
Submitted:
23 April 2024
Posted:
24 April 2024
You are already at the latest version
Abstract
BUB1 is overexpressed in most human solid cancers including breast cancer. Higher BUB1 levels are associated with poor prognosis especially in patients with triple-negative breast cancer (TNBC). Women with TNBC often develop resistance to chemotherapy and radiotherapy, which are still the mainstay of treatment for TNBC. Our previous studies demonstrated that BUB1 kinase inhibitor (BAY1816032) reduced tumor cell proliferation and significantly enhanced radiotherapy efficacy in TNBC. In this study, we evaluated the effectiveness of BAY1816032 with a PARP inhibitor (olaparib), platinum agent (cisplatin), and microtubule poison (paclitaxel) alone or in combination with radiotherapy using cytotoxicity and clonogenic survival assays. BUB1 inhibitor sensitized BRCA1/2 wild-type SUM159 and MDA-MB-231 cells to olaparib, cisplatin and paclitaxel synergistically (combination index; CI
Keywords:
TNBC
; BAY1816032
; BUB1
; PARP
; olaparib
; cisplatin
; paclitaxel
; radiation
; chemoradiation
Introduction
Triple-negative breast cancer (TNBC) is a subtype of breast cancer characterized by the absence of human epidermal growth factor receptor 2 (HER2), progesterone receptor (PR), and estrogen receptor (ER) [1]. TNBC is more difficult to cure because it lacks these receptors which are frequently targeted for treatment [2,3]. TNBC accounts for approximately 10-15% of all breast cancer cases [4]. The recurrence risk of TNBC is higher in the first few years following diagnosis and it tends to be more aggressive [5]. While surgery, chemotherapy and/or radiotherapy are the mainstays of treatment, intrinsic or acquired resistance results in poor clinical outcome [6,7,8].
Currently, the frontline standard chemotherapy, composed of anthracyclines, alkylating agents, and taxanes, is commonly used to treat high-risk and locally advanced TNBC. Chemotherapeutic agents induce cell death by directly or indirectly causing DNA damage [9]. Cancer cells acquire resistance to chemotherapy by enhancing DNA damage responses. Targeting DNA repair pathway can increase the tumor sensitivity to chemotherapies in TNBC [10]. For example, about 15% of TNBC patients harbor germline mutations in BRCA1 or BRCA2 making them susceptible to targeted agents such as PARP inhibitors [11]. However, chemotherapy options for patients without these mutations are currently limited. DNA damage is a critical determinant of radiation-induced cell death [12]. Radiation induces both single strand breaks (SSB) and double strand breaks (DSB). The ability of cells to recognize and respond to DSB is fundamental in determining the sensitivity (or resistance) of cells to radiation [13]. Following DNA damage, cell cycle checkpoints are activated to block cell cycle progression and prevent propagation of cells with damaged DNA [14]. DSB repair is comprised of two major and mechanistically distinct processes: non-homologous end-joining (NHEJ) and homologous recombination (HR). Both DNA damage repair and cell cycle checkpoints are positively regulated by several kinases and inhibition of these kinases results in enhanced radiotherapy efficacy. TNBC have higher expression of cell-cycle related growth-regulatory molecules [15]. The molecular drivers of therapeutic resistance are complex and include increased drug resistance due to drug efflux, chemotherapy and radiation resistance due to enhanced DNA repair, senescence escape, epigenetic changes, tumor heterogeneity, abnormal tumor microenvironment, epithelial-to-mesenchymal transition (EMT) and/or changes in cell metabolism [16].
Multiple kinases, including BUB1, positively regulate both DNA damage repair and cell cycle checkpoints. BUB1 is a G2/M cell cycle checkpoint kinase which perform several functions including accurate chromosomal segregation during cell division [17,18,19]. BUB1 dysregulation or mutations can lead to chromosomal instability and are associated with several types of cancers including TNBC [20,21]. BUB1 is overexpressed in most solid cancers. BUB1 transcripts are significantly higher in breast cancer cell lines and in high-grade primary breast cancer tissues compared to normal mammary epithelial cells, or in normal breast tissues [22,23]. Moreover, high BUB1 expression (transcript) correlates with extremely poor outcome in breast cancer [24,25]. Increased expression of BUB1 is associated with resistance to DNA-damaging agents (i.e., chemotherapy and radiotherapy) [26]. In previous studies, we showed that biochemical or genomic ablation of BUB1 was cytotoxic in TNBC and sensitized TNBC to radiation [27]. We also demonstrated earlier that BUB1's kinase activity exploits TGF-β signaling to drive aggressive cancer phenotypes including cell migration, invasion, and EMT [28]. BUB1 has been identified as a potential therapeutic target for improving the effectiveness of chemo-, radio-, and targeted therapies [29]. Pharmacological inhibition of BUB1 sensitized cancer cells to taxanes, ATR, or PARP inhibitors and significantly reduced tumor xenografts [30]. However, a role for BUB1 in improving the efficacy of radiation or chemoradiation in TNBC was not evaluated.
Materials and Methods
Co-Expression Studies Using cBioPortal Database
Breast cancer dataset (METABRIC, 2509 samples) from the cBio Cancer Genomics Portal (http://cbioportal.org/) was used to check the correlation between mRNA expression levels (Illumina HT-12 v3 microarray, 1866 samples) of BUB1 (query gene) and BRCA1, BRCA2, PARP1, PARP2, PARP3 and TP53. cBioPortal mRNA expression data are computed by comparing the relative expression of a particular gene in a tumor sample to the gene's expression distribution in a reference population of samples [31]. This cohort’s mRNA expression correlations were examined using Spearman’s test. Correlation plots were displayed along with the regression line and estimated significance (P values).
Chemicals
BUB1 kinase inhibitor, BAY1816032 (Catalog No. HY-103020) was obtained from MedChemExpress. Olaparib/AZD2281 (Catalog No. CT-A2281) and paclitaxel (Catalog No. CT-0502) were obtained from Chemietek, and cisplatin (PHR1624-200MG) was sourced from Millipore Sigma.
Cell Lines and Cell Culture
SUM159 cell line was obtained from Sofia Merajver (University of Michigan). The HAM’S F-12 media (Catalog No. 31765305, Thermo Fisher Scientific) supplemented with 5% FBS, 10 mM HEPES, 1 μg/ml Hydrocortisone, 6 μg/ml Insulin, and 1% Penicillin-Streptomycin was used to grow SUM159 cells. DMEM media (Catalog No. 30-2002, ATCC) supplemented with 10% FBS and 1% Penicillin-Streptomycin was used to culture MDA-MB-231 cells which were obtained from the American Type Culture Collection (ATCC). HCC1937 cells were grown in RPMI-1640 media supplemented with 10% FBS, and 1% Penicillin-Streptomycin. All cell lines were maintained at 37 °C in a 5% CO2 incubator and passaged at 70% confluence. Cells were routinely tested for Mycoplasma contamination using the MycoAlert PLUS kit (Lonza, Cat. No. LT07-705).
Drug Treatment and Radiation
BUB1 inhibitor (BUB1i) BAY1816032, AZD2281 (Olaparib), cisplatin, and paclitaxel were dissolved in DMSO (20 mM BUB1i, 10 mM olaparib, 20 mM cisplatin, and 10 mM paclitaxel) and stored as single use aliquots at -80 °C. Working concentrations were prepared in serum and supplement-containing medium, and cells were treated with doses ranging from 1 nanomolar (1 nM) to 100 micromolar (100 µM). Cells were treated one hour after drug treatment using a CIX-3 orthovoltage unit (Xstrahl Life Sciences) with a copper filter.
Cell Growth and Viability
SUM159 and MDA-MB-231 were seeded at a density of 2000 cells per well and HCC1937 at a density of 4000 cells per well in 96-well plates. After 24 hours, cells were treated with BAY1816032, AZD2281 (olaparib), cisplatin, and paclitaxel at various concentrations for 72 h. Cytotoxicity was assessed using the alamarBlue cell viability kit (Thermo Fisher Scientific, Cat. No. DAL1100) according to the manufacturer's protocol. Absorbance was measured at 570 nM on a Synergy H1 Hybrid Reader (BioTek Instruments). The values were compared to vehicle/mock treated cells. A non-linear regression best-fit model was used to determine the IC50 values (GraphPad prism V9). Half maximal inhibitory concentration, or IC50, is the half-way point at which the compound completely inhibits a biological or biochemical activity. The combination index (C.I.) is used to determine the degree of drug interaction, and it was calculated from the IC50 by using the formula: C.I = IC50(A+B)/IC50(A) + IC50(A+B)/IC50(B). Here, IC50(A) and IC50(B) are the IC50 values obtained from each drug separately. IC50 (A + B) is the IC50 value of both drugs in combination. C.I > 1 implies antagonism, C.I = 1 entails additivity while C.I < 1 indicates synergistic effect. Significance between two groups were estimated using Student’s t-test (GraphPad Prism V9). The findings are presented as mean ± standard error of the Mean (SEMs). All experiments were performed in triplicates and were done at least three times. P < 0.05 was considered statistically significant.
Clonogenic Survival Assay
Cells were plated in 6-well plates at different cell densities in appropriate culture media and were maintained overnight at 37 °C. TNBC cell lines were then treated with BAY1816032 alone or in combination with AZD2281 (olaparib), cisplatin, or paclitaxel and were irradiated (0, 2, 4 Gy) after one hour. The cells were fixed and stained with methanol and crystal violet after being allowed to grow for 7-15 days or until visible colonies formed. A clone of 50 or more cells was considered a colony. The radiation enhancement ratio (rER) was estimated in MS Excel using the formula: D bar of varying inhibitor concentrations / D bar of vehicle (DMSO). The above formula indicates the radiation dose required to produce a certain level of cell killing in the absence of the inhibitor (DMSO/vehicle) divided by the radiation dose required to produce the same level of cell killing in the presence of the inhibitor. Radiation sensitization was defined as rER > 1, whilst radiation resistance or protection was defined as rER < 1. All experiments were performed in triplicates and were conducted at least three times.
Results
Correlation of BUB1 mRNA Expression with BRCA1/2, PARP1/2/3 and TP53 in Breast Cancer
To determine the correlation between BUB1 and the genes associated with DNA damage response or repair pathways such as BRCA1, BRCA2, PARP1, PARP2, PARP3, and TP53 [32,33], we used the cBioPortal database to perform a co-expression analysis in breast cancer datasets. Spearman’s test showed a significant positive correlation between mRNA expression of BUB1 and BRCA1 (0.331, p = 4.5e-49), BRCA2 (0.123, p = 1.10e-7), PARP1 (0.390, p = 1.12e-68), and PARP2 (0.371, p = 5.67e-62) genes in breast cancer (Figure 1A–D). A significant negative correlation was observed for PARP3 (-0.489, p = 1.61e-112) (Figure 1E). TP53 showed positive correlation (6.68e-3, p = 0.773) but it was not significant (Figure 1F).
Antiproliferative Effects of Single Agent Olaparib, Cisplatin, and Paclitaxel in TNBC Cell Lines
Our previous studies indicated that BUB1 inhibition significantly reduced proliferation of TNBC cells but did not affect breast epithelial cells [34]. In this study, we assessed the effects of PARP inhibitor (olaparib/AZD2281) and chemotherapeutic agents (cisplatin and paclitaxel) in TNBC cell lines. The cytotoxicity of single agent (IC50) was determined using alamarBlue assay in SUM159 and MDA-MB-231 cells. The cells were treated for 72 h with different concentrations of drugs and absorbance was read. At higher concentrations of the drugs, the decrease in viability was similar for both the cell lines, with MDA-MB-231 generally presenting higher IC50 values than SUM159 for all three drugs. The IC50 for olaparib varied from 21.2 μM (SUM159) to 41.2 μM (MDA-MB-231; Figure 2A,B). Single agent cisplatin IC50 was 1.63 μM in SUM159 while it was 7.14 μM in MDA-MB-231 cells (Figure 2C,D) while single agent paclitaxel IC50 was less than 10 nM for both the cell lines (Figure 2E,F). We estimated single agent IC50 of BUB1 inhibitor BAY1816032 less than 4 μM in these cells [34].
BUB1 Inhibitor Synergistically Sensitizes TNBC to Olaparib, Cisplatin, and Paclitaxel
To assess the effects of BUB1 inhibition on PARP inhibitor sensitivity and chemosensitivity in TNBC cell lines, cell viability was assessed 72 h after exposure to olaparib (1 μM), cisplatin (1 μM), and paclitaxel (1.5 nM) in combination with BAY1816032 (1 μM). Our results demonstrated that BAY1816032 significantly increased cytotoxicity of PARP inhibitor in TNBC cell lines (Figure 3A,B). Consistent with this, inhibition of BUB1 also increased sensitivity to cisplatin (Figure 3C,D) and paclitaxel (Figure 3E,F) compared to cisplatin or paclitaxel alone. BUB1 inhibitor demonstrated strong synergistic effects with PARPi, cisplatin, and paclitaxel with C.I. less than 1 in both the cell lines. This synergism was observed at approximately 10-fold less drug concentrations (than single agents). The C.I. values in SUM159 and MDA-MB-231 for 1 μM olaparib + 1 μM BUB1i were 0.55 and 0.52, 1 μM cisplatin + 1 μM BUB1i were 0.9 and 0.63, and 1.5 nM paclitaxel + 1 μM BUB1i were 0.78 and 0.75, respectively.
Olaparib, Cisplatin, and Paclitaxel Differentially Radiosensitize SUM159 and MDA-MB-231 Cell Lines
We evaluated single agent radiosensitization potential of AZD2281 (olaparib), cisplatin, and paclitaxel in SUM159 and MDA-MB-231 cells before combining with BUB1 inhibitor. The cells were exposed to olaparib (500 nM and 2.5 μM), cisplatin (750 nM and 1 μM) or paclitaxel (1 nM and 1.5 nM) 1h prior to irradiation and clonogenic cell survival assays were performed as described under methods. SUM159 cells were moderately radiosensitized at lower olaparib concentration (rER = 1.45; 500 nM) whereas MDA-MB-231 showed marginal radiosensitization (rER = 1.06; Figure 4A,B). Both the cell lines demonstrated increased radiosensitization at non-toxic increased concentration (2.5 μM; rER = 1.71 and 1.40). Although there was moderate radiosensitization with cisplatin (rER = 1.27 and 1.13; Figure 4C,D) and paclitaxel (rER = 1.16 and 1.15; Figure 4E,F) at the doses tested, the radiosensitization by single agent olaparib was much stronger in SUM159 and MDA-MB-231.
BUB1 Inhibitor Enhances the Radiation Sensitization by Olaparib, Cisplatin and Paclitaxel in SUM159 and MDA-MB-231 Cell Lines
Individually, olaparib, cisplatin and paclitaxel reduced the colony formation ability in SUM159 and MDA-MB-231 cells indicative of radiation sensitization (Figure 5A–F, red curves). BUB1 inhibitor BAY1816032 moderately sensitized these cells to radiation (Figure 5A–F, blue curves). However, combining BUB1i with olaparib (Figure 5A,B), cisplatin (Figure 5C,D) and paclitaxel (Figure 5E,F) led to enhanced radiosensitization as seen by significant increase in the rER (Figure 5A–F, magenta curves).
The greatest reduction in the surviving fraction with BUB1i and olaparib combination was observed in SUM159 (rER 1.65; Figure 5A), while most reduction with BUB1i and cisplatin combination was in MDA-MB-231 cells (rER 1.65; Figure 5D). BUB1 inhibitor with paclitaxel significantly increased radiation sensitization in both the cell lines (rER 1.53 in SUM159 and rER 1.67 in MDA-MB-231; Figure 5E,F). This data confirms that BUB1 inhibition potentiates the cytotoxic effects of olaparib, cisplatin and paclitaxel with radiation in TNBC cell lines.
BUB1 Inhibitor BAY1816032 Sensitizes BRCA Mutant TNBC Cell Line to PARP Inhibitor and Radiation
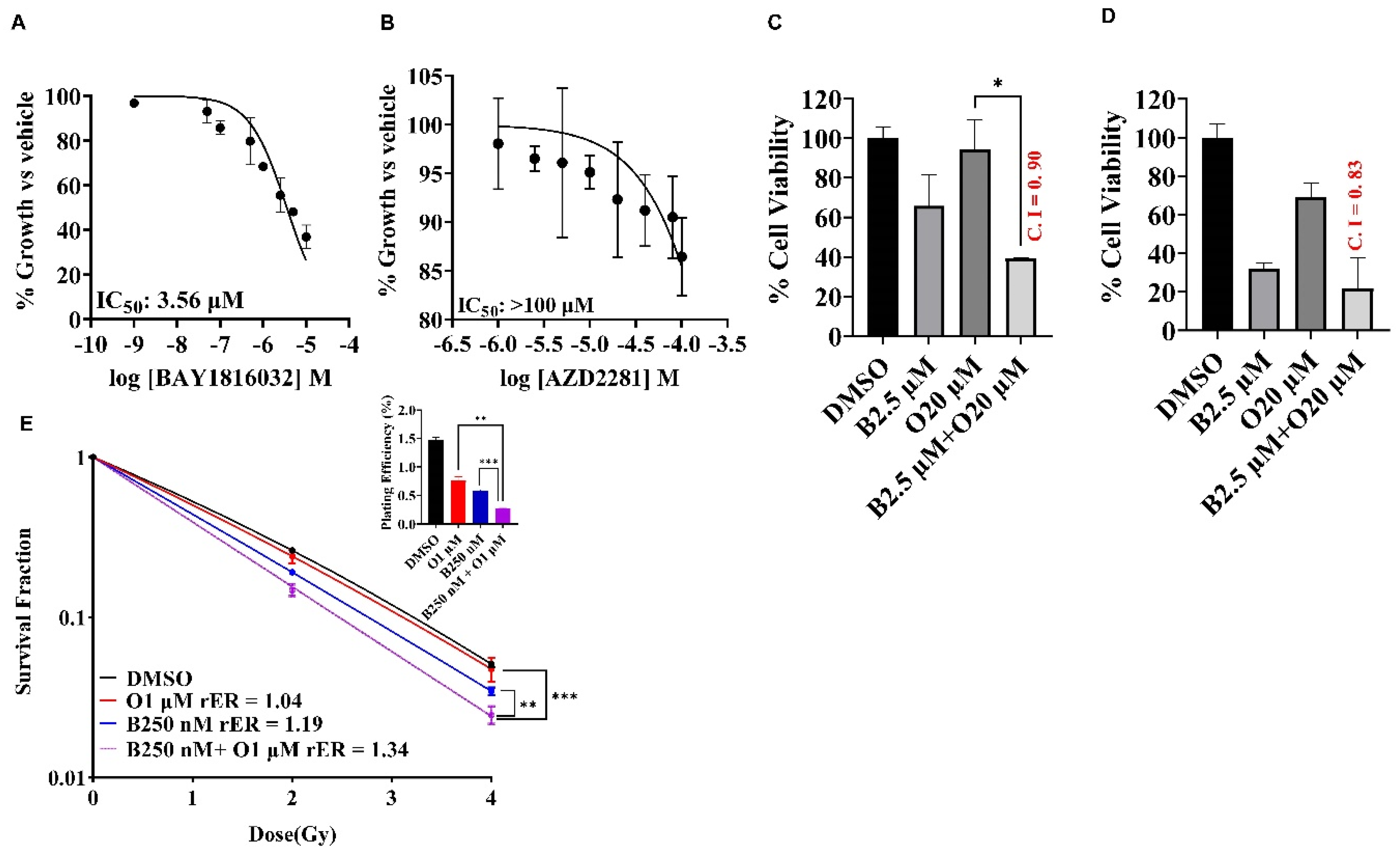
PARP inhibitors induce synthetic lethality in BRCA1/2 mutated cancers by selectively targeting tumor cells that fail to repair DSB. Since BUB1 regulates DSB repair, we examined the potential benefits of combining BUB1i with PARPi (olaparib) in a TNBC cell line that carry BRCA mutations (HCC1937). Single agent BUB1 inhibitor BAY1816032 yielded IC50 at 3.56 μM (Figure 6A) while single agent olaparib IC50 was estimated to be close to/higher than 100 μM (Figure 6B). We then investigated the efficacy of BUB1i in combination with olaparib in HCC1937 cells to check if the BRCA mutant cell line can become sensitive to olaparib in presence of BUB1i. Our data indicates that BUB1i sensitized HCC1937 cells to olaparib synergistically (CI < 1) at 5-fold lower olaparib concentrations than IC50 (Figure 6C). Inclusion of radiation further potentiated the synergistic effects of BUB1i with olaparib (CI 0.83, Figure 6D). We next performed clonogenic survival assay to reconfirm the synergistic effect of the trimodality treatment in these cells. BUB1 inhibitor BAY1816032 demonstrated statistically significant radiation sensitization with 100-fold less olaparib concentration than the IC50 in these cells (Figure 6E).
Discussion
In this study, we assessed the efficacy of BUB1 kinase inhibitor in combination with a PARP inhibitor (olaparib), platinum agent (cisplatin), and paclitaxel and radiation in TNBC. We observed that BUB1 sensitized TNBC to cisplatin, PARP inhibitor olaparib, and paclitaxel and improved the radiation-mediated cytotoxicity of these agents. Interestingly, we also demonstrate that BUB1i sensitized BRCA mutated TNBC cell line to olaparib in combination with radiation. Overall, our results provide evidence that targeting BUB1 with PARP inhibitor, cisplatin or paclitaxel with radiation would be a novel approach for effectively treating TNBC.
TNBC is the most aggressive type of breast cancer which generally occur in younger women, particularly those of African ancestry, and is difficult to cure using adjuvant therapy only [35]. The molecular drivers of therapeutic resistance in TNBC [36,37] are complex and may include increased drug efflux, enhanced DNA repair, senescence escape, epigenetic changes, tumor heterogeneity, abnormal tumor microenvironment or epithelial-to-mesenchymal transition. Germline mutations of BRCA1, BRCA2, and TP53 genes encoding important components of the DNA-damage response (DDR) are associated with high-risk of TNBC [38]. Among several PARP family members, PARP1, PARP2, and PARP3 also play vital roles in DNA damage and repair processes [39] which may contribute to the anti-tumor activity of PARP inhibitors [40]. In this study, all the TNBC predisposition genes showed significantly positive correlation with BUB1, except PARP3 and TP53 at mRNA level (Figure 1). PARPi therapeutic effectiveness is thought be higher in tumors that harbor germline or somatic BRCA mutations than in BRCA wt tumors. BRCA mutations or inherent tumor sensitivity to platinum agents are interpreted as signs of deficiency in DSB repair by HR and favorable response to PARP inhibitors. However, clinical benefit from these agents is not uniform across all BRCA-mutated or platinum-responsive patients. Contrary, a small number of patients with platinum resistant or BRCA wt tumors get benefitted from PARPi. Therefore, identification and validation of additional reliable biomarkers will help select patients that will benefit from PARPi-based therapies in the absence of BRCA mutations or platinum sensitivity.
As TNBC accounts for about 30% of breast cancer-associated deaths with lack of specific treatment targets [41], we envision that identification of novel molecular targets would improve TNBC outcome. Molecularly targeted agents can enhance chemoradiation sensitivity [13]. Given BUB1’s strong correlation to aggressiveness and different classes of drugs [23] and our observation that BUB1 inhibition sensitizes TNBC to radiation and lung cancers to chemoradiation [27,42,43,44], we rationalized that combining BUB1 inhibitor would provide strong chemoradiation sensitization in TNBC. Although the effectiveness of BUB1 inhibitor BAY1816032 was evaluated with PARPi, cisplatin and paclitaxel in a prior study [30], the combination with cisplatin resulted in antagonistic effects and BUB1's potential role in improving the efficacy of chemoradiation in TNBC was not assessed.
As PARP inhibitors, taxanes and platinum compounds are suitable treatment options for TNBC [30,45], we first investigated the cytotoxic effects of single agent AZD2281 (olaparib), cisplatin, or paclitaxel on TNBC cell lines (SUM159, MDA-MB-231). TNBC cells demonstrated high IC50 (20-40 µM) for olaparib (Figure 2) which is in line with earlier findings that TNBC cells are resistant to single agent PARPi [46]. SUM159 and MDA-MB-231 cells were modestly sensitive to cisplatin and highly sensitive to paclitaxel (Figure 2). This is consistent with a previous study which reported that BL (basal-like) subtypes was more sensitive to cisplatin than to MSL (mesenchymal stem-like) and LAR (luminal androgen receptor) [36].
Our data demonstrate synergistic effects with BUB1 inhibitor BAY1816032 in combination with cisplatin, paclitaxel and PARPi (olaparib) (Figure 3). Our results expand the finding by Siemeister et al. [30] on BUB1 inhibition sensitizing TNBC to paclitaxel and olaparib. However, our results are opposite to their findings with cisplatin. This could be due to the limited number of drug concentrations and combinations of BAY1816032 and cisplatin in their assays [30]. PARP inhibitors radiosensitize TNBC in preclinical models [47] and have been safe and effective in high-risk TNBC patients in clinical studies [48,49]. We tested whether BUB1 inhibitor (i.e., molecularly targeted agent) would further enhance the radiosensitization by PARPi (Figure 4). Indeed, BUB1i significantly increased radiosensitization by olaparib in SUM159 and MDA-MB-231 cells (Figure 5). Not surprisingly, BUB1i also enhanced radiosensitization by cisplatin and paclitaxel (Figure 5). These findings consolidate a role for BUB1 as a molecular target for increasing the efficacy of radiotherapy [50] and are supported by encouraging results with WEE1 inhibitors that have been combined with radiation in clinical trials with promising results [51,52].
Since PARP inhibitors have recently been approved for early-stage disease and for BRCA mutations in the metastatic setting, it is worth considering how the efficacy of PARP inhibitors can be improved [53]. This study provides evidence that BUB1i increases the sensitivity of PARPi (olaparib) in BRCA mutant TNBC cells (Figure 6). This effect was compounded when combined with radiation (Figure 6) similar to what been suggested by Kawanishi [54] and Feng [55] for BRCA mutant TNBC. Although recent studies have demonstrated that PARP inhibitor sensitivity does not depend on BRCA mutations [56], different cell lines have differential sensitivity/resistance to PARP inhibitors. Our findings demonstrate that BUB1i can sensitize different TNBC cell lines irrespective of BRCA mutation status to PARPi (Figure 3 and Figure 6). In the current study, we used a single PARP inhibitor olaparib which inhibits both PARP1 and PARP2. It would be interesting to evaluate the effect of BUB1i with additional PARP inhibitors that inhibit either PARP1 alone (talazoparib), PARP 1/2 both equally (niraparib) or PARP1/2/3 (rucaparib) [57]. Our findings are further support by observations that CDK1 inhibitor [58] or androgen receptor inhibitors [59] increase PARPi sensitivity in breast cancers while DNAPK inhibitor are effective at sensitizing TNBC to PARPi and IR [60].
PARP1/2 proteins usually detect SSB and recruit factors to repair the SSB. PARPi causes either PARP trapping on DNA break sites that lead to replication fork collapse and cell death (especially in BRCA mutant cell lines [61]) or PARPi can upregulate NHEJ and reduce HR leading to genomic instability and cell killing [62]. Although the current studies were limited in their scope and we did not explore the mechanism of BUB1 mediated sensitization, we speculate that BUB1i sensitizes with PARP inhibitors because PARPi can increase the dependency on NHEJ for which BUB1 is critical. Platinum agents (cisplatin, carboplatin) form DNA adducts that causes DNA replication errors leading from SSB to DSB, cell-cycle arrest in G1-S and ultimately cell death [63,64]. Increased platinum-DNA adduct repair has been shown to be associated with cisplatin resistance [65]. Similarly, paclitaxel blocks depolarization of microtubules, leading to improper chromosome segregation, G2/M cell-cycle arrest [66,67] resulting in apoptotic cell-death [68,69]. BUB1 not only regulates cell-cycle but it also regulates DNA damage response through NHEJ. TP53 is mutated in majority of TNBC [70]. BRCA deletion has been shown to cause changes in the level/mutations in p53 and BUB1 [71,72,73] suggesting that these may be regulated by the same mechanism, thus BUB1 inhibition could sensitize TNBC tumors to chemotherapy, even in p53 mutant background. Therefore, our findings in this study, that BUB1 inhibition increases cytotoxicity of paclitaxel, cisplatin or olaparib with radiation in TNBC cell lines could be due to BUB1’s ability to target multiple pathways. Our findings strongly support nomination of BUB1 as a potential biomarker and a therapeutic target for chemoradiosensitization in TNBC.
Conclusions: The data presented here demonstrate that BUB1 inhibition sensitizes TNBC to PARP inhibitor and radiation irrespective of the BRCA mutation status. Moreover, inhibition of BUB1 synergistically sensitizes TNBC cell lines to cisplatin and paclitaxel with radiotherapy. Our studies nominate BUB1 as a novel molecular target for improving chemoradiation efficacy in TNBC.
Author contributions
SS: performed all the experiments with help from ST. SS also drafted the manuscript with inputs from all other authors. FS, BM, and EW helped with data analysis and gave feedback on the study. SN conceptualized and directed the study. SN also edited the final manuscript.
Funding
This work was supported by NCI R21 (1R21CA252010-01A1), HFHS Research Administration Start up, HFHS Proposal Development Award and HFHS-Rad Onc Start Up, Game on Cancer award to Shyam Nyati. We also thank HFCI for providing Translational Oncology Postdoctoral Fellowship to Sushmitha Sriramulu.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data availability
All the relevant data is already presented in the manuscript. Any additional data will be available upon request to the corresponding author.
Conflict of interest
SS, ST, SB, SN: no COI, FS: Varian Medical Systems Inc - Honorarium and travel reimbursement for lectures and talks, Varian Noona – Member of Medical Advisory Board - Honorarium (no direct conflict), BM: Research support from Varian, ViewRay, and Philips (no direct conflict). EW: Genentech research support for clinical trials.
References
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Research 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Wu, Y.; Ma, F.; Kaklamani, V.; Xu, B. Advances in medical treatment of breast cancer in 2022. Cancer Innovation 2023, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Niwińska, A.; Murawska, M.; Pieńkowski, T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Medical Oncology 2013, 30, 388. [Google Scholar] [CrossRef] [PubMed]
- Newton, E.E.; Mueller, L.E.; Treadwell, S.M.; Morris, C.A.; Machado, H.L. Molecular Targets of Triple-Negative Breast Cancer: Where Do We Stand? Cancers 2022, 14, 482. [Google Scholar] [CrossRef] [PubMed]
- Corrales-Guerrero, S.; Cui, T.; Castro-Aceituno, V.; Yang, L.; Nair, S.; Feng, H.; et al. Inhibition of RRM2 radiosensitizes glioblastoma and uncovers synthetic lethality in combination with targeting CHK1. Cancer Lett 2023, 570, 216308. [Google Scholar] [CrossRef]
- Pesch, A.M.; Chandler, B.C.; Michmerhuizen, A.R.; Carter, H.M.; Hirsh, N.H.; Wilder-Romans, K.; et al. Bcl-xL inhibition radiosensitizes PIK3CA/PTEN wild-type triple negative breast cancers with low Mcl-1 expression. Cancer Res Commun 2022, 2, 679–693. [Google Scholar] [CrossRef]
- Neal, J.A.; Sugiman-Marangos, S.; VanderVere-Carozza, P.; Wagner, M.; Turchi, J.; Lees-Miller, S.P.; et al. Unraveling the complexities of DNA-dependent protein kinase autophosphorylation. Mol Cell Biol 2014, 34, 2162–2175. [Google Scholar] [CrossRef]
- Jin, J.; Tao, Z.; Cao, J.; Li, T.; Hu, X. DNA damage response inhibitors: An avenue for TNBC treatment. Biochim Biophys Acta Rev Cancer 2021, 1875, 188521. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer☆. Annals of Oncology 2021, 32, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol 2013, 3, 113. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Parsels, L.A.; Maybaum, J.; Lawrence, T.S. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov 2014, 4, 280–291. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: a therapeutically targetable double-edged sword. Journal of Experimental & Clinical Cancer Research 2016, 35, 153. [Google Scholar] [CrossRef]
- Speers, C.; Tsimelzon, A.; Sexton, K.; Herrick, A.M.; Gutierrez, C.; Culhane, A.; et al. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin Cancer Res 2009, 15, 6327–6340. [Google Scholar] [CrossRef] [PubMed]
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; et al. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Y.; Shu, H.J.; Oncel, D.; Chen, S.; Yu, H.T. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell 2004, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Y.; Sun, Y.X.; Harley, S.E.; Zou, H.; Yu, H.T. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 18012–18017. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Tang, Z. Bub1 multitasking in mitosis. Cell Cycle 2005, 4, 262–265. [Google Scholar] [CrossRef]
- Williams, G.L.; Roberts, T.M.; Gjoerup, O.V. Bub1: escapades in a cellular world. Cell Cycle 2007, 6, 1699–1704. [Google Scholar] [CrossRef]
- Zhu, L.J.; Pan, Y.; Chen, X.Y.; Hou, P.F. BUB1 promotes proliferation of liver cancer cells by activating SMAD2 phosphorylation. Oncol Lett 2020, 19, 3506–3512. [Google Scholar] [CrossRef]
- Yuan, B.; Xu, Y.; Woo, J.H.; Wang, Y.; Bae, Y.K.; Yoon, D.S.; et al. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res 2006, 12, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Cicirò, Y.; Ragusa, D.; Sala, A. Expression of the checkpoint kinase BUB1 is a predictor of response to cancer therapies. Scientific Reports 2024, 14, 4461. [Google Scholar] [CrossRef]
- Dai, H.; van't Veer, L.; Lamb, J.; He, Y.D.; Mao, M.; Fine, B.M.; et al. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Research 2005, 65, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Cai, J.H.; Wang, C.C.N. Identification of Key Prognostic Genes of Triple Negative Breast Cancer by LASSO-Based Machine Learning and Bioinformatics Analysis. Genes 2022, 13, 902. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Inamoto, T.; Tsujino, T.; Matsui, Y.; Konuma, T.; Nishimura, K.; et al. Increased BUB1B/BUBR1 expression contributes to aberrant DNA repair activity leading to resistance to DNA-damaging agents. Oncogene 2021, 40, 6210–6222. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Li, P.; Brown, S.L.; Siddiqui, F.; Movsas, B.; et al. Abstract 689: BUB1 interferes with the repair of radiation-induced DNA damage to radiosensitize triple-negative breast cancer. Cancer Research 2024, 84 (6_Supplement), 689. [Google Scholar] [CrossRef]
- Nyati, S.; Schinske-Sebolt, K.; Pitchiaya, S.; Chekhovskiy, K.; Chator, A.; Chaudhry, N.; et al. The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-β signaling. Sci Signal 2015, 8, ra1. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Duan, X.; Xiao, Y.; Yuan, M.; Zhao, Z.; Cui, X.; et al. BUB1 Is Identified as a Potential Therapeutic Target for Pancreatic Cancer Treatment. Front Public Health 2022, 10, 900853. [Google Scholar] [CrossRef]
- Siemeister, G.; Mengel, A.; Fernández-Montalván, A.E.; Bone, W.; Schröder, J.; Zitzmann-Kolbe, S.; et al. Inhibition of BUB1 Kinase by BAY 1816032 Sensitizes Tumor Cells toward Taxanes, ATR, and PARP Inhibitors In Vitro and In Vivo. Clin Cancer Res 2019, 25, 1404–1414. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J Hematol Oncol 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Li, P.; Brown, S.L.; Siddiqui, F.; Movsas, B.; et al. BUB1 inhibition radiosensitizes triple-negative breast cancer by targeting the DNA-damage repair pathways. Cancer Research 2023, 83 (7_Supplement), 2816. [Google Scholar] [CrossRef]
- Kyndi, M.; Sorensen, F.B.; Knudsen, H.; Overgaard, M.; Nielsen, H.M.; Overgaard, J.; et al. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: the Danish Breast Cancer Cooperative Group. J Clin Oncol 2008, 26, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; et al. Cisplatin-resistant triple-negative breast cancer subtypes: multiple mechanisms of resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Sun, L.; Gai, J.; Cao, Y.; Zhang, S. Exploring the resistance mechanism of triple-negative breast cancer to paclitaxel through the scRNA-seq analysis. PLoS ONE 2024, 19, e0297260. [Google Scholar] [CrossRef]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J Natl Cancer Inst 2018, 110, 855–862. [Google Scholar] [CrossRef]
- Langelier, M.F.; Riccio, A.A.; Pascal, J.M. PARP-2 and PARP-3 are selectively activated by 5' phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 2014, 42, 7762–7775. [Google Scholar] [CrossRef]
- Brown, J.S.; O'Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Li, J.; Qiu, J.; Han, J.; Li, X.; Jiang, Y. Tumor Microenvironment Characterization in Breast Cancer Identifies Prognostic Pathway Signatures. Genes 2022, 13, 1976. [Google Scholar] [CrossRef] [PubMed]
- Thoidingjam, S.; Sriramulu, S.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Gadgeel, S.; et al. Abstract 1115: Enhancing lung cancer sensitivity to chemotherapy, radiation, and chemo-radiation through inhibition of a mitotic checkpoint kinase BUB1. Cancer Research 2024, 84 (6_Supplement), 1115. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Li, P.; Brown, S.L.; Siddiqui, F.; Movsas, B.; et al. Abstract 2816: BUB1 inhibition radiosensitizes triple-negative breast cancer by targeting the DNA-damage repair pathways. Cancer Research 2023, 83 (7_Supplement), 2816. [Google Scholar] [CrossRef]
- Thoidingjam, S.; Sriramulu, S.; Siddiqui, F.; Movsas, B.; Gadgeel, S.; Nyati, S. Abstract 6116: Ablation of mitotic checkpoint kinase BUB1 sensitizes lung adenocarcinoma to different classes of chemotherapy, radiation, and chemo-radiation. Cancer Research 2023, 83 (7_Supplement), 6116. [Google Scholar] [CrossRef]
- Singh, D.D.; Yadav, D.K. TNBC: Potential Targeting of Multiple Receptors for a Therapeutic Breakthrough, Nanomedicine, and Immunotherapy. Biomedicines 2021, 9, 876. [Google Scholar] [CrossRef] [PubMed]
- Marijon, H.; Lee, D.H.; Ding, L.; Sun, H.; Gery, S.; de Gramont, A.; et al. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed Pharmacother 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Michmerhuizen, A.R.; Pesch, A.M.; Moubadder, L.; Chandler, B.C.; Wilder-Romans, K.; Cameron, M.; et al. PARP1 Inhibition Radiosensitizes Models of Inflammatory Breast Cancer to Ionizing Radiation. Mol Cancer Ther 2019, 18, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Loap, P.; Loirat, D.; Berger, F.; Rodrigues, M.; Bazire, L.; Pierga, J.-Y.; et al. Concurrent Olaparib and Radiotherapy in Patients With Triple-Negative Breast Cancer: The Phase 1 Olaparib and Radiation Therapy for Triple-Negative Breast Cancer Trial. JAMA Oncology 2022, 8, 1802–1808. [Google Scholar] [CrossRef] [PubMed]
- Bellon, J.R.; Chen, Y.-H.; Rees, R.; Taghian, A.G.; Wong, J.S.; Punglia, R.S.; et al. A Phase 1 Dose-Escalation Trial of Radiation Therapy and Concurrent Cisplatin for Stage II and III Triple-Negative Breast Cancer. International Journal of Radiation Oncology*Biology*Physics 2021, 111, 45–52. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Nyati, S. Molecular targets that sensitize cancer to radiation killing: From the bench to the bedside. Biomed Pharmacother 2022, 158, 114126. [Google Scholar] [CrossRef]
- Kong, A.; Good, J.; Kirkham, A.; Savage, J.; Mant, R.; Llewellyn, L.; et al. Phase I trial of WEE1 inhibition with chemotherapy and radiotherapy as adjuvant treatment, and a window of opportunity trial with cisplatin in patients with head and neck cancer: the WISTERIA trial protocol. BMJ Open 2020, 10, e033009. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J Clin Oncol 2019, 37, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: progress made and future hopes. NPJ Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Fujita, M.; Karasawa, K. Combining Carbon-Ion Irradiation and PARP Inhibitor, Olaparib Efficiently Kills BRCA1-Mutated Triple-Negative Breast Cancer Cells. Breast Cancer 2022, 16, 11782234221080553. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.Y.; Speers, C.; Liu, M.; Jackson, W.C.; Moon, D.; Rinkinen, J.; et al. Targeted radiosensitization with PARP1 inhibition: optimization of therapy and identification of biomarkers of response in breast cancer. Breast Cancer Res Treat 2014, 147, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J Clin Med 2020, 9, 940. [Google Scholar] [CrossRef]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int J Mol Sci 2021, 22, 4203. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Cai, Y.; Peng, R.; Wu, G.; Shi, Y.; Jiang, W. The CDK1 inhibitor RO3306 improves the response of BRCA-pro fi cient breast cancer cells to PARP inhibition. International Journal of Oncology 2014, 44, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Jang, H.; Kim, S.; Lee, K.H.; Kim, D.K.; Suh, K.J.; et al. Androgen Receptor Inhibitor Enhances the Antitumor Effect of PARP Inhibitor in Breast Cancer Cells by Modulating DNA Damage Response. Mol Cancer Ther 2018, 17, 2507–2518. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun 2019, 10, 5065. [Google Scholar] [CrossRef]
- Ji, W.; Weng, X.; Xu, D.; Cai, S.; Lou, H.; Ding, L. Non-small cell lung cancer cells with deficiencies in homologous recombination genes are sensitive to PARP inhibitors. Biochem Biophys Res Commun 2020, 522, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O'Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J Exp Pharmacol 2021, 13, 303–328. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer PJ JSW, Hamilton T C Cisplatin and its analogues In: DeVita V T HS, Rosenberg S A editor. Cancer: Principles and Practice of Oncology. Philadelphia Lippincott-Raven 1997. p. 467-83.
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proceedings of the National Academy of Sciences of the United States of America 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.B. Taxol (paclitaxel): mechanisms of action. Ann Oncol 1994, 5 (Suppl. 6), S3–S6. [Google Scholar] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed]
- Dibra, D.; Moyer, S.M.; El-Naggar, A.K.; Qi, Y.; Su, X.; Lozano, G. Triple-negative breast tumors are dependent on mutant p53 for growth and survival. Proceedings of the National Academy of Sciences 2023, 120, e2308807120. [Google Scholar] [CrossRef]
- Lee, H.; Trainer, A.H.; Friedman, L.S.; Thistlethwaite, F.C.; Evans, M.J.; Ponder, B.A.; et al. Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol Cell 1999, 4, 1–10. [Google Scholar] [CrossRef]
- Liu, J.; Adhav, R.; Miao, K.; Su, S.M.; Mo, L.; Chan, U.I.; et al. Characterization of BRCA1-deficient premalignant tissues and cancers identifies Plekha5 as a tumor metastasis suppressor. Nat Commun 2020, 11, 4875. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Yu, H.; Deng, C.X. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 17108–17113. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
BUB1 expression demonstrate positive correlation with BRCA1, BRCA2, PARP1, and PARP2 genes. mRNA expression plots showing correlation of BUB1 vs. (A) BRCA1, (B) BRCA2, (C) PARP1, (D) PARP2, (E) PARP3, and (F) TP53 in Breast cancer (METABRIC, 2509 samples) from cBioPortal.
Figure 1.
BUB1 expression demonstrate positive correlation with BRCA1, BRCA2, PARP1, and PARP2 genes. mRNA expression plots showing correlation of BUB1 vs. (A) BRCA1, (B) BRCA2, (C) PARP1, (D) PARP2, (E) PARP3, and (F) TP53 in Breast cancer (METABRIC, 2509 samples) from cBioPortal.
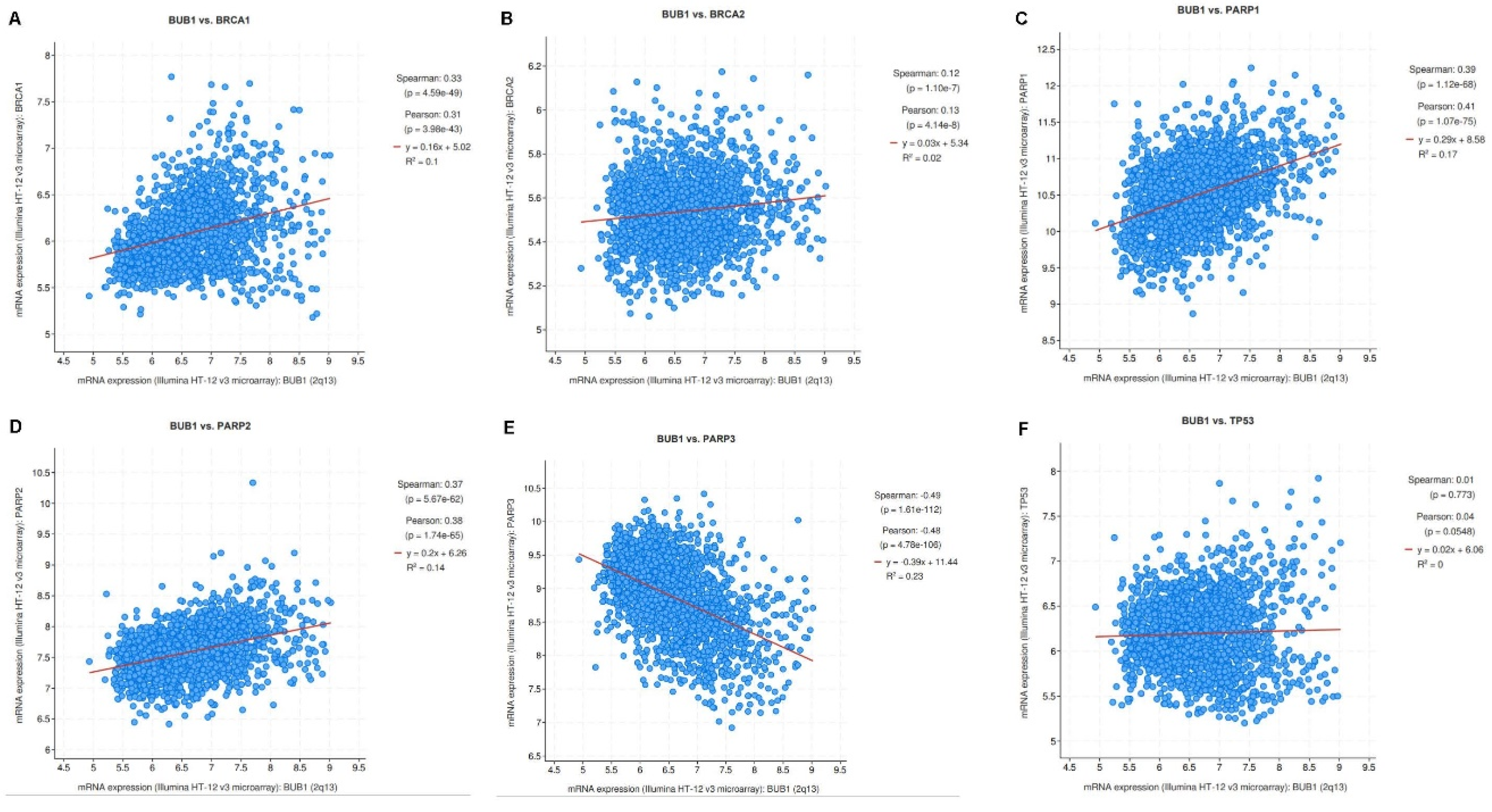
Figure 2.
Cytotoxicity of anti-cancer agents on SUM159 and MDA-MB-231 cells by alamarBlueTM assay. IC50 of single agents in SUM159 (A,C,E) and MDA-MB-231 cells (B,D,F) were 21.2 μM and 41.2 μM (AZD2281/olaparib, panels A and B), 1.63 μM and 7.14 μM (cisplatin, panels C and D), 4.7 nM and 6.2 nM (paclitaxel, panels E and F).
Figure 2.
Cytotoxicity of anti-cancer agents on SUM159 and MDA-MB-231 cells by alamarBlueTM assay. IC50 of single agents in SUM159 (A,C,E) and MDA-MB-231 cells (B,D,F) were 21.2 μM and 41.2 μM (AZD2281/olaparib, panels A and B), 1.63 μM and 7.14 μM (cisplatin, panels C and D), 4.7 nM and 6.2 nM (paclitaxel, panels E and F).
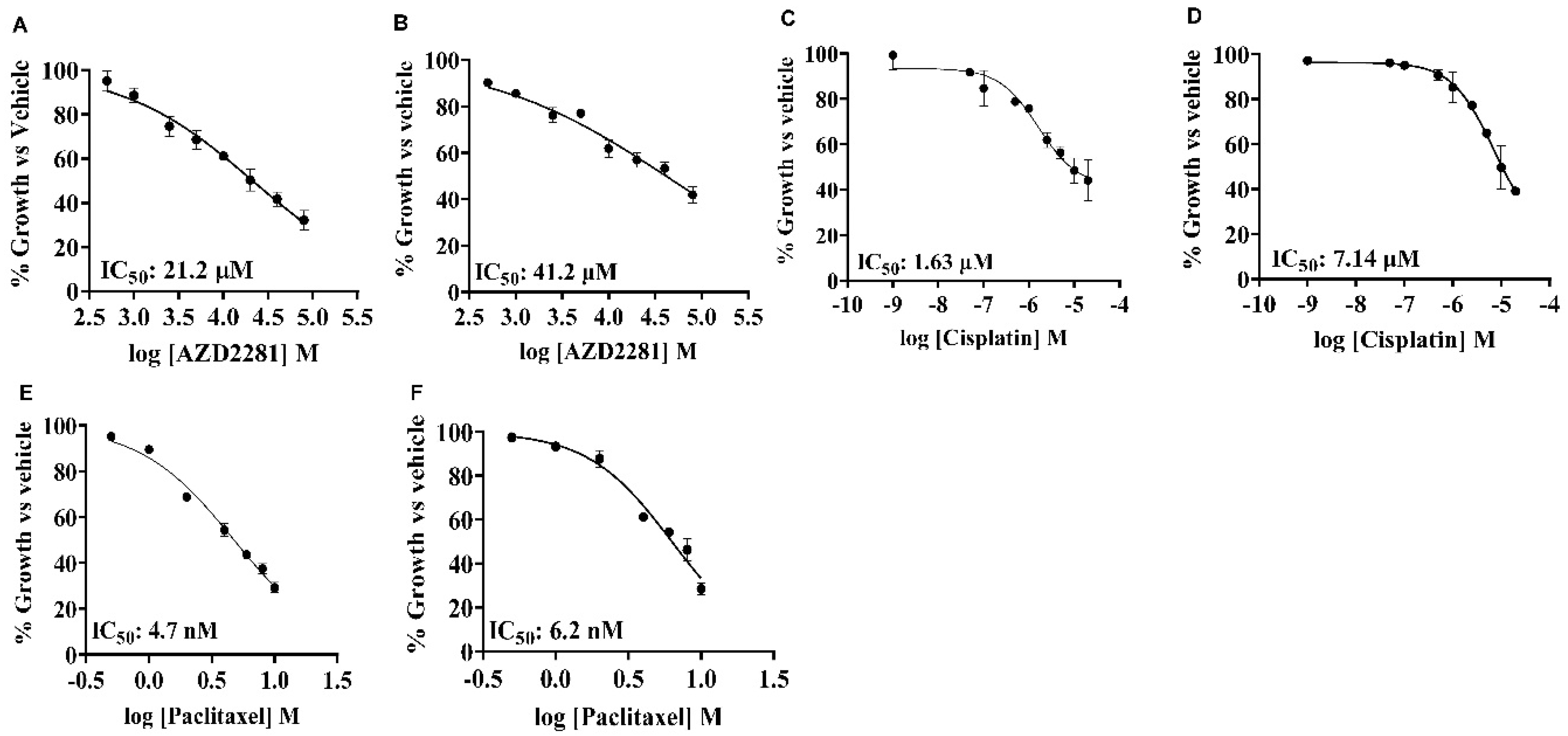
Figure 3.
BUB1 kinase inhibitor synergistically sensitizes TNBC cells to olaparib, cisplatin, and paclitaxel. SUM159 (A, C, E) and MDA-MB-231 (B, D, F) cells were treated with AZD2281 (1 µM), Cisplatin (1 µM), and Paclitaxel (1.5 nM) in combination with BAY1816032 (1 µM). BUB1 inhibitor BAY1816032 showed synergistic effects (C.I. < 1) with all three classes of drugs. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Figure 3.
BUB1 kinase inhibitor synergistically sensitizes TNBC cells to olaparib, cisplatin, and paclitaxel. SUM159 (A, C, E) and MDA-MB-231 (B, D, F) cells were treated with AZD2281 (1 µM), Cisplatin (1 µM), and Paclitaxel (1.5 nM) in combination with BAY1816032 (1 µM). BUB1 inhibitor BAY1816032 showed synergistic effects (C.I. < 1) with all three classes of drugs. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
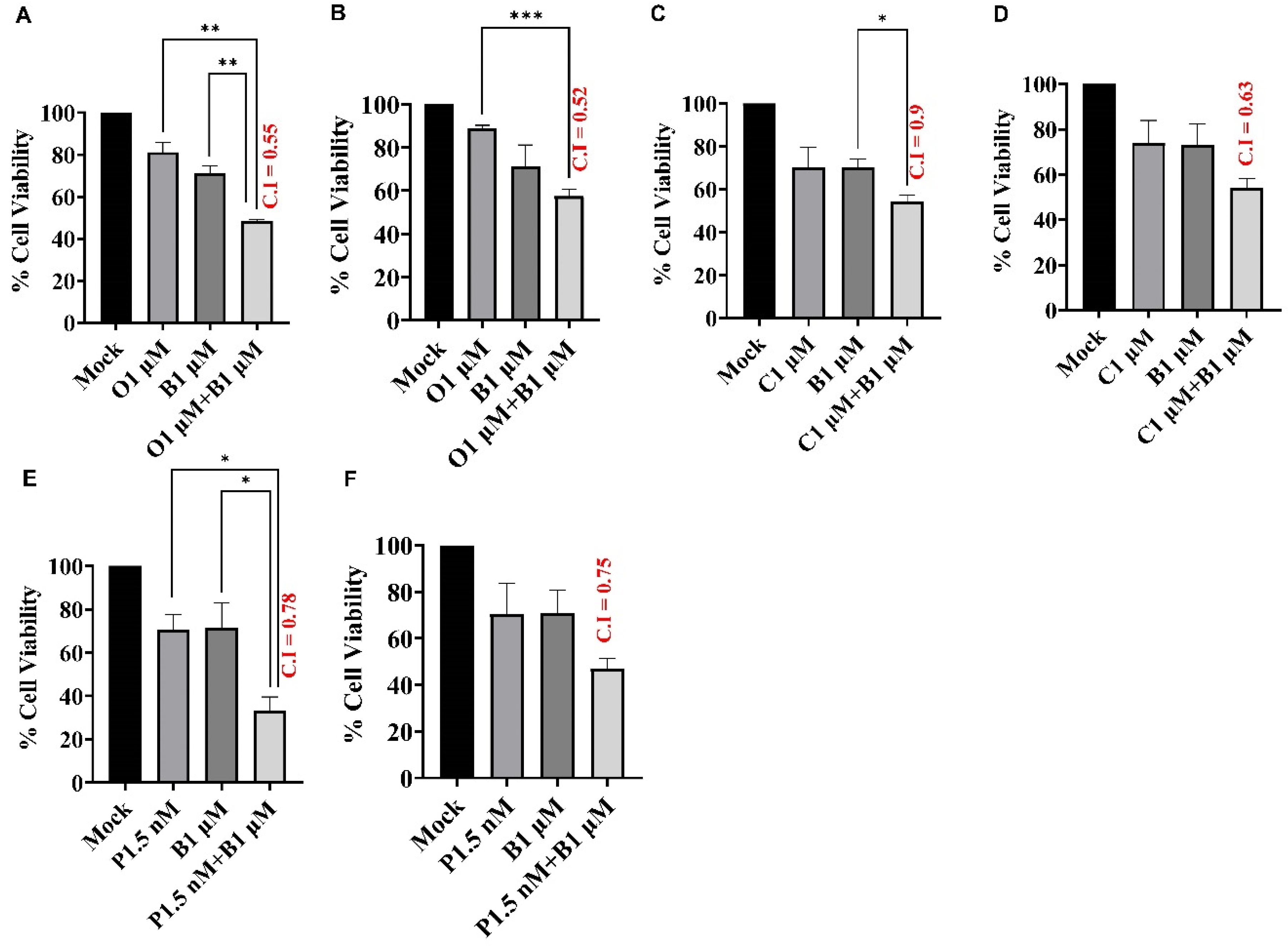
Figure 4.
Olaparib, cisplatin, and paclitaxel radiosensitize SUM159 and MDA-MB-231 cells. SUM159 (A,C,E) and MDA-MB-231 (B,D,F) cells were treated with single agents AZD2281, Cisplatin, and Paclitaxel followed by ionizing radiation and clonogenic survival was estimated. Radiation enhancement radio (rER > 1) indicates radiosensitization by single agent drugs. Plating efficiency graphs were plotted at 2 Gy. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001.
Figure 4.
Olaparib, cisplatin, and paclitaxel radiosensitize SUM159 and MDA-MB-231 cells. SUM159 (A,C,E) and MDA-MB-231 (B,D,F) cells were treated with single agents AZD2281, Cisplatin, and Paclitaxel followed by ionizing radiation and clonogenic survival was estimated. Radiation enhancement radio (rER > 1) indicates radiosensitization by single agent drugs. Plating efficiency graphs were plotted at 2 Gy. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001.
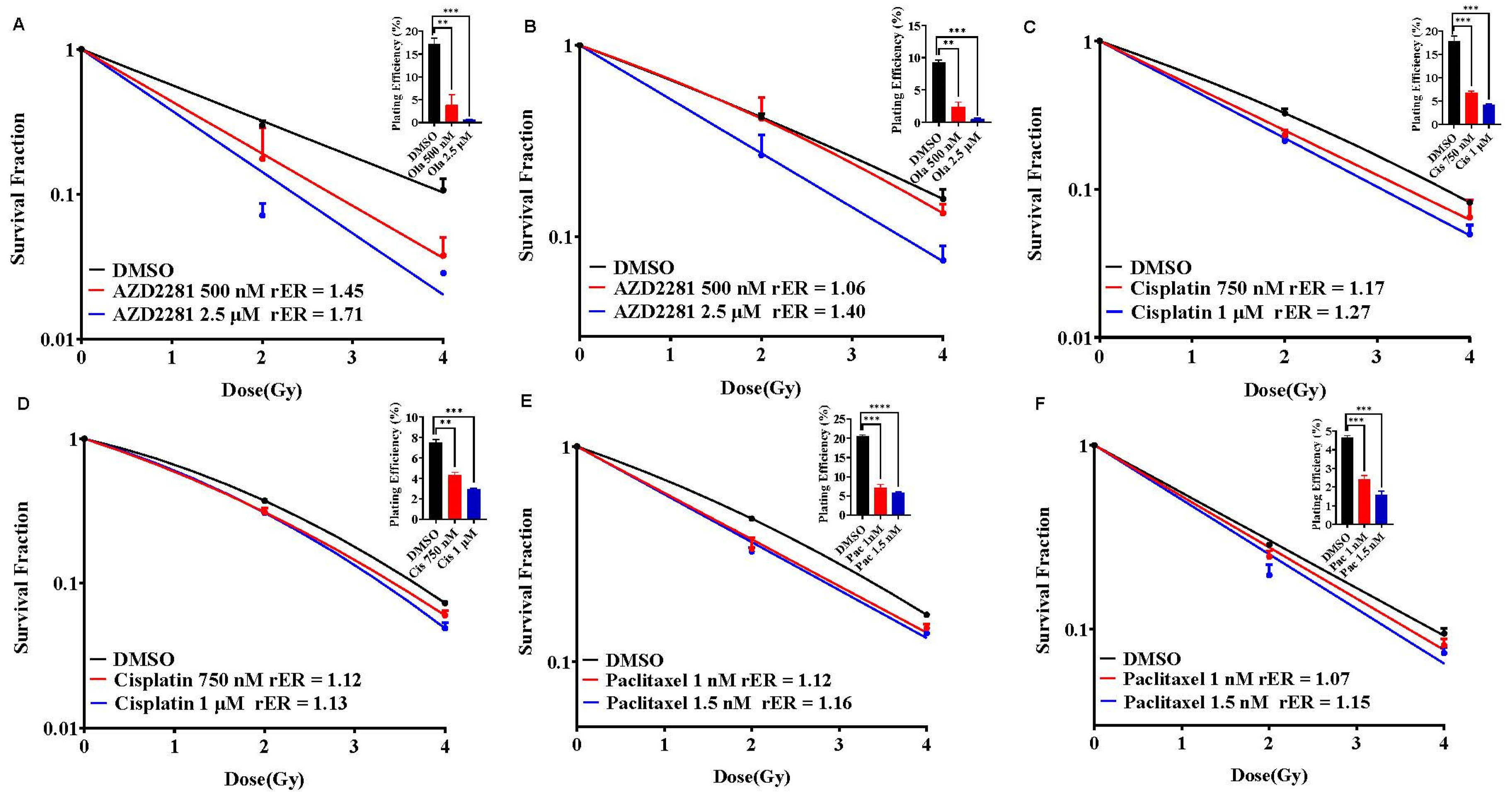
Figure 5.
BAY1816032 enhances radiosensitization potential of olaparib, cisplatin, and paclitaxel in SUM159 and MDA-MB-231 cells. SUM159 (A, C, E) and MDA-MB-231 (B, D, F) cells were treated with BUB1 inhibitor BAY1816032 in combination with olaparib (A, B), cisplatin (C, D) or paclitaxel (E, F) followed by ionizing radiation. Cells were allowed to form colonies and radiation enhancement ratio (rER) was estimated. BUB1 inhibitor synergistically increases rER of olaparib, cisplatin, and paclitaxel. Plating efficiency graphs were plotted at 2 Gy. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001.
Figure 5.
BAY1816032 enhances radiosensitization potential of olaparib, cisplatin, and paclitaxel in SUM159 and MDA-MB-231 cells. SUM159 (A, C, E) and MDA-MB-231 (B, D, F) cells were treated with BUB1 inhibitor BAY1816032 in combination with olaparib (A, B), cisplatin (C, D) or paclitaxel (E, F) followed by ionizing radiation. Cells were allowed to form colonies and radiation enhancement ratio (rER) was estimated. BUB1 inhibitor synergistically increases rER of olaparib, cisplatin, and paclitaxel. Plating efficiency graphs were plotted at 2 Gy. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001.
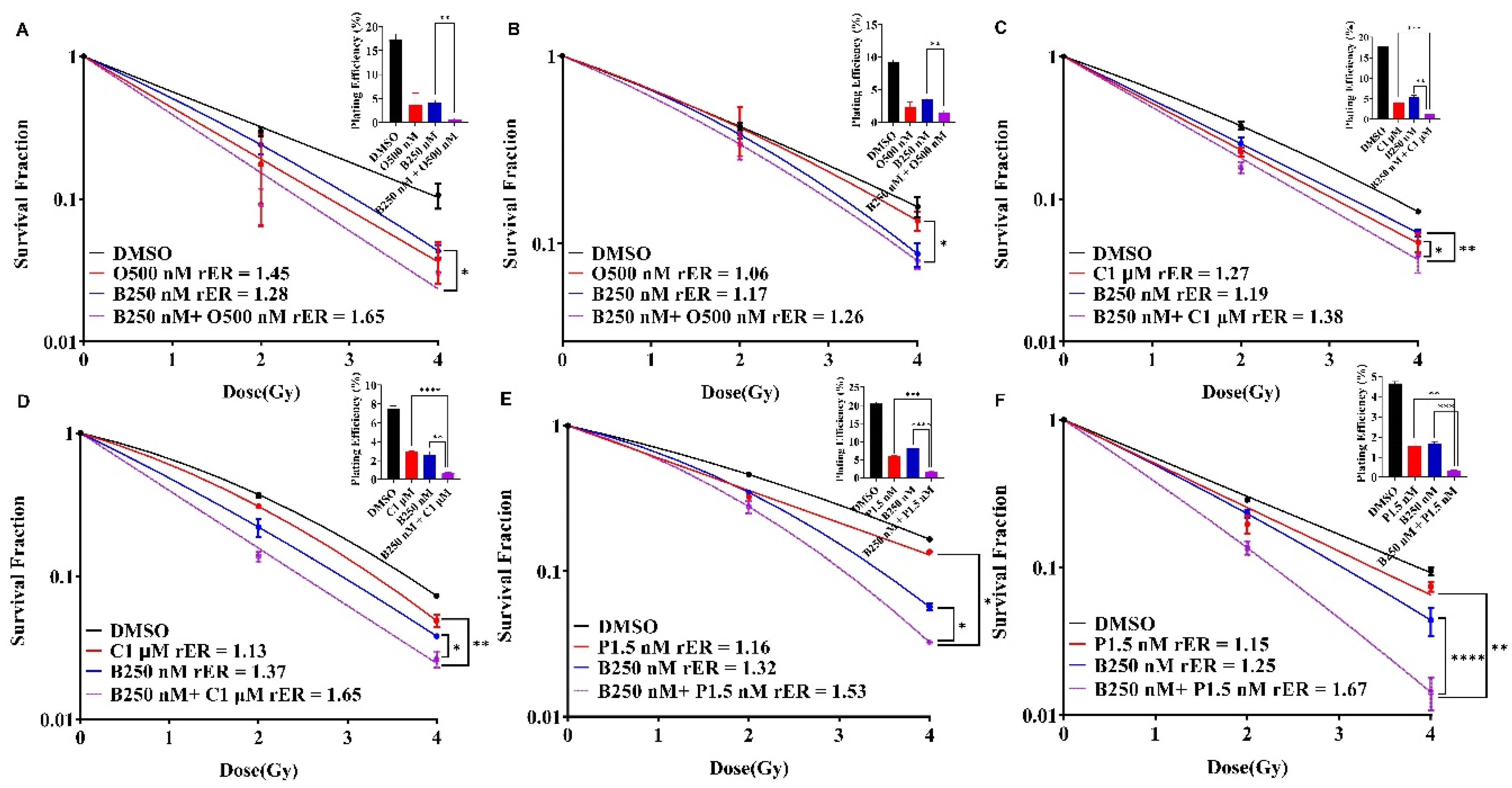
Figure 6.
BAY1816032 sensitizes BRCA mutant TNBC cell line HCC1937 to PARP inhibitor and combination of these two inhibitors enhance radiosensitization in HCC1937 cells. Inhibitory effects of BAY1816032 (A) and Olaparib (B) at 72 h on BRCA mutant HCC1937 cell line using alamarBlue assay. IC50 values of BAY1816032 and AZD2281 (Olaparib) in HCC1937 cell line were 3.56 μM and >100 μM, respectively. (C) BAY1816032 sensitizes HCC1937 cells to olaparib synergistically (CI < 1). (D) BUB1i mediated olaparib sensitization is enhanced with radiation (4 Gy). (E) HCC1937 cells were treated with BUB1 inhibitor and olaparib at lower concentration than IC50 ([BAY1816032] = 250 nM, [AZD2281] = 1 μM) and irradiated. Cells were allowed to form colonies and radiation enhancement (rER) was estimated. BUB1i significantly increased the radiosensitization by olaparib. Plating efficiency graphs were plotted at 2 Gy. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Figure 6.
BAY1816032 sensitizes BRCA mutant TNBC cell line HCC1937 to PARP inhibitor and combination of these two inhibitors enhance radiosensitization in HCC1937 cells. Inhibitory effects of BAY1816032 (A) and Olaparib (B) at 72 h on BRCA mutant HCC1937 cell line using alamarBlue assay. IC50 values of BAY1816032 and AZD2281 (Olaparib) in HCC1937 cell line were 3.56 μM and >100 μM, respectively. (C) BAY1816032 sensitizes HCC1937 cells to olaparib synergistically (CI < 1). (D) BUB1i mediated olaparib sensitization is enhanced with radiation (4 Gy). (E) HCC1937 cells were treated with BUB1 inhibitor and olaparib at lower concentration than IC50 ([BAY1816032] = 250 nM, [AZD2281] = 1 μM) and irradiated. Cells were allowed to form colonies and radiation enhancement (rER) was estimated. BUB1i significantly increased the radiosensitization by olaparib. Plating efficiency graphs were plotted at 2 Gy. P-values were defined as * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



