
Submitted:
24 April 2024
Posted:
24 April 2024
You are already at the latest version
Abstract
In this work, we explore the intrinsic disorder status of the three members of the synuclein family of proteins - α-, β-, and γ-synucleins. We also analyzed the peculiarities of the amino acid sequences and modeled 3D structures of the human synuclein family members to find potential effects of pathological mutations. Furthermore, we conducted a comparative sequence-based analysis of the synuclein proteins from various evolutionary distant species and evaluated their levels of intrinsic disorder using a set of commonly used bioinformatics tools. Next, we conducted a detailed functional disorder analysis of the proteins in the interactomes of the human synuclein family members using various web-based disorder analysis and prediction tools, such as RIDAO, D2P2 and FuzDrop. We identify that the interactome of human α-synuclein has relatively higher levels of intrinsic disorder, as compared to the interactomes of human β- and γ- synucleins. These observations highlight the critical importance of intrinsic disorder of human α-synuclein and its interactors in neuronal processes as compared to other proteins of the synuclein family.
Keywords:
α-synuclein
; β-synuclein
; γ-synuclein
; intrinsically disordered protein
; liquid-liquid phase separation
; Parkinson’s disease
; protein-protein interactions
1. Introduction
The synuclein family of proteins comprising of α-, β-, and γ-synucleins plays a critical role in synaptic regulation [1,2] The proteins of the synuclein family are primarily expressed in the vertebrate neuronal tissues, and in humans, they have been found to be associated with various neurodegenerative diseases, such as Parkinson’s disease (PD) [1,2]. All three family members were shown to be mostly disordered in the purified form in vitro [3,4,5,6,7,8,9], and the intrinsically disordered nature of α-synuclein was verified in cellulo [10,11,12,13,14,15,16]. However, at interaction with lipid membranes, the synuclein proteins can undergo disorder-to-order transitions and exhibit an α-helical lipid-bound structure, peculiarities of which have been well-studied due to the analysis of the pathological mutations causing toxicity related to the development of the early onset of PD [1,2].
Of the three synuclein proteins, α-synuclein, has been the most studied due to its higher abundance in the brain and because of the discovery of its link to the pathogenesis of PD and later to the development of many other neurodegenerative diseases collectively known as synucleinopathies [17,18,19,20,21,22,23]. In fact, as of March 31, 2024, Web of Science database contained 30,697 papers dedicated to this protein, a remarkable two-fold increase in comparison with the results of the analogous literature analysis reported in 2017 [1]. This strong attention of the researchers to this protein is determined by its important roles in the pathogenesis of neurodegenerative diseases. Although α-synuclein has been originally found to be accumulated in the Lewy bodies (LBs) and Lewy neurites (LNs), which are specific pathological hallmarks in the PD cases, later misbehavior of this protein has also been linked to multiple other neurodegenerative diseases, such as Alzheimer’s disease, Down’s syndrome [1] and many other synucleinopathies [17,18,19,20,21,22,23]. In fact, some of the other maladies associated with α-synuclein misbehavior include neurodegeneration with brain iron accumulation type 1 (NBIA1), pure autonomic failure, Down’s syndrome, amyotrophic lateral sclerosis-parkinsonism-dementia complex of Guam (Guam ALS/PDC), multiple system atrophy (MSA), and several LB disorders (that, in fact, might represent a clinical continuum [24]), such as sporadic and familial PD, dementia with Lewy bodies (DLB), diffuse Lewy body disease (DLBD), the Lewy body variant of Alzheimer’s disease (LBVAD), and PD dementia (PDD) [25,26,27,28,29,30,31,32,33].
α-Synuclein aggregation leading to the formation of various oligomers, amorphous aggregates, and amyloid-like fibrils is one of its critical features, which is can be affected by the variety of means [1,34,35,36]. It was indicated that synucleinopathies represent the α-synuclein-related brain amyloidoses, as selectively vulnerable neurons and glia in different affected brain regions are characterized by the presence of common pathological intracellular inclusions containing α-synuclein, formation of which correlates with the degeneration of the afflicted brain regions leading to the onset and progression of the clinical symptoms of these diseases [17,18,23,26,33,37,38,39]. Accumulation of α-synuclein-containing inclusions was detected in the dorsal motor vagal and solitary nuclei, locus coeruleus, parabrachial nuclei, pedunculopontine and raphe nuclei, periaqueductal gray, prepositus hypoglossal, substantia nigra, reticular formation, and ventral tegmental area, and demonstrated the presence of LN in brainstem fiber tracts and the existence of LBs and LNs in cranial nerve nuclei, premotor oculomotor, precerebellar and vestibular brainstem nuclei [40,41,42]. Furthermore, the α-synuclein deposition-related pathological processes were shown to spread transneuronally along anatomical pathways [42], supporting the notion of the prion-like propagation of the pathological spread within the affected brain during the disease progression (e.g., as described by the Braak’s staging criteria of PD [43,44]).
Recent research also suggested that α-synuclein can form polymorphic structures under certain conditions [1]. Moreover, both the monomeric and the polymorphic forms of α-synuclein are amenable to various post-translational modifications (PTMs), providing means for further increase in structural and functional diversity of this protein. Furthermore, the capability of α-synuclein to form different high-molecular weight assemblies was linked to the ability of this protein to trigger different synucleinopathies [45], as demonstrated by the direct observation of the induction of different synucleinopathies after injection of the different α-synuclein aggregated forms (oligomers, ribbons, and fibrils) in rat brain [46].
Additionally, several pathological mutations of α-synuclein associated with the early onset of PD have been found to increase the aggregation potential of this protein in neurodegenerative diseases [47,48,49,50,51,52,53]. A change of alanine residue 53 to threonine in α-synuclein results in mutation A53T which has been found to accelerate fibril formation, thus increasing the chances of inconsistent interactions [1,47]. Another mutation is A30P, which is caused by the replacement of alanine at position 30 by proline. A30P has been found to reduce the binding of α-synuclein to vesicles [47]. Another mutation that has been well studied is E46K, where glutamic acid at position 46 is replaced with lysine [47]. This mutation increases the binding of α-synuclein to liposomes and shows similar effects as A53T. Histidine 50 to glutamine substitution (H50Q) represents another α-synuclein mutation associated with the familial PD [54,55]. This mutation was predicted to perturb the same amphipathic α-helix as the previously described pathogenic mutations [55]. It was shown that H50Q was able to enhance the aggregation, secretion, and toxicity of α-synuclein, suggesting that this mutation may play a role in the extracellular toxicity of this protein [56].
Besides its astonishing multipathogeneity, α-synuclein has also been shown to present remarkable multifunctionality, exhibiting a wide range of highly diversified biological functions, ranging from control of the neuronal survival [57], regulation of the neuronal apoptotic response [58], and protection of neurons from various apoptotic stimuli [58], to metal binding [59,60,61,62] and interaction with pesticides and herbicides [63,64,65], to fatty acid binding [57] and interaction with plasma membranes leading to the formation of membrane channels or modification of membrane activity [66], to synaptic vesicle release and trafficking [57] and positive and negative regulation of neurotransmitter release [67], to association with mitochondria causing mitochondrial dysfunction [66], to regulation of various enzymes and transporters [57], to and to promiscuous interaction with hundreds of unrelated proteins and other binding partners [57,68,69,70]. To be able to possess its multifunctionality, α-synuclein structure is expected to be pliable enough to accommodate such features, and indeed it expresses in the form of an intrinsically disordered protein [1,34,35,36]. Such a diverse set of unrelated functions prompted interest among the researchers into exploring the various interactions of α-synuclein with other proteins and their roles in various degenerative diseases. An interesting question pertaining to the functionality of α-synuclein is the prevalence of intrinsic disorder in its interactome.
In contrast, β-synuclein has been understudied (actually, according to the Web of Science database, as of March 31, 2024, there are 463 papers dedicated to this protein) due to its relative scarcity in the neuronal tissues as compared with α-synuclein, which is estimated to account up to 1% of the total protein in soluble cytosolic brain fractions [71]. However, β-synuclein is typically co-expressed with α-synuclein and acts as a molecular chaperone to inhibit α-synuclein aggregation [72]. Recent research also linked β-synuclein to various neurodegenerative diseases sparking interest into the functions of this protein [72]. β-Synuclein has been found to be critical in the reduction of α-synuclein aggregation-induced toxicity [36,72]. In addition, β-synuclein also regulates synaptic function and dopamine transmission through various structural changes [35].
γ-Synuclein is expressed primarily in the peripheral nervous system in contrast to α- and β-synucleins [73]. Similar to β-synuclein, γ-synuclein has been relatively understudied due to its lesser abundance as compared to the other members of the synuclein family (as of March 31, 2024, there are 498 papers dedicated to this protein in the Web of Science database). γ-Synuclein has been found to be linked to breast and ovarian cancer [73]. However, specific γ-synuclein mutations have also been found in various neurodegenerative diseases, such as Alzheimer’s raising speculation regarding its role in the detection and potential treatment of such diseases.
One of the basic premises of modern protein science is the recognition and acceptance of the existence of intrinsically disordered proteins (IDPs) and hybrid proteins with intrinsically disordered regions (IDRs) [74,75,76,77,78], which are abundantly present in nature [75]. These biologically active proteins that do not have unique 3D structures as a whole or in part exist as dynamic conformational ensembles [77,79,80,81,82,83,84], which, at the global level, can be collapsed-disordered (molten globule-like), partially collapsed-disordered (pre-molten globule-like) or extended-disordered (coil-like) [85,86]. In a more general view, IDPs are characterized by a highly dynamic, complex, and mosaic structure with the multi-level spatiotemporal heterogeneity, where different parts of a protein can be ordered or disordered to a different degree [87,88]. Since ordered and differently disordered protein regions might have well-defined and specific functions, these spatiotemporal heterogeneity of IDPs/IDRs defines their multifunctionality [89]. Therefore, IDPs/IDRs represent structurally and functionally heterogeneous complex systems that operate within the frames of the protein structure-function continuum model [89,90,91,92,93]. Functional repertoire of IDPs, which are typically engaged in recognition, regulation, signaling, and in control of various biological pathways and processes [94,95,96], complements functions of ordered proteins [97,98,99,100]. The structural flexibility of IDPs/IDRs also determines the variety of ways that can be used to regulate and control their functions [87,101,102,103], with one of the important regulatory means being a variety of post-translational modifications (PTMs) [104,105]. Furthermore, structural pliability and the capability of IDPs/IDRs to be involved in weak multivalent interactions define the broad involvement of these proteins in the biological liquid-liquid phase separation (LLPS) that forms the molecular mechanism of the biogenesis of various membrane-less organelles (MLO) and biomolecular condensates [89,106,107,108]. Finally, many IDPs are involved in various human diseases [57,84,94,97,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127].
The intrinsically disordered nature of the synuclein family of proteins and their link to various cellular structures and processes observed in the norm and neurodegenerative diseases prompted research into the synuclein family. The functional and structural diversity of these proteins introduces various challenges in the determination of the complete function of the synuclein family. Moreover, the interactions of these proteins with other proteins, which may or may not be intrinsically disordered introduces additional challenges in the study of neurodegenerative disease. In this work, we explore and compare the sequence and structure of the human synuclein family with that of species from other classes. We attempt to determine the similarity of the synuclein family across species to aid in establishing the function of the proteins. Further, we also conduct a detailed disorder analysis of the proteins of the human synuclein family. Due to the wide variety of interacting proteins in the interactomes of the synuclein family, we perform a detailed disorder analysis of the interacting proteins exhibiting the highest disorder.
2. Results and Discussion
2.1. Intrinsic Disorder Status of Members of Human Synuclein Family
The amino acid sequences of all the synucleins analyzed in this study are listed in the Supplementary Table S1. Figure 1, Figure 2 and Figure 3 represents the results of the intrinsic disorder-centric analysis of human α-, β-, and γ-synucleins, which consist of 140, 134, and 127 amino acids, respectively. It was emphasized that among of the characteristic features of human synucleins is the presence of acidic stretches within their C-terminal regions, whereas within their 87 N-terminal residues, they possess a degenerative KTKEGV repeat that defines the hydrophobic variability of their sequences with a periodicity of 11 amino acids, which is characteristic of the amphipathic helices [128]. Although human α- and β-synucleins share 78% identical residues including conserved C-termini containing three identically placed tyrosine residues, β-synuclein lacks 11 residues (residues 73–83) within its middle region [19]. There is 60% sequence similarity between human α- and γ-synucleins, with γ-synuclein lacking the tyrosine-rich C-terminal signature of α- and β-synucleins [19].
Analysis of these figures provide compelling evidence of the highly disordered nature of all three members of human synuclein family. Originally, interest of the researchers to human α-synuclein was promoted by finding a relation of aggregation of this protein to the pathogenesis of Parkinson’s disease (PD), which is recognized as the most common aging-related movement disorder and the second most common neurodegenerative disease after Alzheimer’s disease (AD). It is estimated that ~1.5 million Americans are affected by PD. Sporadic (or idiopathic) forms of this disease account for about 95% of the PD patients [129,130]. The probability of sporadic PD development increases with age, with only a small percentage of patients diagnosed before the age of 50 [131]. The prevalence of PD is much greater among those who are at least 65 years old [132]. Approximately 1% of the population at 65–70 years of age is affected by PD, whereas the number of PD patients increases to 4–5% in 85-year-olds [133]. In addition to the sporadic form, multiple familial forms of the PD are associated with the mutations in a number of genes. These hereditary forms account for ~4% of PD patients, who develop early-onset disease before the age of 50 [134,135]. The pathological hallmarks of PD is the id the presence of the cytosolic filamentous inclusions known as Lewy bodies (LBs) and Lewy neurites (LNs) in surviving dopaminergic neurons within the substantia nigra [8,9]. These inclusions that contain aggregated forms of α-synuclein, can also be found in other parts of brain [136] and are associated with the pathogenesis of various synucleinopathies [25,26,27,28,29,30,31,32,33] characterized by the presence of the common pathologic inclusions composed of aggregated α-synuclein, which are deposited in selectively vulnerable neurons and glia [17,18,23,38]. Finding α-synuclein in LBs and LNs [32,37], as well as the existence of the specific missense mutations in the SNCA gene, corresponding to the A30P, E46K, and A53T substitutions in the α-synuclein protein in autosomal dominant early-onset forms of PD [137,138,139], and a link of other early-onset PD forms to the hyper-expression of wild type α-synuclein due to the gene duplication/triplication [140,141,142] strongly implicated α-synuclein in the PD pathogenesis.
The α-synuclein sequence is assumed to contain three functional regions: the N-terminal region (residues 1−60) contains four 11-amino acid imperfect repeats with a conserved motif (KTKEGV, residues 10-15, 21-26, 32-37, and 43-48); the central region (residues 61−95) that contains three additional repeats (residues 58-63, 69-74, and 80-85) and is known as a highly amyloidogenic non-Aβ component of AD plagues (NAC) region that was found in amyloid plaques associated with AD [118]; and the highly charged C-terminal region (residues 96−140) which is involved in protein-protein interactions. Note that the N-terminal and central regions comprise a lipid-binding domain. Detailed experimental analysis of purified α-synuclein in vitro provided strong evidence of the highly disordered nature of this protein [3,4,6,143]. However, it was also indicated that structure of α-synuclein does not represent a random coil, but is characterized by the presence of transient long-range contacts within the protein [9,144,145,146].
In agreement with experimental data, Figure 1A,B show that human α-synuclein is predicted to be highly disordered by most computational tools utilized in this study. Furthermore, Figure 1B shows that the C-terminal region of this protein contains two molecular recognition features (MoRFs, which are disordered regions that can undergo binding-induced folding at interaction with specific partners) (residues 87-94 and 111-140), and the entire protein is heavily decorated by multiple PTMs, clearly indicating the crucial functional role of its intrinsic disorder. Figure 1C shows that human α-synuclein is characterized by a high liquid-liquid phase separation (LLPS) potential. Its probability of spontaneous liquid-liquid phase separation (pLLPS) value of 0.6249 exceeds the threshold of 0.6 indicating that the α-synuclein can act as a droplet-driver capable of undergoing LLPS spontaneously [147]. Furthermore, the C-terminal region of this protein contains a long droplet-promoting region (DPR, residues 101-140), which also includes an aggregation hot-spot (residues 115-123), which is defined as a region that is capable of promoting the conversion of the liquid-like condensed state into a solid-like amyloid state [148]. These predicted LLPS potential of human α-synuclein is in line with the experimentally demonstrated capability of this protein to undergo LLPS [149,150,151,152,153].
Curiously, Figure 1D shows that human α-synuclein is expected to contain multiple regions with context-dependent interactions (residues 3-13, 15-75, 77-92, 94-105, and 115-123), i.e., regions exhibiting ordered or disordered binding modes depending on the cellular context (environment, sub-cellular localization, partners, and PTMs). These regions are capable to be engaged in the multiplicity of binding modes in the cellular context-dependent manner [154]. Data shown in Figure 1B,D indicate that human α-synuclein is predisposed to be a promiscuous binder, as its almost entire sequence can act as potential binding platform. In line with this conjecture, Figure 1E shows that α-synuclein can be engaged in interaction with 356 proteins forming a very dense protein-protein interaction network, 357 members of which are connected by 7,316 interactions. This network is characterized by the average node degree of 41 and average local clustering coefficient of 0.639. Since the expected number of edges in a random set of proteins of the same size and degree distribution drawn from the genome is 2,946, this α-synuclein-centric network has significantly more interactions than what would be expected (its PPI enrichment p-value is < 1.0e-16). Five most enriched biological processes, molecular functions and cellular components (as per Gene Ontology annotations) of the members of this network, as well as most enriched local STRING network clusters, and KEGG pathways are listed in Table 1.
Figure 1F demonstrated 3D structure of human α-synuclein modeled by AlphaFold. According to this model, α-synuclein does not have a compact core, with the only structured element predicted in this protein being a long α-helix spanning residues 1-91. This is a rather unrealistic structure, as long α-helices typically cannot exist in isolation, as they need to be stabilized by interactions either with the compact protein core or via binding to specific partners, such as other proteins, nucleic acids or membranes. Therefore, it is likely that in this case, AlphaFold predicts a 3D structure of a bound form of α-synuclein. In fact, comprehensive experimental analysis of purified α-synuclein in vitro using a multitude of techniques sensitive to different levels of proteins structural organization revealed that this protein is highly disordered [3,4,6,143]. Although transient long-range interactions were observed within this protein [9,144,145,146] solution NMR analysis did not show the presence any stable structural elements in the unbound form of this protein. However, this protein has been shown to adopt secondary structure of mostly helical nature upon interaction with the negatively charged small, unilamellar vesicles (SUVs) or detergent micelle surfaces [3,5,155,156], and α-helical structure was induced in this protein in the presence of lipids [157] and organic solvents [158]. Furthermore, binding of α-synuclein to a micelle of the detergent sodium lauroyl sarcosinate (SLAS) was shown to be accompanied by the disorder-to-order transition resulting in the formation of two antiparallel micelle-bound α-helices (residues 1-31 and 41-91) [159]. In agreement with this NMR-EPR based study, solution NMR analysis of the micelle-bound form of α-synuclein revealed the presence of the two anti-parallel curved α-helices (residues 3-37 and 45-92) connected via an extended but well-ordered linker [160].
Similar to α-synuclein, human β-synuclein is predicted to contain high levels of intrinsic disorder (see Figure 2). The major difference between these two proteins is the lack of 11 residues (residues 73–83) within the middle region of β-synuclein [19]. As a result, the overall percent disordered residues (as per PONDR® VSL2 analysis) decreases from 90.71% in α-synuclein to 87.31% in β-synuclein. On the contrary, the average prediction score increased from 0.7199 in α-synuclein to 0.7342 in β-synuclein (see Figure 2A). Figure 2B shows that human β-synuclein, being predicted as mostly disordered by all the tools included into the D2P2-based analysis, is expected to have three MoRFs (residues 1-9, 65-89, and 100-134), indicating that intrinsic disorder plays a crucial role in its interactability. Furthermore, function of β-synuclein can be modulated by various PTMs. At the same time, this protein has lost the capability to undergo spontaneous LLPS (its pLLPS of 0.5427 is below the threshold of 0.6) together with the aggregation hot spot. However, β-synuclein still can act as a droplet client, since it has a long DPR (residues 95-134) at its C-terminal tail (see Figure 2C). As per Figure 2D, human β-synuclein contains four regions with context-dependent interactions (residues 8-19, 21-58, 78-87, and 92-98). Therefore, this protein is also expected to act as a highly promiscuous binder. The idea is supported by Figure 2E showing the β-synuclein-centered PPI network generated by STRING, which contains 85 nodes connected by 715 edges. The average node degree of this network is 16.8, and its averaged local clustering coefficient is 0.682. Furthermore, this network has significantly more interactions than expected (715 vs. 143), being characterized by the PPI enrichment p-value of < 1.0e-16. Five most enriched biological processes, molecular functions, and cellular components (as per Gene Ontology annotations) of the members of this network, as well as most enriched local STRING network clusters, and KEGG pathways are listed in Table 1. Among functional differences of the members of the α-synuclein- and β-synuclein-centered PPI networks is a remarkable change in the KEGG pathways from exclusively disease-oriented pathways in α-synuclein-centered network (PD, ALS, AD, Prion disease, and Huntington’s disease) to the Synaptic vesicle cycle, PD, Nicotine addiction, Serotonergic synapse, and Insulin secretion pathways in the β-synuclein-centered PPI network.
Similar to α-synuclein, human β-synuclein was shown experimentally to be extensively disordered [6,8,9,72], with β-synuclein being somewhat more disordered than α-synuclein [6]. These experimental observations are supported by the results of our computational analysis. Figure 2F represents the AlphaFold-generated 3D structural model of human β-synuclein, showing the presence of a single, long, horse-shoe-like α-helix (residues 2-80). Solution NMR analysis of this protein in the unbound form revealed that its residual structure was shown to noticeably differ from that of α-synuclein, with the helical propensity of β-synuclein being clearly reduced between residues 66 and 83 [9]. This difference in the residual structure of the unbound state was shown to propagate to its micelle-bound form, as the NMR analysis revealed that although the lipid-binding domain of β-synuclein, which is missing 11 residues, remains predominantly helical in the micelle-bound form and preserves the break around position 42, it is characterized by a dramatic decrease in the stability of the helical structure within the 65-83 region [8].
Figure 3 shows that human γ-synuclein (which is different from other members of the human synuclein family by the absence of the tyrosine-rich C-terminal signature [19]) is also predicted as highly disordered protein. In fact, it seems that it is the most disordered member of the family, since its overall percent disordered residues (as per PONDR® VSL2 analysis) is 100% and its average prediction score is 0.8328 (see Figure 3A). Figure 3B represents the functional disorder profile of human γ-synuclein generated by the D2P2 platform and also shows the high prevalence of disorder in this protein, which is also expected to have three MoRFs (residues 1-10, 68-77, and 87-97) and several PTMs. As per FuzDrop analysis (see Figure 3C), γ-synuclein is not expected to undergo spontaneous LLPS but can serve as a droplet client, and also contains an aggregation hot-spot (residues 94-106). These features make this protein closer to α-synuclein than to β-synuclein. This hypothesis is supported by experimental analyses that revealed the closer structural similarity of these two proteins [6,9,161]. The decreased aggregation potential of γ-synuclein in comparison with that of α-synuclein was attributed to an increased α-helical propensity in the amyloid-forming region that is critical for α-synuclein fibrillation, suggesting that increased structural stability in this region may protect against γ-synuclein aggregation [161]. Figure 3D shows the presence of four regions with context-dependent interactions (residues 4-66, 70-75, 83-89, and 94-106). Two of these regions overlap with MoRFs. Figure 3E represents the γ-synuclein centered PPI network containing 32 nodes and 117 edges. Although this network is the smallest one among the synuclein family members, it is still has significantly more interactions than expected (117 vs. 46). It is characterized by the PPI enrichment p-value of < 1.0e-16, average node degree of 7.31, and high average local clustering coefficient of 0.752. Five most enriched biological processes, molecular functions, and cellular components (as per Gene Ontology annotations) of the members of this network, as well as most enriched local STRING network clusters, and KEGG pathways are listed in Table 1. Finally, Figure 3F represents 3D model of human γ-synuclein generated by AlphaFold. In line with all other data discussed in this section, this structural model is very similar to that generated for α-synuclein, where a single long α-helix (residues 2-91) is observed.
2.2. Effect of Familial Point Mutations on the Intrinsic Disorder Propensity of α-Synuclein
It is known that the residual structure in α-synuclein is affected by the familial PD missense mutations. There are at least six such mutations: A53T [138], A30P [162], E46K [163] H50Q [54,55], G51D [164,165], and A53E [166]. To understand how these point mutations associated with the early onset familial cases of PD affect propensity of α-synuclein for intrinsic disorder, we analyzed the corresponding sequences of the wild type protein (WT), as well as A30P, E46K, H50Q, G51D, A53T, and A53E mutants using PONDR® VSL2. Results of this analysis are shown in Figure 4A, whereas Figure 4B represents the “difference disorder spectra” calculated by the subtracting of the wild type per-residue disorder propensities from the corresponding data for the mutants. The use of “difference disorder spectra” simplify the understanding of the effects of mutations, as positive (negative) peaks in these plots show regions in mutant proteins with the increased (decreased) local disorder propensity relative to the wild type protein. Since with the exception for G51D, all “difference disorder spectra” contain positive peaks, the disease-associated mutations A30P, E46K, H50Q, A53T, and A53E caused some increase in the local disorder propensity. On the other hand, local intrinsic disorder propensity is deceased in the G51D mutant.
Figure 5 illustrates the effect of these mutations on the propensity of human α-synuclein for spontaneous LLPS. Although the droplet-promoting region is located within the C-terminal region of this protein and although all the mutations are located within the N-terminal region, the A30P, E46K, H50Q, G51D, A53T, and A53E mutations show noticeable effects on the LLPS potential of this protein. In fact, based on their propensity of spontaneous liquid-liquid phase separation, pLLPS, these forms of α-synuclein can be arranged in the following order: A53T (PLLPS = 0.6416) > A30P (PLLPS = 0.6413) > A53E (PLLPS = 0.6350) > WT (PLLPS = 0.6249) > H50Q (PLLPS = 0.6165) > E46K (PLLPS = 0.5730) > G51D (PLLPS = 0.5153). Based on these observations, one can hypothesize that the capability of α-synuclein to undergo spontaneous LLPS can be eliminated by point mutations E46K and G51D. Since formation of LLPS is considered as a step preceding the fibril formation, these data indicates that aggregation potential of α-synuclein is modulated by mutations. In agreement with these suppositions, these mutations associated with the early onset of PD were experimentally shown to differently modulate α-synuclein functions and aggregation propensity. A30P mutation promoted fast formation of non-fibrillar aggregate (such as oligomers or protofibrils) and not fibrils [48,167]. Two other PD mutants, A53T and E46K, were characterized by the accelerated fibrillation [48,49,168,169]. Similarly, the α-synuclein aggregation and fibrillation were dramatically accelerated by the H50Q mutant [56]. On the other hand, significant reduction in the α-synuclein oligomerization and fibrillation rates was induced by the G51D and A53E mutations, with the G51D mutant forming amorphous aggregates [165,170], and with the A53E mutant being able to slowly form very thin amyloid fibrils [170,171,172].
2.3. Intrinsic Disorder Potential of α-, β-, and γ-Synucleins from other Species
To check if the propensity for intrinsic disorder is an evolutionary conserved feature of the members of synuclein family, we analyzed disorder propensity in a variety evolutionary distinct species, such as Macaca fascicularis, Mus musculus, Monodelphis domestica, Tachyglossus aculeatus, Gallus gallus, Pelodiscus sinensis, Xenopus laevis, and Erpetoichthys calabaricus (see Figure 6). In other words, our analysis encompassed mammals including a marsupial and an egg laid monotreme, a bird, a reptile, an amphibian, and a fish. Amino acid sequences of α- (where available), β-, and γ-synucleins from these species were used for the multiple sequence alignments and per-residue disorder analysis. We did not find sequences of α-synucleins from Monodelphis domestica and Tachyglossus aculeatus, and therefore these proteins were not included in subsequent analyses. Amino acid sequences of all proteins used in these analyses are shown in Supplementary Table S1.
Figure 7 represents results of multiple sequence alignments of these proteins conducted using Clustal Omega [173] and shows remarkable sequence similarity of these intrinsically disordered proteins. In fact, percent of sequence identity of human protein with the α-synucleins from other species ranged from 76.3% (Erpetoichthys calabaricus) and 98.57% (Macaca fascicularis) (see Supplementary Figure S1). In the case of human β-synuclein, percent of sequence identity ranged from 61.07% (Monodelphis domestica) to 97.74% (Mus musculus) (see Supplementary Figure S2). Finally, human γ-synuclein was shown to have highest (96.06%) and lowest percent of sequence identity (61.34%) with Macaca fascicularis and Erpetoichthys calabaricus, respectively (see Supplementary Figure S3).
Furthermore, the global multiple sequence alignment of all 25 synuclein proteins selected for the analysis revealed that these proteins as a group have relatively high sequence similarity that was ranging from 30.00% to 98.57% (see Supplementary Figure S4). Based on these observations, it was not surprising to find that the members of synuclein family are characterized by rather strong conservation of their within-group per-residue disorder profiles (see Figure 8). This analysis indicated that proteins with high levels of intrinsic disorder can be characterized by a remarkable evolutionary conservation.
2.4. Functional Disorder Analysis of Human Proteins Engaged in Interaction with Members of Synuclein Family
At the next stage, we checked the prevalence of intrinsic disorder in human proteins involved in interactions with α-, β-, and γ-synucleins. PPI networks generated for individual proteins are shown in Figures 1E, 2E, and 3E, whereas a global PPI network centered at all three synucleins is shown in Figure 9. This network was generated using the confidence level of 0.45 as a minimum required interaction score. The network includes 469 proteins involved in 10,731 interactions, which significantly exceed the 4,889 interactions expected to happen in a random set of proteins of the same size and degree distribution drawn from the genome. The average node degree of this network is 45.8, whereas its average local clustering coefficient is 0.585. Five most enriched biological processes, molecular functions and cellular components (as per Gene Ontology annotations) of the members of this network, as well as most enriched local STRING network clusters, and KEGG pathways are listed in Table 1.
Next, we compared the levels of intrinsic disorder in all these interactomes with the disorder status of all proteins in human brain. Results of this analysis are shown in Figure 10, which clearly indicates that all analyzed protein sets contain noticeable levels of intrinsic disorder. Figure 10A summarizes the results of this analysis in a form of the PONDR® VSL2 score vs. PONDR® VSL2 (%) plot. Based on the results of these analyses, proteins can be classified using the percent of predicted intrinsically disorder residues (PPIDR; i.e., percent of residues with the disorder score of 0.5 or higher). Here, a PPIDR value of less than 10% is taken to correspond to a highly ordered protein, PPIDR between 10% and 30% is ascribed to moderately disordered protein, and PPIDR greater than 30% corresponds to a highly disordered protein [174,175]. In addition to PPIDR, average disorder score (ADS) was calculated for each query protein as a protein length-normalized sum of all the per-residue disorder scores. The resulting MDS values can be used for protein classification as highly ordered (MDS < 0.15), moderately disordered of flexible (MDS between 0.15 and 0.5), and highly disordered (MDS ≥ 0.5). Figure 10B represents the results of global disorder analysis in the form of the ΔCH-ΔCDF plot that can be used for further classification of proteins as mostly ordered, molten globule-like or hybrid, or highly disordered based on their positions within the resulting CH-CSD phase space [109,176,177,178]. The results of the corresponding classification are summarized in Table 2. This analysis revealed that although proteins in the joint α-β-γ synuclein interactome and especially proteins interacting with human α-synuclein are somewhat less disordered than protein in the human brain proteome, interactors of β- and especially γ-synuclein are noticeably more disordered. In fact, as per PONDR® VSL2 analysis, all proteins interacting with β- and γ-synucleins are moderately or highly disordered.
Figure 11A and Table 2 provide further illustration for this observation and also shows that, on average, most of proteins in these various sets are classified as moderately or highly disordered, emphasizing the potential importance of intrinsic disorder for functionality of these proteins.
Next, we took a look at the interactability of different proteins from the joint α-β-γ synuclein interactome and compared the corresponding node degree of these proteins with their disorder status. Results of this analysis are shown in Figure 11B. In this network, almost half of proteins (207 of 467, 44.3%) are involved in more than 47 interactors each, indicating that these proteins can be considered as hubs. These hub proteins are characterized by the mean node degree of 76±41 and mean PPID of 37.8±22.8%. Curiously, 60 proteins with the least number of interactors (with 10 or less partners each) were characterized by the mean node degree of 6.0±2.7 and the mean PPID of 51.4±25.9%. On the hand, 60 most connected proteins were characterized by the mean node degree of 123±43 and the mean PPID of 43.1±21.4%. Curiously, 60 most disordered proteins in this dataset had the mean node degree of 44.2±60.6 and the mean PPID of 87.5±8.4%, whereas 60 most ordered proteins of this set were characterized by the mean node degree of 43.7±32.0 and the mean PPID of 11.1±11.6%. These data taken together indicated that generally proteins with lower disorder levels are expected to be engaged in a bit more of interactions. However, the situation is changed if one compares 20 most ordered (PPID of 7.5±1.8%) with 20 most disordered proteins (PPID of 96.6±3.9%), as their mean node degrees were of 40.0±19.5 and 57.0±92.8.
We also looked for a correlation between the overall disorder status, interactability, and LLPS predisposition of human proteins in the joint α-β-γ synuclein interactome. Results of this analysis are summarized in Figure 12 showing the corresponding outputs in the form of the 3D plot. This analysis revealed that proteins predicted by FuzDrop as droplet drivers (i.e., possessing the pLLPS ≥ 0.6) are on average more disordered than proteins which are not capable of spontaneous liquid-liquid phase separation. In fact, 130 proteins with the pLLPS ≥ 0.6 were characterized by the mean PPIDR of 66.3±19.5%, whereas remaining 337 proteins from the joint human α-β-γ synuclein interactome were characterized by the mean PPIDR of 31.4±18.4%. On the other hand, LLPS drivers and non-drives did not show noticeable difference in their within network interactivity: within the joint human α-β-γ synuclein interactome, their corresponding mean node degrees were 40.4±49.3% (drivers) and 48.0±35.6% (non-drivers), respectively. Comparative analysis of the 130 most disordered proteins revealed that they are characterized by the mean PPIDR of 74.5±14.0%, have mean node degree of 41.7±48.2 and mean pLLPS of 0.753±0.295. Remaining 337 proteins are characterized by the mean PPIDR of 28.3±12.4%, have mean node degree of 47.4±35.2 and mean pLLPS of 0.311±0.229. Comparative analysis of the 130 most connected proteins with the mean node degree of 90.8±45.6 revealed that they are characterized by the mean PPIDR of 38.4±22.2% and the mean pLLPS of 0.379±0.273. Remaining less interactive human proteins in the joint α-β-γ synuclein interactome have the mean node degree of 28.6±16.3, the mean PPIDR of 42.2±25.2%, and the mean pLLPS of 0.456±0.331.
2.5. Functionality of Disorder in 11 most Disordered Proteins from the Joint α-β-γ Synuclein Interactome
Results of the PONDR® VSL2-based analysis of intrinsic disorder predisposition of the proteins from the joint α-β-γ synuclein interactome revealed that among the 467 members of this set, 144 (i.e., 30.8%) were mostly disordered, being predicted to have PPIDR of at least 50%. Furthermore, 24 of these proteins had the PPIDR exceeding 90%. In other words, these almost entirely disordered proteins accounted for 5.1% of the whole joint α-β-γ synuclein interactome or constituted 16.7% of the mostly disordered set of α-β-γ interactors. Furthermore, 9 proteins (MT3, CHMP2B, NRGN, CPLX1, CPLX2, NUCKS1, SNCG, MBP, and CAST) were predicted to be completely disordered (they heave PPIDR of 100%). In agreement with these observations, PONDR® VL3 (a tool specifically designed for finding long disordered regions and fully disordered proteins) confirmed 100% disorder status of these proteins, and predicted four more proteins (MAPT, HEMGN, H1-2, and SNCA) to have PPIDR of 100%. Since the disorder-centric functionality of SNCA (α-synuclein) and SNCG (γ-synuclein) was already introduced, the sections below provides brief description of the 11 remaining completely disordered protein.
2.5.1. MT3 (Metallothionein-3; UniProt ID: P25713; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.9952)
Metallothionein-3 (MT3) is one of the major intracellular zinc-binding proteins that play a number of important regulatory roles in the uptake, distribution, storage, and release of zinc [179]. In mammals, the family of metallothioneins includes four members, with specific tissue distributions, where MT1 and MT2 are found in all organs, whereas MT-3 is expressed mainly in the brain and MT-4 is mostly found in the stratified squamous epithelial tissues [179,180,181]. MT3 is also known as human neuronal growth inhibitory factor (hGIF), since it is known to inhibit the outgrowth of embryonic cortical neurons [182]. Based on the analysis of the native MT3 purified from human brain, it was established that a single molecule of this protein contains seven metal ions: three Zn2+ and four Cu+ ions, which are bound in the form of homo metal-thiolate clusters to two specific domains, a cooper-binding N-terminal β-domain (residues 1-30) and a zinc-binding C-terminal α-domain (residues 31-68) [183]. The neuron inhibitory activity of the MT3/hGIH is driven by the Cys6-Pro7-Cys8-Pro9 motif located within the β-domain of this protein, with a crucial role being played by its two proline residues, as their substitution entirely abolishes the activity of this domain [184,185,186].
Importantly, it was established that MT3 is deficient in Alzheimer’s disease brain [187], as well as in other neurodegenerative disease, such as multiple-system atrophy, Parkinson’s disease, progressive supranuclear palsy and amyotrophic lateral sclerosis [188,189,190], with the reduced levels of this protein in the subset of reactive astrocytes in lesioned areas associated with the aforementioned diseases being correlated with the neuronal loss [191]. Altogether, MT3 was reported as a multifunctional player in the control of cellular processes and diseases [190]. In fact, MT3 is not only responsible for maintaining the homeostasis of copper and zinc in cells and acts as a neuronal growth-inhibitory factor, but also plays a role in protection of cells from the oxidative stress and regulates a broad spectrum of cellular processes, such as cell growth and differentiation [190,192].
Structural information is available for the metal-bound forms of the α-domain (e.g., [193]), whereas no sufficient long- and medium-range Nuclear Overhauser effect (NOE) signals are available for the NMR-based structural determination of the β-domain of hGIF due to extensive internal dynamics [193,194]. Almost no structural information is available for the highly dynamic apo-MT3, which was shown to exist in a compact conformation (likely resembling a molten globule form) under the physiological conditions [195].
MT3 is a 68-residue-long protein with very unusual amino acid composition: it does not have any arginine, asparagine, histidine, isoleucine, leucine, phenylalanine, tryptophan, or tyrosine residues, but includes 20 (29.4%) cysteine residues, 8 (11.8%) of each glutamic acid and lysine residues, as well as 7 (10.3%) of each alanine and serine residues. Because of this high cysteine content, many disorder predictors do not classify MT3 as a disordered protein, since cysteines are typically considered as strongest order-promoting residues. However, Figure 13A shows that PONDR® VSL2 identifies this protein as completely disordered. Although both short and long forms of the IUPred classifier showed PPIDR of 0%, the use of the context-dependent mode of IUPred2A predictor [196] revealed that the entire protein represents a redox-sensitive region that is expected to be completely ordered in the oxidized form and completely disordered in the reduced form (see Figure 13B). Figure 13C represents the AlphaFold modeled 3D structure of human MT3 and shows that this proteins almost does not contain regular secondary structure elements. MT3-centered PPI network generated by STRING is shown in Figure 13D, which illustrates that this protein forms a densely connected network containing 415 proteins connected by 25,163 interactions (the expected number of edges is 3,897). Network is characterized by the average node degree of 121, an average local clustering coefficient of 0.668, and PPI enrichment p-value: < 1.0e-16. Among members of this network are α-synuclein (SNCA) and β-synuclein (SNCB) involved in interaction with MT3 and 156 and 212 other proteins. In line with these observations, MT3 was shown to co-localize with α-synuclein glial cytoplasmic inclusions (GCIs), which are multiple system atrophy-related intracytoplasmic inclusion bodies found in the oligodendrocytes [197]. Finally, human MT3 was predicted to have a low pLLPS of 0.3717, indicating that this protein is not capable of spontaneous LLPS and cannot act as droplet-driver. Since MT3 also does not have DPRs, it also cannot operate as a droplet-client.
2.5.2. CHMP2B (Charged Multivesicular Body Protein 2b; UniProt ID: Q9UQN3; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.8144)
As it follows from its name, charged multivesicular body protein 2b (CHMP2B) is involved in the formation of multivesicular bodies (MVBs). This 213 residue-long protein is evolutionary conserved and represents a core component of the Endosomal Sorting Complex Required For Transport III (ESCRT-III) machinery that plays crucial role in the MVB biogenesis and sorting of the endosomal cargo [198], as well controls a number of other fundamental cellular processes, such as autophagy [199], cytokinesis [200,201], endo/lysosomal repair [202] and viral exocytosis [203]. Importantly, mutations in CHMP2B (I29V, T104N, D148Y, Q165X, M178V, and Q206H [204], as well as CHMP2BIntron5 mutation leading to the production of the truncated form of the protein with missing C-terminal residues 179-213 [205]) are linked to the pathogenesis of frontotemporal dementia (FTD) associated with frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) [206,207]. Since there is a significant clinical, genetic, and neuropathological overlap between ALS and FTD, represent a continuum of a single ALS-FTD spectrum disorder [198]. Furthermore, Parkinsonian syndrome was described in familial FTD in families with mutations in the CHMP2B, as well as chromosome 9 open reading frame 72 (C9ORF72), fused in sarcoma (FUS), microtubule-associated protein tau (MAPT), progranulin (PRGN), transactive DNA-binding protein (TARDBP), and valosin-containing protein (VCP) [208] genes.
CHMP2B includes two N-terminal coiled-coil regions (residues 1–50 and 120–150) and a C-terminal MIT-interacting motif (MIM, residues 201-211) critical for interacting with vacuolar protein sorting-associated protein 4 (Vps4) and other proteins containing microtubule interacting and transport (MIT) domains [198,209]. It was shown that CHMP2B can self-polymerize into helical complexes (likely via coiled-coil regions) capable of deforming membranes [210]. However, the polymerization is typically autoinhibited via interaction between the MIM-containing acidic C-terminus and basic N-terminus [210]. Importantly, binding of Vps4 to the MIM of CHMP2B releases autoinhibition of the protein thereby and initiating its polymerization [210].
Despite crucial importance of this protein for various physiological and pathological processes and conditions, structural information on human CHMP2B is limited to the NMR structure of the MIM motif (residues 195-213) bound to human VPS4B (PDB ID: 2JQK [211]). Figure 14A,B provide a logical explanation of this phenomenon by showing that human CHMP2B is expected to be mostly disordered. However, this disorder could be of functional importance, as CHMP2B is predicted to have 6 MoRFs (residues 1-7, 54-69, 96-101, 141-151, 162-180, and 206-213), with the last MoRF overlapping with MIM motif. In other words, 67 of 213 residues of CHMP2B (31.5%) form disorder-based binding platforms, indicating that this protein can be a promiscuous binder. This hypothesis is supported by Figure 14C showing the CHMP2B-centered PPI network that includes 139 nodes (proteins) connected by 2,694 edges (interactions) and is characterized by the average node degree of 38.8 and average local clustering coefficient of 0.732. With expected number of edges of 425, this network has significantly more interactions than expected (PPI enrichment p-value: < 1.0e-16). One of the members of this CHMP2B-centered PPI network, is α-synuclein, which itself is involved in interaction with 46 CHMP2B interactors. Importantly, the FuzDrop-based analysis revealed that CHMP2B cannot undergo spontaneous LLPS but act as a droplet client, since although it is characterized by the probability of spontaneous liquid-liquid phase separation below the 0.6 threshold (pLLPS = 0.4588), it has two droplet-promoting regions (DRPs, residues 107-118 and 184-198).
2.5.3. NRGN (Neurogranin, UniProt ID: Q92686; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.8643)
Neurogranin (NRGN), a 78-residue-long multifunctional protein from the calpacitin family, which is also known as b50-immunoreactive C kinase substrate (BICKS), RC3, and P17, is involved in the plasticity and regeneration of synapse mediated by the calcium- and calmodulin-signaling pathways [212]. This protein is preferentially found in the perikarya and dendrites of advanced differentiated neurons, as well as in the neuronal nuclei of the cerebral cortex [212]. Similar to another member of the calpacitin protein family, growth-associated protein-43 (GAP-43), NRGN is involved in long-term potentiation (LTP) and the elaboration of pre- and postsynaptic structures . The protein got its name “neurogranin” based on the fact that it is typically expressed in granule-like structures in pyramidal cells of the hippocampus and cortex [213]. This forebrain-enriched, postnatal-onset, thyroid hormone-dependent protein is known to serve as a selective substrate for protein kinase C (PKC) [214]. Unphosphorylated NRGN/EC3 interacts with calmodulin (CaM) in Ca2+-dependent manner [215] and plays a role in adult neural plasticity and neonatal synaptogenesis, being involved in the Ca2+-mediated second messenger cascades [216]. It was hypothesized that “a Ca2+-"sensitive," bimodal interaction between RC3 and CaM regulates the transduction of postsynaptic Ca2+ fluxes into physiological responses through the modulation of Ca2+/CaM availability” [216]. Interaction between NRGN and CaM is driven by the IQ motif (residues 26-47) containing the PKC target residue Ser36, phosphorylation of which abrogates NRGN CaM interaction [217,218].
Deregulation of this protein is linked to the pathogenesis of multiple neurological and mental diseases, such as Alzheimer’s disease (AD), acute ischemic stroke (AIS), Creutzfeldt–Jakob disease (CJD), depression, first episode psychosis (FEP), Huntington disease (HD), mild cognitive impairment (MCI), neuro-HIV, neurosyphilis (NS), Parkinson’s disease (PD), traumatic brain injury (TBI), and schizophrenia [212]. For example, both AD and MCI are characterized by high NRGN levels in cerebrospinal fluid (CSF) [219,220,221,222,223], the CSF levels of this protein were shown to correlate with the cognitive decline in AD [224], and higher CSF NRGN levels were shown to positively correlate with the higher scores of tau tangles pathology and Aβ neuritic plaques [225]. In the progressive MCI group, accelerated cognitive deterioration was shown to correlate with the elevated CSF NRGN levels [223]. In CJD as well, highly elevated NRGN levels were found in CSF [226]. On the contrary, the CSF NRGN levels were significantly decreased in PD, PD with MCI, and PD with dementia (PDD) [225]. Similarly, NRGN was shown to be one of the most robustly down-regulated genes in HD [227,228]. Curiously, NRGN together with α- and β-synuclein, as well as visinin-like protein 1 (VILIP-1) and neuronal pentraxin 2 are considered now as fluid AD biomarkers [229,230], with neurogranin, α-synuclein, and β-synuclein being considered as potential biomarkers for synaptic dysfunction in neurodegenerative diseases [231,232].
Multiparametric experimental analysis of the NRGN fragment (residues 28-43) corresponding to the CaM binding IQ motif and containing Ser36 residue targeted by PKC revealed that in aqueous solution, this peptide existed preferentially in the random coil state but underwent transition to a-helical form in the presence of sodium dodecyl sulfate (SDS) micelles or organic solvents [233]. Using triple resonance NMR techniques it was shown that in the unbound form, the full-length rat NRGN is mostly unfolded in the unbound form and contains a residual structure in the form of the nascent local α-helical region between residues 25-42 [234]. In line with these observations, Figure 15A,B shows that human NRGN is predicted as mostly disordered protein containing four MoRFs (residues 1-6, 14-24, 26-47, and 64-72), one of which overlaps with the CaM binding IQ motif (see Figure 15A) and is predicted as an α-helix by AlphaFold (Figure 15B). In other words, 48 residues (61.5%) of this protein are expected to be engaged in disorder-based protein-protein interactions, which can be controlled by PTMs (see Figure 15A). Therefore, it is not surprising to find that NRGN forms a very dense PPI network containing 418 nodes connected by 25,430 edges (an expected number of edges is 8,111) (see Figure 15C). An average node degree of this network is very high, as, on average, each member is expected to interact with 122 in-network partners. Furthermore, this network contains 82 members that interact with more than 200 partners each, and 212 members have at least 122 partners each. Importantly, human neurogranin was predicted to have a very high probability of spontaneous liquid-liquid phase separation (pLLPS = 0.9722) and possess a long C-terminally located DPR (residues 38-78), which also includes an aggregation hot-spot (residues 38-48), suggesting that this protein is capable of spontaneous LLPS and can potentially drive the aggregation of condensates.
2.5.4. CPLX1 (Complexin-1; UniProt ID: O14810; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.8819)
Complexin-1 (CPLX1 or CPX1, also known as synaphin-2) is a member of a family of two closely related proteins (complexins 1 and 2) that were originally discovered as proteins interacting with SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) [235,236,237]. The soluble and insoluble forms of complexins are enriched in synapses [237,238,239], where they may act as negative regulators of neurotransmitter release [238,240] at a step immediately preceding vesicle fusion [241]. Interaction of complexins (together with synaptotagmins) with SNAREs regulates conformational changes within the SNAR proteins associated with the Ca2+-triggered exocytosis [242]. This study also revealed that for synaptotagmin-Ca2+ to trigger synaptic fusion, the conformational switch from open to closed in complexin is required [242].
Similar to many other intrinsically disordered proteins (IDPs), complexins are characterized by broad multifunctionality. This led Justine A. Lottermoser and Jeremy S. Dittman to prompted to state in their recent review: “Complexin is one of the key synaptic proteins for which a simple mechanistic understanding is still lacking. Living up to its name, this small protein has been associated with a variety of roles differing between synapses and between species, but little consensus has been reached on its fundamental modes of action… Unlike other core fusion machinery proteins, Cpx function and its conservation across animal taxa has proven more difficult to elucidate.” [243]. In relation to the subject of this study, it was shown that changes in the brain levels of CPLX1 are associated with the α-synuclein pathology in mouse brain [244].
Despite its diminutive size (human CPLX1 contains 134 residues), this protein has four functional domains [243,245]: N-terminal domain (NT, residues 1-28), which is involved in interaction with SNAP25 and membrane binding as well as may support the fusogenic activity of CPLX1 [245,246,247,248,249,250]; the accessory helix domain (AH, residues 29-47) required for CPLX1-driven inhibition of fusion [245,250]; central helical domain (CH, residues 48-69), responsible for tight binding to the assembled SANARE proteins and required for all known CPLX1 functions [251,252,253]; and the poorly conserved C-terminal domain (CT, residues 70-134) involved in membrane interactions required for the proper localization of CPLX1 relative to SNAP and syntaxin-1 and related to membrane fusion[243,254,255,256,257,258,259].
Solution NMR analysis revealed that purified recombinant rat complexin-1 lacks a tertiary structure but contains a conserved α-helical middle region, where a stable α-helix is found in 29–64 region, whereas residues 65–86 contain a substantial but lower population of α-helix [251]. In line with these observations, Figure 16A,B show that a highly disordered human CPLX1 contains 6 MoRFs (residues 1-20, 51-57, 64-72, 84-89, 100-105, and 115-134), three of which overlap with the aforementioned helical regions. STRING-generated PPI-network centered at human CPLX1 includes 378 proteins connected by 14,779 interactions and is characterized by average node degree of 78.2 and an average local clustering coefficient of 0.604 (see Figure 16C). Based on the outputs of FuzDrop, human complexin-1 is characterized by the pLLPS of 0.9678, has three DPRs (residues 1-35, 42-69, 86-104 38-78), and five aggregation hot-spot (residues 30-35, 49-55, 62-68, 86-91, and 94-99), suggesting that this protein is capable of spontaneous LLPS and can potentially drive the aggregation of condensates.
2.5.5. CPLX2 (Complexin-2; UniProt ID: Q6PUV4; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.9135)
Complexin-2 is a second member of the human complexin family. These proteins share 84.3% of sequence identity and show sequence similarity of 91.8%. Therefore, it is not surprising that CPLX2 was shown to interact with the SNARE complex and thereby regulate the Ca2+-triggered fusion between vesicles and the plasma membrane [260]. However, although CPLX1 is preferentially expressed in the brain, CPLX2 is found in the brain and in some secretory cells [236,237], including pancreatic secretory cells [261] and peripheral mast cells [262], where it participates in the Ca2+-dependent degranulation through syntaxin 3 [262]. Furthermore, CPLX2 can be expressed in B lymphocytes and regulates secretion of immunoglobulin in antibody-secreting cells [263]. It was also shown that CPLX2 participates in docking, locking and unlocking of different SNARE complexes during sperm capacitation and induced acrosomal exocytosis [264]. Immunocytochemical analyses of the frontal cortex of HD patients revealed significant reduction in the CPLX2 levels in comparison with the HD presymptomatic patients, which seemed to correlate with the pathological grade of the disease [265].
Comparison of the data inFigure 16 and Figure 17 indicates that in line with their high sequence similarity, human CPLX1 and CPLX2 possess similar levels of disorder. Being highly disordered, human CPLX2 has 5 MoRFs (residues 1-21, 65-72, 85-94, 97-104, and 115-134) and several PTMs. Figure 17C represents the PPI-network centered at the human CPLX2. This STRING-generated network includes 348 proteins connected by 17,425 interactions. It is characterized by an average node degree of 100 and an average local clustering coefficient of 0.649. Finally, FuzDrop analysis showed that human CPLX2 is a bit more prone to spontaneous LLPS and CPLX1, as its pLLPS is 0.9811. It has one long DPR (residues 1-110) that covers more than 82% of its sequence) and two aggregation hot spots (residues 59-66 and 83-109).
2.5.6. NUCKS1 (Nuclear Ubiquitous Casein and Cyclin-Dependent Kinase Substrate 1; UniProt ID: Q9H1E3; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.9879)
Nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 is a 243-residue-long chromatin-associated protein that is involved in DNA repair by homologous recombination (HR, a DNA repair pathway critical for tumor suppression) and chromosome stability [266]. This protein is known to bind to the double-stranded DNA (dsDNA) and also can interact with the secondary DNA structures, such as D-loop structures [266]. NUCKS1 is highly expressed in a variety of malignant tumors, such as breast cancer [267,268], hepatocellular carcinoma [269,270], ovarian cancer [271], gastric cancer [272], and cervical squamous cell carcinoma [273], and is believed to function as an oncogen [274]. This protein was shown to promote the progression of colorectal cancer by activating the PI3K/AKT/mTOR signaling pathway [274]. In osteosarcoma, NUCKS1 elevates asparagine synthesis by transcriptionally upregulating asparagine synthetase (ASNS) expression, thereby promoting osteosarcoma progression and metastasis [275]. In lung adenocarcinoma, upregulation of NUCKS1 is associated with a poor prognosis [276].
In HIV-1 infection, NUCKS1 acts as a Tat activator and plays a crucial role in HIV-1 replication by enhancing the Tat-mediated viral transcription on the HIV-1 LTR promoter [277]. This protein also can serve as a biomarker of metabolic disease, since the NUCKS protein levels are inversely correlated with body mass index in humans [278]. Some genetic variants in NUCKS1 are associated with the sporadic Parkinson’s disease in Han Chinese [279]. Being located within the PARK16 gene locus, which possibly regulates the PD risk, NUCKS1 represents a potential PD susceptibility biomarker [280]. Genetic polymorphism of NUCKS1 is associated with the susceptibility of adolescent idiopathic scoliosis [281].
NUCKS1, being similar to the HMG (high-mobility group) protein family, is one of the most modified proteins in the mammalian proteome [278]. In fact, it was shown that the NUCKS1 protein can be phosphorylated at ~25 different residues [282,283]. It was also shown that in solution, NUCKS1 does not contain defined structure and shows a very low content of α-helical and β-structural, containing instead a relatively high proportion of β-turns [284].
In agreement with the aforementioned experimental structural analysis of human human NUCKS1 Figure 18A,B shows that this protein is predicted to be almost completely disordered. It contains 5 MoRFs (residues 1-27, 32-40, 89-113, 124-157, and 170-197) and is heavily decorated by numerous various PTMs (see Figure 18A). According to AlphaFold, it almost does not contain elements of ordered secondary structure. The exception is given by residues 13-19 and 96-108, which show some helical propensity and overlap with two MoRFs (Figure 18B). Based on the results of STRING analysis, NUCKS1 is involved in formation of a PPI network containing 366 proteins connected by 9,275 interactions (see Figure 18C). Since an expected number of edges is 5,028, this NUCKS1-centered network has significantly more interactions than expected (PPI enrichment p-value < 1.0e-16). An average node degree of this network is 50.7, and its average local clustering coefficient is 0.51. FuzDrop analysis revealed that human NUCKS1 has a probability of spontaneous liquid-liquid phase separation of 0.9945, with entire sequence acting as one long DPR. Furthermore, this protein has an impressive set of aggregation hot-spots that are located at residues 8-13, 20-27, 31-40, 54-72, 79-110, 115-128, 135-140, 144-159, 184-190, and 205-213.
2.5.7. MBP (Myelin Basic Protein; UniProt ID: P02686; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.8706)
Myelin basic protein (MBP) together with proteolipid protein (PLP) are two major protein components of the myelin sheath of the central nervous system (CNS) [285,286,287,288], which is “formed by membranes that extend from oligodendrocytes and that wrap concentrically (up to a hundred times) around nerve fibers, thereby insulating them and facilitating rapid transmission of nerve impulses” [289]. Deficiencies in myelin assembly and structure are associated with various neurological diseases [288,289,290]. For example, MBP immunoreactivity was found in the core of LBs in the brainstem, cingulate cortex and sympathetic ganglia of the PD and dementia with LBs patients [291]. Multiple system atrophy (MSA) pathogenesis is linked in part to the dysfunction of α-synuclein and myelin protein [292].
In human, differential splicing of a single mRNA transcripts generates four MBP isoforms: 21.5, 20.2, 18.5, and 17.2 kDa [289]. Although MBP exists in the multiple isoforms, in human, one of the most abundant proteins of the myelin sheath is known as “classic” 18.5 kDa isoform [289,293]. All isoforms of MBP are IDPs [289,294,295]. The intrinsically disordered nature of this protein made it non-crystallizable. In fact, in their comprehensive search for a suitable composition of a crystallization medium, Jan Sedzik and Daniel A. Kirschner tried 4600 different conditions but failed to induce MBP crystallization [296]. Based on these observations, the authors concluded: “as long as its random coil flexibility is not suppressed, 18.5 kDa MBP and possibly also its isoforms will remain preeminent examples of proteins that cannot be crystallized” [296].
Figure 19A,B shows that human MBP is predicted to be almost completely disordered. It contains 10 MoRFs that covers 73% of its sequence (residues 1-16, 30-52, 59-67, 70-101, 116-139, 141-185, 198-209, 218-229, 240-255, and 258-292) and is heavily modified by phosphorylation and methylation (see Figure 19A). According to AlphaFold, human MBP contains very limited amount of the elements of ordered secondary structure. In fact, there are two short α-helical segments (residues 171-180 and 218-228), one of which is included into the MoRF spanning residues 141-185, and another overlaps with 218-229 MoRF (Figure 19B). STRING-generated PPI network centered at MBP includes 422 proteins connected by 19,398 interactions (see Figure 19C). An expected number of interactions for a random set of proteins of the same size and degree distribution drawn from the genome is 8,971, indicating that this network has significantly more interactions than expected (PPI enrichment p-value < 1.0e-16). An average node degree of this network is 91.9, and its average local clustering coefficient is 0.646. FuzDrop analysis revealed that human MBP is characterized by the has pLLPS of 0.9903 and contains 4 DPRs (residues 1-89, 135-145, 165-219, and 228-304) and 7 aggregation hot-spots (residues 36-46, 179-184, 192-200, 208-218, 232-238, 259-267, and 278-298).
2.5.8. CAST (Calpastatin; UniProt ID: P20810; PPIDRPONDR® VSL2 = 100.0%; ADS PONDR® VSL2 = 0.9547)
Calpastatin (also known as Calpain inhibitor or Sperm BS-17 component) is a 708-residue-long protein acting as a specific inhibitor of a Ca2+-dependent cysteine protease calpain. The interest to CAST is determined by its ability to act as a specific endogenous protein inhibitor modulating calpain activity. Due to their critical involvement in apoptosis, aging, and neurodegeneration (e.g., AD pathogenesis), non-lysosomal cysteine proteases calpains are studied very well (as of April 2024, there were more than 10,720 papers dedicated to calpain in PubMed). One of the peculiar features of the proteolytic activity of calpains is their dependence on the tertiary structure features of the protein substrates rather than their specific primary amino acid motifs. As a result, “Calpains cleave at highly selective sites within a substrate rather than breaking down proteins into small fragments or amino acids” [297]. This feature helps calpains to regulate specific enzymes (such as Ca2+-dependent kinases and phosphatases, calcineurin, calcium ATPase, Ca2+-dependent cyclic nucleotide phosphodiesterase, tyrosine hydroxylase, CAMP-dependent protein kinases, phosphorylase kinase, and glycogen synthase) by specific cleavage between a regulatory and catalytic domains both in Ca2+-dependent and Ca2+-independent manner [297]. In human, there are more than a dozen of calpain isoforms, some with multiple splice variants [298,299]. Depending on their calcium requirements for physiological functions, calpains are grouped into two major types, μcalpain (μCANP or Calpain I) and mcalpain (mCANP or Calpain II) that have optimal activities at calcium concentrations in the low micromolar or nearly millimolar levels, respectively [300,301,302,303]. Furthermore, calpains have a crucial Ca2+ level-dependent transition from regulators to destroyers [297], acting at the physiologic calcium levels as coordinators of a multitude of signaling pathways that control diverse intracellular proteins and organelles at the membrane-cytoskeleton interface [304,305,306] or as vicious destructors capable of cleavage of more than half of the cell’s protein pools in 1 hour [307].
Calpastatins represent a family of isoforms derived from a single CAST gene by alternative mRNA splicing [308], PTMs (preferentially phosphorylation) [309], and proteolysis [310]. These isoforms shows tissue-specific distribution and range in molecular mass from 7- to 140 kDa [297]. Typical CAST contains four equivalent inhibitory domains (I, II, III, and IV), each capable of inhibiting a separate calpain molecule [311]. As a result, calpastatin was defined as a multiheaded inhibitor capable of inhibiting more than one calpain molecule [312]. In the canonical form of human CAST, these inhibitory domains are located at residues 137-277, 278-326, 427-563, and 564-708. Each of these inhibitory domains contain three conserved subdomains, A, B, and C, which are located at residues 170-222, 304-356, 446-499, and 583-636, and are primarily responsible for the calpain inhibition. Inhibition of calpain is potentiated by the subdomains A and C that interact with the enzyme in a Ca2+-dependent fashion [313,314].
Combined analysis of the inhibitory domain I of human CAST using 1H-NMR and circular dichroism (CD) revealed that this domain did not have any ordered structure in solution [315]. Similarly, a comprehensive structural characterization of pig calpastatin domain I revealed that at neutral pH, this domain is in an expanded and flexible conformation without secondary and tertiary structures [316]. A full NMR assignment of the CAST inhibitory domain I (residues 137-277) revealed that although this domain is mostly disordered in the unbound form, it retains some residual transient structure [317]. In fact, regions with helical propensity were found within all three subdomains of domain I, residues 18-25 within subdomain A (residues 12-30), 51-59 and 68-75 within subdomain B (residues 50-70), and residues 91-104 within the subdomain C (residues 87-105) [317].
Our bioinformatics analysis of human CAST provides strong support to the idea that this important protein has very high levels of intrinsic disorder. Figure 20A shows that CAST is predicted as mostly disordered by all the predictors included in D2P2 platform. Furthermore, this protein is predicted to have 20 MoRFs covering 64.5% of its sequence (residues 1-33, 39-52, 64-89, 103-124, 132-141, 150-166, 182-215, 226-246, 258-270, 286-316, 333-347, 366-379, 397-444, 476-488, 491-499, 502-551, 564-581, 593-629, 647-662, and 673-685) and a multitude of various PTMs (see Figure 20A). AlphaFold-predicted 3D structural model of this protein includes several short α-helices that do not form a hydrophobic core (Figure 20B). CAST-centered PPI network generated by STRING includes 279 proteins engaged in 9,487 interactions. The averaged node degree of this network is 68, and it has an average local clustering coefficient of 0.66 (see Figure 20C). High levels intrinsic disorder combined with the prevalence of disorder-based interaction sites are likely related to its extremely high probability of spontaneous liquid-liquid phase separation (pLLPS = 0.9989). With its three very long DPRs (residues 1-414, 415-532, and 537-708) and with 22 aggregation hot-spots spread through the entire sequence, CAST is not only expected to be extremely prone for spontaneous LLPS, but also can trigger formation of aggregates within phase-separated droplets.
2.5.9. MAPT (Microtubule-Associated Protein tau; UniProt ID: P10636; PPIDRPONDR® VSL2 = 99.1%; ADS PONDR® VSL2 = 0.8612)
The microtubule-associated protein (MAP) tau is one of a group of MAPs, which, in addition to the presence of various tubulin isoforms subjected to a broad spectrum of different PTMs, are involved in controlling the assembly/disassembly, functionality, morphology, and stability of the essential constituents of the cytoskeleton in eukaryotic cells, microtubules (MTs) [318]. The primary function of tau is MT stabilization via its binding to MTs in a tau phosphorylation-dependent manner [319]. Under pathological conditions, hyperphosphorylation of tau reduces its affinity to MTs causing the abnormal detachment of tau from the MTs that leads to the axonal transport defects [320] and triggers misfolding and aggregation of tau [321,322] that eventually results in the formation of the intracellular filamentous inclusions neurofibrillary tangles (NFTs) found in AD and other neurodegenerative disorders [323]. Therefore, to everyone who is even very superficially familiar with AD, microtubule-associated protein tau does not require special introduction, as this IDP is one of the most studied molecular drivers of AD. This fact is supported by more than 37,500 papers dedicated to this protein found in PubMed (as of April 2024). Importantly, AD is not the only neurodegenerative pathology associated with misbehavior of tau. These neurodegenerative maladies are known as primary tauopathies. They represent a heterogeneous group of disorders that are driven by the misfolding and abnormal aggregation of filamentous tau to form different pathological inclusions [323,324,325,326,327,328]. Accumulation of these inclusions leads to the degeneration within the afflicted brain regions. This gives raise to the specific clinical impairments reflected in a broad spectrum of behavioral, cognitive, and motor symptoms [326,329]. Similar to prion protein, α-synuclein and many other proteins related to the various amyloidosis, tau is capable of formation of the conformationally distinct pathological protein aggregates, or strains. Furthermore, these pathological aggregates can be transmitted between the anatomically connected brain regions, resulting in the spread of pathological protein inclusions [330,331]. Histopathological characteristics, such as the temporal distribution, morphology, and affected cell types for the foundation for subcategorization of tauopathies into several diseases [330]. Furthermore, depending on the tau isoform found in the inclusions, the major tauopathies can be also subdivided into 3R, 4R, and 3R/4R tauopathies [330].
Human tau protein is known to exist as a mixture of multiple isoforms, which are produced by alternative splicing and which differ from each other by the presence or absence of up to 5 of the 15 exons. The longest isoform includes 758 residues and has been chosen as a canonical form. This isoform includes a long IDR (residues 1-573) and a microtubule-binding domain (residues 561-685) possessing four tandem repeats of a conserved tubulin-binding domain (residues 561-591, 592-622, 623-653, 654-685) [332]. The most common isoform of tau is known as Tau-F (also known as Tau-4 or 2N4R [333], which contains 441 residues and is different from the canonical form by missing residues 125-375 and 395-460. Solution NMR analysis revealed that this tau isoform, as well as two shorter isoforms (with 383 and 352 residues) are all typical IDPs with very limited and highly dynamic residual secondary structure [334]. It was also shown that the structure of the 2N4R, being highly dynamic and polymorphic, is characterized by “a distinct domain character and an intricate network of transient long-range contacts important for pathogenic aggregation” [335].
In agreement with these experimental observations, Figure 21 show that the canonical isoform of tau is predicted as a highly disordered protein (Figure 21A) with almost non-existent elements of flexible secondary structure (Figure 21B). Furthermore, in line with the aforementioned crucial dependence of the physiological and pathological behavior of this protein on phosphorylation, Figure 21A shows that human tau (especially its microtubule-binding domain) is heavily decorated by PTMs. Figure 21A also shows that almost entire sequence of this protein can serve as disorder-based platform for protein-protein interactions, suggesting its high binding promiscuity. This hypothesis is supported by Figure 21C showing that tau-centered PPI network includes 337 proteins connected by 5,794 interactions. This STRING-generated network is characterized by the average node degree of 34.4 and the average local clustering coefficient of 0.611. Recently, it was shown that human tau is capable of spontaneous LLPS and preserves it’s mostly disordered nature in the droplet state, with repeat region attaining transient β-hairpin propensity upon LLPS [336,337,338]. It was also pointed out that similar to α-synuclein, fused in sarcoma (FUS), and the transactive response DNA-binding protein of 43 kDa (TDP-43), the biomolecular condensates formed by tau “may undergo liquid-to-solid phase transition thereby maturating to amyloid fibrils, oligomeric species, or amorphous aggregates and contributing to the pathology of several neurodegenerative diseases” [339]. Our sequence-based bioinformatics analysis using the FuzDrop platform revealed that tau is indeed characterized by very high LLPS potential (pLLPS = 0.9985), has four DPRs (residues 1-300, 309-589, 608-622, and 719-739) that includes numerous aggregation hot-spots (residues 2-12, 113-125, 128-133, 139-145, 186-195, 265-278, and 290-295 within the DPR1, residues 309-315, 454-461, 539-546, 562-574, and 578-589 within the DPR2, as well as residues 608-620 and 719-739 within the DPRs 3 and 4, respectively).
Finally, in line with the well-established pathological and functional cross-talks of α-synuclein and tau in the central nervous system (e.g., co-occurrence of α-synuclein and tau aggregates in the post-mortem brains with synucleinopathies and tauopathies and the overlapping clinical symptoms of dementia and parkinsonism [340,341,342,343], molecular interactions and cross-seeding between α-synuclein and tau in neurodegenerative diseases [344,345,346], as well as functional cooperation of α-synuclein and tau during proper brain development via maintenance of progenitor cells [347]), STRING analysis indicated that the tau-centered PPI network includes α- and β-synucleins that interact with 161 and 17 members of this network, respectively.
2.5.10. HEMGN (Hemogen; UniProt ID: Q9BXL5; PPIDRPONDR® VSL2 = 98.3%; ADS PONDR® VSL2 = 0.8304)
Hemogen, also known as erythroid differentiation-associated gene protein (EDAG-1, which was previously designated as embryonic development-associated gene 1) or hemopoietic gene protein, or negative differentiation regulator (NDR) protein, is a 484-resodue-long hematopoietic tissue-specific transcription regulator involved, which is specifically expressed in hematopoietic cells and in regulation of proliferation, differentiation, and apoptosis of hematopoietic cells [348,349,350,351,352]. It was shown that alternative promoters and polyadenylation of HEMGN gene leads to the biosynthesis of at least two distinct splicing variants in hematopoietic cells and in round spermatids in the testis, suggesting a role of this protein in spermatogenesis [353]. Furthermore, hemogen was shown to be related to the pathogenesis of erythroleukemia and megakaryoblast leukemia [354], its overexpression is associated with poor prognosis in the de novo acute myeloid leukemia (AML) [352], and it can promoteproliferation and invasion of human thyroid cancer cells by activating MAPK/Erk and AKT signal pathways [355].
No structural information is available for hemogen as of yet. Therefore, the results of the bioinformatics analysis reported in Figure 22 provides unique description of this important protein. Figure 22A shows that hemogen is expected to be mostly disordered by the most predictors utilized in D2P2 and is predicted to contain 17 MoRFs. The fact that this protein contains multiple PTM sites suggests that its activity is controlled by posttranslational modifications. Figure 22B provides further support for the mostly disordered nature of human hemogen and shows that as per AlpgaFold, this protein is expected to have just one N-terminally located α-helix (residues 30-66) which is included into the region necessary for nuclear localization (residues 7-87). Hemogen-centered PPI network generated by STRING includes 373 proteins engaged in 7,813 interactions. The averaged node degree of this network is 41.9, and it has an average local clustering coefficient of 0.52 (see Figure 22C). One of the members of this network is α-synuclein, which is involved in interaction with hemogen and 127 hemogen interactors. Finally, FuzDrop-based analysis showed that human hemogen has a high LLPS potential (pLLPS = 0.9935) and is predicted to have four DPRs (residues 1-31, 41-92, 97-126, and 261-475) that includes numerous aggregation hot-spots (residues 86-92, 120-126, 194-202, 261-288, 291-300, 307-344, 348-358, 392-401, 404-414, 442-251, and 454-464) mostly concentrated with the C-terminally located longest DPR. However, DPRs 2 and 3 have short aggregation hot-spots as well.
2.5.11. H1.2 (Histone H1.2; UniProt ID: P16403; PPIDRPONDR® VSL2 = 97.7%; ADS PONDR® VSL2 = 0.8947)
Histone H1.2 is a 213-residue long linker histone that is responsible for condensation of the nucleosome chains into the higher-order structured chromatin fibers. It is one of the seven H1 variant found in human somatic cells (H1.1 to H1.5, H1.0, and H1X) [356,357,358]. It is believed that H1 variants are specifically distributed among different cell lines [358,359,360,361] and have different functional repertoire, being able to act as general repressors and also possessing variant-specific functional diversity in chromatin regulation [356,357,358]. It was emphasized that the proportions of H1 variants present in a specific (i.e., H1 cell complement) differ between the cell types and are also dynamically changed throughout differentiation and cancer [358]. In fact, chromatin structural defects are known to be caused by the compromised H1 content [362]. A resent super-resolution microscopy-based analysis of several human cell lines revealed that somatic H1 histones are characterized by the differential nuclear distribution [358]. The authors pointed out: “H1.2, H1.3, H1.5 and, to a lesser extent, H1.0, are enriched at the nuclear periphery and coincide more with more compacted DNA... Contrarywise, H1X and H1.4 are distributed throughout the nucleus with a significant H1X enrichment in nucleoli” [358]. Based on these observations they concluded that different H1 variants have diverse implications in the genome functionality. It was also emphasized that the chromatin structure can be affected by the H1 variants depletion in a variant-specific manner, with a global chromatin decompaction being triggered by a H1.2 knock-down [358]. Detailed description of the H1.2 functionality and its crucial roles in the maintenance of genome stability, apoptosis, cell cycle regulation, as well as its association with disease, is provided in a recent comprehensive review [363].
Structurally, human H1.2 histone is characterized by the presence of disordered N- and C-terminal regions (residues 1-41 and 92-213, respectively) and a linker histone H1/H5 globular (H15) domain (residues 36-109). In line with these observations, circular dichroism-based analysis of the murine H1.2 linker histone revealed that although its structure was dominated by the random coil, some α-helical components belonging to the stably folded globular domain were also present [364]. Earlier comprehensive bioinformatics analysis 2007 histones from 746 species revealed that all the analyzed members of the histone family are intrinsically disordered proteins and that their copious intrinsic disorder is absolutely necessary for various functions of these proteins [365].
Figure 23A shows that according to the predictive tools assembled into the D2P2 platform, human linker H1.2 histone is predicted to have high levels of intrinsic disorder in the N- and C-terminal regions and also possess a more ordered structure in the central region. This is further supported by Figure 23B showing modeled 3D structure of this protein, where the presence of helical globular domain (residues 40-107) is evident. Similar to other histones, the linker histone H1.2 is heavily decorated by a wide spectrum of different PTMs, such as acetylation, formylation, methylation, PARylation, phosphorylation, and ubiquitination [363,366,367,368,369,370]. Figure 23A supports these observations and show that the entire protein is densely covered by various PTMs. Furthermore, human H1.2 histone is predicted to have 6 MoRFs (residues 42-49, 56-70, 79-94, 103-112, 132-145, and 151-213) (see Figure 23A).
Involvement of this protein in multiple interactions is illustrated by Figure 23C showing the STRING-generated PPI network, where 339 proteins being connected by 16,477 interactions, form a dense interactome with the average node degree of 97.2 and the average local clustering coefficient of 0.622. Importantly, it was established that incubation of the murine H1.2 with the 1-42 Amyloid-β peptide helped the folding of Aβ monomers, promoted the formation of laminar aggregates and thick bundles and stabilized the parallel association of fibrils [364]. Similarly, it was shown that the linker histone H1 (as well as other histones) can interact with human α-synuclein forming a tight 2:1 complex and dramatically accelerating the fibrillation rate of α-synuclein in vitro [371]. Furthermore, in mice exposed to a toxic insult (i.e., injections of the herbicide paraquat), α-synuclein was found in nucleus, where it was co-localized with the histones in the nuclei of nigral neurons [371].
The analysis of the human linker H1.2 histone by FuzDrop revealed that this protein has a very strong predisposition of spontaneous LLPS (it is characterized by the pLLPS of 0.9966). Bothe N- and C-terminal regions of this protein were also predicted as DPRs (residues 1-53 an 112-213, respectively). Furthermore, the human linker H1.2 histone was predicted to have five aggregation hot-spots: residues 21-27, 38-43, 96-103, 112-124, and 161-171. Curiously, with the exception for the last hot-spot, which is located within the last MoRFs, other aggregation hot-spots are mostly positioned between the MoRFs.
3. Materials and Methods
3.1. Overview
In order to facilitate sequence-based and structure-based comparison of synuclein proteins of different species, we utilized web-based computational tools, such as UniProt [372], NCBI Blast, and AlphaFold [373]. To perform disorder based analysis and comparison, we utilized the RIDAO application [374], a computational tool to identify the predicted disorder throughout the amino acid sequence. Further, we also utilized the D2P2 tool [375] and the FuzDrop tool [147,148,376] to examine intrinsic disorder and predict liquid-liquid phase separation (LLPS). We conducted extensive analysis of interacting proteins through the STRING database [377] to enable disorder-based comparison of the human synuclein family with the proteins in their respective interactomes.
3.2. Sequence and Structure-Based Analysis
We utilized the UniProt database [372] to extract the amino acid sequence information for human α-synuclein, β-synuclein and γ-synuclein. UniProt is a database which provides the known amino acid sequences along with additional information regarding the species and protein identity. Utilizing the extracted amino acid sequence, we visualized the predicted 3D structure of the proteins utilizing the AlphaFold platform [373]. AlphaFold is an AI-based computational tool that predicts the 3D structure of a protein given its amino acid sequence. Having analyzed the sequence and structure of the synuclein family of proteins, we utilized the NCBI Blast tool to compare the human synuclein family sequences with that of species from other species. To this end, we selected eight species from different classes of animals as shown in Figure 6. We extracted the amino acid sequences of the three synuclein proteins for each of these species using UniProt and performed sequence-based comparison with the corresponding human synucleins using NCBI Blast. Further, we analyzed the intrinsic disorder of the synucleins of these species using the RIDAO platform.
3.3. Disorder-Based Analysis of the Interactomes of Human Synucleins
Having performed sequence and structure based comparison of human synucleins with the synucleins of various species, we performed detailed intrinsic disorder analysis of the three human synucleins and the proteins in their interactomes. To this end, we utilized the STRING database [377] to identify proteins which are known to interact with human α-, β-, and γ-synucleins. The STRING database assembles information from different sources, such as laboratory experiments, previous research and text mining models. The STRING database takes the query protein sequence as input and provides a network of interacting proteins with varying levels of confidence. The interacting proteins are sorted by confidence with a confidence score of 0.7 or above being termed as high confidence, a score of 0.4 being considered as a medium confidence, and a score of 0.15 and below being taken as low confidence. Additional customization allows us to specify the maximum number of interactors in the first shell (the proteins directly interacting with the target protein). Known 3D structures of interactors and a nature of interactions (known interaction or predicted interaction) are also provided. We specify a maximum of 500 interactors in the first shell to enable extensive search of the interacting proteins.
Further, we predict intrinsic disorder for each of these proteins using the RIDAO platform [374] and, based on the outputs of CH-CDF analysis incorporated into RIDAO, label them as ‘disordered’, ‘mixed’, ‘rare’ or ‘structured’. We select the first 10 most disordered proteins in the interactomes of each of the member of synuclein family and performed a detailed intrinsic disorder analysis with the RIDAO and the D2P2 platforms [375]. Further, we analyze the propensity of these proteins for liquid-liquid phase separation (LLPS) with the FuzDrop computational platform [147,148,376].
4. Conclusions
This work provides a discussion of the sequence-based and structure-based functionality of the proteins of the human synuclein family. Through comparative inter-species sequence-based analysis, various insights regarding the similarity of α-, β-, and γ-synucleins from different species are obtained. Intrinsic disorder analysis demonstrates the presence of disordered, ordered, and mixed members in the human joint α-β-γ interactome. Interestingly, comprehensive disorder analysis reveals the presence of a significant percentage of intrinsically disordered interacting proteins in the interactomes of human α-, β-, and γ-synucleins. The analysis of the liquid-liquid phase separation probability of human synucleins and their interactors provides important insights into the potential roles of intrinsic disorder in organization of synuclein-related MLOme. Finally, we explore the potential functionality of intrinsic disorder in a set of the most disordered members of the joint α-β-γ interactome using a set of bioinformatics tools.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Amino acid sequences of α-, β-, and γ-synucleins analyzed in this study; Figure S1: CLUSTAL O(1.2.4) multiple sequence alignment of alpha-synucleins; Figure S2: CLUSTAL O(1.2.4) multiple sequence alignment of beta-synucleins; Figure S3: CLUSTAL O(1.2.4) multiple sequence alignment of gamma-synucleins; Figure S4: CLUSTAL O(1.2.4) multiple sequence alignment of all synucleins.
Author Contributions
Conceptualization, V.N.U.; methodology, V.N.U.; validation, S.R.V. and V.N.U.; formal analysis, S.R.V. and V.N.U.; investigation, S.R.V. and V.N.U.; data curation, S.R.V. and V.N.U.; writing—original draft preparation, S.R.V. and V.N.U.; writing—review and editing, S.R.V. and V.N.U.; visualization, V.N.U.; supervision, V.N.U. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Uversky, V.N. Looking at the recent advances in understanding alpha-synuclein and its aggregation through the proteoform prism. F1000Res 2017, 6, 525. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2012, 2, a009399. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem 2002, 277, 11970–11978. [Google Scholar] [CrossRef]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Sung, Y.H.; Eliezer, D. Secondary structure and dynamics of micelle bound beta- and gamma-synuclein. Protein Sci 2006, 15, 1162–1174. [Google Scholar] [CrossRef]
- Sung, Y.H.; Eliezer, D. Residual structure, backbone dynamics, and interactions within the synuclein family. J Mol Biol 2007, 372, 689–707. [Google Scholar] [CrossRef]
- Binolfi, A.; Theillet, F.X.; Selenko, P. Bacterial in-cell NMR of human alpha-synuclein: A disordered monomer by nature? Biochem Soc Trans 2012, 40, 950–954. [Google Scholar] [CrossRef]
- Limatola, A.; Eichmann, C.; Jacob, R.S.; Ben-Nissan, G.; Sharon, M.; Binolfi, A.; Selenko, P. Time-Resolved NMR Analysis of Proteolytic alpha-Synuclein Processing in vitro and in cellulo. Proteomics 2018, 18, e1800056. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Schneider, R.; Cantrelle, F.X.; Huvent, I.; Lippens, G. Studying Intrinsically Disordered Proteins under True In Vivo Conditions by Combined Cross-Polarization and Carbonyl-Detection NMR Spectroscopy. Angew Chem Int Ed Engl 2016, 55, 7418–7422. [Google Scholar] [CrossRef] [PubMed]
- Sciolino, N.; Burz, D.S.; Shekhtman, A. In-Cell NMR Spectroscopy of Intrinsically Disordered Proteins. Proteomics 2019, 19, e1800055. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Zhou, L.Z.; Pielak, G.J. Hydrogen exchange of disordered proteins in Escherichia coli. Protein Sci 2015, 24, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef]
- Waudby, C.A.; Camilloni, C.; Fitzpatrick, A.W.; Cabrita, L.D.; Dobson, C.M.; Vendruscolo, M.; Christodoulou, J. In-cell NMR characterization of the secondary structure populations of a disordered conformation of alpha-synuclein within E. coli cells. PLoS ONE 2013, 8, e72286. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Lee, V.M.; Trojanowski, J.Q. Synucleinopathies: Clinical and pathological implications. Arch Neurol 2001, 58, 186–190. [Google Scholar] [CrossRef]
- Goedert, M. Filamentous nerve cell inclusions in neurodegenerative diseases: Tauopathies and alpha-synucleinopathies. Philos Trans R Soc Lond B Biol Sci 1999, 354, 1101–1118. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Parkinson’s disease and other alpha-synucleinopathies. Clin Chem Lab Med 2001, 39, 308–312. [Google Scholar] [CrossRef]
- Goedert, M.; Falcon, B.; Clavaguera, F.; Tolnay, M. Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr Neurol Neurosci Rep 2014, 14, 495. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J Parkinsons Dis 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci 2000, 920, 16–27. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1998, 249, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 1998, 251, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.P.; Power, J.H.; Blumbergs, P.C.; Blessing, W.W. Multiple-system atrophy: A new alpha-synuclein disease? Lancet 1998, 352, 547–548. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ 1998, 5, 832–837. [Google Scholar] [CrossRef]
- Takeda, A.; Mallory, M.; Sundsmo, M.; Honer, W.; Hansen, L.; Masliah, E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol 1998, 152, 367–372. [Google Scholar]
- Lucking, C.B.; Brice, A. Alpha-synuclein and Parkinson’s disease. Cell Mol Life Sci 2000, 57, 1894–1908. [Google Scholar] [CrossRef]
- Arawaka, S.; Saito, Y.; Murayama, S.; Mori, H. Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology 1998, 51, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Matsumoto, K.; Takayama, K.; Yoshimoto, M.; Takahashi, H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett 1997, 239, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb Perspect Med 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. Synucleinopathies and tauopathies. In Basic Neurochemistry; Elsevier: 2012; pp. 829–843.
- Surguchov, A.; Surguchev, A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Lee, V.M. Parkinson’s disease and related alpha-synucleinopathies are brain amyloidoses. Ann N Y Acad Sci 2003, 991, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Lundvig, D.; Lindersson, E.; Jensen, P.H. Pathogenic effects of alpha-synuclein aggregation. Brain Res Mol Brain Res 2005, 134, 3–17. [Google Scholar] [PubMed]
- Kosaka, K. Lewy bodies in cerebral cortex, report of three cases. Acta Neuropathol (Berl) 1978, 42, 127–134. [Google Scholar] [CrossRef]
- Kosaka, K.; Mehraein, P. Dementia-Parkinsonism syndrome with numerous Lewy bodies and senile plaques in cerebral cortex. Arch Psychiatr Nervenkr 1979, 226, 241–250. [Google Scholar] [CrossRef]
- Seidel, K.; Mahlke, J.; Siswanto, S.; Kruger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.T.; Wicht, H.; Korf, H.W.; et al. The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol 2015, 25, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.; Bagic, A. Olfactory pathogenesis of idiopathic Parkinson disease revisited. Mov Disord 2008, 23, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Brooks, P.L.; Hazrati, L.N.; Lang, A.E. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol Commun 2013, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J Parkinsons Dis 2015, 5, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Spillantini, M.G. Physiological and pathological properties of alpha-synuclein. Cell Mol Life Sci 2007, 64, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T., Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39, 2552–2563. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci U S A 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J Biol Chem 2014, 289, 21856–21876. [Google Scholar] [CrossRef]
- Dev, K.K.; Hofele, K.; Barbieri, S.; Buchman, V.L.; van der Putten, H. Part II: Alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 2003, 45, 14–44. [Google Scholar] [CrossRef] [PubMed]
- da Costa, C.A.; Ancolio, K.; Checler, F. Wild-type but not Parkinson’s disease-related ala-53 --> Thr mutant alpha -synuclein protects neuronal cells from apoptotic stimuli. J Biol Chem 2000, 275, 24065–24069. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar] [CrossRef]
- Ahmad, A.; Burns, C.S.; Fink, A.L.; Uversky, V.N. Peculiarities of copper binding to alpha-synuclein. J Biomol Struct Dyn 2012, 29, 825–842. [Google Scholar] [CrossRef]
- Carboni, E.; Lingor, P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics 2015, 7, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Bower, K.; Fink, A.L. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: Implications for Parkinson’s disease. Neurotoxicology 2002, 23, 527–536. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett 2001, 500, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Maturana, M.G.; Pinheiro, A.S.; de Souza, T.L.; Follmer, C. Unveiling the role of the pesticides paraquat and rotenone on alpha-synuclein fibrillation in vitro. Neurotoxicology 2015, 46, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Cali, T.; Szabo, I.; Brini, M. Alpha-synuclein at the intracellular and the extracellular side: Functional and dysfunctional implications. Biol Chem 2017, 398, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, M.; Chieregatti, E. Mechanisms of alpha-synuclein action on neurotransmission: Cell-autonomous and non-cell autonomous role. Biomolecules 2015, 5, 865–892. [Google Scholar] [CrossRef]
- Uversky, V.N. Alpha-synuclein misfolding and neurodegenerative diseases. Curr Protein Pept Sci 2008, 9, 507–540. [Google Scholar] [CrossRef]
- Payton, J.E.; Perrin, R.J.; Clayton, D.F.; George, J.M. Protein-protein interactions of alpha-synuclein in brain homogenates and transfected cells. Brain Res Mol Brain Res 2001, 95, 138–145. [Google Scholar] [CrossRef]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins interacting with both a-synuclein and DJ-1. Mol Cell Proteomics 2006. [Google Scholar]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Hayashi, J.; Carver, J.A. beta-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Liu, Y.E.; Jia, T.; Wang, M.; Liu, J.; Xiao, G.; Joseph, B.K.; Rosen, C.; Shi, Y.E. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res 1997, 57, 759–764. [Google Scholar] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform 2000, 11, 161–171. [Google Scholar] [PubMed]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in thirty five hundred proteomes from viruses and the three domains of life. J Biomol Struct Dyn In press. 2012. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac Symp Biocomput 1998, 473–484. [Google Scholar]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Biol 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J Mol Graph Model 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem Sci 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Daughdrill, G.W.; Pielak, G.J.; Uversky, V.N.; Cortese, M.S.; Dunker, A.K. Natively disordered proteins. In Handbook of Protein Folding; Buchner, J., Kiefhaber, T., Eds.; Wiley-VCH, Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 271–353. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim Biophys Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z. The protein trinity--linking function and disorder. Nat Biotechnol 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J 2015, 282, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int J Mol Sci 2016, 17, 1874. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog Mol Biol Transl Sci 2019, 166, 1–17. [Google Scholar] [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Multi-functionality of proteins involved in GPCR and G protein signaling: Making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell Mol Life Sci 2019, 76, 4461–4492. [Google Scholar] [CrossRef]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS Journal 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 2005, 18, 343–384. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys J 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Vucetic, S.; Xie, H.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res 2007, 6, 1899–1916. [Google Scholar] [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res 2007, 6, 1882–1898. [Google Scholar] [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res 2007, 6, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem Rev 2014, 114, 6561–6588. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem Rev 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Uversky, V.N. Multitude of binding modes attainable by intrinsically disordered proteins: A portrait gallery of disorder-based complexes. Chem Soc Rev 2011, 40, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Hsu, W.L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci 2014, 23, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol 2017, 44, 18–30. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv Colloid Interface Sci 2017, 239, 97–114. [Google Scholar] [CrossRef]
- Uversky, V.N. Recent Developments in the Field of Intrinsically Disordered Proteins: Intrinsic Disorder–Based Emergence in Cellular Biology in Light of the Physiological and Pathological Liquid–Liquid Phase Transitions. Annual Review of Biophysics 2021, 50, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst 2008, 4, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.H.; Suk, J.E.; Kim, D.H.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K.H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J Biol Chem 2000, 275, 29426–29432. [Google Scholar] [CrossRef] [PubMed]
- Adkins, J.N.; Lumb, K.J. Intrinsic structural disorder and sequence features of the cell cycle inhibitor p57Kip2. Proteins 2002, 46, 1–7. [Google Scholar] [CrossRef]
- Chang, B.S.; Minn, A.J.; Muchmore, S.W.; Fesik, S.W.; Thompson, C.B. Identification of a novel regulatory domain in Bcl-X(L) and Bcl-2. EMBO J 1997, 16, 968–977. [Google Scholar] [CrossRef]
- Campbell, K.M.; Terrell, A.R.; Laybourn, P.J.; Lumb, K.J. Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry 2000, 39, 2708–2713. [Google Scholar] [CrossRef]
- Sunde, M.; McGrath, K.C.; Young, L.; Matthews, J.M.; Chua, E.L.; Mackay, J.P.; Death, A.K. TC-1 is a novel tumorigenic and natively disordered protein associated with thyroid cancer. Cancer Res 2004, 64, 2766–2773. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 1984, 122, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J 1985, 4, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Balin, B.J.; Otvos, L., Jr.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, K.E.; Dalton, A.J.; McLachlan, C.; Wen, G.Y.; Wisniewski, H.M. Alzheimer’s disease in Down’s syndrome: Clinicopathologic studies. Neurology 1985, 35, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Shattuck lecture--neurodegenerative diseases and prions. N Engl J Med 2001, 344, 1516–1526. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Polyglutamine diseases: Protein cleavage and aggregation. Curr Opin Neurobiol 1999, 9, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry 2006, 45, 10448–10460. [Google Scholar] [CrossRef]
- Uversky, V.N. Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res 2008, 5, 260–287. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Midic, U.; Xie, H.; Xue, B.; Vucetic, S.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genomics 2009, 10 Suppl 1, S7. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci 2009, 14, 5188–5238. [Google Scholar] [CrossRef] [PubMed]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Protein disorder in the human diseasome: Unfoldomics of human genetic diseases. PLoS Computational Biology In press. 2008. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. Bioessays 2009, 31, 328–335. [Google Scholar] [CrossRef] [PubMed]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M. Is the cause of Parkinson’s disease environmental or hereditary? Evidence from twin studies. Adv Neurol 2003, 91, 133–142. [Google Scholar] [PubMed]
- Farrer, M.J. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat Rev Genet 2006, 7, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci 1999, 22, 123–144. [Google Scholar] [CrossRef]
- Moghal, S.; Rajput, A.H.; D’Arcy, C.; Rajput, R. Prevalence of movement disorders in elderly community residents. Neuroepidemiology 1994, 13, 175–178. [Google Scholar] [CrossRef]
- Fahn, S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci 2003, 991, 1–14. [Google Scholar] [CrossRef]
- Mizuno, Y.; Hattori, N.; Kitada, T.; Matsumine, H.; Mori, H.; Shimura, H.; Kubo, S.; Kobayashi, H.; Asakawa, S.; Minoshima, S.; et al. Familial Parkinson’s disease. Alpha-synuclein and parkin. Adv Neurol 2001, 86, 13–21. [Google Scholar]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am J Epidemiol 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 1996, 55, 259–272. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Singleton, A.; Gwinn-Hardy, K.; Sharabi, Y.; Li, S.T.; Holmes, C.; Dendi, R.; Hardy, J.; Crawley, A.; Goldstein, D.S. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 2004, 127, 768–772. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Kachergus, J.; Forno, L.; Lincoln, S.; Wang, D.S.; Hulihan, M.; Maraganore, D.; Gwinn-Hardy, K.; Wszolek, Z.; Dickson, D.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 2004, 55, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Morar, A.S.; Olteanu, A.; Young, G.B.; Pielak, G.J. Solvent-induced collapse of alpha-synuclein and acid-denatured cytochrome c. Protein Sci 2001, 10, 2195–2199. [Google Scholar] [CrossRef]
- Bussell, R., Jr.; Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J Biol Chem 2001, 276, 45996–46003. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A 2005, 102, 1430–1435. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci U S A 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Sequence Determinants of the Aggregation of Proteins Within Condensates Generated by Liquid-liquid Phase Separation. J Mol Biol 2022, 434, 167201. [Google Scholar] [CrossRef]
- Hardenberg, M.C.; Sinnige, T.; Casford, S.; Dada, S.T.; Poudel, C.; Robinson, E.A.; Fuxreiter, M.; Kaminksi, C.F.; Kaminski Schierle, G.S.; Nollen, E.A.A.; et al. Observation of an alpha-synuclein liquid droplet state and its maturation into Lewy body-like assemblies. J Mol Cell Biol 2021, 13, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Mo, X.; Wang, J.; Ye, X.; Yu, H.; Liu, Y. alpha-Synuclein phase separation and amyloid aggregation are modulated by C-terminal truncations. FEBS Lett 2022, 596, 1388–1400. [Google Scholar] [CrossRef]
- Huang, S.; Xu, B.; Liu, Y. Calcium promotes alpha-synuclein liquid-liquid phase separation to accelerate amyloid aggregation. Biochem Biophys Res Commun 2022, 603, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. alpha-Synuclein aggregation nucleates through liquid-liquid phase separation. Nat Chem 2020, 12, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Sawner, A.S.; Ray, S.; Yadav, P.; Mukherjee, S.; Panigrahi, R.; Poudyal, M.; Patel, K.; Ghosh, D.; Kummerant, E.; Kumar, A.; et al. Modulating alpha-Synuclein Liquid-Liquid Phase Separation. Biochemistry 2021, 60, 3676–3696. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Fuxreiter, M. Protein condensation diseases: Therapeutic opportunities. Nat Commun 2022, 13, 5550. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Sudhof, T.C. A broken alpha -helix in folded alpha -Synuclein. J Biol Chem 2003, 278, 15313–15318. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fink, A.L. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem 2003, 278, 16873–16877. [Google Scholar] [CrossRef] [PubMed]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational behavior and aggregation of alpha-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.N.; Jao, C.C.; Hegde, B.G.; Langen, R.; Ulmer, T.S. A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc 2010, 132, 8657–8668. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Singh, V.K.; Jia, Z.; Forman-Kay, J.D. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: Implications for fibrillation. Protein Sci 2006, 15, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. alpha-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 2013, 125, 753–769. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 2014, 35, 2180–e2181. [Google Scholar] [CrossRef]
- Lemkau, L.R.; Comellas, G.; Kloepper, K.D.; Woods, W.S.; George, J.M.; Rienstra, C.M. Mutant protein A30P alpha-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J Biol Chem 2012, 287, 11526–11532. [Google Scholar] [CrossRef]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T., Jr. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef]
- Pandey, N.; Schmidt, R.E.; Galvin, J.E. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp Neurol 2006, 197, 515–520. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J Neurochem 2014, 131, 859–867. [Google Scholar] [CrossRef]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates alpha-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Giasson, B.I. The A53E alpha-synuclein pathological mutation demonstrates reduced aggregation propensity in vitro and in cell culture. Neurosci Lett 2015, 597, 43–48. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Uversky, V.N. Analyzing IDPs in interactomes. In Intrinsically Disordered Proteins, Kragelund, B.B., Skriver, K., Eds.; Humana New York, NY, 2020; Volume Methods in Molecular Biology, pp. 895–945.
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: Intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol Biol 2011, 77, 205–223. [Google Scholar] [CrossRef]
- Xue, B.; Oldfield, C.J.; Van, Y.Y.; Dunker, A.K.; Uversky, V.N. Protein intrinsic disorder and induced pluripotent stem cells. Mol Biosyst 2012, 8, 134–150. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput 2012, 128–139. [Google Scholar]
- Vasak, M.; Hasler, D.W. Metallothioneins: New functional and structural insights. Curr Opin Chem Biol 2000, 4, 177–183. [Google Scholar] [CrossRef]
- Quaife, C.J.; Findley, S.D.; Erickson, J.C.; Froelick, G.J.; Kelly, E.J.; Zambrowicz, B.P.; Palmiter, R.D. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry 1994, 33, 7250–7259. [Google Scholar] [CrossRef]
- Moffatt, P.; Denizeau, F. Metallothionein in physiological and physiopathological processes. Drug Metab Rev 1997, 29, 261–307. [Google Scholar] [CrossRef]
- Ding, Z.C.; Zheng, Q.; Cai, B.; Ni, F.Y.; Yu, W.H.; Teng, X.C.; Gao, Y.; Liu, F.; Chen, D.; Wang, Y.; et al. Study on structure-property-reactivity-function relationship of human neuronal growth inhibitory factor (hGIF). J Inorg Biochem 2008, 102, 1965–1972. [Google Scholar] [CrossRef]
- Bogumil, R.; Faller, P.; Binz, P.A.; Vasak, M.; Charnock, J.M.; Garner, C.D. Structural characterization of Cu(I) and Zn(II) sites in neuronal-growth-inhibitory factor by extended X-ray absorption fine structure (EXAFS). Eur J Biochem 1998, 255, 172–177. [Google Scholar] [CrossRef]
- Sewell, A.K.; Jensen, L.T.; Erickson, J.C.; Palmiter, R.D.; Winge, D.R. Bioactivity of metallothionein-3 correlates with its novel beta domain sequence rather than metal binding properties. Biochemistry 1995, 34, 4740–4747. [Google Scholar] [CrossRef]
- Hasler, D.W.; Jensen, L.T.; Zerbe, O.; Winge, D.R.; Vasak, M. Effect of the two conserved prolines of human growth inhibitory factor (metallothionein-3) on its biological activity and structure fluctuation: Comparison with a mutant protein. Biochemistry 2000, 39, 14567–14575. [Google Scholar] [CrossRef]
- Romero-Isart, N.; Jensen, L.T.; Zerbe, O.; Winge, D.R.; Vasak, M. Engineering of metallothionein-3 neuroinhibitory activity into the inactive isoform metallothionein-1. J Biol Chem 2002, 277, 37023–37028. [Google Scholar] [CrossRef]
- Uchida, Y.; Takio, K.; Titani, K.; Ihara, Y.; Tomonaga, M. The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron 1991, 7, 337–347. [Google Scholar] [CrossRef]
- Vasak, M.; Meloni, G. Mammalian Metallothionein-3: New Functional and Structural Insights. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef]
- Jiang, Z.; Shen, B.; Xiang, J. Metal-dependent interactions of metallothionein-3 beta-domain with amyloid-beta peptide and related physiological implications. J Inorg Biochem 2019, 196, 110693. [Google Scholar] [CrossRef]
- Koh, J.Y.; Lee, S.J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol Brain 2020, 13, 116. [Google Scholar] [CrossRef]
- Uchida, Y. Growth-inhibitory factor, metallothionein-like protein, and neurodegenerative diseases. Biol Signals 1994, 3, 211–215. [Google Scholar] [CrossRef]
- Howells, C.; West, A.K.; Chung, R.S. Neuronal growth-inhibitory factor (metallothionein-3): Evaluation of the biological function of growth-inhibitory factor in the injured and neurodegenerative brain. FEBS J 2010, 277, 2931–2939. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Q.; Cai, B.; Li, H.; Sze, K.H.; Huang, Z.X.; Wu, H.M.; Sun, H. Solution structure and dynamics of human metallothionein-3 (MT-3). FEBS Lett 2006, 580, 795–800. [Google Scholar] [CrossRef]
- Oz, G.; Zangger, K.; Armitage, I.M. Three-dimensional structure and dynamics of a brain specific growth inhibitory factor: Metallothionein-3. Biochemistry 2001, 40, 11433–11441. [Google Scholar] [CrossRef]
- Yuan, A.T.; Korkola, N.C.; Stillman, M.J. Apo-metallothionein-3 cooperatively forms tightly compact structures under physiological conditions. J Biol Chem 2023, 299, 102899. [Google Scholar] [CrossRef]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Pountney, D.L.; Dickson, T.C.; Power, J.H.; Vickers, J.C.; West, A.J.; Gai, W.P. Association of metallothionein-III with oligodendroglial cytoplasmic inclusions in multiple system atrophy. Neurotox Res 2011, 19, 115–122. [Google Scholar] [CrossRef]
- Ugbode, C.; West, R.J.H. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis 2021, 147, 105144. [Google Scholar] [CrossRef]
- Rusten, T.E.; Stenmark, H. How do ESCRT proteins control autophagy? J Cell Sci 2009, 122, 2179–2183. [Google Scholar] [CrossRef]
- Bhutta, M.S.; McInerny, C.J.; Gould, G.W. ESCRT function in cytokinesis: Location, dynamics and regulation by mitotic kinases. Int J Mol Sci 2014, 15, 21723–21739. [Google Scholar] [CrossRef]
- Caballe, A.; Martin-Serrano, J. ESCRT machinery and cytokinesis: The road to daughter cell separation. Traffic 2011, 12, 1318–1326. [Google Scholar] [CrossRef]
- Radulovic, M.; Schink, K.O.; Wenzel, E.M.; Nahse, V.; Bongiovanni, A.; Lafont, F.; Stenmark, H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 2018, 37. [Google Scholar] [CrossRef]
- Lata, S.; Schoehn, G.; Solomons, J.; Pires, R.; Gottlinger, H.G.; Weissenhorn, W. Structure and function of ESCRT-III. Biochem Soc Trans 2009, 37, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Krasniak, C.S.; Ahmad, S.T. The role of CHMP2B(Intron5) in autophagy and frontotemporal dementia. Brain Res 2016, 1649, 151–157. [Google Scholar] [CrossRef]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Bugiani, O. The many ways to frontotemporal degeneration and beyond. Neurol Sci 2007, 28, 241–244. [Google Scholar] [CrossRef]
- Urwin, H.; Ghazi-Noori, S.; Collinge, J.; Isaacs, A. The role of CHMP2B in frontotemporal dementia. Biochem Soc Trans 2009, 37, 208–212. [Google Scholar] [CrossRef]
- Siuda, J.; Fujioka, S.; Wszolek, Z.K. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat Disord 2014, 20, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Babst, M.; Wendland, B.; Estepa, E.J.; Emr, S.D. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J 1998, 17, 2982–2993. [Google Scholar] [CrossRef] [PubMed]
- Bodon, G.; Chassefeyre, R.; Pernet-Gallay, K.; Martinelli, N.; Effantin, G.; Hulsik, D.L.; Belly, A.; Goldberg, Y.; Chatellard-Causse, C.; Blot, B.; et al. Charged multivesicular body protein 2B (CHMP2B) of the endosomal sorting complex required for transport-III (ESCRT-III) polymerizes into helical structures deforming the plasma membrane. J Biol Chem 2011, 286, 40276–40286. [Google Scholar] [CrossRef] [PubMed]
- Stuchell-Brereton, M.D.; Skalicky, J.J.; Kieffer, C.; Karren, M.A.; Ghaffarian, S.; Sundquist, W.I. ESCRT-III recognition by VPS4 ATPases. Nature 2007, 449, 740–744. [Google Scholar] [CrossRef]
- Xiang, Y.; Xin, J.; Le, W.; Yang, Y. Neurogranin: A Potential Biomarker of Neurological and Mental Diseases. Front Aging Neurosci 2020, 12, 584743. [Google Scholar] [CrossRef]
- Represa, A.; Deloulme, J.C.; Sensenbrenner, M.; Ben-Ari, Y.; Baudier, J. Neurogranin: Immunocytochemical localization of a brain-specific protein kinase C substrate. J Neurosci 1990, 10, 3782–3792. [Google Scholar] [CrossRef]
- Chen, S.J.; Klann, E.; Gower, M.C.; Powell, C.M.; Sessoms, J.S.; Sweatt, J.D. Studies with synthetic peptide substrates derived from the neuronal protein neurogranin reveal structural determinants of potency and selectivity for protein kinase C. Biochemistry 1993, 32, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Baudier, J.; Deloulme, J.C.; Van Dorsselaer, A.; Black, D.; Matthes, H.W. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. J Biol Chem 1991, 266, 229–237. [Google Scholar] [PubMed]
- Gerendasy, D.D.; Herron, S.R.; Watson, J.B.; Sutcliffe, J.G. Mutational and biophysical studies suggest RC3/neurogranin regulates calmodulin availability. J Biol Chem 1994, 269, 22420–22426. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, B.; Morley, P.; Whitfield, J. Ca2+-calmodulin and protein kinase Cs: A hypothetical synthesis of their conflicting convergences on shared substrate domains. Trends Neurosci 1999, 22, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Gerendasy, D. Homeostatic tuning of Ca2+ signal transduction by members of the calpacitin protein family. J Neurosci Res 1999, 58, 107–119. [Google Scholar] [CrossRef]
- Fyfe, I. Alzheimer disease: Neurogranin in the CSF signals early Alzheimer disease and predicts disease progression. Nat Rev Neurol 2015, 11, 609. [Google Scholar] [CrossRef]
- Hellwig, K.; Kvartsberg, H.; Portelius, E.; Andreasson, U.; Oberstein, T.J.; Lewczuk, P.; Blennow, K.; Kornhuber, J.; Maler, J.M.; Zetterberg, H.; et al. Neurogranin and YKL-40: Independent markers of synaptic degeneration and neuroinflammation in Alzheimer’s disease. Alzheimers Res Ther 2015, 7, 74. [Google Scholar] [CrossRef]
- Kester, M.I.; Teunissen, C.E.; Crimmins, D.L.; Herries, E.M.; Ladenson, J.H.; Scheltens, P.; van der Flier, W.M.; Morris, J.C.; Holtzman, D.M.; Fagan, A.M. Neurogranin as a Cerebrospinal Fluid Biomarker for Synaptic Loss in Symptomatic Alzheimer Disease. JAMA Neurol 2015, 72, 1275–1280. [Google Scholar] [CrossRef]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol 2016, 73, 561–571. [Google Scholar] [CrossRef]
- Portelius, E.; Zetterberg, H.; Skillback, T.; Tornqvist, U.; Andreasson, U.; Trojanowski, J.Q.; Weiner, M.W.; Shaw, L.M.; Mattsson, N.; Blennow, K.; et al. Cerebrospinal fluid neurogranin: Relation to cognition and neurodegeneration in Alzheimer’s disease. Brain 2015, 138, 3373–3385. [Google Scholar] [CrossRef]
- Kvartsberg, H.; Duits, F.H.; Ingelsson, M.; Andreasen, N.; Ohrfelt, A.; Andersson, K.; Brinkmalm, G.; Lannfelt, L.; Minthon, L.; Hansson, O.; et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement 2015, 11, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Olsson, B.; Hoglund, K.; Cullen, N.C.; Kvartsberg, H.; Andreasson, U.; Zetterberg, H.; Sandelius, A.; Shaw, L.M.; Lee, V.M.Y.; et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: Relation to clinical phenotypes and neuropathology. Acta Neuropathol 2018, 136, 363–376. [Google Scholar] [CrossRef]
- Blennow, K.; Diaz-Lucena, D.; Zetterberg, H.; Villar-Pique, A.; Karch, A.; Vidal, E.; Hermann, P.; Schmitz, M.; Ferrer Abizanda, I.; Zerr, I.; et al. CSF neurogranin as a neuronal damage marker in CJD: A comparative study with AD. J Neurol Neurosurg Psychiatry 2019, 90, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 2006, 15, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Runne, H.; Kuhn, A.; Wild, E.J.; Pratyaksha, W.; Kristiansen, M.; Isaacs, J.D.; Regulier, E.; Delorenzi, M.; Tabrizi, S.J.; Luthi-Carter, R. Analysis of potential transcriptomic biomarkers for Huntington’s disease in peripheral blood. Proc Natl Acad Sci U S A 2007, 104, 14424–14429. [Google Scholar] [CrossRef] [PubMed]
- Lista, S.; Santos-Lozano, A.; Emanuele, E.; Mercuri, N.B.; Gabelle, A.; Lopez-Ortiz, S.; Martin-Hernandez, J.; Maisto, N.; Imbimbo, C.; Caraci, F.; et al. Monitoring synaptic pathology in Alzheimer’s disease through fluid and PET imaging biomarkers: A comprehensive review and future perspectives. Mol Psychiatry 2024. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Lu, Q.; Kang, H.; Suridjan, I.; Kollmorgen, G.; Wild, N.; Deming, Y.; Van Hulle, C.A.; Anderson, R.M.; Zetterberg, H.; et al. CSF metabolites associated with biomarkers of Alzheimer’s disease pathology. Front Aging Neurosci 2023, 15, 1214932. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Gobom, J.; Sjodin, S.; Brinkmalm, G.; Ashton, N.J.; Svensson, J.; Johansson, P.; Portelius, E.; Zetterberg, H.; Blennow, K.; et al. Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement (Amst) 2021, 13, e12179. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, T.; Blandino, V.; Maniscalco, L.; Matranga, D.; Graziano, F.; Guajana, F.; Agnello, L.; Lo Sasso, B.; Gambino, C.M.; Giglio, R.V.; et al. Biomarkers Related to Synaptic Dysfunction to Discriminate Alzheimer’s Disease from Other Neurological Disorders. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Chien, W.J.; Arunkumar, A.I. Conformation of a protein kinase C substrate NG(28-43), and its analog in aqueous and sodium dodecyl sulfate micelle solutions. Biophys J 1997, 72, 554–566. [Google Scholar] [CrossRef]
- Ran, X.; Miao, H.H.; Sheu, F.S.; Yang, D. Structural and dynamic characterization of a neuron-specific protein kinase C substrate, neurogranin. Biochemistry 2003, 42, 5143–5150. [Google Scholar] [CrossRef]
- Ishizuka, T.; Saisu, H.; Odani, S.; Abe, T. Synaphin: A protein associated with the docking/fusion complex in presynaptic terminals. Biochem Biophys Res Commun 1995, 213, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Missler, M.; Li, C.; Sudhof, T.C. Complexins: Cytosolic proteins that regulate SNAP receptor function. Cell 1995, 83, 111–119. [Google Scholar] [CrossRef]
- Takahashi, S.; Yamamoto, H.; Matsuda, Z.; Ogawa, M.; Yagyu, K.; Taniguchi, T.; Miyata, T.; Kaba, H.; Higuchi, T.; Okutani, F.; et al. Identification of two highly homologous presynaptic proteins distinctly localized at the dendritic and somatic synapses. FEBS Lett 1995, 368, 455–460. [Google Scholar] [CrossRef]
- Ono, S.; Baux, G.; Sekiguchi, M.; Fossier, P.; Morel, N.F.; Nihonmatsu, I.; Hirata, K.; Awaji, T.; Takahashi, S.; Takahashi, M. Regulatory roles of complexins in neurotransmitter release from mature presynaptic nerve terminals. Eur J Neurosci 1998, 10, 2143–2152. [Google Scholar] [CrossRef]
- Yamada, M.; Saisu, H.; Ishizuka, T.; Takahashi, H.; Abe, T. Immunohistochemical distribution of the two isoforms of synaphin/complexin involved in neurotransmitter release: Localization at the distinct central nervous system regions and synaptic types. Neuroscience 1999, 93, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Itakura, M.; Misawa, H.; Sekiguchi, M.; Takahashi, S.; Takahashi, M. Transfection analysis of functional roles of complexin I and II in the exocytosis of two different types of secretory vesicles. Biochem Biophys Res Commun 1999, 265, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Reim, K.; Mansour, M.; Varoqueaux, F.; McMahon, H.T.; Sudhof, T.C.; Brose, N.; Rosenmund, C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 2001, 104, 71–81. [Google Scholar] [CrossRef]
- Krishnakumar, S.S.; Radoff, D.T.; Kummel, D.; Giraudo, C.G.; Li, F.; Khandan, L.; Baguley, S.W.; Coleman, J.; Reinisch, K.M.; Pincet, F.; et al. A conformational switch in complexin is required for synaptotagmin to trigger synaptic fusion. Nat Struct Mol Biol 2011, 18, 934–940. [Google Scholar] [CrossRef]
- Lottermoser, J.A.; Dittman, J.S. Complexin Membrane Interactions: Implications for Synapse Evolution and Function. J Mol Biol 2023, 435, 167774. [Google Scholar] [CrossRef]
- Gispert, S.; Kurz, A.; Brehm, N.; Rau, K.; Walter, M.; Riess, O.; Auburger, G. Complexin-1 and Foxp1 Expression Changes Are Novel Brain Effects of Alpha-Synuclein Pathology. Mol Neurobiol 2015, 52, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Reim, K.; Chen, X.; Chao, H.T.; Deng, H.; Rizo, J.; Brose, N.; Rosenmund, C. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol 2007, 14, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Hobson, R.J.; Liu, Q.; Watanabe, S.; Jorgensen, E.M. Complexin maintains vesicles in the primed state in C. elegans. Curr Biol 2011, 21, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Hu, Z.; Fenz, K.M.; Fernandez, J.; Dittman, J.S. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 2011, 21, 97–105. [Google Scholar] [CrossRef]
- Lai, Y.; Choi, U.B.; Zhang, Y.; Zhao, M.; Pfuetzner, R.A.; Wang, A.L.; Diao, J.; Brunger, A.T. N-terminal domain of complexin independently activates calcium-triggered fusion. Proc Natl Acad Sci U S A 2016, 113, E4698–E4707. [Google Scholar] [CrossRef] [PubMed]
- Zdanowicz, R.; Kreutzberger, A.; Liang, B.; Kiessling, V.; Tamm, L.K.; Cafiso, D.S. Complexin Binding to Membranes and Acceptor t-SNAREs Explains Its Clamping Effect on Fusion. Biophys J 2017, 113, 1235–1250. [Google Scholar] [CrossRef] [PubMed]
- Bera, M.; Ramakrishnan, S.; Coleman, J.; Krishnakumar, S.S.; Rothman, J.E. Molecular determinants of complexin clamping and activation function. Elife 2022, 11. [Google Scholar] [CrossRef]
- Pabst, S.; Hazzard, J.W.; Antonin, W.; Sudhof, T.C.; Jahn, R.; Rizo, J.; Fasshauer, D. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem 2000, 275, 19808–19818. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tomchick, D.R.; Kovrigin, E.; Arac, D.; Machius, M.; Sudhof, T.C.; Rizo, J. Three-dimensional structure of the complexin/SNARE complex. Neuron 2002, 33, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.E.; Weninger, K.; Ernst, J.; Chu, S.; Brunger, A.T. Single-molecule studies of synaptotagmin and complexin binding to the SNARE complex. Biophys J 2005, 89, 690–702. [Google Scholar] [CrossRef]
- Malsam, J.; Seiler, F.; Schollmeier, Y.; Rusu, P.; Krause, J.M.; Sollner, T.H. The carboxy-terminal domain of complexin I stimulates liposome fusion. Proc Natl Acad Sci U S A 2009, 106, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Kaeser-Woo, Y.J.; Yang, X.; Sudhof, T.C. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci 2012, 32, 2877–2885. [Google Scholar] [CrossRef] [PubMed]
- Wragg, R.T.; Snead, D.; Dong, Y.; Ramlall, T.F.; Menon, I.; Bai, J.; Eliezer, D.; Dittman, J.S. Synaptic vesicles position complexin to block spontaneous fusion. Neuron 2013, 77, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Wragg, R.T.; Dittman, J.S.; Eliezer, D. Membrane curvature sensing by the C-terminal domain of complexin. Nat Commun 2014, 5, 4955. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Lai, Y.; Li, X.; Wang, M.; Leitz, J.; Hu, Y.; Zhang, Y.; Choi, U.B.; Cipriano, D.; Pfuetzner, R.A.; et al. C-terminal domain of mammalian complexin-1 localizes to highly curved membranes. Proc Natl Acad Sci U S A 2016, 113, E7590–E7599. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.C.; Wu, L.; Mandal, T.; Swift, M.; Zhang, Z.; Alaghemandi, M.; Wu, Z.; Bradberry, M.M.; Deo, C.; Lavis, L.D.; et al. The complexin C-terminal amphipathic helix stabilizes the fusion pore open state by sculpting membranes. Nat Struct Mol Biol 2022, 29, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Trimbuch, T.; Rosenmund, C. Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci 2016, 17, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, M.A.; Thomas, D.D.; Groblewski, G.E. Complexin 2 modulates vesicle-associated membrane protein (VAMP) 2-regulated zymogen granule exocytosis in pancreatic acini. J Biol Chem 2010, 285, 35558–35566. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, S.; Nakanishi, M.; Hirashima, N. Complexin II facilitates exocytotic release in mast cells by enhancing Ca2+ sensitivity of the fusion process. J Cell Sci 2005, 118, 2239–2246. [Google Scholar] [CrossRef]
- Tsuru, E.; Oryu, K.; Sawada, K.; Nishihara, M.; Tsuda, M. Complexin 2 regulates secretion of immunoglobulin in antibody-secreting cells. Immun Inflamm Dis 2019, 7, 318–325. [Google Scholar] [CrossRef]
- Tsai, P.S.; Brewis, I.A.; van Maaren, J.; Gadella, B.M. Involvement of complexin 2 in docking, locking and unlocking of different SNARE complexes during sperm capacitation and induced acrosomal exocytosis. PLoS ONE 2012, 7, e32603. [Google Scholar] [CrossRef] [PubMed]
- DiProspero, N.A.; Chen, E.Y.; Charles, V.; Plomann, M.; Kordower, J.H.; Tagle, D.A. Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol 2004, 33, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Parplys, A.C.; Zhao, W.; Sharma, N.; Groesser, T.; Liang, F.; Maranon, D.G.; Leung, S.G.; Grundt, K.; Dray, E.; Idate, R.; et al. NUCKS1 is a novel RAD51AP1 paralog important for homologous recombination and genome stability. Nucleic Acids Res 2015, 43, 9817–9834. [Google Scholar] [CrossRef] [PubMed]
- Symonowicz, K.; Dus-Szachniewicz, K.; Wozniak, M.; Murawski, M.; Kolodziej, P.; Osiecka, B.; Jurczyszyn, K.; Ziolkowski, P. Immunohistochemical study of nuclear ubiquitous casein and cyclin-dependent kinase substrate 1 in invasive breast carcinoma of no special type. Exp Ther Med 2014, 8, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Drosos, Y.; Kouloukoussa, M.; Ostvold, A.C.; Grundt, K.; Goutas, N.; Vlachodimitropoulos, D.; Havaki, S.; Kollia, P.; Kittas, C.; Marinos, E.; et al. NUCKS overexpression in breast cancer. Cancer Cell Int 2009, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.Y.; Kim, Y.B.; Woo, J.H.; Kim, D.K.; Yeo, M.; Yang, S.J.; Yang, K.S.; Soon, S.K.; Wang, H.J.; Kim, B.W.; et al. Identification of NUCKS1 as a putative oncogene and immunodiagnostic marker of hepatocellular carcinoma. Gene 2016, 584, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Li, X.; Bao, H.; Li, G.; Li, N.; Li, H.; Dou, J. NUCKS1 Acts as a Promising Novel Biomarker for the Prognosis of Patients with Hepatocellular Carcinoma. Cancer Biother Radiopharm 2023, 38, 720–725. [Google Scholar] [CrossRef]
- Shi, C.; Qin, L.; Gao, H.; Gu, L.; Yang, C.; Liu, H.; Liu, T. NUCKS nuclear elevated expression indicates progression and prognosis of ovarian cancer. Tumour Biol 2017, 39, 1010428317714631. [Google Scholar] [CrossRef]
- Huang, Y.K.; Kang, W.M.; Ma, Z.Q.; Liu, Y.Q.; Zhou, L.; Yu, J.C. NUCKS1 promotes gastric cancer cell aggressiveness by upregulating IGF-1R and subsequently activating the PI3K/Akt/mTOR signaling pathway. Carcinogenesis 2019, 40, 370–379. [Google Scholar] [CrossRef]
- Gu, L.; Xia, B.; Zhong, L.; Ma, Y.; Liu, L.; Yang, L.; Lou, G. NUCKS1 overexpression is a novel biomarker for recurrence-free survival in cervical squamous cell carcinoma. Tumour Biol 2014, 35, 7831–7836. [Google Scholar] [CrossRef]
- Zhu, L.L.; Shi, J.J.; Guo, Y.D.; Yang, C.; Wang, R.L.; Li, S.S.; Gan, D.X.; Ma, P.X.; Li, J.Q.; Su, H.C. NUCKS1 promotes the progression of colorectal cancer via activating PI3K/AKT/mTOR signaling pathway. Neoplasma 2023, 70, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Ji, R.; He, H.; Li, N.; Han, C.; Han, J.; Li, X.; Zhang, L.; Wang, Y.; Zhao, W. NUCKS1, a LINC00629-upregulated gene, facilitated osteosarcoma progression and metastasis by elevating asparagine synthesis. Cell Death Dis 2023, 14, 489. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Xu, J.; Zhao, R.; Qi, Y.; Ji, Y.; Ma, K. Upregulation of NUCKS1 in Lung Adenocarcinoma is Associated with a Poor Prognosis. Cancer Invest 2021, 39, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Choi, B.S.; Kim, S.S.; Roh, T.Y.; Park, J.; Yoon, C.H. NUCKS1, a novel Tat coactivator, plays a crucial role in HIV-1 replication by increasing Tat-mediated viral transcription on the HIV-1 LTR promoter. Retrovirology 2014, 11, 67. [Google Scholar] [CrossRef]
- Qiu, B.; Han, W.; Tergaonkar, V. NUCKS: A potential biomarker in cancer and metabolic disease. Clin Sci (Lond) 2015, 128, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, L.; Lu, Z.J.; Sun, X.Y.; Li, J.Y.; Peng, R. Association of three candidate genetic variants in RAB7L1/NUCKS1, MCCC1 and STK39 with sporadic Parkinson’s disease in Han Chinese. J Neural Transm (Vienna) 2016, 123, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Seth, P.K. Functional association between NUCKS1 gene and Parkinson disease: A potential susceptibility biomarker. Bioinformation 2019, 15, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xia, C.; Sun, W.; Qin, X.; Qiu, Y.; Zhu, Z. Genetic Polymorphism of NUCKS1 Is Associated With the Susceptibility of Adolescent Idiopathic Scoliosis. Spine (Phila Pa 1976) 2017, 42, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Ostvold, A.C.; Holtlund, J.; Laland, S.G. A novel, highly phosphorylated protein, of the high-mobility group type, present in a variety of proliferating and non-proliferating mammalian cells. Eur J Biochem 1985, 153, 469–475. [Google Scholar] [CrossRef]
- Maelandsmo, G.M.; Ostvold, A.C.; Laland, S.G. Phosphorylation of the high-mobility-group-like protein P1 by casein kinase-2. Eur J Biochem 1989, 184, 529–534. [Google Scholar] [CrossRef]
- Ostvold, A.C.; Norum, J.H.; Mathiesen, S.; Wanvik, B.; Sefland, I.; Grundt, K. Molecular cloning of a mammalian nuclear phosphoprotein NUCKS, which serves as a substrate for Cdk1 in vivo. Eur J Biochem 2001, 268, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, E.J.; Scherer, S.S. On the molecular architecture of myelinated fibers. Histochem Cell Biol 2000, 113, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J.; Mierzwa, A.; Shroff, S. Molecular architecture of myelinated nerve fibers: Leaky paranodal junctions and paranodal dysmyelination. Neuroscientist 2013, 19, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Tzimourakas, A.; Giasemi, S.; Mouratidou, M.; Karagogeos, D. Structure-function analysis of protein complexes involved in the molecular architecture of juxtaparanodal regions of myelinated fibers. Biotechnol J 2007, 2, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Ishiyama, N.; Hill, C.M.; Bates, I.R.; Libich, D.S.; Fares, C. Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron 2004, 35, 503–542. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.M.; Schardt, A.; Nave, K.A. Membrane traffic in myelinating oligodendrocytes. Microsc Res Tech 2001, 52, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Tanji, K.; Mori, F.; Kimura, A.; Kakita, A.; Wakabayashi, K. Immunoreactivity of myelin-associated oligodendrocytic basic protein in Lewy bodies. Neuropathology 2019, 39, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.H.; Halliday, G.M.; Kim, W.S. Exploring myelin dysfunction in multiple system atrophy. Exp Neurobiol 2014, 23, 337–344. [Google Scholar] [CrossRef]
- Smith, R. The basic protein of CNS myelin: Its structure and ligand binding. J Neurochem 1992, 59, 1589–1608. [Google Scholar] [CrossRef]
- Hill, C.M.; Bates, I.R.; White, G.F.; Hallett, F.R.; Harauz, G. Effects of the osmolyte trimethylamine-N-oxide on conformation, self-association, and two-dimensional crystallization of myelin basic protein. J Struct Biol 2002, 139, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.M.; Haines, J.D.; Antler, C.E.; Bates, I.R.; Libich, D.S.; Harauz, G. Terminal deletion mutants of myelin basic protein: New insights into self-association and phospholipid interactions. Micron 2003, 34, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Sedzik, J.; Kirschner, D.A. Is myelin basic protein crystallizable? Neurochem Res 1992, 17, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Saito, K.I.; Grynspan, F.; Griffin, W.R.; Katayama, S.; Honda, T.; Mohan, P.S.; Shea, T.B.; Beermann, M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease. Ann N Y Acad Sci 1994, 747, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, K.K. The calpain family and human disease. Trends Mol Med 2001, 7, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Hata, S.; Kawabata, Y.; Sorimachi, H. Structure, activation, and biology of calpain. Diabetes 2004, 53 (Suppl. 1), S12–S18. [Google Scholar] [CrossRef]
- Mellgren, R.L.; Rozanov, C.B. Calpain II-dependent solubilization of a nuclear protein kinase at micromolar calcium concentrations. Biochem Biophys Res Commun 1990, 168, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.K.; Dasgupta, S.; Banik, N.L.; Hogan, E.L. Regulation of the calcium-activated neutral proteinase (CANP) of bovine brain by myelin lipids. Biochim Biophys Acta 1990, 1038, 195–198. [Google Scholar] [CrossRef]
- Saido, T.C.; Shibata, M.; Takenawa, T.; Murofushi, H.; Suzuki, K. Positive regulation of mu-calpain action by polyphosphoinositides. J Biol Chem 1992, 267, 24585–24590. [Google Scholar] [CrossRef]
- Salamino, F.; De Tullio, R.; Mengotti, P.; Viotti, P.L.; Melloni, E.; Pontremoli, S. Site-directed activation of calpain is promoted by a membrane-associated natural activator protein. Biochem J 1993, 290 ( Pt 1) Pt 1, 191–197. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohno, S. Calcium activated neutral protease--structure-function relationship and functional implications. Cell Struct Funct 1990, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Murachi, T. Intracellular regulatory system involving calpain and calpastatin. Biochem Int 1989, 18, 263–294. [Google Scholar] [PubMed]
- Nixon, R.A. Calcium-activated neutral proteinases as regulators of cellular function. Implications for Alzheimer’s disease pathogenesis. Ann N Y Acad Sci 1989, 568, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Quackenbush, R.; Vitto, A. Multiple calcium-activated neutral proteinases (CANP) in mouse retinal ganglion cell neurons: Specificities for endogenous neuronal substrates and comparison to purified brain CANP. J Neurosci 1986, 6, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Ma, H.; Takano, E.; Yang, H.Q.; Hatanaka, M.; Maki, M. Molecular diversity in amino-terminal domains of human calpastatin by exon skipping. J Biol Chem 1992, 267, 8437–8442. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Ishida-Takahashi, A.; Takahashi, C.; Takano, E.; Murachi, T.; Hatanaka, M. Phosphorylation and subcellular distribution of calpastatin in human hematopoietic system cells. J Biol Chem 1991, 266, 3968–3972. [Google Scholar] [CrossRef]
- Nakamura, M.; Inomata, M.; Imajoh, S.; Suzuki, K.; Kawashima, S. Fragmentation of an endogenous inhibitor upon complex formation with high- and low-Ca2+-requiring forms of calcium-activated neutral proteases. Biochemistry 1989, 28, 449–455. [Google Scholar] [CrossRef]
- Emori, Y.; Kawasaki, H.; Imajoh, S.; Minami, Y.; Suzuki, K. All four repeating domains of the endogenous inhibitor for calcium-dependent protease independently retain inhibitory activity. Expression of the cDNA fragments in Escherichia coli. J Biol Chem 1988, 263, 2364–2370. [Google Scholar] [CrossRef]
- Wendt, A.; Thompson, V.F.; Goll, D.E. Interaction of calpastatin with calpain: A review. Biol Chem 2004, 385, 465–472. [Google Scholar] [CrossRef]
- Takano, E.; Ma, H.; Yang, H.Q.; Maki, M.; Hatanaka, M. Preference of calcium-dependent interactions between calmodulin-like domains of calpain and calpastatin subdomains. FEBS Lett 1995, 362, 93–97. [Google Scholar] [CrossRef]
- Tompa, P.; Mucsi, Z.; Orosz, G.; Friedrich, P. Calpastatin subdomains A and C are activators of calpain. J Biol Chem 2002, 277, 9022–9026. [Google Scholar] [CrossRef] [PubMed]
- Uemori, T.; Shimojo, T.; Asada, K.; Asano, T.; Kimizuka, F.; Kato, I.; Maki, M.; Hatanaka, M.; Murachi, T.; Hanzawa, H.; et al. Characterization of a functional domain of human calpastatin. Biochem Biophys Res Commun 1990, 166, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Tanaka, N.; Kataoka, M.; Takano, E.; Maki, M. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim Biophys Acta 1997, 1342, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Kovacs, D.; Tompa, P.; Perczel, A. Local structural preferences of calpastatin, the intrinsically unstructured protein inhibitor of calpain. Biochemistry 2008, 47, 6936–6945. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Brunden, K.R.; Huryn, D.M.; Trojanowski, J.Q.; Lee, V.M.; Smith, A.B., 3rd. Microtubule stabilizing agents as potential treatment for Alzheimer’s disease and related neurodegenerative tauopathies. J Med Chem 2012, 55, 8979–8996. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhang, B.; Lee, V.M.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol 2005, 109, 5–13. [Google Scholar] [CrossRef]
- Kuret, J.; Congdon, E.E.; Li, G.; Yin, H.; Yu, X.; Zhong, Q. Evaluating triggers and enhancers of tau fibrillization. Microsc Res Tech 2005, 67, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Kuret, J.; Chirita, C.N.; Congdon, E.E.; Kannanayakal, T.; Li, G.; Necula, M.; Yin, H.; Zhong, Q. Pathways of tau fibrillization. Biochim Biophys Acta 2005, 1739, 167–178. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu Rev Neurosci 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A 1997, 94, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Trojanowski, J.Q. Neurodegenerative tauopathies: Human disease and transgenic mouse models. Neuron 1999, 24, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Hodges, J.R.; Snowden, J.S.; Mackenzie, I.R.; Neumann, M.; Mann, D.M.; Dickson, D.W. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011, 122, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res Bull 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu Rev Pathol 2019, 14, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Kneynsberg, A.; Combs, B.; Christensen, K.; Morfini, G.; Kanaan, N.M. Axonal Degeneration in Tauopathies: Disease Relevance and Underlying Mechanisms. Front Neurosci 2017, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Uemura, M.T.; Luk, K.C.; Lee, V.M.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and alpha-Synuclein. Trends Mol Med 2020, 26, 936–952. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol 2019, 76, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Drewes, G.; Trinczek, B.; Illenberger, S.; Biernat, J.; Schmitt-Ulms, G.; Meyer, H.E.; Mandelkow, E.M.; Mandelkow, E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem 1995, 270, 7679–7688. [Google Scholar] [CrossRef]
- Clark, L.N.; Poorkaj, P.; Wszolek, Z.; Geschwind, D.H.; Nasreddine, Z.S.; Miller, B.; Li, D.; Payami, H.; Awert, F.; Markopoulou, K.; et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A 1998, 95, 13103–13107. [Google Scholar] [CrossRef]
- Narayanan, R.L.; Durr, U.H.; Bibow, S.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Automatic assignment of the intrinsically disordered protein Tau with 441-residues. J Am Chem Soc 2010, 132, 11906–11907. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Reddy, J.G.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Residue-specific identification of phase separation hot spots of Alzheimer’s-related protein tau. Chem Sci 2019, 10, 6503–6507. [Google Scholar] [CrossRef] [PubMed]
- Ukmar-Godec, T.; Wegmann, S.; Zweckstetter, M. Biomolecular condensation of the microtubule-associated protein tau. Semin Cell Dev Biol 2020, 99, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Savastano, A.; Singh, P.; Mukhopadhyay, S.; Zweckstetter, M. Liquid-liquid phase separation of tau: From molecular biophysics to physiology and disease. Protein Sci 2021, 30, 1294–1314. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Zweckstetter, M. Phase separation of the microtubule-associated protein tau. Essays Biochem 2022, 66, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Li, C.; Meng, L.; Tian, Y.; He, M.; Yuan, X.; Zhang, G.; Zhang, Z.; Xiong, J.; Chen, G.; et al. Tau accelerates alpha-synuclein aggregation and spreading in Parkinson’s disease. Brain 2022, 145, 3454–3471. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener 2019, 14, 23. [Google Scholar] [CrossRef]
- Jin, M.; Wang, S.; Gao, X.; Zou, Z.; Hirotsune, S.; Sun, L. Pathological and physiological functional cross-talks of alpha-synuclein and tau in the central nervous system. Neural Regen Res 2024, 19, 855–862. [Google Scholar] [CrossRef]
- Kayed, R.; Dettmer, U.; Lesne, S.E. Soluble endogenous oligomeric alpha-synuclein species in neurodegenerative diseases: Expression, spreading, and cross-talk. J Parkinsons Dis 2020, 10, 791–818. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, S.; Ma, X.; Jia, C.; Liu, Z.; Huang, C.; Liu, C.; Li, D. Structural basis of the interplay between alpha-synuclein and Tau in regulating pathological amyloid aggregation. J Biol Chem 2020, 295, 7470–7480. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Sorrentino, Z.; Weinrich, M.; Giasson, B.I.; Chakrabarty, P. Differential cross-seeding properties of tau and alpha-synuclein in mouse models of tauopathy and synucleinopathy. Brain Commun 2020, 2, fcaa090. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fu, Y.; Miyata, T.; Matsumoto, S.; Shinoda, T.; Itoh, K.; Harada, A.; Hirotsune, S.; Jin, M. Functional Cooperation of alpha-Synuclein and Tau Is Essential for Proper Corticogenesis. J Neurosci 2022, 42, 7031–7046. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xu, W.X.; Wang, S.Y.; Zhan, Y.Q.; Jiang, Y.; Cai, W.M.; Yang, X.M. Isolation and Characterization of EDAG-1, A Novel Gene Related to Regulation in Hematopoietic System. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2001, 33, 641–646. [Google Scholar]
- Yang, L.V.; Nicholson, R.H.; Kaplan, J.; Galy, A.; Li, L. Hemogen is a novel nuclear factor specifically expressed in mouse hematopoietic development and its human homologue EDAG maps to chromosome 9q22, a region containing breakpoints of hematological neoplasms. Mech Dev 2001, 104, 105–111. [Google Scholar] [CrossRef]
- Liu, C.C.; Chou, Y.L.; Ch’ang, L.Y. Down-regulation of human NDR gene in megakaryocytic differentiation of erythroleukemia K562 cells. J Biomed Sci 2004, 11, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Zhan, Y.Q.; Xu, C.W.; Xu, W.X.; Wang, S.Y.; Lv, J.; Zhou, Y.; Yue, P.B.; Chen, B.; Yang, X.M. EDAG regulates the proliferation and differentiation of hematopoietic cells and resists cell apoptosis through the activation of nuclear factor-kappa B. Cell Death Differ 2004, 11, 1299–1308. [Google Scholar] [CrossRef]
- An, L.L.; Li, G.; Wu, K.F.; Ma, X.T.; Zheng, G.G.; Qiu, L.G.; Song, Y.H. High expression of EDAG and its significance in AML. Leukemia 2005, 19, 1499–1502. [Google Scholar] [CrossRef]
- Yang, L.V.; Heng, H.H.; Wan, J.; Southwood, C.M.; Gow, A.; Li, L. Alternative promoters and polyadenylation regulate tissue-specific expression of Hemogen isoforms during hematopoiesis and spermatogenesis. Dev Dyn 2003, 228, 606–616. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, W.X.; Zhan, Y.Q.; Li, C.Y.; Xu, C.W.; Zheng, H.; Li, F.F.; Yang, X.M.; Wang, S.Y. [Expression of EDAG-1 gene in human leukemia and lymphoma cell lines]. Ai Zheng 2004, 23, 1238–1243. [Google Scholar]
- Chen, D.L.; Hu, Z.Q.; Zheng, X.F.; Wang, X.Y.; Xu, Y.Z.; Li, W.Q.; Fang, H.S.; Kan, L.; Wang, S.Y. EDAG-1 promotes proliferation and invasion of human thyroid cancer cells by activating MAPK/Erk and AKT signal pathways. Cancer Biol Ther 2016, 17, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Millan-Arino, L.; Izquierdo-Bouldstridge, A.; Jordan, A. Specificities and genomic distribution of somatic mammalian histone H1 subtypes. Biochim Biophys Acta 2016, 1859, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 2018, 19, 192–206. [Google Scholar] [CrossRef]
- Salinas-Pena, M.; Rebollo, E.; Jordan, A. Imaging analysis of six human histone H1 variants reveals universal enrichment of H1.2, H1.3, and H1.5 at the nuclear periphery and nucleolar H1X presence. Elife 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Lailler, N.; Zhang, Y.; Kumar, A.; Uppal, K.; Liu, Z.; Lee, E.K.; Wu, H.; Medrzycki, M.; Pan, C.; et al. High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet 2013, 9, e1003417. [Google Scholar] [CrossRef]
- Izzo, A.; Kamieniarz-Gdula, K.; Ramirez, F.; Noureen, N.; Kind, J.; Manke, T.; van Steensel, B.; Schneider, R. The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep 2013, 3, 2142–2154. [Google Scholar] [CrossRef]
- Millan-Arino, L.; Islam, A.B.; Izquierdo-Bouldstridge, A.; Mayor, R.; Terme, J.M.; Luque, N.; Sancho, M.; Lopez-Bigas, N.; Jordan, A. Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res 2014, 42, 4474–4493. [Google Scholar] [CrossRef]
- Serna-Pujol, N.; Salinas-Pena, M.; Mugianesi, F.; Le Dily, F.; Marti-Renom, M.A.; Jordan, A. Coordinated changes in gene expression, H1 variant distribution and genome 3D conformation in response to H1 depletion. Nucleic Acids Res 2022, 50, 3892–3910. [Google Scholar] [CrossRef]
- Lai, S.; Jia, J.; Cao, X.; Zhou, P.K.; Gao, S. Molecular and Cellular Functions of the Linker Histone H1.2. Front Cell Dev Biol 2021, 9, 773195. [Google Scholar] [CrossRef]
- Roque, A.; Sortino, R.; Ventura, S.; Ponte, I.; Suau, P. Histone H1 Favors Folding and Parallel Fibrillar Aggregation of the 1-42 Amyloid-beta Peptide. Langmuir 2015, 31, 6782–6790. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Mizianty, M.J.; Xue, B.; Kurgan, L.; Uversky, V.N. More than just tails: Intrinsic disorder in histone proteins. Mol Biosyst 2012, 8, 1886–1901. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Kruger, S.; Mann, M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics 2007, 6, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, X.; Taghizadeh, K.; Dong, M.; Dedon, P.C. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc Natl Acad Sci U S A 2007, 104, 60–65. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Mann, M. Nepsilon-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res 2008, 36, 570–577. [Google Scholar] [CrossRef]
- Weiss, T.; Hergeth, S.; Zeissler, U.; Izzo, A.; Tropberger, P.; Zee, B.M.; Dundr, M.; Garcia, B.A.; Daujat, S.; Schneider, R. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 2010, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Schneider, R. The role of linker histone H1 modifications in the regulation of gene expression and chromatin dynamics. Biochim Biophys Acta 2016, 1859, 486–495. [Google Scholar] [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- UniProt_Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci 2022, 31, e4496. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Hatos, A.; Tosatto, S.C.E.; Vendruscolo, M.; Fuxreiter, M. FuzDrop on AlphaFold: Visualizing the sequence-dependent propensity of liquid-liquid phase separation and aggregation of proteins. Nucleic Acids Res 2022, 50, W337–W344. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Functional disorder analysis of human α-synuclein (UniProt ID: P37840). A, Multiparametric disorder analysis of the protein using RIDAO. The outputs of PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, IUPred long, and IUPred short are shown by black, red, green, pink, blue, and yellow lines, respectively. Mean disorder profile (or mean disorder prediction, MDP) calculated as an average of outputs of these six predictors is shown by dashed dark pink line, whereas error distribution are shown as light pink shadow. In this per-residue disorder analysis, a disorder score was assigned to each residue. A residue with disorder score equal to or above 0.5 is considered as disordered and a residue with disorder score below 0.5 is predicted as ordered. Residues/regions with disorder scores between 0.15 and 0.5 were considered as ordered but flexible. The corresponding thresholds are shown by solid (0.5) and long-dashed lines (0.15). B. Functional disorder profile generated for α-synuclein by the D2P2 database showing the outputs of several disorder predictors such as VLXT, VSL2b, PrDOS, IUPred and Espritz. The colored bar highlighted by blue and green shade represents the disorder prediction, yellow zigzagged bars shows positions of MoRFs, whereas colored circles in the bottom of the plot show the positions of predicted PTMs, such as phosphorylation (red circles marked P), sumoylation (green circles marked S), acetylation (yellow circles marked A), glycosylation (orange circles marked G), and ubiquitylation (violet circles marked U). C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP, for human α-synuclein. D. The FuzDrop-generated plot of the multiplicity of binding modes showing positions of regions that can sample multiple binding modes in the cellular context (sub-cellular localization, partners, posttranslational modifications)-dependent manner. E. Protein-protein interaction network generated by STRING. This PPI network was generated using the minimum required interaction score of 0.5 and adjusting the value of a maximum number of interactors in the first shell to 500. Network nodes represent individual proteins and edges represent protein-protein interaction for shared function, with types of Interactions; the blue line represents curated databases, black line for co-expression, and green line for gene neighborhood. F. 3D structural model is predicted through AlphaFold. The structure is colored according to the per-residue model confidence score (pLDDT) ranging from orange to blue, where fragments of structure with very high (pLDDT > 90), confident (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50) pLDDT scores are shown by blue, cyan, yellow and orange colors, respectively.
Figure 1.
Functional disorder analysis of human α-synuclein (UniProt ID: P37840). A, Multiparametric disorder analysis of the protein using RIDAO. The outputs of PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, IUPred long, and IUPred short are shown by black, red, green, pink, blue, and yellow lines, respectively. Mean disorder profile (or mean disorder prediction, MDP) calculated as an average of outputs of these six predictors is shown by dashed dark pink line, whereas error distribution are shown as light pink shadow. In this per-residue disorder analysis, a disorder score was assigned to each residue. A residue with disorder score equal to or above 0.5 is considered as disordered and a residue with disorder score below 0.5 is predicted as ordered. Residues/regions with disorder scores between 0.15 and 0.5 were considered as ordered but flexible. The corresponding thresholds are shown by solid (0.5) and long-dashed lines (0.15). B. Functional disorder profile generated for α-synuclein by the D2P2 database showing the outputs of several disorder predictors such as VLXT, VSL2b, PrDOS, IUPred and Espritz. The colored bar highlighted by blue and green shade represents the disorder prediction, yellow zigzagged bars shows positions of MoRFs, whereas colored circles in the bottom of the plot show the positions of predicted PTMs, such as phosphorylation (red circles marked P), sumoylation (green circles marked S), acetylation (yellow circles marked A), glycosylation (orange circles marked G), and ubiquitylation (violet circles marked U). C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP, for human α-synuclein. D. The FuzDrop-generated plot of the multiplicity of binding modes showing positions of regions that can sample multiple binding modes in the cellular context (sub-cellular localization, partners, posttranslational modifications)-dependent manner. E. Protein-protein interaction network generated by STRING. This PPI network was generated using the minimum required interaction score of 0.5 and adjusting the value of a maximum number of interactors in the first shell to 500. Network nodes represent individual proteins and edges represent protein-protein interaction for shared function, with types of Interactions; the blue line represents curated databases, black line for co-expression, and green line for gene neighborhood. F. 3D structural model is predicted through AlphaFold. The structure is colored according to the per-residue model confidence score (pLDDT) ranging from orange to blue, where fragments of structure with very high (pLDDT > 90), confident (90 > pLDDT > 70), low (70 > pLDDT > 50), and very low (pLDDT < 50) pLDDT scores are shown by blue, cyan, yellow and orange colors, respectively.
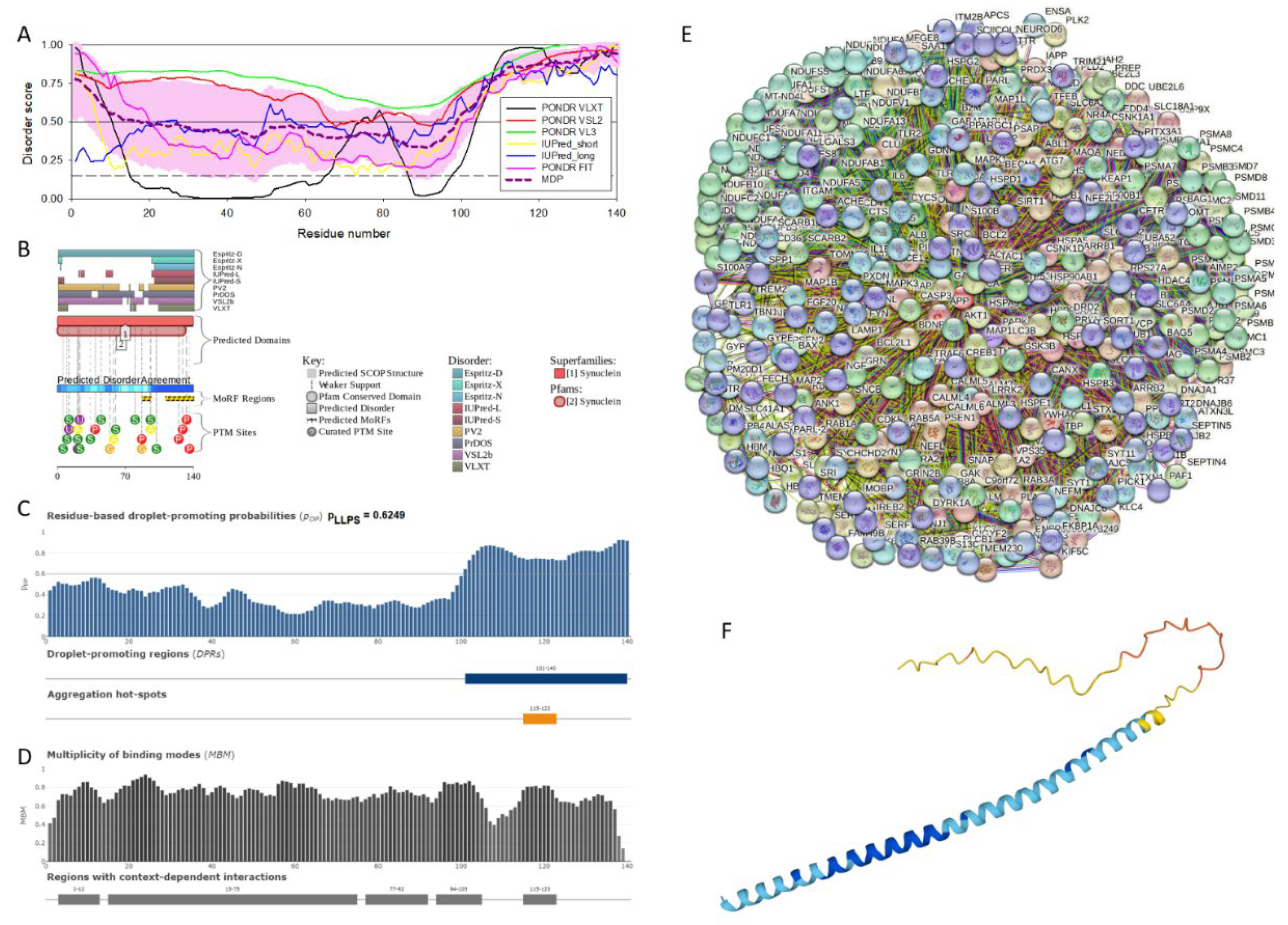
Figure 2.
Functional disorder analysis of human β-synuclein (UniProt ID: Q16143). A, Multiparametric disorder analysis of the protein using RIDAO. B. Functional disorder profile generated for human β-synuclein by the D2P2 database. Colored circles in the bottom of the plot show the localization of PTMs, such as phosphorylation (red circles marked P) and acetylation (yellow circles marked A). C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. D. The FuzDrop-generated plot of the multiplicity of binding modes. E. Protein-protein interaction network generated by STRING. This PPI network was generated using the minimum required interaction score of 0.4 (medium confidence) and adjusting the value of a maximum number of interactors to 500. F. 3D structural model is predicted through AlphaFold.
Figure 2.
Functional disorder analysis of human β-synuclein (UniProt ID: Q16143). A, Multiparametric disorder analysis of the protein using RIDAO. B. Functional disorder profile generated for human β-synuclein by the D2P2 database. Colored circles in the bottom of the plot show the localization of PTMs, such as phosphorylation (red circles marked P) and acetylation (yellow circles marked A). C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. D. The FuzDrop-generated plot of the multiplicity of binding modes. E. Protein-protein interaction network generated by STRING. This PPI network was generated using the minimum required interaction score of 0.4 (medium confidence) and adjusting the value of a maximum number of interactors to 500. F. 3D structural model is predicted through AlphaFold.
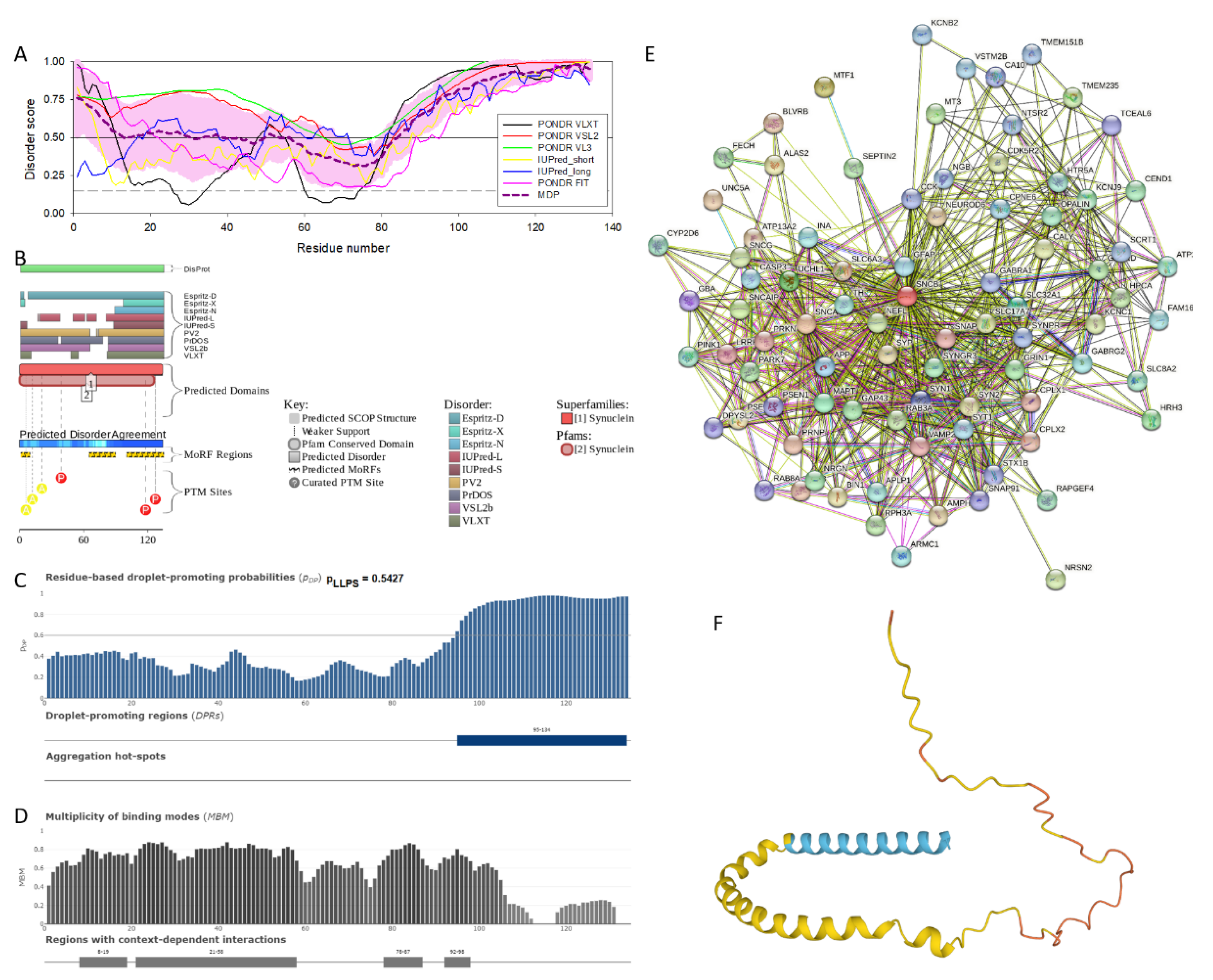
Figure 3.
Functional disorder analysis of human γ-synuclein (UniProt ID: O76070). A, Multiparametric disorder analysis of the protein using RIDAO. B. Functional disorder profile generated for human γ-synuclein by the D2P2 database. Colored circles in the bottom of the plot show the localization of PTMs, such as phosphorylation (red circles marked P) and acetylation (yellow circles marked A). C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. D. The FuzDrop-generated plot of the multiplicity of binding modes. E. Protein-protein interaction network generated by STRING. This PPI network was generated using the minimum required interaction score of 0.4 (medium confidence) and adjusting the value of a maximum number of interactors to 500. F. 3D structural model is predicted through AlphaFold.
Figure 3.
Functional disorder analysis of human γ-synuclein (UniProt ID: O76070). A, Multiparametric disorder analysis of the protein using RIDAO. B. Functional disorder profile generated for human γ-synuclein by the D2P2 database. Colored circles in the bottom of the plot show the localization of PTMs, such as phosphorylation (red circles marked P) and acetylation (yellow circles marked A). C. The FuzDrop-generated plot showing the sequence distribution of the residue-based droplet-promoting probabilities, pDP. D. The FuzDrop-generated plot of the multiplicity of binding modes. E. Protein-protein interaction network generated by STRING. This PPI network was generated using the minimum required interaction score of 0.4 (medium confidence) and adjusting the value of a maximum number of interactors to 500. F. 3D structural model is predicted through AlphaFold.
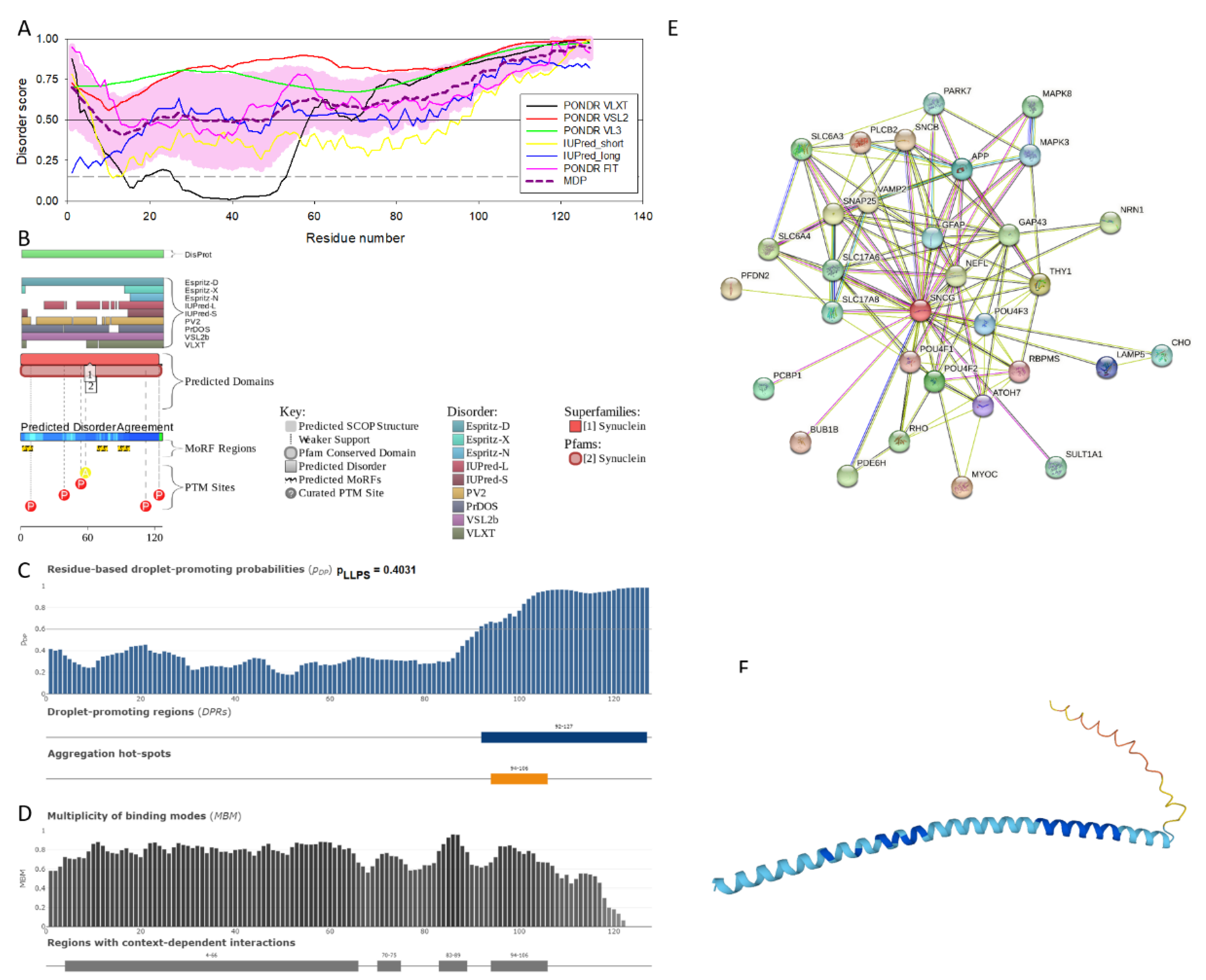
Figure 4.
Effect of the missense point mutations associated with the familial cases of PD (A30P, E46K, H50Q, G51D, A53T, and A53E) on the intrinsic disorder propensity of human α-synuclein. A. Per-residue disorder profiles generated by PONDR® VSL2. B. Difference disorder spectra calculated by subtracting mutant profiles from that of the wild type protein.
Figure 4.
Effect of the missense point mutations associated with the familial cases of PD (A30P, E46K, H50Q, G51D, A53T, and A53E) on the intrinsic disorder propensity of human α-synuclein. A. Per-residue disorder profiles generated by PONDR® VSL2. B. Difference disorder spectra calculated by subtracting mutant profiles from that of the wild type protein.
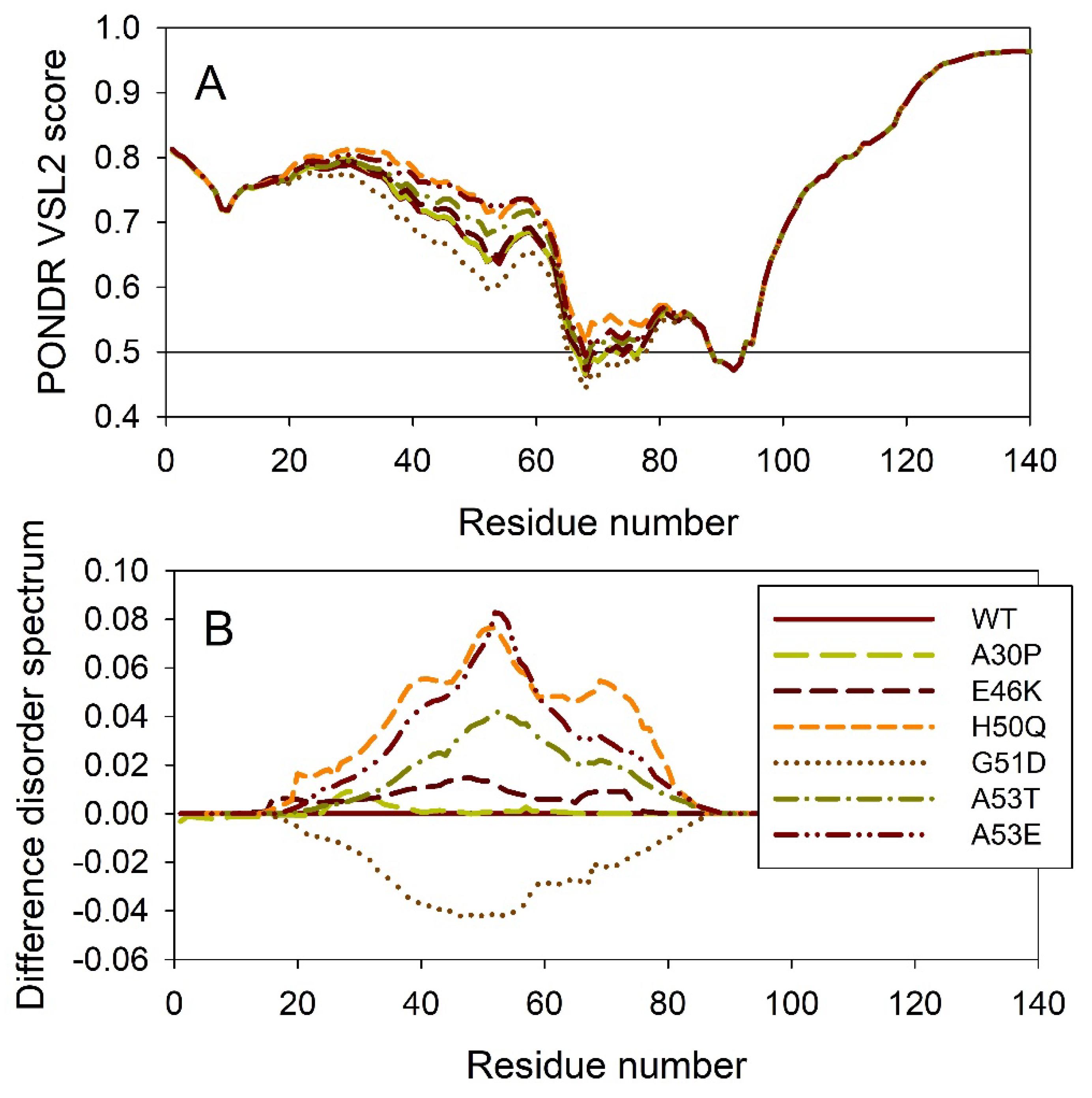
Figure 5.
Effect of the missense point mutations associated with the familial cases of PD (A30P, E46K, H50Q, G51D, A53T, and A53E) on intrinsic disorder propensity of human α-synuclein. A. Per-residue droplet-promoting probabilities (pDP) evaluated by FuzDrop. B. Difference pDP spectra calculated by subtracting mutant profiles from that of the wild type protein.
Figure 5.
Effect of the missense point mutations associated with the familial cases of PD (A30P, E46K, H50Q, G51D, A53T, and A53E) on intrinsic disorder propensity of human α-synuclein. A. Per-residue droplet-promoting probabilities (pDP) evaluated by FuzDrop. B. Difference pDP spectra calculated by subtracting mutant profiles from that of the wild type protein.
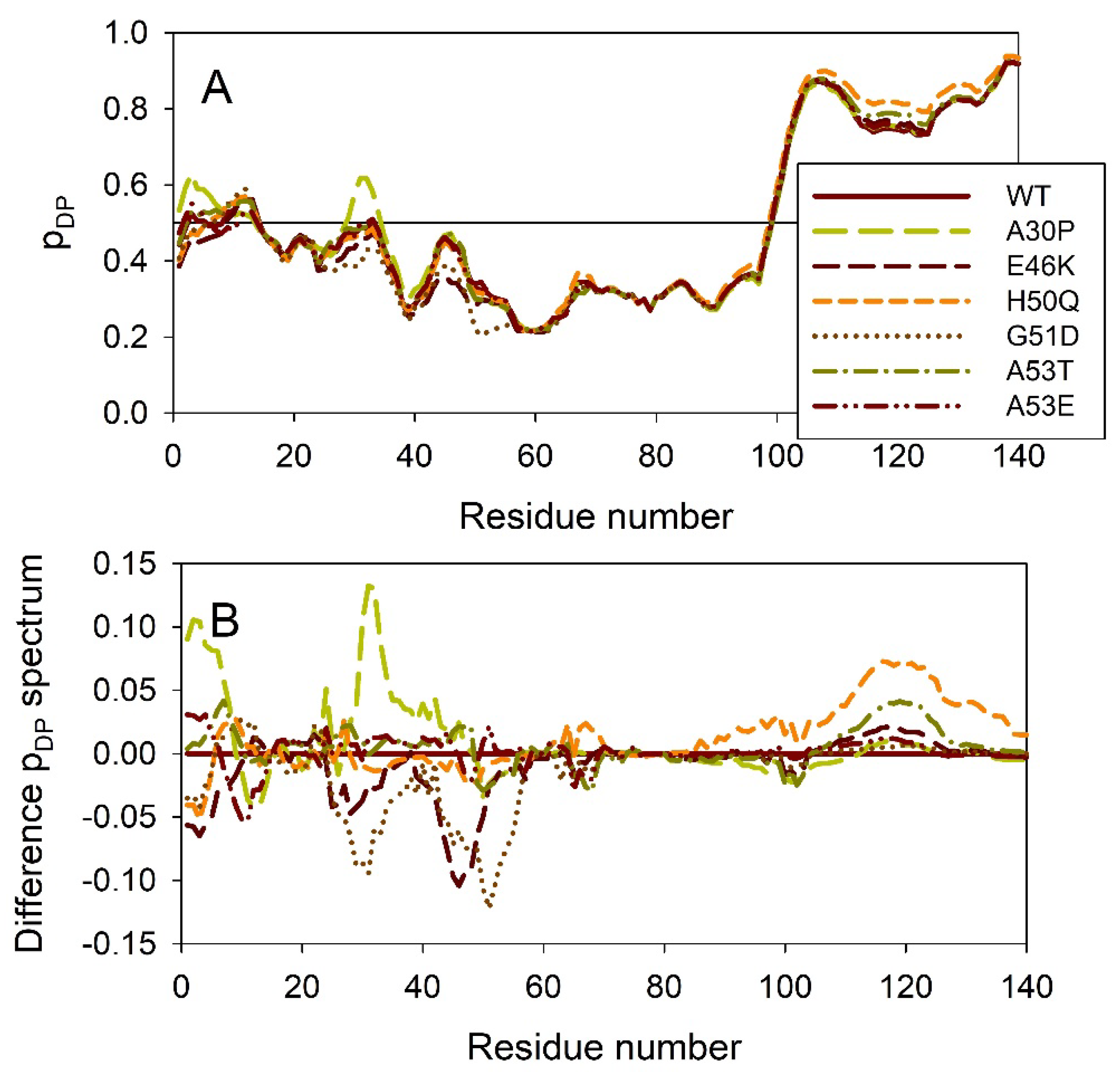
Figure 6.
Species selected for the analysis of conservation of intrinsic disorder in the members of synuclein family.
Figure 6.
Species selected for the analysis of conservation of intrinsic disorder in the members of synuclein family.

Figure 7.
Multiple sequence alignements of α- (A), β- (B), and γ-synucleins (C) conducted by Clustal Omega using default parameters.
Figure 7.
Multiple sequence alignements of α- (A), β- (B), and γ-synucleins (C) conducted by Clustal Omega using default parameters.
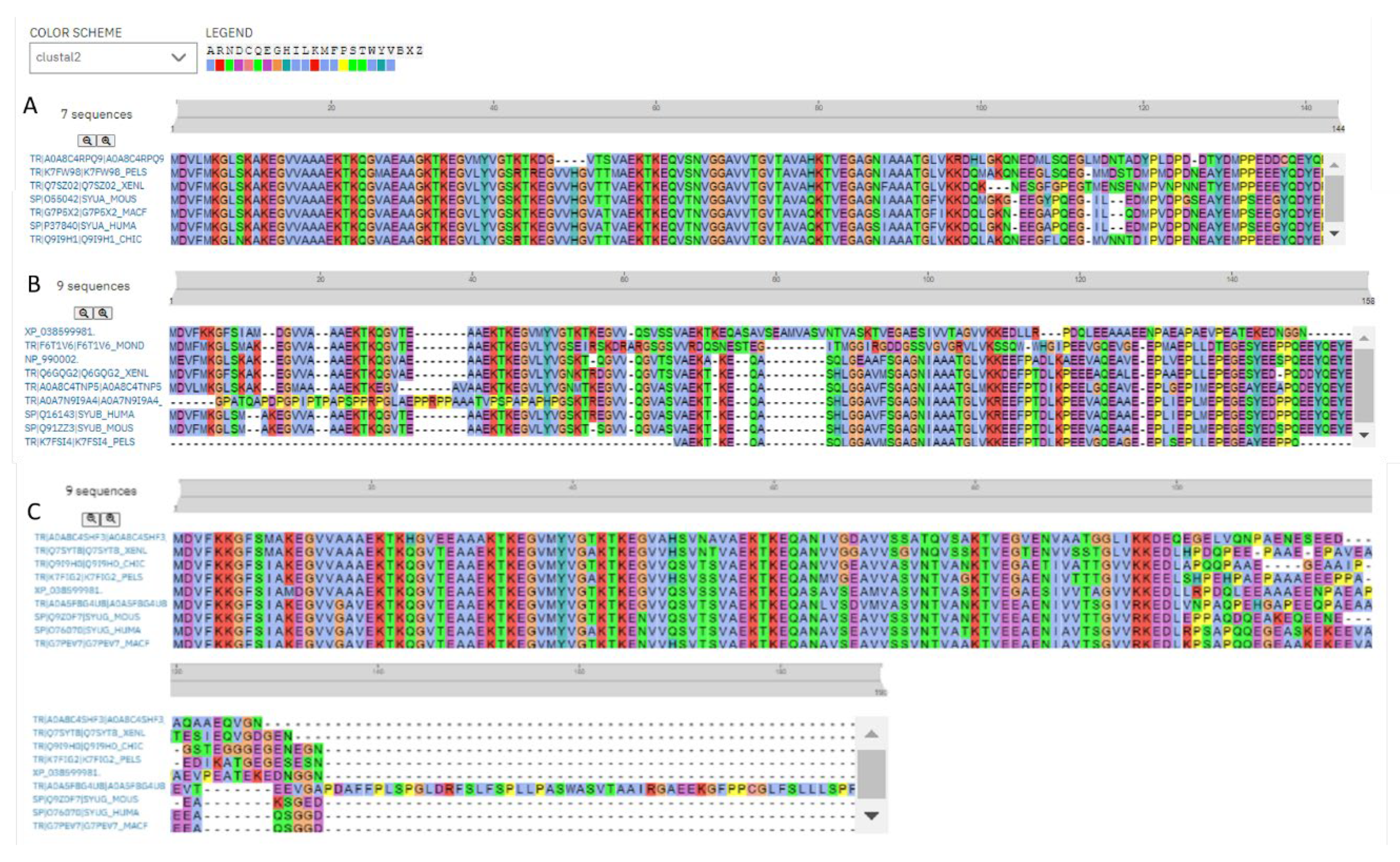
Figure 8.
Conservation of the peculiarities of the per-residue intrinsic disorder propensity among the members of synuclein family. Disorder profiles were generated for α- (A), β- (B), and γ-synucleins (C) by PONDR® VSL2.
Figure 8.
Conservation of the peculiarities of the per-residue intrinsic disorder propensity among the members of synuclein family. Disorder profiles were generated for α- (A), β- (B), and γ-synucleins (C) by PONDR® VSL2.
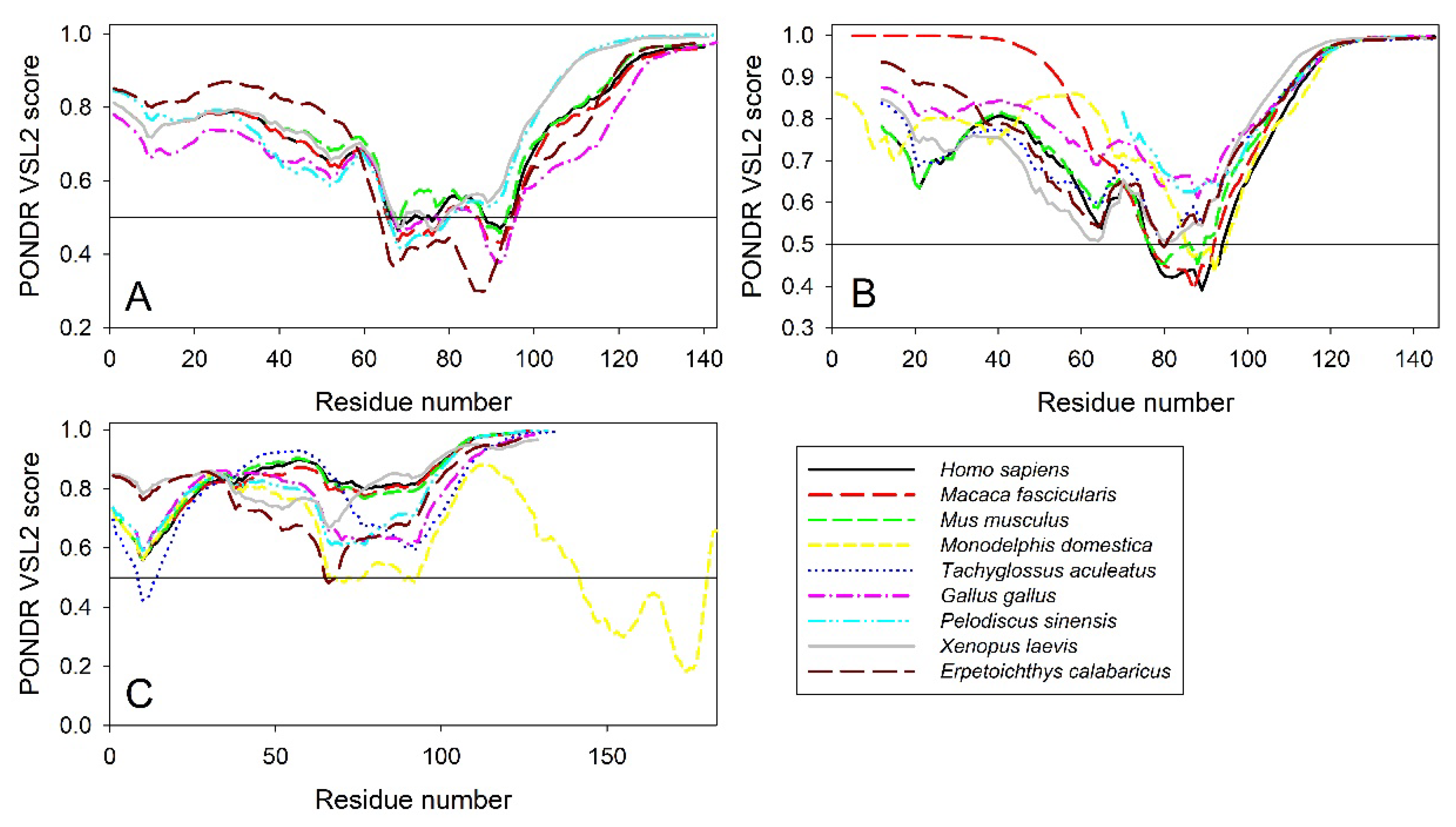
Figure 9.
STRING-generated PPI network centered at human α-, β-, and γ-synucleins.

Figure 10.
Evaluation of the global disorder status of 10,611 proteins from the human brain proteome (gray circles), as well as in interactomes of individual human synucleins and the global interactome centered at the three synucleins, with corresponding data shown by differently colored circles. A. PONDR® VSL2 score vs. PONDR® VSL2 (%) plot. Here, each point corresponds to a query protein coordinates of which are evaluated from the corresponding PONDR® VSL2 data as its average disorder score (ADS) and percent of the predicted intrinsically disordered residues (PPIDR). Color blocks are used to visualize proteins based on the accepted classification, with red, pink/light pink, and blue/light blue regions containing highly disordered, moderately disordered, and ordered pro-teins, respectively (see the text). Dark blue or pink regions correspond to the regions, where PPIDR agrees with ADS, whereas areas in which only one of these criteria applies are shown by light blue or light pink. B. CH-CDF plot, where the coordinates for a query protein are calculated as the average distance of its CDF curve from the CDF boundary (X axis) and its distance from the CH boundary. Protein classification is based on the quadrant, where it is located: Q1, protein predicted to be ordered by both predictors. Q2, protein predicted to be ordered to by CH-plot and disordered by CDF. Q3, protein predicted to be disordered by both predictors. Q4, protein predicted to be disordered by CH-plot and ordered by CDF.
Figure 10.
Evaluation of the global disorder status of 10,611 proteins from the human brain proteome (gray circles), as well as in interactomes of individual human synucleins and the global interactome centered at the three synucleins, with corresponding data shown by differently colored circles. A. PONDR® VSL2 score vs. PONDR® VSL2 (%) plot. Here, each point corresponds to a query protein coordinates of which are evaluated from the corresponding PONDR® VSL2 data as its average disorder score (ADS) and percent of the predicted intrinsically disordered residues (PPIDR). Color blocks are used to visualize proteins based on the accepted classification, with red, pink/light pink, and blue/light blue regions containing highly disordered, moderately disordered, and ordered pro-teins, respectively (see the text). Dark blue or pink regions correspond to the regions, where PPIDR agrees with ADS, whereas areas in which only one of these criteria applies are shown by light blue or light pink. B. CH-CDF plot, where the coordinates for a query protein are calculated as the average distance of its CDF curve from the CDF boundary (X axis) and its distance from the CH boundary. Protein classification is based on the quadrant, where it is located: Q1, protein predicted to be ordered by both predictors. Q2, protein predicted to be ordered to by CH-plot and disordered by CDF. Q3, protein predicted to be disordered by both predictors. Q4, protein predicted to be disordered by CH-plot and ordered by CDF.
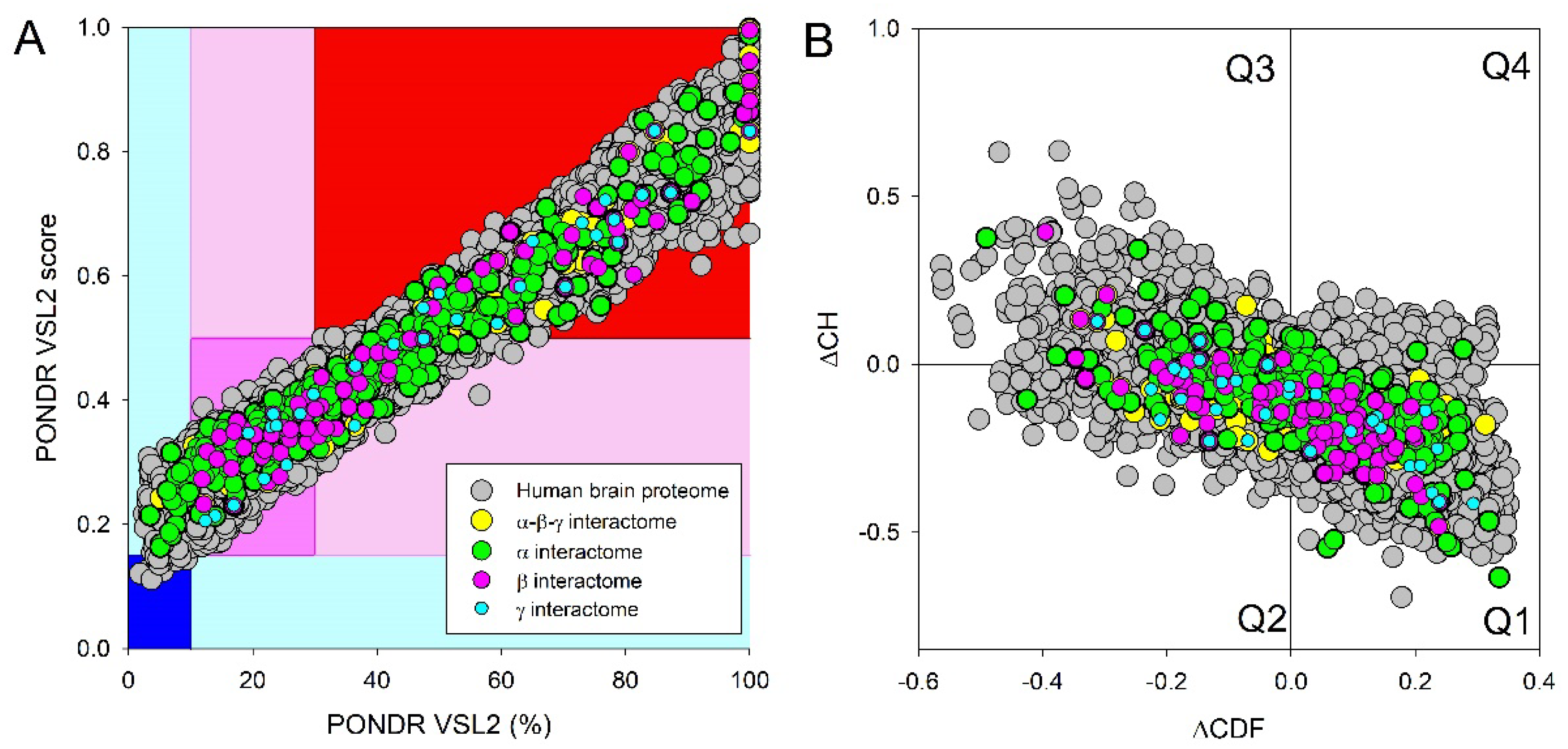
Figure 11.
Disorder analysis of proteins interacting with human synucleins. A. Global disorder analysis of proteins in human brain proteome, and four synuclein interactomes. Global disorder status of these five protein sets was evaluated by computing their mean PPIDs and average disorder scores generated by PONDR® VSL2. B. Correlation between the number of interactions and intrinsic disorder level of human proteins in the joint α-β-γ synuclein interactome. A vertical solid line represents the average node degree of this network (which is 45.8). Two verical solid lines represent two disorder boundaries of 15% and 30%.
Figure 11.
Disorder analysis of proteins interacting with human synucleins. A. Global disorder analysis of proteins in human brain proteome, and four synuclein interactomes. Global disorder status of these five protein sets was evaluated by computing their mean PPIDs and average disorder scores generated by PONDR® VSL2. B. Correlation between the number of interactions and intrinsic disorder level of human proteins in the joint α-β-γ synuclein interactome. A vertical solid line represents the average node degree of this network (which is 45.8). Two verical solid lines represent two disorder boundaries of 15% and 30%.
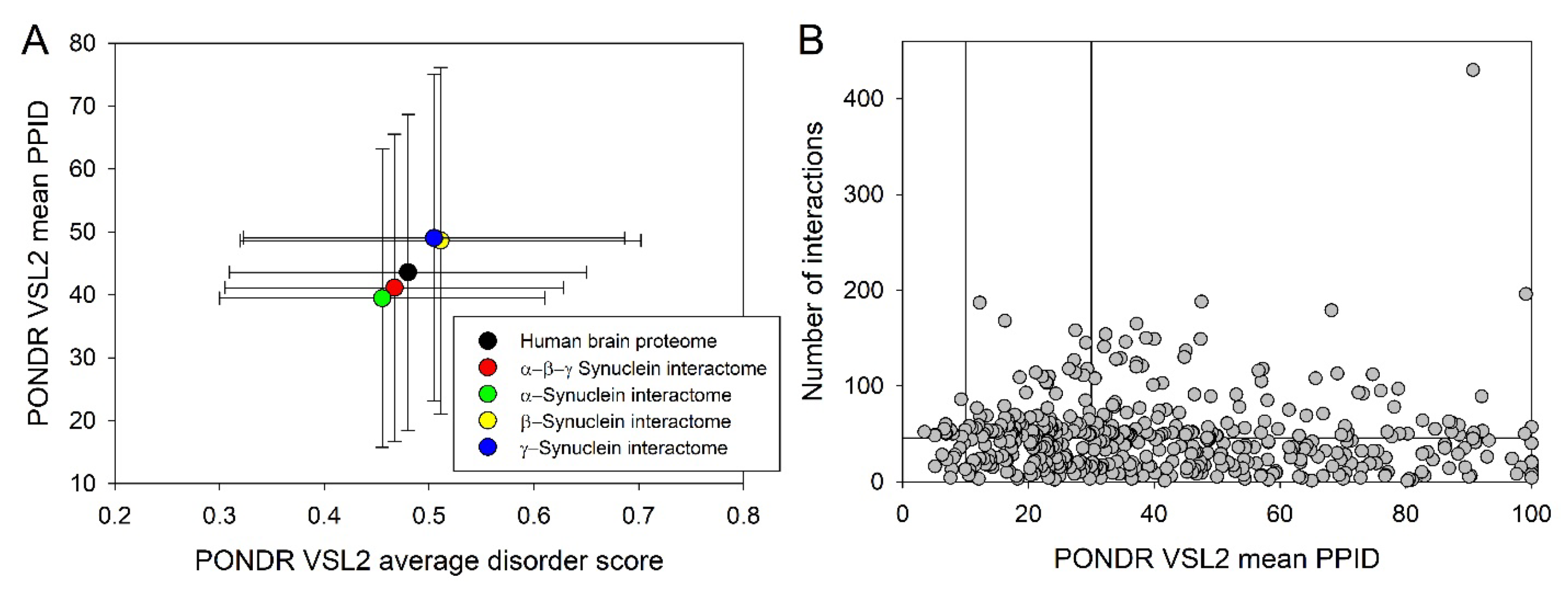
Figure 12.
A correlation between the overall disorder status (PONDR VSL2, %), interactability (node degree), and LLPS predisposition (pLLPS) of 467 human proteins in the joint α-β-γ synuclein interactome.
Figure 12.
A correlation between the overall disorder status (PONDR VSL2, %), interactability (node degree), and LLPS predisposition (pLLPS) of 467 human proteins in the joint α-β-γ synuclein interactome.
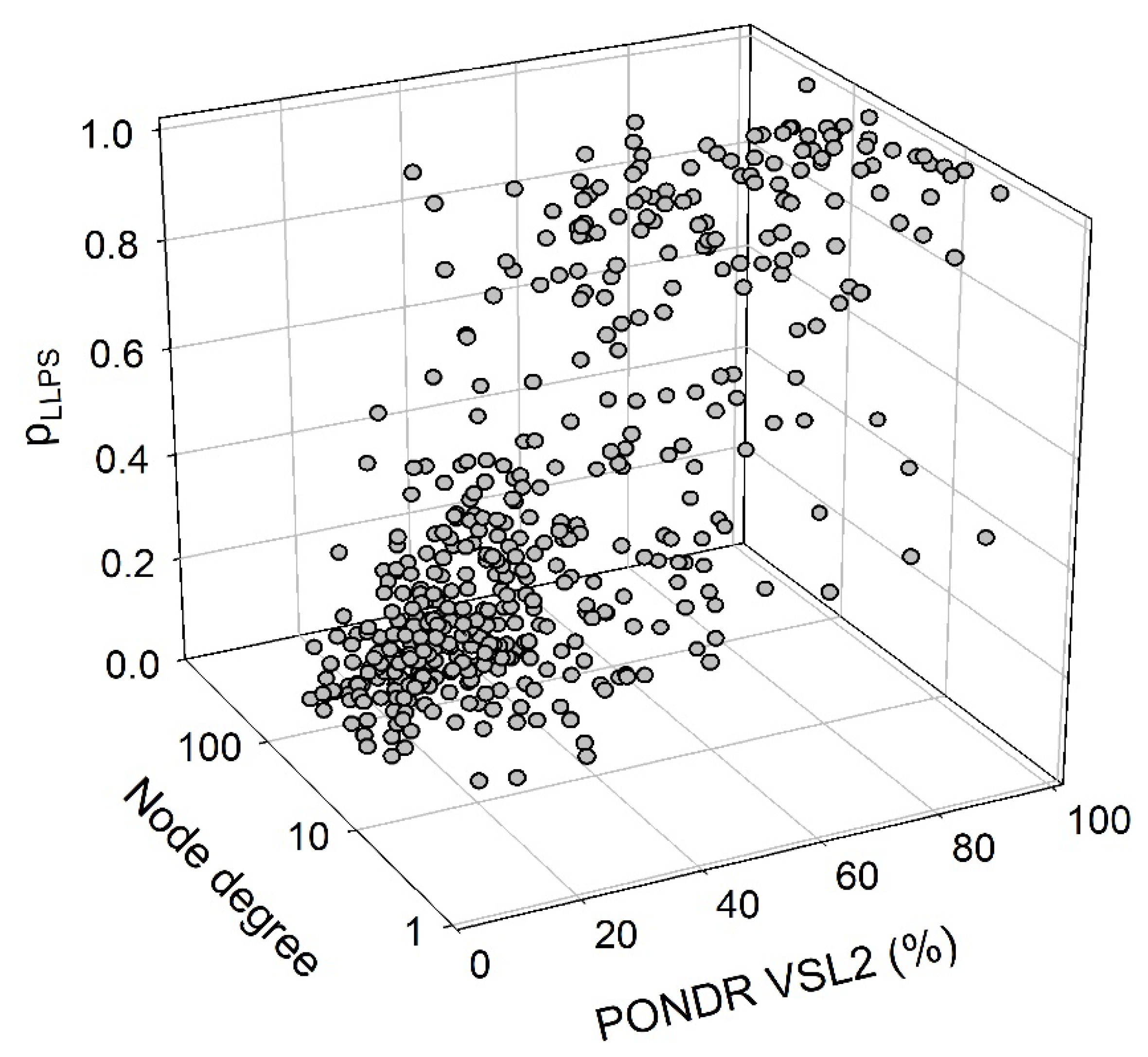
Figure 13.
Functional disorder analysis of human Metallothionein-3 (UniProt ID: P25713). A. Functional disorder profile generated by D2P2. B. Redox sensitive disorder profile generated by IUPred2A. C. 3D structural model generated by AlphaFold. S. CHMP2B-centered PPI network generated by STRING. This network was generated using medium confidence of 0.185.
Figure 13.
Functional disorder analysis of human Metallothionein-3 (UniProt ID: P25713). A. Functional disorder profile generated by D2P2. B. Redox sensitive disorder profile generated by IUPred2A. C. 3D structural model generated by AlphaFold. S. CHMP2B-centered PPI network generated by STRING. This network was generated using medium confidence of 0.185.
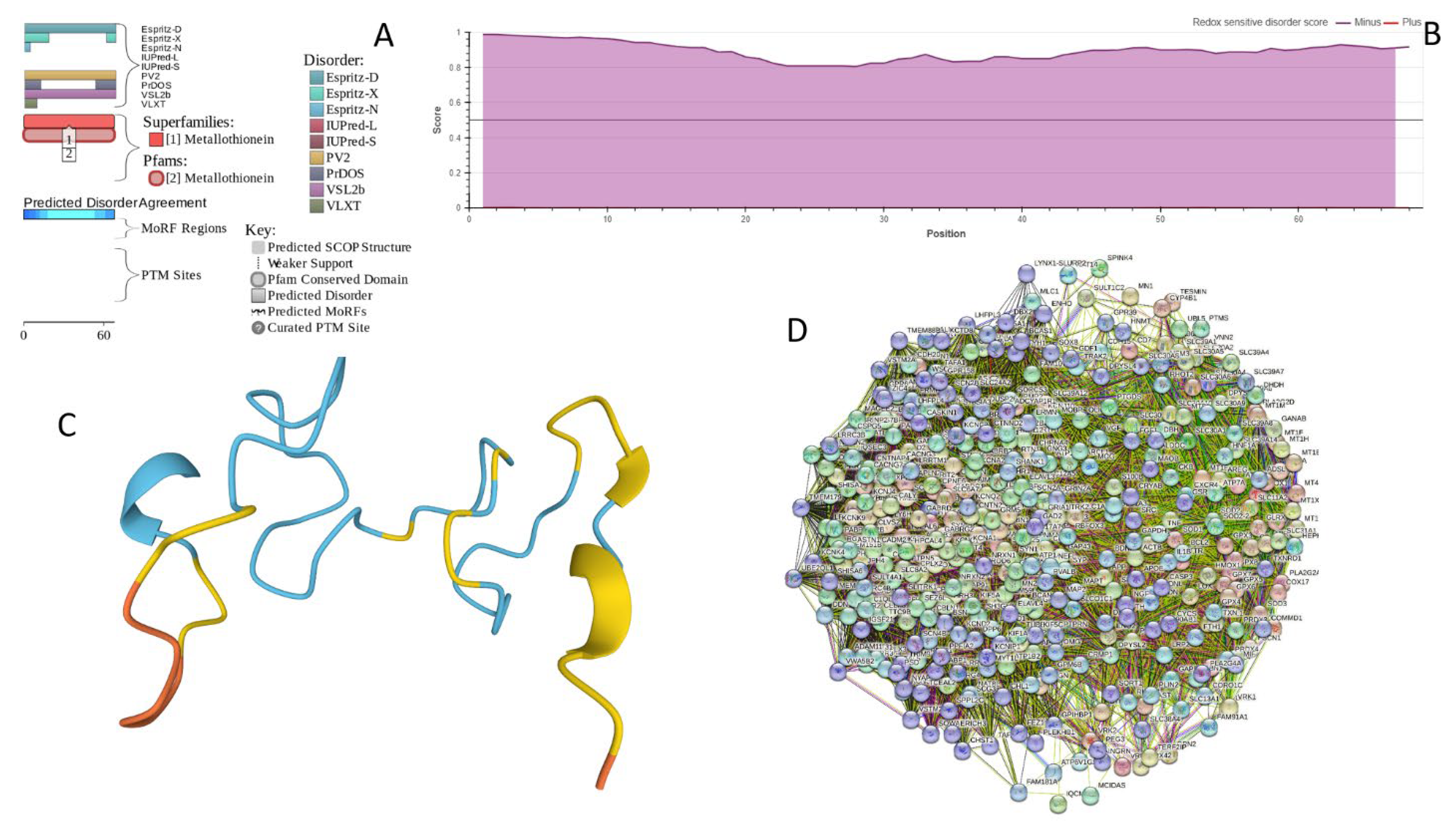
Figure 14.
Functional disorder analysis of human CHMP2B (Q9UQN3). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CHMP2B-centered PPI network generated by STRING. This network was generated using medium confidence of 0.4.
Figure 14.
Functional disorder analysis of human CHMP2B (Q9UQN3). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CHMP2B-centered PPI network generated by STRING. This network was generated using medium confidence of 0.4.
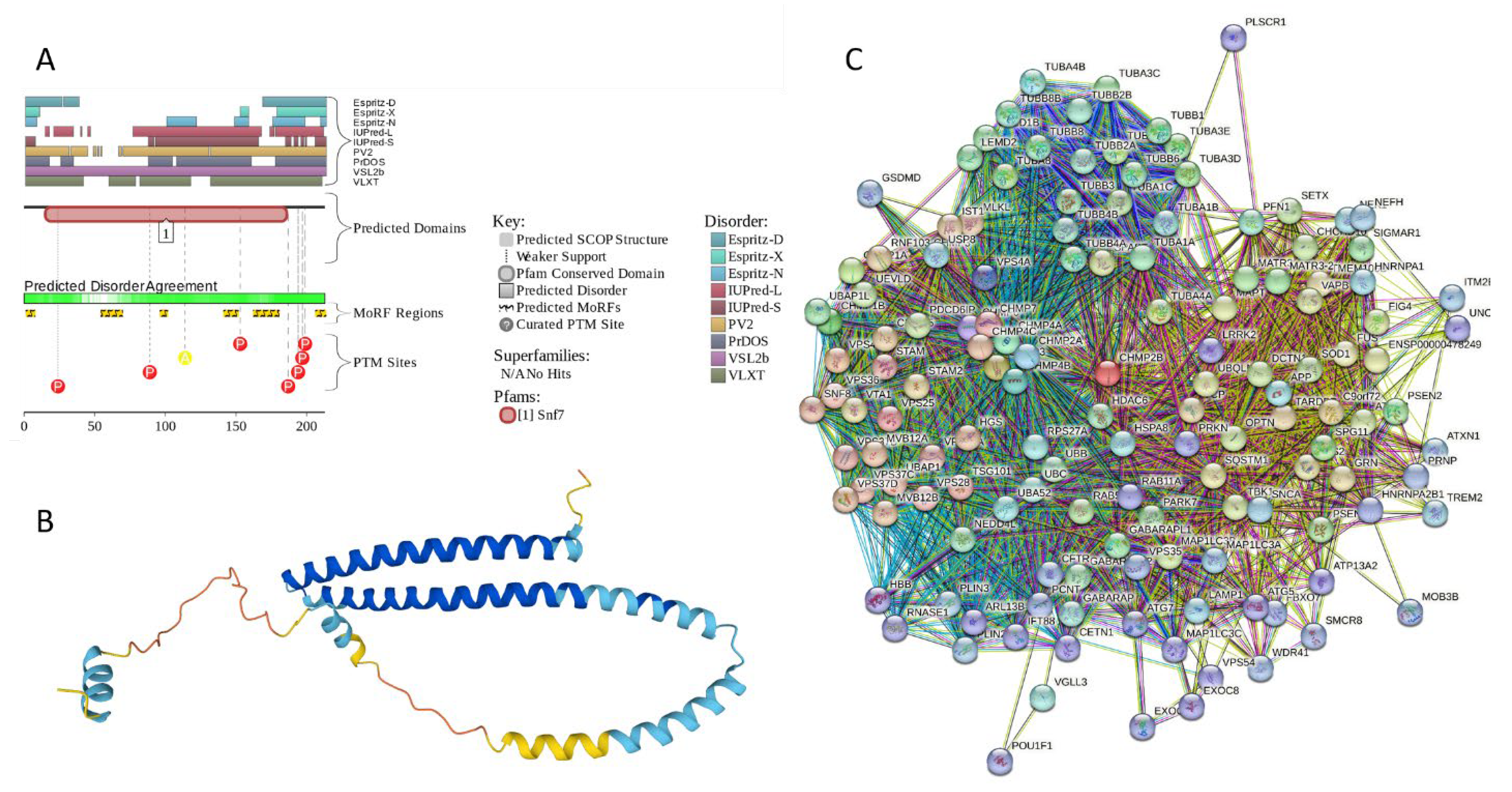
Figure 15.
Functional disorder analysis of human neurogranin (UniProt ID: Q92686). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. NRGN-centered PPI network generated by STRING. This network was generated using low confidence of 0.15 for minimum required interaction score.
Figure 15.
Functional disorder analysis of human neurogranin (UniProt ID: Q92686). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. NRGN-centered PPI network generated by STRING. This network was generated using low confidence of 0.15 for minimum required interaction score.
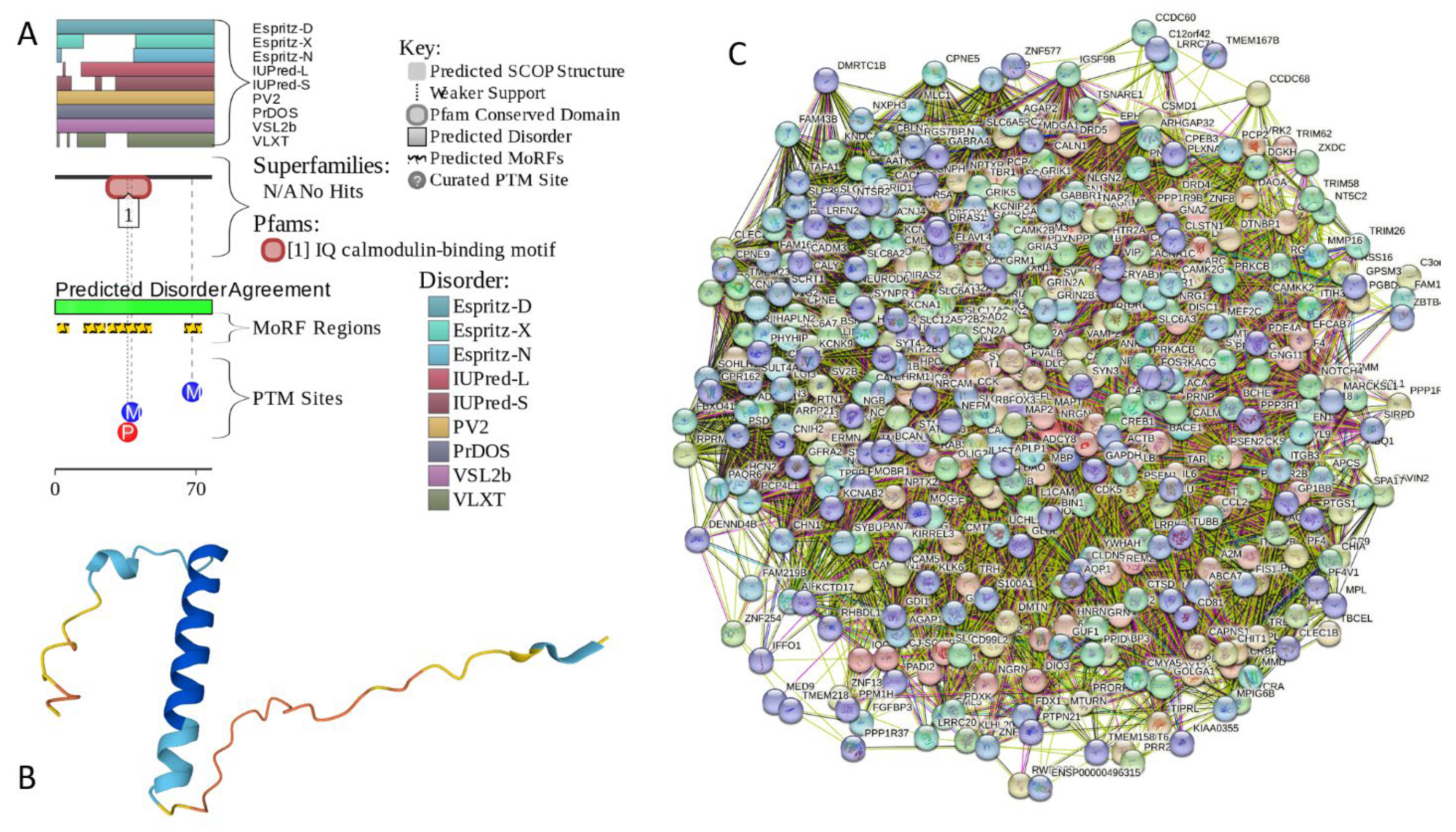
Figure 16.
Functional disorder analysis of human complexin-1 (UniProt ID: O14810). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CPLX1-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.225 for minimum required interaction score.
Figure 16.
Functional disorder analysis of human complexin-1 (UniProt ID: O14810). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CPLX1-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.225 for minimum required interaction score.
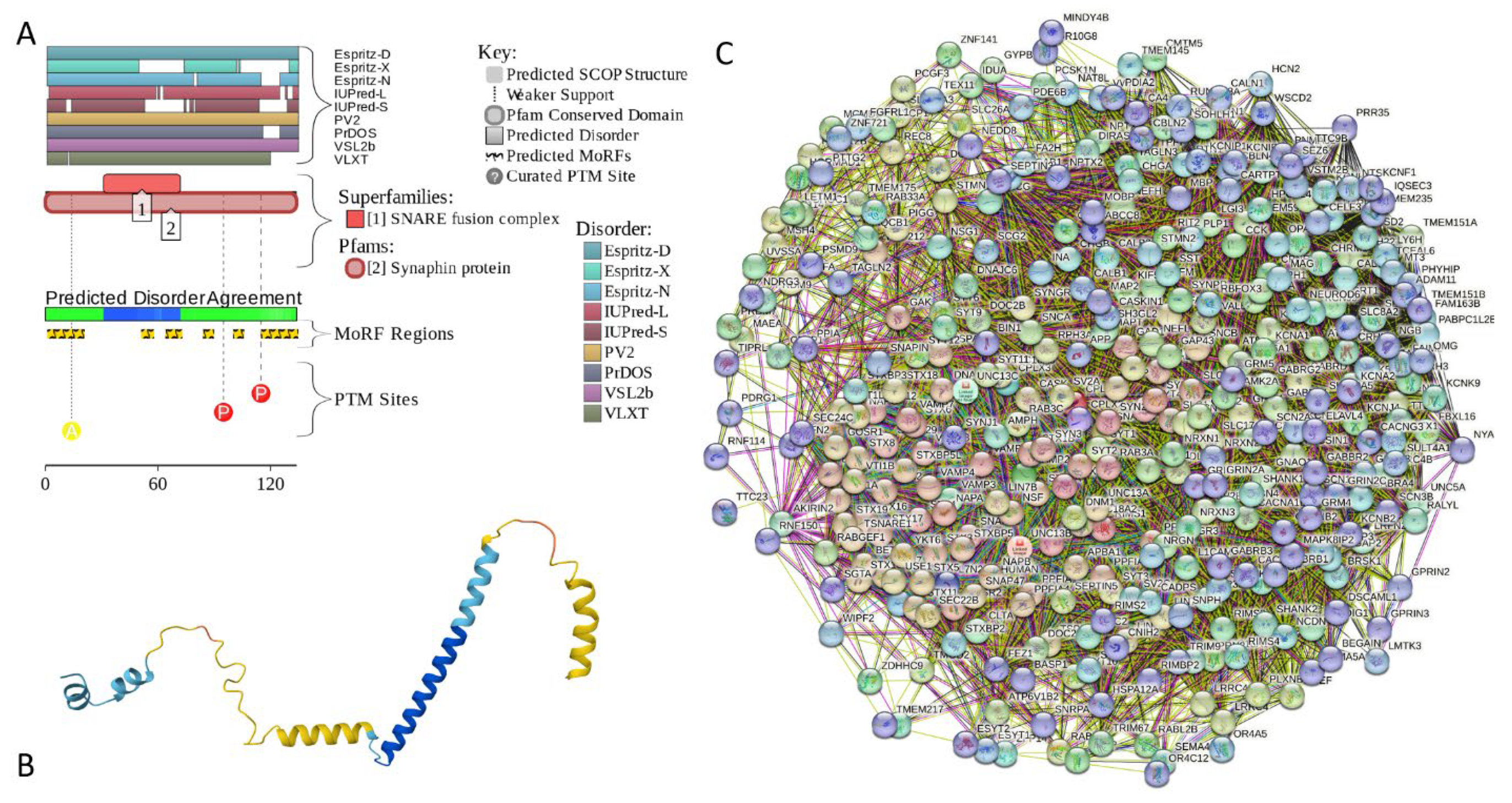
Figure 17.
Functional disorder analysis of human complexin-2 (UniProt ID: Q6PUV4). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CPLX2-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.225 for minimum required interaction score.
Figure 17.
Functional disorder analysis of human complexin-2 (UniProt ID: Q6PUV4). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CPLX2-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.225 for minimum required interaction score.
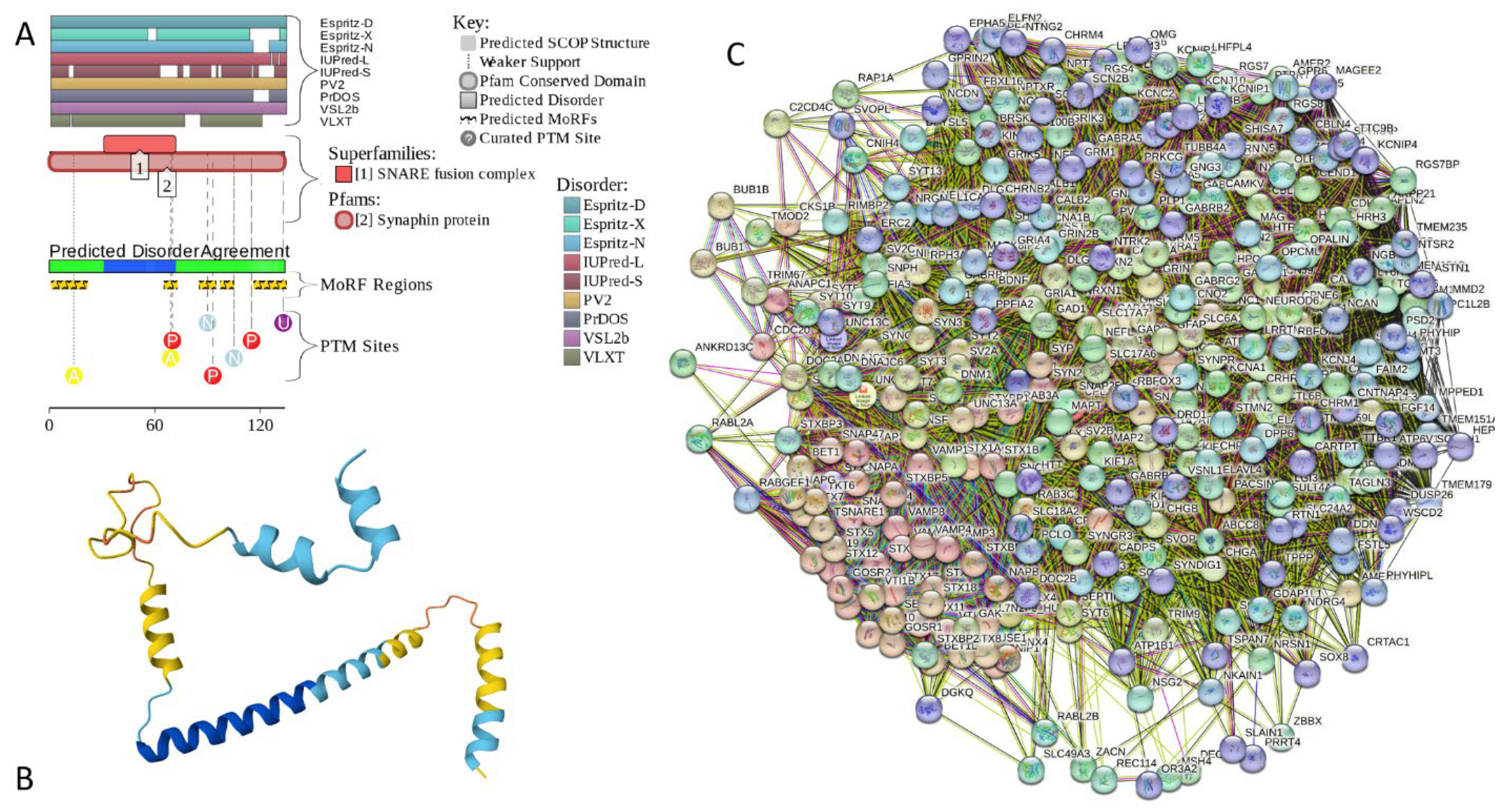
Figure 18.
Functional disorder analysis of human NUCKS1 (UniProt ID: Q9H1E3). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. NUCKS1-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.200 for minimum required interaction score.
Figure 18.
Functional disorder analysis of human NUCKS1 (UniProt ID: Q9H1E3). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. NUCKS1-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.200 for minimum required interaction score.
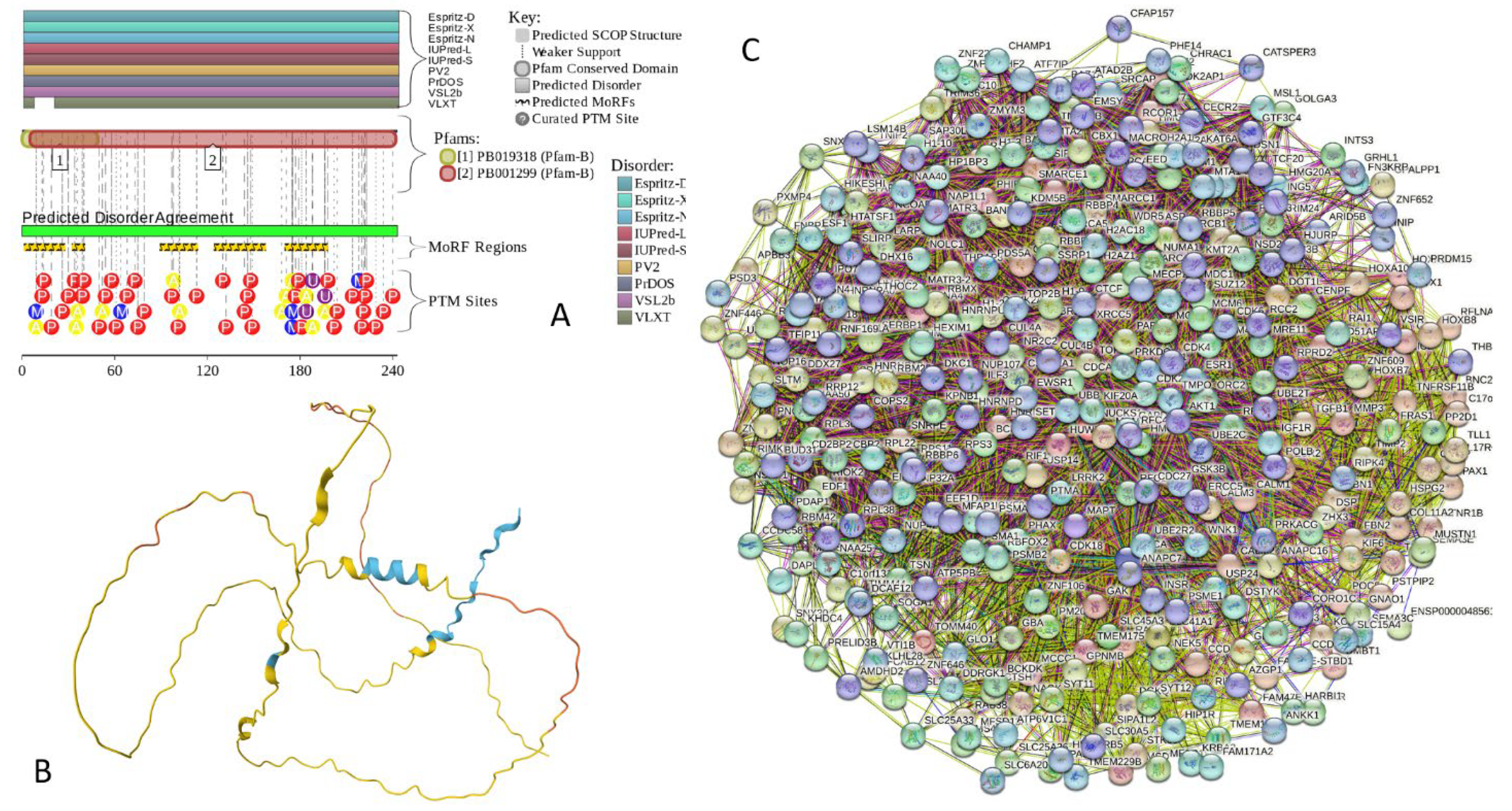
Figure 19.
Functional disorder analysis of human MBP (UniProt ID: P02686). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. MBP-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.300 for minimum required interaction score.
Figure 19.
Functional disorder analysis of human MBP (UniProt ID: P02686). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. MBP-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.300 for minimum required interaction score.
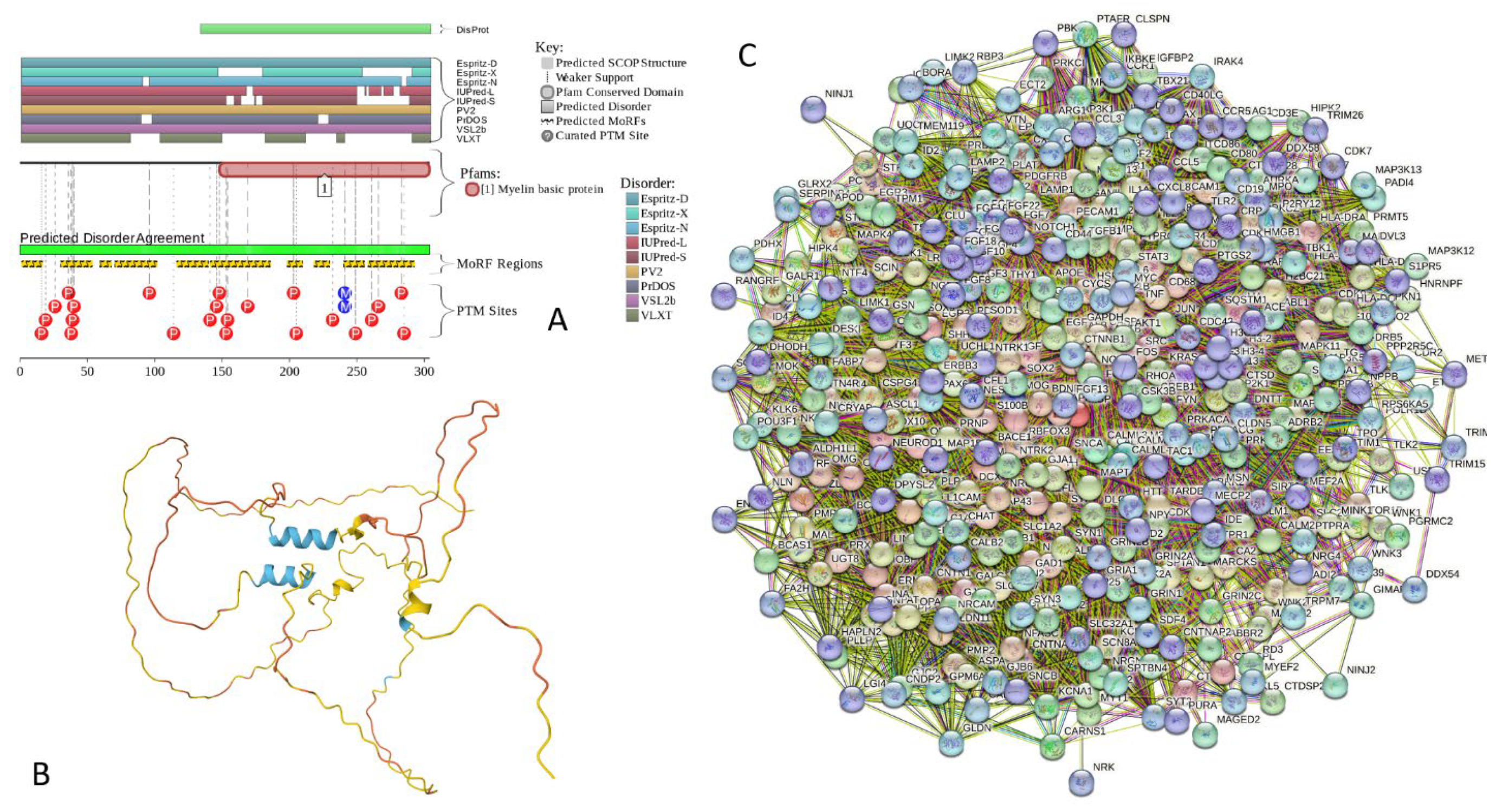
Figure 20.
Functional disorder analysis of human Calpastatin (UniProt ID: P20810). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CAST-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.200 for minimum required interaction score.
Figure 20.
Functional disorder analysis of human Calpastatin (UniProt ID: P20810). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. CAST-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.200 for minimum required interaction score.
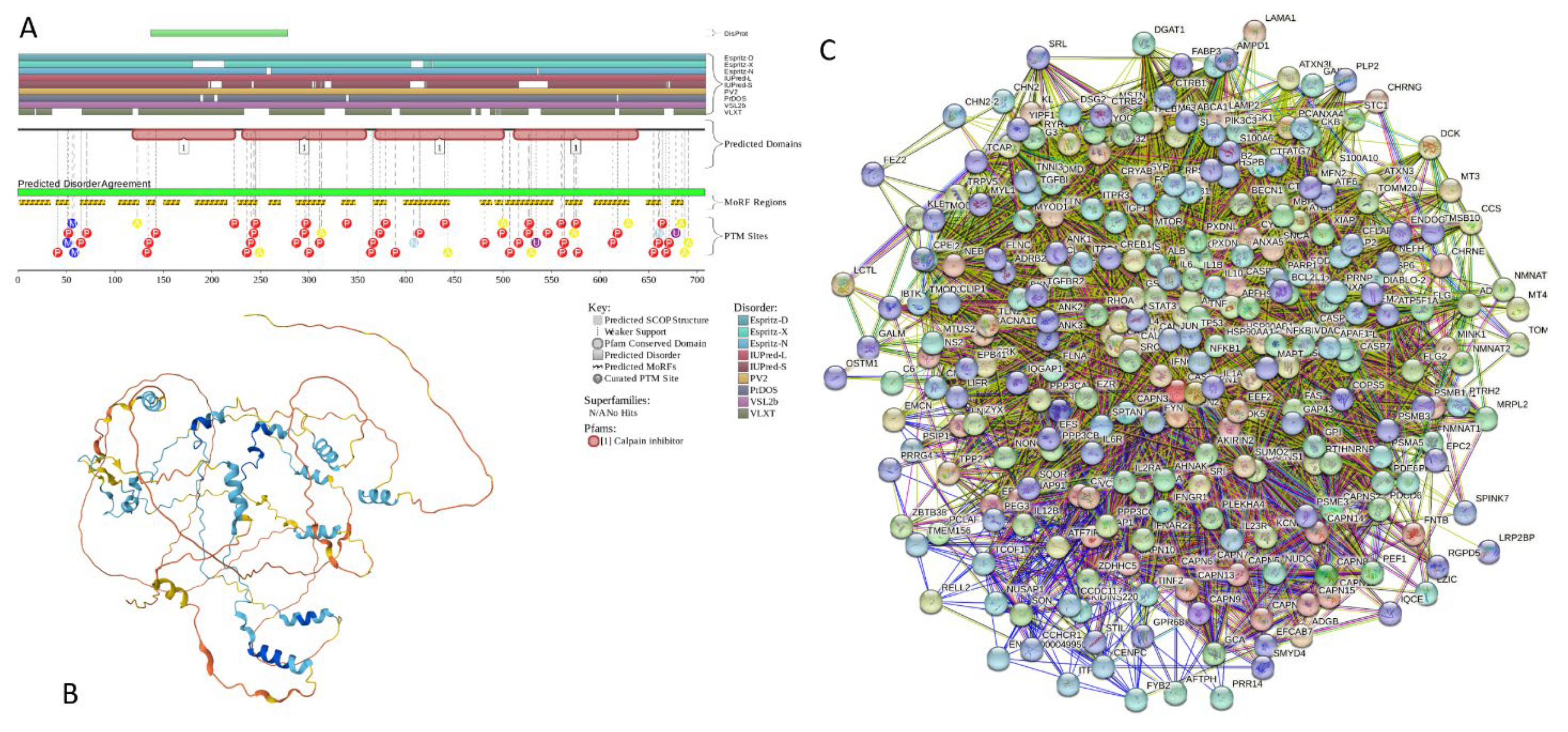
Figure 21.
Functional disorder analysis of human MAPT (UniProt ID: P10636). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. MAPT-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.500 for minimum required interaction score.
Figure 21.
Functional disorder analysis of human MAPT (UniProt ID: P10636). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. MAPT-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.500 for minimum required interaction score.
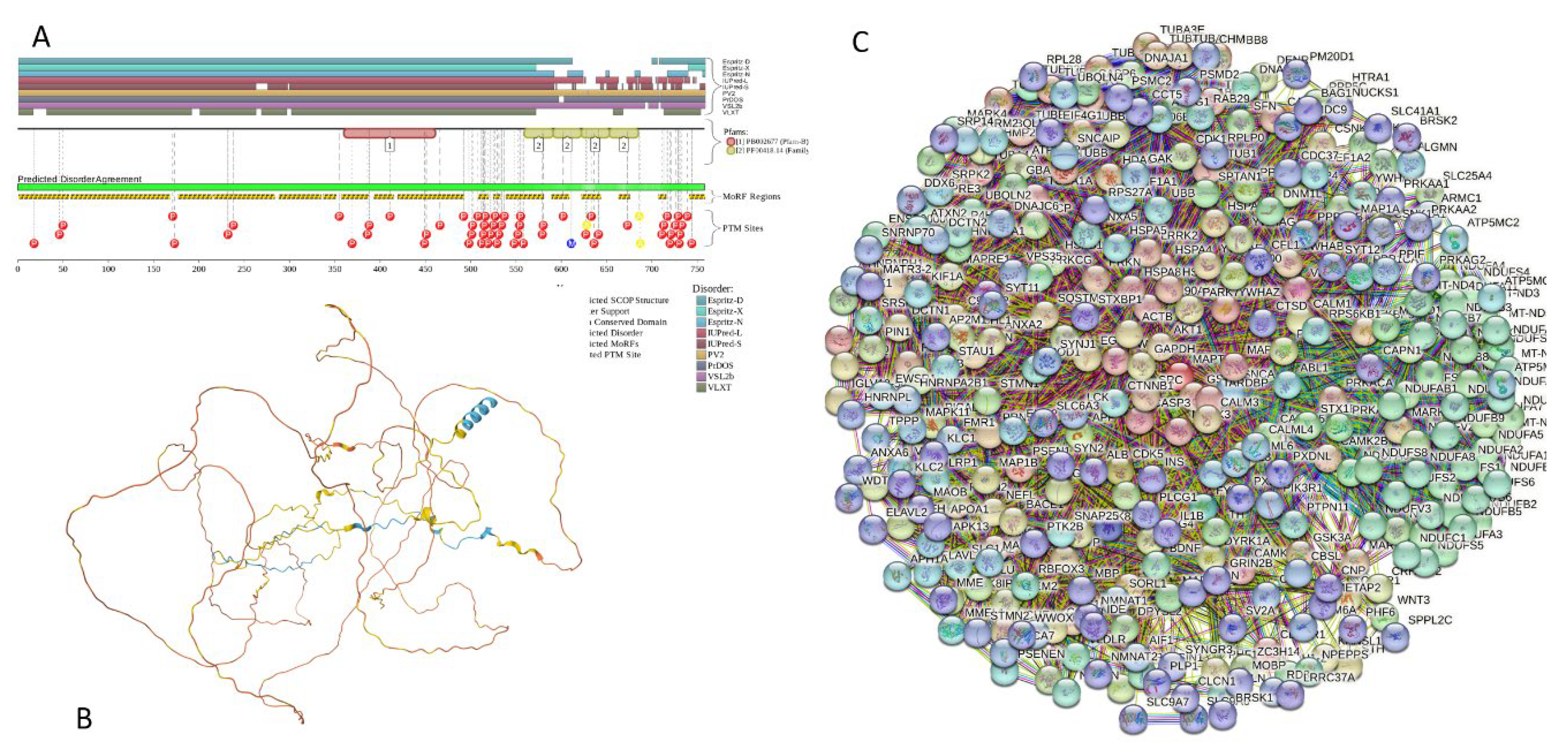
Figure 22.
Functional disorder analysis of human Hemogen (UniProt ID: Q9BXL5). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. HEMGN-centered PPI network generated by STRING. This network was generated using the low confidence of 0.150 for minimum required interaction score.
Figure 22.
Functional disorder analysis of human Hemogen (UniProt ID: Q9BXL5). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. HEMGN-centered PPI network generated by STRING. This network was generated using the low confidence of 0.150 for minimum required interaction score.
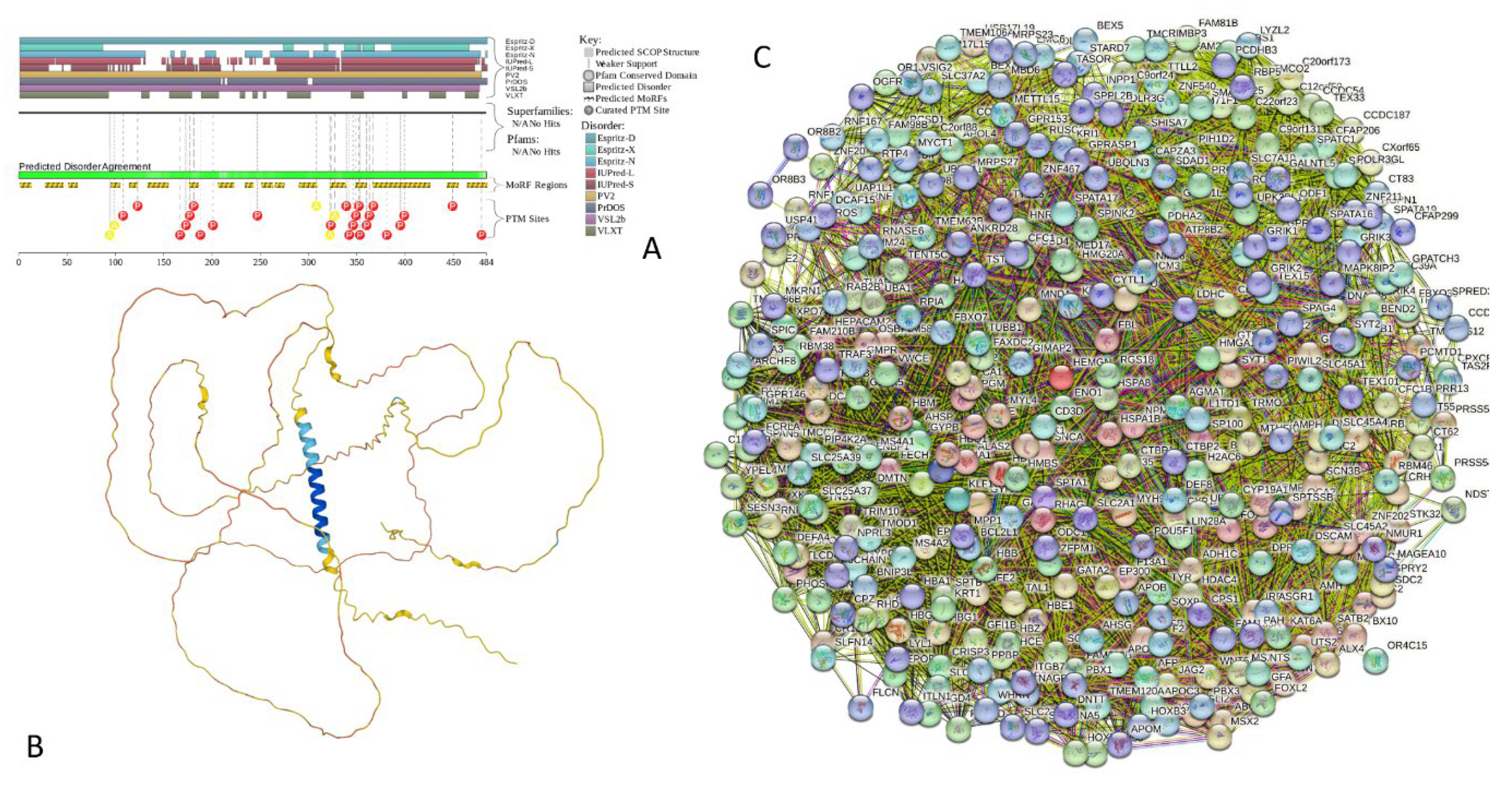
Figure 23.
Functional disorder analysis of human Histone H1.2 (UniProt ID: P16403). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. H1.2-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.250 for minimum required interaction score.
Figure 23.
Functional disorder analysis of human Histone H1.2 (UniProt ID: P16403). A. Functional disorder profile generated by D2P2. B. 3D structural model generated by AlphaFold. C. H1.2-centered PPI network generated by STRING. This network was generated using the custom confidence value of 0.250 for minimum required interaction score.
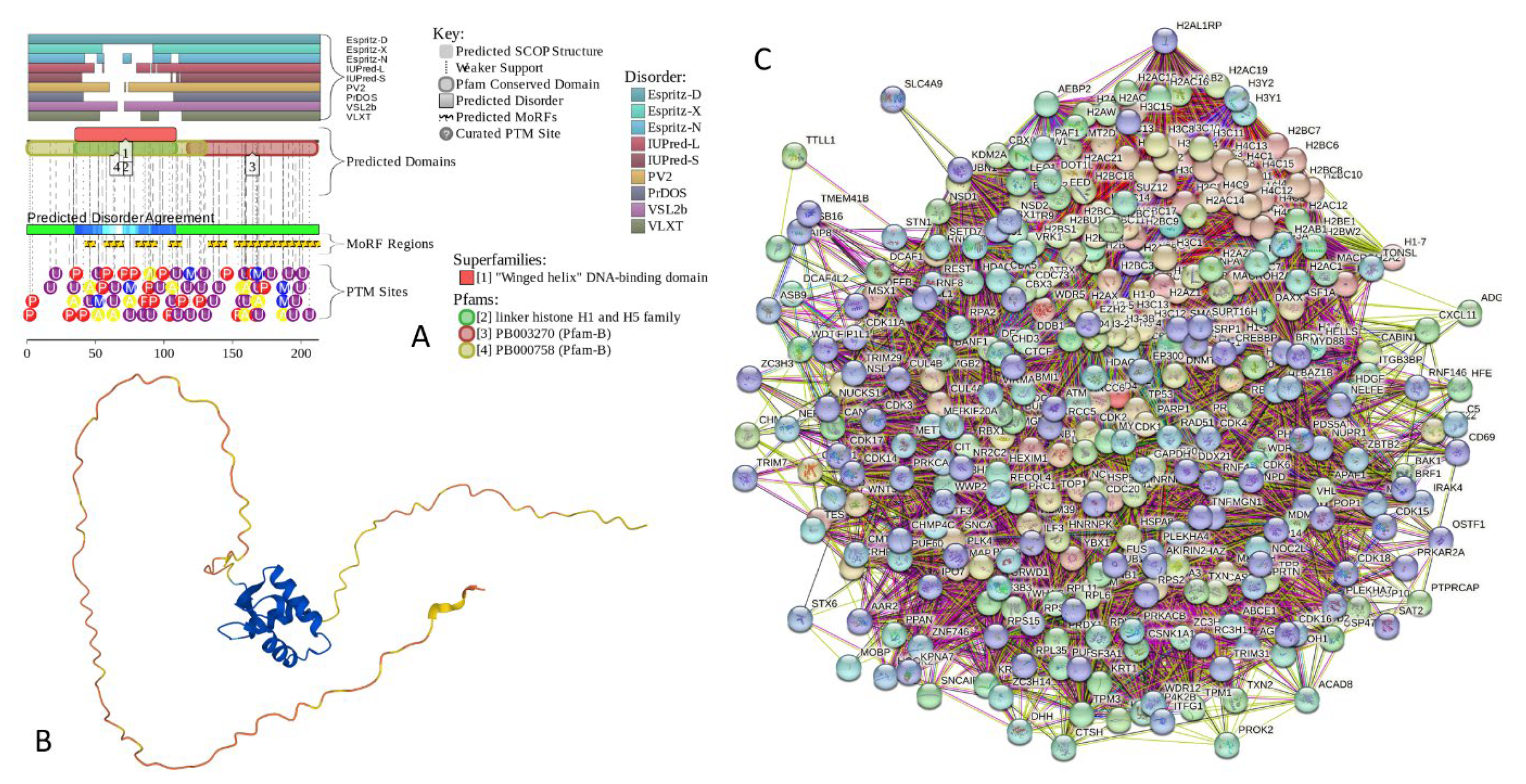

Table 1.
Functional enrichment of the networks centered at human α-, β-, and γ-synucleins.
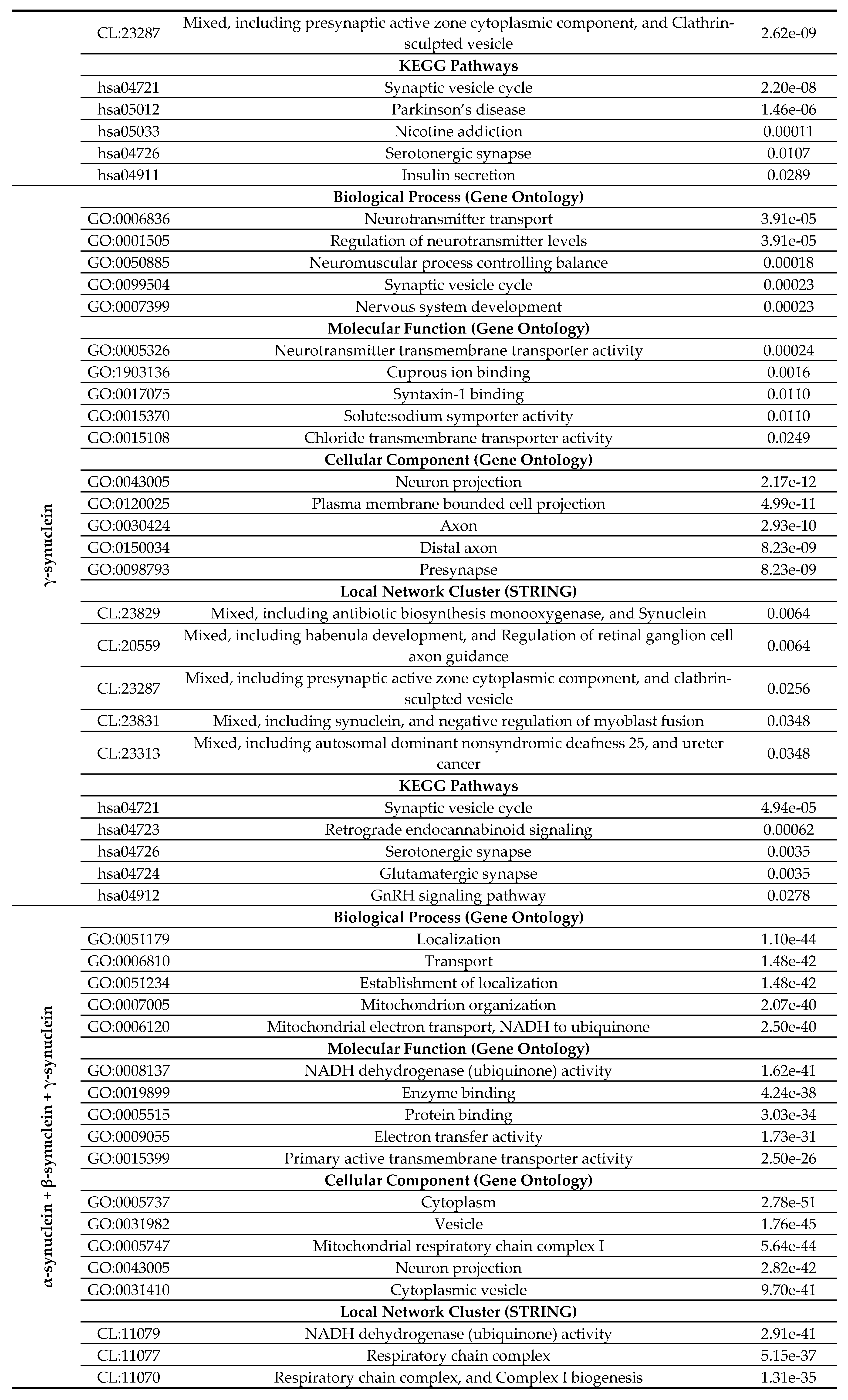

Table 2.
Distribution of human synuclein-iteracting proteins among different disorder catgores.
| Dataset | Protein number | PONDR® VSL2 score vs. PONDR® VSL2 (%) plot | CH-CDF plot | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Blue | Cyan | Dark pink | Pink | Red | Q1 | Q2 | Q3 | Q4 | ||
| Human brain proteome | 10,611 | 15 (0.15%) | 411 (3.87%) | 3,593 (33.86%) |
2,335 (22.00%) |
4,257 (40.12%) | 6,203 (58.5%) | 2,938 (27.7%) | 1,193 (11.2%) |
277 (2.6%) |
| Joint α-β-γ interactome | 467 | 0 (0.0%) |
22 (4.7%) | 172 (36.8%) |
110 (23.6%) |
163 (34.9%) |
292 (62.5%) |
105 (22.5%) |
61 (13.1%) |
9 (1.9%) |
| α-Synuclein interactome | 356 | 0 (0.0%) |
20 (5.6%) |
135 (37.9%) |
89 (25.0%) |
112 (31.5%) |
234 (65.7%) |
65 (18.3%) |
48 (13.5%) |
9 (2.5%) |
| β-Synuclein interactome | 85 | 0 (0.0%) |
0 (0.0%) |
30 (35.3%) |
19 (22.3%) |
36 (32.4%) |
48 (56.5%) |
26 (30.6%) |
11 (12.9%) |
0 (0.0%) |
| γ-Synuclein interactome | 32 | 0 (0.0%) |
0 (0.0%) |
12 (37.5%) |
4 (12.5%) |
16 (50.0%) |
14 (43.75%) |
14 (43.75%) |
4 (12.5%) |
0 (0.0%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



