
Submitted:
21 April 2024
Posted:
22 April 2024
You are already at the latest version
Abstract
Advanced oxidation processes, including photocatalysis, have been proven effective at organic dye degradation. Tailored porous materials with regulated pore size, shape and morphology offer a sustainable solution to the water pollution problem, by acting as support materials to grafted photocatalytic nanoparticles (NPs). This research investigates the influence of pore and particle sizes of photocatalytic MICROSCAFS®, on the degradation of methyl orange (MO) in aqueous solution (10 mg/L). Photocatalytic MICROSCAFS® are made of binder-less supported P25 TiO2 NPs within MICROSCAFS®, which are silica-titania microspheres with controlled size and intercon-nected macroporosity, synthesized by an adapted sol-gel method that involves a polymeriza-tion-induced phase separation process. Photocatalytic experiments were done both in batch and flow reactors, these latter ones targeting real-life conditions. Photocatalytic degradation of 87% in 2 hours (batch) and 29% in 5 hours (flow), was achieved, using a calibrated solar light simulator (1 sun) and a photocatalyst/pollutant mass ratio of 23. This study introduces a novel flow kinetic model which provides the modeling and simulation of the photocatalytic MICROSCAFS® per-formance. A scavenger study was performed enabling in-depth mechanistic understanding. Fi-nally, the transformation products resulting from the MO photocatalytic degradation were elu-cidated by high-resolution mass spectrometry experiments and subjected to in-silico toxicity as-sessment.
Keywords:
microspheres
; macroporosity
; sol-gel
; titania
; heterogeneous photocatalysis
; kinetics
1. Introduction
Water pollution is one of the biggest ecological problems that humanity currently faces. Synthetic dyes are used in many industries, such as textile processing, pharmaceutical sector, and food production, and approximately 80% of the resulting wastewaters are released untreated into the environment [1].
Water remediation is therefore one of the priorities in present-day scientific research. Among the explored methods for this purpose, heterogeneous photocatalysis offers many advantages, such as low cost, high flexibility of the process, the use of natural energy sources, relatively low quantity of required catalyst and, depending on the photocatalyst nature, high physical stability and recyclability of the materials employed, which make it an environmentally friendly technique [2,3,4]. However, the typical nanometric size of the photocatalysts, when used unsupported, in the powder form, makes complex their removal after the reaction, which is a significant drawback of this technique, being currently among the major limitations for the application of photocatalysis in a real-life scenario.
One solution for facilitating the photocatalysts recovery is their immobilization, preferably by grafting, into inert supports of relatively greater proportions. Binder-less processes enable to overpass issues regarding the decrease of reactive sites due to hydrophobic effects and low surface area that result from organic or inorganic binders [5]. They are also preferred to achieve stable supported photocatalysts.
Undoped TiO2 NPs, such as Aeroxide P25, is still a viable photocatalyst in today’s standards, due to its low cost and high commercial availability. TiO2 NPs have been supported in multiple materials like biochar [6], silica spheres [7], natural clays [8], cellulose nanofibrils [9], poly(methyl methacrylate) (PMMA) [10] and polyvinyl alcohol (PVA) films [11], and fly ash [12] to degrade MO. Additionally, TiO2 aerogel was deposited over silica-coated bacterial nanocellulose scaffolds with relatively fast photocatalytic degradation of methylene blue using UV-Vis light in a continuous flow reactor [13].
The majority of reported photocatalytic experiments is still made in batch conditions, which have some drawbacks if a real-life application is envisioned, like finite volume and non-continuous processing. Seldomly reported, the photocatalysts are susceptible to mechanical stress and eventual fracture caused by the stirring in the batch reactor, generating smaller particles and higher surface area, which overestimates the photocatalytic performance, and jeopardizes the claimed easy removal of the photocatalysts after a test. Also, porosity data, like specific surface area and pore diameter, are not properly discussed or even totally or partially missing in some papers in the literature [14,15,16,17,18,19,20,21,22], neglecting possible synergies and effects granted by the catalytic supports, in what regards e.g. mass transfer. Concerning the particle size, most of the reported microspherical supports are usually very small ranging from 0.5 to 3 µm [18,19,20,21,23,24,25] which may impose problems and extra complexity and costs in a real-life application, in what regards the set-up and recovery aspects.
Lucchini et al. [9] have employed a continuous-flow reactor design, with total recirculation to a tank, consisting of a cylindrical PMMA cell topped by a quartz window, where the simulated sunlight impinges at a constant irradiance. The reactor chamber was filled with a cellulose nanofibrils (CNF)-based monolith loaded with synthesized TiO2 NPs for the degradation of MO and paracetamol in liquid solution, which were made to circulate in a tube system connected to the bottom of the chamber and the tank. Then, in our previous study reported by Marques et al. [7], we applied the same reactor design and procedure using photocatalytic silica microspheres, instead of a CNF monolith. These silica microspheres, namely SiO2 MICROSCAFS®, were for the first time applied as supports for synthesized Trizma® functionalized TiO2 NPs. Their inorganic nature provides them high thermal and chemical stability.
Herein, we study in detail the effect of pore and particle sizes of supported photocatalytic systems, called photocatalytic MICROSCAFS®, in batch and continuous flow-type reactors, on the photocatalytic degradation of MO in aqueous solution (10 mg/L). The MICROSCAFS® in the present work are made of a multicomponent oxide composition, silica-titania (SiO2-TiO2 at ca. 80%-20% (molar)) and were obtained by sol-gel reaction inside the water droplets of a water-in-oil emulsion (W/O). As before, the tailored interconnected macroporosity of the MICROSCAFS® was achieved through polymerization-induced phase separation [26,27,28,29]. The resulting MICROSCAFS® were loaded with well-known, commercial P25 TiO2 NPs using a simple binder-less process in which the NPs were at first physically entrapped inside the porosity of the MICROSCAFS®, forming a layer on the surface and inner pores of the full particle, followed by a covalent binding promoted by heat treatment. Therefore, photocatalytic MICROSCAFS®, in this paper, are composed of MICROSCAFS® with immobilized photocatalytic P25 NPs. These latter ones are the active phase of our photocatalytic system. A similar preparation was followed in our previous paper, including additional gold nanoparticles, but, contrary to the present paper, only addressed a batch reactor for photocatalysis experiments [30]. Moreover, a new kinetic model for the flow reactor is herein proposed, and the solar light-driven degradation of MO using the photocatalytic MICROSCAFS® is simulated using the proposed model and compared with experimental results. Finally, the MO degradation is followed by liquid chromatography-tandem high resolution mass spectrometry (LC-HRMS/MS) to identify and characterize its by-products, thus providing a deeper understanding of the degradation process.
2. Results & Discussion
2.1. Characterization of the MICROSCAFS® before and after Heat Treatment and Photocatalyst (P25 TiO2 NPs) Loading
MICROSCAFS® of silica-titania composition are herein shown to be a versatile type of materials that can be obtained with high reproducibility via a relatively simple two-step, fast and low energy, adapted sol-gel process. Their final characteristics, like pore and particle diameter, and mechanical properties have been recently reported to be easily tuned by changing the synthesis parameters [27]. In this work, we aim to assess the effect of pores size and particles diameter on the photocatalytic performance of P25 TiO2 NPs loaded MICROSCAFS®.
All the MICROSCAFS® particle size distributions before and after calcination (HT) are shown in Figure 1, obtained from the SEM images present in Figure S3. The average particle diameter tends to increase with the quantity of GPTMS, and the span or the dispersion was found to decrease, particularly when using +25% GPTMS (Table S1). GPTMS precursor slows the condensation reaction of the titania and silica precursors due to steric hindrance caused by the relatively big aliphatic chain. This might provide a steady formation of siloxanes and Si-O-Ti bonds inside the water droplets of the emulsion, producing particles with lower size dispersions. The diameters of S- particles were mostly within the range of 20 to 40 µm, which was expected due to the sieving procedure applied in this sample. As expected, after the first HT, most of the samples decreased in size, with sample P- being the only exception. Despite a non-linear change in the average diameter, the particle diameter mode did not change, meaning that the HT had no significant effect on its size, and the difference in the distribution might be due to sampling heterogeneity. This fact is linked to the lower porosity of the P- samples, demonstrated below, so that there are less pores to collapse during calcination.
Optical microscopy photographs of the P25 TiO2 NPs loaded and unloaded MICROSCAFS® used in this work are presented in Figure S4. A concentration of P25 TiO2 NPs equal to ~23 wt% was achieved in most samples, except for sample S-/HT/P25, which achieved 22 wt%, and sample P-/HT/P25, where such concentration corresponds to 25 wt%, being very close to the nominal concentration at 26 wt%. The different colors of the MICROSCAFS® after the HT are a direct result of the internal porosity of the samples, dictated by the different content of TEOS and GPTMS (Figure S4). Spheres with smaller pore sizes, namely P- sample, exhibit a darker color because of the entrapment, inside the pore network, of generated gases resulting from organics degradation.
The porosity exhibited by the dried, heat-treated and P25 TiO2 NPs loaded MICROSCAFS® was analyzed using SEM, MIP (Figure 2) and N2 adsorption-desorption isotherms (Figure S5). A higher quantity of GPTMS precursor yielded spherical particles with significantly greater macropore size and cumulative pore volume (Figure 2a), which means that the domains of the separated phases, oxide-based xerogel-rich phase (containing Si-O-Si, Si-O-Ti and Ti-O-Ti bonds) and water-rich phase, are larger. The presence of GPTMS simultaneously affected the phase separation and condensation processes, without requiring any phase separation inducer [27]. The N2 adsorption isotherms of all the dried MICROSCAFS® were of type III (IUPAC classification) further corroborating, in the present case, their macroporous morphology, exhibiting specific surface areas (SSA) that ranged from 4.19 to 46.10 m2/g (Figure S5a). Samples S- and P0 exhibit the largest SSA, suggesting the presence of mesopores, which is in agreement with the findings from MIP (Figure 2a). They were both prepared with the same amount of GPTMS, however sample S- shows less macropores, probably because of the larger content of surfactants (10 g), which might contribute to the disruption of the phase separation due to lower interfacial tension, breaking the skeleton particles as the macropores domains were forming. The presence of Pluronic® P123 is also responsible for the formation of mesopores. Additionally, this sample is found to consist of smaller particles with a rough and fragmented morphology (Figure S3 and S4). As expected, after the HT at 900 °C for 30 min, the pore diameters and cumulative pore volumes, as well as the SSA, decreased in almost all the samples (Figure 2b), suggesting the occurrence of some sintering or pore collapse, however, the presence of meso and macropores is retained (Figure S5b). On the other hand, the loading with P25 TiO2 NPs resulted in the appearance of a peak at the mesoporosity range, between 30 and 50 nm (Figure 2c), and an increment of the SSA (Figure S5c) and did not significantly affect macroporosity. The employed P25 TiO2 NPs have an average diameter of about 27.4 ± 0.9 nm (Figure S6), so that the interparticle spaces contribute to the mesoporosity exhibited by the P25 TiO2 NPs loaded MICROSCAFS®.
As for elemental composition, EDS data in Table S2 revealed that P+ sample is the one with a Si and Ti at. % closer to the nominal Si and Ti precursor molar ratio of 80/20. Also, it is clear that MICROSCAFS® present higher Ti/Si at. % ratios after loading with the P25 TiO2 NPs, as expected. Interestingly, P+ sample, the one exhibiting large interconnected macroporosity, has less loading of P25 TiO2 NPs, given by the lower Ti/Si at. % ratio, compared to P- and P0 samples, for the same loading conditions. This might be due to the facilitated flow of the P25 TiO2 NPs dispersion throughout the large pores, and therefore, less clogging of the pores, which may be beneficial for the wastewater flow throughout the MICROSCAFS® in the photocatalytic tests.
The FTIR spectra of the MICROSCAFS® after drying at 45 °C (Figure S7a,b) contain multiple bands characteristic of the oxides network, but also of other organic compounds from the reactional mixture, including some residues of precursors (alkoxides), surfactant and decalin. The band at 1267 cm-1 is characteristic of the epoxy group of the GPTMS [31,32], however the peaks at 906 and 850 cm−1, ascribed to C–O and C–O–C stretching of the GPTMS’ epoxy (oxirane) are hard to detect because they might be hidden by the intense band at 915 cm-1 (Si-OH, or Si-O- from Si-O-Ti units). After the heat treatment at 900 °C (Figure S7c,d), the organic groups completely disappeared. The intensity of the band at around 934 cm-1, which includes the silanol (OH groups) vibrations, becomes lower, probably because of the OH elimination during heat treatment and some degree of phase separation, i.e. Si-O-Ti conversion to Si-O-Si and Ti-O-Ti links. An intense band at 1065 cm-1 is related to Si-O-Si asymmetric stretching vibration. After loading with P25 TiO2 NPs (Figure S7e,f) the I(Ti-O-Ti, 450 cm-1)/I(Si-O-Si, 1065 cm-1) ratio increased in all samples (Table S3), as expected, meaning that higher amount of titania is present in the photocatalytic MICROSCAFS®. Again, the higher the pore size (P+/HT/P25), the lower the amount of Ti (TiO2), corroborating the EDS results (Table S2).
2.2. Photocatalytic Studies on the P25 TiO2 NPs Loaded MICROSCAFS®
Photolysis of MO resulted in no degradation over the 2 hours of light exposure, whereas the photocatalytic P25 TiO2 NPs (non-supported) were able to fully degrade the pollutant in the same period (Figure 3a). No adsorption was detected for the photocatalytic NPs, nor for the heat-treated unloaded MICROSCAFS®. Also, these latter ones exhibited no significant photocatalytic activity (Figure 3a), which means that TiO2 NPs are the active phase of our photocatalytic system.
Regarding the loaded MICROSCAFS®, photocatalytic degradation data using the batch reactor showed that overall, larger pores proved to be better, with sample P+/HT/P25 degrading 87% of the MO dye in 2 hours of exposure to simulated solar light (Figure 3b). Larger interconnected pores improved the accessibility and the diffusion of both the reactants and products to and from the active sites, speeding up the reaction and promoting the photocatalytic degradation of MO species. Also, similar results are achieved for P0/HT/P25 and S-/HT/P25, which reveals that the difference in size of these particles does not play an important role in the photocatalytic performance.
The MO UV-Vis spectra had two major absorption bands, around 464 and 271 nm, which are from the aromatic ring in the MO molecule (Figure 3c) [33]. UV-Vis absorption spectra intensity at ~464 nm decreased as the reaction proceeded during light exposure, suggesting some degree of degradation. The characteristic orange color of the solution clearly faded over time with the exposure to the simulated solar light, proving the photocatalytic MICROSCAFS®’s efficacy (Figure 3d).
The presence of silica in the MICROSCAFS® hindered the absorption of the photocatalyst (Figure S8). Its effect was higher for sample P+/HT/P25 sample which had the highest amount of Si atoms by EDS (Table S2). Silicon dioxide is known for its wide bandgap of ca. 9 eV, which plays the effect of an energetic barrier for the solar radiation (in particular visible light), thus not generating excited electrons and holes, which inhibits photocatalytic activity [34]. On the other hand, the photocatalytic activity exhibited by sample P+/HT/P25 was the highest, demonstrating that other factors such as the presence of wider and interconnected pores, played an important role in the achievement of photocatalytic activity. The presence of pores has been reported to increase the absorbance of the light in e.g. aerogels [35]. In this case, it has been reported that the pore network traps the photons, which then diffuse over a few millimeters and thus increase the probability of the light to be absorbed. We propose that a similar effect might be happening with our MICROSCAFS®. Compared to a dense bead, the macroporous network extends the pathway of the photons, increasing their probability to be absorbed by P25 TiO2 NPs located in the inner surface of the pores, thus yielding higher photocatalytic activities.
Regarding the tests in the continuous flow reactor, the same parameters (pH, irradiance, temperature, and photocatalyst/pollutant mass ratio) were employed and indeed, the same tendency was observed as in the batch reactor: the greater the interconnected pores size (P+/HT/P25), the higher the pollutant (MO) degradation. MO suffers a 29% degradation within 6 hours (Figure 4a) using P+/HT/P25 as photocatalytic MICROSCAFS®.
It should be noted that the reaction was significantly slower than that in the batch reactor, which is explained by the different reactor designs. First, the flow reactor allows for a much smaller exposure area (3.14 cm2) to solar light than the batch reactor (12.57 cm2). In the flow reactor, the pollutant solution passes under the quartz glass, inside the chamber, going through the thin layer of porous photocatalytic MICROSCAFS® (Figure S9). Despite the thin layer of MICROSCAFS®, of only ca. 1.5 mm thickness, the particles at the top, closer to the quartz glass, are more exposed to the solar light than the particles beneath, whereas in the batch reactor, all the particles are constantly exposed to the light, in a free flow fashion. In contrast, in batch, all the MO solution is in permanent contact with the light and photocatalyst over the full extent of the reaction. Moreover, the fraction of the MO solution exposed to the solar light in the flow set-up, and therefore partially degraded, returns to the intercalated tank, that contains MO solution in a higher concentration. On the other hand, the continuous flow system is somewhat similar to a real case scenario, and greatly facilitates and enables the recyclability of the photocatalytic MICROSCAFS®, preserving their mechanical integrity during numerous cycles (does not involve stirring) and their easy removal/collection. Figure S10 shows samples P-/HT/P25 and P+/HT/P25 after a photocatalytic test in flow and batch set-ups. Compared to the SEM images of Figure S3, we can see that the denser (stiffer [27]) sample, P-/HT/P25, is fully preserved whatever the set-up employed for photocatalysis, whereas the largely porous P+/HT/P25 MICROSCAFS® are fully preserved after the continuous flow tests but tend to fracture when stirring at a high rate is applied. The higher compressibility, or flexibility, of the P+/HT/P25 spheres did not influence the photocatalytic efficiency because the same trend occurred both in batch and in flow. Sample S-/HT/P25 was not studied in the flow reactor due to its relatively small particle diameter, which clogged the filter employed in the experiment.
As observed in Figure 4b, the final degradation of MO with P+/HT/P25 increased slightly after the first cycle and stayed relatively stable during the subsequent five tested flow cycles. The UV-Vis absorbance spectra in Figure 4c and d were very similar from the 2nd cycle onward, whereas in the 1st cycle, not only the 271 nm band absorbance is higher, but it is shifted to lower wavelengths, suggesting a (minor) release of TiO2 NPs, probably those in excess and not covalently bonded to the MICROSCAFS® surface. Also, the lower MO degradation achieved in the 1st cycle might have been due to some congestion of the pores of the P+/HT/P25 sample (Figure 4b), which after the 1st cycle does not happen anymore due to the release of the NPs in excess. In this sense, a better flow throughout the MICROSCAFS® and more access to active sites is promoted, leading to the observed better performance after the 1st cycle. On the other hand, we cannot disregard variability issues, and such difference in MO degradation for the various cycles could be simply within the error of the analyses. Finally, the flow reactor was more suitable for the recyclability studies than the batch reactor since the same particles stayed inside the reactor in every cycle without the need for any separation process, like centrifugation or filtration, which avoided the loss of some photocatalyst mass inherent to these separation processes.
2.3. Kinetics Studies
The kinetics of MO degradation was assessed using a small laboratory batch reactor with stirring, in which the approximation to perfect mixing inside the reactor, is considered very reasonable. The MO concentration evolution with the reaction time (t) is fitted by Equation 1, stated in the Materials and Methods section. The results of the corresponding adjustments are shown in Figure 5, where the quality of all the fittings reinforces the fact that the degradation of MO occurs according to a first-order reaction (A → B).
It is worth noting that the P+/HT/P25 sample shows the highest reproducibility among all the photocatalytic MICROSCAFS®. This is possibly due to the large pore size, which possibly enabled well-distributed and dispersed photocatalytic NPs and ensured a good flow of the pollutant solution, increasing the contact between active sites and pollutant species and avoiding the formation of concentration gradients.
Regarding the continuous flow set-up, whose diagram is shown in Figure 11, and taking into account the considerations well-described in the Materials and Methods section, the modeling and fitting of the MO concentration evolution with the reaction time (t) was carried-out, using Equations 2 and 3.
The best adjusted models of the flow reactor are displayed in Figure 6 in triplicate for the photocatalytic MICROSCAFS® with different porosities.
Table 1 lists the average kinetic rate constant values, obtained from experiments in batch conditions, and the average apparent flow (adjusted) kinetic rate constant values, obtained from experiments in flow conditions for the photocatalysts under study. The constant obtained from the batch reaction studies (min-1) was multiplied by the batch reactor volume (50 mL) and divided by the mass of active phase to be comparable with the apparent flow kinetic rate constants (expressed in mL min-1 mg-1). In this way the kinetic constants better characterize the activity of the catalyst.
The MO degradation graphs from the flow reactor modeling (Figure 6) clearly show that the adjusted model is able to describe very well the experimental data during the timeframe of the experiment (6 hours of reaction). In this case, the fitting process shows that the apparent velocity constant values, kapp, are lower than those (k) obtained at the batch experiment (Table 1), which is also in agreement with Figure 3b and Figure 4a.
Since the reaction in study is a photocatalytic heterogeneous transformation, the reactant has to be adsorbed onto the active sites of the solid surface of the photocatalyst and, simultaneously, the available light and effective irradiance at the solid surface is also essential for the reaction to proceed. There is probably a lower efficiency of mass transfer of the reactant from the fluid to the solid surface and a less favored interaction of the radiation with the active sites of the catalyst in the continuous flow set-up, when compared to the batch set-up. The photocatalytic MICROSCAFS® are densely packed inside the flow reactor chamber, whereas, in batch, they are free-flowing and well dispersed, facilitating their exposure to solar radiation. Also, the surface area exposed to the simulated solar light is much less than in the batch set-up. Thus, despite the benefit of the flow set-up for real-life applications, the observed reaction rate for the photocatalytic heterogeneous transformation is expected to be reduced, when compared to the batch set-up. A design optimization of the reactor targeting a larger surface area of exposure to solar light might approximate the flow reactor performance to that of the batch reactor.
A full degradation of the MO species present in solution was achieved for ca. 50 hours using our continuous flow set-up, which is slightly less than the model (Figure 7). This fact suggests that other effects not considered in the model might become relevant, particularly the effect of MO solution volume, which is made to decrease along the experiment due to removing aliquots, to ca. 13% of the initial volume, and possibly some evaporation. Additionally, the circular shape of the reactor chamber might be less favorable than, for instance, the well-known tubular reactors, these latter ones promoting better contact between the solid photocatalyst and liquid pollutant solution. Therefore, the probability for concentration gradients generation inside our flow reactor chamber might be considerable. For longer reaction times these effects might be significant, having a cumulative effect.
Table 2 compiles various supported photocatalyst systems in the literature and the corresponding rate constant values factorized to the reactor volume and mass of the active phase. The relevant experimental conditions are described as well. We have only taken into consideration literature works where the photocatalytic MO degradation was carried out using supported TiO2 NPs [6,7,8,9,10,11,12,36].
We found that the kapp achieved in the current work, for continuous flow experiments, was higher than that achieved for SiO2 MICROSCAFS® ((4.19 ± 0.7) × 10-3 vs. 1.36 × 10-3) [7], even for a smaller photocatalyst/pollutant mass ratio (23 vs. 33). It is suggested that the small extra amount of TiO2 already present in the SiO2-TiO2 MICROSCAFS® of the current work might contribute to a more efficient photocatalytic process, as well as the larger interconnected macropores which have a size distribution peaked at ~1.95 µm (almost 20 times higher than the reported for SiO2 MICROSCAFS®). Also, as an extra variable, SiO2 MICROSCAFS® had been loaded with synthesized TiO2 NPs (anatase) and not P25 TiO2 NPs, contrary to SiO2-TiO2 MICROSCAFS® where P25 TiO2 NPs were employed. Ljubas et al. have reported higher photocatalytic activity achieved by P25 TiO2 than anatase [37]. Compared to a work which used a similar continuous flow reactor and cellulose nanofibrils supports [9], the MICROSCAFS® are still behind, possibly because of their lower transparency to the UV-Vis radiation. However, in what regards chemical and mechanical resistance and durability, MICROSCAFS®, due to their inorganic nature, are expected to display a better performance.
Notably, the present work fits the experimental photocatalytic behavior in flow using the equations for the specific employed continuous flow set-up, which is a more accurate methodology to calculate kapp, than using a batch reactor as a model. Another best practice of the present work is the type of solar source and conditions employed. The 1 sun AM 1.5G reference spectrum was used as standard for the photocatalytic tests in our work. Also, full experimental conditions under which the photocatalytic tests were carried out were revealed, with special attention for the active area of the device, the calibration protocol and properties of the illumination including spectral irradiance and intensity. Other works herein referred for comparison used unfiltered Hg lamps or even UV lamps, not mimicking sunlight, which does not really comply with the critical need of using solar light in a real application, to minimize energy consumption. Some other works [8,36] used Xe lamps without filters, which emit more UV radiation than the present work lamp, which might explain the higher degradations and photocatalytic activities reported for TiO2 NPs. P25 TiO2 when immobilized on a PVA film [11] has demonstrated higher activity than the present work, however the photocatalyst/pollutant mass ratio was three times higher, a UV radiation source was used and pH value was not disclosed, which is known to greatly affect the photocatalysis activity, with acidic media being more favorable in this case. For instance, works [6,8,10,12,36], where a batch set-up was employed, did not refer experimental conditions, such as the irradiance, which is crucial to compare results. Nevertheless, it is worth noting that the present work still yielded higher reaction rate constant than some UV-irradiated batch systems [10,12]. Systematic computational investigations on TiO2 NPs [38], have revealed that thermal annealing leads to amorphization (disorder) of the anatase NPs´ surface, inducing valence band edge broadening and higher photoactivities. In our work, besides thermal annealing at 500 °C, P25 TiO2 NPs are grafted to the silica-titania MICROSCAFS® and, therefore, in very close contact with amorphous, low coordination, titania species existent in the MICROSCAFS®. This, together with the wide and interconnected macropores characteristic of the MICROSCAFS®, might suggest a reason for the relevant photocatalytic MICROSCAFS® activity, achieved in the present work, when exposed to solar radiation, which includes only a very small fraction of UV radiation.
2.4. Study of the By-Products from Photocatalytic MO Degradation
The transformation products (TPs) formed during the photocatalytic degradation of MO using sample P0/HT/P25 were analyzed by LC-HRMS/MS. Figure 8 shows the formation of MO TPs over irradiation time in the presence of the photocatalytic MICROSCAFS® P0/HT/P25 using a batch (Figure 8a) and flow (Figure 8b) reactor. The analysis was carried out up to a solar exposure time of 120 min (batch) and 360 min (continuous flow), which corresponds to a partial MO degradation at 72 % and 27 %, respectively (by UV-Vis).
Five by-products were identified in total, whose chemical structure is included in Table 3, in agreement with a degradation mechanism proposed in the literature [39,40,41,42]. More specifically, five by-products were found using the batch reactor, where a more extended MO degradation process was achieved, and three of them using the flow reactor. The proposed ionic formulas for the TPs (Table 3) were supported by tandem mass spectrometry experiments, and the fragmentation pathways, for the precursor ions of MO and their TPs, are presented in SI (Figures S11 – S16). TP320-a and TP320-b are isomers, although showing different fragmentation patterns (Figures S12 and S16). These isomers are formed because the MO molecular structure contains two benzene groups, and the addition of a hydroxyl radical can occur in two positions.
Figure 8a shows that MO degradation (in the batch reactor) starts with its partial conversion to the compounds TP290 and TP320-a, following a similar pattern to a previously reported one [30], whose amount starts to decrease after around 60 min of exposure to simulated solar light. The formation of these by-products, TP320-a and TP290, occurs due to the addition of a hydroxyl radical at a benzene group of MO, and to the cleavage of a methyl in the dimethylamine group of MO, respectively. After 30 min of irradiation the formation of the compounds TP306 and TP320-b were detected, due to the cleavage of a methyl in the dimethylamine group of TP320-a, and isomerization of TP320-a, respectively. In the meantime, the loss of both methyl units of the dimethylamine group of MO occurs, and the formation of the compound TP276 was observed. It should be noted that the formation of the compounds TP320-b and TP276 was in a very small amount. Although it was not possible to remove all the TPs, a decrease was observed after around 60 min of irradiation. After 120 min of irradiation, we still identify residual MO species, which were expected because of the incomplete MO degradation at this stage, together with the just mentioned 5 TPs in solution. Therefore, a longer irradiation time will be necessary to achieve the complete degradation of both MO and by-products.
As for the flow reactor results (Figure 8b), it should be noted that only the initial stages of MO degradation are being analyzed, i.e. until 27% MO degradation. Indeed, MO degradation was found to be slower than in batch conditions, which might explain the lower number of TPs (TP290, TP320-a and TP276) identified in this case. So, it is found to begin with MO´s conversion to compound TP290, followed by the formation of compound TP320-a after 120 min of irradiation. After 150 min of irradiation compound TP276 appears in a very small amount. After 360 min of irradiation, which corresponds to 27% MO degradation (by UV-Vis), we still have TPs and MO species in solution, as expected, suggesting that a longer irradiation period is required for the complete degradation of MO. Comparing the same level of MO degradation at 27%, determined by UV-Vis spectroscopy, i.e. 30 min (batch) and 360 min (flow), a similar trend is observed, with the same TPs being identified, except for TP306 (that appears just in batch).
Finally, regarding toxicity of the TPs, Ecological Structure Activity Relationships (ECOSAR) prediction was carried out, and the results are presented in Table 4 [46]. As can be seen all the TPs are harmless for fish and for green algae (except TP276) in terms of acute toxicity. Regarding chronic toxicity for fish species, TP290 remains harmless, TP320-a, TP320-b and TP306 are harmful, whereas the by-product found in residual quantity (TP276) is toxic. For green algae, TP320-b, TP306 and TP290 are harmless, while the remaining TPs are harmful, as MO is. It should be stressed that the by-product found in higher amount, TP290, is a non-toxic (harmless) by-product for fish, daphnid and green algae, in what regards both acute and chronic toxicity.
2.5. Scavenger’s Study on the Photocatalytic MICROSCAFS® and Photocatalysis Mechanistic Study
To go further in this study and to analyze whether the MO photocatalytic degradation takes place via oxygen radical species, such as •O2−, •OH or via direct positive holes (h+) or electron (e-) transfer, radical scavengers were added to the MO aqueous solution to remove the corresponding reactive species and compared with the photocatalytic reaction without scavenger (Figure 9). In particular, BQ, EtOH and EDTA were used as •O2−, •OH and h+ scavengers, respectively. Considering the photocatalytic performance results discussed above, the P+/HT/P25 sample was chosen for this study. The obtained results revealed that using EtOH, an •OH scavenger, no significant differences in the pollutant removal were observed. On the other hand, the most pronounced photodegradation suppression can be seen when BQ was added to the MO solution. The addition of this well-known •O2− scavenger, during MO photocatalytic degradation, reduced by around 49% the MO removal (absolute value). On the other hand, the degradation of MO was enhanced in the presence of EDTA. When this h+ scavenger was added to the system an increase by 22% (absolute value) in the MO removal was obtained comparing to the case without scavengers. Similar results were already published by other authors, such as Liu et. al. [43] who used EDTA as a scavenger in the degradation of dyes. This obtained result can be justified because EDTA acts both as a h+ scavenger and also an e- donor [44]. In this sense, the recombination of the e-/h+ was reduced, since the EDTA was used as a scavenger to quench h+, and consequently more e- can react with the O2 present at the surface to produce •O2−. Furthermore, we could see that the superoxide radicals, •O2−, are the main oxidant species involved in this photocatalytic process. Therefore, in the presence of EDTA we have more e- in the system able to provide O2/O2•− reduction in the conduction band (CB), enhancing the MO degradation under solar irradiation. Therefore, we can conclude that MO degradation does not take place directly through •OH and h+, but through •O2− which are the main oxidant species involved in this photocatalytic process.
A mechanism for the solar light activation of the P+/HT/P25 MICROSCAFS® is proposed in Figure 10. When the MICROSCAFS® are exposed to solar radiation with energy enough to promote the photogeneration of charge carriers, e- and h+ will be generated in the CB and valence band (VB), respectively. The excited electrons in the CB will react with the adsorbed O2 and reduce them into •O2−. This superoxide radical plays an active role in the photodegradation of MO as concluded by the scavenger’s study, and also contributes to a longer lifetime of the charge carriers. Furthermore, the •O2− can also produce H2O2 derived from the O2 photo-reduction that also enhances the photodegradation of MO. On the other hand, in the VB the photogenerated h+ react with either the adsorbed H2O or OH−, resulting in the formation of •OH radicals. These reactive species do not play an active role in this photodegradation system; however, they are responsible for the formation of two TPs detected by HRMS, the isomers TP320-a and TP320-b, formed due to the hydroxylation of MO. In our previous work we also observed a hydroxylation of the antidepressant amitriptyline forming three isomers, during their photocatalytic degradation, due to the addition of hydroxyl radicals in different positions on the amitriptyline molecule [45]. Similar results were also published, during the photodegradation of several compounds, such as sulfaclozine, sulfonamides and sulfachloropyridazine, reporting that the photocatalytic degradation of the studied pollutants starts with their hydroxylation, due to the addition of •OH to the aniline ring [46,47,48,49].
3. Materials and Methods
3.1. Materials
Tetraethyl orthosilicate (TEOS, 98%, Sigma-Aldrich, USA), (3-glycidyloxypropyl) trimethoxy silane (GPTMS, Xiameter OFS-6040, >98.5%, kindly supplied by Dow, USA), titanium (IV) isopropoxide (TiPOT, 98%, Acros Organics, USA), glacial acetic acid (≥99%, Fisher Chemical, USA), decahydronaphthalene (decalin, mixture of cis and trans isomers, 98%, Merck, Germany), sorbitan monooleate nonionic biodegradable surfactant (Span® 80, HLB: 4.3, Merck, Germany), Pluronic® P-123 nonionic surfactant, a copolymer comprising poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) in an alternating linear fashion (HLB: 8, Sigma-Aldrich, USA), ammonia 25% aqueous solution (Chem-Lab, Belgium), Aeroxide® P25 Degussa TiO2 NPs (Acros Organics, USA), methyl orange (MO, Fisher Chemical, USA), p-benzoquinone (BQ, Merck, Germany), ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA, Panreac) and ethanol (EtOH, ≥99.8%, Fisher Chemical, USA).
3.2. Synthesis of the MICROSCAFS®
MICROSCAFS® were synthesized using the same method recently published by our research group [27,50]. It consisted of two main steps: step 1 – silane precursors hydrolysis and simultaneous complexation of the titania precursor, and step 2 – alkaline condensation in the water droplets of the emulsion medium and MICROSCAFS® formation.
In step 1, 16.3 mL of TEOS, 13.8 mL of GPTMS, and 11.6 mL of HCl 0.28 M (aq. solution) were mixed in a closed container under constant vigorous stirring (hydrolysate solution), for 65 minutes. In parallel, 10 mL of TiPOT and 8 mL of glacial acetic acid were mixed in a different container and left under stirring at room temperature (RT) for 45 minutes.
In step 2, the mixture of both hydrolysate solution and chelated TiPOT solution (silica-titania precursor solution) was transferred to a three-neck round bottom flask, which contained a W/O emulsion, at 50 °C. The emulsion consists of 45 g of water, 114 mL of decahydronaphthalene, and 6 mL of Span® 80 previously mixed using a high energy dispersing instrument IKA T18 digital ULTRA-TURRAX® (IKA, Germany) at 13 000 rpm for 10 minutes. This was followed by the addition of 16 mL of ammonia 25% aqueous solution at 600 rpm of mechanical stirring. Finally, after 1 hour and 30 minutes, the formed MICROSCAFS® were collected through vacuum assisted filtration with acetone and then dried at 45 °C overnight. The MICROSCAFS® synthesis was relatively straightforward and quick (2 hours and 35 minutes) and it required no more than 50 °C to reach solid porous microspheres.
Four different MICROSCAFS® were prepared and studied in this work. Sample “P0” followed the previous procedure as described and was considered the reference in this paper. Samples “P-” and “P+” were synthesized using lower and higher volume of GPTMS than the reference, respectively. Sample “S-” was synthesized using a higher volume of Span® 80 together with another surfactant, Pluronic® P-123, both added to the water phase of the emulsion. Table 5 lists the four different samples of study, and corresponding variable synthesis parameters.
3.3. Preparation of the Photocatalytic MICROSCAFS®
All the four samples of MICROSCAFS® were used as a scaffolding or support to commercial Aeroxide® P25 Degussa TiO2 NPs using a wet impregnation method adapted from [7], forming the photocatalytic MICROSCAFS®. First, all the MICROSCAFS® were heat-treated for 30 min at 900 °C, to confer an inorganic nature to the support material and provide higher mechanical, chemical and thermal stability during the final application. These particles are indicated by the acronym “HT” throughout this paper. Sample S-/HT was sieved and the fraction between 25-45 µm of diameter was collected and used in the subsequent studies. All the other samples were sieved <354 µm to ensure the absence of any agglomerate of MICROSCAFS® in the tests. Then, 372 mg of MICROSCAFS® were placed in 2.1 mL of a 62.5 mg mL-1 aqueous dispersion of P25 TiO2 NPs in a small glass vial, targeting at a loading of 26wt% relative to the total weight of MICROSCAFS® and P25 TiO2 NPs. This dispersion was submitted to ultrasound sonication for 2 minutes, dried at 60 °C for 15 h, and finally subjected to a heat treatment at 500 °C for 1 hour. P25 TiO2 NPs are driven into the pores of the MICROSCAFS® by capillary action, and chemical bonds (e.g. Si-O-Ti) are formed during the subsequent heat treatment. These particles are indicated by the acronym “P25” throughout this paper.
3.4. Characterization
The morphology and the elemental composition of the MICROSCAFS® were evaluated through scanning electron microscopy (SEM) images and energy-dispersive X-ray spectroscopy (EDS) data acquired using a Phenom ProX G6 benchtop SEM (ThermoScientific, USA). Internal porosity was observed on purposedly broken particles. Prior to observation, a 15 nm gold–palladium layer was sputtered onto the samples using a turbomolecular pumped coater Q150T ES from Quorum Technologies (Lewes, UK). The MICROSCAFS® diameter was measured manually using the ImageJ software as previously reported [27]. The transmission electron microscopy (TEM) images of the P25 TiO2 NPs (shown in supplementary information) were obtained using a microscope (H8100, Hitachi, Japan) operating at 300 kV.
Dried, P25 TiO2 NPs loaded and unloaded MICROSCAFS®’ porosity was assessed by mercury intrusion porosimetry (MIP) using an Autopore IV 9500 Mercury Porosimeter (Micromeritics, USA) and N2 adsorption/desorption isotherms using an ASAP 2010 adsorption analyzer (Micromeritics, USA), in the same fashion as in [27].
The chemical structure of the samples was assessed through Fourier-transform infrared spectroscopy (FTIR) using a Spectrum Two spectrometer (PerkinElmer, USA) coupled with a universal attenuated total reflectance (ATR) accessory (PerkinElmer, USA). The spectra were obtained at 4 cm−1 resolution, and 8 scans of data accumulation.
The UV-Vis diffuse-reflectance spectra (UV-Vis DRS) was measured by a V-750 UV-Vis spectrophotometer (JASCO, USA) equipped with an integrating sphere and converted in absorption units (FKM) using the Kubelka-Munk function, which is related to the diffuse reflectance by the expression FKM(R)=(1-R)2/2R [51,52,53].
3.5. Photocatalytic Tests
The photocatalytic MICROSCAFS® were tested in a batch and continuous flow reactor. The batch reactor was a 100 mL jacketed vessel connected to a water recirculatory bath at 19 °C. In each batch test 50 mg of photocatalytic MICROSCAFS® were dispersed in 50 mL of a MO aqueous solution with an initial MO concentration of 10 mg/L and pH of 7, under magnetic stirring. The system was kept in the darkness for 1 hour, to study possible adsorption, followed by illumination during 2 hours with a solar lamp. For this purpose, a Newport 94011A-ES solar simulator (Newport Corporation, USA) was employed, with a 100 W Xe lamp with reflector (Newport Corporation, USA), that produces a 3.8 cm x 3.8 cm collimated beam. This solar simulator includes an AM1.5G air mass filter which provides Class A spectral performance based on current applicable standards. The solar lamp’s distance from the solution’s surface was adjusted to correspond to the irradiance of 1 sun (1000 W/m2). The simulated solar light goes inwards the reactor through a quartz window of 4 cm diameter (12.57 cm2) placed at the top of the reactor. The transmission spectrum of the quartz window is shown in the Supporting Information (Figure S1). It transmits at ca. 92% from 380 to 900 nm. Aliquots of 1 mL were taken every 20 minutes during the dark phase and every 15 minutes during the light phase and centrifuged. The MO solution concentration at each specific time was then determined by measuring the absorbance at 464 nm using a JASCO V-750 UV-Vis spectrophotometer (JASCO, USA). All the experiments were conducted for three replicates at 19 °C, and the mean results were reported.
Additionally, photocatalytic experiments (in batch) were carried out using distinct radical scavengers. EtOH was employed as •OH scavenger in the photocatalysis reaction, EDTA was chosen as h+ quencher, and BQ was added to the system as •O2− scavenger. The MO photocatalytic experiments were performed using the same conditions described above while using separately 0.5 mM of each scavenger.
The flow tests were carried out using a set-up built to allow a continuous flow with recirculation to an intercalated tank (Figure S2). The photoreactor employed was adapted from the one previously reported [7,9]. This reactor comprises three separate acrylic pieces that, when assembled, form a chamber (solar reactor) where the photocatalytic MICROSCAFS® sample can be placed, and the solution can circulate throughout the sample. The top of this chamber displays a quartz window of 2 cm of diameter (3.14 cm2) and transmission spectrum shown in Figure S1, on which the solar light will impinge, and the back contains the inlet and outlet of the circulating solution. 200 mg of photocatalytic MICROSCAFS® were inserted and compressed inside this chamber, around ~1.5 mm thick, resulting in a chamber volume of 0.47 cm3. We placed a filter paper (1318 from FILTER-LAB®, Spain) at the back of the chamber, where the inlet and outlet of the circulating solution were placed, to prevent the MICROSCAFS® from exiting the chamber. The reactor was connected to a tank with two 78 cm long TYGON® hoses (Hirschmann Laborgeräte, Germany), and 200 mL of MO solution (10 mg/L) were made to circulate at a constant flow at 10 mL/min with the help of a Rotarus® standard 50 peristaltic pump (Hirschmann Laborgeräte, Germany) during the tests. The reactor was exposed to a constant 1000 W/m2 (1 sun) at the exterior surface of the quartz window over 6 hours, after 1 hour of darkness, to account for any possible adsorption. The concentration of the pollutant was checked every 30 minutes by measuring the absorbance at 464 nm of 1 mL MO aq. solution aliquots. The same solar simulator and reference solar cell from the batch tests were used. All the experiments were conducted for three replicates at 25 °C, and the mean results were reported.
In the stability (recyclability) tests, six consecutive cycles were performed, by simply replacing, in the stirred tank, the degraded MO solution by a new one at 10 mg/L, and the corresponding MO degradation was recorded to examine the stability and reusability of the photocatalytic MICROSCAFS® at a catalyst/pollutant mass ratio of 23, pH of 7, under solar light (1 sun) exposure and 25 °C.
3.6. Kinetics Modeling of the Photocatalytic Performance
The kinetics for MO photocatalytic degradation was analyzed in batch and flow conditions. By assuming that it follows a first order transformation, with the reaction rate given by r = kCA, the MO concentration evolution with the reaction time (t) can be described by Equation 1:
where k is the kinetic rate constant, CA is the MO concentration inside the reactor at time t under light irradiation, and CA0 is the initial MO concentration before irradiation. The batch reactor is known to allow a good efficiency of mass transfer from the fluid to the surface of the solid photocatalyst, where the transformation takes place; for heterogeneous catalysis to occur, the MO pollutant dissolved in the liquid medium must contact and be adsorbed on the active sites of the photocatalyst.
The constant k was determined by fitting Equation 1 to the experimental points of CA as a function of time, using the sum of the square’s deviations minimization method.
To model the experimental MO photodegradation system using our continuous flow set-up (continuous flow with recirculation to an intercalated tank), the following process flow diagram, shown in Figure 11, was employed, exhibiting the small continuous flow chamber (solar reactor) (2) with complete recirculation. The reactor, exposed to the solar simulator (3), is connected to the stirred tank (4) with 200 mL of volume in a closed circuit pumped by a peristaltic pump (1) at a constant volumetric flow rate of 10 mL/min.
Figure 11.
Process flow diagram of the flow reactor. (1) Peristaltic pump; (2) continuous flow chamber (solar reactor, side view) containing the photocatalytic MICROSCAFS®; (3) solar simulator (Xe lamp), and (4) stirred tank.
Figure 11.
Process flow diagram of the flow reactor. (1) Peristaltic pump; (2) continuous flow chamber (solar reactor, side view) containing the photocatalytic MICROSCAFS®; (3) solar simulator (Xe lamp), and (4) stirred tank.
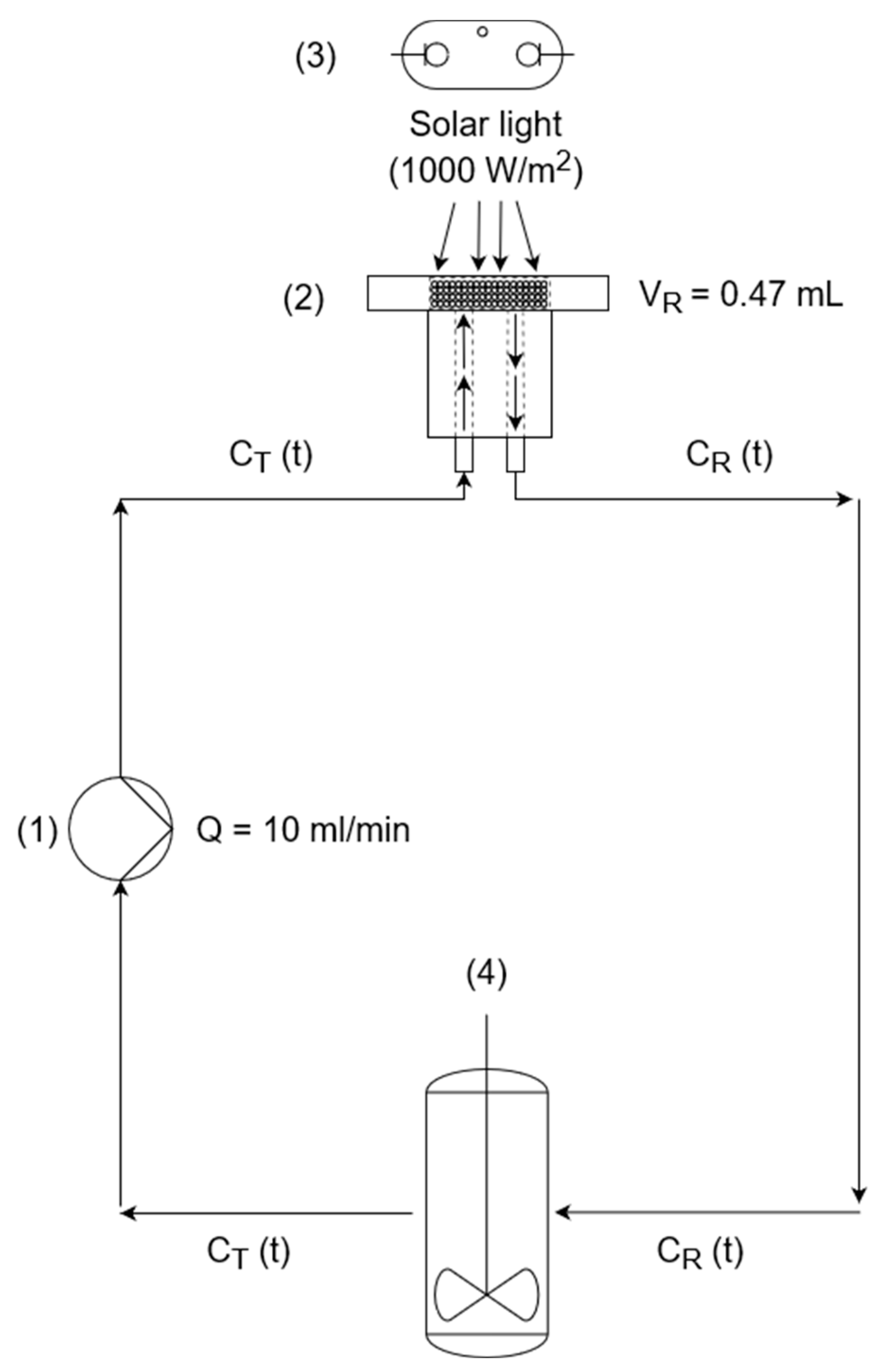
Homogeneous conditions were considered inside the solar reactor (chamber), which is a reasonable approximation when the conversion of the reactant, for each pass of a volume element of fluid through the reactor, is low. The inlet concentration of the solar reactor is the same as the outlet concentration of the tank (CT(t)), while the inlet concentration of the tank is equal to the outlet concentration of the solar reactor (CR(t)), both changing over time.
After the peristaltic pump starts, the MO solution enters the reactor and flows throughout the photocatalytic MICROSCAFS® interparticle spaces and internal pores. We consider that the flow solar reactor works in differential conditions, at constant flow and almost negligible concentration gradient inside, since the pollutant solution quickly flows through it (~2.8 s as mean residence time). The pollutant fluid stream leaving the reactor is mixed with the solution that remains behind in the dark, in the stirred tank (Figure 11).
The mass balance equations describing the transient behavior of the reactor, and of the tank where no reaction occurs, are given by Equations 2 and 3 respectively.
where VR is the volume of the flow reactor in L, VT is the tank volume in L, Q is the volumetric flow in L/min, CR(t) is the MO concentration in the reactor (mg/L), CT(t) is the MO concentration in the tank (mg/L) and W is the mass of the photocatalytic MICROSCAFS® placed inside the reactor (200 mg). The kinetic rate constant, k, is the variable parameter of this mathematical model.
The simultaneous resolution (integration) of Equations 2 and 3 was accomplished using the Euler method, allowing the determination of the evolution of CR(t) and CT(t). Several integrations were carried out until the best value of k is reached (kapp), yielding the lowest sum of the squared deviations between the experimental and model CT(t) values.
3.7. Analytical Methods for Photocatalysis By-Products Determination
The transformation products (TPs) formed during the photocatalytic degradation of MO were analyzed by LC-HRMS/MS. The MO solutions were analyzed on an UHPLC Elute system interfaced with a QqTOF Impact II mass spectrometer equipped with an ESI source, operating in the negative mode (Bruker Daltonics, Germany). Chromatographic separation was carried out under gradient conditions using a RF-C18 Kinetex column 100 Å (150 mm × 2.1 mm, 2.6 μm particle size, Phenomenex). Detailed settings on LC-HRMS/MS settings were described in our previous work [30]. The TPs peak areas (A) were normalized with respect to MO at t = 0 minutes (A/A0), and their variation as a function of irradiation time is shown in 3D bar graphs. The TPs are identified by HRMS based on their accurate m/z values released as deprotonated molecules ([M-H]−), considering the accuracy and precision of the measurement parameters such as error (ppm) and mSigma. The molecular formulas were validated by extracting the ionic chromatograms from the raw data, and accurate mass isotopic patterns and fragmentation paths were evaluated, supporting the respective proposed chemical structures. Detailed equipment specifications and experimental protocol for calibration, data acquisition and processing are described elsewhere [54].
Lastly, the toxicity of the TPs was assessed by in silico predictions using the free available tool Ecological Structure Activity Relationship (ECOSAR v1.11) to estimate ecotoxicity. From the ECOSAR prediction of toxicity, three different organisms were selected, fish, daphnid and green algae, for the acute and chronic ecotoxicity. Detailed specifications on toxicity prediction by ECOSAR are described in our previous work [55] for a different system and other chemical pollutants.
4. Conclusions
Silica-titania MICROSCAFS® with pore sizes at different scales, and different particle size diameters were successfully developed with high reproducibility using an adapted sol-gel method, involving polymerization-induced phase separation. They were loaded with commercial photocatalytic P25 TiO2 NPs, whose immobilization within the MICROSCAFS® was promoted by HT at 500 °C. MICROSCAFS® characteristics, mainly macropore size, directly affected the photocatalytic performance in what regards the organic dye MO degradation. MICROSCAFS® with greater interconnected pore sizes exhibited higher photocatalytic activity both in batch and in flow set-ups. Their engineered macroporous network favors mass transport, and therefore the flow of the pollutant solution throughout the pores, confers higher accessibility of the pollutant molecules to the active sites, and provides conditions to spread the photons´ pathway inside the pore channels. The size of the particles appears not to have such a relevant effect on the photocatalytic performance. MICROSCAFS® are herein shown to be a viable solution for environmental remediation (e.g. wastewater purification), using straightforward flow conditions, does not requiring any separation step, like centrifugation or filtration, which otherwise would be very expensive on an industrial scale, or of high complexity in remote areas such as rural villages. A modeling study of our flow set-up was developed and tested, which yields more accurate results and successfully predicts the reaction evolution. There are other works from the literature that have reported higher degradation rates than the present work, however the employed photocatalyst/pollutant ratio was generally higher and/or they used UV light (instead of solar light) which gives rise to high photo-efficiency, or relevant information on the experimental conditions was missing. Our process used solar light as the light source, which has only ~5% UV. Finally, the design of a new larger scale flow reactor, with higher photocatalyst/pollutant/light contact, is promising to further enhance photocatalytic activity and allowing the application of this system in an industrial scale, providing a viable solution for the global water pollution problem.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Transmission spectrum of the silica quartz window employed in the continuous flow reactor, and also to cover the batch reactor; Figure S2: Continuous flow reactor with total recirculation to a tank. 1 – Solar simulator;.2 – Sample chamber (solar reactor); 3 – Peristaltic pump; 4 – MO solution stirred tank; Figure S3: SEM images of the MICROSCAFS® used for the particle size distributions, before (left column) and after (right column) being heat treated at 900 °C for 30 minutes; Figure S4: Optical microscopy photographs of the MICROSCAFS® in three subsequent stages: 1 (first column) – dried MICROSCAFS®; 2 - (second column) MICROSCAFS® after being heat treated at 900 °C for 30 minutes; 3 (third column) – MICROSCAFS® after being loaded with the P25 TiO2 NPs; Figure S5. N2 adsorption-desorption isotherms of the MICROSCAFS® (a) dried at 45 °C, (b) heat-treated at 900 °C and (c) loaded with P25 TiO2 NPs; Figure S6: (a) TEM image of the P25 TiO2 NPs; (b) their respective particle size distribution; Figure S7. ATR-FTIR spectra of all the MICROSCAFS® (a, c and e) and respective magnification in the range of 1500 to 400 cm-1 (b, d and f); Figure S8: Kubelka-Munk transformed UV-Vis DRS absorption spectra of the P25 TiO2 NPs loaded MICROSCAFS® and P25 TiO2 NPs; Figure S9: Side and top views of the continuous flow reactor’s sample chamber; Figure S10: SEM images of the P-/HT/P25 (left column) and P+/HT/P25 (right column) photocatalytic MICROSCAFS® after one cycle in batch and in flow; Figure S11: Proposed fragmentation mechanism for the precursor ion m/z 304.0770, tR9.5 min, assigned to the deprotonated molecule of MO; Figure S12. Proposed fragmentation mechanism for the precursor ion m/z 320.0712, tR10.9 min, attributed to the deprotonated molecule of TP320-a; Figure S13: Proposed fragmentation mechanism for the precursor ion m/z 306.0562, tR9.0 min, attributed to the deprotonated molecule of TP306; Figure S14: Proposed fragmentation mechanism for the precursor ion m/z 290.0614, tR7.8 min, attributed to the deprotonated molecule of TP290; Figure S15: Proposed fragmentation mechanism for the precursor ion m/z 276.0453, tR5.8 min, attributed to the deprotonated molecule of TP276; Figure S16: Proposed fragmentation mechanism for the precursor ion m/z 320.0712, tR3.9 min, assigned to the deprotonated molecule of TP320-b; Table S1: Particle size distribution data of the treated and non-heat-treated MICROSCAFS®; Table S2: EDS atomic concentration data of the MICROSCAFS®. Ti/Si is the atomic % ratio; Table S3: FTIR peak intensity ratio ;
Author Contributions
Conceptualization, Mário Vale, Beatriz Barrocas, José M. Lopes and Ana C. Marques; methodology, Mário Vale, Beatriz Barrocas, José M. Lopes and Ana C. Marques; validation, Mário Vale, Beatriz Barrocas, José M. Lopes and Ana C. Marques; investigation, Mário Vale, Beatriz Barrocas, and M. Conceição Oliveira; resources, Ana C. Marques and M. Conceição Oliveira; writing—original draft preparation, Mário Vale and Beatriz Barrocas; writing—review and editing, Mário Vale, Beatriz T. Barrocas, Rita Serôdio, M. Conceição Oliveira, José M. Lopes, and Ana C. Marques; visualization, Mário Vale and Beatriz T. Barrocas; supervision, José M. Lopes and Ana C. Marques; project administration, Ana C. Marques; funding acquisition, M. Conceição Oliveira and Ana C. Marques. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundação para a Ciência e a Tecnologia (FCT) through the CERENA institution (Strategic Project FCT-UIDB/04028/2020), the PhD fellowships (SFRH/BD/138717/2018 and COVID/BD/152946/2022), the project http://doi.org/10.54499/PTDC/EQU-EQU/1056/2020 (SOLAR2CLEAN—Engineered solar light driven photocatalytic systems for wastewater purification), as well as the project RNEM-LISBOA-01–0145-FEDER-022125-IST, Centro de Química Estrutural (grant numbers UIDB/00100/2020, UIDP/00100/2020) and Institute of Molecular Sciences (grant LA/P/0056/2020).
Data Availability Statement
Data available on the Zenodo platform under the following DOI: https://doi.org/10.5281/zenodo.10613584.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors want to thank Prof. Markus Niederberger and his team (MFM group, ETH Zurich) for the continuous-flow set-up, which served as an inspiration for the one employed in this work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lin, J.; Ye, W.; Xie, M.; Seo, D.H.; Luo, J.; Wan, Y.; Van der Bruggen, B. Environmental Impacts and Remediation of Dye-Containing Wastewater. Nat Rev Earth Environ 2023, 4, 785–803. [Google Scholar] [CrossRef]
- Saravanan, A.; Deivayanai, V.C.; Kumar, P.S.; Rangasamy, G.; Hemavathy, R. V; Harshana, T.; Gayathri, N.; Alagumalai, K. A Detailed Review on Advanced Oxidation Process in Treatment of Wastewater: Mechanism, Challenges and Future Outlook. Chemosphere 2022, 308, 136524. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, A.; Iervolino, G. Synthesis and Application of Innovative and Environmentally Friendly Photocatalysts: A Review. Catalysts 2022, 12, 1074. [Google Scholar] [CrossRef]
- Rauf, M.A.; Ashraf, S.S. Fundamental Principles and Application of Heterogeneous Photocatalytic Degradation of Dyes in Solution. Chemical Engineering Journal 2009, 151, 10–18. [Google Scholar] [CrossRef]
- Park, S.; Choi, G.R.; Lee, J.C.; Kim, Y.C.; Oh, D.; Cho, S.; Lee, J.-H. Organic and Inorganic Binder-Coating Properties for Immobilization of Photocatalytic ZnO Nanopowders. Research on Chemical Intermediates 2010, 36, 819–825. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, A. Cornstalk Biochar-TiO2 Composites as Alternative Photocatalyst for Degrading Methyl Orange. Environmental Science and Pollution Research 2023, 30, 31923–31934. [Google Scholar] [CrossRef]
- Marques, A.C.; Vale, M.; Vicente, D.; Schreck, M.; Tervoort, E.; Niederberger, M. Porous Silica Microspheres with Immobilized Titania Nanoparticles for In-Flow Solar-Driven Purification of Wastewater. Global Challenges 2021, 5, 2000116. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wang, D.; Wei, C.; Zhang, X.; Liu, Z.; Feng, P.; Ou, X.; Qiang, Y.; Garcia, H.; Niu, J. A Comparative Photocatalytic Study of TiO2 Loaded on Three Natural Clays with Different Morphologies. Appl Clay Sci 2019, 183, 105352. [Google Scholar] [CrossRef]
- Lucchini, M.A.; Lizundia, E.; Moser, S.; Niederberger, M.; Nyström, G. Titania-Cellulose Hybrid Monolith for In-Flow Purification of Water under Solar Illumination. ACS Appl Mater Interfaces 2018, 10, 29599–29607. [Google Scholar] [CrossRef]
- Stewart, B.D.; Andrews, L.G.; Pelletier, B.S.; Daly, C.A.; Boyd, J.E. Porous PMMA-Titania Composites: A Step towards More Sustainable Photocatalysis. Journal of Water Process Engineering 2015, 8, 179–185. [Google Scholar] [CrossRef]
- Lei, P.; Wang, F.; Gao, X.; Ding, Y.; Zhang, S.; Zhao, J.; Liu, S.; Yang, M. Immobilization of TiO2 Nanoparticles in Polymeric Substrates by Chemical Bonding for Multi-Cycle Photodegradation of Organic Pollutants. J Hazard Mater 2012, 227–228, 185–194. [Google Scholar] [CrossRef]
- Ökte, A.N.; Karamanis, D.; Tuncel, D. Dual Functionality of TiO2-Flyash Nanocomposites: Water Vapor Adsorption and Photocatalysis. Catal Today 2014, 230, 205–213. [Google Scholar] [CrossRef]
- Almeida da Silva, T.C.; Marchiori, L.; Oliveira Mattos, B.; Ullah, S.; Barud, H. da S.; Romano Domeneguetti, R.; Rojas-Mantilla, H.D.; Boldrin Zanoni, M.V.; Rodrigues-Filho, U.P.; Ferreira-Neto, E.P.; et al. Designing Highly Photoactive Hybrid Aerogels for In-Flow Photocatalytic Contaminant Removal Using Silica-Coated Bacterial Nanocellulose Supports. ACS Appl Mater Interfaces 2023, 15, 23146–23159. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Dong, H.; Tapa, A.R.; Shamsi, J.; Shayeh, J.S.; Trokourey, A.; Liu, B.; Zhao, X.; Xie, Y. NaBr-Assisted Aqueous Synthesis of Perovskite-Embedded PbBr(OH) Hierarchical Nanostructures for Dye Photodegradation. ACS Appl Nano Mater 2023, 6, 2538–2548. [Google Scholar] [CrossRef]
- Wu, J.; Ke, K.; Qin, N.; Lin, E.; Kang, Z.; Bao, D. Magnetically Retrievable Fe3O4@SiO2@ZnO Piezo-Photocatalyst: Synthesis and Multiple Catalytic Properties. J Colloid Interface Sci 2023, 636, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Li, N.; Zuo, S.; Shen, W.; Wu, M.; Li, Q.; Shi, M.; Ma, J. Piezo-Photocatalysis over Phase-Engineered MoSe2 Modified Bi2WO6 Hierarchical Microspheres: Utilizing Piezoelectric Effect to Enhance Photocatalytic Performance. Ceram Int 2022, 48, 37242–37252. [Google Scholar] [CrossRef]
- Wang, J.; Sgarzi, M.; Němečková, Z.; Henych, J.; Licciardello, N.; Cuniberti, G. Reusable and Antibacterial Polymer-Based Nanocomposites for the Adsorption of Dyes and the Visible-Light-Driven Photocatalytic Degradation of Antibiotics. Global Challenges 2022, 6, 2200076. [Google Scholar] [CrossRef]
- Imran, M.; Ashraf, W.; Hafiz, A.K.; Khanuja, M. Synthesis and Performance Analysis of Photocatalytic Activity of ZnIn2S4 Microspheres Synthesized Using a Low-Temperature Method. ACS Omega 2022, 7, 22987–22996. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Jin, K.; Li, X.; Wang, R.; Zhao, Y.; Wang, H. Bi Nanosphere-Decorated Oxygen-Vacancy BiOBr Hollow Microspheres with Exposed (110) Facets to Enhance the Photocatalytic Performance for the Degradation of Azo Dyes. New Journal of Chemistry 2022, 46, 12410–12418. [Google Scholar] [CrossRef]
- Kim, J.; Jo, S.; Lee, W.; Lim, J.; Seung Lee, T. Moving Photocatalyst of a Titanium Dioxide-Based Micromotor Asymmetrically Decorated with Conjugated Polymer Dots. Mater Des 2022, 219, 110743. [Google Scholar] [CrossRef]
- Li, X.; Raza, S.; Liu, C. Preparation of Titanium Dioxide Modified Biomass Polymer Microspheres for Photocatalytic Degradation of Rhodamine-B Dye and Tetracycline. J Taiwan Inst Chem Eng 2021, 122, 157–167. [Google Scholar] [CrossRef]
- Liang, H.; Wang, S.; Lu, Y.; Ren, P.; Li, G.; Yang, F.; Chen, Y. Highly Efficient and Cheap Treatment of Dye by Graphene-Doped TiO2 Microspheres. Water Science and Technology 2020, 83, 223–232. [Google Scholar] [CrossRef]
- He, J.; Liu, Q.; Zhang, Y.; Zhao, X.; Zhang, G.; Xiao, B.; Fu, K. In Situ Synthesis of the Mesoporous C–TiO2 Microspheres Derived from Partial Hydrolysis Tetrabutyl Titanate for Enhanced Photocatalytic Degradation under Visible Light. Mater Res Bull 2023, 161, 112168. [Google Scholar] [CrossRef]
- Feng, X.; Gu, L.; Wang, N.; Pu, Q.; Liu, G. Fe/N Co-Doped Nano-TiO2 Wrapped Mesoporous Carbon Spheres for Synergetically Enhanced Adsorption and Photocatalysis. J Mater Sci Technol 2023, 135, 54–64. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, X.; Guo, R.; Li, X.; Peng, Y.; Zhao, X.; Pu, X. Hollow Mesoporous G-C3N4/Ag2CrO4 Photocatalysis with Direct Z-Scheme: Excellent Degradation Performance for Antibiotics and Dyes. Sep Purif Technol 2021, 270, 118797. [Google Scholar] [CrossRef]
- Vale, M.; Loureiro, M. V.; Ferreira, M.J.; Marques, A.C. Silica-Based Microspheres with Interconnected Macroporosity by Phase Separation. J Solgel Sci Technol 2020, 95, 746–759. [Google Scholar] [CrossRef]
- Vale, M.; Orišková, S.; Mariquito, A.; Reis, L.; Pinto, M.; Marques, A.C. Multicomponent Oxide Microspheres with Designed Macroporosity (MICROSCAFS®): A Customized Platform for Chemicals Immobilization. RSC Adv 2023, 13, 12951–12965. [Google Scholar] [CrossRef]
- Loureiro, M. V.; Vale, M.; De Schrijver, A.; Bordado, J.C.; Silva, E.; Marques, A.C. Hybrid Custom-Tailored Sol-Gel Derived Microscaffold for Biocides Immobilization. Microporous and Mesoporous Materials 2018, 261, 252–258. [Google Scholar] [CrossRef]
- Marques, A.C.; Vale, M. Macroporosity Control by Phase Separation in Sol-Gel Derived Monoliths and Microspheres. Materials 2021, 14, 4247. [Google Scholar] [CrossRef]
- Fernandes, S.M.; Barrocas, B.T.; Nardeli, J.V.; Montemor, M.F.; Maçoas, E.; Oliveira, M.C.; de Carvalho, C.C.C.R.; Lauria, A.; Niederberger, M.; Marques, A.C. Maximizing Photocatalytic Efficiency with Minimal Amount of Gold: Solar-Driven TiO2 Photocatalysis Supported by MICROSCAFS® for Facile Catalyst Recovery. J Environ Chem Eng 2024, 12, 112043. [Google Scholar] [CrossRef]
- Loureiro, M. V.; Ciriminna, R.; Lourenço, M.J.; Santos, L.F.; De Schrijver, A.; Bordado, J.C.; Pagliaro, M.; Marques, A.C. Organically-Modified Silica Based Microspheres for Self-Curing Polyurethane One Component Foams. Microporous and Mesoporous Materials 2017, 244, 244–250. [Google Scholar] [CrossRef]
- Marques, A.C.; Loureiro, M. V.; Lourenço, M.J.; De Schrijver, A.; Bordado, J.C. Amino Surface Functionalized Microcapsules as Curing Agents for Polyurethane Foams. Materials and Manufacturing Processes 2017, 32, 1304–1309. [Google Scholar] [CrossRef]
- Nagar, N.; Devra, V. A Kinetic Study on the Degradation and Biodegradability of Silver Nanoparticles Catalyzed Methyl Orange and Textile Effluents. Heliyon 2019, 5, e01356. [Google Scholar] [CrossRef]
- Benz, D.; Van Bui, H.; Hintzen, H.T.; Kreutzer, M.T.; van Ommen, J.R. Mechanistic Insight into the Improved Photocatalytic Degradation of Dyes for an Ultrathin Coating of SiO2 on TiO2 (P25) Nanoparticles. Chemical Engineering Journal Advances 2022, 10, 100288. [Google Scholar] [CrossRef]
- Luna, A.L.; Matter, F.; Schreck, M.; Wohlwend, J.; Tervoort, E.; Colbeau-Justin, C.; Niederberger, M. Monolithic Metal-Containing TiO2 Aerogels Assembled from Crystalline Pre-Formed Nanoparticles as Efficient Photocatalysts for H2 Generation. Appl Catal B 2020, 267, 118660. [Google Scholar] [CrossRef]
- Williams, P.A.; Ireland, C.P.; King, P.J.; Chater, P.A.; Boldrin, P.; Palgrave, R.G.; Claridge, J.B.; Darwent, J.R.; Chalker, P.R.; Rosseinsky, M.J. Atomic Layer Deposition of Anatase TiO2 Coating on Silica Particles: Growth, Characterization and Evaluation as Photocatalysts for Methyl Orange Degradation and Hydrogen Production. J Mater Chem 2012, 22, 20203–20209. [Google Scholar] [CrossRef]
- Ljubas, D.; Smoljanić, G.; Juretić, H. Degradation of Methyl Orange and Congo Red Dyes by Using TiO2 Nanoparticles Activated by the Solar and the Solar-like Radiation. J Environ Manage 2015, 161, 83–91. [Google Scholar] [CrossRef]
- Morales-García, Á.; Macià Escatllar, A.; Illas, F.; Bromley, S.T. Understanding the Interplay between Size, Morphology and Energy Gap in Photoactive TiO2 Nanoparticles. Nanoscale 2019, 11, 9032–9041. [Google Scholar] [CrossRef]
- Comparelli, R.; Fanizza, E.; Curri, M.L.; Cozzoli, P.D.; Mascolo, G.; Passino, R.; Agostiano, A. Photocatalytic Degradation of Azo Dyes by Organic-Capped Anatase TiO2 Nanocrystals Immobilized onto Substrates. Appl Catal B 2005, 55, 81–91. [Google Scholar] [CrossRef]
- Kokilavani, S.; Alaraidh, I.A.; Okla, M.K.; Chandran, P.; Mohebaldin, A.; Soufan, W.; AL-ghamdi, A.A.; Abdel-Maksoud, M.A.; AbdElgawad, H.; Thomas, A.M.; et al. Efficient Photocatalytic Degradation of Methyl Orange and Malachite Green by Ag3PO4 Decorated BiOBr Nanoflower under Visible Light: Performance Evaluation, Mechanism Insights and Toxicology of the by-Products. J Alloys Compd 2022, 909, 164703. [Google Scholar] [CrossRef]
- Putri, R.A.; Safni, S.; Jamarun, N.; Septiani, U.; Kim, M.-K.; Zoh, K. Kinetics Studies on Photodegradation of Methyl Orange in the Presence of C-N-Codoped TiO2 Catalyst. Egypt J Chem 2019, 62, 563–575. [Google Scholar] [CrossRef]
- Baiocchi, C.; Brussino, M.C.; Pramauro, E.; Prevot, A.B.; Palmisano, L.; Marcı̀, G. Characterization of Methyl Orange and Its Photocatalytic Degradation Products by HPLC/UV–VIS Diode Array and Atmospheric Pressure Ionization Quadrupole Ion Trap Mass Spectrometry. Int J Mass Spectrom 2002, 214, 247–256. [Google Scholar] [CrossRef]
- Liu, T.; Wang, L.; Lu, X.; Fan, J.; Cai, X.; Gao, B.; Miao, R.; Wang, J.; Lv, Y. Comparative Study of the Photocatalytic Performance for the Degradation of Different Dyes by ZnIn2S4: Adsorption, Active Species, and Pathways. RSC Adv. 2017, 7, 12292–12300. [Google Scholar] [CrossRef]
- Mills, A.; Williams, G. Methyl Orange as a Probe of the Semiconductor–Electrolyte Interfaces in CdS Suspensions. J. Chem. Soc., Faraday Trans. 1 1987, 83, 2647–2661. [Google Scholar] [CrossRef]
- Osawa, R.A.; Barrocas, B.T.; Monteiro, O.C.; Conceição Oliveira, M.; Florêncio, M.H. Photocatalytic Degradation of Amitriptyline, Trazodone and Venlafaxine Using Modified Cobalt-Titanate Nanowires under UV–Vis Radiation: Transformation Products and in Silico Toxicity. Chemical Engineering Journal 2019, 373, 1338–1347. [Google Scholar] [CrossRef]
- Ismail, L.; Rifai, A.; Ferronato, C.; Fine, L.; Jaber, F.; Chovelon, J.-M. Towards a Better Understanding of the Reactive Species Involved in the Photocatalytic Degradation of Sulfaclozine. Appl Catal B 2016, 185, 88–99. [Google Scholar] [CrossRef]
- Kim, K.-S.; Kam, S.K.; Mok, Y.S. Elucidation of the Degradation Pathways of Sulfonamide Antibiotics in a Dielectric Barrier Discharge Plasma System. Chemical Engineering Journal 2015, 271, 31–42. [Google Scholar] [CrossRef]
- Dirany, A.; Sirés, I.; Oturan, N.; Özcan, A.; Oturan, M.A. Electrochemical Treatment of the Antibiotic Sulfachloropyridazine: Kinetics, Reaction Pathways, and Toxicity Evolution. Environ Sci Technol 2012, 46, 4074–4082. [Google Scholar] [CrossRef]
- Fabiańska, A.; Białk-Bielińska, A.; Stepnowski, P.; Stolte, S.; Siedlecka, E.M. Electrochemical Degradation of Sulfonamides at BDD Electrode: Kinetics, Reaction Pathway and Eco-Toxicity Evaluation. J Hazard Mater 2014, 280, 579–587. [Google Scholar] [CrossRef]
- Vale, M.; Marques, A.C. Mechanistic Study of the Formation of Multicomponent Oxide Porous Microspheres (MICROSCAFS®) by Cryo-Scanning Electron Microscopy. Gels 2023, 9, 704. [Google Scholar] [CrossRef]
- Landi, S.; Segundo, I.R.; Freitas, E.; Vasilevskiy, M.; Carneiro, J.; Tavares, C.J. Use and Misuse of the Kubelka-Munk Function to Obtain the Band Gap Energy from Diffuse Reflectance Measurements. Solid State Commun 2022, 341, 114573. [Google Scholar] [CrossRef]
- Makuła, P.; Pacia, M.; Macyk, W. How To Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV–Vis Spectra. J Phys Chem Lett 2018, 9, 6814–6817. [Google Scholar] [CrossRef] [PubMed]
- Barrocas, B.; Chiavassa, L.D.; Conceição Oliveira, M.; Monteiro, O.C. Impact of Fe, Mn Co-Doping in Titanate Nanowires Photocatalytic Performance for Emergent Organic Pollutants Removal. Chemosphere 2020, 250, 126240. [Google Scholar] [CrossRef] [PubMed]
- Barrocas, B.T.; Oliveira, M.C.; Nogueira, H.I.S.; Fateixa, S.; Monteiro, O.C. Ruthenium-Modified Titanate Nanowires for the Photocatalytic Oxidative Removal of Organic Pollutants from Water. ACS Appl Nano Mater 2019, 2, 1341–1349. [Google Scholar] [CrossRef]
- Osawa, R.; T. Barrocas, B.; C. Monteiro, O.; Oliveira, M.C.; Florêncio, M.H. Photocatalytic Degradation of Cyclophosphamide and Ifosfamide: Effects of Wastewater Matrix, Transformation Products and in Silico Toxicity Prediction. Science of The Total Environment 2019, 692, 503–510. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Particle size distributions of the MICROSCAFS® before (left) and after HT (right) at 900 °C during 30 min.
Figure 1.
Particle size distributions of the MICROSCAFS® before (left) and after HT (right) at 900 °C during 30 min.
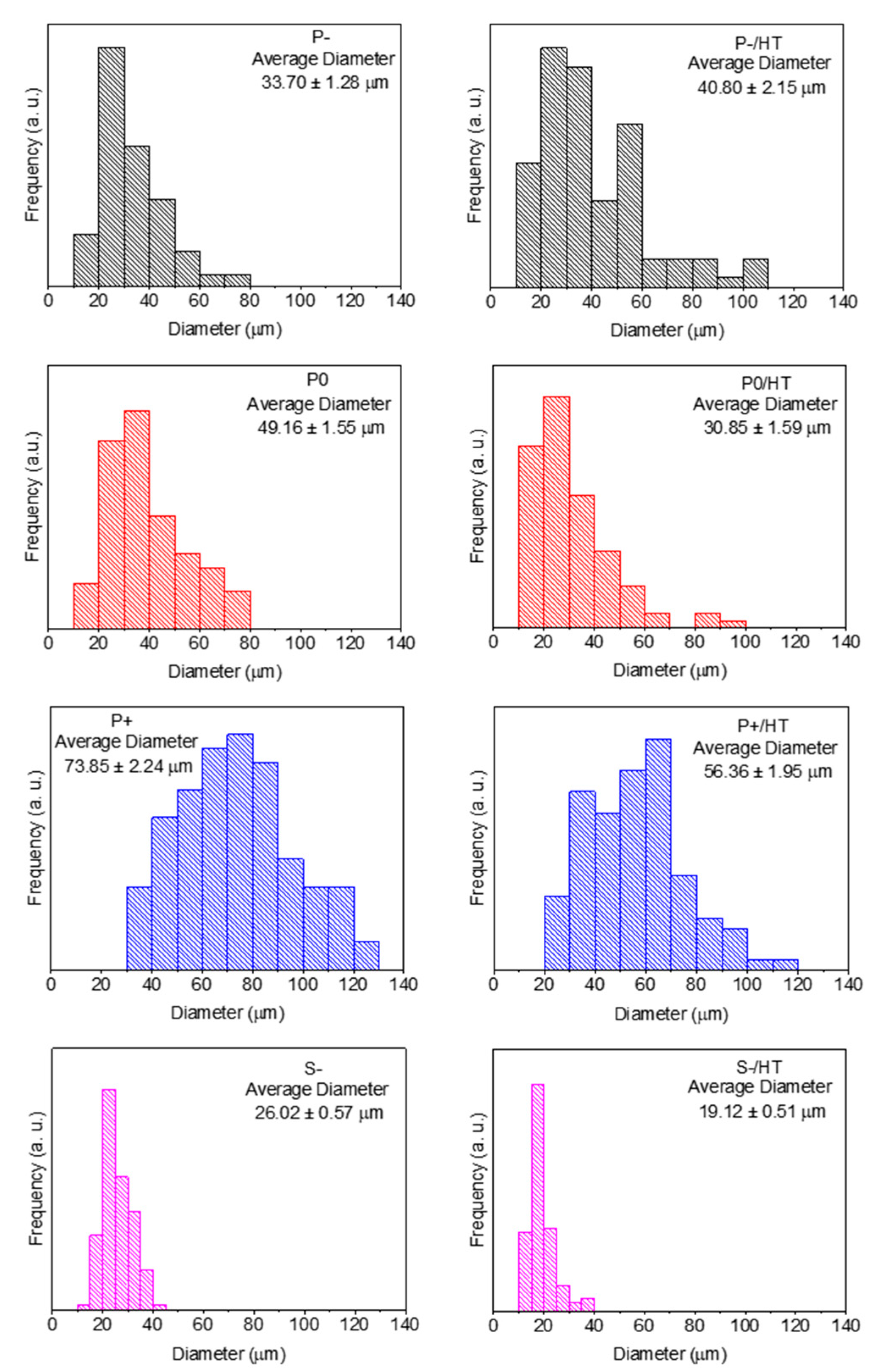
Figure 2.
SEM images of the internal porosity (left), and corresponding MIP pore size distributions (right) of the MICROSCAFS® (a) dried at 45 °C, (b) heat-treated at 900 °C and (c) loaded with P25 TiO2 NPs and heat-treated at 500 °C.
Figure 2.
SEM images of the internal porosity (left), and corresponding MIP pore size distributions (right) of the MICROSCAFS® (a) dried at 45 °C, (b) heat-treated at 900 °C and (c) loaded with P25 TiO2 NPs and heat-treated at 500 °C.
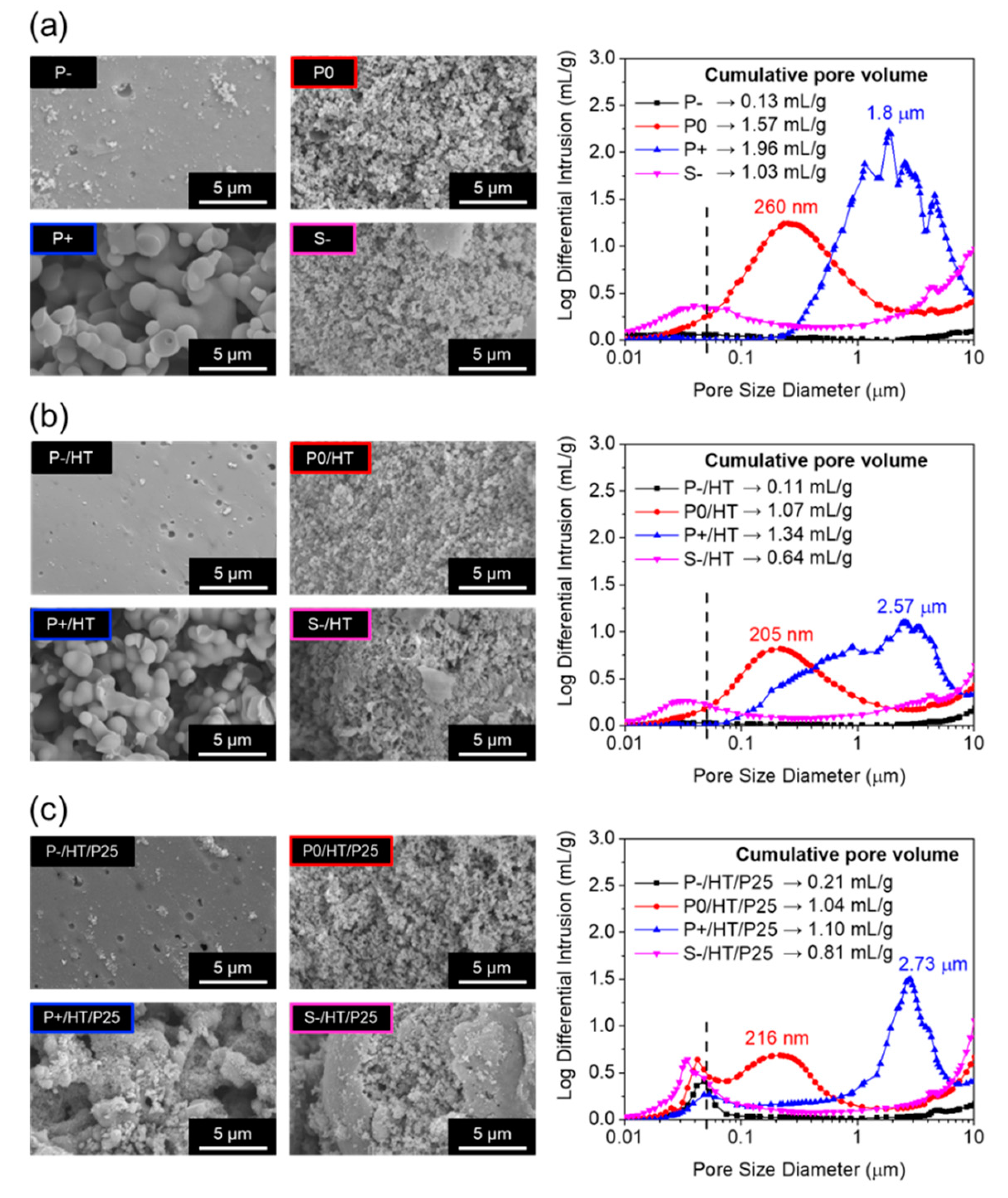
Figure 3.
(a) Plots of C/C0 versus time of the heat-treated unloaded MICROSCAFS®, i.e. without photocatalyst (P-/HT, P0/HT and P+/HT), P25 TiO2 NPs (13 mg) and photolysis of the MO dye; (b) Plots of C/C0 versus time of the photocatalytic P25 TiO2 NPs loaded MICROSCAFS®; (c) UV-vis spectra of MO solution aliquots taken during the light phase of a P+/HT/P25 experiment; (d) Photos of MO solution aliquots at the starting of the illumination and after 120 minutes when using the P+/HT/P25. Experiments done in batch conditions at 19 °C, pH = 7, mass(TiO2 NPs)/mass(MO) = 23, 50 mL of 10 mg/L MO aq. solution, 50 mg of P25 TiO2 NPs loaded MICROSCAFS® (11 – 12.5 mg TiO2), irradiance = 1000 W m−2 (1 sun).
Figure 3.
(a) Plots of C/C0 versus time of the heat-treated unloaded MICROSCAFS®, i.e. without photocatalyst (P-/HT, P0/HT and P+/HT), P25 TiO2 NPs (13 mg) and photolysis of the MO dye; (b) Plots of C/C0 versus time of the photocatalytic P25 TiO2 NPs loaded MICROSCAFS®; (c) UV-vis spectra of MO solution aliquots taken during the light phase of a P+/HT/P25 experiment; (d) Photos of MO solution aliquots at the starting of the illumination and after 120 minutes when using the P+/HT/P25. Experiments done in batch conditions at 19 °C, pH = 7, mass(TiO2 NPs)/mass(MO) = 23, 50 mL of 10 mg/L MO aq. solution, 50 mg of P25 TiO2 NPs loaded MICROSCAFS® (11 – 12.5 mg TiO2), irradiance = 1000 W m−2 (1 sun).
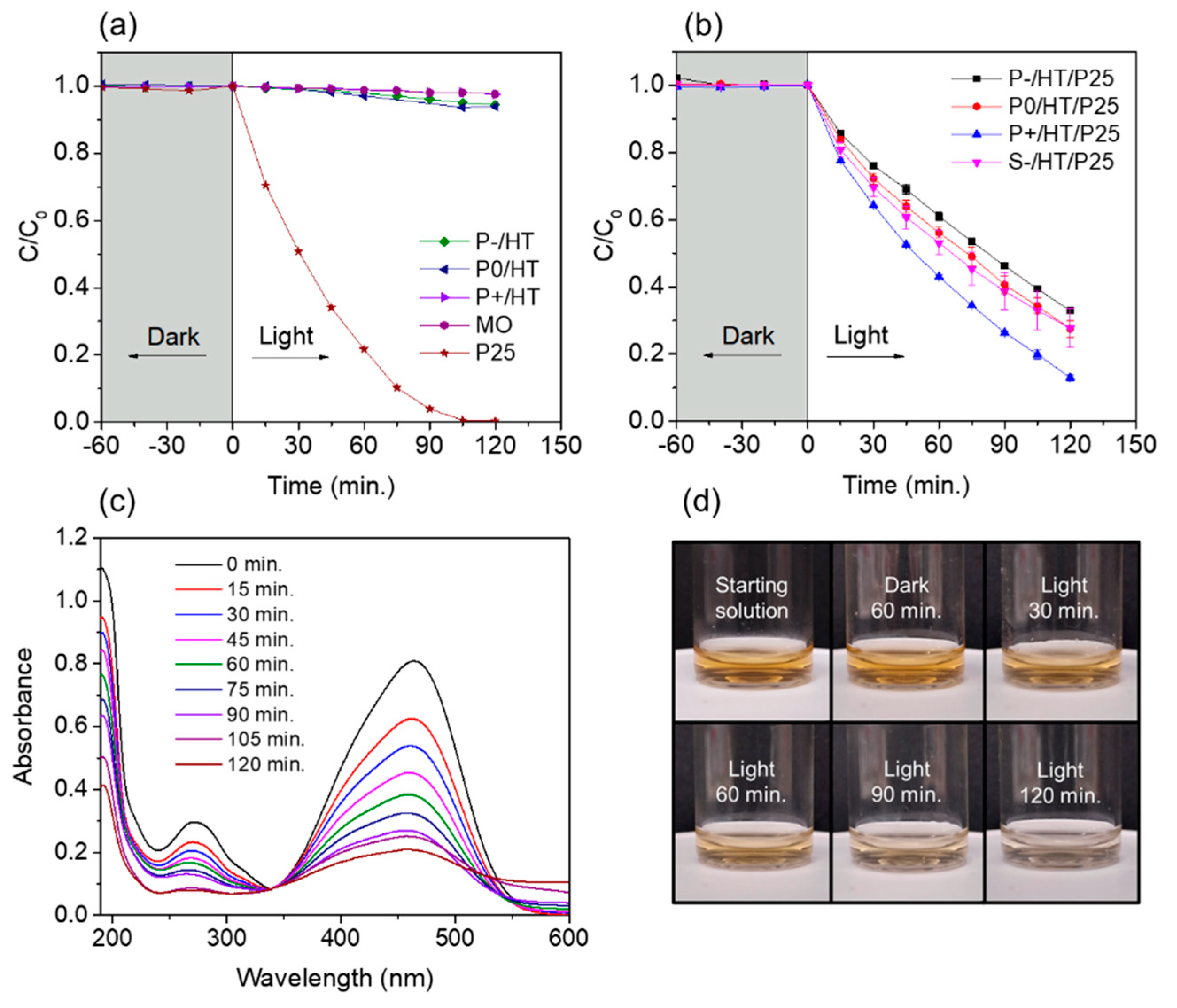
Figure 4.
(a) Plots of C/C0 versus time of the P25 TiO2 NPs loaded MICROSCAFS® using a continuous flow set-up; (b) MO degradation evolution achieved for 6 consecutive cycles with solar light irradiation for 6 hours using sample P+/HT/P25; (c) UV-Vis spectra of the final MO solution after each cycle in the range of 190 to 600 nm; (d) UV-Vis spectra of the final MO solution after each cycle in the range of 230 to 300 nm. Experiments done in flow conditions at 19 °C, pH = 7, mass (P25 TiO2 NPs)/mass(MO) = 23, 200 mL of 10 mg/L MO aq. solution, 200 mg of P25 TiO2 NPs loaded MICROSCAFS® (46-50 mg TiO2), volumetric flow = 10 mL min−1, irradiance = 1000 W m−2 (1 sun).
Figure 4.
(a) Plots of C/C0 versus time of the P25 TiO2 NPs loaded MICROSCAFS® using a continuous flow set-up; (b) MO degradation evolution achieved for 6 consecutive cycles with solar light irradiation for 6 hours using sample P+/HT/P25; (c) UV-Vis spectra of the final MO solution after each cycle in the range of 190 to 600 nm; (d) UV-Vis spectra of the final MO solution after each cycle in the range of 230 to 300 nm. Experiments done in flow conditions at 19 °C, pH = 7, mass (P25 TiO2 NPs)/mass(MO) = 23, 200 mL of 10 mg/L MO aq. solution, 200 mg of P25 TiO2 NPs loaded MICROSCAFS® (46-50 mg TiO2), volumetric flow = 10 mL min−1, irradiance = 1000 W m−2 (1 sun).
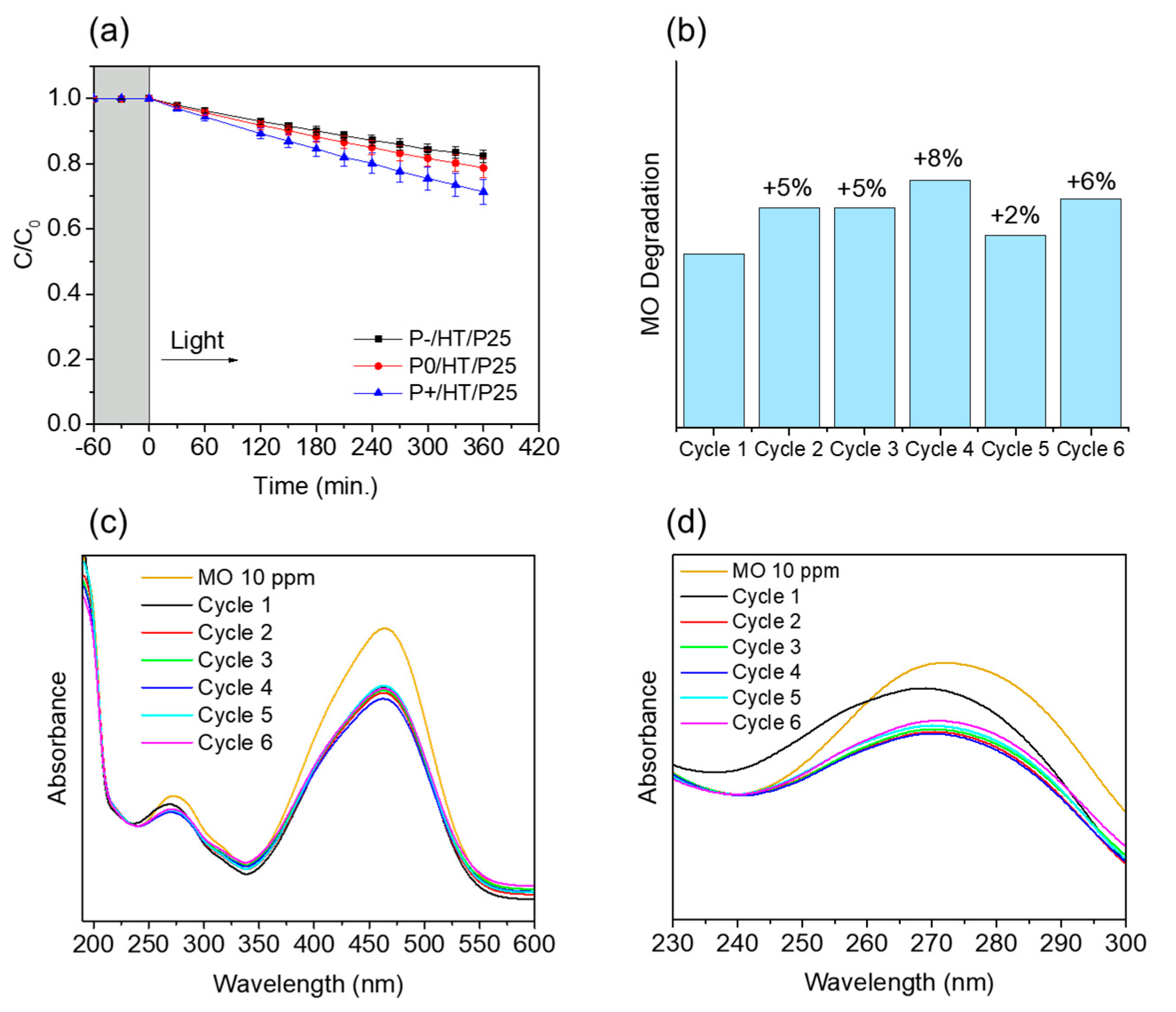
Figure 5.
Adjusted kinetic models in batch using the photocatalytic MICROSCAFS® with different porosities and size. Kinetic rate constant values (k) are indicated for each case.
Figure 5.
Adjusted kinetic models in batch using the photocatalytic MICROSCAFS® with different porosities and size. Kinetic rate constant values (k) are indicated for each case.
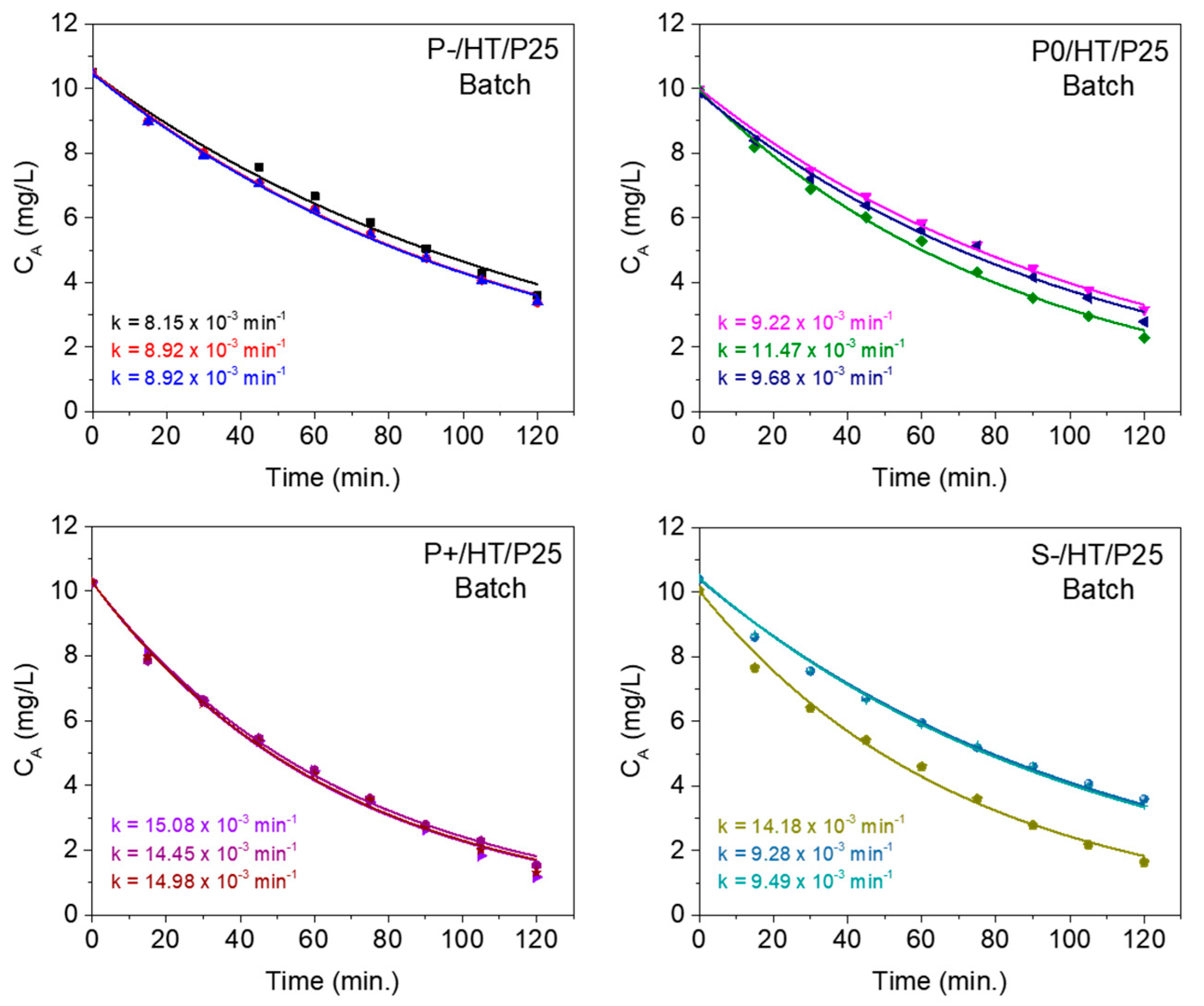
Figure 6.
Adjusted kinetic models of the flow reactor using the photocatalytic MICROSCAFS® with different porosities, and a constant tank volume of 200 mL. Apparent flow (adjusted) kinetic rate constant values (kapp) are indicated for each case.
Figure 6.
Adjusted kinetic models of the flow reactor using the photocatalytic MICROSCAFS® with different porosities, and a constant tank volume of 200 mL. Apparent flow (adjusted) kinetic rate constant values (kapp) are indicated for each case.
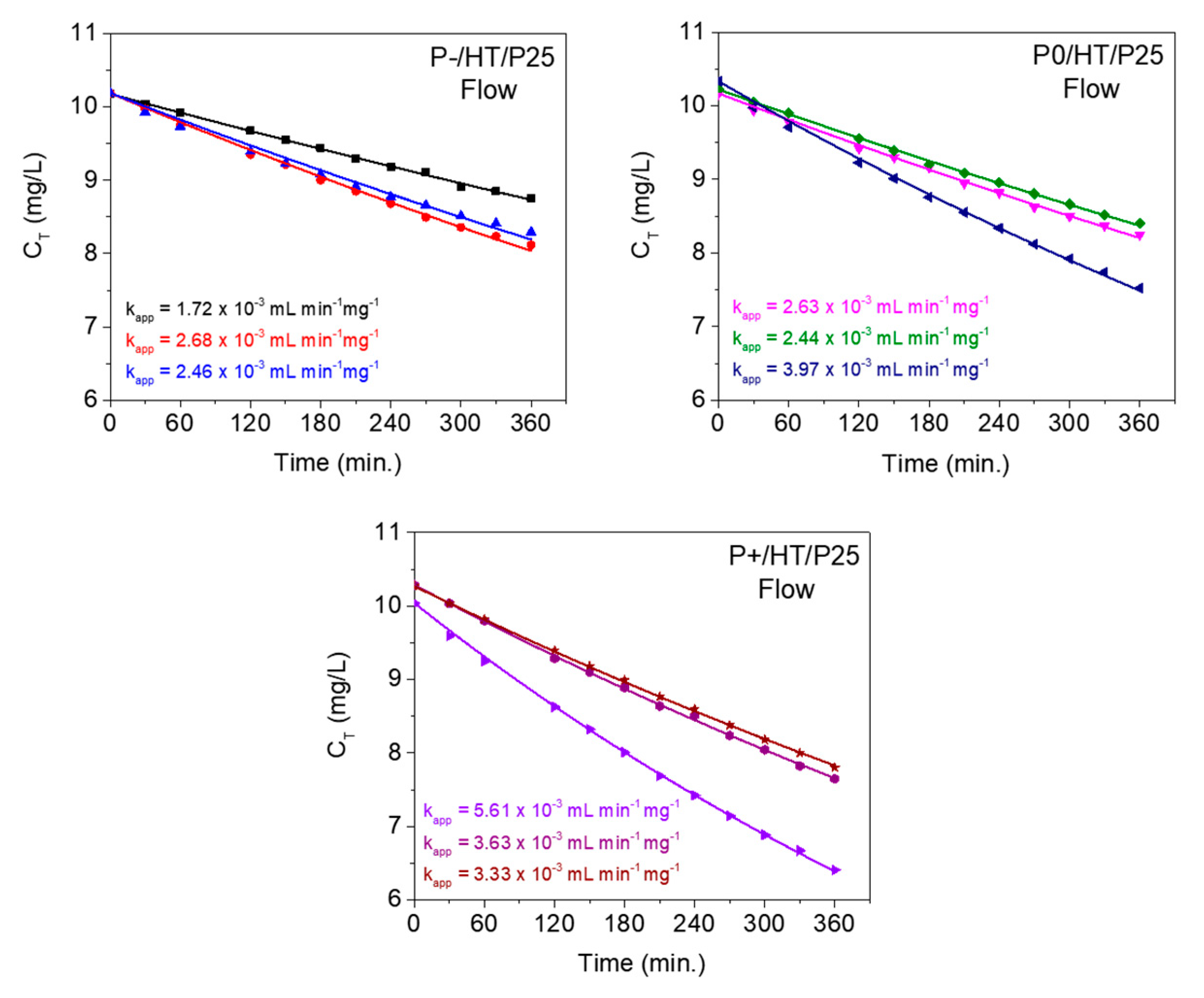
Figure 7.
Adjusted kinetic model of the complete MO degradation using the continuous flow reactor.
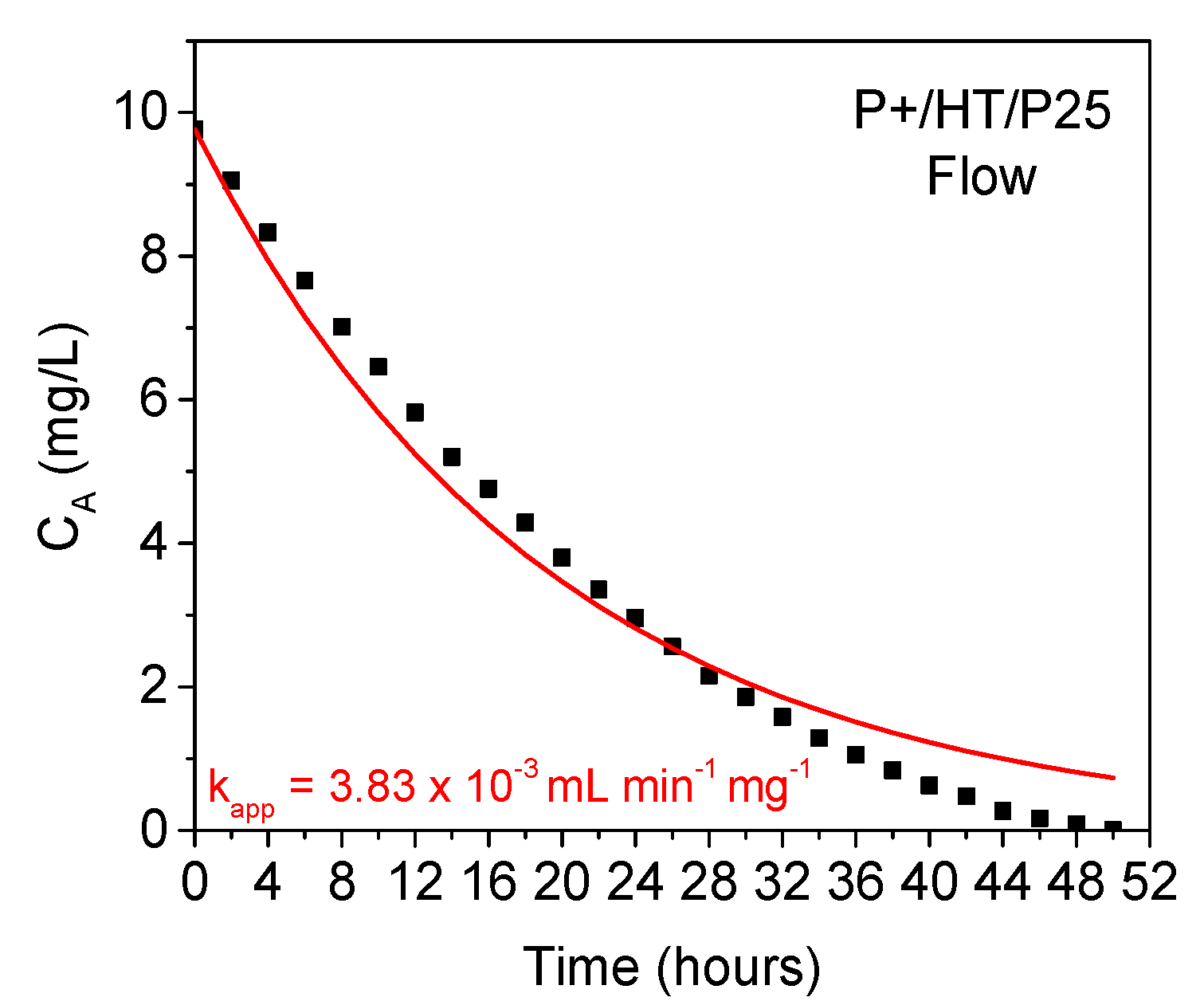
Figure 8.
TPs identification of MO photocatalytic degradation, during 120 and 360 min using P0/HT/P25 MICROSCAFS® on the (a) batch and (b) flow reactor, respectively. The MO degradation values (%) indicated were obtained by UV-Vis spectroscopy.
Figure 8.
TPs identification of MO photocatalytic degradation, during 120 and 360 min using P0/HT/P25 MICROSCAFS® on the (a) batch and (b) flow reactor, respectively. The MO degradation values (%) indicated were obtained by UV-Vis spectroscopy.
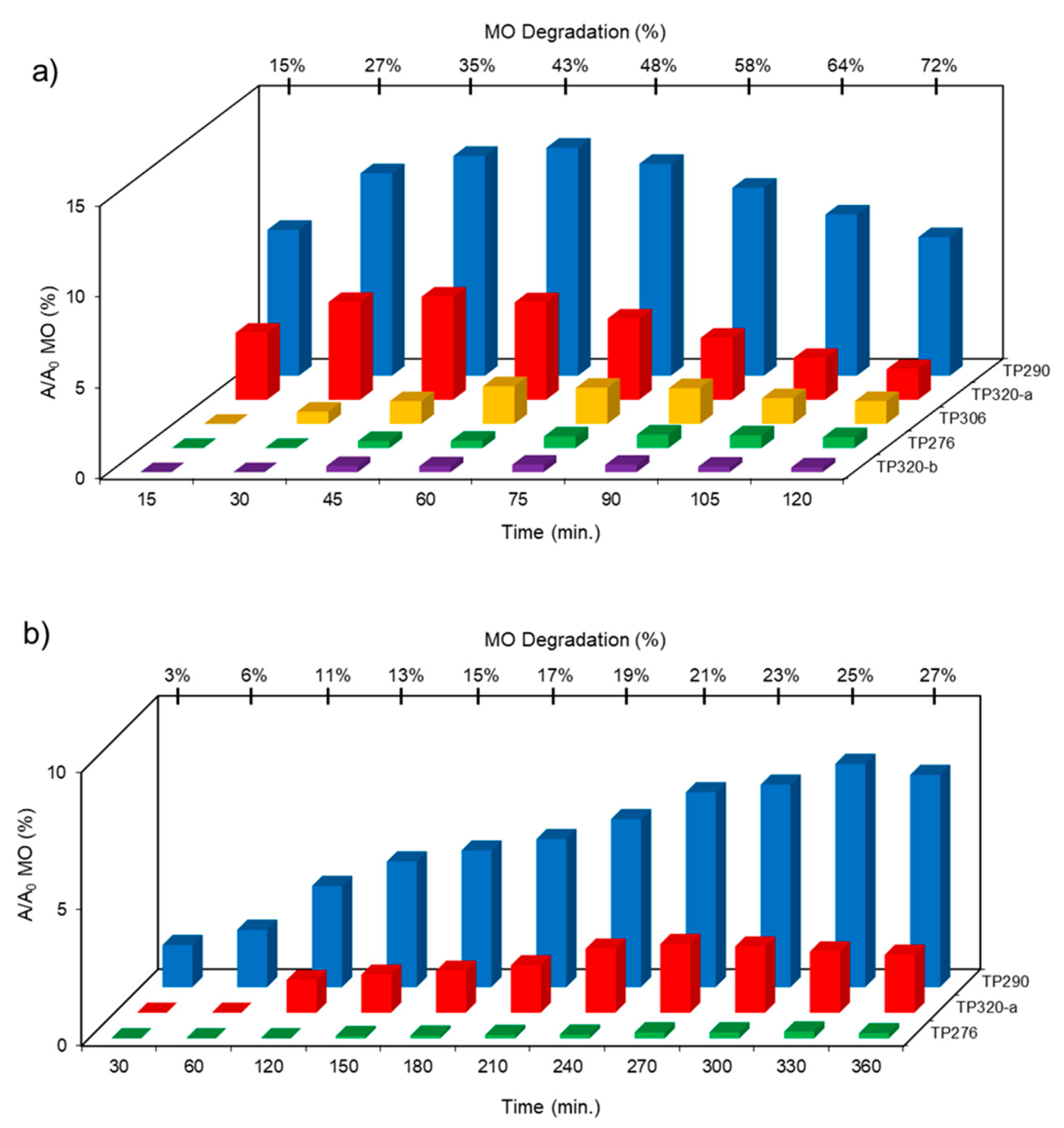
Figure 9.
MO degradation evolution after 120 min of solar irradiation using photocatalytic MICROSCAFS® P+/HT/P25, when in the presence of selected scavengers: EtOH, BQ and EDTA to quench •OH, •O2−, and h+, respectively.
Figure 9.
MO degradation evolution after 120 min of solar irradiation using photocatalytic MICROSCAFS® P+/HT/P25, when in the presence of selected scavengers: EtOH, BQ and EDTA to quench •OH, •O2−, and h+, respectively.
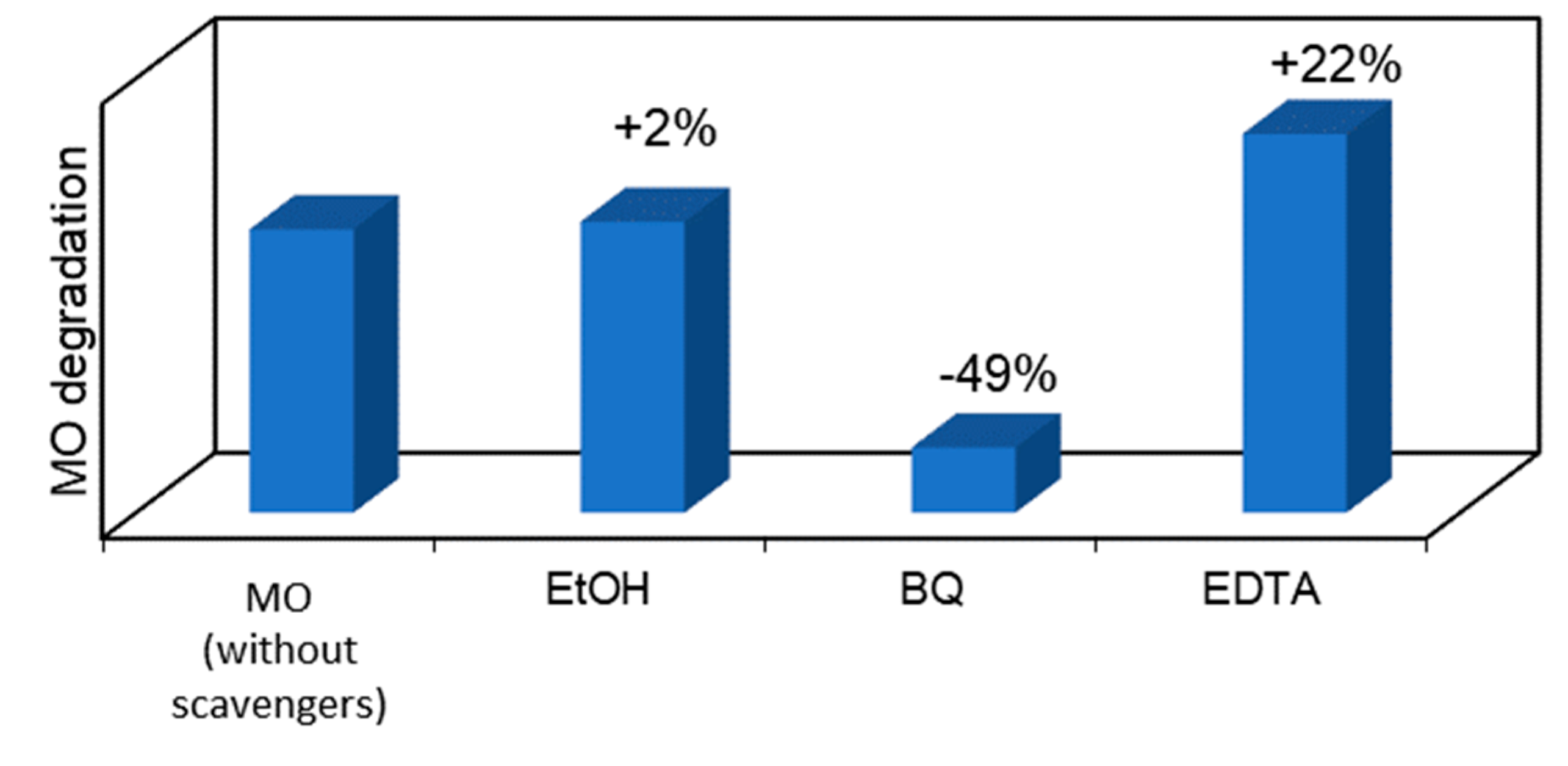
Figure 10.
The photoactivation mechanism proposed for photocatalytic MICROSCAFS® P+/HT/P25, used as catalyst for the photodegradation of MO under solar light irradiation. Photocatalytic MICROSCAFS® P+/HT/P25, are composed of MICROSCAFS® P+/HT with immobilized photocatalytic P25 NPs.
Figure 10.
The photoactivation mechanism proposed for photocatalytic MICROSCAFS® P+/HT/P25, used as catalyst for the photodegradation of MO under solar light irradiation. Photocatalytic MICROSCAFS® P+/HT/P25, are composed of MICROSCAFS® P+/HT with immobilized photocatalytic P25 NPs.
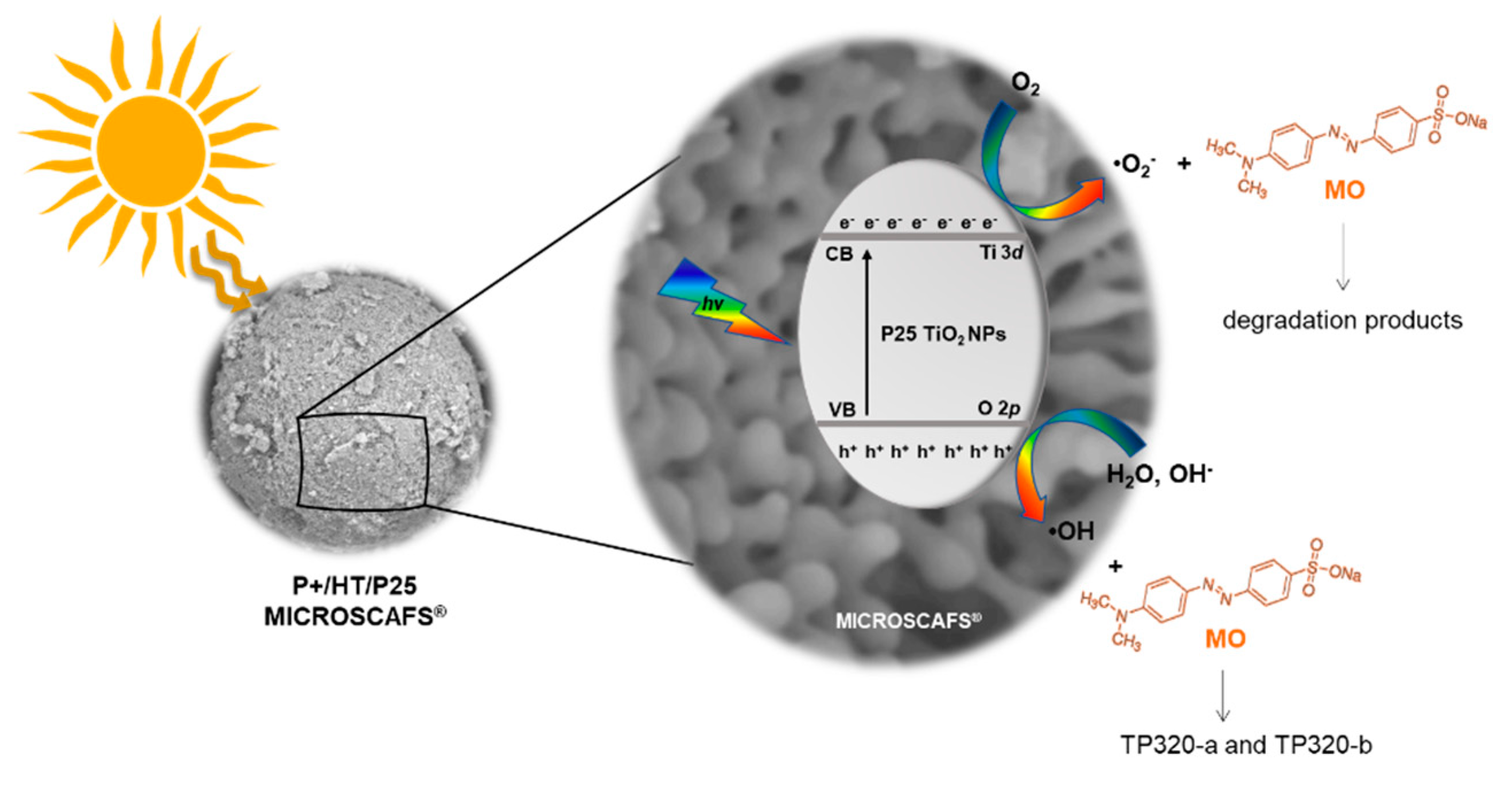

Table 1.
Average kinetic rate constants (batch) and apparent flow reactor rate constants of the photocatalytic MICROSCAFS®.
Table 1.
Average kinetic rate constants (batch) and apparent flow reactor rate constants of the photocatalytic MICROSCAFS®.
| Sample Acronym | Average Kinetic Rate Constant k (mL min-1 mg-1) |
Average Apparent Flow Reaction Rate Constant kapp (mL min-1 mg-1) |
|---|---|---|
| P-/HT/P25 | (3.47 ± 0.1) × 10-2 | (2.28 ± 0.3) × 10-3 |
| P0/HT/P25 | (4.40 ± 0.3) × 10-2 | (3.01 ± 0.5) × 10-3 |
| P+/HT/P25 | (6.45 ± 0.08) × 10-2 | (4.19 ± 0.7) × 10-3 |
| S-/HT/P25 | (4.99 ± 0.7) × 10-2 | n. a. |
Table 2.
Experimental conditions, MO degradation and kinetic rate constants (k or kapp) of supported TiO2 photocatalyst – comparison of the present study with the literature.
Table 2.
Experimental conditions, MO degradation and kinetic rate constants (k or kapp) of supported TiO2 photocatalyst – comparison of the present study with the literature.
| Supported Photocatalyst System | Radiation Source | Reactor Type and pH | Reactor Volume and Initial Concentration of MO Solution | Photocatalyst (Active Phase)/Pollutant Mass Ratio | MO Degradation at 464 nm |
k (Batch) or kapp (Flow) (mL min-1 mg-1) |
Ref., Year |
|---|---|---|---|---|---|---|---|
| P25 TiO2 NPs supported in SiO2-TiO2 MICROSCAFS® (11.5 mg batch; 46 mg flow) (23 wt% loading) |
Solar simulator Xe lamp, 100 W, 1000 W/m2 (1 sun) |
Batch pH=7 | 50 mL, 10 mg/L |
23 | 87% in 120 min. | (6.45 ± 0.08) × 10-2 | Present study |
| Flow pH=7 |
0.47 mL, 10 mg/L |
23 | 29% in 360 min. | (4.19 ± 0.7) × 10-3 | |||
| Commercial anatase TiO2 (11 mg, 2-3 µm) supported in biochar (75% wt% loading) |
Hg lamp, 500 W |
Batch pH = 6.3 |
30 mL, 60 mg/L |
6 | ~99% in 60 min. | 1.39 × 10-1 | [6], 2023 |
| Synthesized anatase TiO2 (66 mg) supported in SiO2 MICROSCAFS®, (22 wt% loading) | Solar simulator Xe lamp, 100 W, 1000 W/m2 (1 sun) |
Flow pH=7 |
100 mL, 20 mg/L |
33 | 30% in 375 min. | 1.36 × 10-3 | [7], 2021 |
| Synthesized anatase TiO2 (2 mg) supported on natural clays (~11 wt% loading) |
Xe lamp, 500 W |
Batch pH=n/d |
20 mL, 10 mg/L |
10 | 90% in 150 min. | 1.20 × 10-1 | [8], 2019 |
| Synthesized anatase TiO2 (19 mg) supported on cellulose nanofibrils (17 wt% loading) | Solar simulator Xe lamp, 100 W, 1000 W/m2 (1 sun) |
Flow pH=7 |
100 mL, 20 mg/L |
10 | 21% in 200 min. | 5.79 × 10-3 | [9], 2018 |
| P25 TiO2 (16 mg) supported in porous PMMA wafers (loading n/d) |
UV-LED | Batch pH =n/d |
60 mL, 10 mg/L |
27 | 28% in ~30 min. | 4.08 × 10-2 | [10], 2015 |
| P25 TiO2 (50 mg) supported on fly ash (25 wt% loading) | Black light lamps, 320-440 nm | Batch pH=8.5 |
200 mL, 3.27 mg/L |
76 | 50% in 170 min. | 6.40 × 10-3 | [12], 2014 |
| Anatase TiO2 layer (~9.8 mg) deposited on 500 nm SiO2 spheres (10 wt% loading) | Xe lamp, 300 W |
Batch pH=n/d |
90 mL, 20 mg/L |
5 | 90% in 120 min. | 1.65 × 10-1 | [36], 2012 |
| P25 TiO2 (10 mg) supported on a PVA film (loading n/d) |
UV 300 nm lamps, 6x8 W, 90 ±10 µW/cm2 |
Batch pH=n/d |
10 mL, 15 mg/L |
67 | ~100% in 300 min. | 1.30 × 10-2 | [11], 2012 |
Table 3.
LC-HRMS/MS identification of MO and their degradation by-products (TPs).
| Compound | Structure | tR (min) |
Proposed empirical formula |
[M-H]− [m/z (Δ ppm) mSigma] |
|---|---|---|---|---|
| MO | 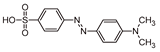 |
9.5 | C14H15N3O3S | [304.0770 (-2.9; 6.5)] |
| TP320-a | 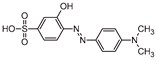 |
10.9 | C14H15N3O4S | [320.0722 (-3.7; 9.2)] |
| TP306 | 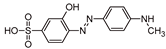 |
9.0 | C13H13N3O4S | [306.0562 (-2.7;12.5)] |
| TP290 | 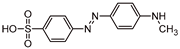 |
7.8 | C13H13N3O3S | [290.0614 (-3.2; 10.5)] |
| TP276 | 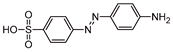 |
5.8 | C12H11N3O3S | [276.0453 (-3.2; 21.6)] |
| TP320-b | 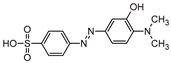 |
3.9 | C14H15N3O4S | [320.0712 (-0.2; 9.7)] |
Table 4.
Toxicological assessment of MO and their TPs by ECOSAR.
| Compound | Acute Toxicity | Chronic Toxicity | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (mg/L) | (mg/L) | ||||||||
| Fish (LC50) | Daphnid (LC50) | Green Algae (EC50) | Fish (ChV) | Daphnid (ChV) | Green Algae (ChV) | ||||
| MO | 1513.99 | 793.35 | 424.07 | 134.62 | 61.88 | 92.88 | |||
| TP320-a | 173.97 | 39.22 | 192.16 | 15.97 | 7.47 | 91.02 | |||
| TP320-b | 592.29 | 97.78 | 512.74 | 49.07 | 18.64 | 245.03 | |||
| TP306 | 433.60 | 76.62 | 395.85 | 36.73 | 14.60 | 188.80 | |||
| TP290 | 4479.49 | 2231.63 | 967.94 | 375.25 | 151.23 | 189.43 | |||
| TP276 | 922.37 | 2.07 | 9.26 | 5.36 | 0.13 | 13.94 | |||
| Harmless - LC50/EC50/ChV > 100 | Harmful - 100 > LC50/EC50/ChV > 10 | Toxic - 10 > LC50/EC50/ChV > 1 | Very toxic - LC50/EC50/ChV < 1 | ||||||
Table 5.
MICROSCAFS®’ synthesis parameters, which suffered variation.
| Sample Acronym | Volume of GPTMS (mL) | Volume of Span® 80 (mL) | Mass of Pluronic® P123 (g) |
|---|---|---|---|
| P- | 10.4 | 6.0 | 0 |
| P0 (reference) | 13.8 | 6.0 | 0 |
| P+ | 17.3 | 6.0 | 0 |
| S- | 13.8 | 7.7 | 2.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



