
Submitted:
22 April 2024
Posted:
23 April 2024
You are already at the latest version
Abstract
High pathogenicity avian influenza (HPAI) H5N1 clade 2.3.4.4b continues to have a substantial impact on wildlife, globally. A recent study has described the putative detection of HPAI in clinically healthy penguins, and those which were fitted with satellite tags reportedly engaged in apparently normal foraging behaviour in the months following sample collection. Herein we investigate the diagnostic approach utilised, and reveal that while the authors likely did detect subtype H5 influenza A virus, the most parsimonious conclusion is that they detected low pathogenicity H5 rather than HPAI H5N1. In response, we have provided an overview key considerations when selecting a diagnostic and outline published diagnostic assays that should be considered for future studies.
Keywords:
avian influenza
; LPAI
; HPAI
; high pathogenicity avian influenza
; panguins
; Antarctica
Introduction
High pathogenicity avian influenza (HPAI) H5N1 clade 2.3.4.4b continues to have a substantial impact on wildlife, globally. This virus entered the Antarctic region in the 2023/24 austral summer, with the first detection on South Georgia Island as early as October 2023 [1], and detections on the Antarctic Peninsula occurring from February and March 2024 (https://scar.org/library-data/avian-flu). In response to the initial elevated risk [2], numerous research teams have undertaken enhanced surveillance efforts through the screening of both apparently healthy as well as from sick and dead animals [1,3,4,5]. Understanding viral spread and its impact on wildlife in Antarctica is critical for further risk assessment, potential mitigation through enhanced biosecurity practises, and in the longer term, conservation impacts and strategies.
Case Study: LPAI or HPAI in Apparently Healthy Infected Penguins?
A preprint by León et al (2024)[4] describes the putative detection of HPAI H5N1 clade 2.3.4.4b in Adelie penguins (Pygoscelis adeliae) and an Antarctic hag (Leucocarbo bransfieldensis) in the Antarctic region. In their survey, they collected cloacal swabs from 115 seabirds of 4 species across 14 locations. All birds were apparently healthy and seven of the Adelie penguins which tested positive had been fitted with Argos satellite tags, and reportedly engaged in apparently normal foraging behaviour in the months following sample collection. The authors interpreted their findings as an indication that Adelie penguins and Antarctic shags could be asymptomatic carriers of HPAI H5N1 clade 2.3.4.4b viruses, potentially playing a role in the spread of these viruses in Antarctica. This interpretation was widely publicized in the international media [6,7,8] and raised interest and debate with the Antarctic scientific community, as it would have important implications for governance and biosafety management of research and tourism operations in the region.
The finding of apparently healthy animals infected with HPAI H5Nx viruses is not unprecedented – Mallards (Anas platyrhynchos) are known to migrate while infected [9,10], and in infection experiments >50% show no clinical disease [11]. However, based on the widespread mortality observed in seabirds [12,13,14], and specifically penguins and cormorants in South America and southern Africa [15,16,17,18,19,20], this finding was surprising.
As both HPAI H5N1 and low pathogenic avian influenza (LPAI) H5N5 [21,22] have now been detected in Antarctica, one hypothesis to explain the findings of the authors pertains to the diagnostic approach used. To address this, we mapped the reported primers to both LPAI H5 and HPAI H5 sequences. We selected two different LPAI sequences, including A/chinstrap penguin/Antarctica/B04/2015(H5N5) (GenBank accession KX458007) as it’s known to circulate on Antarctica, and (Genbank accession OL369953) A/wild duck/New South Wales/M10-13503-MD07/2010(H5) as the LPAI H5 viruses in Australia form a discrete clade and have been circulating on the continent in isolation for decades, to capture LPAI diversity. We also included two HPAI sequences, specifically including A/Brown skua/Bird Island/128288/2023(H5N1) (GISAID accession EPI2780125). Alignments demonstrate that the degenerate sites in the forward primer aligns well with SNP diversity of both LPAI and HPAI sequences. The “test with saved primers” in Geneious Prime v 2023.2.1 indicated only a single mismatch in the forward primer to the alignment: the final base on the 5’ end of the primer is a C, whereas in all 4 sequences this base position is a T. Of note, the current pre-print provides only the forward primer sequence (the reported reverse primer sequence is a duplication of the forward primer). The corrected reverse primer, shared by the authors upon request, has only 1 mismatched base with the LPAI viruses tested, and is a perfect match to the HPAI viruses tested here. Using the corrected reverse primer, we used MFEprimer 3.1 (https://mfeprimer3-1.igenetech.com/) to determine the diversity of viruses to which the forward and reverse primer would bind to LPAI sequences, by selecting the Influenza A virus database in the query, and allowing for 1 mismatch in the 3’ end. In addition to a diversity of HPAI H5 strains, the combination or primers would bind to KX458007 A/chinstrap penguin/Antarctica/B04/2015(H5N5), with a resultant product size of 773bp (Figure 2). It is similarly able to bind to the unique Australian lineage LPAI H5 viruses (e.g. CY096736). Overall, the RT-PCR employed by León et al. (2024) would not have been able to distinguish between HPAI and LPAI H5 strains.
A broader overview of the methodology suggests that the authors utilized an H5 end-point PCR approach with no preliminary matrix detection step, no post PCR confirmation (neither running of a reference gold-standard test and/or post-PCR sequencing), and no positive controls. The authors did, however, repeat the assay to confirm positives. As such, confirmation of the agent behind the positive test samples was not demonstrated.
Taken together, while the authors likely did detect subtype H5 influenza A virus, the most parsimonious conclusion is that they detected LPAI H5N5 rather than HPAI H5N1. In contrast to HPAI, LPAI is known to cause a typically mild to asymptomatic disease outcome upon infection in wild birds [23] and may or may not have a minor effect on bird movement [24,25,26,27]. Unsurprisingly, due to the dramatic increase of surveillance in the Antarctic this season and its known prevalence, teams have reported the detection of LPAI in Antarctica (pers. comm). In cases where pan-influenza assays, or degenerate primers are used, it is important that confirmation work is undertaken. In the case of León et al. (2024), sequencing of the PCR products would likely resolve the outcome rapidly.
Figure 1.
Forward primer (AH5-918F) binding to an alignment comprising two LPAI sequences and two HPAI sequences. Degenerate bases in the primer region match the SNP diversity of the alignment, as indicated by orange boxes. Alignment generated in Geneious Prime.
Figure 1.
Forward primer (AH5-918F) binding to an alignment comprising two LPAI sequences and two HPAI sequences. Degenerate bases in the primer region match the SNP diversity of the alignment, as indicated by orange boxes. Alignment generated in Geneious Prime.
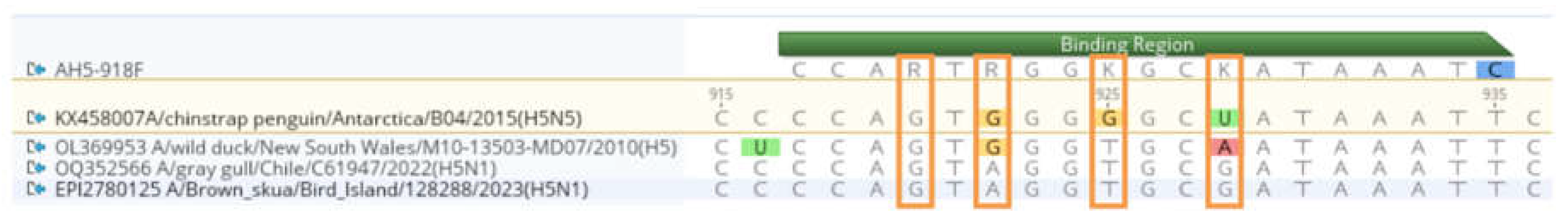
Figure 2.
Binding affinity of forward and reverse primers used in Léon et al. (2024) to KX458007 A/chinstrap penguin/Antarctica/B04/2015(H5N5) generated by MFE Primer 3.1.
Figure 2.
Binding affinity of forward and reverse primers used in Léon et al. (2024) to KX458007 A/chinstrap penguin/Antarctica/B04/2015(H5N5) generated by MFE Primer 3.1.

Considerations for the Detection of HPAI H5 Viruses
In the Antarctic context, disease surveillance has largely been on the onus of researchers in field laboratories rather than by veterinary officers/authorities with testing performed in accredited laboratories. As there is no limitation on diagnostic choice by researchers compared to accredited laboratories, a diversity of HPAI diagnostic assays have been used and are routinely published in literature. The internationally accepted gold standard approaches for influenza A testing are outlined in the World Organisation for Animal Health Terrestrial Manual [28]. Herein we provide some key considerations when selecting an in-field diagnostic approach (Table 1). For simplicity, we will limit our discussion to real-time PCR (qPCR/rRT-PCR) -based diagnostics which are the gold-standard for HPAI virus, and influenza A virus in general, due to their high sensitivity and specificity.
The general diagnostic approach for avian influenza is a multi-step process. First, samples are assayed for the matrix gene, which is highly conserved across influenza A viruses. The most frequently used assays are those developed by Spackman et al (2002) [29] and Nagy et al (2021)[30] . This step is intended as a triage to detect all influenza A virus strains, narrowing down which samples merit further testing. In an outbreak scenario, and/or when HPAI is suspected, this assay may or may not be used but has great utility where infection with non-notifiable subtypes is a possibility. Samples are subsequently tested for H5 (and H7) as these are globally recognised notifiable avian influenza subtypes. There is a diversity of primers/probes available for H5/H7 testing due to the continued requirements for updating in light of viral evolution, and differing country context. Finally, confirmation through sequencing is considered essential.
The diagnostic assay selected should be both sensitive, specific, and fit-for purpose. As influenza A viruses have a rapid rate of evolution, continuously monitoring fitness-for-purpose testing of assay performance using both in silico and in-vitro methodology is critical, and thus laboratories frequently re-evaluate diagnostic panels against new and circulating strains to ensure assays are performing optimally [31]. Due to the presence of both LPAI and HPAI virus strains co-circulating on almost all continents, it is critical that the diagnostic approach used is able to distinguish between LPAI and HPAI viruses as the risk, response and reporting requirements of these pathotypes varies. For example, if a more general H5 test is used, i.e., one that detects multiple lineages of H5 such as employed by León et al (2024), then confirmation of LPAI or HPAI through sequencing is critical. The use of appropriate controls is also essential to enable evaluation of diagnostic assay performance, as well the inclusion of within and between assay performance assessments. For example, to ensure that the H5 detected is indeed HPAI, it is necessary to have both a LPAI H5 and HPAI H5 control in test panels. These positives can include extracted viral RNA, but alternatively can include synthesized sequences.
As outlined by the WOAH terrestrial code [28]: “RNA detection test methodologies should be validated to the WOAH standard (see Chapter 1.1.6 Validation of diagnostic assays for infectious diseases of terrestrial animals) using clinical material to demonstrate the tests as being ‘fit for purpose’ for application in a field diagnostic setting, which may include the use of internal test standards. The control reactions enable greater confidence in the integrity of the molecular reactions, clinical samples and results”.
If a more general H5 test is used, i.e. one that detects multiple lineages of H5, then confirmation through sequencing is critical. Ideally, whole genome sequencing should be performed since it will provide valuable insight into the phylogenetics of the virus, genomic reassortment and adaptive mutations. Even when using assays that are believed to be specific to specific HPAI lineages, confirmation through sequencing is always beneficial, and is particularly warranted in new/atypical contexts – e.g. new hosts, new geographic areas, asymptomatic/aberrant hosts.
Of note is that H5 diagnostics are specific to only one part of the genome (hemagglutinin [HA] gene), and thus don’t provide insight into the neuraminidase (NA) subtype. While currently circulating clade 2.3.4.4b H5 is generally linked to N1, previous 2.3.4.4 lineages were detected with a diversity of NA subtypes, including N6 and N8 [32]. The NA subtype can be distinguished through a specific end point or rRT-PCR N1 assay [33,34], through sequencing the NA gene, or whole genome sequencing.
These considerations are particularly important in the Antarctic context due to the presence of other LPAI viruses, including a LPAI H5N5. Furthermore, due to limited capacity, and in some cases inability to perform sequence confirmation, a robust, sensitive and specific diagnostic assay is critical. Wherever possible samples should be shipped to an appropriate reference laboratory for confirmatory testing wherever the conclusions are in doubt or are not supported by the appropriate scientific diagnostic evidence.
While a number of diagnostic approaches are present in the literature, it would be ideal to align the diagnostic approach selected with that of the relevant national veterinary authority. National reference laboratories have collaborated to define and validate protocols that can be recommended for use [28], and further, confirmation and reporting by national laboratories will ensure the detection is notified to WOAH. Herein we highlight select assays available in the literature which have been recommended in the WOAH terrestrial manual and/or are in used by accredited reference laboratories (Table 2). Importantly, not all validated diagnostic assays are available in the public domain (e.g. [3,35,36]). In general, a combination of assays, as outlined in Slomka et al. 2023 is ideal as to ensure there is no doubt, which is particularly important if no confirmatory sequencing is being performed in the field. Final, confirmatory whole genome sequencing is of the essence to provide deeper insight on the ecology and evolution of these viruses in Antarctic wildlife communities.
Conclusions
Accurate and robust diagnostics play a pivotal role in the timely identification and management of HPAI. The selection of appropriate diagnostic tools is crucial, ensuring sensitivity, specificity, and rapidity in detecting HPAI viral strains. Molecular techniques such as rRT-PCR offer high sensitivity and specificity, facilitating early detection and containment efforts. However, challenges persist in deploying these diagnostics in field settings, and in the Antarctic setting the onus is on researchers in field laboratories, rather than veterinary officials and accredited laboratories to respond, potentially leading misunderstandings around caveats and limitations and misinterpretations of the results.
Surveillance results from the Antarctic region reported in the literature are of high interest, and can have significant impacts on both scientific and tourist activities in the region, including site closures and cessation of research activities, including important long-term ecological studies. As recommended by Dewar et al (2023)[2] and the World Organisation for Animal Health[37], it is essential that researchers:
- Use validated and accredited methods to confirm the presence of the virus (whether in a field or reference laboratory),
- Provide an absolute definition of the presence of either HPAI or LPAI strains wherever detection of these pathogens are reported
- Submit all results to the World Organisation for Animal Health and the SCAR HPAI monitoring database to assist in the global monitoring and surveillance of the virus in the region and around the globe.
- Undertake full genome sequencing of any positive samples to enable genomic surveillance to assist with understanding of virus movement into and within the region and to identify any potential mutations. Submit genome sequences to a publicly-available database (e.g. GenBank) in a reasonable timeline.

Table 2.
Diagnostic approaches recommended in the WOAH terrestrial manual and/or are in use by accredited H5 reference laboratories.
Table 2.
Diagnostic approaches recommended in the WOAH terrestrial manual and/or are in use by accredited H5 reference laboratories.
| Reference | Target | Assay type | Confirmation required? | Select examples of use |
|---|---|---|---|---|
| Spackman et al (2002) [29] | 99bp region of the M Highly conserved to detect all avian influenza viruses |
rRT-PCR | Yes, H5 diagnostic and/or sequencing | 2001 citations. Detection of clade 2.3.4.4b in farmed mink in Spain [38] and wild birds in North America [39] |
| Nagy et al. (2021)[30] | 149bp region of the M Highly conserved to detect all influenza A viruses |
rRT-PCR | Yes, H5 diagnostic and/or sequencing | Detection of clade 2.3.4.4b in Gannets in UK [13] , in carnivores in Finland [40]. Is integrated into combination HA, NA, M test by Slomka et al (2023)[33] |
| Hassan et al. (2022)[41] which is updated from Hoffmann et al. (2016)[42] | All HA and NA subtypes, M. Designed to detect all avian influenza viruses |
Multiplexed rRT-PCR | Yes, sequencing to reveal H5 lineage. | Detection of clade 2.3.4.4b in Sandwich Terns in Germany [43] and in Grey Seals in Europe [44] |
| Slomka et al. (2007)[45], updated from Spackman et al (2002) [29] | 229 bp of HA segment in the HA2 region Designed to detect all H5 viruses |
rRT-PCR | Yes, sequencing to reveal H5 lineage. | Detection of clade 2.3.4.4b in Sandwich Terns in the Netherlands [46] . Integrated into combination HA, NA, M test by Slomka et al (2023)[33] |
| Slomka et al (2012)[47] | 191bp of HA segment across HA cleavage site. |
rRT-PCR | No, but best practice. | Detection of clade 2.3.4.4b in mammals in a rehabilitation centre [48] . Integrated into combination HA, NA, M test by Slomka et al (2023)[33] |
| James et al. (2022)[49] modified from an unpublished protocol based on Naguib et al. (2017)[50] | 109bp region of HA cleavage site. Designed to be specific to 2020/21 clade 2.3.4.4b H5Nx viruses Confirmed not to detect LPAI |
rRT-PCR | No, but best practice. | Detection of clade 2.3.4.4b in birds in South Georgia Island and the Falkland (Malvinas) Islands[1], Gannets in the UK[13], ducks in Botswana [51] |
| Naguib et al. (2017)[50] | 109-161bp of HA segment in the HA1 region Designed to discriminate gs/GD clades 2.2.1.2, 2.3.2.1 and 2.3.4.4 and LPAI |
Multiplexed rRT-PCR | No, but best practice. | Detection of clade 2.3.4.4b in Swedish wild birds and poultry [52], and a novel 2.3.4.4 H5N8 reassortant in Germany [53] |
| Fereidouni et al. (2009)[54] | 126-250bp of NA segment | End point RT-PCR | Yes | Detection of clade 2.3.4.4b in carnivores in Finland [40], emergence 2.3.4.4b in wild birds in South Korea[55] |
| James et al. (2018)[34] updated from Hoffman (2016) [42] | ~150bp of NA segment | rRT-PCR | Yes | Detection of clade 2.3.4.4b in ducks in Botswana [51], in birds in South Georgia Island and the Falkland (Malvinas) Islands[1]. Integrated into combination HA, NA, M test by Slomka et al (2023)[33] |
References
- Bennison, A.; Byrne, A.M.; Reid, S.M.; Lynton-Jenkins, J.G.; Mollett, B.; Sliva, D.D.; Peers-Dent, J.; Finlayson, K.E.; Hall, R.; Blockley, F.; et al. Detection and spread of high pathogenicity avian influenza virus H5N1 in the Antarctic Region. bioRxiv 2023. [Google Scholar]
- Dewar, M.; Wille, M.; Gamble, A.; Vanstreels, R.; Boulinier, T.; Smith, A.; Varsani, A.; Ratcliffe, N.; Black, J.; Lynnes, A. The risk of avian influenza in the Southern Ocean: A practical guide for operators interacting with wildlife. Antarctic Science 2023. [Google Scholar] [CrossRef]
- Bennet, B. Confirmation of highly pathogenic avian influenza (HPAI) H5N1 associated with an unexpected mortality event in South Polar Skuas (Stercorarius maccormicki) during 2023-2024 surveillance activities in Antarctica. bioRxiv, 2024. [Google Scholar]
- León, F.; Bohec, C.L.; Pizarro, E.J.; Baille, L.; Cristofari, R.; Houstin, A.; Zitterbart, D.P.; Barriga, G.; Poulin, E.; Vianna, J.A. Highly Pathogenic Avian Influenza A (H5N1) Suspected in penguins and shags on the Antarctic Peninsula and West Antarctic Coast. bioRxiv 2024. [Google Scholar]
- Lisovski, S. No evidence for highly pathogenic avian influenza H5N1 (clade 2.3.4.4b) in the Antarctic region due the austral summer 2022/23. bioRxiv, 2023. [Google Scholar]
- The Guardian. ‘Cautious optimism’ as penguins test positive for bird flu but show no symptoms. (2024). Available online: https://www.theguardian.com/environment/2024/mar/2026/bird-flu-asymptomatic-penguins-adelie-penguins-antarctic.
- INACH. Chile detecta casos positivos de gripe aviar en pingüinos, cormoranes y skuas en la Antártica. (2024). Available online: https://www.inach.cl/chile-detecta-casos-positivos-de-gripe-aviar-en-pinguinos-cormoranes-y-skuas-en-la-antartica/.
- Reuters. Antarctic scientists warn of bird flu spread as penguin cases confirmed. (2024). Available online: https://www.reuters.com/business/healthcare-pharmaceuticals/antarctic-scientists-warn-bird-flu-spread-penguin-cases-confirmed-2024-2003-2014/.
- Lv, X.; et al. Highly pathogenic avian influenza A(H5N8) clade 2.3.4.4b viruses in satellite-tracked wild ducks, Ningxia, China, 2020. Emerg. Infect. Dis. 2022, 28, 1039–1042. [Google Scholar] [CrossRef]
- Teitelbaum, C.S.; et al. North American wintering mallards infected with highly pathogenic avian influenza show few signs of altered local or migratory movements. Sci Rep-Uk 2023, 13, 14473. [Google Scholar] [CrossRef]
- Spackman, E.; Pantin-Jackwood, M.J.; Lee, S.A.; Prosser, D. The pathogenesis of a 2022 North American highly pathogenic clade 2.3.4.4b H5N1 avian influenza virus in mallards (Anas platyrhynchos). Avian Pathology 2023, 52, 219–228. [Google Scholar] [CrossRef]
- Banyard, A.C. Continued expansion of high pathogenicity avian influenza H5 in wildlife in South America and incursions in to the Antarctic Region. OFFLU statement, 2023. Available online: https://www.offlu.org/wp-content/uploads/2023/2012/OFFLU-wildlife-statement-no.-II.pdf.
- Lane, J.V.; Jeglinski, J.W.; Avery-Gomm, S.; Ballstaedt, E.; Banyard, A.C.; Barychka, T.; Brown, I.H.; Brugger, B.; Burt, T.V.; Careen, N.; et al. High pathogenicity avian influenza (H5N1) in Northern Gannets: Global spread, clinical signs, and demographic consequences. Ibis 2023. [Google Scholar] [CrossRef]
- Knief, U. Highly pathogenic avian influenza causes mass mortality in Sandwich tern (Thalasseus sandvicensis) breeding colonies across northwestern Europe. bioRxiv, 2023. Available online: https://www.biorxiv.org/content/10.1101/2023.1105.1112.540367v540361.
- Molini, U.; et al. Avian influenza H5N8 outbreak in African Penguins (Spheniscus demersus), Namibia, 2019. Journal of Wildlfe Diseases 2020, 56, 214–218. [Google Scholar] [CrossRef]
- Roberts, L.C. Descriptive Epidemiology of and Response to the High Pathogenicity Avian Influenza (H5N8) Epidemic in South African Coastal Seabirds, 2018. Transbound Emerg Dis, 2708458; https://doi.org/2708410.2701155/2702023/2708458. [Google Scholar]
- Abolnik, C. The Molecular Epidemiology of Clade 2.3.4.4B H5N1 High Pathogenicity Avian Influenza in Southern Africa, 2021–2022. Viruses, 2023; 15, 1383, https://doi.org/1310.3390/v15061383. [Google Scholar]
- Muñoz, G. Stranding and Mass Mortality in Humboldt Penguins (Spheniscus humboldti), Associated to HPAIV H5N1 Outbreak in Chile. Preventative Veterinary Medicine, 2024. [Google Scholar] [CrossRef]
- Roberts, L.C. Vaccination of African penguins (Spheniscus demersus) against high-pathogenicity avian influenza. VetRecord, 2023. [Google Scholar] [CrossRef]
- Molini, U.; et al. Highly pathogenic avian influenza H5N1 virus outbreak among Cape cormorants (Phalacrocorax capensis) in Namibia, 2022. Emerging Microbes & Infections 2023, 12, 2167610. [Google Scholar]
- Barriga, G.P.; et al. Avian influenza virus H5 strain with North American and Eurasian Genes in an Antarctic Penguin. Emerg. Infect. Dis. 2016, 22, 2221–2223. [Google Scholar] [CrossRef]
- Hurt, A.C.; Su, Y.C.F.; Aban, M.; Peck, H.; Lau, H.; Baas, C.; Deng, Y.-M.; Spirason, N.; Ellström, P.; Hernandez, J.; et al. Evidence for the Introduction, Reassortment, and Persistence of Diverse Influenza A Viruses in Antarctica. J. Virol. 2016, 90, 9674–9682. [Google Scholar] [CrossRef]
- Kuiken, T. Is low pathogenic avian influenza virus virulent for wild waterbirds? Proceedings. Biological sciences 2013, 280, 20130990, doi: 20130910.20131098/rspb.20132013.20130990. [Google Scholar]
- Bengtsson, D.; Avril, A.; Gunnarsson, G.; Elmberg, J.; Söderquist, P.; Norevik, G.; Tolf, C.; Safi, K.; Fiedler, W.; Wikelski, M.; et al. Movements, Home-Range Size and Habitat Selection of Mallards during Autumn Migration. PLOS ONE 2014, 9, e100764. [Google Scholar] [CrossRef]
- van Toor, M.L.; Avril, A.; Wu, G.; Holan, S.H.; Waldenström, J. As the Duck Flies—Estimating the Dispersal of Low-Pathogenic Avian Influenza Viruses by Migrating Mallards. Front. Ecol. Evol. 2018, 6. [Google Scholar] [CrossRef]
- van Dijk, J. et al. Weak negative associations between avian influenza virus infection and movement behaviour in a key host species, the mallard Anas platyrhynchos. Oikos 10, 1293–1303.
- van Gils, J.A.; Munster, V.J.; Radersma, R.; Liefhebber, D.; Fouchier, R.A.; Klaassen, M. Hampered Foraging and Migratory Performance in Swans Infected with Low-Pathogenic Avian Influenza A Virus. PLOS ONE 2007, 2, e184. [Google Scholar] [CrossRef]
- World Organisation for Animal Health. Chapter 3.3.4. AVIAN INFLUENZA (INCLUDING INFECTION WITH HIGH PATHOGENICITY AVIAN INFLUENZA VIRUSES) WOAH Terrestrial Manual, 2025, p 28 pages.
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a Real-Time Reverse Transcriptase PCR Assay for Type A Influenza Virus and the Avian H5 and H7 Hemagglutinin Subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef]
- Nagy, A. et al. A universal RT-qPCR assay for "One Health" detection of influenza A viruses. Plos One 16 (2021).
- Laconi, A.; et al. Detection of avian influenza virus: a comparative study of the in silico and in vitro performances of current RT-qPCR assays. Sci Rep-Uk, 2020; 10, 8441. [Google Scholar]
- Xie, R.; Edwards, K.M.; Wille, M.; Wei, X.; Wong, S.-S.; Zanin, M.; El-Shesheny, R.; Ducatez, M.; Poon, L.L.M.; Kayali, G.; et al. The episodic resurgence of highly pathogenic avian influenza H5 virus. Nature 2023, 622, 810–817. [Google Scholar] [CrossRef]
- Slomka, M.J.; Reid, S.M.; Byrne, A.M.P.; Coward, V.J.; Seekings, J.; Cooper, J.L.; Peers-Dent, J.; Agyeman-Dua, E.; de Silva, D.; Hansen, R.D.E.; et al. Efficient and Informative Laboratory Testing for Rapid Confirmation of H5N1 (Clade 2.3.4.4) High-Pathogenicity Avian Influenza Outbreaks in the United Kingdom. Viruses 2023, 15, 1344. [Google Scholar] [CrossRef]
- James, J.; Slomka, M.J.; Reid, S.M.; Thomas, S.S.; Mahmood, S.; Byrne, A.M.P.; Cooper, J.; Russell, C.; Mollett, B.C.; Agyeman-Dua, E.; et al. Development and Application of Real-Time PCR Assays for Specific Detection of Contemporary Avian Influenza Virus Subtypes N5, N6, N7, N8, and N9. Avian Dis. 2018, 63, 209–218. [Google Scholar] [CrossRef]
- Ulloa, M.; et al. Mass mortality event in South American sea lions correlated to highly pathogenic avian influenza (HPAI) H5N1 outbreak in Chile. 2023; 43, 8–18. [Google Scholar]
- Ariyama, N.; et al. Emergence and rapid dissemination of highly pathogenic avian influenza virus H5N1 clade 2.3.4.4b in wild birds, Chile. bioRxiv (2023).
- World Organisation for Animal Health. Wildlife under threat as avian influenza reaches Antarctica. 2024. Available online: https://www.woah.org/en/wildlife-under-threat-as-avian-influenza-reaches-antarctica/.
- Agüero, M.; Monne, I.; Sánchez, A.; Zecchin, B.; Fusaro, A.; Ruano, M.J.; Arrojo, M.d.V.; Fernández-Antonio, R.; Souto, A.M.; Tordable, P.; et al. Highly pathogenic avian influenza A(H5N1) virus infection in farmed minks, Spain, October 2022. Eurosurveillance 2023, 28, 2300001–10. [Google Scholar] [CrossRef]
- Alkie, T.N.; Lopes, S.; Hisanaga, T.; Xu, W.; Suderman, M.; Koziuk, J.; Fisher, M.; Redford, T.; Lung, O.; Joseph, T.; et al. A threat from both sides: Multiple introductions of genetically distinct H5 HPAI viruses into Canada via both East Asia-Australasia/Pacific and Atlantic flyways. Virus Evol. 2022, 8, veac077. [Google Scholar] [CrossRef] [PubMed]
- Tammiranta, N.; Isomursu, M.; Fusaro, A.; Nylund, M.; Nokireki, T.; Giussani, E.; Zecchin, B.; Terregino, C.; Gadd, T. Highly pathogenic avian influenza A (H5N1) virus infections in wild carnivores connected to mass mortalities of pheasants in Finland. Infect. Genet. Evol. 2023, 111, 105423. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.E.; Ahrens, A.K.; Ali, A.; El-Kady, M.F.; Hafez, H.M.; Mettenleiter, T.C.; Beer, M.; Harder, T. Improved Subtyping of Avian Influenza Viruses Using an RT-qPCR-Based Low Density Array: ‘Riems Influenza a Typing Array’, Version 2 (RITA-2). Viruses 2022, 14, 415. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Hoffmann, D.; Henritzi, D.; Beer, M.; Harder, T.C. Riems influenza a typing array (RITA): An RT-qPCR-based low density array for subtyping avian and mammalian influenza a viruses. Sci Rep-Uk, 2016; 6, 27211. [Google Scholar]
- Pohlmann, A.; Stejskal, O.; King, J.; Bouwhuis, S.; Packmor, F.; Ballstaedt, E.; Hälterlein, B.; Hennig, V.; Stacker, L.; Graaf, A.; et al. Mass mortality among colony-breeding seabirds in the German Wadden Sea in 2022 due to distinct genotypes of HPAIV H5N1 clade 2.3.4.4b. J. Gen. Virol. 2023, 104, 001834. [Google Scholar] [CrossRef] [PubMed]
- Mirolo, M.; Pohlmann, A.; Ahrens, A.K.; Kühl, B.; Rubio-Garcìa, A.; Kramer, K.; Meinfelder, U.; Rosenberger, T.; Morito, H.L.; Beer, M.; et al. Highly pathogenic avian influenza A virus (HPAIV) H5N1 infection in two European grey seals ( Halichoerus grypus ) with encephalitis. Emerg. Microbes Infect. 2023, 12, e2257810. [Google Scholar] [CrossRef] [PubMed]
- Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; McNally, A.; Essen, S.; Brown, I.H. Validated H5 Eurasian Real-Time Reverse Transcriptase–Polymerase Chain Reaction and Its Application in H5N1 Outbreaks in 2005–2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Rijks, J.M.; Leopold, M.F.; Kühn, S.; Veld, R.I.; Schenk, F.; Brenninkmeijer, A.; Lilipaly, S.J.; Ballmann, M.Z.; Kelder, L.; de Jong, J.W.; et al. Mass Mortality Caused by Highly Pathogenic Influenza A(H5N1) Virus in Sandwich Terns, the Netherlands, 2022. Emerg. Infect. Dis. 2022, 28, 2538–2542. [Google Scholar] [CrossRef]
- Slomka, M.J.; To, T.L.; Tong, H.H.; Coward, V.J.; Hanna, A.; Shell, W.; Pavlidis, T.; Densham, A.L.E.; Kargiolakis, G.; Arnold, M.E.; et al. Challenges for accurate and prompt molecular diagnosis of clades of highly pathogenic avian influenza H5N1 viruses emerging in Vietnam. Avian Pathol. 2012, 41, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Floyd, T.; Banyard, A.C.; Lean, F.Z.; Byrne, A.M.; Fullick, E.; Whittard, E.; Mollett, B.C.; Bexton, S.; Swinson, V.; Macrelli, M.; et al. Encephalitis and Death in Wild Mammals at a Rehabilitation Center after Infection with Highly Pathogenic Avian Influenza A(H5N8) Virus, United Kingdom. Emerg. Infect. Dis. 2021, 27, 2856–2863. [Google Scholar] [CrossRef]
- James, J.; Seekings, A.H.; Skinner, P.; Purchase, K.; Mahmood, S.; Brown, I.H.; Hansen, R.D.; Banyard, A.C.; Reid, S.M. Rapid and sensitive detection of high pathogenicity Eurasian clade 2.3.4.4b avian influenza viruses in wild birds and poultry. J. Virol. Methods 2022, 301, 114454. [Google Scholar] [CrossRef]
- Naguib, M.M.; Graaf, A.; Fortin, A.; Luttermann, C.; Wernery, U.; Amarin, N.; A Hussein, H.; Sultan, H.; Al Adhadh, B.; Hassan, M.K.; et al. Novel real-time PCR-based patho- and phylotyping of potentially zoonotic avian influenza A subtype H5 viruses at risk of incursion into Europe in 2017. Eurosurveillance 2017, 22, 15–26. [Google Scholar] [CrossRef]
- Letsholo, S.L.; James, J.; Meyer, S.M.; Byrne, A.M.P.; Reid, S.M.; Settypalli, T.B.K.; Datta, S.; Oarabile, L.; Kemolatlhe, O.; Pebe, K.T.; et al. Emergence of High Pathogenicity Avian Influenza Virus H5N1 Clade 2.3.4.4b in Wild Birds and Poultry in Botswana. Viruses 2022, 14, 2601. [Google Scholar] [CrossRef]
- Grant, M. et al. Highly Pathogenic Avian Influenza (HPAI H5Nx, Clade 2.3.4.4.b) in Poultry and Wild Birds in Sweden: Synopsis of the 2020-2021 Season. Veterinary Sciences 9 (2022).
- King, J.; Schulze, C.; Engelhardt, A.; Hlinak, A.; Lennermann, S.-L.; Rigbers, K.; Skuballa, J.; Staubach, C.; Mettenleiter, T.C.; Harder, T.; et al. Novel HPAIV H5N8 Reassortant (Clade 2.3.4.4b) Detected in Germany. Viruses 2020, 12, 281. [Google Scholar] [CrossRef]
- Fereidouni, S.; Starick, E.; Grund, C.; Globig, A.; Mettenleiter, T.; Beer, M.; Harder, T. Rapid molecular subtyping by reverse transcription polymerase chain reaction of the neuraminidase gene of avian influenza A viruses. Veter- Microbiol. 2009, 135, 253–260. [Google Scholar] [CrossRef]
- Sagong, M.; Lee, Y.; Song, S.; Cha, R.M.; Lee, E.; Kang, Y.; Cho, H.; Kang, H.; Lee, Y.; Lee, K. Emergence of clade 2.3.4.4b novel reassortant H5N1 high pathogenicity avian influenza virus in South Korea during late 2021. Transbound. Emerg. Dis. 2022, 69, E3255–E3260. [Google Scholar] [CrossRef]
Table 1.
Consideration for the selection or design of PCR assays for the detection of 2.3.4.4b HPAI H5N1.
Table 1.
Consideration for the selection or design of PCR assays for the detection of 2.3.4.4b HPAI H5N1.
| Consideration | Reason | What to do |
|---|---|---|
| Assay is sensitive | Assay can detect influenza A viruses from samples adequately. | > Select an assay that is well validated > Select an assay that is frequently assessed against new strains containing mutations within primer and probe binding regions > Known well-characterised positive controls (should fall into known Ct value range) |
| Assay is specific to target (clade 2.3.4.4b HPAI H5N1) | Both LPAI H5 and HPAI H5 co-circulate, so imperative to distinguish as risk, response, notification pathway differs depending on the result. | > Select an assay that is well validated > Use both LPAI H5 and HPAI H5 controls > Sequencing of either the HA PCR products, or whole genome sequencing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



