacene Ph4H2N14HEPT, because its synthetic availability allows direct comparison of its properties especially during protonation and deprotonation. Transformations such as tautomerisation and oxidation, although known to occur in lower pyrazinacene analogues, have not been so far observed in Ph4H2N14HEPT. While oxidation of Ph4H2N14HEPT is thought to be precluded by its extensive electronic delocalization, Ox (Ph4N14HEPT) may also be too reactive to be observed by usual methods. While Ox appears to be computationally stable with a smaller HOMO-LUMO gap as expected for more highly conjugated chromophores, it has an irregular molecular orbital structure, which might also suggest reasons for its instability under normal conditions. This situation can be addressed by observing H2N14HEPT under different conditions such as adsorbed at a metal interface [61]. Lower analogues of oxidized pyrazinacenes are known to be stable when adsorbed at Cu(111) surfaces although for H2N14HEPT these measurements are made difficult by its large molecular weight and tendency to decompose when heated at temperatures used for sublimation.
For deprotonation, our computational regime effectively reproduces spectral data providing a basis for us to investigate electronic properties of those species. MA and DA have absorbances and emission in the near infrared (NIR) making this chromophore highly suitable for bio-imaging applications possibly as a probe of local pH during intracellular processes. Deprotonation obliterates protic tautomeric processes but introduces other possibilities such as intramolecular electron transfer as indicated by the tendency for HOMO and LUMO orbitals to be localized on different regions of the molecule (see Figure 5b,d). For tautomerisation of Ph4H2N14HEPT, our calculations indicate that the tautomer T0 having a central dihydropyrazine ring is the most stable, while dihydropyrazine ring shifts away from center lower the HOMO-LUMO gap and red-shift the main UV-vis absorption bands. Amongst the protonated tautomers, the most stable are those where the protonated ring is one or two rings away from the remote rings (e.g., T0-H2; see Figure 11). An increase in the proton-hydrogen distance is correlated with a HOMO-LUMO gap decrease, as well as an intensity decrease and a red-shifting effect in simulated UV-vis spectra. Removing the protons on the reduced rings also leads to a red shift but the effect is smaller. Spectral predictions for protonated states obtained by computational methods used here were not accurate although shifts were predicted. We are currently assessing the reasons for this and hope to be able to identify the factors involved. Regardless, protonation of Ph4H2N14HEPT provides an interesting route to affect tautomer identity or to vary the spectroscopic output of the molecules.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Bond length averages for optimised structures; Figure S1: Calculated electronic absorption spectra for tautomer T1 and the monoprotonated tautomers T1-H1, T1-H2, and T1-H3; Figure S2: Calculated electronic absorption spectra for tautomer T2 and the monoprotonated tautomers T2-H0, T2-H1, T2-H2, and T2-H3. Coordinates of the calculated structures (.mol file) openable by Mercury.
Author Contributions
Conceptualization, A.C., S.K., G.J.R., J.-F.H. and J.P.H.; methodology, A.C., S.K. and J.-F.H.; software, A.C.; formal analysis, A.C., S.K., G.J.R., J.-F.H. and J.P.H.; investigation, A.C. and S.K.; data curation, A.C. and S.K.; writing—original draft preparation, A.C., S.K., G.J.R., J.-F.H. and J.P.H.; writing—review and editing, A.C., S.K., G.J.R., J.-F.H. and J.P.H.; visualization, A.C. and J.P.H.; supervision, J.-F.H. and J.P.H.; project administration, G.J.R., J.-F.H. and J.P.H.; funding acquisition, J.-F.H, J.P.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partly funded by GENCI (Grand Equipment National de Calcul Intensif) for HPC resources, grant number A0050807367.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
This work was partly supported by World Premier International Research Center Initiative (WPI Initiative), MEXT, Japan. A.C., S.K., and J.-F.H. are grateful to GENCI (Grand Equipment National de Calcul Intensif) for HPC resources (Grant A0050807367).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Campbell, R.B.; Robertson, J.M.; Trotter, J. The Crystal and Molecular Structure of Pentacene. Acta Cryst. 1961, 14, 705–711. [Google Scholar] [CrossRef]
- Robertson, J.M.; Sinclaire, V.C.; Trotter, J. The Crystal and Molecular Structure of Tetracene. Acta Cryst. 1961, 14, 697–704. [Google Scholar] [CrossRef]
- Watanabe, M.; Chang, Y.J.; Liu, S.-W.; Chao, T.-H.; Goto, K.; Islam, M.M.; Yuan, C.-H.; Tao, Y.-T.; Shinmyozu, T.; Chow, T.J. The Synthesis, Crystal Structure and Charge-Transport Properties of Hexacene. Nat. Chem. 2012, 4, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Goetz, K.P.; Li, Z.; Ward, J.W.; Bougher, C.; Rivnay, J.; Smith, J.; Conrad, B.R.; Parkin, S.R.; Anthopoulos, T.D.; Salleo, A.; Anthony, J.E.; Jurchescu, O.D. Effect of Acene Length on Electronic Properties in 5-, 6-, and 7-Ringed Heteroacenes. Adv. Mater. 2011, 23, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, H.F.; Toenshoff, C. The Longest Acenes. Chem. Rec. 2015, 15, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Pascal, R.A. Twisted Acenes. Chem. Rev. 2006, 106, 4809–4819. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayana, A.N.; Ong, A.; Chi, C. Modification of Acenes for n-Channel OFET Materials. J. Mater. Chem. C 2018, 6, 3551–3563. [Google Scholar] [CrossRef]
- Müller, M.; Ahrens, L.; Brosius, V.; Freudenberg, J.; Bunz, U.H.F. Unusual Stabilization of Larger Acenes and Heteroacenes. J. Mater. Chem. C 2019, 7, 14011–14034. [Google Scholar] [CrossRef]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, Oligoacenes, and Their Buckminsterfullerene Derivatives: The Bricks and Mortar of Organic Electronics, Chem. Rev. 2004, 104, 4891–4945. [Google Scholar] [CrossRef]
- Anthony, J.E. The Larger Acenes: Versatile Organic Semiconductors. Angew. Chem. Int. Ed. 2008, 47, 452–483. [Google Scholar] [CrossRef]
- Zade, S.S.; Bendikov, M. Reactivity of Acenes: Mechanisms and Dependence on Acene Length. J. Phys. Org. Chem. 2012, 25, 452–461. [Google Scholar] [CrossRef]
- Clar, E. The Aromatic Sextet, New York, NY: Wiley, 1972.
- Tönshoff, C.; Bettinger, H.F. Pushing the Limits of Acene Chemistry: The Recent Surge of Large Acenes. Chem. Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef]
- Jancarik, A.; Levet, G.; Gourdon, A. A Practical General Method for the Preparation of Long Acenes. Chem. Eur. J. 2019, 25, 2366–2374. [Google Scholar] [CrossRef]
- Urgel, J.I.; Mishra, S.; Hayashi, H.; Wilhelm, J.; Pignedoli, C.A.; Di Giovannantonio, M.; Widmer, R.; Yamashita, M.; Hieda, N.; Ruffieux, P.; Yamada, H.; Fasel, R. On-Surface Light-Induced Generation of Higher Acenes and Elucidation of their Open-Shell Character. Nat. Commun. 2019, 10, 861. [Google Scholar] [CrossRef]
- Miklík, D.; Mousavi, S.F.; Burešova, Z.; Middleton, A.; Matsushita, Y.; Labuta, J.; Ahsan, A.; Buimaga-Iarinca, L.; Karr, P.A.; Švec, P.; Bureš, F.; Richards, G.J.; Mori, T.; Ariga, K.; Wakayama, Y.; Morari, C.; D’Souza, F.; Jung. T.A.; Hill, J.P. Pyrazinacenes Exhibit On-Surface Oxidation-State-Dependent Conformational and Self-Assembly Behaviours. Commun. Chem. 2021, 4, 29. [Google Scholar] [CrossRef]
- Švec, P.; Webre, W.A.; Richards, G.J.; Labuta, J.; Wakayama, Y.; Miklík, D.; Karr, P.A.; Mori, T.; Ariga, K.; D’Souza, F.; Hill, J.P. Phenanthroline-Fused Pyrazinacenes: One-Pot Synthesis, Tautomerization and a Ru(II)(2,2’-Bipy)2 Derivative. Eur. J. Inorg. Chem. 2018, 2018, 2541–2548. [Google Scholar] [CrossRef]
- Richards, G.J.; Cador, A.; Yamada, S.; Middleton, A.; Webre, W.A.; Labuta, J.; Karr, P.A.; Ariga, K.; D’Souza, F.; Kahlal, S.; Halet, J.-F.; Hill, J.P. Amphiprotism-Coupled Near-Infrared Emission in Extended Pyrazinacenes Containing Seven Linearly Fused Pyrazine Units. J. Am. Chem. Soc. 2019, 141, 19570–19574. [Google Scholar] [CrossRef]
- Richards, G.J.; Hill, J.P.; Subbaiyan, N.K.; D’Souza, F.; Karr, P.A.; Elsegood, M.R.J.; Teat, S.J.; Mori, T.; Ariga, K. Pyrazinacenes: Aza Analogues of Acenes, J. Org. Chem. 2009, 74, 8914–8923. [Google Scholar] [CrossRef]
- Richards, G.J.; Hill, J.P. The Pyrazinacenes. Acc. Chem. Res. 2021, 54, 3228–3240. [Google Scholar] [CrossRef]
- Bunz, U.H.F.; Engelhart, J.U. The Palladium Way to N-Heteroacenes. Chem. Eur. J. 2016, 22, 4680–4689. [Google Scholar] [CrossRef]
- Kang, F.; Yang, J.; Zhang, Q. Recent Progress in Pyrazinacenes Containing Nonbenzenoid Rings: Synthesis, Properties and Applications. J. Mater. Chem. C 2022, 10, 2475–2483. [Google Scholar] [CrossRef]
- Richards, G. J.; Hill, J. P.; Mori, T.; Ariga, K. Putting the ‘N’ in Acene: Pyrazinacenes and their Structural Relatives. Org. Biomol. Chem. 2011, 9, 5005–5017. [Google Scholar] [CrossRef] [PubMed]
- Bunz, U.H.F.; Freudenberg, J. N-Heteroacenes and N-Heteroarenes as N-Nanocarbon Segments. Acc. Chem. Res. 2019, 52, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Anthony, J.E. Functionalized Acenes and Heteroacenes for Organic Electronics. Chem. Rev. 2006, 106, 5028–5048. [Google Scholar] [CrossRef] [PubMed]
- Gang, L.; Miao, J.; Cao, J.; Liu, B.; Zhang, Q. Preparation and Photoelectrochemical Behavior of 1,4,6,8,11,13-Hexazapentacene (HAP). Chem. Commun. 2014, 50, 7656–7658. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.-Y.; Wang, Z.; Zhang, Q. Azaacenes as Active Elements for Sensing and Bio Applications. J. Mater. Chem. B 2016, 4, 7060–7074. [Google Scholar] [CrossRef]
- Fleischauer, J.; Zahn, S.; Beckert, R.; Grummt, U.-W.; Birckner, E.; Görls, H. A Way to Stable, Highly Emissive Fluorubine Dyes: Tuning the Electronic Properties of Azaderivatives of Pentacene by Introducing Substituted Pyrazines. Chem. Eur. J. 2012, 18, 4549–4557. [Google Scholar] [CrossRef]
- Tykwinski, R.R. Synthesis of Unsymmetrical Derivatives of Pentacene for Materials Applications. Acc. Chem. Res. 2019, 52, 2056–2069. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Houk, K.N. Nitrogen-Rich Oligoacenes: Candidates for n-Channel Organic Semiconductors. J. Am. Chem. Soc. 2007, 129, 1805–1815. [Google Scholar] [CrossRef]
- Bendikov, M.; Duong, H.M.; Starkey, K.; Houk, K.N.; Carter, E.A.; Wudl, F. Oligoacenes: Theoretical Prediction of Open-Shell Singlet Diradical Ground States. J. Am. Chem. Soc. 2004, 126, 7416–7417. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Y.; Cai, X. First-Principles Investigation of Electronic and Charge Transport Properties of Pyrazinacenes and Their Structural Relatives. Chem. Phys. Lett. 2021, 772, 138595. [Google Scholar] [CrossRef]
- Scipioni, R.; Hill, J.P.; Richards, G.J.; Boero, M.; Mori, T.; Ariga, K.; Ohno, T. Tautomers of Extended Reduced Pyrazinacenes: A Density-Functional-Theory Based Study. Phys. Chem. Chem. Phys. 2011, 13, 2145–2150. [Google Scholar] [CrossRef]
- Scipioni, R.; Boero, M.; Richards, G.J.; Hill, J.P.; Ohno, T.; Mori, T.; Ariga, K. Tautomerism in Reduced Pyrazinacenes, J. Chem. Theory Comput. 2010, 6, 517–525. [Google Scholar] [CrossRef]
- Miao, S.; Brombosz, S.M.; Schleyer, P.v.R.; Wu, J.I.; Barlow, S.; Marder, S.R.; Hardcastle, K.I.; Bunz, U.H.F. Are N,N-Dihydrodiazatetracene Derivatives Antiaromatic? J. Am. Chem. Soc. 2008, 130, 7339–7344. [Google Scholar] [CrossRef]
- Hele, T.J.H.; Fuemmeler, E.G.; Sanders, S.H.; Kumarasamy, E.; Sfeir, M.Y.; Campos, L.M.; Ananth, N. Anticipating Acene-Based Chromophore Spectra with Molecular Orbital Arguments. J. Phys. Chem. A 2019, 123, 2527–2536. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef]
- Portella, G.; Poater, J.; Bofill, J.M.; Alemany, P.; Solà, M. Local Aromaticity of [n]Acenes, [n]Phenacenes, and [n]Helicenes (n = 1–9). J. Org. Chem. 2005, 70, 2509–2521. [Google Scholar] [CrossRef]
- Wu, J.I.; Wannere, C.S.; Mo, Y.; Schleyer, P.v.R.; Bunz, U.H.F. 4n π Electrons but Stable: N,N-Dihydrodiazapentacenes. J. Org. Chem. 2009, 74, 4343–4349. [Google Scholar] [CrossRef]
- Benkyi, I.; Tapavicza, E.; Fliegl, H.; Sundholm, D. Calculation of Vibrationally Resolved Absorption Spectra of Acenes and Pyrene. Phys. Chem. Chem. Phys. 2019, 21, 21094–21103. [Google Scholar] [CrossRef]
- Blight, B.A.; Hunter, C.A.; Leigh, D.A.; McNab, H.; Thomson, P.L.T. An AAAA-DDDD Quadruple Hydrogen-Bond Array. Nat. Chem. 2011, 3, 244–248. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas, Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phy. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Gaussian 16, Revision A.03, Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.V.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, J.L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.J.; Heyd, J.J.; Brothers, E.N.; Kudin, K.N.; Staroverov, V.N.; Keith, T.A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.P.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Millam, J.M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B.; Fox, D.J.; Gaussian, Inc., Wallingford CT, 2016.
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis, Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality of H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- EMSL Basis Set Library, https://bse.pnl.gov/bse/portal.
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comp. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K. L.; Didier, B. T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T. L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- NBO 6.0. Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Bohmann, J. A.; Morales, C. M.; Landis, C. R.; Weinhold, F.; Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013, http://nbo6.chem.wisc.edu.
- Runge, E.; Gross, E. K. U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Adamo, C.; Jaquemin, D. The Calculations of Excited-State Properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Swizard, revision 4.5. Gorelsky, S. I., http://www.sg-chem.net/.
- AOMix: Program for Molecular Orbital Analysis, version 6.5. Gorelsky, S.I.; 2019, http://www.sg-chem.net.
- Gorelsky, S. I.; Lever, A. B. P. Electronic Structure and Spectra of Ruthenium Diimine Complexes by Density Functional Theory and INDO/S. Comparison of the Two Methods, J. Organomet. Chem. 2001, 635, 187–196. [Google Scholar] [CrossRef]
- Eisenhut, F.; Kühne, T.; Garcia, F.; Fernández, S.; Guitián, E.; Pérez, D.; Trinquier, G.; Cuniberti, G.; Joachim, C.; Peña, D.; Moresco, F. Dodecacene Generated on Surface: Reopening of the Energy Gap. ACS Nano 2020, 14, 1011–1017. [Google Scholar] [CrossRef]
Figure 2.
Chemical structures of tetradecaazaheptacenes studied. (a) X-ray crystal structure of tBu8Ph4H2N14HEPT showing the reduced dihydropyrazine ring located at the central ring [18]. (b) The oxidized product, Ox. (c) Structures of the monoanion, MA, and dianion, DA. (d) Tautomers of Ph4H2N14HEPT (T0, T1, T2, T3) available by concerted double proton shifts (other possible tautomers are neglected here). (e) Structures of selected protonated tautomers of Ph4H2N14HEPT.
Figure 2.
Chemical structures of tetradecaazaheptacenes studied. (a) X-ray crystal structure of tBu8Ph4H2N14HEPT showing the reduced dihydropyrazine ring located at the central ring [18]. (b) The oxidized product, Ox. (c) Structures of the monoanion, MA, and dianion, DA. (d) Tautomers of Ph4H2N14HEPT (T0, T1, T2, T3) available by concerted double proton shifts (other possible tautomers are neglected here). (e) Structures of selected protonated tautomers of Ph4H2N14HEPT.
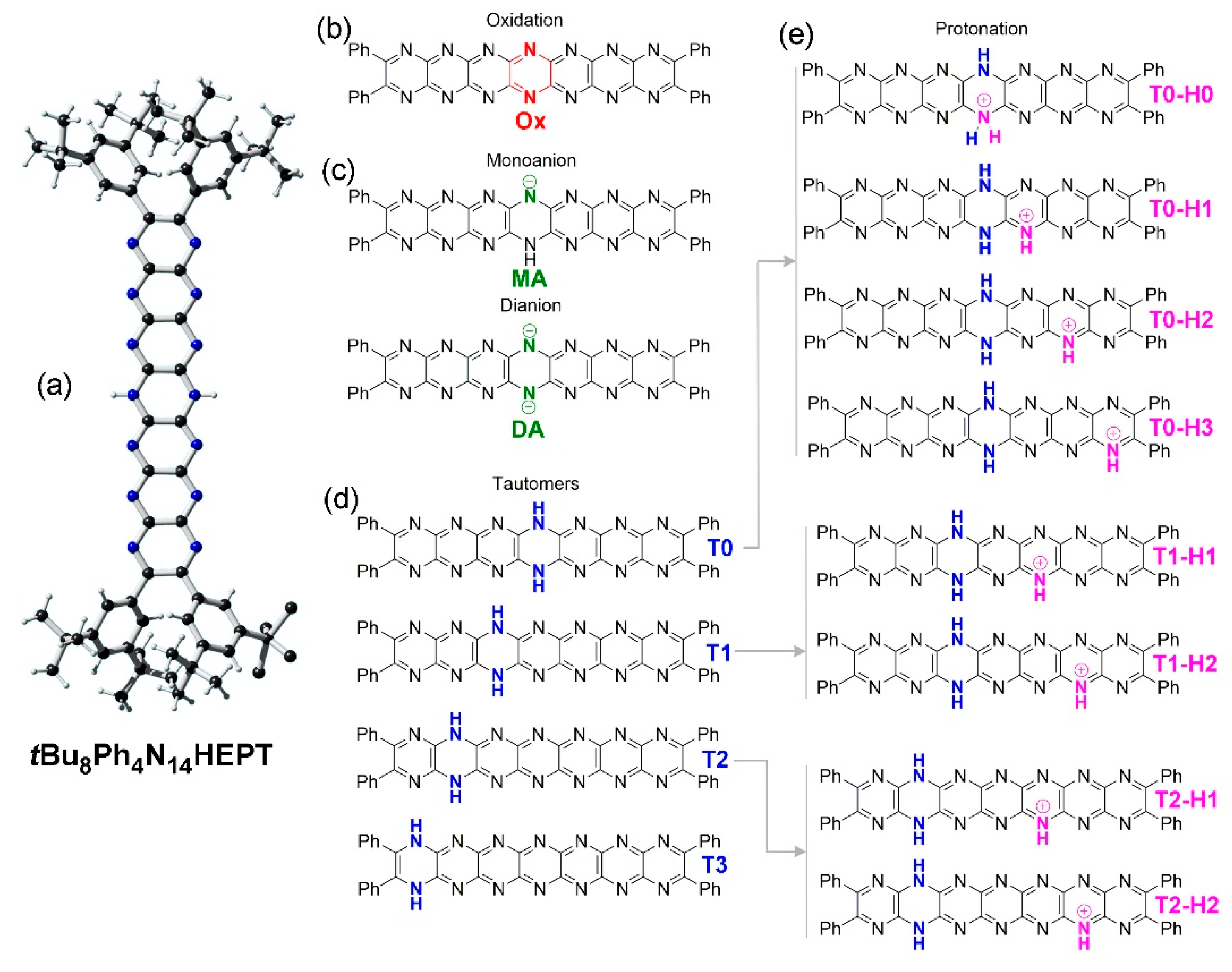
Figure 3.
Comparison of experimental (X-ray) and computed structures. (a) X-ray crystal structure of tBu8Ph4H2N14HEPT [18] (tBu groups not shown for clarity) with pertinent C-N bond distances (Å). End-on view (at left) shows the highly planar N14-acene core. (b) Optimized structure of T0 (Ph4H2N14HEPT) with C-N bond distances (Å). End-on view (at right) shows a (slightly) torsionally distorted acene core.
Figure 3.
Comparison of experimental (X-ray) and computed structures. (a) X-ray crystal structure of tBu8Ph4H2N14HEPT [18] (tBu groups not shown for clarity) with pertinent C-N bond distances (Å). End-on view (at left) shows the highly planar N14-acene core. (b) Optimized structure of T0 (Ph4H2N14HEPT) with C-N bond distances (Å). End-on view (at right) shows a (slightly) torsionally distorted acene core.
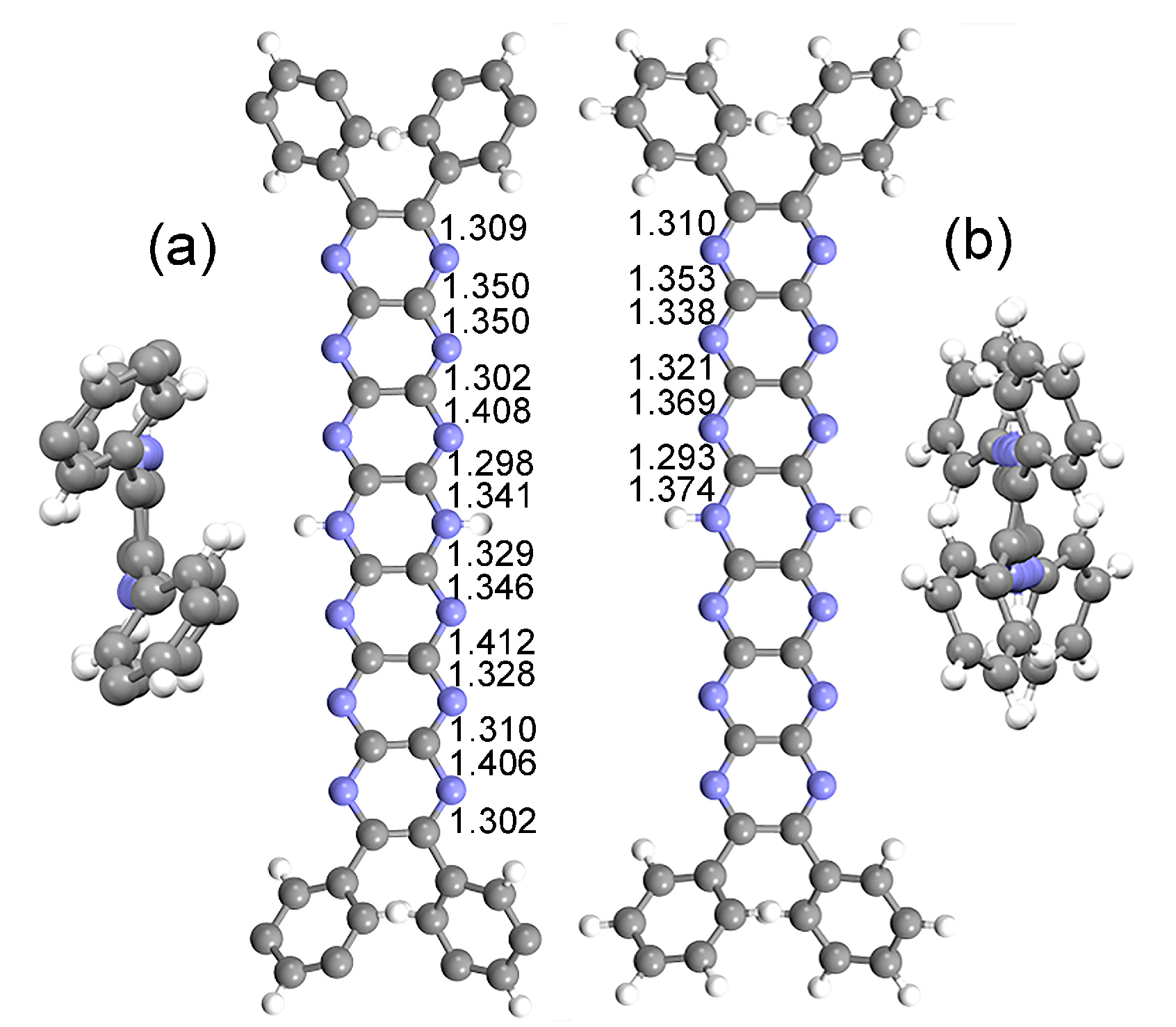
Figure 4.
Variations in electronic structures associated with oxidation of T0 to Ox. (a) Structure of T0 including NICS values. (b) MO diagram of T0. (c) Structure of Ox including NICS values. (d) Structures of HOMOs and LUMOs for Ox. Contour isodensity values: ± 0.02 (e/bohr3)1/2.
Figure 4.
Variations in electronic structures associated with oxidation of T0 to Ox. (a) Structure of T0 including NICS values. (b) MO diagram of T0. (c) Structure of Ox including NICS values. (d) Structures of HOMOs and LUMOs for Ox. Contour isodensity values: ± 0.02 (e/bohr3)1/2.
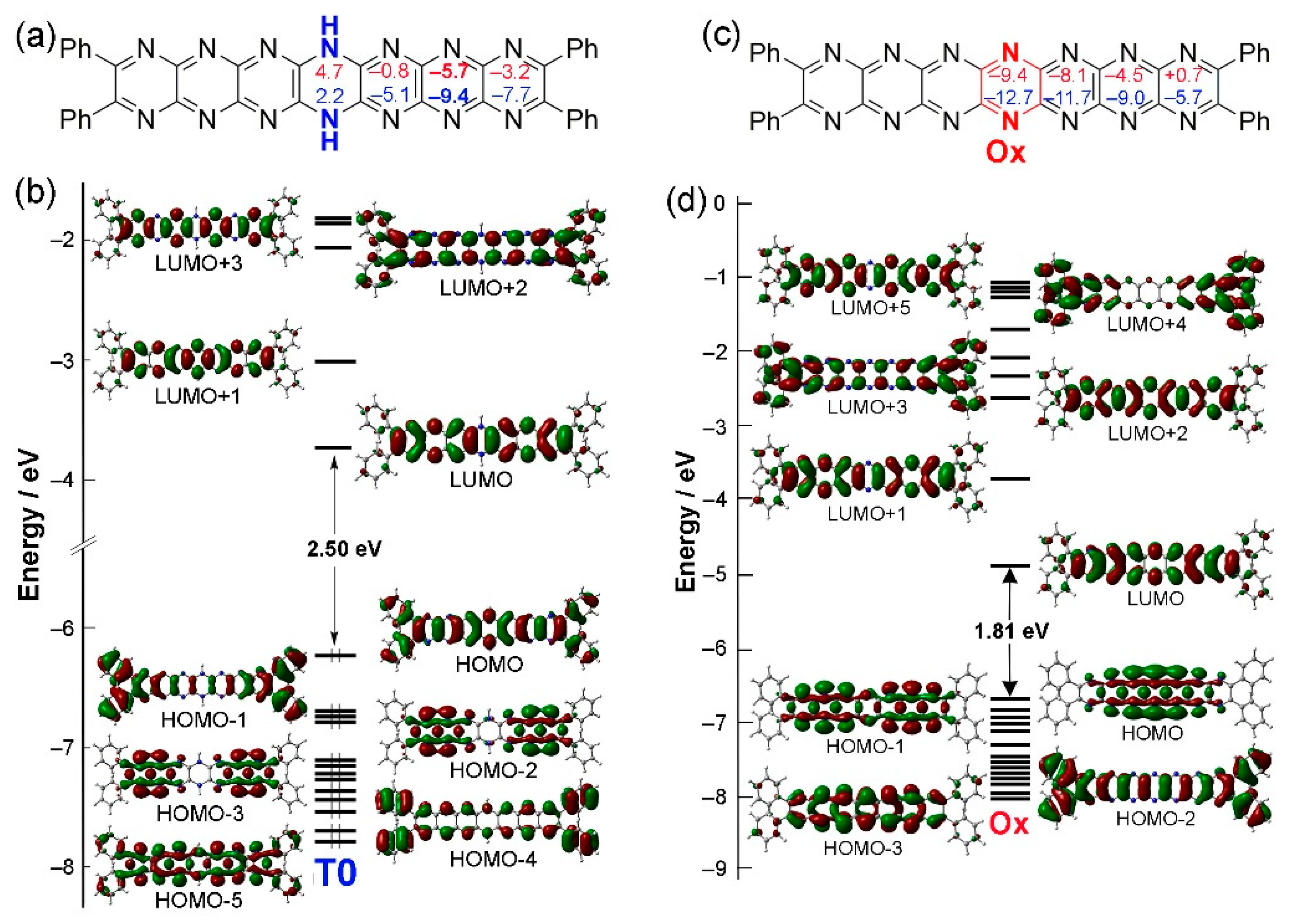
Figure 5.
Electronic structures of deprotonated forms of T0, monanion (MA) and dianion (DA). (a) Structure of MA including NICS values. (b) MO diagram of MA. (b) Structure of DA including NICS values. (d) MO diagram of DA. Contour isodensity values: ± 0.02 (e/bohr3)1/2.
Figure 5.
Electronic structures of deprotonated forms of T0, monanion (MA) and dianion (DA). (a) Structure of MA including NICS values. (b) MO diagram of MA. (b) Structure of DA including NICS values. (d) MO diagram of DA. Contour isodensity values: ± 0.02 (e/bohr3)1/2.
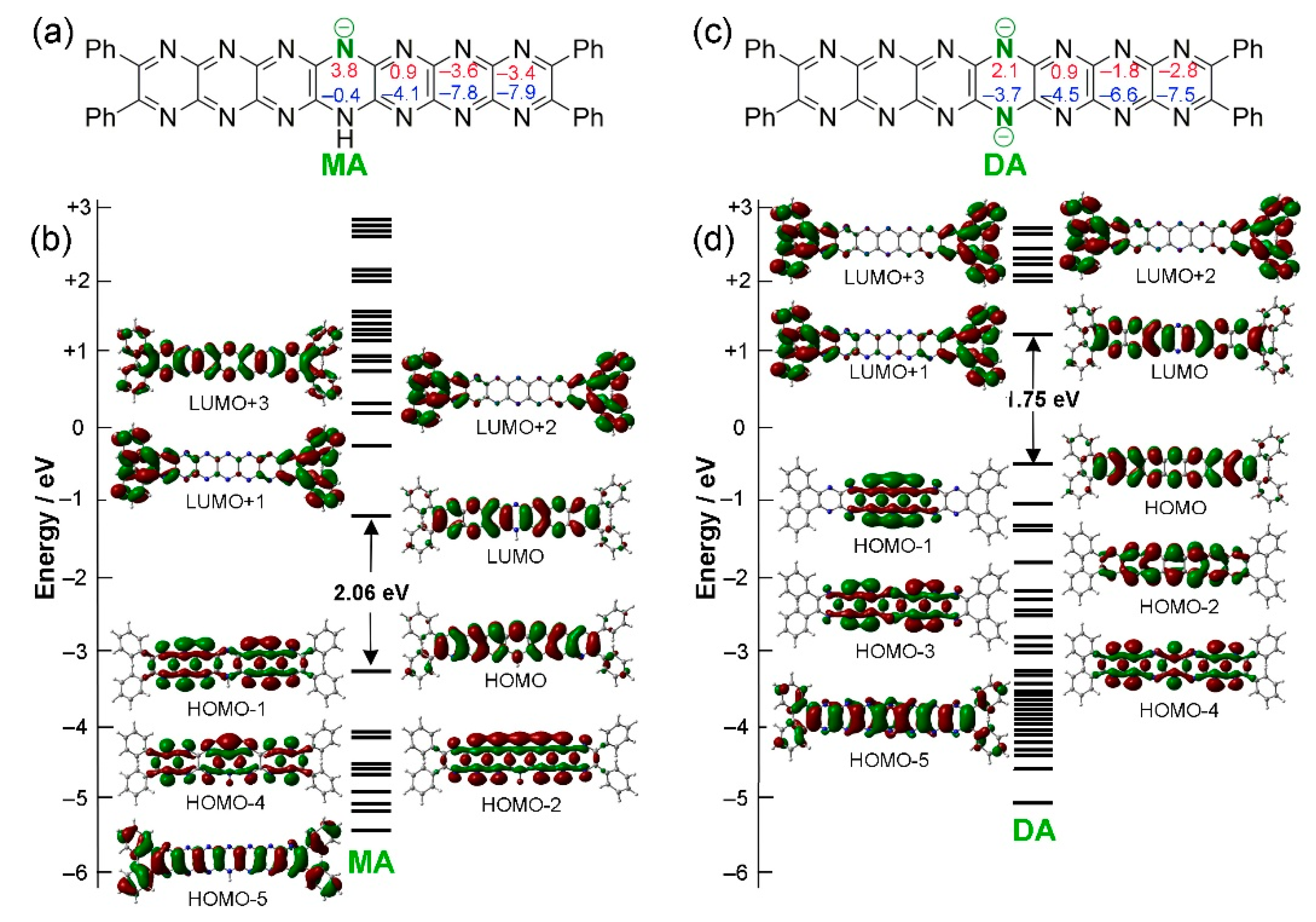
Figure 6.
(a) Computed electronic absorption (UV-vis) spectra of T0, MA, DA and Ox. (b) Experimentally observed UV-vis spectra of tBu8Ph4H2N14HEPT, [Ph4HN14HEPT]–, and [Ph4N14HEPT]2– [18].
Figure 6.
(a) Computed electronic absorption (UV-vis) spectra of T0, MA, DA and Ox. (b) Experimentally observed UV-vis spectra of tBu8Ph4H2N14HEPT, [Ph4HN14HEPT]–, and [Ph4N14HEPT]2– [18].
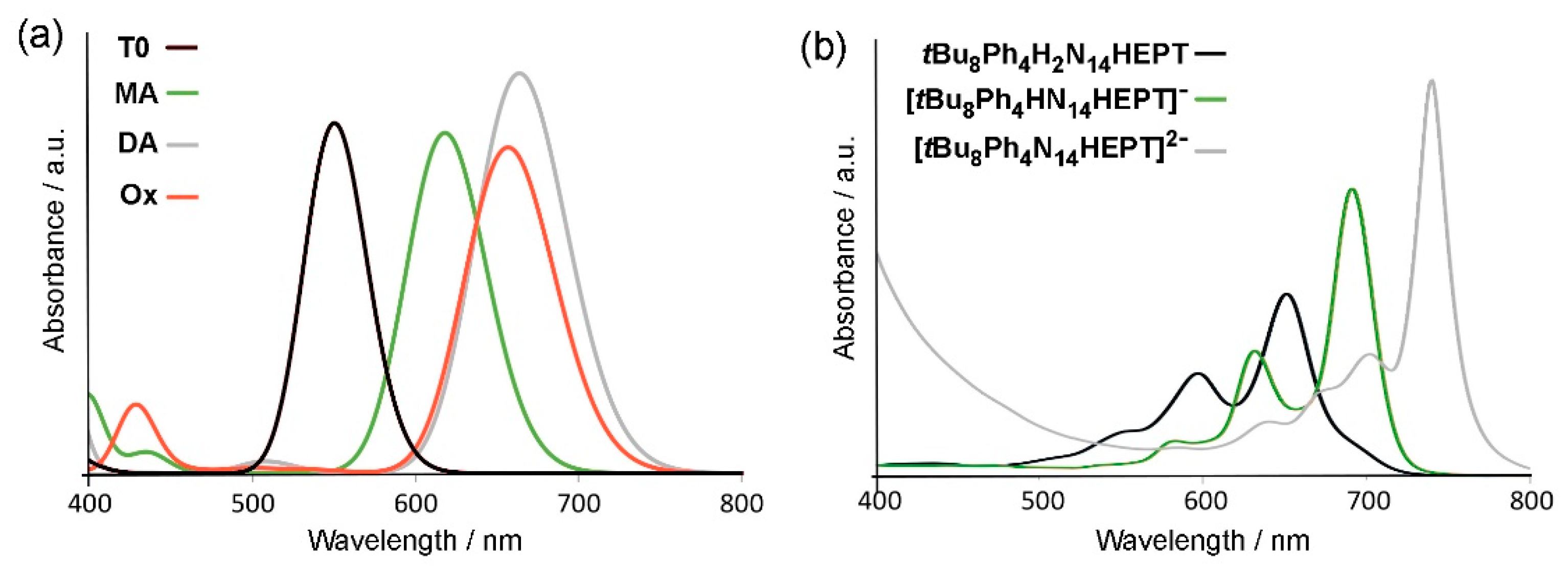
Figure 7.
Experimentally observable examples of tautomerism in pyrazinacenes. (a) Proton shift in the monoanion of Ph2H2N8TET-CN2 observed directly using low temperature 1H nuclear magnetic resonance [34]. (b) N-alkylation of a tautomer mixture leads to indirect observation of tautomers in Phen-H2N8PENT-CN2 [17]. These examples also reveal the inaccessibility of the terminal pyrazine groups in pyrazinacenes as also found here computationally.
Figure 7.
Experimentally observable examples of tautomerism in pyrazinacenes. (a) Proton shift in the monoanion of Ph2H2N8TET-CN2 observed directly using low temperature 1H nuclear magnetic resonance [34]. (b) N-alkylation of a tautomer mixture leads to indirect observation of tautomers in Phen-H2N8PENT-CN2 [17]. These examples also reveal the inaccessibility of the terminal pyrazine groups in pyrazinacenes as also found here computationally.
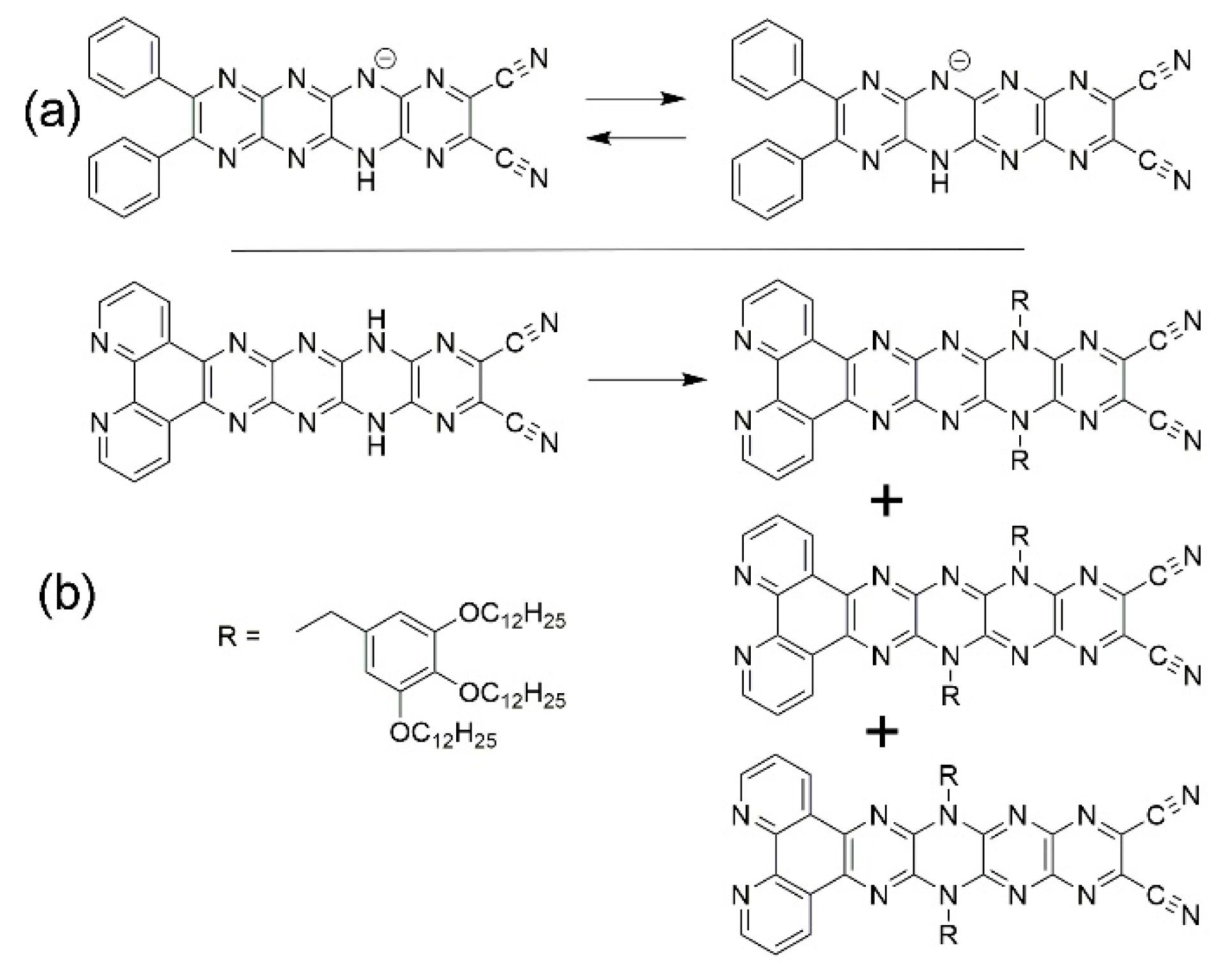
Figure 8.
Calculated electronic absorption spectra for the tautomers T0–T3. Note the stepwise red shift.
Figure 8.
Calculated electronic absorption spectra for the tautomers T0–T3. Note the stepwise red shift.
Figure 9.
The three series of monoprotonated tautomers. Left: Tautomer-0 (T0) with numbering scheme for protonation site. Right (top): Tautomer-1 (T1), (bottom): Tautomer-2 (T2).
Figure 9.
The three series of monoprotonated tautomers. Left: Tautomer-0 (T0) with numbering scheme for protonation site. Right (top): Tautomer-1 (T1), (bottom): Tautomer-2 (T2).
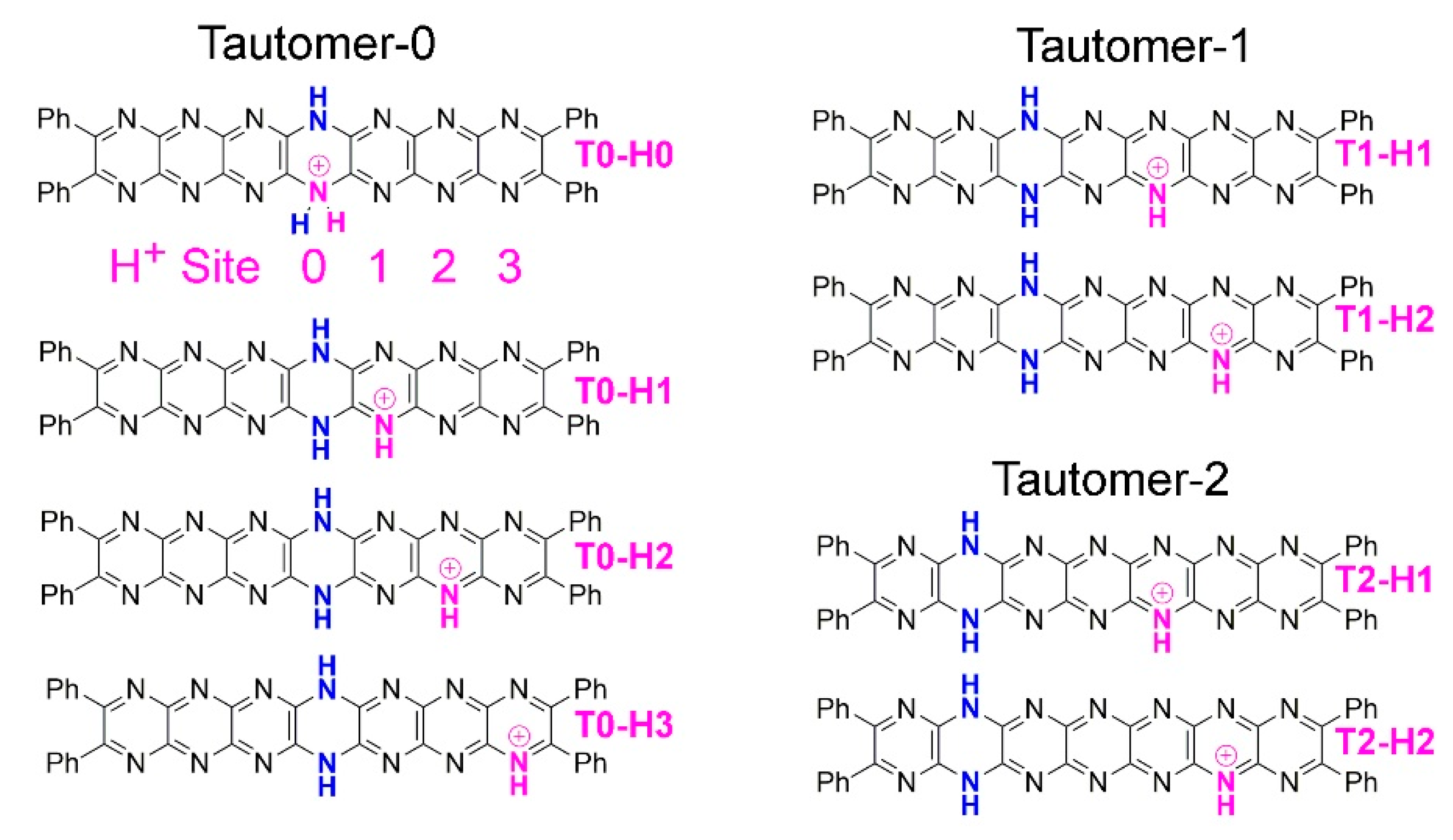
Figure 10.
Charges on the rings of the neutral tautomers (a) T0, (b) T1, (c) T2, (d) T3. Possible sites of protonation indicated in pink in (a).
Figure 10.
Charges on the rings of the neutral tautomers (a) T0, (b) T1, (c) T2, (d) T3. Possible sites of protonation indicated in pink in (a).
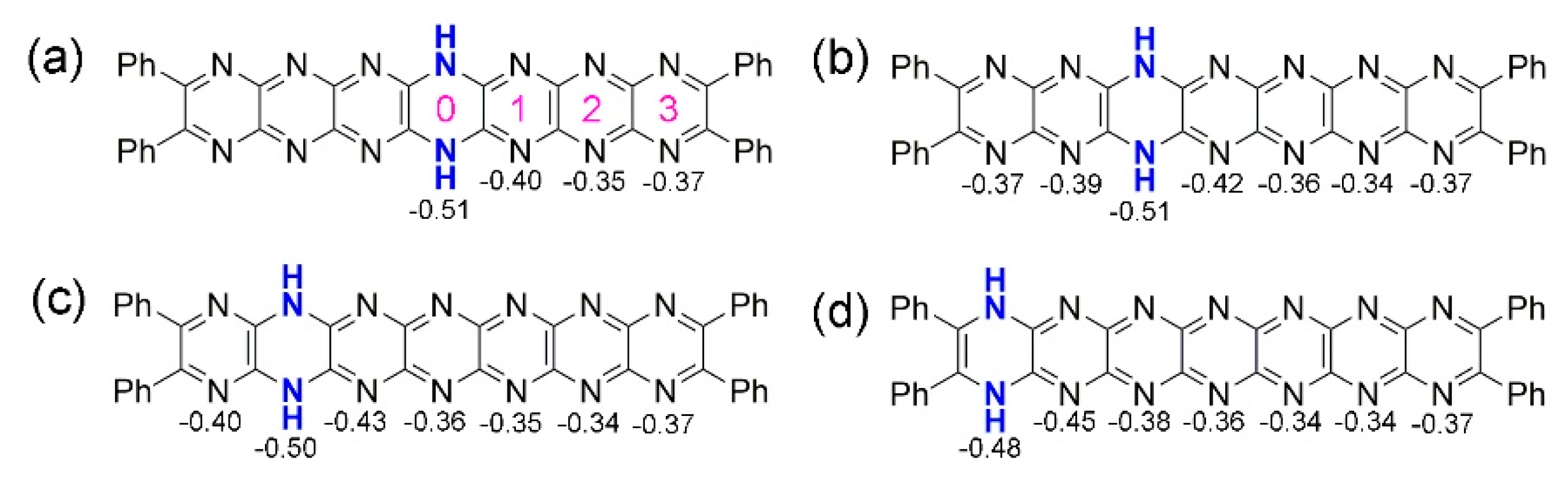
Figure 11.
Protonated structure T0-H2. (a) Structure of T0-H2 including NICS values. (b) MO diagram of T0-H2. Contour isodensity values: ± 0.02 (e/bohr3)1/2.
Figure 11.
Protonated structure T0-H2. (a) Structure of T0-H2 including NICS values. (b) MO diagram of T0-H2. Contour isodensity values: ± 0.02 (e/bohr3)1/2.
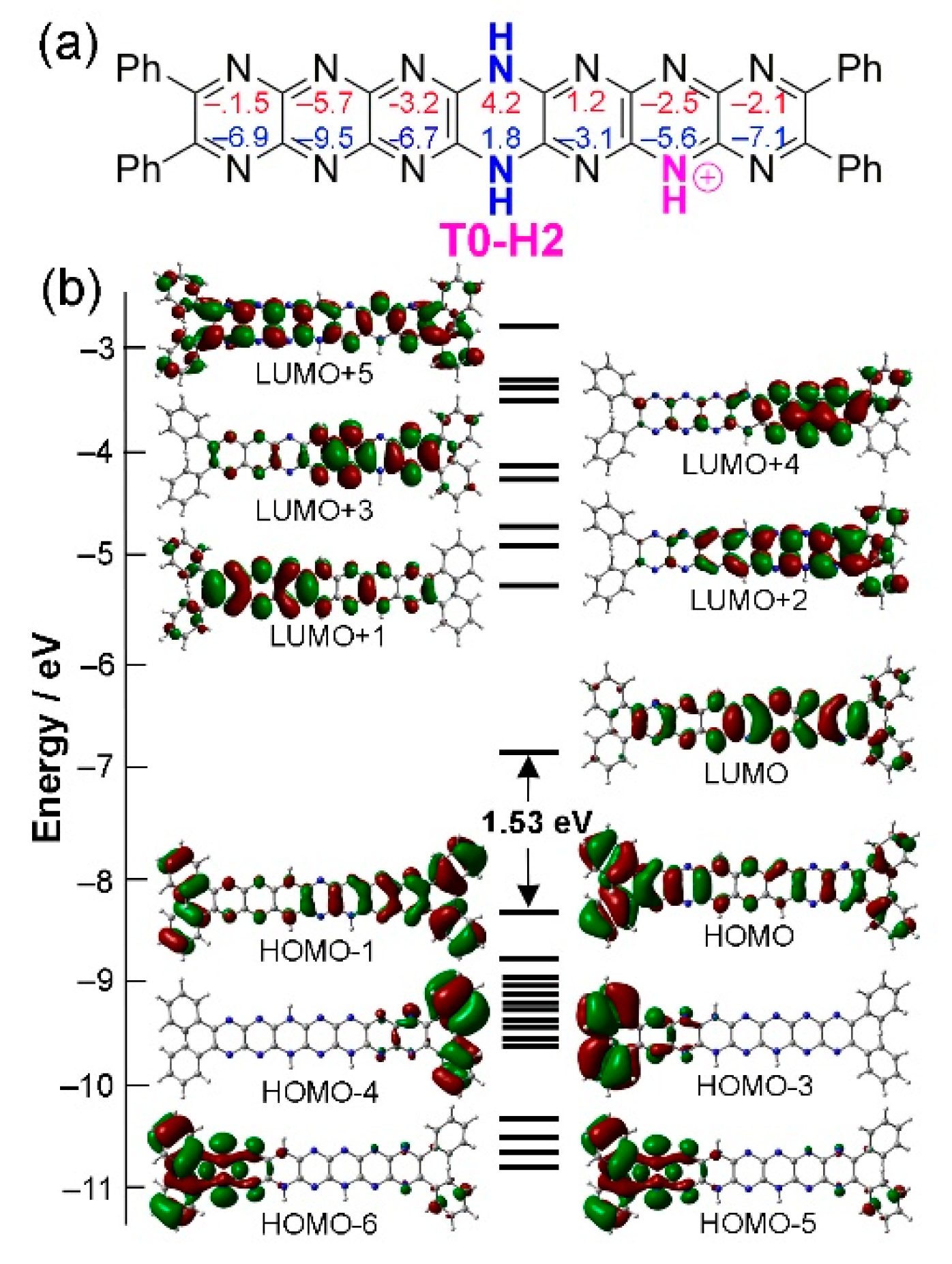
Figure 12.
(a) Electronic absorption spectra of T0 and some of its monoprotonated tautomers. (b) Electronic absorption spectra obtained during titration of tBu8Ph4H2N14HEPT with trifluoroacetic acid (TFA). Panel (b) adapted with permission from G. J. Richards et al., J. Am. Chem. Soc. 2019, 141, 19570. Copyright 2019 American Chemical Society.
Figure 12.
(a) Electronic absorption spectra of T0 and some of its monoprotonated tautomers. (b) Electronic absorption spectra obtained during titration of tBu8Ph4H2N14HEPT with trifluoroacetic acid (TFA). Panel (b) adapted with permission from G. J. Richards et al., J. Am. Chem. Soc. 2019, 141, 19570. Copyright 2019 American Chemical Society.
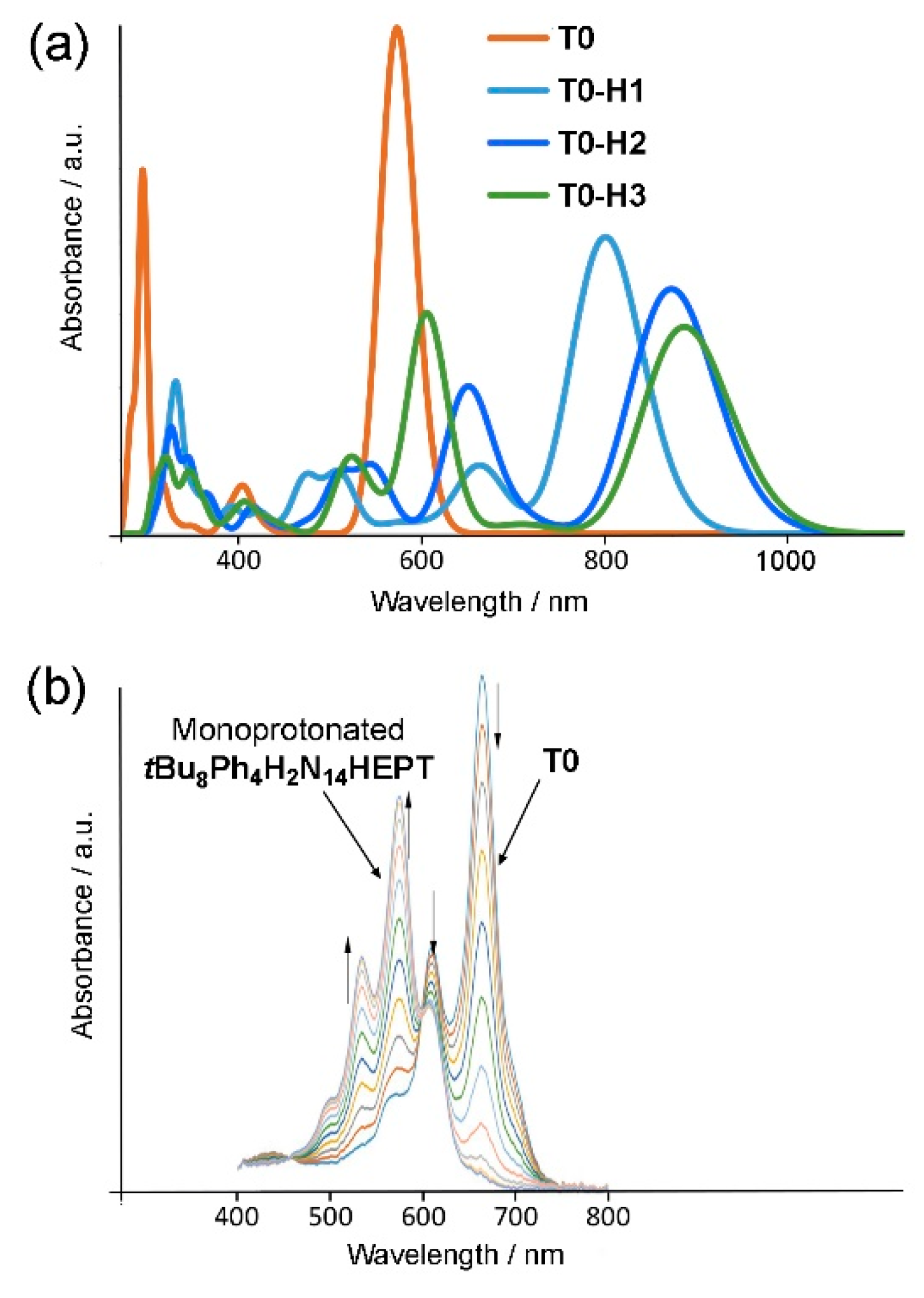
Figure 13.
Nucleus independent chemical shifts (NICS(0,1)) vs. ring number for T0 (Ph4H2N14HEPT), oxidized and anionic species. (a) NICS(0) and (b) NICS(1) values for T0, Ox, MA and DA. (c) NICS(0) and (d) NICS(1) values for T0-T3 tautomers.
Figure 13.
Nucleus independent chemical shifts (NICS(0,1)) vs. ring number for T0 (Ph4H2N14HEPT), oxidized and anionic species. (a) NICS(0) and (b) NICS(1) values for T0, Ox, MA and DA. (c) NICS(0) and (d) NICS(1) values for T0-T3 tautomers.
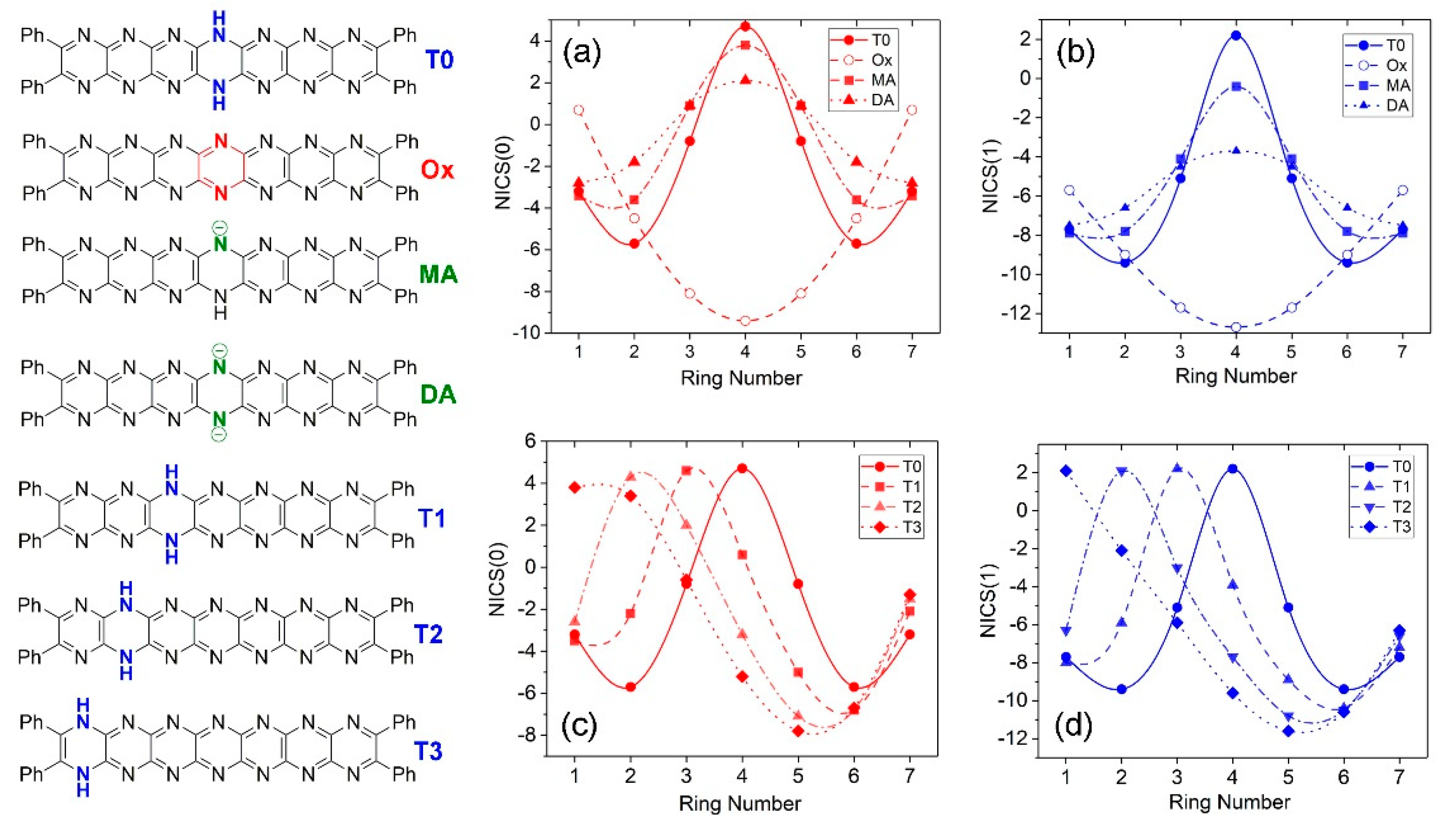
Figure 14.
Nucleus independent chemical shifts (NICS(0 (red), 1(blue))) vs. ring number for T0 protonated tautomer isomers. (a) NICS(0) and (b) NICS(1) values for T0-H0 – T0-H3. (c) NICS(0) and (d) NICS(1) values for T1-H1 and T1-H2 isomers. (c) NICS(0) and (d) NICS(1) values for T1-H1 and T1-H2 isomers. (e) NICS(0) and (f) NICS(1) values for T2-H1 and T2-H2 isomers.
Figure 14.
Nucleus independent chemical shifts (NICS(0 (red), 1(blue))) vs. ring number for T0 protonated tautomer isomers. (a) NICS(0) and (b) NICS(1) values for T0-H0 – T0-H3. (c) NICS(0) and (d) NICS(1) values for T1-H1 and T1-H2 isomers. (c) NICS(0) and (d) NICS(1) values for T1-H1 and T1-H2 isomers. (e) NICS(0) and (f) NICS(1) values for T2-H1 and T2-H2 isomers.
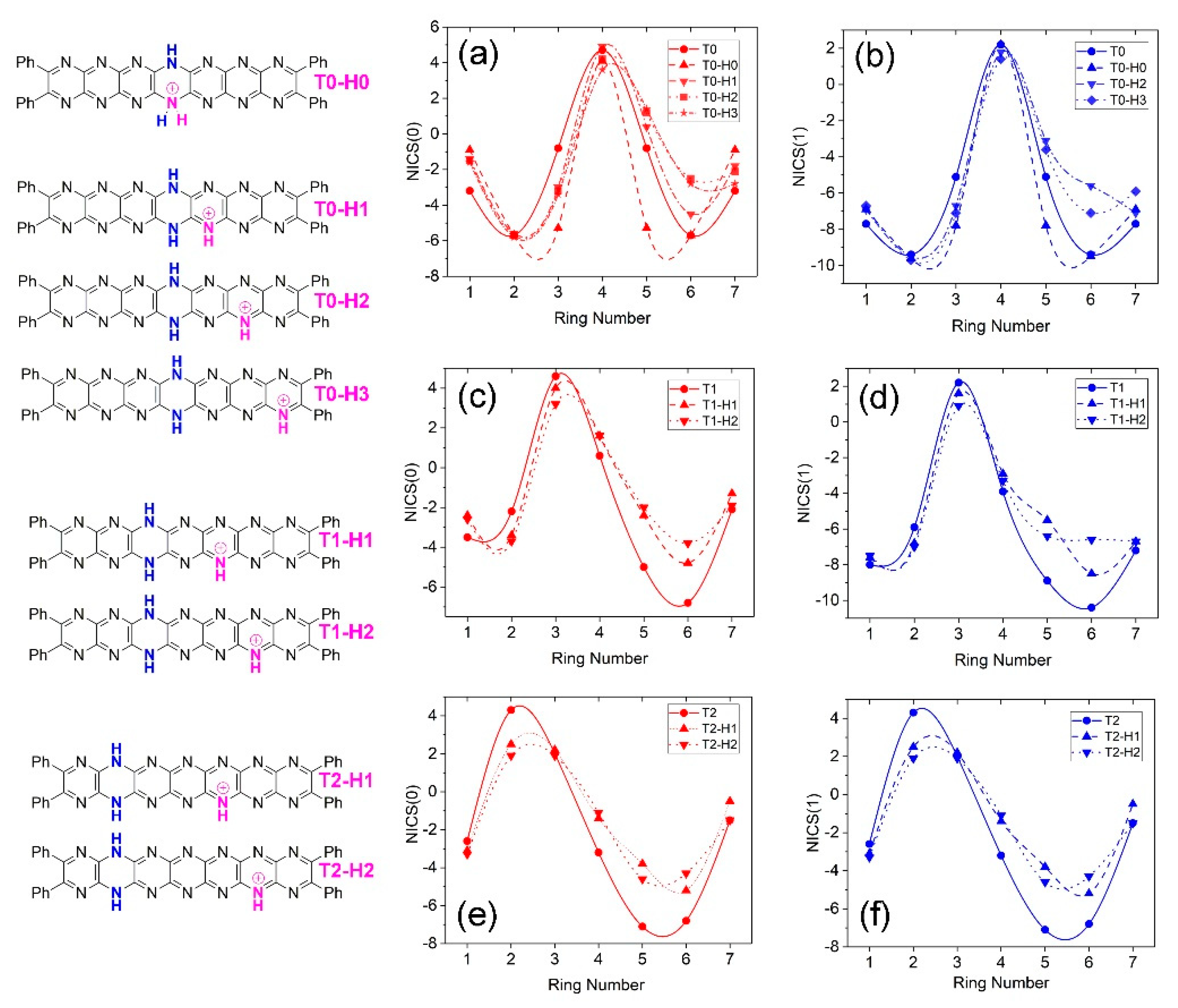

Table 1.
Bond length averages for XRD and optimized structures.
| Average bond length (Å) | X-ray structure (tBu8Ph4H2N14HEPT) | Optimized structure (T0 (Ph4H2N14HEPT)) |
|---|---|---|
| C-N | 1.348 | 1.337 |
| Acene C-C | 1.439 | 1.448 |
| Ph C-C | 1.393 | 1.392 |
| N-H | 0.880 | 1.013 |
Table 2.
TD-DFT results for Ox in gas phase. Results for compound T0 are given for comparison. f values are oscillator strengths.
Table 2.
TD-DFT results for Ox in gas phase. Results for compound T0 are given for comparison. f values are oscillator strengths.
| Compound | HOMO-LUMO gap (eV) |
λabs (nm), Eabs (eV) |
fabs | transition |
λem (nm), Eem (eV) |
fem |
|---|---|---|---|---|---|---|
| T0 | 2.50 | 550 (2.25) | 2.39 | HOMO→LUMO(98%) | 585 (2.12) | 2.42 |
| Ox (singlet) | 1.81 | 657 (1.89) | 2.23 | HOMO-2→LUMO(98%) |
Table 3.
TD-DFT results for the anionic compounds in gas phase. The results of compound T0 are given for comparison. f values are oscillator strengths.
Table 3.
TD-DFT results for the anionic compounds in gas phase. The results of compound T0 are given for comparison. f values are oscillator strengths.
| Compound | HOMO-LUMO gap (eV) |
λabs (nm), Eabs (eV) |
fabs | transition |
λem (nm), Eem (eV) |
fem |
|---|---|---|---|---|---|---|
| T0 | 2.50 | 550 (2.25) | 2.39 | HOMO→LUMO(98%) | 585 (2.12) | 2.42 |
| MA | 2.06 | 618 (2.01) | 2.33 | HOMO→LUMO(99%) | 654 (1.90) | 1.95 |
| DA | 1.75 | 664 (1.87) | 2.73 | HOMO→LUMO(100%) | 695 (1.78) | 1.83 |
Table 4.
HOMO-LUMO gaps and relative energies of the four tautomers. Energies are relative to compound T0.
Table 4.
HOMO-LUMO gaps and relative energies of the four tautomers. Energies are relative to compound T0.
| Compound | HOMO-LUMO gap (eV) | E (kcal.mol-1)a | E0 (kcal.mol-1)b | G (kcal.mol-1) |
|---|---|---|---|---|
| T0 | 2.499 | 0.000 | 0.000 | 0.000 |
| T1 | 2.388 | +2.592 | +2.503 | +2.454 |
| T2 | 2.204 | +11.566 | +10.236 | +13.409 |
| T3 | 2.029 | +31.629 | +31.058 | +30.512 |
aΔE is the total electronic energy. bΔE0 is the zero-point corrected energy.
Table 5.
TD-DFT results (gas phase) for the four tautomers studied. Oscillator strengths are given as f values.
Table 5.
TD-DFT results (gas phase) for the four tautomers studied. Oscillator strengths are given as f values.
| Compound |
λabs (nm), Eabs (eV) |
fabs | transition |
λem (nm), Eem (eV) |
fem |
|---|---|---|---|---|---|
| T0 | 550 (2.25) | 2.39 | HOMO→LUMO (98%) | 585 (2.12) | 2.42 |
| T1 | 569 (2.18) | 2.19 | HOMO→LUMO (98%) | 647 (1.92) | 1.84 |
| T2 | 606 (2.05) | 2.07 | HOMO→LUMO (98%) | 690 (1.80) | 1.75 |
| T3 | 628 (1.97) | 2.28 | HOMO→LUMO (98%) |
Table 6.
HOMO-LUMO gaps and relative energies of the eight studied protonated compounds. The compounds are arranged in series T0, T1, T2 shown in Figure 9 where site of protonation is denoted by -HX as also shown in Figure 9. Energies are relative to T1-H2.
| Compound | HOMO-LUMO gap (eV) | ΔE (kcal.mol-1)a | ΔE0 (kcal.mol-1)b | ΔG (kcal.mol-1) |
|---|---|---|---|---|
| T0-H0 | 2.355 | +51.212 | +50.571 | +48.799 |
| T0-H1 | 1.763 | +17.952 | +17.461 | +17.195 |
| T0-H2 | 1.565 | +2.932 | +2.908 | +2.847 |
| T0-H3 | 1.529 | +4.485 | +4.188 | +4.518 |
| T1-H1 | 1.531 | +1.952 | +1.906 | +1.806 |
| T1-H2 | 1.403 | 0.000 | 0.000 | 0.000 |
| T2-H1 | 1.432 | +4.242 | +4.045 | +3.988 |
| T2-H2 | 1.349 | +6.027 | +5.753 | +5.643 |
aΔE is the total electronic energy. bΔE0 is the zero-point corrected energy.
Table 7.
TD-DFT results for the eight studied protonated compounds in gas phase. The compounds are arranged in the series defined in the series defined in Error! Reference source not found.. The results of compound T0 are given for comparison. f values are oscillator strengths.
Table 7.
TD-DFT results for the eight studied protonated compounds in gas phase. The compounds are arranged in the series defined in the series defined in Error! Reference source not found.. The results of compound T0 are given for comparison. f values are oscillator strengths.
| Compound | HOMO-LUMO gap (eV) |
λabs (nm), Eabs (eV) |
fabs | Transition |
λem (nm), Eem (eV) |
fem |
|---|---|---|---|---|---|---|
| Tautomer-0 (Ph4N14Ph4H2) | 2.50 | 550 (2.25) | 2.39 | HOMO→LUMO (+98%) | 585 (2.12) | 2.42 |
| T0-H0 | 2.36 | Too unstable to be considered (+50 kcal.mol-1) | ||||
| T0-H1 | 1.76 | 777 (1.60) | 1.40 | HOMO→LUMO (100%) | ||
| T0-H2 | 1.57 | 849 (1.46) | 1.16 | HOMO→LUMO (99%) | ||
| T0-H3 | 1.53 | 863 (1.44) | 0.98 | HOMO→LUMO (99%) | ||
| T1-H1 | 1.53 | 853 (1.45) | 1.46 | HOMO→LUMO (99%) | 1002 (1.24) | 1.35 |
| T1-H2 | 1.40 | 907 (1.37) | 1.27 | HOMO→LUMO (99%) | 1071 (1.16) | 1.26 |
| T2-H1 | 1.43 | 891 (1.39) | 1.43 | HOMO→LUMO (99%) | ||
| T2-H2 | 1.35 | 932 (1.33) | 1.28 | HOMO→LUMO (99%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



