
Submitted:
25 April 2024
Posted:
25 April 2024
You are already at the latest version
Abstract
The effect of H2S with high concentrations on catalytic dry reforming of methane (DRM) process has seldom been focused on previously. Herein, a thermodynamic analysis of the DRM reactions in the presence of H2S with the concentration varying in 0-20 vol% was conducted firstly. Several typical types of catalysts including MgO, NiO/MgO and LaNiO3 in different states were prepared for executing DRM at 800 C, 0.1 MPa in the feed of 20 vol% CO2 and 20 vol% CH4 balanced with N2. The catalytic performance of each catalyst for DRM process under conditions of absence and presence of H2S was compared. A promotion effect of increasing H2S concentration on both the conversions of CO2 and CH4 and the molar yields of CO and H2 was observed on all the catalysts especially the MgO and the pristine NiO/MgO. While a significant decline in catalytic activity of either the reduced NiO/MgO or the reduced LaNiO3 catalyst after adding H2S, moderate reactant conversions still sustained. The results of process analysis and catalyst structure characterization suggest that H2S participation can contribute to the increment in CO2 and CH4 conversion, and active S-adsorbed species may play the key role of catalysis in the reactions involving H2S.
Keywords:
Sour natural gas
; Dry reforming of methane
; High H2S concentration
; Promotion effect
; MgO-based catalyst
; S-adsorbed species
1. Introduction
The energy shortage and climate change are currently two major global issues of concern to people. Natural gas is one of the most wildly used clean energy sources in the world, and its production output of ca. 4×1012 m3 was reached in the year of 2022. Along with continuous extraction of natural gas, unfortunately, large quantities of sour natural gas containing high percentage of both CO2 and H2S come out of the ground. The data extracted from some high-sulfur gas fields given in Table 1 provide the typical cases of CO2 and H2S content in natural gas worldwide [1].
Taking advantage of available catalytic dry reforming of methane (DRM) technology, the natural gas industry realm can not only diminish CO2 emission, but also produce high value-added organic chemicals via synthesis gas [2,3,4]. However, the coexistence of H2S in feedstock makes the application of catalytic DRM technology exceedingly challenging, since H2S is generally a notorious poison of catalysts.
So far, researchers have made tremendous efforts to address the deactivation of catalysts caused by H2S, with the aim of attaining high efficient catalysts that can tolerate H2S. For example, Appari et al. [5] developed a detailed kinetic model for biogas steam reforming in the absence and presence of H2S having 20-108 ppm over a Ni-based catalyst. As stated, the model predicted a high sulfur coverage near the reactor inlet during the initial stages of poisoning. When the reaction proceeded further, the sulfur coverage increased towards the reactor outlet. Low-temperature operation could render the catalyst completely deactivated, while high-temperature operation could mitigate sulfur adsorption and slow down the catalyst deactivation. Gaillard et al. [6] investigated the effect of trace (50 ppm) H2S on DRM process over Mo- and Ni-based catalysts and dissected the mechanism of catalyst deactivation. They found that carbon accumulation was the main cause of the deactivation of Mo-based catalysts, whereas H2S poisoning should be responsible chiefly for the activity loss of Ni-based catalysts. The effect of H2S on dry reforming of biogas was studied by Chein et al. [7]. The experimental results showed that the presence of H2S led to a severe deactivation of Ni-based catalysts. The bimetallic Pt-Ni catalysts outperformed the monometallic Ni-based catalysts, but deactivation still emerged. Akansu et al. [8] explored the effect of Mn doping on the activity and the H2S tolerance of Ni-based catalysts in dry reforming of biogas. As reported, both the monometallic Ni and the bimetallic Ni-Mn catalysts exhibited stable activity in the absence of H2S. In the presence of 2 ppm H2S, the activities of the two types of Ni-based catalysts decreased gradually with reacting time, and Mn doping did not improve the resistance of the catalysts to H2S. More recently, Gao et al. [9] inquired into the single and synergistic effects of H2S and NH3 on Ni-based catalysts in biogas dry reforming. They stated that trace amount of H2S caused severe catalyst deactivation, in contrast to NH3, which had a slight inhibitory effect. In the condition of H2S and NH3 coexistence, the catalyst activity decreased more rapidly than when a single component was present.
Meanwhile, there are occasionally research reports that addition of H2S can promote catalytic DRM process. Fidalgo et al. [10] investigated the effect of a small quantity (0.5-1.0 vol%) of H2S present in the feedstock on DRM over activated carbon and Ni based catalysts. As disclosed, for the carbon catalyst, the conversions of both CH4 and CO2 were noticeably increased by the addition of H2S to the reactants. For the Ni-based catalysts, on the contrary, significant deactivation was observed after adding H2S. The influence of trace amount of H2S on commercial methane reforming catalysts during hydrogen production at different temperatures (650 - 850 °C) was plumbed by Chattanathan et al. [11]. They noticed that even with the addition of 0.5 mol% H2S, the conversions of CH4 and CO2 were significantly dropped from 67% and 87% to 19% and 22%, respectively. However, there was a slight increase in the conversions of both CH4 and CO2 when the H2S concentration was raised from 1.0 mol% to 1.5 mol%. Recently, Jin et al. exerted an in situ observation of the promoting effect of H2S on the formation of efficient MoS2 catalyst for CH4/CO2 reforming, and found that introducing H2S further improved the catalytic reactivity by >10% [12].
Although the relevant research was very limited, the findings of promotion effect of H2S on the catalytic process of dry reforming is undoubtedly exciting. H2S is ubiquitous either as primary impurity or as a hydrogenolysis product of organic sulfurs in fossil fuels and biomass processing. To avoid affecting catalyst performance, removing H2S from feedstock as much as possible has long been an arduous task. On the other hand, with the worldwide emphasis on hydrogen energy, it has been realized that H2S is also a valuable source of H2. Utilizing H2S to produce H2 rather than just acquiring sulfur through Claus processes has been put on the agenda. However, rare attention has been paid to the possible reactions occurring directly among H2S, CO2 and CH4 of sour natural gas, which can in effect generate beneficial products including H2.
Herein, we will focus on the catalytic DRM reactions in the presence of H2S with high concentration and compare the catalyst performance in the condition of with and without H2S. Several typical DRM catalyst materials have been elaborately investigated in the reaction system including MgO, NiO/MgO, and LaNiO3 of different chemical states. A promotion effect arising from the reactions of H2S with CO2 and CH4 has been discovered on all the catalysts, which can bring the conversions of CO2 and CH4 and yields of CO and H2 in the presence of H2S to reaching a meaningful level of production in industry. A relatively long-term stability of the reduced NiO/MgO catalyst has been achieved with little carbon deposition and sulfur poisoning. The possible promotion mechanisms of high concentration H2S have been discussed on the basis of a thermodynamic analysis and the characterization of the catalyst structures before and after the reactions. S-adsorbed species formed by dissociation of H2S on the surface of catalysts has been suggested to be crucial for initiating the reactions among H2S, CO2 and CH4. Moreover, the promoting effect of H2S can even occur on the MgO support, manifesting its universality. To the best of our knowledge, there seems to be no report up to now to reveal the promotion effect of H2S in the process of catalytic dry reforming. The results and deduction of the present study may be conducive to guide rational processing and high-value utilization of sour natural gas.
2. Materials and Methods
2.1. Catalyst Preparation
(1) MgO The powdery MgO of analytical purity purchased from Sinopharm Chemical Reagent Co. Ltd in Shanghai was put into a Muffle furnace and heated at a rate of 2 ℃/min to 800 ℃ and maintained for 8 h. After the completion of calcination, the sample of MgO was naturally cooled to room temperature. Prior to be tested as a catalyst, the MgO sample needed to be cast in a mold, and the pellets formed were broken and sieved into granules in size of 60-80 mesh.
(2) NiO/MgO The sample of NiO/MgO was prepared by incipient wetness impregnation method, in which NiO loading was set as 20 wt%. The precursor of NiO was Ni(NO3)2 (analytical purity, Sinopharm Chemical Reagent Co. Ltd in Shanghai) solution and the powdery MgO obtained above was used as the support. Being aged for 4 h, the impregnated sample was dried in an oven at 110 ℃ for 6 h, and then calcined in a Muffle furnace at a rate of 2 ℃/min to 800 ℃ and maintained for 4 h. After cooling down, the sample was also granulated. The pristine NiO/MgO catalyst having the size of 60-80 mesh was thus obtained to be ready for use. The real NiO loading of NiO/MgO prepared was detected to be 20.67 wt% (16.24 wt% Ni) by means of an inductively coupled plasma optical emission spectrometry (ICP-OES) using a Perkin Elmer Optima 4300 dual view model.
For the preparation of reduced NiO/MgO catalyst, the sample of pristine NiO/MgO granules was put into a fixed bed reactor and heated in a flow of 10 vol% H2/N2 mixture at a rate of 2 ℃/min to 600 ℃ and kept for 4 h. The gas hourly space velocity (GHSV) during reduction was set around 10000 h-1. After that, the sample was cooled down and purged using N2 for 2 h before being fetched out from the reactor.
(3) LaNiO3 A citrate sol-gel method was adopted for the synthesis of LaNiO3 according to the procedure descried in the reference [13]. Briefly, nickel nitrate (Ni(NO3)2), lanthanum nitrate (La(NO3)3, analytical purity, Sinopharm Chemical Reagent Co. Ltd in Shanghai) and citric acid were used as the precursors and their molar ratio was fitted to be 1:1:3. The solutions of Ni(NO3)2 and La(NO3)3 formed separately were mixed firstly in a beaker, which was placed in a water bath at 80 ℃. Along with continuously stirring the mixture, the citric acid solution was added into the beaker drop by drop. A green transparent colloid was formed afterwards, which gradually became a sticky gel under the constant stirring. The gel was dried in an oven at 120 ℃ for 24 h, and the dried gel after cooling was then crushed into pieces. For calcining the dried gel in the Muffle furnace the temperature was programed to be 500 ℃ at a rate of 3 ℃/min for 3 h, and then to 800 ℃ at a rate of 5 ℃/min for 4 h. After cooling, the sample of LaNiO3 was also molded and sieved. The reduction of LaNiO3 was fulfilled following the same steps for NiO/MgO reduction recorded above.
2.2. Catalytic DRM Test and Reaction Gas Composition Analysis
The catalytic performance test of a variety of catalysts used for DRM process was executed in a fix-bed tubular quartz reactor under atmospheric pressure in the condition of either absence or presence of H2S. The flow chart of the apparatus for test was provided in Figure S1 of Supporting Information. In a typical procedure for reaction test, 0.6 mL catalyst granules (60-80 mesh) were filled in the middle of the reactor. A gas flow of N2 with high purity was purged through the reactor until the temperature of the reactor reached a set value. After that, a mixture gas composed of 20 vol% CO2 and 20 vol% CH4 balanced with N2 at a total flow 100 mL/min was introduced to attain an assigned GHSV of 10000 h-1.
The compositions of the mixture gas fore-and-aft the reactor were monitored constantly, including CO2, CH4, H2S, CO, COS, CS2, H2O, H2 and N2. Two gas chromatographic (GC) instruments both with thermal conductive detectors (Clause 680, PerkinElmer Instruments, USA) were used for routine composition analysis. GC-1 was mounted in series with PQ and 5A columns using H2 as carrier gas to separate in succession CO2, H2O, H2S, COS, N2, CH4, CO and CS2 in case all existing. A schematic diagram of different components flowing through two GC columns separately and the corresponding switching time of six-way sampling valve is illustrated in Figure S2 for the convenience of understanding the unique GC analysis method. The GC peak order of all the components is displayed in Figure S3 for reference. Another GC (GC-2) was equipped with a 5A column and adopted N2 as carrier gas for H2 detection. An additional GC with flame photometric detector was ready for SO2 detection, which was fixed with a PQs column using H2 as the carrier gas. The elemental sulfur when formed could be captured downstream of the reactor onto the wall of glass vessel immersed in an ice-bath. Liquid water could be also removed by this glass vessel. The tail gas was discharged in the end after passing through secondary ethanolamine washing bottles to absorb the toxic and harmful substances.
It needs to be pointed out that N2 as an inert gas did not take part in any reactions, and its flow rate entering and leaving the reactor was constant. What’s more, the composition of N2 could be determined through GC analysis. Therefore, N2 was used as the standard substance for quantifying the concentration and flow rate of other components contained in the reaction system. The content of various substances relative to N2 was further utilized for calculating mass balance of C, H, O, and S before and after the reaction. In the present study, good mass balances for C and S with both less than 5% deviation are acquired because of few elemental carbon and sulfur formation during reaction and meanwhile smooth discharge of other carbon- and sulfur-contained products from the reactor outlet. For the H and O, however, probable due to the condensation of H2O in the pipeline, there is a mass loss gap before and after the reaction. Nevertheless, this does not hinder the quantification and calculation of changes in important components other than H2O for catalytic performance evaluation.
The conversions of CO2, CH4 and H2S, and the molar yields of CO, H2, COS, H2O and CS2 are defined respectively as follows,
where Fi,in and Fi,out refer to the molar flow rate of the component of i at inlet and outlet respectively of the DRM reactor. Because the product of COS should come from the equimolar conversion of H2S and CO2, and the molar flow rate of H2S is smaller than that of CO2 in the feedstock throughout, the COS molar yield is educed on the basis of H2S feed flow rate. So does the CS2 molar yield. Since the collection and quantification of H2O generated with reaction time on stream is unachievable, the molar flow rate of H2O is derived from the mass balance of oxygen element between the inlet and outlet flow.
2.3. Catalyst Characterization
(1) N2 cryo-physisorption measurement (BET) The textural measurement of the sample which had in advance been degassed in vacuum at 200 °C for 6 h was performed at -196 °C on a Micromeretics ASAP 2020 apparatus using N2 as adsorbate. Specific surface area of the sample was calculated with Brunauer-Emmet-Teller (BET) equation, and pore volume between 1.7 and 300.0 nm in diameter was determined from the N2 desorption isotherm using the Barrett-Joyner-Halenda (BJH) method.
(2) X-Ray diffraction (XRD) The crystal phases of the catalysts were detected by means of XRD using a Rigaku Smartlab 9 X-ray diffractometer with an operating voltage of 40 kV and an operating current of 40 mA. The X-ray light source was a Cu target with a scanning range of 10°-90° and a scanning speed of 10°/min. The diffraction peaks were identified by comparing the results of corresponding materials with the standard JCPDF cards.
(3) X-ray photoelectron spectroscopy (XPS) XPS was used to analyze the elemental valence information on the surface of the catalysts, which was carried out using an Escalab 250Xi photoelectron spectrometer from Thermo Scientific, U.K. The main parameters were: monochromatic, micro-focused Al-K-Alpha radiant light source (1486.6 eV), and analyzer flux energy of 20 eV. During the measurement, a full-spectrum scan of the sample was performed first to collect information, followed by a fine-spectrum scan for the target element. The obtained data were analyzed by Thermo Avantage software, and the binding energy was corrected using the C 1s peak at 284.8 eV, and the background was subtracted by Shirley's method.
3. Results and discussion
3.1. Thermodynamic Analysis of the Effect of H2S Concentration on DRM Reactions
Thermodynamic equilibrium analysis of the reactions involving CO2, CH4 and H2S as feedstock was conducted firstly by Aspen Plus simulation to ascertain the theoretical basis for the participation of H2S in reactions. Considering that the reaction system involves polar gases reacting at high temperature and low pressure, the NRTL property equation was chosen and a Gibbs isobaric and isothermal reactor was used. The reaction conditions for simulation were assumed as 20% CO2, 20% CH4 and H2S of 0-15% all in volume balanced with N2 at the temperature of 400-1000 °C and pressure of 0.1 MPa in a Gibbs isobaric and isothermal reactor. Nine possible products including H2, H2O, C, C2H6, CO, COS, CS2, S2, and SO2 all in gas state excepting solid C were supposed to form in the reaction system. The results are shown in Figure 1, where (a) and (b) display the changes in the equilibrium conversions of three reactants and the molar fractions of the products with reaction temperature, respectively. Because the molar fractions of C, C2H6 and SO2 are all below the order of 10-6, these three products will not be taken into account thereafter.
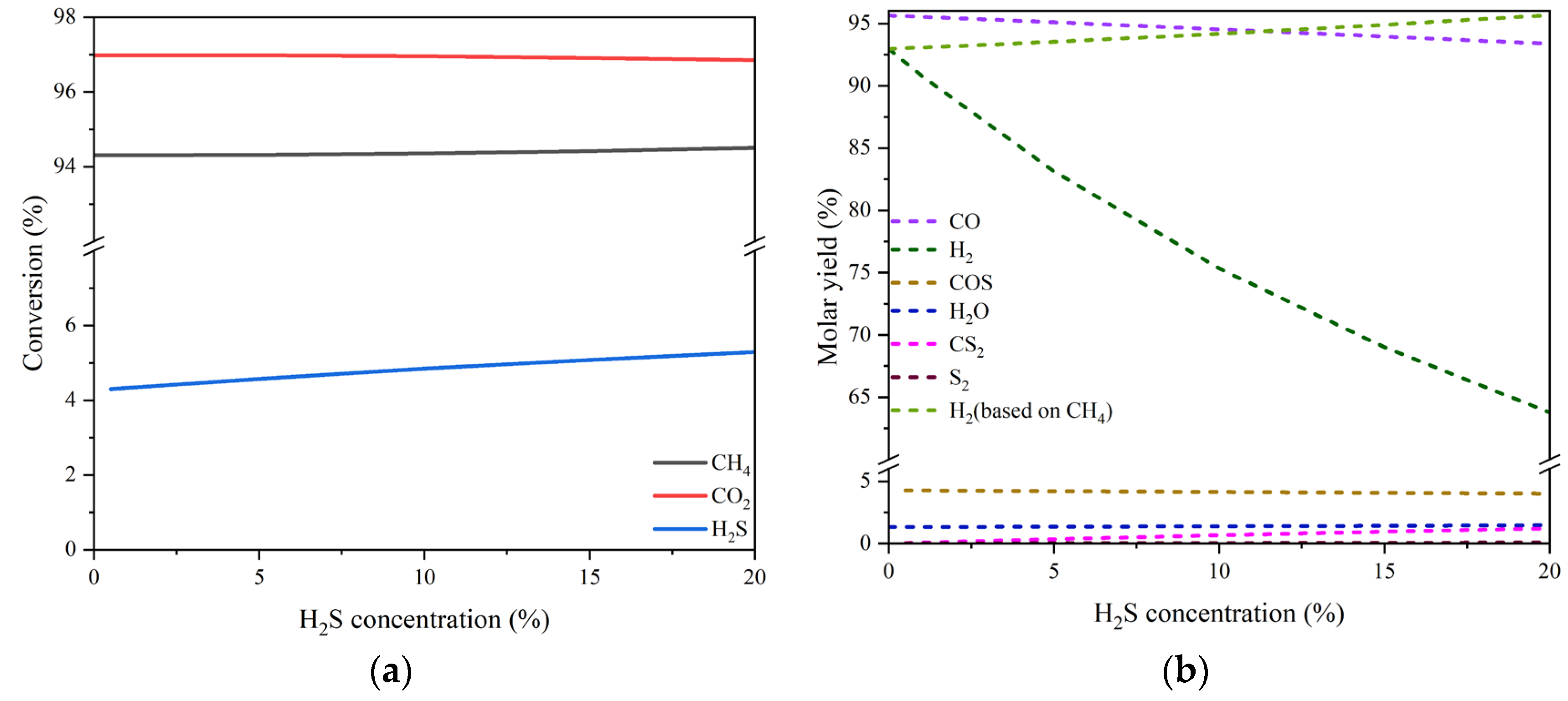
As can be seen, there is a slight fluctuation for the equilibrium conversion of H2S, which varies between 4.8% - 8.7% in the temperature range of 400-1000 °C, although that of either CO2 or CH4 increases significantly from ca. 5% to nearly 100% with temperature rising. Meanwhile, the molar fractions of both CO and H2 overwhelm other products in the whole temperature range especially when higher than 800 °C. The byproduct COS and H2O each accounts for a small proportion. Their individual maximum fraction does not exceed 1.1% and 1.9%, appearing around 400 °C and 600 °C, respectively. The quantity of CS2 and S2 is even more trivial.
The equilibrium conversions of the three reactants and the molar yields of the products changing with H2S concentration (0-20 vol%) at the temperature of 800 °C are drawn in Figs. 2 (a) and (b), respectively. There is a marginal growth of the H2S conversion with the increase of H2S concentration. Nevertheless, for the conversions of both CO2 and CH4, almost no impacts of H2S concentration can be perceived. On the other hand, the molar yield of CO, one of the dominant products, declines from 95.65% to 93.38%, and the molar yield of H2, the other dominant product, drops sharply from 92.97% to 63.78%. It should be noted, however, that the yield of H2 could increase linearly from 92.97% to 95.67% as presented in Figure 2(b) if it is calculated on the basis of feed CH4 only just like when there is no H2S present. This suggests that there would be a potential promotion effect of H2S concentration on H2 production. With respect to the other products, the molar yields of COS and H2O are scarcely affected by the wave of H2S concentration with their values maintained separately around 4.25% and 2.33%. The byproduct CS2 seems to have an obvious increase, but in fact, its maximal molar yield is still very low. For S2, the effect of H2S concentration is not worth mentioning either. The results of thermodynamic analysis tell that high concentrations of H2S couldn’t inhibit the conversions of both CO2 and CH4; instead, they are able to increase the production of H2.
Figure 2.
Simulation results of the equilibrium conversion of reactant (a) and the molar yield of product changing with H2S concentration (b). Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and H2S (0-20%) in volume balanced with N2, at the temperature of 800 °C and pressure of 0.1 MPa, in a Gibbs isobaric and isothermal reactor. Note: the starting point of H2S concentration for the curve of S-containing substance is greater than zero. .
Figure 2.
Simulation results of the equilibrium conversion of reactant (a) and the molar yield of product changing with H2S concentration (b). Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and H2S (0-20%) in volume balanced with N2, at the temperature of 800 °C and pressure of 0.1 MPa, in a Gibbs isobaric and isothermal reactor. Note: the starting point of H2S concentration for the curve of S-containing substance is greater than zero. .
3.2. Effect of H2S Concentration on DRM over Different Catalysts
(1) MgO catalyst
Different catalytic materials were tested to judge whether the effect of H2S concentration on DRM is distinguishable from one another. MgO, an alkaline earth oxide, generally has a good adsorption ability towards acidic gases such as H2S and CO2. Furthermore, MgO can be used as a support to disperse various active phases, which can not only promote activation of acidic reactants but also prevent coke formation from hydrocarbon cracking. As Hu et al. claimed in their early research [14], Ni/MgO solid solution catalysts could display excellent dry reforming performance without coke formation and metal sintering. The latest work reported by Feng et al. [15] believed that the addition of MgO in Ni/Al2O3 catalysts could improve the alkalinity of the support and enhance the metal-support interaction, both of which are beneficial to the reaction of DRM.
In view of the MgO properties, we firstly examined the performance of MgO during DRM reactions in the absence and in the presence of H2S with different concentration. Figure 3(a) shows the conversions of CO2 and CH4 and the molar yields of CO, H2 and H2O in the temperature range of 400-800 °C in the absence of H2S. The reactant mixture gas is composed of 20% CO2 and 20% CH4 in volume balanced with N2 at the pressure of 0.1 MPa and the GHSV of 10000 h-1. It can be inspected that the conversions of CO2 and CH4 are merely 4.09% and 0.92%, respectively, even at the temperature as high as 800 °C, indicating the poor catalytic performance of MgO for DRM. The main products are H2O and CO, despite the fact that their productions are very low. In the meantime, the formation of H2 can hardly be observed in the whole temperature range.
For the DRM proceeding in the presence of 15 vol% H2S in addition to 20 vol% CO2 and 20 vol% CH4, the variations in the conversions of CO2 and CH4 and H2S as well are displayed in Figure 3(b). Surprisingly, the conversions of both CO2 and CH4 increase significantly in comparison with those obtained at the same temperatures but in the absence of H2S. At 800 °C, the conversion of CO2 can reach 36.30% and that of CH4 is 19.36%, which are about 9 and 21 times the corresponding conversions under the reaction conditions without H2S, respectively. The comparison between the reactant conversions under the conditions without and with H2S discloses apparently the promotion effect of H2S addition. Unlike the monotonic increases in CO2 and CH4 conversions, the conversion of H2S exhibits a change like volcanic curve with the rise of reaction temperature. The summit of H2S conversion is 14.30%, appearing at 600 °C.
Figure 3.
On the MgO catalyst, (a) conversions of CO2 and CH4 and molar yields of CO, H2 and H2O changing with temperature in the absence of H2S, (b) conversions of CO2, CH4 and H2S changing with temperature in the presence of 15 vol% H2S, (c) molar yields of CO, H2, H2O and COS changing with temperature in the presence of 15 vol% H2S, (d) conversions of CO2, CH4 and H2S changing with H2S concentration at 800 °C. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and H2S (0 - 15%) in volume balanced with N2, at the temperature of 400 - 800 °C, the pressure of 0.1 MPa and the gas hourly space velocity of 10000 h-1. The dotted line in (a) refers to the molar yield of product.
Figure 3.
On the MgO catalyst, (a) conversions of CO2 and CH4 and molar yields of CO, H2 and H2O changing with temperature in the absence of H2S, (b) conversions of CO2, CH4 and H2S changing with temperature in the presence of 15 vol% H2S, (c) molar yields of CO, H2, H2O and COS changing with temperature in the presence of 15 vol% H2S, (d) conversions of CO2, CH4 and H2S changing with H2S concentration at 800 °C. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and H2S (0 - 15%) in volume balanced with N2, at the temperature of 400 - 800 °C, the pressure of 0.1 MPa and the gas hourly space velocity of 10000 h-1. The dotted line in (a) refers to the molar yield of product.
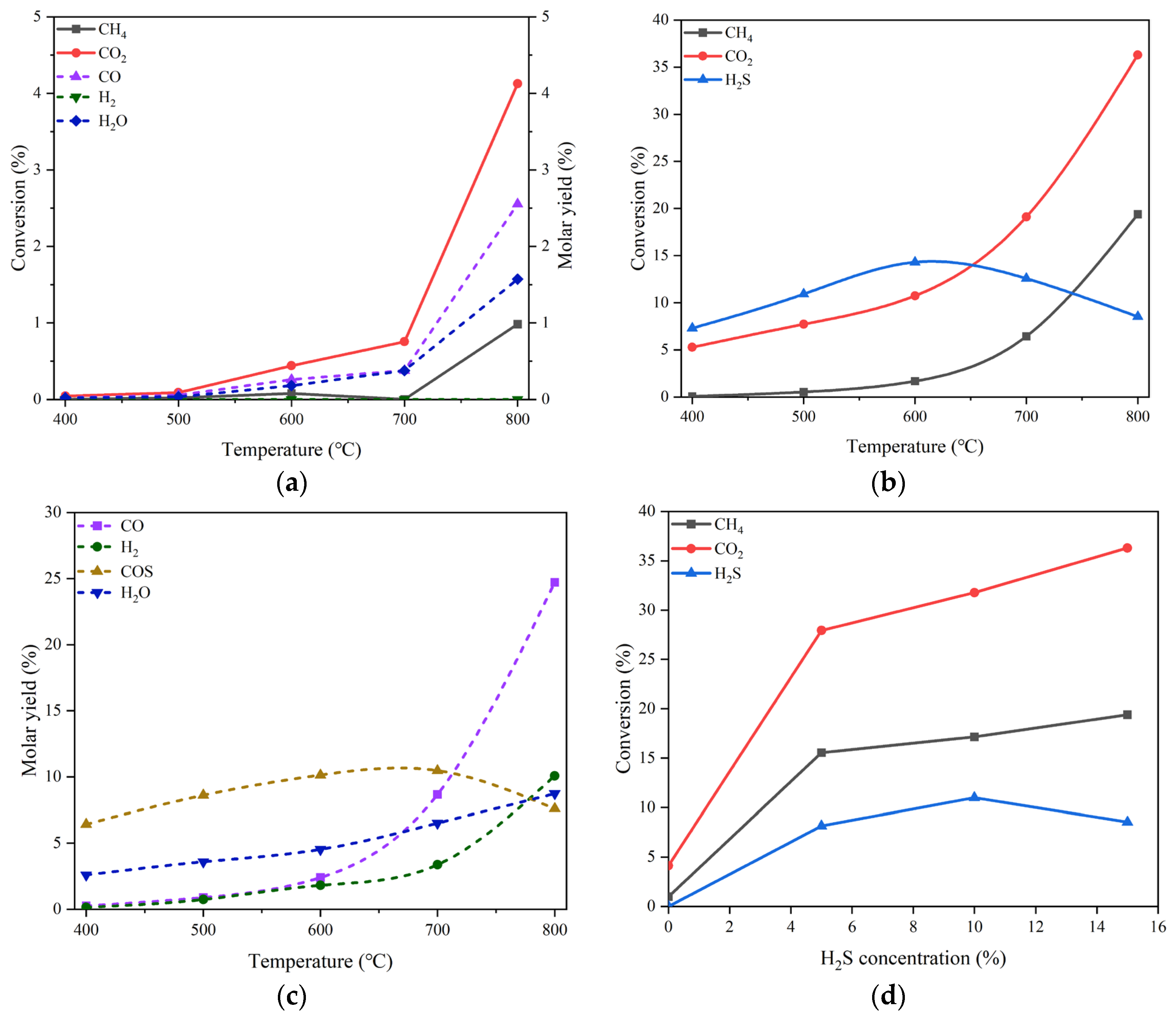
The effect of H2S addition can be further evidenced by the boost in product molar yield. As illustrated in Figure 3(c), the molar yields of the products such as CO, H2, and H2O at 800 °C are all growing remarkably as compared with the counterpart results shown in Figure 3(a). In particular, the production of H2 can achieve the molar yield of 10.07%, being second only to that of CO; the latter is 24.7%. Additionally, a new product, COS was observed, which has a maximal molar yield of 10.47% at 700 °C. The influence of H2S concentration on the conversion of each reactant can be ascertained from the curve trend shown in Figure 3(d). For CO2 and CH4, there are unequivocally positive effects of H2S concentration on their conversions. Regarding H2S transformation, there appears to be an optimal concentration of about 10 vol% H2S to achieve the largest conversion of 10.99%.
(2) NiO/MgO catalyst in pristine and reduced form
As mentioned early, the catalysts based on Ni/MgO are frequently used in the processes of DRM due to their outstanding catalytic performances. Herein, we prepared the NiO/MgO catalyst for DRM and used it in either pristine or reduced form to explore the sensitivity of the chemical state of active metal on the H2S-induced effect. The catalytic performances of the pristine NiO/MgO catalyst in the absence of H2S and in the presence of 15 vol% H2S at different temperatures are depicted in Figure 4(a, b). By comparing between the results of NiO/MgO in Figure 4(a, b) and those of MgO in Figure 3(a, b), one can be aware of that the variation trends with temperature rising of either reactant conversions or product molar yields are very similar for the two catalyst types, whatever in the absence or in the presence of H2S. Nonetheless, on the NiO/MgO catalyst, there are perceptible increments of both reactant conversions and product molar yields. This proves the superior behaviors of NiO/MgO to the simple MgO catalyst. Furthermore, at 800 °C, the conversion of CO2 and the molar yield of CO are 42.10% and 33.10% respectively on the NiO/MgO catalyst in the presence of H2S, which are not only higher than the results recorded in the same conditions on the simple MgO catalyst but also much higher than the corresponding values gained in the absence of H2S. Obviously, the promotion effect of H2S has also been confirmed on the pristine NiO/MgO catalyst.
Figure 4.
(a) Conversion of CO2 and CH4 and molar yield of CO, H2 and H2O changing with temperature in the absence of H2S on the pristine NiO/MgO catalyst, (b) conversion of CO2, CH4 and H2S and molar yield of CO, H2, H2O and COS changing with temperature in the presence of 15 vol% H2S on the pristine NiO/MgO catalyst, (c) conversion of CO2 and CH4 and molar yield of CO, H2 and H2O changing with temperature in the absence of H2S on the reduced NiO/MgO catalyst, (d) conversion of CO2, CH4 and H2S and molar yield of CO, H2, H2O and COS changing with temperature in the presence of 15 vol% H2S on the reduced NiO/MgO catalyst, (e) comparison in conversion of CO2, CH4 and H2S changing with H2S concentration at 800 °C between the pristine and the reduced NiO/MgO catalysts, (f) comparison in molar yield of CO, H2, H2O and COS changing with H2S concentration at 800 °C between the pristine and the reduced NiO/MgO catalysts. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and 0-15% H2S in volume balanced with N2, at the temperature of 400-800 °C, the pressure of 0.1 MPa and the GHSV of 10000 h-1. The dotted line in (a-d) refers to the molar yield of product.
Figure 4.
(a) Conversion of CO2 and CH4 and molar yield of CO, H2 and H2O changing with temperature in the absence of H2S on the pristine NiO/MgO catalyst, (b) conversion of CO2, CH4 and H2S and molar yield of CO, H2, H2O and COS changing with temperature in the presence of 15 vol% H2S on the pristine NiO/MgO catalyst, (c) conversion of CO2 and CH4 and molar yield of CO, H2 and H2O changing with temperature in the absence of H2S on the reduced NiO/MgO catalyst, (d) conversion of CO2, CH4 and H2S and molar yield of CO, H2, H2O and COS changing with temperature in the presence of 15 vol% H2S on the reduced NiO/MgO catalyst, (e) comparison in conversion of CO2, CH4 and H2S changing with H2S concentration at 800 °C between the pristine and the reduced NiO/MgO catalysts, (f) comparison in molar yield of CO, H2, H2O and COS changing with H2S concentration at 800 °C between the pristine and the reduced NiO/MgO catalysts. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and 0-15% H2S in volume balanced with N2, at the temperature of 400-800 °C, the pressure of 0.1 MPa and the GHSV of 10000 h-1. The dotted line in (a-d) refers to the molar yield of product.
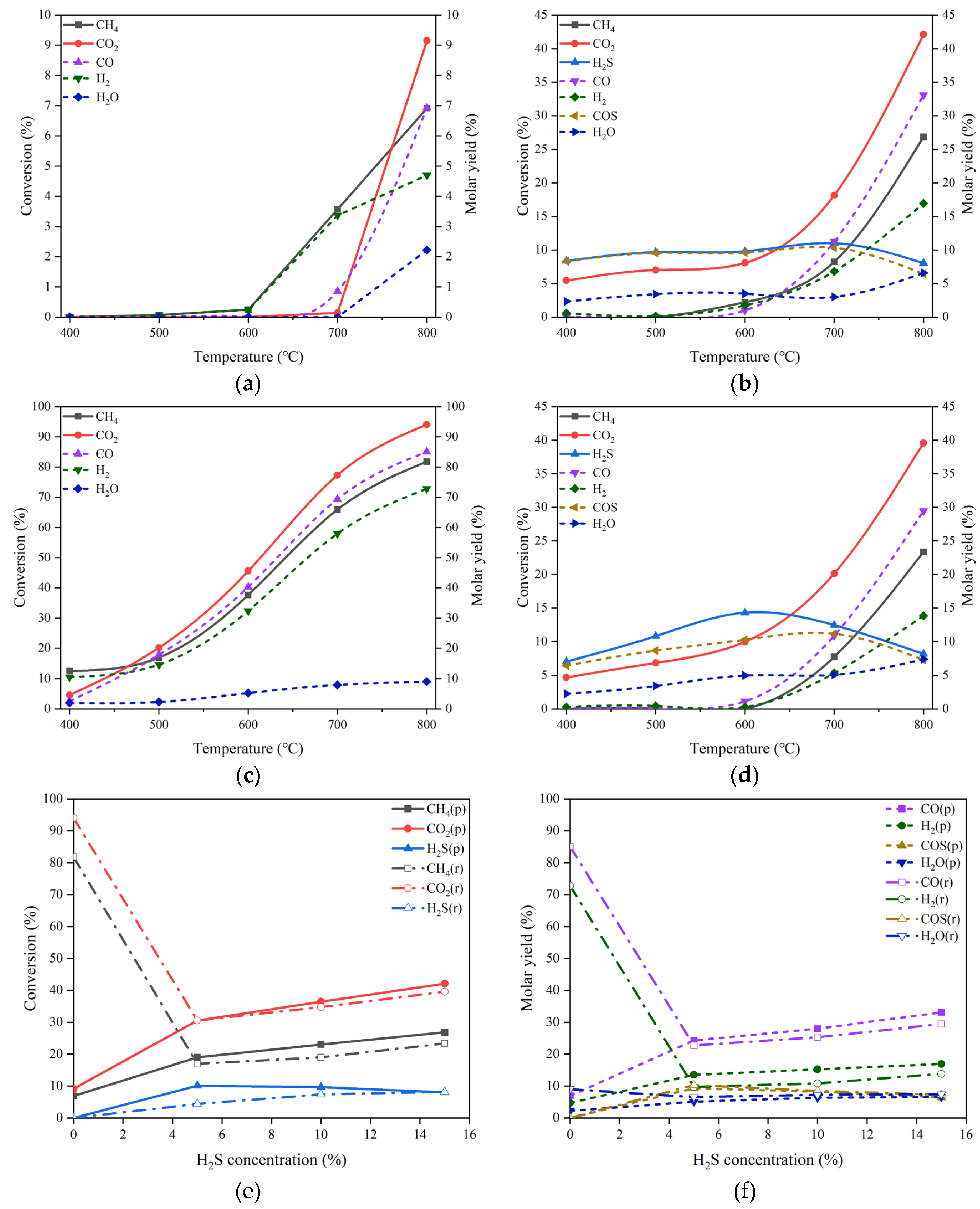
For the reduced NiO/MgO catalyst, the catalytic performances in the reaction conditions without and with 15 vol% H2S at different temperatures are demonstrated in Figs. 4 (c) and (d), respectively. It is interesting that the conversions of both CO2 and CH4 and the molar yields of both CO and H2 are improved greatly on the catalyst of NiO/MgO after reduction as shown in Figure 4(c) by comparing with the corresponding results on the pristine catalyst in Figure 3(a). At 800 °C, the conversion of CO2 and the molar yield of CO are 94.05% and 85.04% respectively, which are comparable to the data reported in the literatures [14,16]. The results manifest the importance of the chemically reduced state of the active metal phase in the NiO/MgO catalyst for the reaction of DRM. In the condition of 15 vol% H2S presence, the changes of reactant conversions and product molar yields with the temperature in Figure 4(d) are virtually identical to those presented on the pristine NiO/MgO catalyst in Figure 4(b). However, in comparison with the results gained in the absence of H2S in Figure 4(c), noticeable drops in the conversions of CO2 and CH4 and the molar yields of CO and H2 can be found, suggesting an inhibition effect of H2S on the catalytic activity of the reduced NiO/MgO catalyst for DRM.
The differentiation in the H2S concentration influence on the catalytic performances between the pristine and the reduced NiO/MgO catalysts can be surveyed in Figure 4(e, f). On the reduced NiO/MgO catalyst, as displayed in Figure 4(e), the trends of the reactant conversions with H2S concentration are basically the same for the two catalyst forms, excepting that there are sharp declines in the CO2 and CH4 conversions when the reaction atmosphere is altered from none of H2S to containing H2S. Further increasing H2S concentration can benefit the conversions of CO2 and CH4 over both of the NiO/MgO catalysts, resembling the circumstance occurring over the MgO catalyst. The changes of the product molar yields with H2S concentration shown in Figure 4(f) are consistent with the paces of reactant conversions. Additionally, the pristine NiO/MgO catalyst performs to some extent better than the reduced one in the reaction system containing H2S, inferring that the catalytic performance may be sensitive to subtle variation in the surface of catalyst.
(3) LaNiO3 derived catalyst
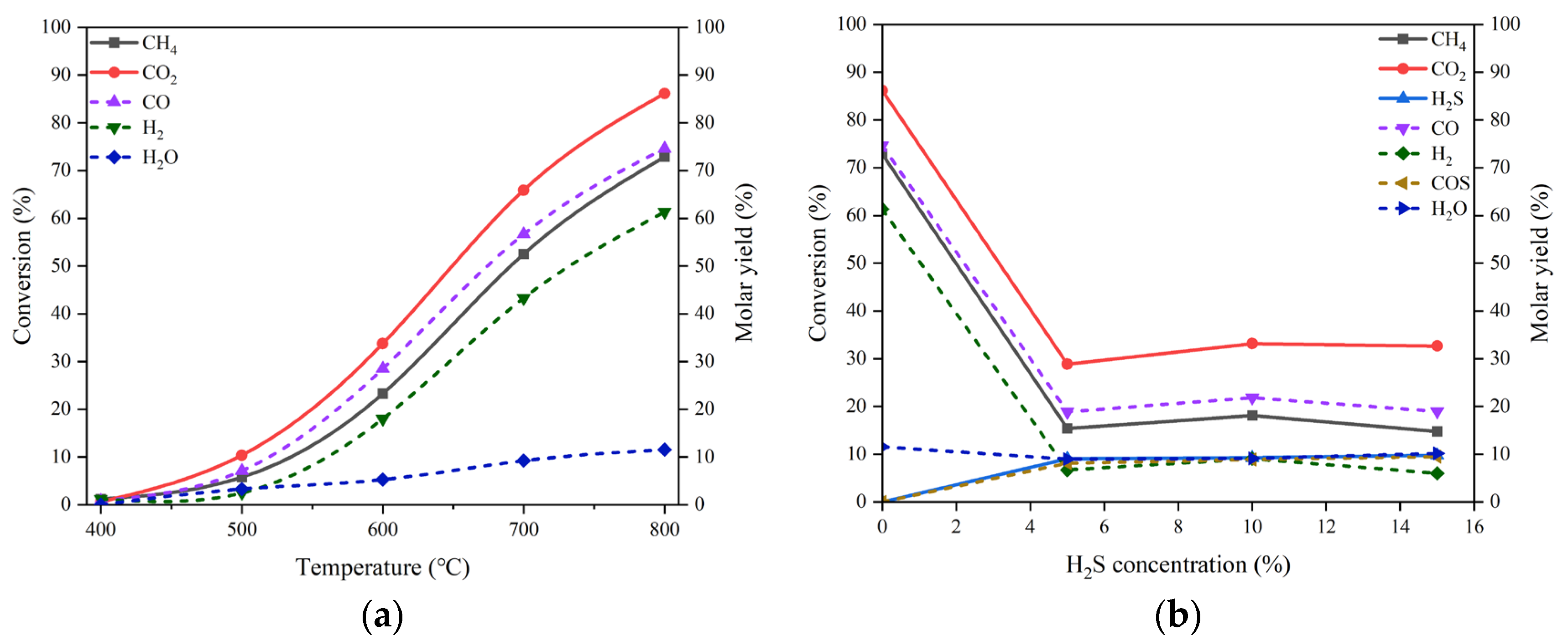
LaNiO3 is the most widespread Ni-based perovskite oxide used as a precursor for DRM, due to the stable performance of its derived catalyst during reaction, in contrast with conventional Ni-based catalysts [17,18,19]. We also prepared the material of LaNiO3 and reduced it to form Ni-based active phase before running the process of DRM. The results of CO2 and CH4 conversions and the molar yields of CO and H2 at different temperatures in the condition of no H2S addition are curved in Figure 5(a). High activity of LaNiO3-derived catalyst is achieved as expected. In the presence of H2S, as shown in Figure 5(b), the changes in the conversions of CO2 and CH4 as well as H2S with the H2S concentration are somewhat analogous to those appearing over the reduced NiO/MgO catalyst. The addition of H2S suppresses to a certain extent the activity of the LaNiO3-derived catalyst for CO2 and CH4 transformation; however, a partial revival of catalytic performance can be effectuated by increasing the H2S concentration.
Figure 5.
(a) Conversion of CO2 and CH4 and molar yield of CO, H2 and H2O changing with temperature in the absence of H2S on the reduced LaNiO3 catalyst, (b) conversion of CO2, CH4 and H2S and molar yield of CO, H2, H2O and COS changing with H2S concentration at 800 °C. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and H2S (0 - 15%) in volume balanced with N2, at the temperature of 400 - 800 °C, the pressure of 0.1 MPa and the GHSV of 10000 h-1. The dotted line refers to the molar yield of product.
Figure 5.
(a) Conversion of CO2 and CH4 and molar yield of CO, H2 and H2O changing with temperature in the absence of H2S on the reduced LaNiO3 catalyst, (b) conversion of CO2, CH4 and H2S and molar yield of CO, H2, H2O and COS changing with H2S concentration at 800 °C. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and H2S (0 - 15%) in volume balanced with N2, at the temperature of 400 - 800 °C, the pressure of 0.1 MPa and the GHSV of 10000 h-1. The dotted line refers to the molar yield of product.
3.3. Discussion
As revealed above, the promotion effect on catalytic performance arising from the addition of high concentration H2S can be observed on all of the materials investigated including the MgO, the NiO/MgO reduced or not, and the reduced LaNiO3 catalysts, implying that there must be a similar reason behind. After comprehensive deliberation of the product composition detected and their possible formation routes through consulting our previous study and other researches published [20,21,22,23], five independent reactions and their thermodynamic functions have been figured out, as listed in Table 2 below.
It is apparent that the pathway through which H2S participates in the reaction may not only embody in the Eq. (10), but also in the Eqs. (12) and (13) probably. As a matter of fact, traces of CS2 and S2 had been searched out occasionally during reaction test in the collected liquid samples and on the reactor outlet wall, respectively, affirming the possibility of the occurrence of the Eqs. (12) and (13). According to the thermodynamic analysis results relevant to CS2 and S2 formation given in Figure 2(b), the yields of CS2 and S2 are exactly very low in theory. Therefore, it is reasonable for the negligible amount of CS2 and S2 gathered. In contrast, COS is one of critical products formed on all the catalysts, which much likely arises from H2S transformation with CO2. Moreover, the molar yield of COS in practical is generally higher than the theoretical value. The consequence hints that the reaction between H2S and CO2 may take priority in virtue of selective catalysis over other CO2 reactions, especially that proceeding following the Eq. (9), while the latter is thermodynamically more favorable. The fall-off in conversions of CO2 and CH4 after adding H2S to the systems of the reduced NiO/MgO and LaNiO3 catalysts points to the alteration of catalytic active phases, thus causing a decrease in activity of the two catalysts for the direct reaction between CO2 and CH4. On the other hand, the happening of diverse reactions of H2S with CO2 and CH4 can multiple the transformation pathways of both CO2 and CH4, and high H2S concentration is definitely promotive to the conversion of CO2 and CH4. This should be the principal reason for the promotion effect of high concentration H2S in the catalytic DRM reaction system.
The reaction results of the reactant mixture of 20 vol% CO2, 20 vol% CH4, and 15 vol% H2S on different catalysts tested at 800 °C are summarized in Table 3. The pristine NiO/MgO catalyst performs prominently among all in terms of reactant conversion and product yield. The conversions of CO2 and CH4 in the presence of H2S can sustain at a meaningful level that can be applicable to industry, in spite of being somewhat lower than those obtained over the reduced NiO/MgO and LaNiO3 catalysts in the absence of H2S. In particular, the catalysts can at least partially overcome the impact of activity reduction caused by H2S addition.
It is worthy of mentioning that there was no detectable decline in catalytic activity of any catalyst with reaction time on stream throughout the entire testing period in the condition of H2S presence. As a matter of fact, we measured the stability of reduced NiO/MgO catalyst for longer than 60 h in feed gas of 20 vol% CO2, 20 vol% CH4 and 15 vol% H2S at 700 °C. The conversions of the three reactants changing with time on stream are supplied in Figure S4 of Supporting Information. The conversion curves are considerably flat, implying that the deactivation typically resulting from coke formation or H2S poisoning in such kind of reaction system may slightly happen to the catalyst.
In order to understand the distinctive role of each catalyst played in the reactions, a series of characterization of the catalysts was implemented by use of BET, XRD and XPS techniques, etc. The sample labelled as “used” refers to the catalyst having undergone the DRM reactions at 800 °C for 1 h in the presence of 15 vol% H2S. The BET measurement results of the catalyst samples before and after the reactions are listed in Table 4, and the corresponding cryogenic N2 adsorption-desorption isotherms and pore size distribution profiles are displayed in Figs. S5 and S6 respectively. The catalysts of the MgO, the pristine and reduced NiO/MgO, and the reduced LaNiO3 differ greatly from one another in their specific surface area, pore volume and average pore diameter. After completing the reactions at 800 °C for 1 h in the presence of H2S, almost all the catalysts alter few with respect to their textural features in comparison with their fresh states, reflecting the high tolerance of the catalyst structures to the H2S-containing reactive atmosphere at high temperature. For LaNiO3, there are obvious changes in N2 adsorption-desorption isotherm and pore size distribution profile shown in Figs. S5 and S6 respectively. It must be originated from the transformation of the crystal structure of LaNiO3 in the process of reaction, as will be revealed in the discussion of XRD results in the following text. Nevertheless, probably due to its inherently small specific surface area, the surface area of LaNiO3 becomes more trivial after experiencing reactions. Associating with the catalytic performance of these catalysts in H2S atmosphere as supplied in Table 3, the texture property of catalyst seems to give an impact on the catalytic reactions to some extent. High specific surface area of catalyst might be helpful to the conversion of the reactants.
The XRD spectra of the three types of the catalysts in various states are spread out in Figure 6(a-c). As shown in Figure 6(a), the two samples of the MgO before and after the reactions involving H2S are virtually identical in the appearance of characteristic peaks of crystal phases. Both of them can be assigned to the standard MgO of PDF #45-0946, indicating that there is little influence of H2S on the crystal structure of MgO. For the pristine NiO/MgO, its characteristic peaks on XRD spectrum in Figure 6(b) also look to be the same as those of MgO. This situation should be associated with the formation of solid solution of NiO and MgO. As revealed in the literatures, pure phase solid solution of brucite can be easily formed from mixture of NiO and MgO by isomorphous substitution, resulting in almost identical XRD pattern of NiO/MgO to that of MgO, due to the fact that bivalent cations of Ni2+(0.62 Å) and Mg2+(0.69 Å) have close ion radius [24,25,26]. The formation of solid solution can favor the homogeneous Ni distribution in the NiO/MgO catalyst, despite of a NiO loading as high as 20 wt%. Incidentally, such a high NiO loading is fairly profitable to achieve better catalytic performance in dry reforming process, as already evidenced by a few researchers [24,27]. On the spectrum of the reduced NiO/MgO sample, there appears a small protrusion at the foot of the chief peak of MgO at 2θ of 42.916°, which position is basically in concordance with the dominant peak at 44.507°of metallic Ni belong to PDF #04-0850. Making a comparison between the pristine and reduced NiO/MgO, one can notice the approximately consistent XRD spectra for the two catalysts both after the reactions involving H2S. A few of characteristic peaks of NiS emerge beside those of MgO, manifesting that the sulfidation of Ni and/or NiO to NiS occurred during the reactions. The sulfidation phenomenon also exists on the catalyst of reduced LaNiO3 as shown in Figure 6(c), where the phases of NiS and La2O2S were transformed from the reduced LaNiO3 after the reactions in the presence of H2S. In consideration of the relatively close reaction results over different catalysts, it can be deduced that the catalytic performance does not depend greatly on the crystal phase of catalyst whether it is MgO, or NiS or La2O2S yet.
To deeply explore the active phase or sites on catalyst surface for the reactions, XPS investigation was carried out. The full-range spectra of various catalysts in different states are exhibited in Figure 7(a, b). The characteristic peaks of the elements belong to the catalyst composition of MgO and NiO/MgO such as Mg, Ni and O can be easily discovered in Figure 7(a). However, the signal of S 2p within 160-170 eV, which usually represents the existence of S element on catalyst, can hardly be probed on the surface of MgO used. The S 2p signal is also very faint for the used NiO/MgO catalyst, regardless of NiO/MgO was whether reduced or not before the reactions. In contrast, on the used LaNiO3-reduced catalyst the peak of S 2p can be distinctly observed in addition to the peaks of La, Ni and O, etc. Taking into account more S-containing crystal species as La2O2S and NiS in the used LaNiO3-reduced catalyst than that in the used NiO/MgO catalyst, which owns NiS only, it is thus explainable for the higher peak intensity of S 2p of the former catalyst on XPS spectrum. Since the active sites for DRM reactions over Ni-based catalysts are generally associated with metallic Ni species, the chemical states of Ni involved in related NiO/MgO and LaNiO3 catalysts were accordingly resolved, respectively, through deconvolution of the peaks of Ni 2p and 3p on fine-range spectra. The deconvolution results of Ni 2p1/2 at 854.68 eV and Ni 2p3/2 at 872.18 eV for NiO/MgO catalyst in different states are illustrated in Figure 7(c), showing that there is a slight variation of the proportions of Ni2+ and Ni3+ after the catalysts experienced the reactions. This is not a surprise because the Ni valence in NiS phase should be approximate to that in NiO. Nevertheless, the deconvolution results of Ni2+ at 66.38 eV and Ni3+ at 67.88 eV both assigned to Ni 3p of LaNiO3 catalyst in Figure 7(d) unveil a significant difference in Ni chemical valance between the catalysts in pristine and in used states. The species of Ni3+ was overwhelming in the pristine LaNiO3, which evolved partially to Ni2+ when the LaNiO3 was subjected to the reactions in the presence of H2S. The ratio of Ni2+ to Ni3+ in the used LaNiO3-reduced catalyst is considerably comparable to that in the used NiO/MgO catalyst, in concert with the common formation of NiS in both catalysts. Therefore, the chemical state of Ni in the two types of catalysts during the reactions may be much the same.
It should be emphasized that there is no any active Ni species on the MgO catalyst. As discussed hereinabove, the catalytic performance of the MgO catalyst in the condition of H2S presence is in no sense inferior to the two types of catalysts containing Ni species. This raises a question of what is the active center on the MgO catalyst for the reactions. In our early study, the catalytic activity of MgO catalyst was also found to be competitive to that of NiO/MgO and NiO/γ-Al2O3 catalysts during the reactions between CO2 and H2S at 800 °C, and consequentially free radicals initiated on the catalyst surface were supposed to dominate the reactions [20]. In recent research of Wang et al., the key active sites were identified as sulfur species (S*) that are dynamically bound to metal cations during the reactions between CH4 and H2S at high temperatures, which was believed to be common for all the catalysts tested covering a wide range of oxides such as MgO, γ-Al2O3 and IUPAC group 4-6 metal-oxide-derived materials [28,29]. Being inspired by these viewpoints, we speculate that the adsorbed S species, which are formed ineluctably through H2S dissociation on the surface of catalyst regardless of catalyst type [28,29,30], may be the key to initiate the activation of CO2 and CH4 in the presence of H2S. A schematic diagram of the possible reaction mechanisms that may occur separately on MgO and NiO/MgO catalysts in the presence of H2S is drawn as the examples and illustrated in Figure 8(a, b). Regardless of whether MgO is loaded with Ni, its cations are supposed to be able to dissociate H2S, thus promoting the conversion of CO2 and CH4.
Comparing with the high catalytic activity of metallic Ni active sites for DRM reactions, however, the catalytic capability of S-adsorbed species may fall into a disadvantage. Due to the inferior activity of S-adsorbed species along with the vanishment of metallic Ni active sites, the reduction in CO2 and CH4 conversions is inevitable in the presence of H2S. Despite all this, the addition of H2S with high concentration can undoubtedly promote the conversions by means of H2S participating the reactions with both CO2 and CH4. Moreover, the deviation in catalytic performance among the three types of catalysts, although weak, still could provide us with clues to improve catalyst. Developing a catalyst having large specific surface area, which can persist at high temperature to afford abundant active S-adsorbed species, may be beneficial to the DRM reactions in the presence of H2S.
4. Conclusions
In the present study, the effect of H2S concentration on DRM over different catalysts has been investigated. The thermodynamic simulation results of the reaction mixture of 20 vol% CO2, 20 vol% CH4 and (0-20 vol%) H2S in N2 show that the equilibrium conversions of both CO2 and CH4 can hardly be affected by the addition of H2S in the temperature range of 400-1000 °C, as the reaction between CO2 and CH4 has a significant thermodynamic advantage over the reactions involving H2S. The equilibrium molar yield of H2 based on CH4 has a slight lift when the H2S concentration increases from 0 to 20 vol% at 800 °C, though its value drops obviously after counting all the hydrogen sources including CH4 and H2S.
On the catalysts with lower catalytic activity for DRM at 800 °C, such as the MgO and the pristine NiO/MgO catalysts, the addition of H2S significantly increases the conversions of CO2 and CH4 and the yields of CO and H2, and the conversion of reactants and the yield of products can be further increased with the increase of H2S concentration. To the contrary, on the catalysts with high catalytic activity for DRM, such as the reduced NiO/MgO and LaNiO3 catalysts, the addition of small amount of H2S severely inhibits their catalytic performance. However, when further increasing the concentration of H2S, e.g., to 15 vol%, the conversions of CO2 and CH4 and the yields of CO and H2 can climb up again. The byproduct COS emerges in all the catalytic systems involving H2S.
The results of process analysis and catalyst structural characterization reveal the countertrend occurrence of the reactions involving H2S over all the catalysts tested. High H2S concentration is promotive to the transformation of either CO2 or CH4 through the reactions of H2S with CO2 and CH4. In the condition of H2S presence, the catalytic performance of different catalysts doesn’t deviate significantly from each other, and a considerable stable activity of catalyst has been achieved. The active S-adsorbed species on the surface of catalysts rather than traditional active metallic Ni sites have been speculated to be responsible for the moderate conversion of CO2 and CH4 in the presence of H2S at high temperature. The obtained results are of great significance for the direct catalytic conversion of sour natural gas to produce valuable chemicals.
References
- G. Xiong; Y. Hu; J. Fu; C. Chen; C. Yang; X. Yan; J. Jing; F. Gao; G. Chen; Q. Liu; J. He; X. Gao. New progress and development direction of high-sulfur natural gas purification technology. Nat. Gas Ind. 2023, 43, 34-48 (in Chinese). [CrossRef]
- Y. Wang; L. Yao; S. Wang; D. Mao; C. Hu. Low-temperature catalytic CO2 dry reforming of methane on Ni-based catalysts: A review. Fuel Process. Technol. 2018, 169, 199-206. [CrossRef]
- Q. Liu; Y. Liu; N. Zhou; P. Zhang; Z. Liu; E.I. Vovk; Y.-A. Zhu; Y. Yang; K. Zhu. Realization of high-pressure dry methane reforming by suppressing coke deposition with Co-Rh intermetallic clusters. Appl. Catal. B-Environ. 2023, 339, 123102 (1-13). [CrossRef]
- Z. Alipour; V.B. Borugadda; H. Wang; A.K. Dalai. Syngas production through dry reforming: A review on catalysts and their materials, preparation methods and reactor type. Chem. Eng. J. 2023, 452, 139416 (1-18). [CrossRef]
- S. Appari; V.M. Janardhanan; R. Bauri; S. Jayanti; O. Deutschmann. A detailed kinetic model for biogas steam reforming on Ni and catalyst deactivation due to sulfur poisoning. Appl. Catal. A-Gen. 2014, 471, 118-125. [CrossRef]
- M. Gaillard; M. Virginie; A.Y. Khodakov. New molybdenum-based catalysts for dry reforming of methane in presence of sulfur: A promising way for biogas valorization. Catal. Today 2017, 289, 143-150. [CrossRef]
- R. Chein; Z.W. Yang; H2S effect on dry reforming of biogas for syngas production. Int. J. Energy Res. 2019, 43, 3330-3345. [CrossRef]
- H. Akansu; H. Arbag; H.M. Tasdemir; S. Yasyerli; N. Yasyerli; G. Dogu. Nickel-based alumina supported catalysts for dry reforming of biogas in the absence and the presence of H2S: Effect of manganese incorporation. Catal. Today 2022, 397-399, 37-49. [CrossRef]
- Y. Gao; J. Jiang; Y. Meng; T. Ju; S. Han. Influence of H2S and NH3 on biogas dry reforming using Ni catalyst: a study on single and synergetic effect. Front. Env. Sci. Eng. 2022, 17, 32. [CrossRef]
- B. Fidalgo; N. Muradov; J.A. Menéndez. Effect of H2S on carbon-catalyzed methane decomposition and CO2 reforming reactions. Int. J. Hydrog. Energy 2012, 37, 14187-14194. [CrossRef]
- S.A. Chattanathan; S. Adhikari; M. McVey; O. Fasina. Hydrogen production from biogas reforming and the effect of H2S on CH4 conversion. Int. J. Hydrog. Energy 2014, 39, 19905-19911. [CrossRef]
- R. Pereñíguez; V.M. González-DelaCruz; J.P. Holgado; A. Caballero. Synthesis and characterization of a LaNiO3 perovskite as precursor for methane reforming reactions catalysts. Appl. Catal. B-Environ. 2010, 93, 346-353. [CrossRef]
- G. Jin; K. Li; L .Zhang; Y.M. Luo; D.K.Chen; D.D. He. In situ observation of the promoting effect of H2S on the formation of efffcient MoS2 catalyst for CH4/CO2 reforming, Sep. Purif. Technol. 2023, 308, 122883 (1-12). [CrossRef]
- Y.H. Hu; E. Ruckenstein. Conment on “Dry reforming of methane by stable Ni-Mo nanocatalysts on single-crystalline MgO”. Science 2020, 368, eabb5459. [CrossRef]
- X. Feng; Y. Zhao; S. Liu; K. Wang; B. Liu; Q. Zhang; H. Wang; Y. Zhao; J. Liu; P. Zhang; L. Gao. Flower-like hollow Ni0.5/xMgO-Al2O3 catalysts with excellent stability for dry reforming of methane: The role of Mg addition, Fuel 2024, 358, 130029 (1-11). [CrossRef]
- K. Taira. Dry reforming reactions of CH4 over CeO2/MgO catalysts at high concentrations of H2S, and behavior of CO2 at the CeO2-MgO interface J. Catal. 2022, 407, 29-43. [CrossRef]
- A.G. Georgiadis; N.D. Charisiou; M.A. Goula. A mini-review on lanthanum–nickel-based perovskite-derived catalysts for hydrogen production via the dry reforming of methane (DRM). Catalysts 2023, 13, 1357 (1-32). [CrossRef]
- A.G. Georgiadis; G.I. Siakavelas; A.I. Tsiotsias; N.D. Charisiou; B. Ehrhardt; W. Wang; V. Sebastian; S.J. Hinder; M.A. Baker; S. Mascotto; M.A. Goula. Biogas dry reforming over Ni/LnOx-type catalysts (Ln = La, Ce, Sm or Pr). Int. J. Hydrog. Energy. 2023, 48, 19953-19971. [CrossRef]
- J. Niu; H. Liu; Y. Jin; B. Fan; W. Qi; J. Ran. A density functional theory study of methane activation on MgO supported Ni9M1 cluster: role of M on C-H activation. Front. Chem. Sci. Eng. 2022, 16, 1485-1492. [CrossRef]
- [20] H. Su; Y. Li; P. Li; Y. Chen; Z. Zhang; X. Fang. Simultaneous recovery of carbon and sulfur resources from reduction of CO2 with H2S using catalysts. J. Energy Chem. 2016, 25, 10-116. [CrossRef]
- H. Wang; J. Wu; Z. Xiao; Z. Ma; P. Li; X. Zhang; H. Li; X. Fang. Sulfidation of MoO3/γ-Al2O3 towards a highly efficient catalyst for CH4 reforming with H2S. Catal. Sci. Technol. 2021, 11, 1125-1140. [CrossRef]
- W. Taifan; J. Baltrusaitis. Minireview: direct catalytic conversion of sour natural gas (CH4 + H2S + CO2) components to high value chemicals and fuels. Catal. Sci. Technol. 2017, 7, 2919-2929. [CrossRef]
- E. Spatolisano; G. de Guido; L.A. Pellegrini; V. Calemma; A.R. de Angelis; M. Nali. Hydrogen sulphide to hydrogen via H2S methane reformation: Thermodynamics and process scheme assessment. Int. J. Hydrog. Energy. 2022, 47, 15612-15623. [CrossRef]
- Y.H. Hu; E. Ruckenstein. An optimum NiO content in the CO2 reforming of CH4 with NiO/MgO solid solution catalysts. Catal. Lett. 1996, 36, 145-149. [CrossRef]
- S.P. Jiang; Y. Lu; S.P. Wang; Y.J. Zhao; X.B. Ma. Insight into the reaction mechanism of CO2 activation for CH4 reforming over NiO-MgO: A combination of DRIFTS and DFT study. Appli. Surf. Sci. 2017, 416, 59-68. [CrossRef]
- T.T. Zhang; Z.X. Liu; Y.A. Zhu; Z.C. Liu; Z.J. Sui; K.K. Zhu; X.G. Zhou. Dry reforming of methane on Ni-Fe-MgO catalysts: Influence of Fe on carbon-resistant property and kinetics. Appl. Catal. B-Environ. 2020, 264, 118497 (1-17). [CrossRef]
- M. Ahadzadeh; S.M. Alavi; M. Rezaei; E. Akbari. Propane dry reforming over highly active NiO-MgO solid solution catalyst for synthesis gas production, Mol. Catal. 2022, 524, 112325 (1-11). [CrossRef]
- Y. Wang; X.M. Chen; H. Shi; J.A. Lercher. Catalytic reforming of methane with H2S via dynamically stabilized sulfur on transition metal oxides and sulfides. Nat. Catal. 2023, 6, 204-214. [CrossRef]
- Y. Wang; W.R. Zhao; X.F. Chen; Y.J. Ji; X.L. Zhu; X.M. Chen; D.H. Mei; H. Shi; J.A. Lercher. Methane-H2S reforming catalyzed by carbon and metal sulfide stabilized sulfur dimers. J. Am. Chem. Soc. 2024, 146, 8630-8640. [CrossRef]
- C. Zheng; H. Zhao. Exploring the microscopic reaction mechanism of H2S and COS with CuO oxygen carrier in chemical looping combustion. Fuel Process. Technol. 2020, 205, 106431 (1-10). [CrossRef]
Figure 1.
Simulation results of change in the conversion of reactant (a) and the molar fraction of product (b) with reaction temperature. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and 15% H2S in volume balanced with N2, at the temperature range of 400-1000 °C and pressure of 0.1 MPa, in a Gibbs isobaric and isothermal reactor.
Figure 1.
Simulation results of change in the conversion of reactant (a) and the molar fraction of product (b) with reaction temperature. Reaction conditions: feed gas composed of 20% CO2, 20% CH4 and 15% H2S in volume balanced with N2, at the temperature range of 400-1000 °C and pressure of 0.1 MPa, in a Gibbs isobaric and isothermal reactor.
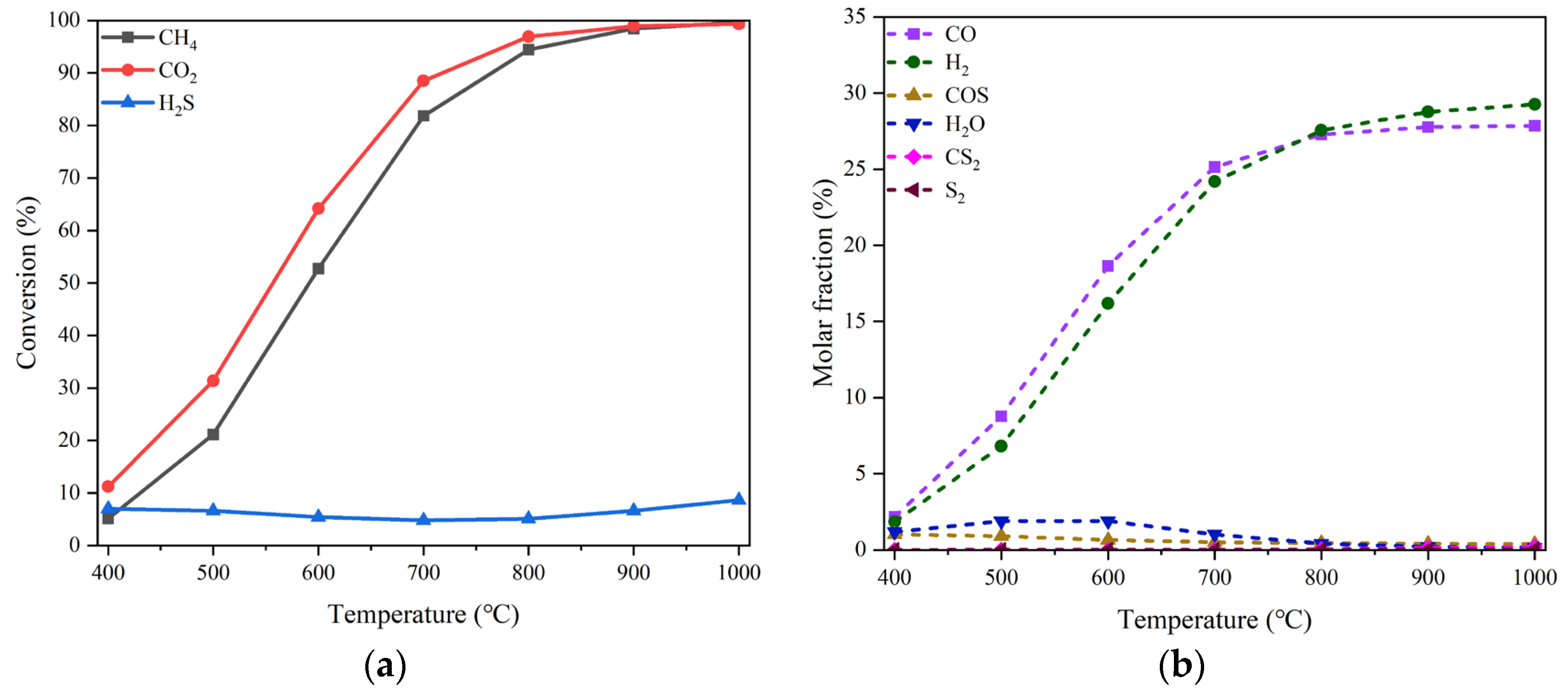
Figure 6.
XRD spectra of the catalysts of MgO, NiO/MgO and LaNiO3 in different states.
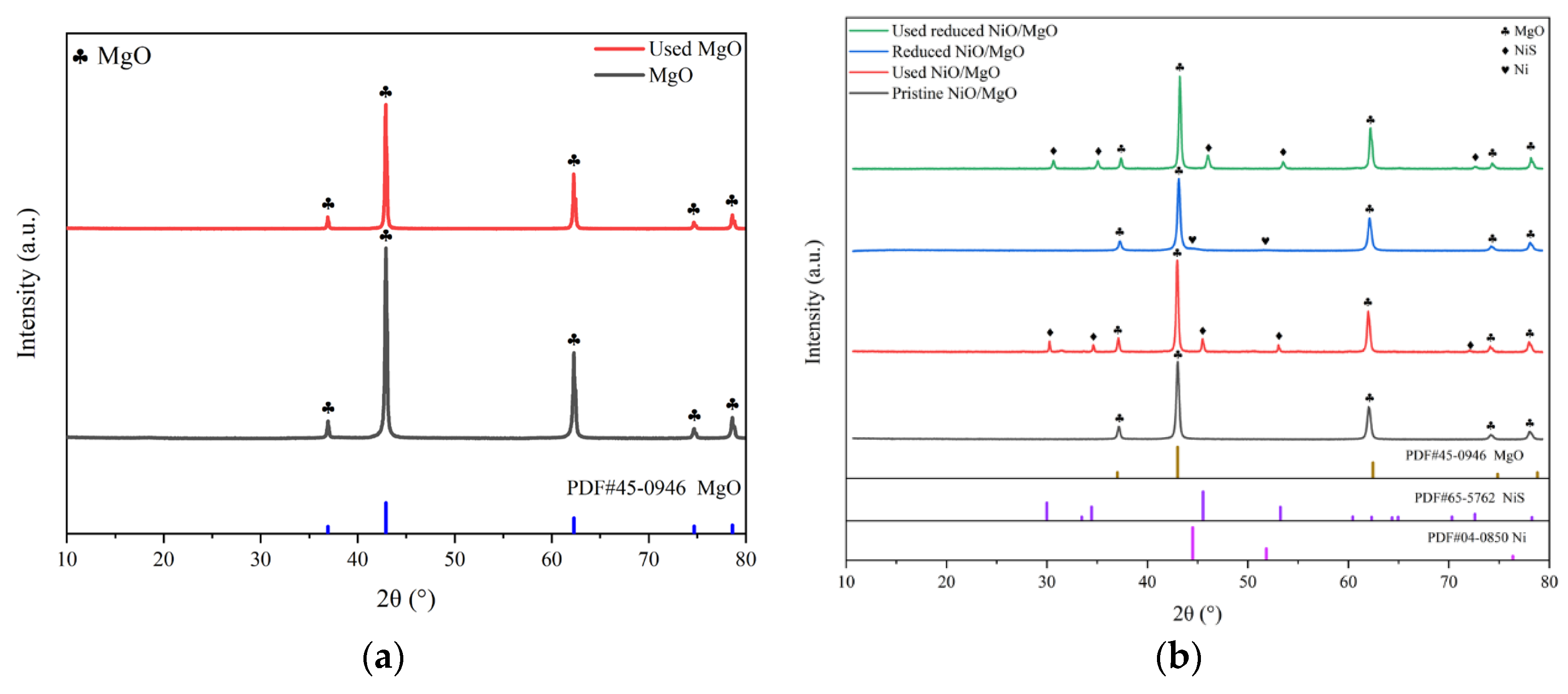
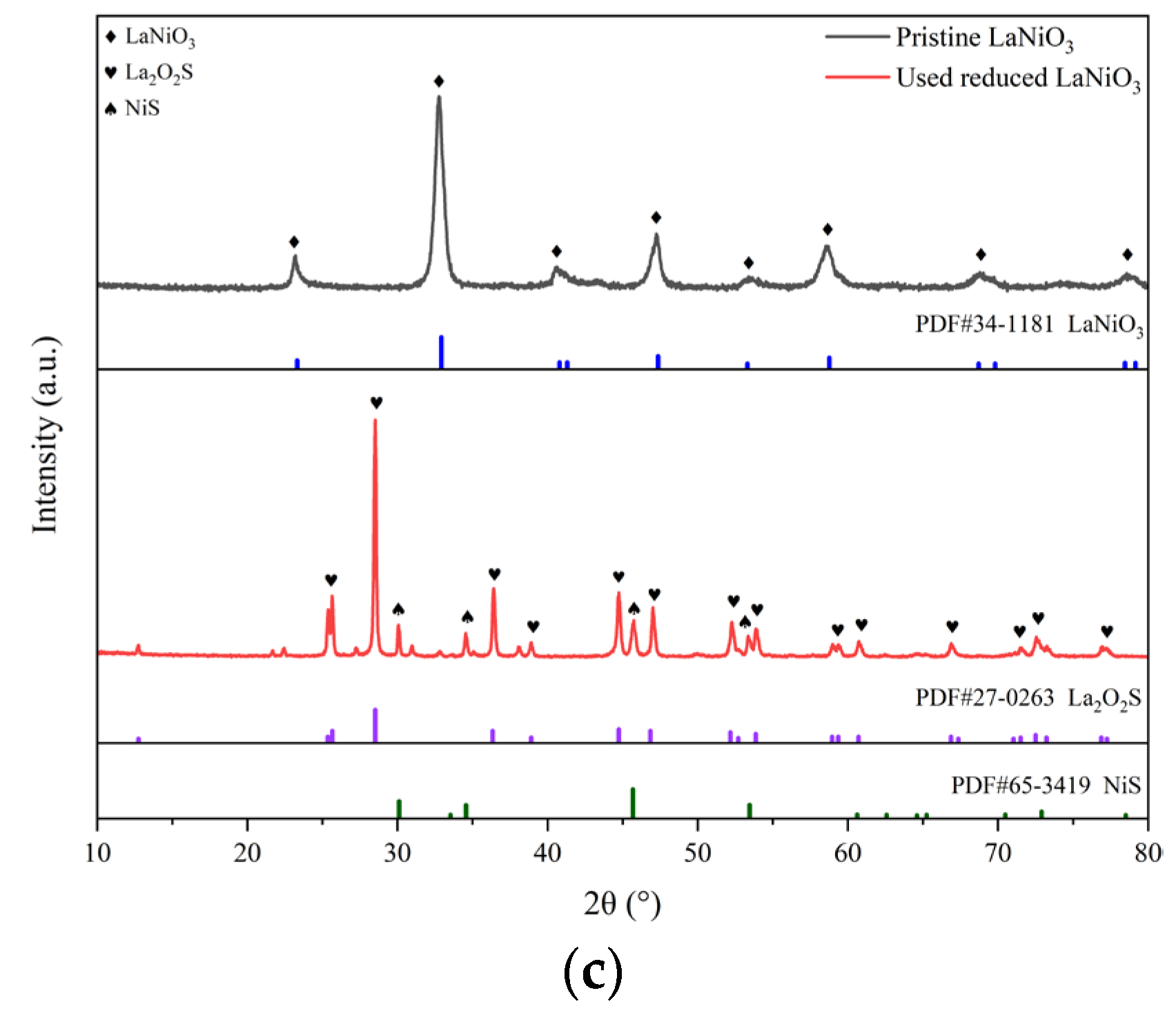
Figure 7.
XPS spectra of three types of catalysts in different states. (a), wide spectrum of NiO/MgO, (b) wide spectrum of LaNiO3, (c) narrow spectrum of Ni 2p of NiO/MgO, and (d) narrow spectrum of Ni 3p of LaNiO3.
Figure 7.
XPS spectra of three types of catalysts in different states. (a), wide spectrum of NiO/MgO, (b) wide spectrum of LaNiO3, (c) narrow spectrum of Ni 2p of NiO/MgO, and (d) narrow spectrum of Ni 3p of LaNiO3.
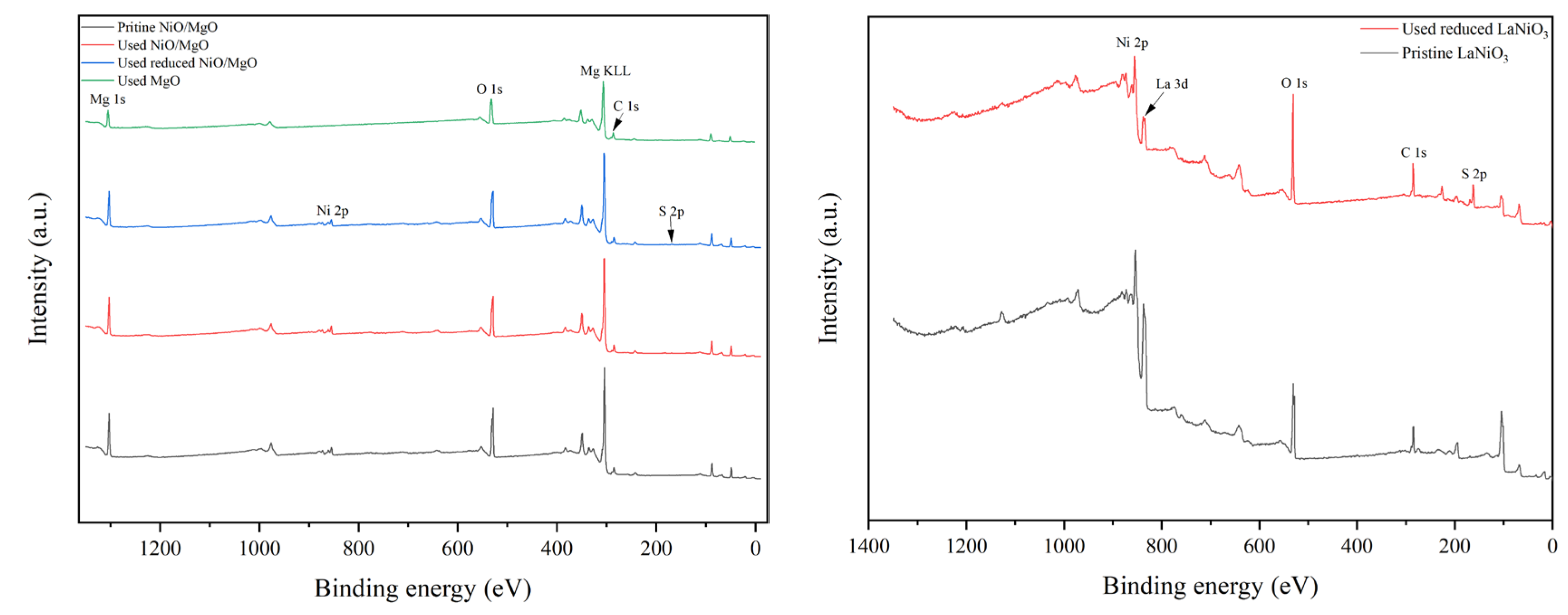
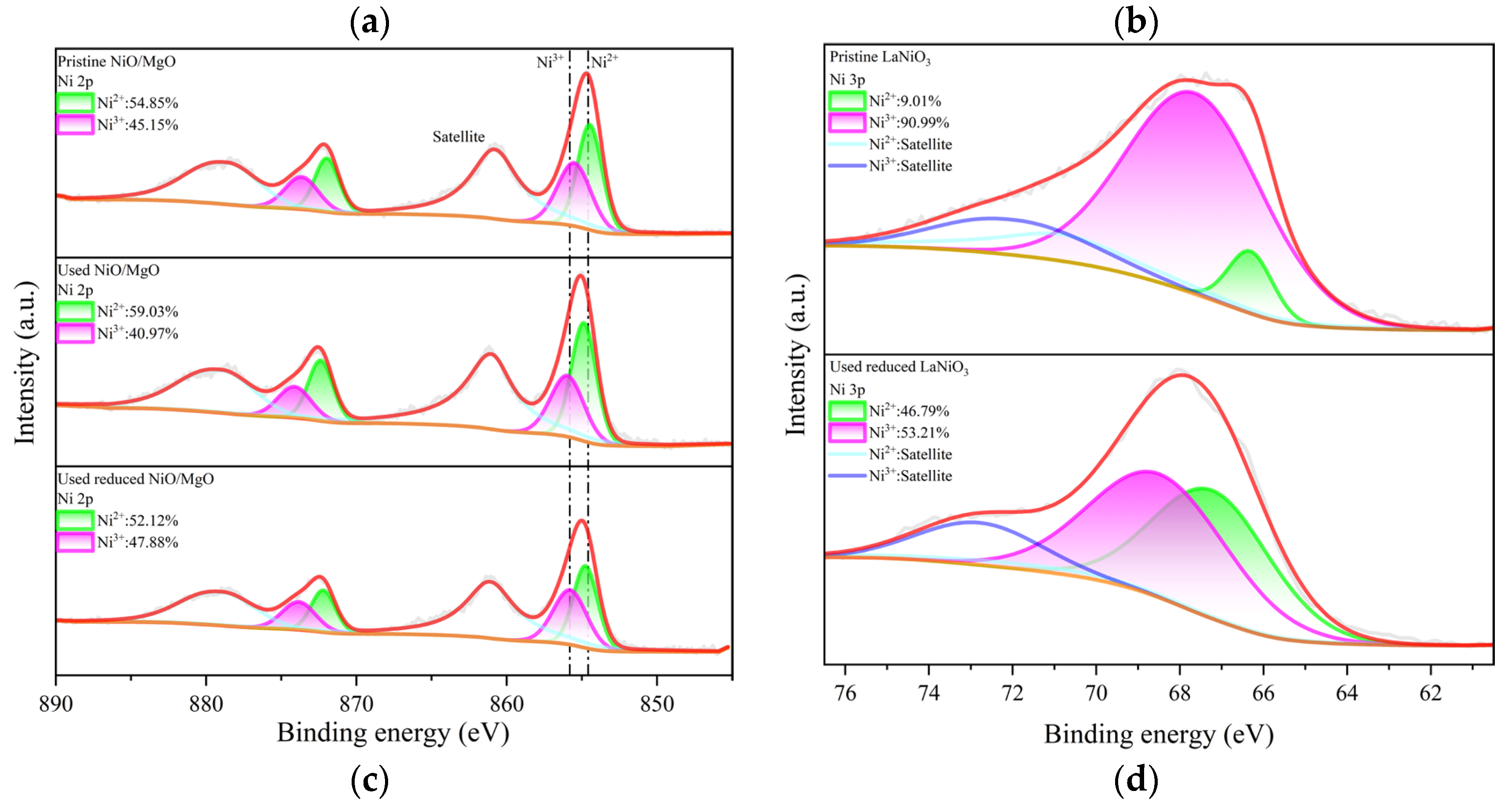
Figure 8.
Schematic diagram of the reaction mechanism possibly occurring on (a) MgO and (b) NiO/MgO catalysts in the presence of H2S.
Figure 8.
Schematic diagram of the reaction mechanism possibly occurring on (a) MgO and (b) NiO/MgO catalysts in the presence of H2S.
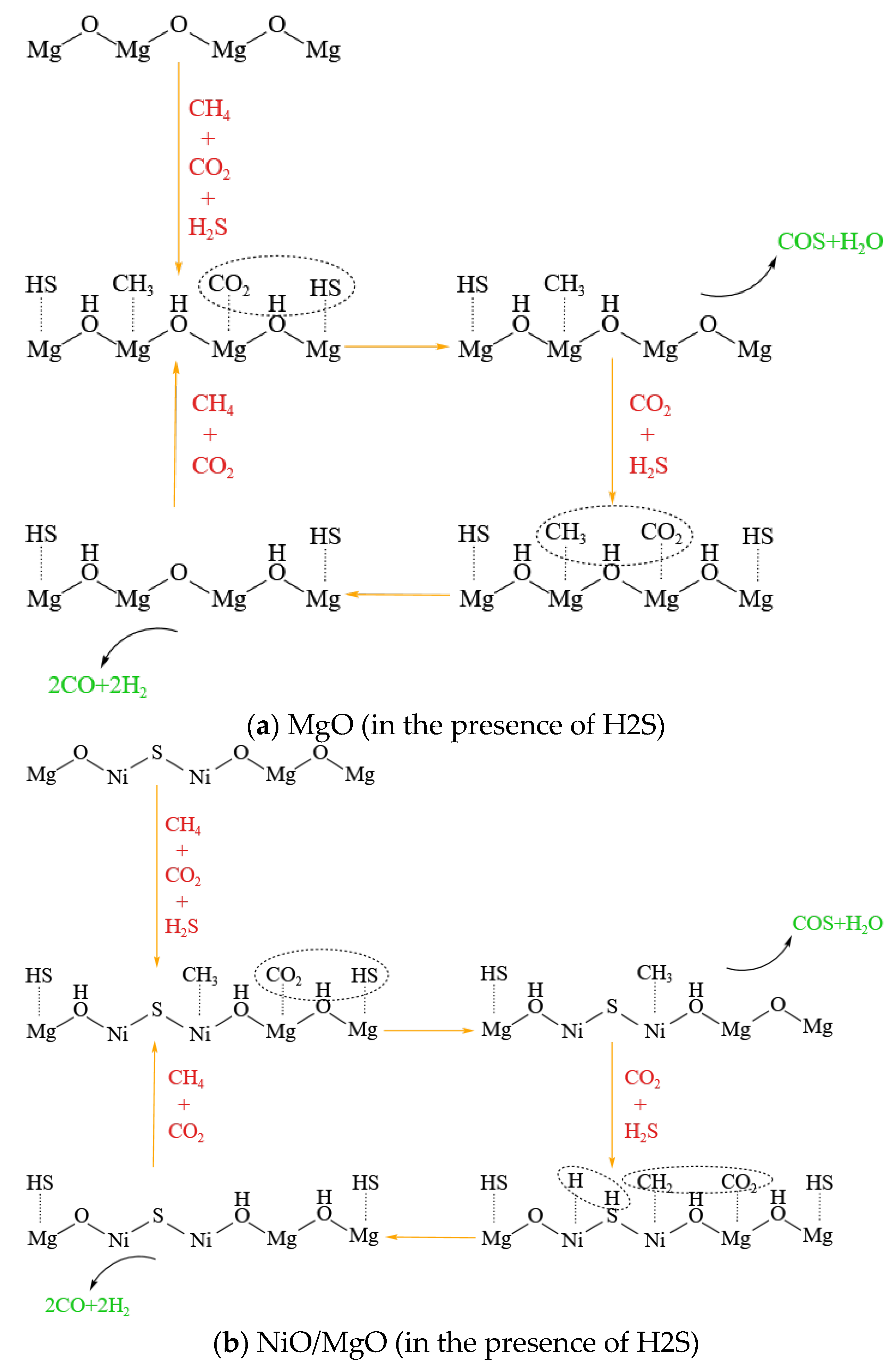

Table 1.
CO2 and H2S concentration data of some gas fields worldwide [1].
Table 1.
CO2 and H2S concentration data of some gas fields worldwide [1].
| Gas field | CO2 (vol%) | H2S (vol%) | Country |
|---|---|---|---|
| East Crossfield | 12.0 | 36.0 | Canada |
| BEC | 40.0 | 21.0 | America |
| Lacq | 9.3 | 15.6 | France |
| Zechstein | 20.0 - 50.0 | 15.0 - 20.0 | Germany |
| Astrakhan | 15.0 | 25.0 | Russia |
| Puguang | 8.6 | 15.2 | China |
Table 2.
Independent reactions involved in DMR process having H2S addition.
| No. | Reaction equation | ΔH(1073.15 K)/kJ⋅mol-1 | ΔG(1073.15 K)/kJ⋅mol-1 |
|---|---|---|---|
| (9) | CO2 + CH4 ↔ 2H2 + 2CO | 260.4 | -45.0 |
| (10) | CO2 + H2S ↔ H2O + COS | 34.4 | 31.6 |
| (11) | CO2 + H2 ↔ H2O + CO | 34.0 | 0.7 |
| (12) | CH4 + 2H2S ↔ 4H2 + CS2 | 261.0 | 29.6 |
| (13) | H2S ↔ H2 + 1/2S2 | 90.3 | 37.5 |
Table 3.
Conversions of three reactants and molar yields of main products on various catalysts at 800 °C and 0.1 MPa.
Table 3.
Conversions of three reactants and molar yields of main products on various catalysts at 800 °C and 0.1 MPa.
| Catalyst sample | CO2 conversion (%) | CH4 conversion (%) | H2S conversion (%) | CO molar yield (%) | H2 molar yield (%) |
|---|---|---|---|---|---|
| MgO | 36.30 | 19.39 | 8.52 | 24.72 | 10.07 |
| Pristine NiO/MgO | 42.10 | 26.86 | 8.03 | 33.10 | 16.93 |
| Reduced NiO/MgO | 34.76 | 19.03 | 7.39 | 25.33 | 10.85 |
| Reduced LaNiO3 | 28.86 | 15.38 | 9.06 | 18.88 | 6.71 |
Table 4.
BET results of the catalysts before and after reaction.
| Catalyst sample | Specific surface area/m2⋅g-1 | Pore volume/cm3⋅g-1 | Average pore diameter/nm |
|---|---|---|---|
| MgO | 16.20 | 0.055 | 13.7 |
| Used MgO | 15.66 | 0.047 | 11.9 |
| Pristine NiO/MgO | 40.12 | 0.30 | 30.3 |
| Used NiO/MgO | 45.67 | 0.46 | 40.5 |
| Reduced NiO/MgO | 43.28 | 0.40 | 35.5 |
| Used reduced NiO/MgO | 42.10 | 0.37 | 35.0 |
| Pristine LaNiO3 | 4.21 | 0.025 | 24.0 |
| Used reduced LaNiO3 | 3.28 | 0.017 | 20.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



