
Submitted:
25 April 2024
Posted:
25 April 2024
You are already at the latest version
Abstract
Systemic amyloidosis is a multisystem illness characterized by fibrillary protein deposition, which causes dysfunction in the afflicted organ systems. Its diagnosis is frequently delayed since the disease's symptoms are unpredictable and non-specific. Its two main variants are light chain (AL) amyloidosis and transthyretin-related ATTR amyloidosis, which has both a sporadic (wildtype, ATTRwt) and hereditary (mutated, ATTRv) subtype. We discuss a case of Amyloidosis emerging in the kidneys and heart as a result of chronic inflammation caused by failure to treat TB, as well as its typical radiological characteristics, clinical presentation, imaging and instrumental evaluations, and therapeutic plan. This case report serves as a good reminder to medical personnel to ensure thorough patient follow-up in order to avoid extremely unusual and late complications resulting from treatment default.
Keywords:
amyloidosis
; Congo red stain
; nephrotic syndrome
; heart failure
Introduction
Amyloidosis is a rare condition in which a protein called amyloid accumulates in organs. The amyloid accumulation can impair organ function. The heart, kidneys, liver, spleen, neurological system, and digestive tract might all be impacted.
Certain types of amyloidosis coexist with other disorders. These types of illnesses may improve with therapy for other conditions. Some kinds of amyloidosis can result in life-threatening organ failure.
Chemotherapy with potent cancer-fighting medicines is one option for treatment. Other drugs can help lower amyloid production and alleviate symptoms. Some individuals may benefit from organ or stem cell transplants [1].
Nephrotic syndrome (NS) is characterized by high proteinuria (more than 40 mg/m^2 per hour) and hypoalbuminemia (less than 30 g/L), leading to hyperlipidemia, edema, and other problems. It is produced by increased permeability of the damaged basement membrane of the renal glomerulus. It is caused by an anomaly in glomerular permeability, which can be primary with a kidney-specific illness or secondary to congenital infections, diabetes, systemic lupus erythematosus, neoplasia, or particular medication usage [2].
The current study diagnoses Amyloidosis as a result of persistent inflammation caused by a past tuberculosis infection, which is accompanied by nephrotic proteinuria and restrictive cardiomyopathy.
Case Presentation
Clinical Presentation
A 55-year-old woman with a history of pulmonary tuberculosis 20 years ago was admitted to the Casualty/Emergency ward for gradual onset of generalized oedema since 3 months and dyspnea on exertion (NYHA Grade 3) since 10 days. The patient was diagnosed with a case of pulmonary tuberculosis 20 years ago and was initiated on anti-tuberculosis therapy. She defaulted the treatment after 2 months and has been off-treatment since then. She had no other significant past medical history or Family history. She was admitted with the present conditions of oedema and breathlessness to the Intensive Care Unit.
General Examination
On physical examination the patient was well oriented to time, place and person during the examination. She had an average build, adequate nutrition, severe pallor in the lower palpebral conjunctiva, bilateral pitting pedal edema that extended up to the knees, and face puffiness. There were no signs of clubbing, cyanosis, icterus, or lymphadenopathy.
The general examination revealed a normal temperature, heart rate of 80 beats per minute, blood pressure of 118/78 mm Hg, and SpO2 of 92% on room air. breathing assessment revealed a breathing rate of 26/min and slight respiratory distress. Auscultation revealed restricted air entry in the right lower zone, as well as fine crepitations in both lower zones. S1 and S2 were found without any abnormal heart sounds during a cardiovascular assessment. During the Central Nervous System assessment, the patient was conscious and oriented to person, place, and time. The pupil reacted to light bilaterally. Power in both upper limbs was 5/5, lower limbs were 5/5, and all limbs had normal tone. The cranial nerve tests were all normal. The plantar reflex was elicited in both legs. Her per Abdominal examination was normal.
Laboratory Investigations
On admission, following investigations were performed, which revealed:

Table 1.
Blood Examination.
| Test | Observed Value | Reference Range |
|---|---|---|
| Haemoglobin | 7.4 g/dl | (12-16) |
| WBC | 8.96 kU/L | (4.0-11.0) |
| RBC | 4.19 * 106/ul | (4.5-5.5) |
| Hematocrit | 43.3% | (40-50) |
| Platelet counts | 317 kU/L | (150-400) |
| Neutrophile | 85 % | (49-74) |
| Lymphocyte | 10% | (26-46) |
| Monocyte | 03% | (2-12) |
| Eosinophil | 02% | (0-5) |
| Basophil | 00% | (0-2) |
Table 2.
Investigations for oedema.
| Test | Observed Value |
|---|---|
| Total protein | 5.8 g/dl |
| Serum Albumin | 2.0 g/dl |
| Serum Globulin | 3.8 g/dl |
| Albumin: Globulin ratio | 0.52 |
| 24 hours Urinary protein | 3.8 g |
| Urine routine microscopic examination | +++ |
Table 3.
Liver Function Test.
| Test | Observed Value | Reference Range |
|---|---|---|
| SGPT serum | 55 U/L | (10-49) |
| SGOT serum | 24 U/L | (0-34) |
| Alkaline Phosphatase Serum | 58 U/L | (45-129) |
| Total Bilirubin | 0.46 mg/dl | (0.3-1.2) |
| Direct Bilirubin | 0.10 mg/dl | (0-0.3) |
| Indirect Bilirubin | 0.36 mg/dl | - |
Table 4.
Renal Function & Electrolytes Test.
| Test | Observed Value | Reference Range |
|---|---|---|
| Blood Urea Nitrogen | 51.0 mg/dl | (15-45) |
| Serum Creatinine | 2.2 mg/dl | (0.5-1.1) |
| Serum Sodium | 118 mmol/L | (132-146) |
| Serum Potassium | 4.3 mmol/L | (3.5-5) |
| Serum Chloride | 112 mmol/L | (98-107) |
Arterial blood gas analysis: mild metabolic acidosis with respiratory compensation and type 1 respiratory failure.
Electrocardiogram (ECG) Report
The patient's ECG revealed low voltage complexes with a sinus tachycardia rhythm and a regular heart rate of 104 beats/minute. There were no substantial ST-T wave alterations or arrhythmias detected. 'There were normal PR intervals and QRS complexes on the ECG. Furthermore, there was no evidence of conduction anomalies.
Radiological Imaging
The patient underwent chest X-ray, USG Abdomen, and transthoracic 2D ECHO [3]. The findings of these scans were as follows:
Chest X-ray: The X-ray results show numerous significant findings. Old calcified granules with fibrotic alterations were present, as well as mediastinal lymphadenopathy. The Costophrenic angles were discovered to be obscured. In addition, the cardiac shadow was enlarged in shape.
An abdominal ultrasound (USG) revealed kidneys with normal echogenicity, greater size, and weak corticomedullary differentiation. There was a mild bilateral pleural effusion. The remainder of the study was normal.
Trans-Thoracic 2D ECHO
The ECHO findings highlight a range of noteworthy features. There was substantial right-sided cardiac dilation, severe global LV hypokinesia, and an ejection fraction of only 20%. In addition, there was a high pulmonary arterial pressure of 65 mm Hg, which indicated pulmonary artery hypertension and restrictive cardiomyopathy.
Diagnosis
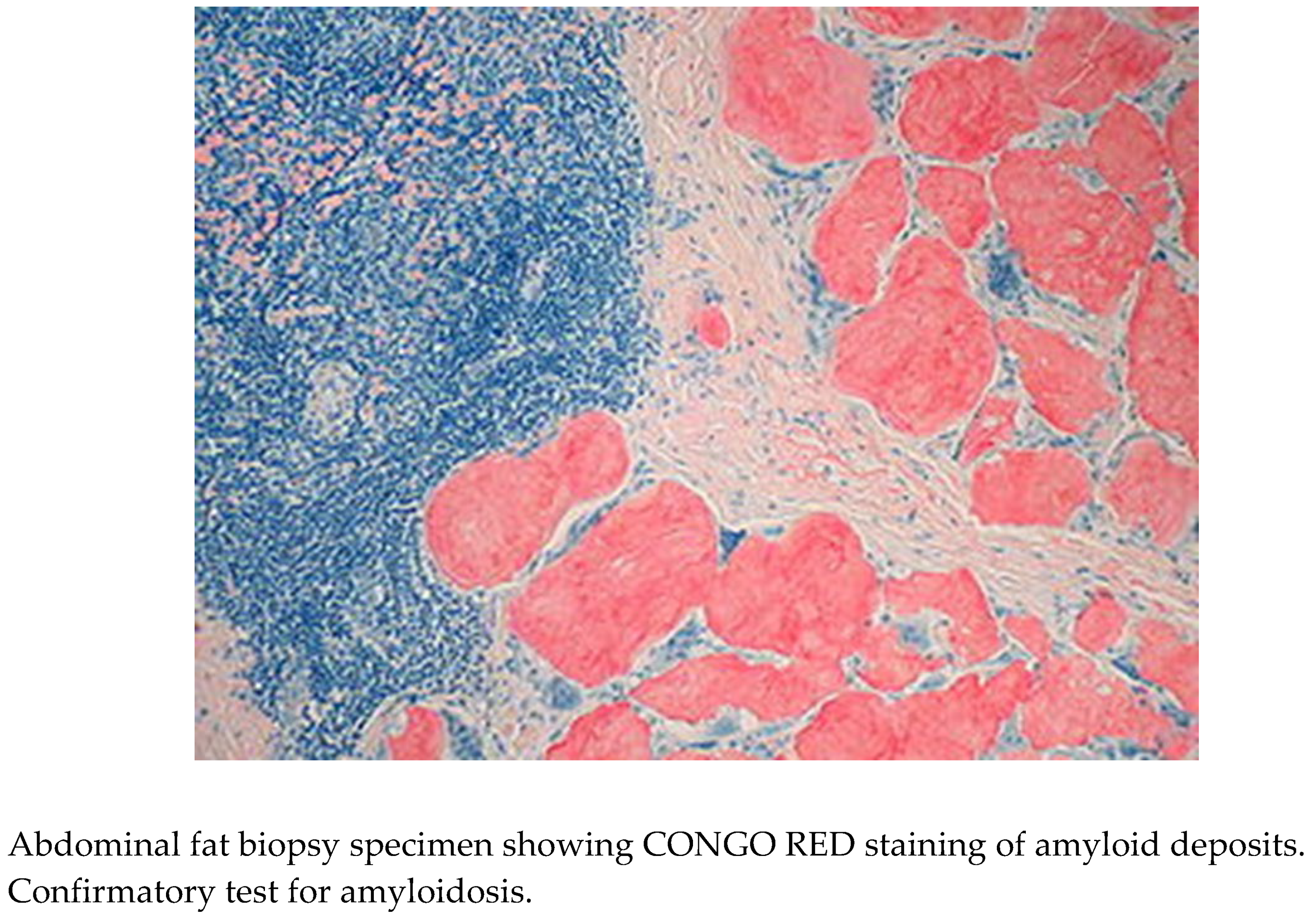
Based on the clinical presentation, clinical history, socioeconomic status, laboratory and radiographic reports, and signs and symptoms, a provisional diagnosis of amyloidosis was made. To corroborate this, a bedside abdominal fat biopsy was taken and sent for histological investigation [4].
Histopathological Examination
The specimen submitted for histological evaluation provided critical information about the nature of the identified anomalies. Congo red staining revealed amyloid plaques in the tissue, confirming the diagnosis of Amyloidosis [4].
Therapeutic Approach
Following admission to the intensive care unit, emergency management was performed to provide the patient with supportive medical therapy, and basic oxygen support was initiated via nasal cannula at a rate of 2l/min.
Given the patient's hypervolemic status and generalized edema affecting her legs and other body parts, she was also started on IV diuretics and fluid restriction to minimize fluid overload.
Due to the heightened risk of danger, in view of anemia, 1 unit of packed cell volume was slowly transfused, with a watch on reactions during her stay.
Due to significant protein loss, particularly albumin, the patient was treated with IV human Albumin (20%) for 5 days. Other treatments employed included ACE inhibitors to reduce proteinuria, calcium acetate, oral bicarbonate, and digitalis. Throughout the indoor stay in the ICU and medical ward, serum creatinine remained stable at 2.0g/dL. A degenerative condition with deposition was suspected based on a state of chronic inflammation predicted by a history of tuberculosis diagnosis and treatment a failure, high kidney size, and a restricted pattern of left ventricular dysfunction.
Discussion
Amyloidosis is a broad category of disorders distinguished by the buildup of insoluble, misfolded proteins in tissues. These proteins can accumulate locally, as seen in Alzheimer's disease, or systemically. The disease's clinical signs are generated by the harmful consequences of aggregated proteins accumulated in tissues. The typical age of diagnosis is in the seventh decade of life, and almost any organ can be damaged. The most common organs affected are the heart, peripheral nerves, kidneys, liver, and gastrointestinal tract, although cardiac, renal, or autonomic nervous system malfunction is commonly the cause of death. The disease's natural history is almost invariably progressive, but it can occasionally be reversed as aggregated protein is turned over [5].
A clinical appearance suggestive of an amyloidosis syndrome and tissue confirmation of Congo red-positive extracellular deposits exhibiting apple green birefringence under polarized light microscopy are necessary for the diagnosis of systemic amyloidosis. The easiest tissue to access for diagnosis has been fat aspiration, yet its yield varies depending on the kind of amyloid, from 15% to 80%. The pathologic examiner's abilities and the amount of fat extracted both affect how sensitive fat aspiration is. Histologically, all kinds of amyloidosis have identical amyloid deposits. As a result, amyloid typing, or the identification of the precursor protein, is a critical step in the diagnostic assessment and has consequences for prognosis, therapy, and diagnosis when performed on tissue samples containing amyloid [6]. In medical practice, amyloid is now classified according to the kind of amyloid protein. The amyloid is referred to as A (for amyloid), followed by an acronym of the protein type: AL (amyloid derived from immunoglobulin light chain), AA (amyloid A/secondary amyloidosis) and ATTR (amyloid derived from transthyretin), etc. [7]. The sensitivity and specificity of typing techniques vary. The most trustworthy technique is mass spectrometry (MS) proteomic-based analysis, which finds the universal amyloid proteins (apolipoprotein A-IV, serum amyloid P, and apolipoprotein E) along with the protein component in the deposit directly. Because MS has a sensitivity and specificity of about 100% and can identify rare or novel types, it is the gold standard for typing [6] AA amyloidosis can be associated with any type of chronic inflammation (e.g., rheumatoid arthritis, inflammatory bowel disease, subacute bacterial endocarditis, tuberculosis).
The kidneys are the first organs to be implicated in AA amyloidosis, followed by hepatomegaly, splenomegaly, and autonomic neuropathy. Excess protein in the urine, or proteinuria, is a common indicator of renal involvement. Usually severe, this illness results in the nephrotic syndrome. Less frequently, the first indication of renal disease brought on by amyloid is growing azotemia, or an excess of urea and other nitrogenous wastes in the blood. Without heart failure, nephrotic syndrome is characterized by an abnormal accumulation of fluid, such as edema in the legs and abdomen. It also involves hypercholesterolemia, or an excess of cholesterol in the blood, which may be quite serious [8]. Cardiomyopathy develops in the latter stages of the disease. Several years of an underlying inflammatory condition with chronic elevated SAA levels usually precede fibril development, while infections might cause AA deposition more quickly.
Signs and symptoms of amyloidosis include fatigue or weakness, weight loss, shortness of breath, swollen feet or legs, and eye bruises, among others [9].
The primary treatment for AA amyloidosis is to treat the underlying inflammatory or infectious condition, as well as to administer medications that decrease the inflammatory state and reduce SAA concentration and amyloid fibril deposition rate. Suppressing serum amyloid A levels to a normal range is the aim of therapy for any kind of treatment. C-reactive protein is a suitable substitute for serum amyloid A testing in cases where it is not available to evaluate the efficacy of treatment [5]. For patients with Familial Mediterranean Fever, colchicine is incredibly helpful in preventing AA amyloidosis. Colchicine can stabilize or reverse non-nephrotic renal disease in people with nephropathy at the time of colchicine initiation; however, it cannot be used in patients with nephrotic syndrome and/or uraemia. It can be used, at larger doses, to prevent disease recurrence following kidney transplantation. As a second-line treatment for Familial Mediterranean Fever and other periodic fever syndromes that are resistant to colchicine, IL-1 blocking with canakinumab, a monoclonal antibody that targets interleukin-1 beta, or anakinra, a recombinant IL-1 receptor antagonist, is advised. Anakinra has been demonstrated to be safe and efficacious for patients with AA amyloidosis of unknown cause.(6)Also, ''Eprodisate,'' a drug that inhibits serum amyloid metabolism, have all been tested and failed. There is no satisfactory treatment for amyloidosis, which is a fatal diagnosis.
Follow up: The patient was started on supportive therapy and AKT to help reduce her symptoms. The patient's relatives were informed of the poor prognosis and outcome of the treatment. The patient's follow-up indicated no more clinically significant problems.
Conclusion
Because amyloidosis is a rare disorder with nonspecific signs and symptoms, doctors often overlook it, leading in underdiagnosis [10]. Furthermore, chronic inflammation caused by underlying illnesses such as TB promotes the deposition of amyloid proteins in the heart and kidneys, leading in restrictive cardiomyopathy and proteinuria, among other symptoms. This study raises medical awareness of the challenges of properly detecting and treating Tuberculosis in India, highlighting the significance of continued medical education and the construction of referral institutions equipped with diagnostic technology and specific medicines. Overall, these measurements may improve patient care, with the objective of attaining improved long-term outcomes such as reduced organ failure and mortality [10].
Funding and Sponsorship
None of the authors are financially interested in any of the products, devices or drugs mentioned in this manuscript.
Ethical Statement
Being a case report study, there were no ethical issues and the IRB was notified about the topic and the case. Still, no formal permission was required as this was a record-based case report. Permission from the patient for the article has been acquired and ensured that their information or identity is not disclosed.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Amyloidosis - Symptoms and causes - Mayo Clinic. (2023, May 13). Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/amyloidosis/symptoms-causes/syc-20353178.
- Tapia C, Bashir K. Nephrotic Syndrome. [Updated 2023 May 29]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470444/.
- Amyloidosis & Kidney Disease. (2024, March 8). National Institute of Diabetes and Digestive and Kidney Diseases. https://www.niddk.nih.gov/health-information/kidney-disease/amyloidosis.
- Facr, W. C. S. J. M. F. (2023, April 13). Amyloidosis Causes, diagnosis, symptoms, treatment, complications. MedicineNet. https://www.medicinenet.com/amyloidosis/article.htm.
- Senecal JB, Abou-Akl R, Allevato P, Mazzetti I, Hamm C, Parikh R, et al. Amyloidosis: a case series and review of the literature. J Med Case Rep. 2023, 7, 184.
- Muchtar E, Dispenzieri A, Magen H, Grogan M, Mauermann M, McPhail ED, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021, 289, 268–292. [CrossRef] [PubMed]
- Picken, M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020, 143, 322–34. [Google Scholar] [CrossRef] [PubMed]
- Amyloidosis - Symptoms, causes, treatment | NORD. (n.d.). National Organization for Rare Disorders. https://rarediseases.org/rare-diseases/amyloidosis/.
- Website, N. (2024, April 8). Amyloidosis. nhs.uk. https://www.nhs.uk/conditions/amyloidosis/.
- Szor RS, Fernandes F, Lino AMM, Mendonça LO, Seguro FS, Feitosa VA, et al. Systemic amyloidosis journey from diagnosis to outcomes: a twelve-year real-world experience of a single center in a middle-income country. Orphanet J Rare Dis. 2022, 17, 425. [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



