Submitted:
24 April 2024
Posted:
25 April 2024
You are already at the latest version
Abstract
Spatial relations between tumor cells and host-infiltrating cells are increasingly important in both basic science and clinical research. In this study we have tested the feasibility of using standard methods of immunohistochemistry (IHC) in a multiplex staining system using a newly developed set of chromogenic substrates for the peroxidase and alkaline phosphatase enzymes. Using this approach we have developed a set of chromogens characterized by 1) providing fine cellular detail, 2) non-overlapping spectral profiles, 3) absence of interactions between chromogens, 4) stable upon storage, and 5) compatible with current standard immunohistochemistry practices. When viewed microscopically under brightfield illumination the chromogens yielded the following colors; red, black, blue, yellow, brown, and green. By selecting compatible color combinations we have shown feasibility for four-color multiplex staining. Depending on the particular type of analysis being performed, visual analysis, without the aid of computer assisted image analysis, was sufficient to differentiate up to four different markers.
Keywords:
Immunohistochemistry
; chromogens
; brightfield
; spatial biology
1. Introduction
There is a substantial body of literature describing the intra-tumor heterogeneity of both tumor and host cells as well as their interactions. Extensive efforts have been made to analyze the various cell types, locations, and associations within the tumor architecture. Such methods have focused primarily on single cell measurements by flow cytometry or nucleic acid sequencing [1,2]. However, such methods based on dissociated cells fail to take into account the spatial relationships between the various cell types.
Discovering the molecular mechanisms affecting cell growth and development is critical in understanding disease processes, and this understanding can ultimately lead to better treatments. For example in tumors there is an intimate relationship between the tumor cells and their surroundings. This can be understood to reflect a critical connection between the tumor cells and their environment. For example, some types of immunotherapy rely upon the treatments ability to interfere with certain interactions between tumor cells and immune cells [3]. It is now apparent that the characterization of the tumor microenvironment and the cell populations residing therein is critical to our understanding of these diseases.
Spatial immunohistochemistry (IHC) has now become a useful tool for analyzing morphological patterns [4,5] and cellular microenvironment [6,7]. While conventional IHC methods using chromogenic labels can capture spatial organization, these methods are limited by the availability of robust chromogens that can be combined together within a single stain to produce multiplex staining images. Therefore, an objective, measureable, and reproducible means of evaluating spatial heterogeneity at the single cell level by IHC remains a challenging goal.
Pathologists have been evaluating cells in tissue sections using classical antibody- and morphology based staining methods for nearly 100 years. In clinical practice these evaluations were frequently performed using chromogenic IHC methods that allowed for the assessment of one or two proteins within a single tissue slide. New technologies are now being introduced such as immunofluorescence using multiple fluorescently labeled antibodies simultaneously [8,9]. However, such technologies also come with new challenges and technical hurdles.
2. Materials and Methods
Sample Preparation
All tissue specimens were obtained from surgical samples as paraffin blocks. Tissues had been formalin-fixed and paraffin-embedded (FFPE) using standard histological methods. Tissue sections were prepared at 4µ, adhered to positively charged microscope slides, and stored at room temperature until time of staining. At the time of staining tissue sections were deparaffinized through graded solutions of xylene and alcohol, and rehydrated in deionized water. After deparaffinization, in order to firmly attach the tissues to the microscope slide during the staining procedure, the slides and the tissues were treated with an adherence promoting reagent (Tissue Glue, Diagnostic Biosystems) according to the manufacturer’s instructions. Deparaffinized tissue sections were then subjected to antigen retrieval by submerging slides in a solution of Tris-EDTA, pH 9.0 and heating in a pressure cooker at 15 psi for 15 minutes. Slides in antigen retrieval solution were allowed to cool at room temperature until the pressure was relieved. Slides were then placed into a Tris-buffered wash solution and cooled at room temperature for five minutes.
Antibodies
Antibodies were screened on tonsil as single stains to determine optimal staining concentraitons. All antibodies were obtained from Diagnostic Biosystems, Pleasanton, CA, and are listed in Table 1.
Reagents
Table 2 lists reagents used. All reagents were obtained from Diagnostic Biosystems.
Chromogens
Table 3 lists all chromogens used. All chromogens were obtained from Diagnostic Biosystems.
Immunohistochemistry
Sequential multiplex staining was used because of its flexibility for using any antibody or enzyme system. The following steps were employed using a manual method at room temperature:
Step Name
1 Primary antibody
2 Rinse
3 Polymer (Enzyme conjugated Secondary antibody)
4 Rinse
5 Substrate/Chromogen
6 Rinse
7 Elution Buffer
Peroxidase Method:
Incubation time for Primary antibodies and Polymers was 20 minutes. Incubation time for Substrate/Chromogens for Horseradish Peroxidase (HRP) was 5 minutes.
Alkaline Phosphatase Method:
Incubation time for Primary antibodies and Polymers was 20 minutes. Incubation time for Substrate/Chromogens for Alkaline Phosphatase (AP) polymers was 10 minutes.
Elution Buffer
Elution buffer was comprised of 0.25% Sodium Dodecyl Sulfate (SDS) in 0.1M Glycine at pH 2.0 (9). Previous experiments had shown that an incubation time of 10 minutes at 50˚ C was sufficient to remove all primary antibodies and secondary polymers from stained slides.
Multiplex Staining
After a single sequence of staining and elution, slides were then re-stained with a different primary antibody using the same sequence as previously described, but using a different colored chromogen. This sequence was repeated multiple times until up to four different chromogens had been applied to the tissues, each chromogen detecting a different antigen based on the specificity of the primary antibody.
Microscopic Evaluation
Slides were mounted using ImmunoHisto-Sealer (Diagnostic Biosystems, Part No.K076 ) followed by a permanent mounting medium, according to manufacturer’s instructions. Mounted slides were viewed using brightfield microscopy at 100X and 400X magnification. Slides were evaluated for specific staining, background staining, and chromogen mixing as indicated by mixed colors. Specific staining was graded on a scale of 0 to 3 in 0.5 grade increments, with 0 indicating no staining, 1 indicating light staining, 2 indicating moderate staining, and 3 indicating strong staining.
3. Results
Antibodies were tested on tonsil tissue as single stains and double stains to determine the optimal concentrations and to evaluate each chromogen for sensitivity, background, and fine cellular detail. Representative images for some of the chromogens are shown in Figure 1. Immunohistochemical staining with pan-cytokeratin on tonsil is shown in Figure 1A – 1D depicting chromogens for green-HRP, blue-HRP, yellow-HRP, and red- AP respectively. These Figures show the expected specific staining of surface and crypt epithelium with minimal background staining of non-epithelial elements. Therefore, these chromogens can be used to identify cells of epithelial origin including most carcinomas. Figures 1E and 1F show staining results for B-lymphocytes in tonsil with chromogens for yellow-HRP-Yellow and red-AP. A typical staining pattern shows an abundance of B-lymphocytes in germinal centers and mantle zones of lymphoid follicles, with a lesser distribution of positive cells within the interfollicular spaces. Figures 1H and 1I show a combination of pan-cytokeratin and B-lymphocyte markers. In Figure 1H pan-cytokeratin is stained with yellow-HRP and B-lymphocytes are stained with blue-HRP. In Figure 1I pan-cytokeratin is stained with green-HRP and B-lymphocytes are stained with yellow-HRP.
Figure 1 Legend. Tonsil stained with chromogens (100X). A. Pan-cytokeratin (AE1/AE3) with green-HRP, B. Pan-cytokeratin (AE1/AE3) with blue-HRP, C. Pan-cytokeratin (AE1/AE3) with yellow-HRP, D. Pan-cytokeratin (AE1/AE3) with red-AP, E. B-cell (CD20) with yellow-HRP, F. B-cell (CD20) with red-AP, G. Macrophage (CD163) with DAB, H. Pan-cytokeratin (AE1/AE3) with yellow-HRP and B-cell (CD20) with blue-HRP, I. Pan-cytokeratin (AE1/AE3) with green-HRP and B-cell (CD20) with yellow-HRP.
Next, chromogens were evaluated for their ability to withstand the elution buffer without fading. The single stains were incubated in elution buffer for 10 minutes at 50˚ C. Photographic images were compared both before and after treatment with the elution buffer. Certain chromogens showed partial loss of signal after incubation with elution buffer as shown below in Table 4.
Because certain chromogens showed a partial loss of staining in the elution buffer, these chromogens were either omitted or used as final stains in a multi-stain sequence. Chromogens showing partial loss of signal included PermalRed-HRP, PermaGreen-HRP, and PermaBlack-HRP.
Using the information gathered from these initial experiments, the optimal panels were constructed. Table 5 shows representative panels, and their corresponding microscopic images are shown in Figure 2Figure 3Figure 4.
Figure 2 shows four-color immunohistochemistry staining on colorectal carcinoma. Tumor cells are stained with yellow-HRP with pan-cytokeratin (AE1/AE3) showing a moderately to well differentiated adenocarcinoma. Staining for Ki67 using green-HRP shows a high Ki67 index indicating aggressive growth potential. The stained nuclei show a basal orientation in some areas of the tumor and loss of polarity in other areas. There is a moderate background of inflammatory cells in the connective tissue between areas of carcinoma. Macrophages (CD163) are stained with DAB and give a brown stain, and B-lymphocytes (CD20) are stained with blue-HRP. Inset A in Figure 2 shows an area of the tumor showing a mixture of both macrophages and lymphocytes.
Figure 3 depicts another moderately differentiated carcinoma showing adenocarcinoma morphology. In this example the carcinoma cells are stained with pan-cytokeratin (AE1/AE3) using red-AP chromogen. Ki67 is stained using green-HRP and shows a moderate to high Ki67 index indicating high growth potential. Nuclei have lost their polarization. Compared to the colorectal carcinoma in Figure 2, this carcinoma displays substantially greater number of inflammatory cells. Macrophages (CD163) are stained brown with DAB, and B-lymphocytes (CD20) are stained blue with blue-HRP. Furthermore, in this area of the tumor there appears to be some separation between the B-cells (Inset A) and the macrophages (Inset B).
Figure 4 depicts an example of another colorectal carcinoma. Staining with red-AP for pan-cytokeratin (AE1/AE3) shows a moderately to well differentiated adenocarcinoma. The tubular structures are widely dispersed throughout an extensive stroma (Inset A). Ki67 labeling with HRP-Green shows a lower Ki67 index compared to the two previous examples of colorectal carcinoma (Figure 2 and Figure 3). There is a moderate scattering of inflammatory cells comprised of macrophages (CD68, brown) and B-lymphocytes (CD79b, blue). Occasional tertiary lymphoid structures are observed and stain positive for B-lymphocytes (Inset B).
Figure 2 Legend. Colorectal carcinoma (100X). Pan-cytokeratin (AE1/AE3) stained with yellow-HRP. Macrophage (CD163) stained with DAB. B-cell (CD20) stained with blue-HRP. Proliferation marker (Ki67) stained with green-HRP. Inset A shows a higher magnification of the selected area.
Figure 3 Legend. Colorectal Carcinoma (100X). Pan-cytokeratin (AE1/AE3) stained with Red-AP. Macrophage (CD163) stained with DAB. B-cell (CD20) stained with blue-HRP. Proliferation marker (Ki67) stained with green-HRP. Inset A shows an area of predominantly B-cells (CD20). Inset B shows an area of predominantly macrophages (CD163). Inset C shows an area of predominantly tumor cells (AE1/AE3) that are also positive for proliferation marker (Ki67).
Figure 4 Legend. Colorectal carcinoma (100X) showing an area of tumor stroma with an extensive inflammatory cell infiltrate. Tumor cells are stained with red-AP for Pan-cytokeratin (AE1/AE3) and green-HRP for proliferation marker (Ki67). Macrophages (CD68) are stained with DAB and B-cells (CD79a) are stained with blue-HRP. Insets A and B show a higher magnification of the selected areas.
4. Discussion
A thorough understanding of the various technologies strengths, weaknesses, and limitations is crucial in answering specific questions regarding tumor heterogeneity and microenvironment. No single technology can provide all answers to all questions. Here we present a new technology based on classical IHC chromogenic methods that can be easily incorporated into most pathology laboratories without modification of existing equipment or practices. This method is based on various chromogenic substrates that provide excellent color separation. Multiplex staining is achieved by sequential IHC with antibody elution performed between each sequential step. Using this approach we have demonstrated the feasibility of identifying up to four different markers with excellent color differentiation. Furthermore, the time required to achieve these results is within a few hours, thus making this method feasible within the typical laboratory daily work flow.
Depending on the specific antibody panel chosen for multiplex staining, the complexity of the information obtained can be overwhelming such that mere visual interpretation by the microscopist may be insufficient to extract all relevant information and interpret all expression patterns. Such situations can certainly benefit from computer-aided image analysis using suitable algorithms [11,12]. In such situations spectrally distinct chromogens are critical for accurate results. In this study we have shown feasibility of a new method for creating multiplex IHC stains that takes advantage of existing methods and extends those methods by introduction of a new set of chromogenic substrates. The implementation of spatial technologies is increasingly important to identify and locate specific cell types and patterns in the context of the tumor microenvironiment, and may well lead to new insights into previously unrecognized biological processes.
5. Conclusions
In this study we have described a method for multiplex staining in tissue samples using standard immunohistochemical methods. This method relies on the development of a new set of chromogens that can be used individually or in combination with existing chromogens to generate multiplex stains. When used in multiplex stains, these chromogens show fine cellular detail, low background, retain their original colors. Chromogens exhibiting sufficient color separation from each other can be combined in such a way that simple visual interpretation of the resulting images is possible. Using this method we have successfully demonstrated four-color immunohistochemistry. Depending on the types of staining patterns generated, visual analysis without the assistance of computer-aided image analysis may be possible.
Author Contributions
Conceptualization, Bipin Gupta and George Yang; methodology, Marc Key; writing—original draft preparation, Marc Key; project administration, Bipin Gupta. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors were employed by, or received funding from Diagnostic Biosystems, Inc.
References
- Lee J, Hyeon DY, Hwang D. Single-Cell Multiomics: Technologies and Data Analysis Methods. Exp Mol Med 2020, 52, 1428–42. [CrossRef] [PubMed]
- Stuart T, Satija R. Integrative Single-Cell Analysis. Nat Rev Genet 2019, 20, 257–72. [CrossRef] [PubMed]
- Topalian SL, Taube JM, Pardoll DM. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367. [Google Scholar]
- Hickey JW, Neumann EK, Radtke AJ, Camarillo JM, Beuschel RT, Albanese A, et al. Spatial Mapping of Protein Composition and Tissue Organization: A Primer for Multiplexed Antibody-Based Imaging. Nat Methods, 2022; 19, 284–95.
- Bolognesi MM, Manzoni M, Scalia CR, Zannella S, Bosisio FM, Faretta M, et al. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. J Histochem Cytochem 2017, 65, 431–44. [Google Scholar]
- Allam M, Cai S, Coskun AF. Multiplex Bioimaging of Single-Cell Spatial Profiles for Precision Cancer Diagnostics and Therapeutics. NPJ Precis Oncol, 2020; 4, 1–14.
- Fl Guillot A, Kohlhepp MS, Bruneau A, Heymann F, Tacke F. Deciphering the Immune Microenvironment on A Single Archival Formalin-Fixed ParaffinEmbedded Tissue Section by An Immediately Implementable Multiplex Fluorescence Immunostaining Protocol. Cancers (Basel), 2020; 12, 1–18.
- Buchwalow IB, Minin EA, Boecker W. A multicolor fluorescence immunostaining technique for simultaneous antigen targeting. Acta Histochem 2005, 107, 143–148. [Google Scholar]
- Lin J-R, Fallahi-Sichani M, Sorger PK. Highly Multiplexed Imaging of Single Cells Using a High-Throughput Cyclic Immunofluorescence Method. Nat Commun 2015, 6, 8390. [CrossRef] [PubMed]
- Pirici, D Mogoanta L, Kumar-Singh S, Piricin I, Margaritescu C, Simionescu C, Radu Stanescu R. Antibody Elution Method for Multiple Immunohistochemistry on Primary Antibodies Raised in the Same Species and of the Same Subtype. J Histochem Cytochem 2009, 57, 567–575. [Google Scholar] [CrossRef]
- Blom S, Paavolainen L, Bychkov D, Turkki R, Mäki-Teeri P, Hemmes A, et al. Systems Pathology by Multiplexed Immunohistochemistry and WholeSlide Digital Image Analysis. Sci Rep 2017, 7, 15580. [Google Scholar] [CrossRef] [PubMed]
- De Smet F, Antoranz Martinez A, Bosisio FM. Next-Generation Pathology by Multiplexed Immunohistochemistry. Trends Biochem Sci 2021, 46, 80–82. [CrossRef] [PubMed]
Figure 1.
Optimization of Chromogens on Tonsil Tissue.
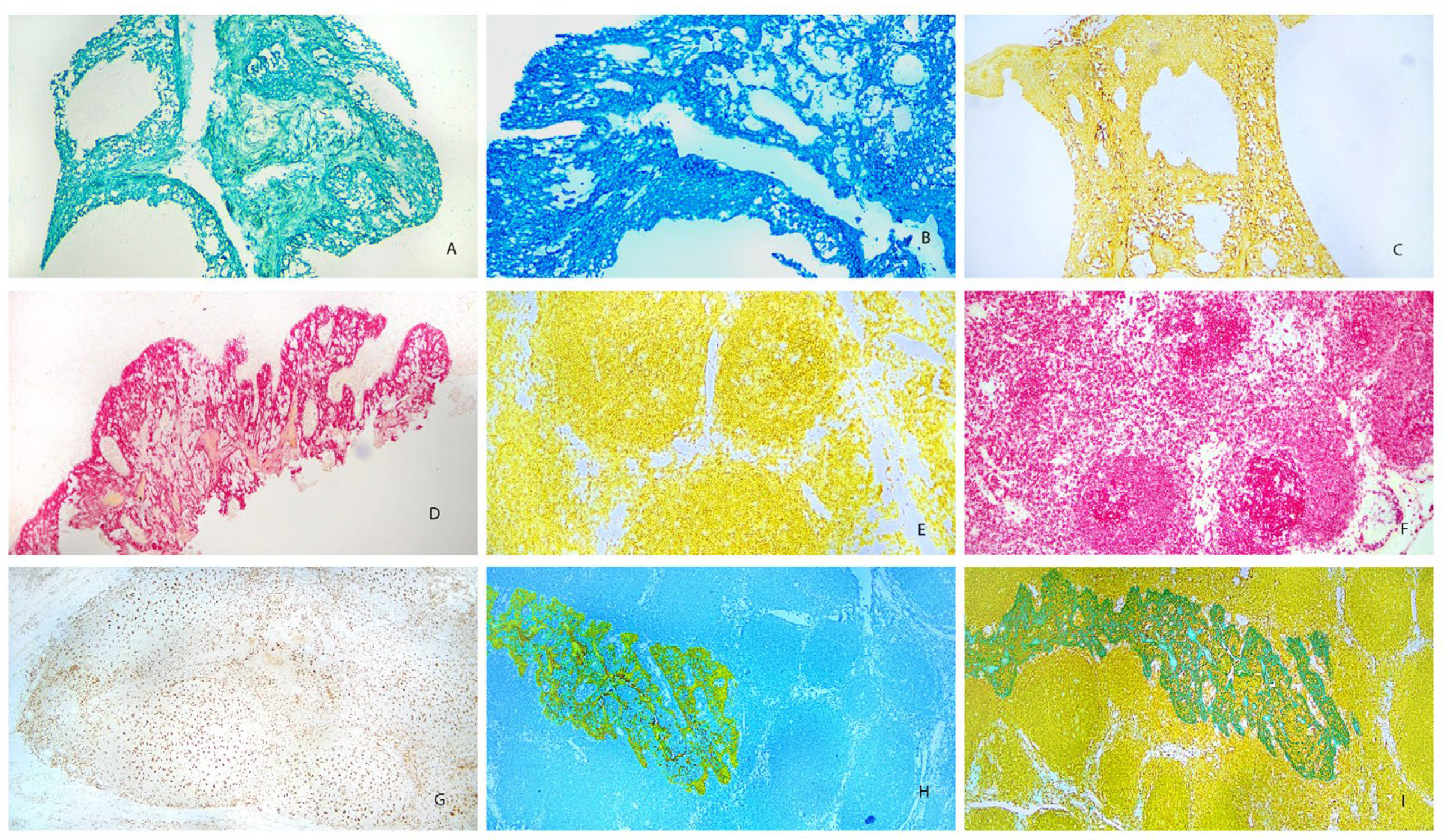
Figure 2.
Four-Plex Stain on Colorectal Carcinoma using Chromogens for HRP.
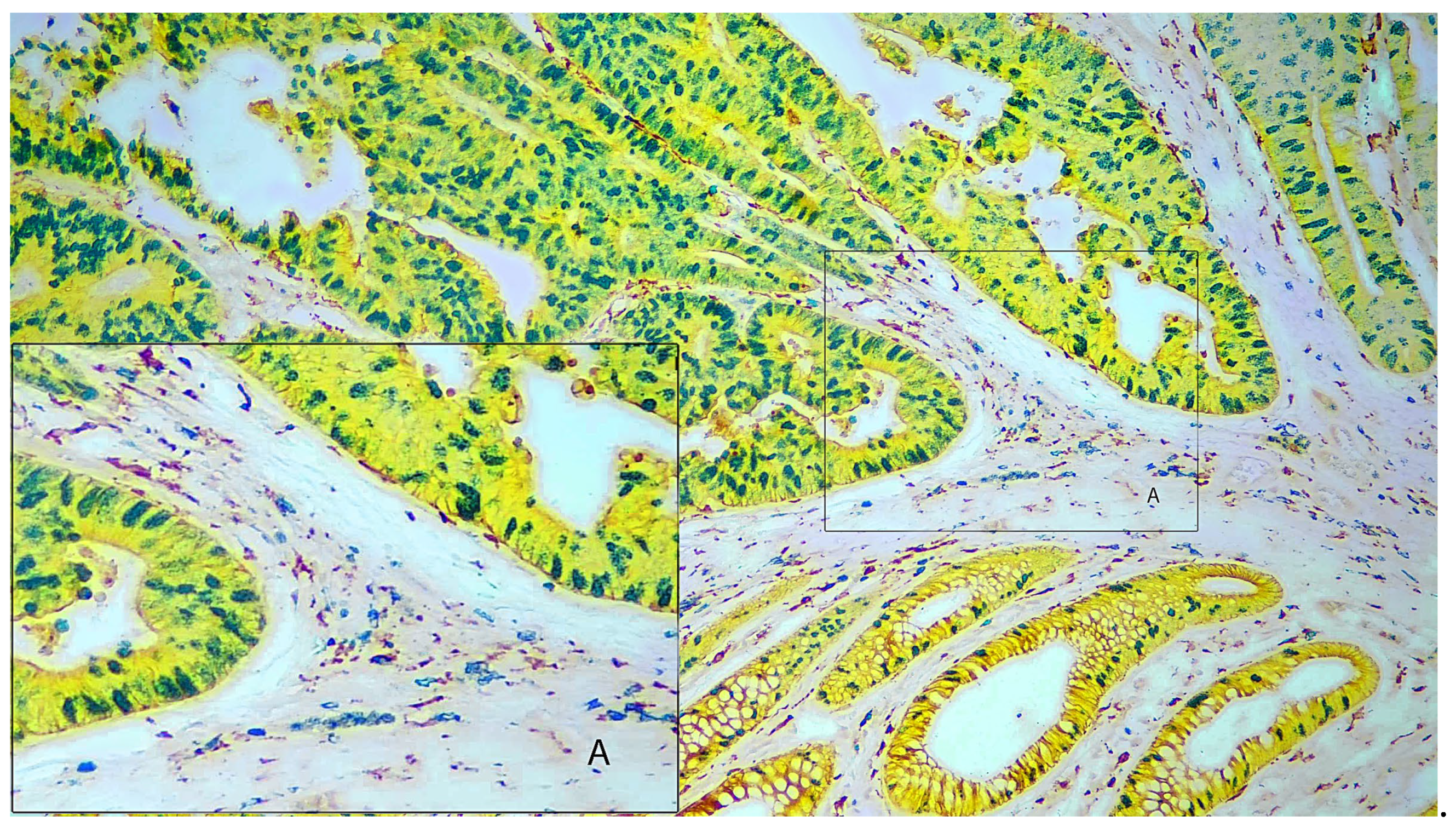
Figure 3.
Four-Plex Stain on Colorectal Carcinoma using Chromogens for AP and HRP.
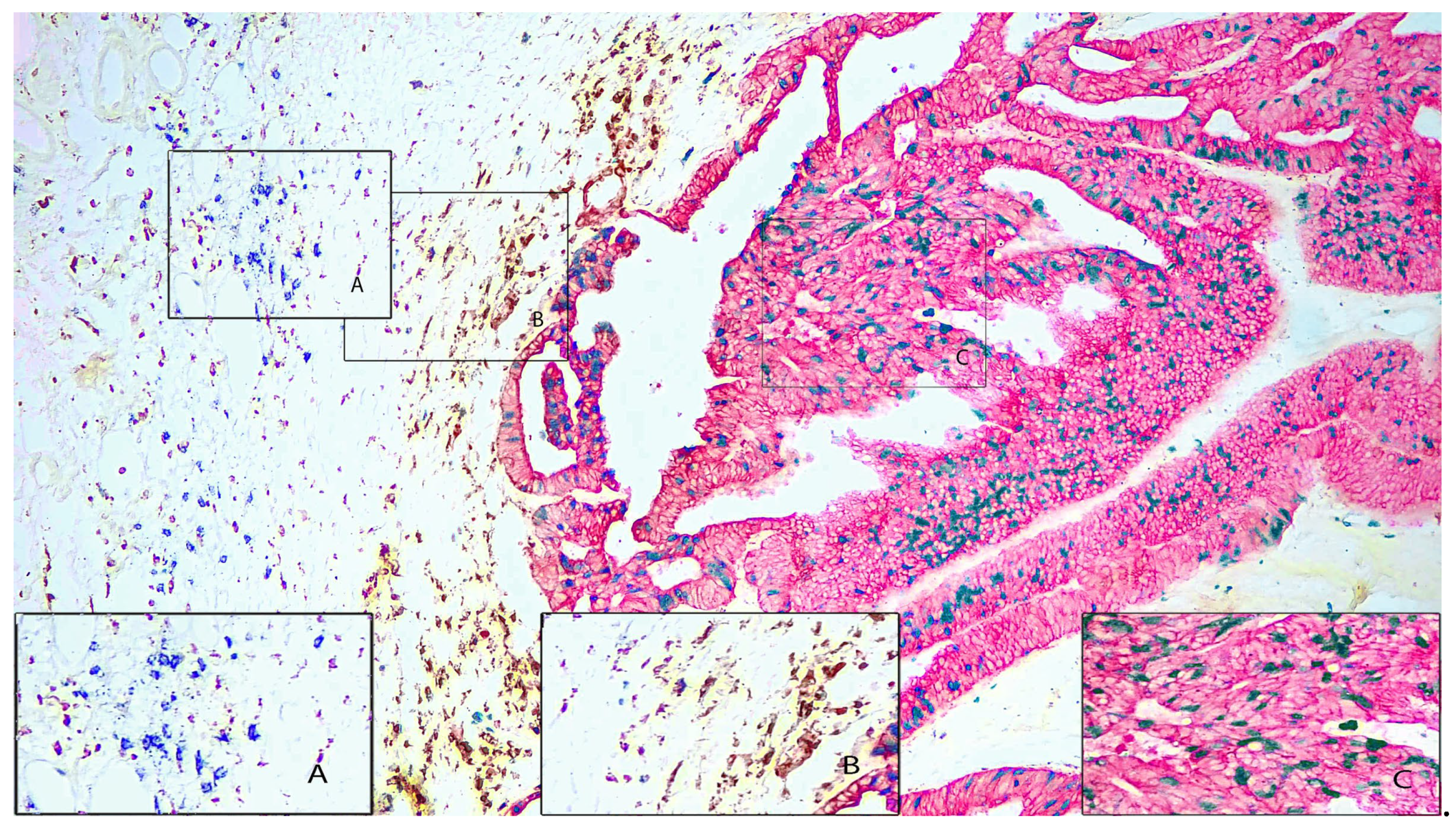
Figure 4.
Four-Plex Stain of Colorectal Carcinoma showing Inflammatory Cells.
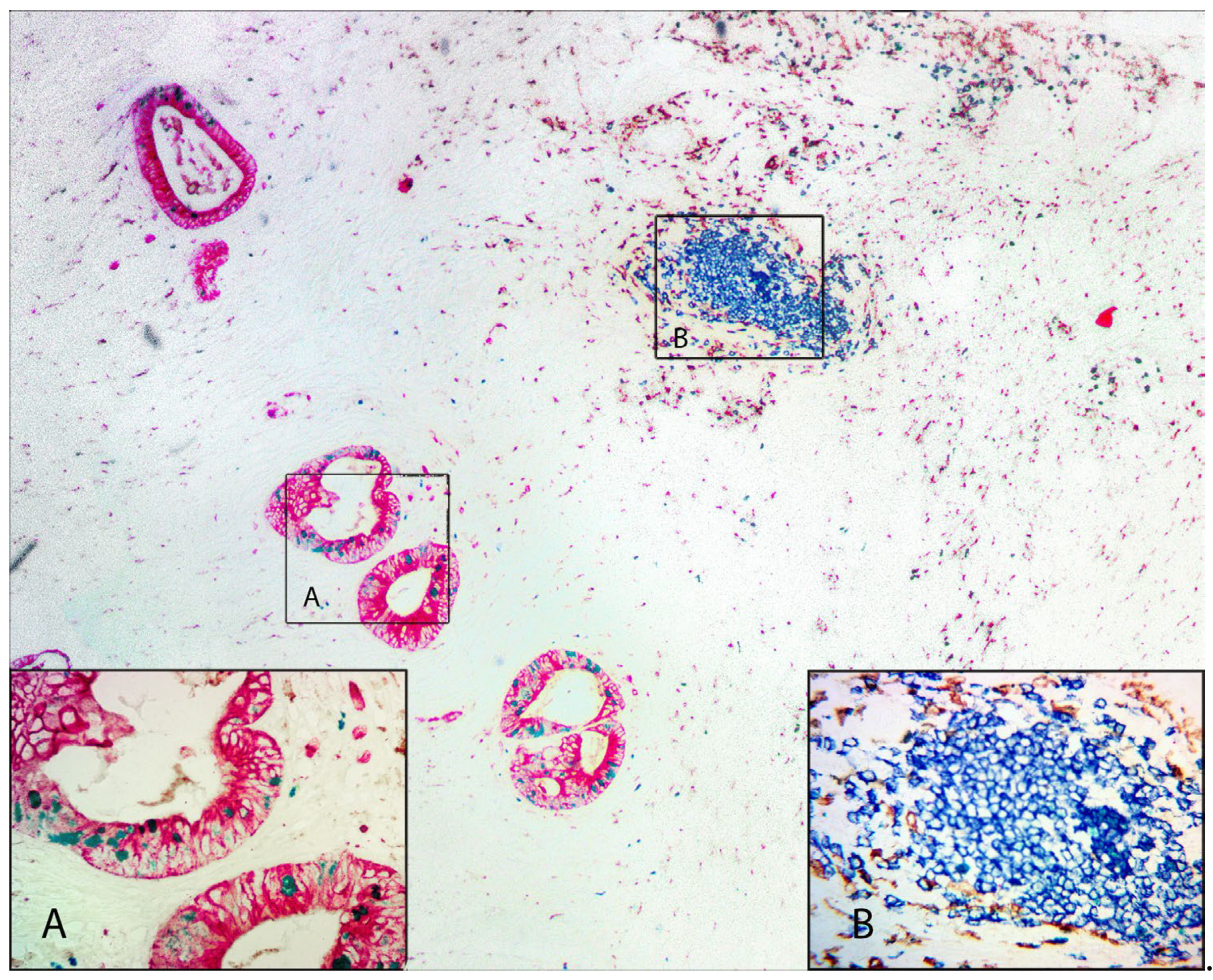

Table 1.
Antibody List for Multiplex Staining.
| Antibody | Species | Clone | Part No. |
|---|---|---|---|
| CD3 | Rabbit | SP7 | RMAB005 |
| CD4 | Rabbit | EP204 | RMAB053 |
| CD5 | Rabbit | SP19 | RMAB11 |
| CD8 | Mouse | 144B | Mob117 |
| CD20 | Mouse | L26 | Mob004 |
| CD68 | Mouse | PGM1 | Mob094 |
| CD79a | Mouse | HM57 | Mob118 |
| CD163 | Mouse | 10D6 | Mob480 |
| Ki67 | Rabbit | SP6 | RMAB004 |
| PD-1 | Mouse | EH33 | Mob573 |
| PD-L1 | Mouse | 405-9A11 | Mob572 |
| Cytokeratin HMW | Mouse | 34BE12 | Mob059 |
| Pan Cytokeratin | Mouse | AE1/AE3 | Mob190 |
Table 2.
Reagent List.
| Name | Part No. |
|---|---|
| Tissue Glue | K096 |
| Wash Buffer | K005 |
| UnoVue anti-mouse HRP | MUHRP-100 |
| UnoVue anti-rabbit HRP | RUHRP-100 |
| UnoVue anti-mouse AP | MUAP-100 |
| UnoVue anti-rabbit AP | RUAP-100 |
| Antigen Retrieval pH 9.0 | K043 |
| Primary Antibody Diluent | K004 |
Table 3.
List of Chromogens.
| Chromogen | Part Number | Enzyme System |
|---|---|---|
| PermaRed-HRP | K075 | Peroxidase |
| PermaBlue-HRP | K063 | Peroxidase |
| PermaGreen HRP | K074 | Peroxidase |
| PermaYellow-HRP | K060 | Peroxidase |
| PermaBlack-HRP | K062 | Peroxidase |
| Stable DAB | K047 | Peroxidase |
| PermaRed-AP | K049 | Alkaline Phosphatase |
Table 4.
Effects of Elution Buffer on Chromogens.
| Chromogen | Effect of Elution Buffer~~~On Chromogen staining |
|---|---|
| PermaRed-HRP | Slight loss of staining |
| PermaBlue-HRP | No effect |
| PermaGreen HRP | Moderate loss of staining |
| PermaYellow-HRP | No effect |
| PermaBlack-HRP | Slight loss of staining |
| Stable DAB | No effect |
| PermaRed-AP | No effect |
Table 5.
Optimal panels for Four-color Immunohistochemistry.
| Sequence | Primary Antibody | Chromogen | Tissue | Image |
|---|---|---|---|---|
| 1 | Pan-cytokeratin (AE1/AE3) | Yellow-HRP | ||
| 2 | Macrophage (CD163) | DAB | Colorectal | Figure 2 |
| 3 | B-Cell (CD20) | Blue-HRP | Carcinoma | |
| 4 | Proliferation marker (Ki67) | Green-HRP | ||
| 1 | Pan-cytokeratin (AE1/AE3) | AP-Red | ||
| 2 | Macrophage (CD163) | DAB | Colorectal | Figure 3 |
| 3 | B-Cell (CD20) | Blue-HRP | Carcinoma | |
| 4 | Proliferation marker (Ki67) | Green-HRP | ||
| 1 | Pan-cytokeratin (AE1/AE3) | AP-Red | ||
| 2 | Macrophage (CD68) | DAB | Colorectal | Figure 4 |
| 3 | B-Cell (CD79a) | Blue-HRP | Carcinoma | |
| 4 | Proliferation marker (Ki67) | Green-HRP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



