
Submitted:
19 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
Sepsis is a life-threatening syndrome triggered by infection and accompanied by high mortality, with antimicrobial resistances (AMRs) further escalating clinical challenges. Rapid and reliable detection of causative pathogens and AMRs are key factors for fast and appropriate treatment, in order to improve outcome in septic patients. However, current sepsis diagnostics based on blood culture is limited by low sensitivity and specificity while current molecular approaches fail to enter clinical routine. Therefore, we developed a Suppression PCR-based selective enrichment sequencing approach (SUPSETS), providing a molecular method combining multiplex suppression PCR with Nanopore sequencing to identify most common sepsis-causative pathogens and AMRs using plasma cell-free DNA. Applying only 1 mL of plasma, we target eight pathogens across three kingdoms and ten AMRs in a proof-of-concept study. SUPSETS was successfully tested on first ten clinical samples and revealed comparable results to clinical metagenomics while clearly outperforming blood culture. Several clinically relevant AMRs could be detected additionally. Furthermore, SUPSETS provided first pathogen and AMR-specific sequencing reads within minutes of starting sequencing, thereby potentially decreasing time to results to 11 - 13 hours and suggesting diagnostic potential in sepsis.
Keywords:
sepsis
; antimicrobial resistances
; cell-free DNA
; precision diagnostics
; Next-Generation sequencing
; suppression PCR
; real-time diagnostics
; nanopore sequencing
1. Introduction
Sepsis, defined as a “life-threatening organ dysfunction caused by a dysregulated host response to infection”, is a major global health threat, with estimated 48.9 mio cases and 11 mio deaths per year worldwide [1,2]. Risk factors including diabetes, obesity, chronic diseases, genetic factors, cancer or age increase probabilities for adverse outcome [3,4,5]. Besides individual’s health implications, sepsis generates significant costs for clinical intervention as well as for long-term follow-up care [6,7]. Numbers of bloodstream infections are rising [8]. On top, increasing incidences of antimicrobial resistances (AMRs) lead to additional threats for septic patients. A meta study from the Antimicrobial Resistance Collaborators revealed that 4.95 mio deaths were associated with bacterial AMRs in 2019 [9]. O’Neill and colleagues estimate that by the year 2050, at least 10 mio deaths worldwide will be attributable to AMRs, exaggerating the need for bloodstream infections and AMR diagnostics [10].
Early and appropriate treatment improves the outcome of septic patients, therefore identifying causative pathogens at the earliest possible time-point is crucial [11]. Currently, culture-based analysis (e.g., blood culture) represents the gold standard to assess pathogenic burden: blood samples are cultivated in culture bottles for several days until a positive result is obtained [12,13]. However, blood culture has several limitations (contamination-prone, antibiotic pre-treatment impacts, volume, time-to-diagnosis, slow growing or uncultivatable specimen) including low sensitivity and specificity [14,15]. Molecular-based methods like standard PCR or qPCR on isolated genomic DNA still fail to prove an impact on clinical utility due to ambiguous results [16,17].
Since first discovered in 1948 by Mandel & Métais [18], cell-free DNA (cfDNA) is increasingly used as biomarker for a wide range of clinical indications, from non-invasive prenatal testing (NIPT) to sepsis as well as cancer [19]. CfDNA has proven to be a highly suitable and dynamic biomarker for pathogen identification in sepsis [20]. Accordingly, Grumaz et al. could show that cfDNA sequencing by Next Generation Sequencing (NGS) resulted in a six times higher sensitivity for pathogen identification than blood culture, with an expert plausibility of 96 % [21]. However, NGS is still associated with limitations regarding costs per sample, high upfront costs for sequencing devices and lack of AMR detection. In contrast, Nanopore sequencing is characterized by low to moderate upfront costs, immediate processing and real-time analysis as well as high flexibility for testing due to small size [22,23]. This enables new diagnostic possibilities, from rapid outbreak surveillance, infection diagnostic or AMR profiling [24,25,26]. Nonetheless, sepsis diagnostic based on Nanopore sequencing remains challenging and only few studies are reported. Previously a workflow for unbiased, real-time cfDNA-sequencing on septic samples was established, which could identify pathogen burden within first hours of sequencing [27]. Extrapolation of results could show that over 90 % of pathogens could be detected with cfDNA Nanopore sequencing. Still, this approach was only applicable for samples with high pathogen burden while also failing to detect AMRs.
With SUPSETS (Suppression PCR-based selective enrichment sequencing) we aimed to establish a diagnostic platform based on a megaplex, highly specific two-step Suppression PCR and subsequent Nanopore sequencing to detect most common sepsis-causative pathogens across three kingdoms and main AMR genes
2. Results
2.1. SUPSETS Procedure
SUPSETS combines selective amplification of target sequences with subsequent Next-Generation Sequencing on randomly fragmented cell-free DNA (cfDNA). We identified species-specific sequences for most common sepsis-causative pathogens and AMRs. After a blood draw, cfDNA is isolated from plasma followed by ligation of suppression adapters to all cfDNA fragments (Figure 1). The GC-rich adapter sequence forces self-annealing, leading to a stem-loop formation upon denaturation, which diminishes unwanted amplification [28,29]. Following adapter ligation, F-primers are used for initial elongation of a fragment, forming a detection template after hybridization to its target sequence. F-primers consist of a universal sequence (US) [30] and a target-specific region at the 3’ end. For n targets, n F-primers are used to minimize primer amount, which enables higher multiplexing. Elongated cfDNA fragments (detection templates) are flanked by the 5' universal sequence and the 3' adapter sequence, allowing an amplification of all detection templates with the same primer pair (US-FW and Ad-rev). Amplified detection templates undergo library preparation for Nanopore sequencing, followed by data analysis for species detection, either after the finished run or in real-time by mapping reads to a target amplicon database.
Candidate primers were tested to determine the most specific and efficient primers (Supplement Figure S1A-D). Exemplary for HSV I, primer candidates were finally tested with 4,000 GE digested and adapter ligated HSV I DNA (Supplement Figure S1D). To mimic patient samples, 6,000 GE of digested and adapter ligated human DNA was spiked in. Primers producing unspecific signals were discarded in favor of HSV I T1 and HSV I T2 primers, which produced strong signals with little background (Supplement Figure S1C,D).
In general, F-Primers were tested for their performance in a Spike-In experiment, where 4,000 GE digested and adapter ligated DNA of each pathogen or AMR plasmid was spiked in 6,000 GE digested and adapter ligated human DNA. A detailed list of used primers per experiment is summarized in Supplement Table S1. Approximately 72,2 % (SD 15,92 %) of reads were of human origin, depending on the Spike-In input (Supplement Table S2). Dependent on the respective pathogen, the number of detected amplicon reads differed from 258 (~0.04 % of all filtered reads per sample) for C. albicans amplicon T3 to 297,783 reads (~9.13 % of all filtered reads per sample) for P. aeruginosa amplicon T2. All target amplicons could be discriminated from background, with the exception for C. albicans amplicon T2 (Figure 2A). The highest proportion of amplicon reads in contrast to all filtered reads was shown for B. frag T1 (~11.67 % of all filtered reads per sample).
To show feasibility of the complex panel in a multiple-pathogen sample, all addressed pathogens and AMRs were spiked into one human DNA sample and SUPSETS was performed with a combined primer panel (= sample “human- & AMR- & pathogen DNA”, used primer see Supplement Table S1). As a negative control, human DNA without any other pathogen spike-in was tested with the same primer mix (sample “human DNA”). All detected amplicons could be clearly distinguished between both samples (Figure 2B). Detected amounts of on target reads exceeded the human DNA only sample by at least more than two orders of magnitude (S. aureus T2) and up to more than three orders of magnitude for most other amplicons. However, primers C. alb T2, AAC(6)-Ib, TEM116, ndm1, OXA48, blaCTX-M-15 V2 and blaKPC-2 V2 could not be detected.
2.2. SUPSETS Validation on Selected Clinical Sepsis Samples
For a first proof-of-concept clinical validation, selected septic samples from Grumaz et al. (2019) [21] were used as a reference for pathogen detection (Supplement Table S3). To see cfDNA amplicon profiles of these samples, sequencing results were mapped to the respective pathogen and AMR reference genome. Exemplarily shown for samples FL_01 and FL_03, no distinct reads could be detected in off target regions, clear signal peaks were found in the selected regions, which started with the primer sequence (Figure 3). While the beginning of the amplicon at the 5' end is clearly defined by the primer, there is no clear terminus at the 3' end. This reflects the randomly fragmented nature of cfDNA, resulting in cfDNA fragments of different length and therefore undefined primer positions of the target region within the fragment (Figure 3).
Samples were analyzed with different primer panels using either a MinION Flongle accompanied with lower sequencing throughput or applied to a MinION Flow Cell (Figure 4). For the MinION Flow Cell samples, a positive signal was considered if at least one read was detected for at least one of the two pathogen targeting primers. Five out of seven pathogens were detected correctly positive by SUPSETS, in contrast, none was identified by blood culture (Figure 4A and Supplement Table S4). Among all pathogen-containing samples (FC_01, FC_02, FC_06), three false positive results were obtained, with non for blood culture. In negative controls (FC_03 to FC_05), comprising post operative samples and healthy controls, one false positive result in sample FC_03 (Figure 4A) could be detected. Accordingly, the best performance was found for E. faecium and B. fragilis targeting primers with exclusively correct positive and negative findings.
For MinION Flongle experiments, four clinical samples were run in one experiment (FL_01 - 04), with sample FL_04 tested additionally as solitary sample (FL_04-single). Overall, SUPSETS detected six out of eight pathogens correctly (Figure 4B and Supplement Table S5), with at least two reads and 0.01 % of all filtered reads considered as a positive result. No false positive signal was obtained and every negative control sample from clinical metagenomics was confirmed negative. In the two samples with multi-pathogen burden, FL_01 and FL_04, two out of three pathogens were detected, resulting in two false negative results. In comparison, only one of eight pathogens was detected correct positve in blood culture (Figure 4B).
For samples FL_01-04, the internal sequencing and PCR control Igκ was used to target human cfDNA (Figure 4C). Human amplicon Igκ received most of the normalized reads, ranging between 6.2 % and 19.1 % of total reads. Amplicons targeting the same pathogen do not necessarily result in comparable percentage of read counts. Exemplarily for sample FL_01, roughly 2.24 % of read counts were received for B. frag T1 and T2, on the one hand. On the other hand, for E. faecium reflecting target regions, only the amplicon E. faec T3 could be detected above the set threshold with 1.5 %. Interestingly, for patient FL_04, differences can be identified regarding signal intensity and results when analyzed as single sample (Figure 4C). While the multiplex sample FL_04 was not tested positive for both E. faecium and E. coli amplicons, FL_4-single has a three times higher percentage normalized read counts, e.g., for E. faec T3 from 0.0034 % to 0.0109 %.
AMRs could not be detected by clinical metagenomic reference analysis from [21]. However, in SUPSETS, several AMRs were detected in selected clinical samples (Figure 4C and Table 1). SUPSETS analysis revealed a potential resistance for tetracycline (tetB) in patients FL_03 and FL_04-single, which was not documented for the corresponding patient before. A vancomycine resistant E. faecium (VRE) was detected for patients FL_01, FL_02 and FC_06. In all these samples, a vancomycine resistance was confirmed by results of other clinical specimen including surgical, abdominal or wound swabs. In patient FL_02, a macrolide resistance against gene ermB was additionally detected. None of the detected AMRs was found by blood culture.
2.3. First Read Detection of SUPSETS within Selected Sepsis Samples
Outcome of septic patients improves when early and targeted therapy is administered, requiring early detection of pathogens [11]. Therefore, we investigated retrospectively at which point in time the first amplicon read corresponding to a pathogen or AMR was detected. Clinical samples FL_04-single and FC_01 were selected for retrospective analysis (Figure 5). Remarkably for sample FL_04-single, all three expected species and AMR targets (E. coli, E. faecium, tetB) could be detected within the first 18 min of sequencing, with both pathogens’ first signals detected after 8 min for E. coli and 14 min for E. faecium, respectively (Figure 5A). Comparable findings were revealed for patient FC_01, where HSV I was first detected after 5 min and E. faecium after 19 min (Figure 5B). Therefore, early detection of pathogenic and AMR burden with real-time analysis is feasible and might reduce the time to diagnosis.
3. Discussion
Sepsis and antimicrobial resistances (AMRs) have both severe health implications, due to inadequate diagnostic options. Since blood culture has severe limitations, several approaches like clinical metagenomics on cfDNA for the identification of pathogens have been published [21,31,32]. Based on these promising results, we aimed for a targeted, more rapid and cost-efficient approach than unbiased clinical metagenomics. Since cfDNA is a challenging molecule due to its random fragmentation, low concentration and short length (Figure 3), we established SUPSETS, a megaplex Suppression PCR-based selective target sequencing for pathogen and antimicrobial resistance detection on cell-free DNA in sepsis (Figure 1).
We first demonstrated feasibility of SUPSETS on artificial samples, which revealed high discriminatory power between target regions. Using SUPSETS we could show that even in a multi-pathogen background comprising 8 different pathogens, all pathogens could be resolved (Figure 2B) in contrast to blood culture analysis [27,33].
Blood culture is already known to be limited with respect to pathogen detection in sepsis. Studies from Grumaz et al. show that in their study over the complete 28-day period, 71 % of samples were positive with clinical metagenomics and only 11 % in blood culture, with an expert plausibility of 96% [21]. We therefore benchmarked our results against clinical metagenomics using samples from this study. With these selected clinical samples, SUPSETS clearly outperforms blood culture results with 11 of 15 correctly detected pathogens (73.3 %) compared to only one correctly positive detected pathogen with blood culture (6.7 %). SUPSETS displays three false positive results in Flow cell experiments, resulting in a specificity of 94.3 % (50 of 53 truly negative detected) (Figure 4A,B). Molecular approaches like SepsiTest reveals a sensitivity and specificity of 66.7 and 94.4 % compared to blood culture, respectively, with additional 15 positive results for SepsiTest and 10 results only positive in blood cultures [34].
Clinical metagenomics cannot detect AMRs reliably, as sequencing depth in most cases is too low [21,27]. Using SUPSETS, we could detect several AMRs in clinical samples in contrast to blood culture. Results were additionally supported by other clinical specimen for vancomycine resistance or for ermB as intrinsic AMR in E. faecium [35]. Resistance findings by SUPSETS might have clinical impact, for instance patient S10 (FL_02) was treated with vancomycin while the status and microbial load was worsening, due to a non-diagnosed VRE infection [21]. Our results provided evidence for adaptation of treatment that might have led to earlier and more targeted treatment.
Early adequate treatment is crucial in sepsis [36]. Blood culture analysis takes up to several days until a potentially positive result, while clinical metagenomics delivers results in up to 48 hours. Within SUPSETS, targeted amplification and Nanopore sequencing allows real-time analysis to reduce time-to-analysis. The retrospective analysis of clinical samples shows that the first pathogen and AMR-assigned reads were detected within few minutes (Figure 5). Mapping to a database containing only the selected target sequences after basecalling would accelerate the SUPSETS’ procedure in contrast to conventional NGS approaches. Several literature approaches already demonstrate the feasibility of real-time analysis [37,38]. A SUPSETS real-time detection of pathogens and AMRs would reduce time-to-diagnosis to approximately 12 h and enable earlier targeted therapy of septic patients at the same day (Supplement Table S6). Protocol optimization could lead to a further decrease in detection time.
Although blood culture is currently much more affordable, study results from clinical metagenomic studies clearly show the advantages of sequencing in terms of time savings and sensitivity [21,33]. Applying SUPSETS, we decrease costs of clinical metagenomics to estimated 160 – 225 € per sample. Also, investments are moderate and therefore in-house analysis by SUPSETS seems manageable (Supplement Table S7). With significantly high costs for a septic patient per day at ICU and more reliable results than blood culture (Figure 4), SUPSETS might be an attractive option with respect to cost savings [7,39].
SUPSETS also exhibits limitations of a targeted approach: Since only selected targets are addressed, pathogens not included into the panel cannot be detected. Since only species-specific primers are required, the tested panel can be quickly expanded and adapted to other targets. Other pathogen and AMR targets should be re-evaluated (OXA-48, KPC-2, ndm-1) and included to increase clinical value. Recently, Oxford Nanopore released information that their chemistry might react sensitive to light, which might have led to decrease in output und quality, particularly for short reads. Since this chemistry was used for Flow Cell experiments, this might explain performance differences between those and Flongle experiments [40]. Taken together, SUPSETS shows great potential in pathogen and AMR detection in septic patients.
4. Materials and Methods
4.1. Ethics Approval and Consent to Participate for Clinical Samples
Septic patient samples from a previously published clinical study were used [21]. The monocentric clinical study (S-097/2013) was conducted in the surgical intensive care unit of Heidelberg University Hospital, Germany (German Clinical Trials Register: DRKS00005463), with study and control patients or their legal representatives signed written informed consent. Results from this previous study were used as a reference for clinical validation of SUPSETS. All study procedures were approved for by the local ethics committee (Ethics Committee of the Medical Faculty of Heidelberg – Trial Code No. S-097/2013). Blood from healthy individuals was acquired commercially from Biomex (Heidelberg, Germany).
4.2. Microbiology and Preparation of Microbial DNA
Frozen samples of bacteria (Enterococcus faecium DSM 20477, Klebsiella pneumoniae DSM 30104, Bacteroides fragilis DSM 2151, Pseudomonas aeruginosa DSM 50071, Staphylococcus aureus DSM 20231) and Candida albicans SC5314 (ATCC MYA-2876) were thawed on ice. 50 µL of each pathogen solution were transferred to 10 mL of prewarmed Brain-Heart-Infusion Broth (Merck Millipore, Darmstadt). After incubating overnight at 37 °C and 150 rpm, microbial DNA extraction was performed with PureLink Genomic DNA kit (Invitrogen, Waltham) according to manufacturer’s advice. Concentration was checked with Qubit 3.0 fluorometer dsDNA High Sensitivity (HS) assay kit (Thermo Fisher, Waltham) and quality control with DNF-488 High Sensitivity Genomic DNA Analysis kit (Fragment Analyzer Automated CE, Agilent Technologies, Santa Clara). Isolated genomic DNA for Escherichia coli strain B and human (both Sigma-Aldrich, Steinheim) were bought and HSV I provided by Dr. Florian Full. The genome sequences of 16 AMRs were taken from The Comprehensive Antibiotic Resistance Database (CARD) [41], reduced to the relevant regions and combined with a DNA linker and a restriction site in one DNA cassette each. These cassettes were combined to four plasmids and ordered at Integrated DNA Technologies GmbH (Munich) (Supplement Figure S2). To mimic cfDNA in length, pathogen DNA was digested with the restriction enzymes AluI and HpyCH4V (both New England Biolabs, Frankfurt) according to the manufacturer’s protocol. Digested DNA was purified with 1.8x AMPure XP beads (Beckman-Coulter, Pasadena) according to the manufacturer’s advice. DNA concentration was checked with Qubit 3.0 fluorometer dsDNA High Sensitivity (HS) assay kit and quality controlled with DNF-474 High Sensitivity NGS Kit (Fragment Analyzer Automated CE, Agilent Technologies).
4.3. Identification of Species- or Genus-Specific Nucleotide Sequences and Primer Design
For the identification of species-specific nucleotide sequences, the NCBI reference genome of the pathogen of choice (Supplement Table S8), excluding plasmids, was used to generate 10 mio Illumina sequencing reads using InSilicoSeq (-model HiSeq; v1.5.0) [42]. These simulated sequencing reads were taxonomically classified using Kraken with the standard prebuilt reference database (v2.1.2) [43]. Only classified reads with a higher confidence score than 0.95 for the pathogen of choice were chosen for further use. 1,000 reads were randomly selected and blasted against an NCBI NR database excluding the pathogen of choice using blastn (-num_alignments 10 -word_size 20 -task blastn -short -evalue 1e-50 -negative_taxids <pathogenTAXID>; v2.13.0) [44,45]. Reads with a match to any other organism were discarded.
For the identification of genus-specific nucleotide sequences, the methodology used previously for species-specific sequences was slightly altered. The initially simulated Illumina sequencing reads were based on the reference genomes of all species belonging to the genus of interest, based on the NCBI taxonomy database (defined with the ‘get_species_taxids’ script from the blast tools). In the blastn analysis of the 1,000 selected nucleotide sequences, the E-value threshold was reduced to 1e-20.
Using Primer3 (libprimer3 release 2.6.1), primers were generated for the identified species-specific or genus-specific nucleotide sequences with the following settings: a minimum amplicon length 50 bp, primer length between 18 to 22 bp, maximum of two repeating bases, no number of unknown bases (N), a melting temperature between 59 °C and 64 °C, and a GC content between 40 % and 60 % [46]. Generated primers were validated against the genus-specific nucleic acid database using local BLAST (using “-taxidlist” blastn option) to ensure specificity for defined TaxIDs. Validation also included a BLAST search against the nt database to exclude hits on non-target genomes. Furthermore, extracted primers were blasted to confirm specificity to the desired genus without hits on other microbial or human sequences within the top hundred hits. Selected primer sequences were ordered at Sigma-Aldrich.
4.4. One Step PCR for Primer Testing
For the evaluation of suitable primers, a one-step PCR with putative US-target-primers was performed. 40,000 genome equivalents (GE) of digested, adapter ligated pathogen DNA was mixed 1 µL dNTP mix (10mM - Sigma Aldrich), with 2.5 µL 10x AmpliTaq Gold 360 buffer, 2.5 µL magnesium chloride (25 mM), 0.25 µL AmpliTaq Gold 360 DNA polymerase (all ThermoFisher, Waltheim), 1µL unique US-target-primer (0.2 µM), 1µL Ad-rev primer (0.2 µM) (both Sigma-Aldrich, Steinheim) and filled up with nuclease-free water up to 25 µL. After mixing, the reaction was incubated for 4 min at 95 °C, followed by 38 cycles of 95 °C for 20 sec, 65 °C for 30 sec and 72 °C for 30 sec. Quality control was performed as described above.
GE were calcualated according to formula:
4.5. SUPSETS Workflow
Suppression adapters were generated from a long (Adapter_long) and short (Adapter_short) oligonucleotide (Sigma-Aldrich, Steinheim) using sequences first published by Matz et al. (1997) [47]. For hybridization, 20 µL of both oligonucleotides were incubated with 10 µL 10x T4 ligase mix (New England Biolabs, Frankfurt) and 50 µL nuclease-free water for 3 min at 95 °C and cooled down overnight at RT.
SUPSETS workflow starts with ligating suppression adapters to blunt-end DNA fragments, either prepared cfDNA or digested genomic DNA. 20 µM suppression adapter were added in a ratio of 1:20 to DNA (600 - 700 ng) together with 400 U of T4 ligase and 10x T4 ligase buffer, adjusted with nuclease-free water to reach a one-time concentration of ligase buffer. The reaction was incubated for 2 h at RT or 16 °C overnight. After heat inactivation at 65 °C for 10 min, adapter-ligated DNA was purified with 1.6 x AMPure XP beads (Beckman Coulter) and the concentration and profile validated as described above.
For elongation, 2.5 µL 10x AmpliTaq Gold 360 buffer was mixed with 2.5 µL magnesium chloride (25 mM), 1 µL dNTP mix (10mM - Sigma Aldrich), 0.25 µL AmpliTaq Gold 360 DNA polymerase and a panel of Fusion primers (F-primers) with final concentration of 0.3 µM per F-primer. The list of F-Primers and used F-Primers per experiment can be found in Supplement Table S9 and S1, respectively. As input, either 40,000 to 4,000 and 6,000 genome equivalents of adapter ligated, digested pathogen or human DNA, respectively, was used and the reaction filled up to 25 µL with nuclease-free water (Equation 1). For clinical samples, a defined volume of 15.92 µL blunt-end cfDNA was used. The reaction was incubated for 5 min at 95 °C, followed by 66 °C for 20 min and 72 °C for 5 min. The elongation product was purified with 1.6x AMPure XP beads to remove excessive F-primers and used for amplification: 13.5 µL of this elongation product was mixed with 2.5 µL 10x AmpliTaq Gold 360 buffer, 2.5 µL magnesium chloride (25 mM), 1 µL dNTP mix, 4.25 µL nuclease-free water, 0.25 µL AmpliTaq Gold 360 DNA polymerase and US-FW and Ad-rev primers (both Sigma-Aldrich, Steinheim), each with final concentration of 0.2 µM. The reaction was incubated for 4 min at 95 °C, followed by 38 cycles of 95 °C for 20 sec, 65 °C for 30 sec and 72 °C for 30 sec and a final elongation of 5 min at 72 °C. The amplification product was purified with 1.6x AMPure XP beads and quality control performed as described above.
Purified amplification products were applied to Nanopore sequencing. For MinION Flow Cell experiments, the Native Barcoding Kit 24 V14 (Oxford Nanopore Technologies, ONT, London) was used according to manufacturer’s advice. For MinION Flongle experiments, the Ligation Sequencing Kit SQK-LSK 109 for sample preparation and the Native Barcoding Expansion Kit EXP-NBD 104 were used according to the manufacturer’s protocol. For the end-prep reaction, 12.5 µL of sample was used as input. After barcoding and pooling of the reaction, native adapter was ligated with 5 µL T4 Ligase. A MinION Flow Cell (R.10.4.1) or Flongle (R.9.4.1) was primed and the DNA sequencing library loaded according to the protocol. Samples for technical validation were sequenced for a total of 24 h. Clinical samples were sequenced the following: FL_01 - 04 for 24 h, FL_04-single for 3 h, FC_01 - 06 for 72 h.
4.6. Analysis of Sequencing Results
After sequencing with MinION Flongle, basecalling and demultiplexing of sequencing data was performed with the Guppy (version 6.5.7 ONT) using the high accuracy model with minimal allowed Phred score equal to 9 (dna_r10.4.1_e8.2_400bps_5khz_hac). Afterwards, adapters and barcodes were trimmed using qcat [45] followed by a quality control using Nanoplot [48]. Next, quality score-based read filtering was performed with a minimum q-score of nine and a minimum length of 50 bp with NanoFilt [48]. Afterwards, the reads were mapped to the corresponding reference genomes using mimimap2 with default settings [49]. For the antibiotic resistances, an artificial mapping reference was generated, where all available AMR nucleotide sequences from CARD were concatenated to a fasta file. Reads with a mapq score below 20 were removed from the mapping output using samtools [50]. Sorted and indexed bam files were then visualized IGV Browser (v2.16.1) [51]. For MinION Flow Cell and further analysis of MinION Flongle experiments, basecalling, demultiplexing, adapter trimming, quality control and quality filtering were performed as described above. Only reads with a q-score of at least 12 (9 for Flongle) and a size range between 100 to 1000 bp were kept. Mapping was performed with minimap2 against an artificial mapping reference consisting of the previously identified target regions for evaluated pathogens, including AMRs and the human genome (hg38) [49]. Data obtained by SUPSETS workflow were evaluated in the context of Illumina-based, clinical metagenomics results previously published [21].
For the retrospective analysis of first received amplicon reads, timestamps for every barcode from Guppy-generated, filtered reads were extracted. 1) Filtered reads were mapped to a reference pathogen genome or to the artificially generated AMR reference using minimap2. 2) Only uniquely-mapped reads and corresponding read-ID were extracted using samtools. 3) The two generated lists were combined to find timestamps of uniquely-mapped reads using a custom perl script. After repeating the described steps 1-3 for every reference genome, results were plotted using a custom R script.
4.7. Clinical Sample Preparation
5. Conclusions
Sepsis and antimicrobial resistances (AMRs) are major global health threats, also due to a lack of appropriate clinical diagnostic tools. Within SUPSETS, we established a platform to bridge the current diagnostic gap by combining a multiplex Suppression PCR with subsequent Nanopore sequencing to detect sepsis pathogens and AMRs. We could show the potential by simultaneous and specific detection of up to eight different pathogens and ten AMRs. A first proof on selected clinical samples outperformed blood culture analysis and delivered comparable results to clinical metagenomic analysis. We could show that SUPSETS can detect pathogenic and AMR signals within minutes after sequencing, opening the field for real-time sequencing analysis to decrease further time-to-diagnosis. Further studies on clinical samples in sepsis or related fields like cancer panel diagnostics or environmental sample assessment have to be done to evaluate SUPSETS. Nonetheless, obtained results suggest that SUPSETS as a diagnostic platform might be a valuable diagnostic in-house solution for the future in the field of sepsis/AMR or related clinical fields.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplement Figure S1: Identification of suitable HSV I primers, Supplement Figure S2: Antibiotic resistance plasmids, Supplement Table S1: Primer set used per experiment, Table Supplement S2: Raw sequencing and mapping data for technical validation, Supplement Table S3: Characteristics of used clinical samples, Supplement Tables S4 and 5: Raw sequencing and mapping data for selected clinical samples, Supplement Table S6: Approximate duration of SUPSETS workflow, Supplement Table S7: Approximate costs and price per sample of SUPSETS, Supplement Table S8: Reference genomes and corresponding TaxIDs, Supplement Table S9: List of primers and sequence.
Author Contributions
Conceptualization, M.S. and K.S.; methodology, M.S., V.E. and L.J.; software, Y.V, V.E and J.M.; validation, V.E. and M.S.; formal analysis, Y.V., V.E., J.M. and M.S.; resources (clinical samples), T.B. and S.O.D.; data curation, J.M. and V.E.; writing - original draft preparation, M.S. and V.E.; writing - review and editing, K.S., M.S. and V.E.; visualization, V.E., M.S. and J.M; supervision, K.S. and M.S.; project administration, K.S.; funding acquisition, K.S. and M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of the Medical Faculty of Heidelberg University (S-097/2013 - 15.04.2013) and conducted in the surgical intensive care unit of Heidelberg University Hospital, Germany (German Clinical Trials Register: DRKS00005463).
Informed Consent Statement
Written informed consent was obtained from all subjects or from their legal representative involved in the study.
Data Availability Statement
Original patient data are unavailable due to ethical considerations, only pseudonymous information was used. The original data presented in the study are openly available in NCBI at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1100967.
Acknowledgments
We want to thank Dr. Florian Full and colleagues from University medicine Freiburg for providing HSV I DNA.
Conflicts of Interest
M.S., V.E., L.J. and K.S filed in for patent for this invention (EP23195471.0). K.S. is a cofounder of Noscendo, a company dedicated to the diagnoses of infections. The other authors declare that they have no conflict of interest.
References
- Singer, M.; Deutschman, C.S.; Seymour, C.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D. V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. The Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Kotfis, K.; Wittebole, X.; Jaschinski, U.; Solé-Violán, J.; Kashyap, R.; Leone, M.; Nanchal, R.; Fontes, L.E.; Sakr, Y.; Vincent, J.L. A Worldwide Perspective of Sepsis Epidemiology and Survival According to Age: Observational Data from the ICON Audit. J Crit Care 2019, 51, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Gonzalez, A.; Spearman, P.W.; Stoll, B.J. Neonatal Infectious Diseases. Evaluation of Neonatal Sepsis. Pediatr Clin North Am 2013, 60, 367–389. [Google Scholar] [CrossRef]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of Severe Sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.F.R.; Huelle, K.; Reinhold, T.; Prescott, H.C.; Gehringer, R.; Hartmann, M.; Lehmann, T.; Mueller, F.; Reinhart, K.; Schneider, N.; et al. Healthcare Utilization and Costs in Sepsis Survivors in Germany–Secondary Analysis of a Prospective Cohort Study. J Clin Med 2022, 11, 1142. [Google Scholar] [CrossRef]
- van den Berg, M.; van Beuningen, F.E.; ter Maaten, J.C.; Bouma, H.R. Hospital-Related Costs of Sepsis around the World: A Systematic Review Exploring the Economic Burden of Sepsis. J Crit Care 2022, 71, 154096. [Google Scholar] [CrossRef]
- Rhee, C.; Klompas, M. Sepsis Trends: Increasing Incidence and Decreasing Mortality, or Changing Denominator? J Thorac Dis 2020, 12, S89. [Google Scholar] [CrossRef]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report And Recommendations. The Review On Antimicrobial Resistance 2016.
- Bloos, F.; Rüddel, H.; Thomas-Rüddel, D.; Schwarzkopf, D.; Pausch, C.; Harbarth, S.; Schreiber, T.; Gründling, M.; Marshall, J.; Simon, P.; et al. Effect of a Multifaceted Educational Intervention for Anti-Infectious Measures on Sepsis Mortality: A Cluster Randomized Trial. Intensive Care Med 2017, 43, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Utility of Blood Culture in Sepsis Diagnostics. Journal of The Academy of Clinical Microbiologists 2016, 18, 74. [Google Scholar] [CrossRef]
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med 2017, 43, 304–377. [Google Scholar] [CrossRef] [PubMed]
- Scheer, C.S.; Fuchs, C.; Gründling, M.; Vollmer, M.; Bast, J.; Bohnert, J.A.; Zimmermann, K.; Hahnenkamp, K.; Rehberg, S.; Kuhn, S.O. Impact of Antibiotic Administration on Blood Culture Positivity at the Beginning of Sepsis: A Prospective Clinical Cohort Study. Clinical Microbiology and Infection 2019, 25, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Brealey, D.; Libert, N.; Abidi, N.E.; O’Dwyer, M.; Zacharowski, K.; Mikaszewska-Sokolewicz, M.; Schrenzel, J.; Simon, F.; Wilks, M.; et al. Rapid Diagnosis of Infection in the Critically Ill, a Multicenter Study of Molecular Detection in Bloodstream Infections, Pneumonia, and Sterile Site Infections. Crit Care Med 2015, 43, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Jupe, J.; Mack, H.; Coleman, T.P.; Lawrence, S.M.; Fraley, S.I. Emerging Technologies for Molecular Diagnosis of Sepsis. Clin Microbiol Rev 2018, 31. [Google Scholar] [CrossRef] [PubMed]
- Kaleta, E.J.; Clark, A.E.; Johnson, D.R.; Gamage, D.C.; Wysocki, V.H.; Cherkaoui, A.; Schrenzel, J.; Wolk, D.M. Use of PCR Coupled with Electrospray Ionization Mass Spectrometry for Rapid Identification of Bacterial and Yeast Bloodstream Pathogens from Blood Culture Bottles. J Clin Microbiol 2011, 49, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P. and M.P. Les Acides Nucléiques Du Plasma Sanguin Chez l’Homme. Available online: https://pubmed.ncbi.nlm.nih.gov/18875018/ (accessed on 26 June 2020).
- Stawski, R.; Stec-Martyna, E.; Chmielecki, A.; Nowak, D.; Perdas, E. Current Trends in Cell-Free DNA Applications. Scoping Review of Clinical Trials. Biology (Basel) 2021, 10, 906. [Google Scholar] [CrossRef]
- Hartwig, C.; Drechsler, S.; Vainshtein, Y.; Maneth, M.; Schmitt, T.; Ehling-Schulz, M.; Osuchowski, M.; Sohn, K. From Gut to Blood: Spatial and Temporal Pathobiome Dynamics during Acute Abdominal Murine Sepsis. Microorganisms 2023, 11, 627. [Google Scholar] [CrossRef]
- Grumaz, S.; Grumaz, C.; Vainshtein, Y.; Stevens, P.; Glanz, K.; Decker, S.O.; Hofer, S.; Weigand, M.A.; Brenner, T.; Sohn, K. Enhanced Performance of Next-Generation Sequencing Diagnostics Compared with Standard of Care Microbiological Diagnostics in Patients Suffering from Septic Shock. Crit Care Med 2019, 47, e394–e402. [Google Scholar] [CrossRef]
- Mikheyev, A.S.; Tin, M.M.Y. A First Look at the Oxford Nanopore MinION Sequencer. Mol Ecol Resour 2014, 14, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Ashton, P.; Calus, S.; Chatt, C.; Gossain, S.; Hawker, J.; Nair, S.; Neal, K.; Nye, K.; Peters, T.; et al. Rapid Draft Sequencing and Real-Time Nanopore Sequencing in a Hospital Outbreak of Salmonella. Genome Biol 2015, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Mwaigwisya, S.; Crossman, L.C.; Doumith, M.; Munroe, D.; Pires, C.; M. Khan, A.; Woodford, N.; Saunders, N.J.; Wain, J.; et al. Identification of Bacterial Pathogens and Antimicrobial Resistance Directly from Clinical Urines by Nanopore-Based Metagenomic Sequencing. Journal of Antimicrobial Chemotherapy 2017, 72, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore Metagenomics Enables Rapid Clinical Diagnosis of Bacterial Lower Respiratory Infection. Nature Biotechnology 2019 37:7 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Grumaz, C.; Hoffmann, A.; Vainshtein, Y.; Kopp, M.; Grumaz, S.; Stevens, P.; Decker, S.O.; Weigand, M.A.; Hofer, S.; Brenner, T.; et al. Rapid Next-Generation Sequencing–Based Diagnostics of Bacteremia in Septic Patients. Journal of Molecular Diagnostics 2020, 22, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Broude, N.E.; Storm, N.; Malpel, S.; Graber, J.H.; Lukyanov, S.; Sverdlov, E.; Smith, C.L. PCR Based Targeted Genomic and CDNA Differential Display. Genet Anal 1999, 15, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Broude, N.E.; Zhang, L.; Woodward, K.; Englert, D.; Cantor, C.R. Multiplex Allele-Specific Target Amplification Based on PCR Suppression. Proc Natl Acad Sci U S A 2001, 98, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Heath, K.E.; Day, I.N.M.; Humphries, S.E. Universal Primer Quantitative Fluorescent Multiplex (UPQFM) PCR: A Method to Detect Major and Minor Rearrangements of the Low Density Lipoprotein Receptor Gene. J Med Genet 2000, 37, 272–280. [Google Scholar] [CrossRef]
- Grumaz, S.; Stevens, P.; Grumaz, C.; Decker, S.O.; Weigand, M.A.; Hofer, S.; Brenner, T.; von Haeseler, A.; Sohn, K. Next-Generation Sequencing Diagnostics of Bacteremia in Septic Patients. Genome Med 2016, 8, 73. [Google Scholar] [CrossRef]
- Huang, Q.; Fu, A.; Wang, Y.; Zhang, J.; Zhao, W.; Cheng, Y. Microbiological Diagnosis of Endophthalmitis Using Nanopore Targeted Sequencing. Clin Exp Ophthalmol 2021, 49, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Brenner, T.; Decker, S.O.; Grumaz, S.; Stevens, P.; Bruckner, T.; Schmoch, T.; Pletz, M.W.; Bracht, H.; Hofer, S.; Marx, G.; et al. Next-Generation Sequencing Diagnostics of Bacteremia in Sepsis (Next GeneSiS-Trial): Study Protocol of a Prospective, Observational, Noninterventional, Multicenter, Clinical Trial. Medicine 2018, 97. [Google Scholar] [CrossRef] [PubMed]
- Nieman, A.E.; Savelkoul, P.H.M.; Beishuizen, A.; Henrich, B.; Lamik, B.; MacKenzie, C.R.; Kindgen-Milles, D.; Helmers, A.; Diaz, C.; Sakka, S.G.; et al. A Prospective Multicenter Evaluation of Direct Molecular Detection of Blood Stream Infection from a Clinical Perspective. BMC Infect Dis 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Marimón, J.M.; Valiente, A.; Ercibengoa, M.; García-Arenzana, J.M.; Pérez-Trallero, E. Erythromycin Resistance and Genetic Elements Carrying Macrolide Efflux Genes in Streptococcus Agalactiae. Antimicrob Agents Chemother 2005, 49, 5069–5074. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Gesten, F.; Prescott, H.C.; Friedrich, M.E.; Iwashyna, T.J.; Phillips, G.S.; Lemeshow, S.; Osborn, T.; Terry, K.M.; Levy, M.M. Time to Treatment and Mortality during Mandated Emergency Care for Sepsis. New England Journal of Medicine 2017, 376, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Malla, S.; Stout, M. Real-Time Selective Sequencing Using Nanopore Technology. Nat Methods 2016, 13, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nature Nanotechnology 2009, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadees, A.A.N.; Al-Khayyat, A.N.; Yaqoob, Q.A. The Outcome of Sepsis Patients Admitted to the Intensive Care Unit: Experience of 100 Cases. Medical Archives 2021, 75, 35. [Google Scholar] [CrossRef] [PubMed]
- Why Do I Need to Put a Light Shield on My Flow Cell? | Oxford Nanopore Technologies Help Center. Available online: https://help.nanoporetech.com/en/articles/8304478-why-do-i-need-to-put-a-light-shield-on-my-flow-cell (accessed on 21 December 2023).
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Gourlé, H.; Karlsson-Lindsjö, O.; Hayer, J.; Bongcam-Rudloff, E. Simulating Illumina Metagenomic Data with InSilicoSeq. Bioinformatics 2019, 35, 521–522. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol 2019, 20. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- GitHub - Nanoporetech/Qcat: Qcat Is a Python Command-Line Tool for Demultiplexing Oxford Nanopore Reads from FASTQ Files. Available online: https://github.com/nanoporetech/qcat (accessed on 22 February 2021).
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--New Capabilities and Interfaces. Nucleic Acids Res 2012, 40. [Google Scholar] [CrossRef]
- Matz, M.; Usman, N.; Shagin, D.; Bogdanova, E.; Lukyanov, S. Ordered Differential Display: A Simple Method for Systematic Comparison of Gene Expression Profiles. Nucleic Acids Res 1997, 25, 2541–2542. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief Bioinform 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Principle and workflow of SUPSETS. SUPSETS workflow depicted from blood draw and cfDNA isolation (step 1), suppression adapter ligation (step 2) and multiplex suppression PCR (step 3 -5) to sequencing and data analysis for pathogen detection (steps 6 and 7). Image created by BioRender.
Figure 1.
Principle and workflow of SUPSETS. SUPSETS workflow depicted from blood draw and cfDNA isolation (step 1), suppression adapter ligation (step 2) and multiplex suppression PCR (step 3 -5) to sequencing and data analysis for pathogen detection (steps 6 and 7). Image created by BioRender.
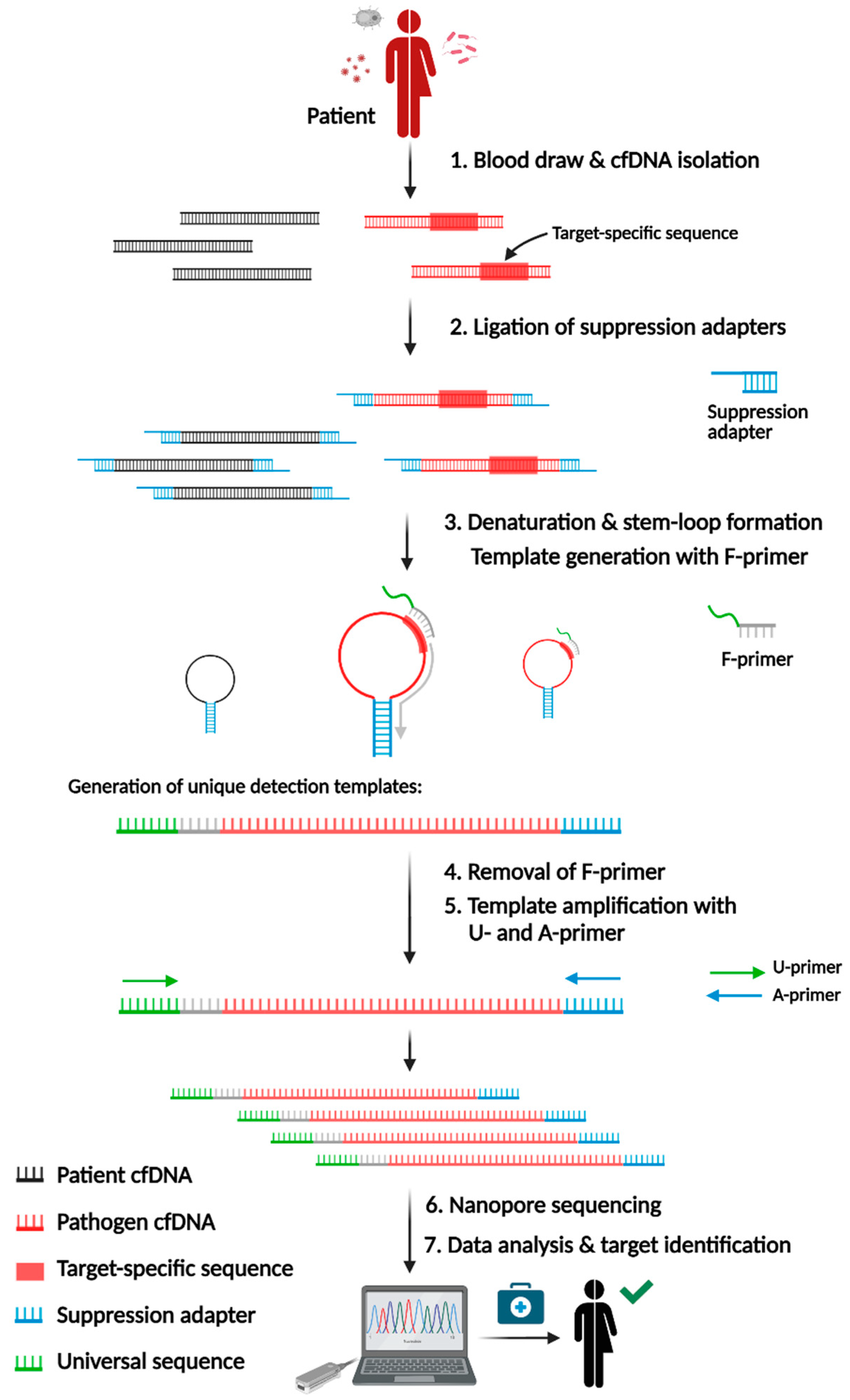
Figure 2.
SUPSETS technical validation on artificial Spike-In samples. A: Technical validation of SUPSETS with single Spike-In samples. 4,000 GE digested and adapter ligated genomic pathogen DNA were spiked in 6,000 GE digested and adapter ligated genomic human DNA and SUPSETS workflow performed with a primer mix containing two primers per tested targeted pathogen, one genus specific Candida primer and one general 16 S targeting primer. Single primers were exchange between experiments to improve performance or added with expanse of the targeted pathogen panel (Supplement Table S1). X-axis: Spike in pathogen per column. Y-axis: Regions targeted by pathogen specific primers. B: Reads per primer related amplicon region for artificial human DNA sample with and without spiked in target DNA. 4,000 GE digested and adapter ligated genomic DNA for each pathogen and AMR were spiked to 6,000 GE digested and adapter ligated genomic human DNA in the human- & AMR- & pathogen DNA sample, incubated with primer mix containing two primers per targeted pathogen, one genus specific Candida primer, one general 16 S targeting primer and 16 primers targeting genes for antimicrobial resistances. For negative control, no pathogen-derived DNA was included in the same experimental setup and only using 6,000 GE digested adapter-ligated human DNA. X-axis shows target regions excluding primers C. alb T2, AAC(6)-Ib, TEM116, ndm1, OXA48, blaCTX-M-15 V2 and blaKPC-2 V2 due to performance lack. Y-axis depicts percentage of normalized read counts. For corresponding primers see Supplement Table S1. Normalized counts=(max(coverage)i ⁄ (∑(i→j)max(coverage)i + ∑human reads); column corrected. i for target region.
Figure 2.
SUPSETS technical validation on artificial Spike-In samples. A: Technical validation of SUPSETS with single Spike-In samples. 4,000 GE digested and adapter ligated genomic pathogen DNA were spiked in 6,000 GE digested and adapter ligated genomic human DNA and SUPSETS workflow performed with a primer mix containing two primers per tested targeted pathogen, one genus specific Candida primer and one general 16 S targeting primer. Single primers were exchange between experiments to improve performance or added with expanse of the targeted pathogen panel (Supplement Table S1). X-axis: Spike in pathogen per column. Y-axis: Regions targeted by pathogen specific primers. B: Reads per primer related amplicon region for artificial human DNA sample with and without spiked in target DNA. 4,000 GE digested and adapter ligated genomic DNA for each pathogen and AMR were spiked to 6,000 GE digested and adapter ligated genomic human DNA in the human- & AMR- & pathogen DNA sample, incubated with primer mix containing two primers per targeted pathogen, one genus specific Candida primer, one general 16 S targeting primer and 16 primers targeting genes for antimicrobial resistances. For negative control, no pathogen-derived DNA was included in the same experimental setup and only using 6,000 GE digested adapter-ligated human DNA. X-axis shows target regions excluding primers C. alb T2, AAC(6)-Ib, TEM116, ndm1, OXA48, blaCTX-M-15 V2 and blaKPC-2 V2 due to performance lack. Y-axis depicts percentage of normalized read counts. For corresponding primers see Supplement Table S1. Normalized counts=(max(coverage)i ⁄ (∑(i→j)max(coverage)i + ∑human reads); column corrected. i for target region.
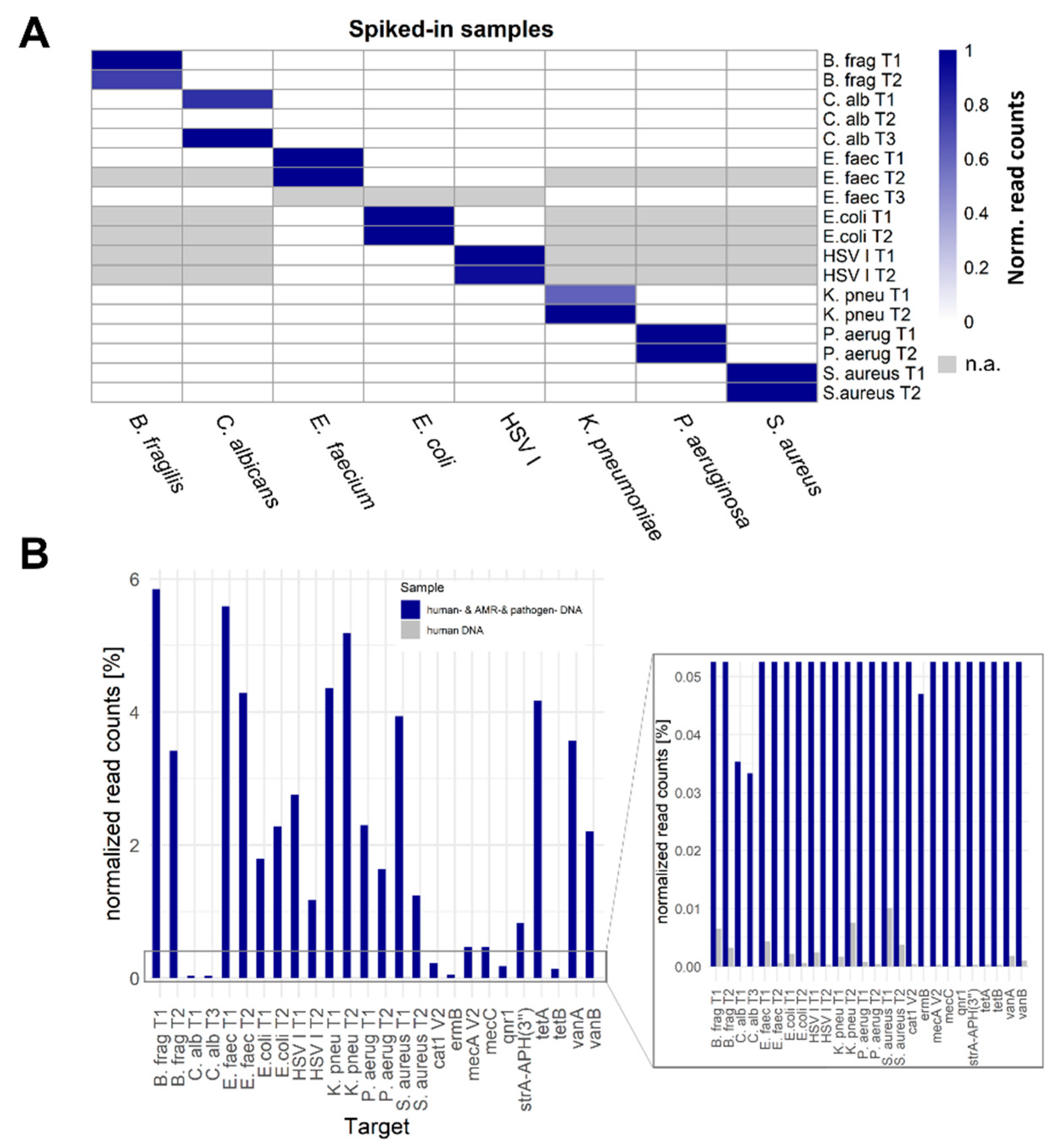
Figure 3.
Appearance of SUPSETS detected cfDNA amplicons. Results of SUPSETS workflow for clinical samples FL_01 and FL_03 were mapped against the respective pathogen or AMR reference genome and visualized in IGV browser. For each amplicon, maximum coverages with a coverage greater than two were counted and visualized. The primer binding site is depicted in blue. Mismatches are displayed in multi color.
Figure 3.
Appearance of SUPSETS detected cfDNA amplicons. Results of SUPSETS workflow for clinical samples FL_01 and FL_03 were mapped against the respective pathogen or AMR reference genome and visualized in IGV browser. For each amplicon, maximum coverages with a coverage greater than two were counted and visualized. The primer binding site is depicted in blue. Mismatches are displayed in multi color.
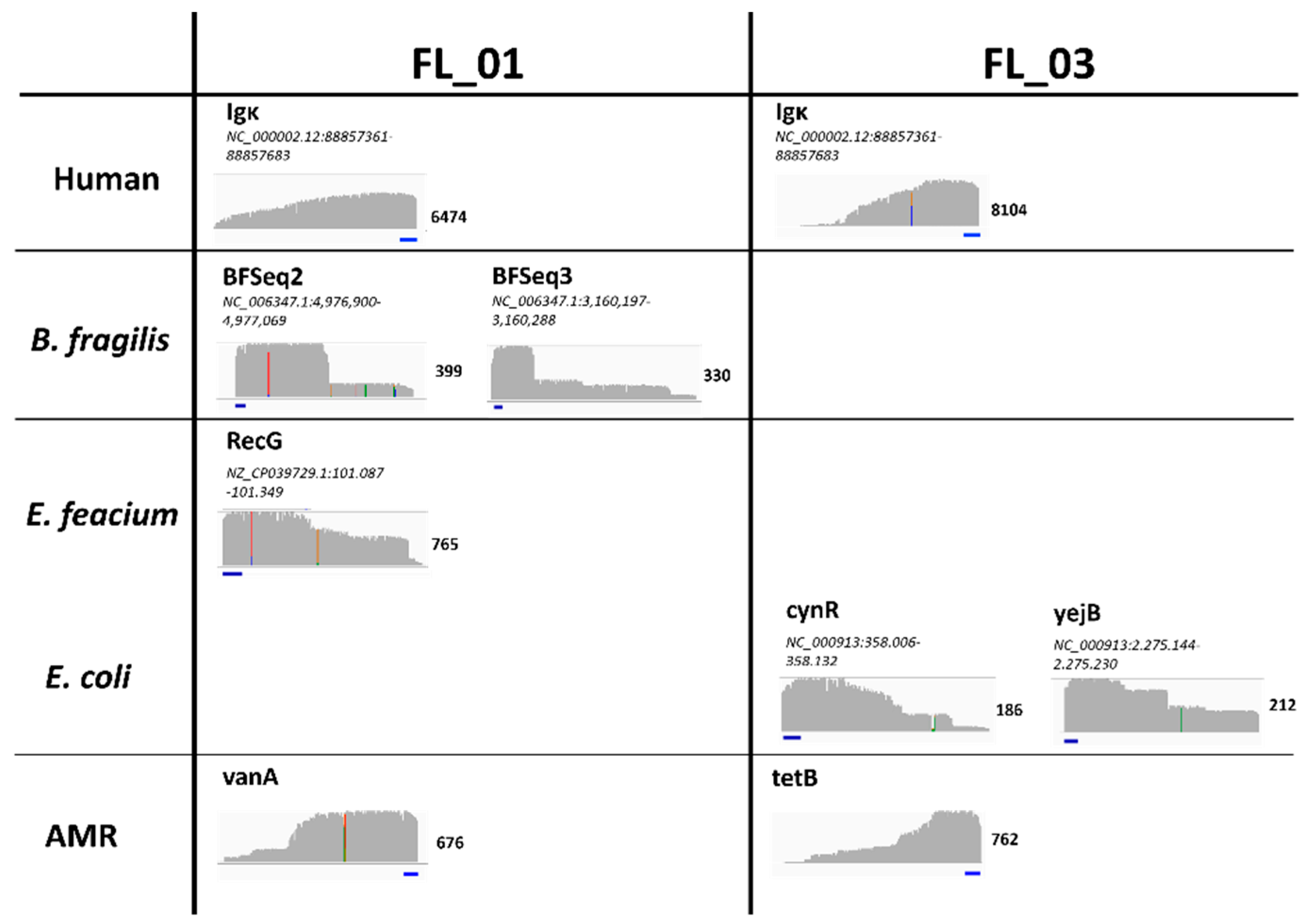
Figure 4.
Validation on selected clinical samples. Septic patient samples were selected according to their pathogen and AMR profile from MIRSI study [21] and applied to SUPSETS workflow. A: Qualitative analysis comparing NGS data (“ground truth”) to SUPSETS experiment on MinION Flow Cell and blood culture results. B: Qualitative analysis comparing NGS data (“ground truth”) to SUPSETS experiment on MinION Flongle and blood culture results. C: Processed reads were normalized to filtered mapped reads (Q-score > 9) for each sample for four selected patient samples within one MinION Flongle sequencing experiment. Samples FL_01 - 04 were sequenced together on one MinION Flongle run for 24 h; FL_04-single indicates a single sample MinION Flongle sequencing experiment for 3 h. The red vertical indicates the threshold of 0.01 % normalized reads. Target amplicons are indicated according the color legends, respectively. Subfigures A and B: True positive hits compared to NGS data are indicated in dark blue, false positve signals in light grey, light blue reflects true negative, dark grey false negative results and white for no data available. * Igκ reads were taken after mapping against the reference genome (hg38).
Figure 4.
Validation on selected clinical samples. Septic patient samples were selected according to their pathogen and AMR profile from MIRSI study [21] and applied to SUPSETS workflow. A: Qualitative analysis comparing NGS data (“ground truth”) to SUPSETS experiment on MinION Flow Cell and blood culture results. B: Qualitative analysis comparing NGS data (“ground truth”) to SUPSETS experiment on MinION Flongle and blood culture results. C: Processed reads were normalized to filtered mapped reads (Q-score > 9) for each sample for four selected patient samples within one MinION Flongle sequencing experiment. Samples FL_01 - 04 were sequenced together on one MinION Flongle run for 24 h; FL_04-single indicates a single sample MinION Flongle sequencing experiment for 3 h. The red vertical indicates the threshold of 0.01 % normalized reads. Target amplicons are indicated according the color legends, respectively. Subfigures A and B: True positive hits compared to NGS data are indicated in dark blue, false positve signals in light grey, light blue reflects true negative, dark grey false negative results and white for no data available. * Igκ reads were taken after mapping against the reference genome (hg38).
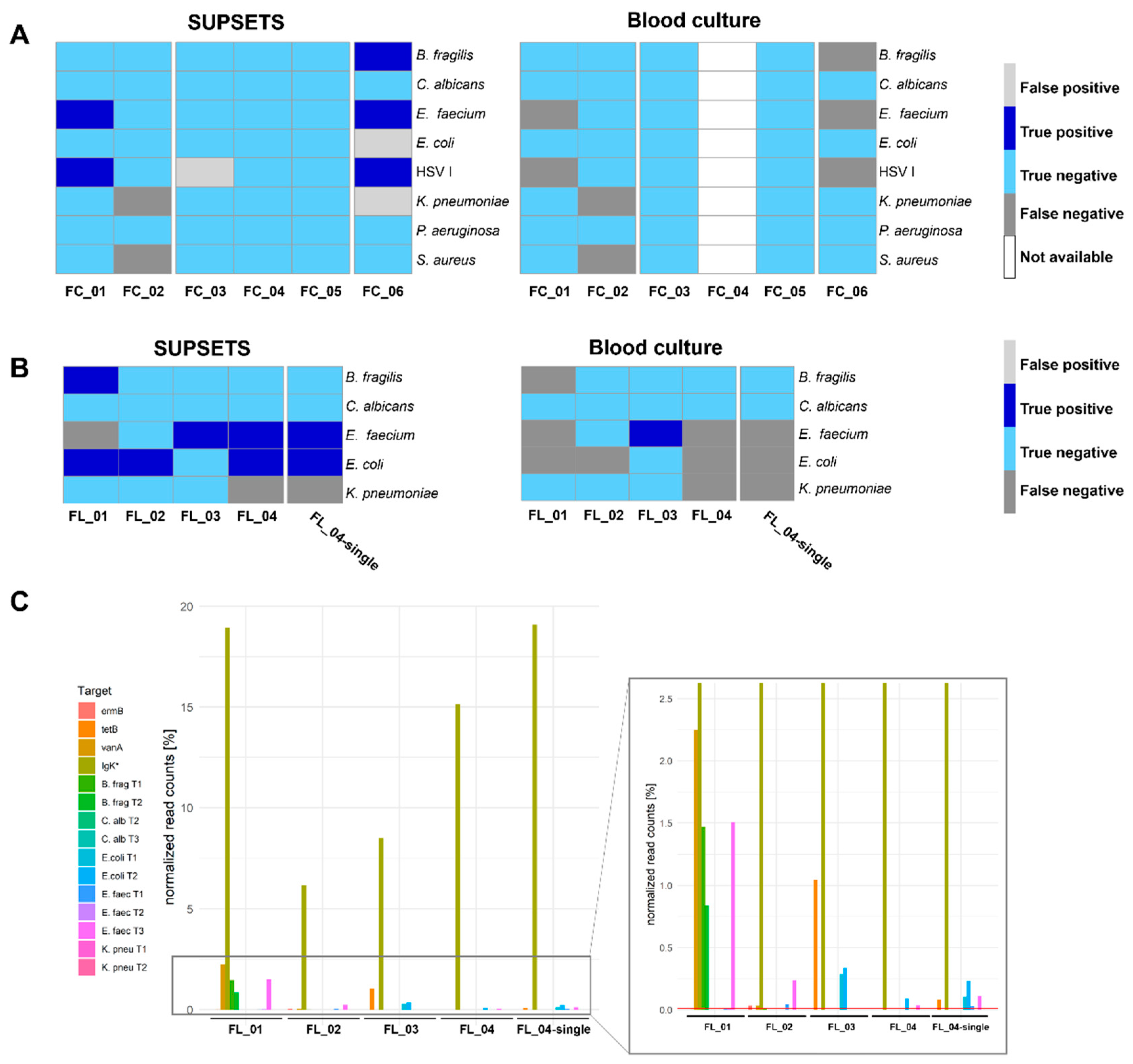
Figure 5.
Real-time detection of first pathogen and AMR reads. For a retrospective analysis, processed reads are shown at the time they were sequenced. Analysis is shown for two different clinical samples A (FL_04-single) and B (FC_01). Arrows with time data indicate the first target hits according to the shape and colored legend, respectively.
Figure 5.
Real-time detection of first pathogen and AMR reads. For a retrospective analysis, processed reads are shown at the time they were sequenced. Analysis is shown for two different clinical samples A (FL_04-single) and B (FC_01). Arrows with time data indicate the first target hits according to the shape and colored legend, respectively.
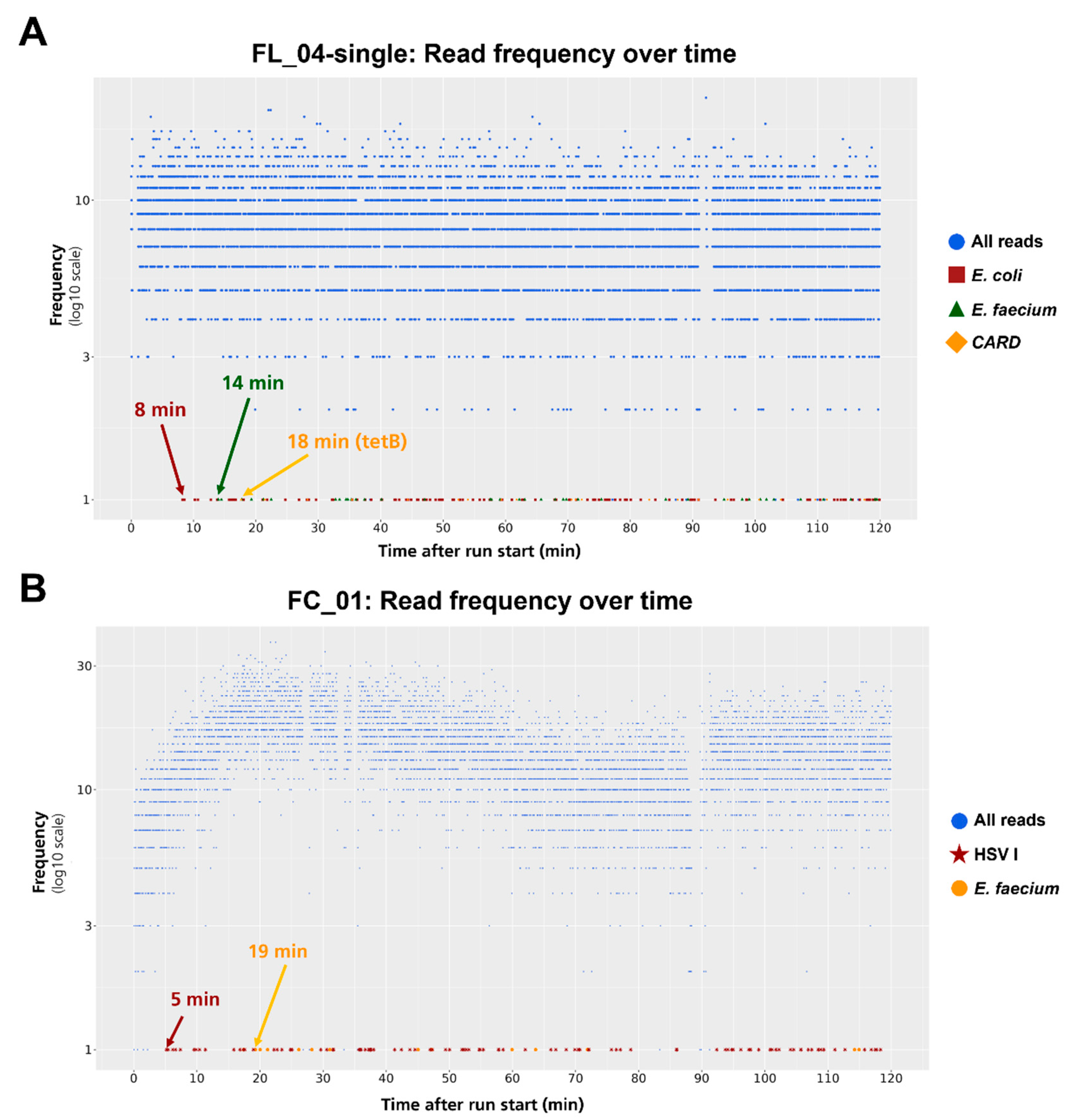

Table 1.
Antimicrobial resistances detected in clinical samples. Resistance genes found with SUPSETS are indicated. Corresponding information about the resistome for blood culture and analysis of other clinical specimens from the patient are derived from Grumaz et al (2019) [21]. -, no resistance found.
Table 1.
Antimicrobial resistances detected in clinical samples. Resistance genes found with SUPSETS are indicated. Corresponding information about the resistome for blood culture and analysis of other clinical specimens from the patient are derived from Grumaz et al (2019) [21]. -, no resistance found.
| Sample | SUPSETS | Blood culture | Clinical specimen |
|---|---|---|---|
| FC_06 | vanA | - | Vancomycine |
| FL_01 | vanA | - | Vancomycine |
| FL_02 | vanA, ermB | - | Vancomycine |
| FL_03 | tetB | - | - |
| FL_04-single | tetB | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



