
Submitted:
26 April 2024
Posted:
28 April 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Exploring therapeutic options is crucial in the ongoing COVID-19 pandemic caused by SARS-CoV-2. Nirmatrelvir, a potent inhibitor targeting the SARS-CoV-2 Mpro, shows promise as an antiviral treatment. Additionally, Ivermectin, a broad-spectrum antiparasitic drug, has demonstrated effectiveness against the virus in laboratory settings. However, its clinical implications are still debated. Using computational methods such as molecular docking and 100 ns molecular dynamics simulations, we investigated how Nirmatrelvir and Ivermectin interact with SARS-CoV-2 Mpro(A). Calculations using density functional theory have been instrumental in elucidating the behavior of isolated molecules, primarily by analyzing the frontier molecular orbitals. Our analysis revealed distinct binding patterns: Nirmatrelvir formed strong interactions with amino acids like MET49, MET165, HIS41, HIS163, HIS164, PHE140, CYS145, GLU166, and ASN142, showing stable binding with a root mean square deviation (RMSD) of around 2.0 Å. On the other hand, Ivermectin interacted with THR237, THR239, LEU271, LEU272, and LEU287, displaying an RMSD of 1.87 Å, indicating enduring interactions. Both ligands stabilized Mpro(A), with Ivermectin showing stability and persistent interactions despite forming fewer hydrogen bonds. These findings offer detailed insights into how Nirmatrelvir and Ivermectin bind to the SARS-CoV-2 main protease, providing valuable information for potential therapeutic strategies against COVID-19.
Keywords:
main protease (Mpro)
; ivermectin
; nirmatrelvir
; SARS-CoV-2
; molecular docking
; molecular dynamics
1. Introduction
Emerging in Wuhan, China, towards the end of 2019, a new acute respiratory illness caused by the novel coronavirus (SARS-CoV-2) was officially named COVID-19. The World Health Organization (WHO) declared it a global pandemic on March 11, 2020 [1]. This virus is known for its high transmissibility. It primarily spreads through human-to-human contact via contaminated respiratory secretions released during coughing, sneezing, or talking [2,3]. When in close proximity, aerosol particles can also transmit the infection, initiating the viral replication process in a new host [4].
Previously, smaller-scale epidemics, such as those in 2002, 2003, and 2012, were linked to the Coronavirus, impacting various hosts, including bats, rodents, mammals, and birds [5,6] and by 2024, just over four years after the initial case in Wuhan, SARS-CoV-2 infections affected nearly 704 million people, with approximately 7 million resulting in fatalities [7]. It is worth noting the well-known challenges in official reporting, which may lead to underestimating these values [8].
SARS-CoV-2 comprises three structural proteins and 16 non-structural proteins, among which the , or , holds a central role in the virus’s replication mechanism, acting as a cysteine protease [9,10] — positioned strategically at the active site of , a cysteine acts like molecular scissors, facilitating the cleavage of viral polyproteins. These lengthy protein chains, synthesized from the viral genome, are broken down into smaller, functionally independent units crucial for viral replication, assembly, and release. Given the vital importance of , various research groups are currently actively engaged in the development of targeted inhibitors, aiming for potential therapeutic applications for COVID-19 [11]. Several promising candidates are in advanced stages of clinical trials, raising optimism about their eventual incorporation into the antiviral arsenal against this persistent pathogen [12].
In this context, Alugubelli and colleagues explored the potential of Boceprevir, a known inhibitor of HCV’s NSP3, as a repurposed treatment for COVID-19 [13]. This compound exhibited promising inhibitory effects on the of SARS-CoV-2, featuring an -ketoamide group as a warhead, along with specific structural elements including a P1 -cyclobutylalanine segment, a P2 dimethyl cyclopropyl proline motif, a P3 test-butyl glycine unit, and a P4 N-terminal start-butyl carbamide. Through modifications at all four positions, 20 inhibitors based on Boceprevir were synthesized [13]. One such derivative, PF-07321332, underwent characterization for its potency against both in vitro and in cellulo, using test tubes and 293T cells, respectively. The study involved analyzing the crystal structures of bound to 10 of these inhibitors, alongside assessing the cytotoxicity and antiviral efficacy of four selected inhibitors.
By substituting the P1 site with a -(S-2-oxopyrrolidin-3-yl)-alanil (Opal) residue and altering the warhead to an aldehyde, a high in vitro potency was achieved [13]. Additionally, the original structural components in P2, P3, and the P4 N-terminal positions of Boceprevir demonstrated superior performance compared to alternative chemical groups tested for their in vitro potency. In crystal structures, all inhibitors formed a covalent adduct with the cysteine at the active site of [13]. Specifically, the Opal P1 residue, P2 dimethylcyclopropylproline, and P4 N-terminal tert-butylcarbamide established robust hydrophobic interactions with , elucidating the high in vitro potency associated with compounds containing these structural features.
Andi and colleagues conducted a study focused on developing novel antivirals and vaccines targeting [14]. Their findings, supported by crystallographic evidence and binding assay data, reveal that three drugs approved for hepatitis C virus treatment, along with two drug-like compounds, establish covalent bonds with the catalytic residue Cys145 within the active site of . Furthermore, molecular docking studies offered additional insights, aiding in the formulation of new antiviral inhibitors for SARS-CoV-2 by leveraging these drugs as lead compounds. It is worth mentioning that numerous other investigations into potential inhibitors have been documented in the literature [15,16,17,18].
The substrate binding site of , a region known for its remarkable conservation across various coronavirus genera, was investigated by Li and colleagues [19]. They specifically examined Nirmatrelvir (PF-07321332), an oral inhibitor developed by Pfizer that targets the of SARS-CoV-2, with demonstrated effectiveness against other coronaviruses as well. Through their analysis of these structures, the researchers unveiled a conserved binding site shared among coronaviruses, offering valuable insights into the mechanism underlying the inhibition of viral replication [19].
Viewing it from this angle, Nirmatrelvir specifically targets [20]. By inhibiting this protease at a critical stage of proteolysis before viral RNA replication, Nirmatrelvir effectively curbs the proliferation of SARS-CoV-2. It has exhibited potent antiviral activity against various human coronaviruses and notable off-target selectivity [20]. Additionally, animal experiments have confirmed its favorable safety profile and demonstrated its capacity to reduce lung viral load in a mouse model engineered to simulate SARS-CoV-2 infection [20]. Preclinical investigations have found no evidence of mutagenic interactions with DNA [21]. Moreover, a phase I clinical trial involving healthy volunteers showed plasma concentrations surpassing the threshold of in vitro cellular antiviral potency [21].
Conversely, Ivermectin is a broad-spectrum antiparasitic medication that initially garnered attention due to its in vitro ability to inhibit SARS-CoV-2 replication, sparking interest in its potential as a COVID-19 treatment [22,23,24]. However, its off-label use for this purpose is advised cautiously. Available in both human and veterinary formulations, Ivermectin is widely used clinically to combat parasites like Onchocerca volvulus (which causes river blindness) and Strongyloides stercoralis. Nonetheless, at elevated doses, it can lead to potentially severe adverse effects such as nausea, vomiting, diarrhea, dizziness, and even fatalities. Large-scale clinical trials investigating its efficacy against COVID-19 have yielded mixed results. While some studies fail to demonstrate significant benefits in reducing symptoms or duration, others suggest potential advantages [25,26]. Consequently, a consensus based on consistent, high-quality evidence regarding Ivermectin’s role in treating COVID-19 has yet to emerge in the literature.
Driven by the potential therapeutic implications, we conducted an in silico study to investigate the interactions between Ivermectin and Nirmatrelvir with . Employing molecular docking, we first examined how Ivermectin binds to , specifically focusing on its interaction with protomer A, denoted as . Subsequently, we conducted molecular dynamics simulations for both Ivermectin and Nirmatrelvir bound to , facilitating a comprehensive analysis of their interaction profiles at a molecular level.
2. Metodology
2.1. Electronic Properties
We investigated the electronic properties of Ivermectin and Nirmatrelvir using density functional theory (DFT) and frontier molecular orbital (FMO) analyses within the Gaussian 16 software suite [27]. Molecular geometries were optimized through the semi-empirical Parameterization Method 6 (PM6) coupled with a steepest descent algorithm [28]. Following optimization, single-point energy calculations utilizing restricted DFT [29] were conducted to determine the energies of the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and their energy gap [30]. Additionally, Mulliken population analysis and electric charge distribution were performed. For a comprehensive DFT analysis, Becke’s three-parameter Lee-Yang-Parr (B3LYP) exchange-correlation functional [31] and the def2-TZVP (valence triple-zeta polarization) basis set [32] were employed.
2.2. Molecular Docking
Ligands and proteins were prepared using AutoDock Tools 1.5.6 (ADT) [33]. Molecular docking simulations were employed to examine the non-covalent binding interactions between the [20] (PDB: 7VH8) complex and Ivermectin. The crystallographic structure from the PDB contained the complex of and the inhibitor Nirmatrelvir. Autodock Vina, integrated into PyRx, was integrated with the genetic algorithm set as the default configuration for the docking simulations. The characterization of interactions between Ivermectin and was facilitated using ChimeraX and Discovery Studio programs. The simulation box encompassing the entire protein had dimensions of , with a center point at (-1.74, 18.92, -26.05) Å. Ligand screening was performed with an accuracy of ±2 Å for ligand positions and ±0.01 Kcal/mol for binding affinities.
2.3. Molecular Dynamics
Molecular dynamics (MD) simulations were conducted using the Nanoscale Molecular Dynamics (NAMD) 2.9 computational package [34] along with the CHARMM36 force field [35], using a time step of 1.0 fs. The model protein/ligand complexes were maintained under constant pressure and temperature conditions within the NPT ensemble at 300 K and 1 atm, respectively. Equilibration occurred throughout 10 ns, followed by a 100 ns production run. To assess the temporal evolution of the system, Root-mean-square deviation (RMSD) and Fluctuation (RMSF) calculations were performed using Visual Molecular Dynamics (VMD) 1.9.1 [36]. Subsequently, re-docking was executed after assembling interactions derived from the initial docking poses.
3. Results
Initially, DFT calculations were performed to understand the behavior of molecules when isolated, particularly concerning the calculated frontier molecular orbitals (HOMO and LUMO). Figure 1 displays, on the left side, the results for HOMO and LUMO of the Ivermectin molecule, while, on the right side, it presents the same characteristics for Nirmatrelvir.
The figure analysis reveals favorable features for the interaction of Ivermectin with its target protein, indicating a potential enhancement of bioactivity. The high HOMO energy of -6.08 eV, coupled with a low LUMO energy of -0.96 eV, results in a significant energy gap of 5.12 eV for Ivermectin. In the case of Nirmatrelvir, the HOMO energy is -6.75 eV, and the LUMO energy is -1.65 eV. Consequently, the energy gap is 5.10 eV. This wide gap shows an efficient charge transfer within Ivermectin, potentially facilitating stable interactions with the target protein. Moreover, the high energy gap implies strong inhibition efficiency due to the minimum energy required for electron removal from the HOMO.
It is observed that ideal inhibitory molecules readily accept and donate electrons, exhibiting an electron-rich character associated with superior inhibition potential. Consistent with these principles, the previously discussed calculated LUMO energy for Ivermectin shows a good electron-accepting capacity, further supporting its potential as an effective inhibitor. Regarding the overall electronic characteristics of both Ivermectin and Nirmatrelvir, we find they are similar in both molecules. This trend implies that Ivermectin, like Nirmatrelvir, can potentially be used as an inhibitor ligand for , hindering the replication process of SARS-CoV-2.
In molecular docking, we prioritized ligand poses exhibiting more favorable binding affinity, as lower G free energy scores indicated, reflecting the most probable and stable binding modes [37]. The docking simulations revealed favorable binding affinities for Ivermectin, with the top-scoring pose presenting a G of -9.0 kcal/mol.
In this perspective, Figure 2 details the ligand- interactions. At the top, we analyze Nirmatrelvir. This complex was obtained from Zhao and colleagues [20]. We observed that the molecule establishes interactions near the N-terminus of , encompassing methionines 49 and 165 (MET), histidine 41 (HIS), phenylalanine 140 (PHE), glycine 143 (GLY), threonine 190 (THR), cysteine 145 (CYS), glutamate 166 (GLU), glutamine 189 and 192 (GLN), and proline 168 (PRO). On the other hand, at the bottom of this figure, Ivermectin, in the reported best energy configuration, occupies a distinct pocket closer to the C-terminus of , interacting with alanine 154 (ALA), aspartic acid 197 (ASP), leucines 286 and 287 (LEU), methionine 176 (MET), and proline 168 (PRO). Remarkably, while Nirmatrelvir and Ivermectin bind to separate pockets near the N- and C-termini, respectively, they converge towards the central region, as evidenced by their shared interaction with proline 168 (PRO). The dominant interactions of Nirmatrelvir involve conventional H-bonds. In contrast, those of Ivermectin are dominated by alkyl and pi-alkyl interactions, showing that Nirmatrelvir interacts more strongly with .
Building upon the molecular docking results for Ivermectin and the Nirmatrelvir/ complex discussed earlier, MD simulations of these systems were conducted to explore the interaction dynamics of both molecules with under ambient temperature and pressure. This aimed to verify their stability in the presence of external factors and their persistence as inhibitors throughout the simulation.
Figure 3 illustrates the temporal evolution of RMSD (top left panel) and RMSF (top right panel) simultaneously for Nirmatrelvir (blue curves) and Ivermectin (red curves) over the 100 ns simulation time. The lower panels depict the number of h-bonds for Nirmatrelvir (left panel) and Ivermectin (right panel). The RMSD values for best pose score for with Nirmatrelvir are 2.0 Å, and with Ivermectin, it is 1.87 Å. These values, hovering around 2 Å, suggest that the reference structures are highly similar to the initial structures, signifying that both ligands remain securely bound to the cavity throughout the simulation time. The RMSD curves exhibit notable similarity, characterized by low fluctuations, showing that Ivermectin has the potential to act as an effective ligand against . Moreover, in the interval between 25 ns and 45 ns of simulation, our results indicate that Nirmatrelvir experiences more pronounced RMSD fluctuations than Ivermectin, followed by increased stability beyond this point. These results imply a more pronounced stability of in complex with Ivermectin. RMSF, on the other hand, analysis was employed to assess the dynamic behavior of the protein when bound to Nirmatrelvir and Ivermectin. Both docking complexes exhibited minimal amino acid fluctuations throughout the 100 ns simulations, as illustrated by their RMSF plots. Thus, both ligands significantly stabilize the protein.
In terms of h-bond numbers, Figure 3 (lower panels) shows that the complex with Ivermectin exhibited superior stabilization over 100 ns compared to the complex with Nirmatrelvir. The temporal progression of hydrogen bond formation between the protein and ligands was also scrutinized to glean insights into molecular interactions. The analysis uncovered that Nirmatrelvir initially formed four hydrogen bonds with the protein, with two and three persisting and alternating throughout the simulation. In contrast, Ivermectin formed two to three initial hydrogen bonds, with only one remaining stable throughout the simulation.
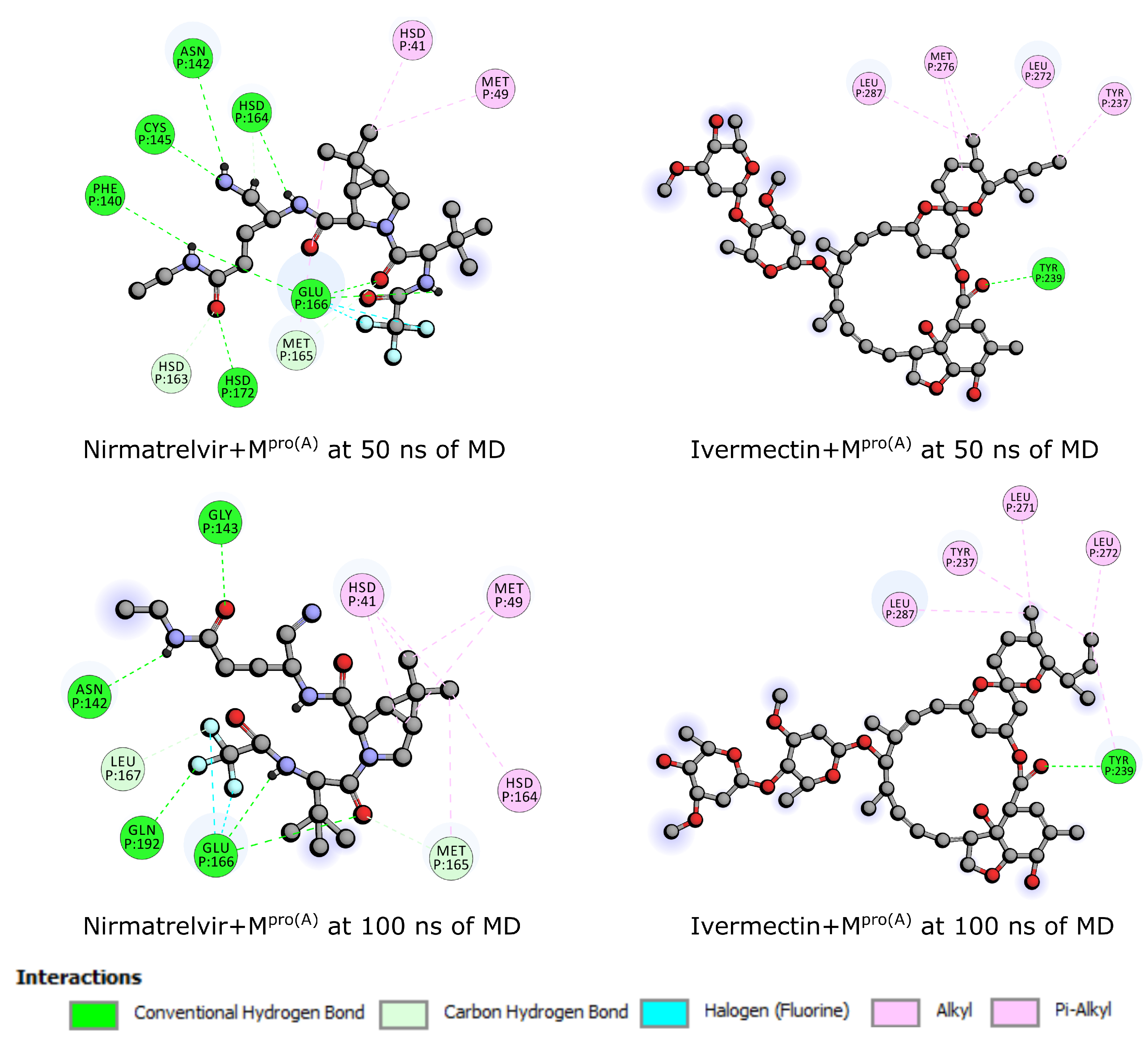
From the above-discussed MD simulations, we emphasize two snapshots at 50 ns and 100 ns (see Figure 4). These visualizations unveil a notable difference in conformational flexibility, wherein both ligands maintain binding to throughout the simulations. However, Nirmatrelvir exhibits a high conformational change compared to Ivermectin.
Finally, Figure 5 delineates the interaction profiles of Nirmatrelvir and Ivermectin with at 50 ns and 100 ns, representing the molecular dynamics snapshots discussed earlier. At the 50 ns mark, Nirmatrelvir (right-upper panel) engages with amino acids, including methionine 49 and 165 (MET), histidine 41, 163, and 164 (HIS), phenylalanine 140 (PHE), cysteine 145 (CYS), glutamate 166 (GLU), and asparagine 142 (ASN). In the subsequent 100 ns interval (left-upper panel), the Nirmatrelvir/ complex maintains interactions with methionine 49 and 165 (MET), histidine 41, glutamate 166 (GLU), asparagine 142 (ASN), glutamine 192 (GLN), and leucine 167 (LEU). Conversely, Ivermectin’s interactions persist over 50 ns and 100 ns (lower panels) with amino acids tyrosine 237 and 239 (THR) and leucine 271, 272, and 287 (LEU). Notably, Ivermectin exhibits a more enduring interaction pattern with the same amino acids over time, while Nirmatrelvir interacts with approximately half of the amino acids at 50 ns compared to 100 ns. Consistent with the docking results, Nirmatrelvir predominantly relies on conventional hydrogen bonds, whereas Ivermectin primarily engages in alkyl and pi-alkyl interactions. Consequently, the more robust nature of hydrogen bonds contributes to Nirmatrelvir forming a higher number of interactions with .
4. Summary and Conclusions
In conclusion, this study aims to investigate the distinct binding modes exhibited by Nirmatrelvir and Ivermectin with the SARS-CoV-2 , with the perspective of understanding the nature of these molecules as inhibitors in the viral replication process. In this regard, Nirmatrelvir demonstrates engagement with the , forming numerous interactions involving amino acids such as methionine 49 and 165 (MET), histidine 41, 163, and 164 (HIS), phenylalanine 140 (PHE), cysteine 145 (CYS), glutamate 166 (GLU), and asparagine 142 (ASN). Through molecular dynamics simulations, the RMSD analysis reveals a stable binding of Nirmatrelvir, with an RMSD value of around 2.0 Å, indicating its secure attachment to the binding pocket over the 100 ns simulation period.
On the other hand, Ivermectin exhibits good stability, maintaining prolonged interactions with specific amino acids, such as tyrosine 237 and 239 (THR) and leucine 271, 272, and 287 (LEU). The RMSD for the Ivermectin- complex is 1.87 Å, reflecting its consistent and secure binding throughout the simulation. It is worth noting that Ivermectin interacts with approximately the same set of amino acids at both 50 ns and 100 ns, demonstrating enduring interaction patterns over time.
The diversified exploration of ligand-protein interactions emphasizes the intricate nature of these binding profiles. Nirmatrelvir, relying on conventional hydrogen bonds, engages in a significant number of stronger interactions with , contributing to its strong binding. However, Ivermectin primarily utilizes alkyl and pi-alkyl interactions, indicating a different binding mode.
Author Contributions
Y.A.O.S., K.S.B., M.L.P.J, and F.L.L.M.: Data curation, Formal analysis, Methodology, Prepared Figures, and Writing – Original draft preparation. J.T.S., R.G., U.L.F., and L.A.R.J.: Conceptualization, Funding acquisition, and Writing – Reviewing and Editing. All authors reviewed the manuscript.
Funding
Brazilian Research Councils CNPq, CAPES, FAPEPI, FAPDF, and FAPESP.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors gratefully acknowledge the financial support from Brazilian research agencies CNPq, CAPES, and FAP-DF. L.A.R.J acknowledges the financial support from Brazilian Research Council FAP-DF grants 00193.00001808/2022-71, 00193-00001247/2021-20, 00193-00000857/2021-14, 00193-00001247/2021-20, and 00193-00000811/2021-97 and CNPq grants 302236/2018-0 and 350176/2022-1. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 88887.691997/2022-00. M.L.P.J acknowledges the financial support from FAP-DF grant 00193-00001807/2023-16. R.G., M.L.P.J, and L.A.R.J acknowledge CENAPAD-SP for providing computational facilities. F.L.L.M. and L.A.R.J. acknowledge General Attorney of the Union - AGU grant 697.935/2019. L.A.R.J. acknowledges Núcleo de Computação de Alto Desempenho (NACAD) for providing the computational facilities through the Lobo Carneiro supercomputer. L.A.R.J. acknowledges the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil) for providing HPC resources of the SDumont supercomputer (URL: http://sdumont.lncc.br), which have contributed to the research results reported within this paper. The authors acknowledge the technical and computational support from the LATITUDE Laboratory, University of Brasília, the TED 01/2019 grant from Advocacia Geral da União (Grant AGU 697.935/2019), the Administrative Council for Economic Defense – CADE (TED 08700.000047/2019-14), the TED 01/2021 grant from the National Social Assistance Secretariat – SNAS/DGSUAS/CGRS, the Dean of Research and Innovation – DPI/UnB, the SISTER City Project – Safe and Real-Time Effective Intelligent Systems for Smart Cities (Grant 625/2022), and the Foundation for Research Support of the Federal District - FAP/DF.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Atzrodt, C.L.; Maknojia, I.; McCarthy, R.D.; Oldfield, T.M.; Po, J.; Ta, K.T.; Stepp, H.E.; Clements, T.P. A Guide to COVID-19: a global pandemic caused by the novel coronavirus SARS-CoV-2. Febs J. 2020, 287, 3633–3650. [Google Scholar] [CrossRef] [PubMed]
- Jayaweera, M.; Perera, H.; Gunawardana, B.; Manatunge, J. Transmission of COVID-19 virus by droplets and aerosols: A critical review on the unresolved dichotomy. Environ. Res. 2020, 188, 109819. [Google Scholar] [CrossRef] [PubMed]
- Louten, J. Virus transmission and epidemiology. Essent. Hum. Virol. 2016, 71. [Google Scholar]
- Asadi, S. Airborne Infectious Disease Transmission Via Expiratory Aerosol Particles and Aerosolized Fomites; University of California: Davis, CA, USA, 2020. [Google Scholar]
- Weiss, R.A.; Sankaran, N. Emergence of epidemic diseases: Zoonoses and other origins. Fac. Rev. 2022, 11. [Google Scholar] [CrossRef]
- Rajeev, R.; Prathiviraj, R.; Kiran, G.S.; Selvin, J. Zoonotic evolution and implications of microbiome in viral transmission and infection. Virus Res. 2020, 290, 198175. [Google Scholar] [CrossRef] [PubMed]
- WORLDOMETER. COVID-19 CORONAVIRUS PANDEMIC. 2024. Available online: https://www.worldometers.info/coronavirus (accessed on 5 March 2024).
- Pellis, L.; Scarabel, F.; Stage, H.B.; Overton, C.E.; Chappell, L.H.; Fearon, E.; Bennett, E.; Lythgoe, K.A.; House, T.A.; Hall, I. Challenges in control of COVID-19: short doubling time and long delay to effect of interventions. Philos. Trans. R. Soc. 2021, 376, 20200264. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2020, 65, 2716–2746. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Perera, L.; Tillekeratne, L.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Agost-Beltrán, L.; de la Hoz-Rodríguez, S.; Bou-Iserte, L.; Rodríguez, S.; Fernández-de-la Pradilla, A.; González, F.V. Advances in the development of SARS-CoV-2 Mpro inhibitors. Molecules 2022, 27, 2523. [Google Scholar] [CrossRef]
- Alugubelli, Y.R.; Geng, Z.Z.; Yang, K.S.; Shaabani, N.; Khatua, K.; Ma, X.R.; Vatansever, E.C.; Cho, C.C.; Ma, Y.; Xiao, J. A systematic exploration of boceprevir-based main protease inhibitors as SARS-CoV-2 antivirals. Eur. J. Med. Chem. 2022, 240, 114596. [Google Scholar] [CrossRef] [PubMed]
- Andi, B.; Kumaran, D.; Kreitler, D.F.; Soares, A.S.; Keereetaweep, J.; Jakoncic, J.; Lazo, E.O.; Shi, W.; Fuchs, M.R.; Sweet, R.M. Hepatitis C virus NS3/4A inhibitors and other drug-like compounds as covalent binders of SARS-CoV-2 main protease. Sci. Rep. 2022, 12, 12197. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.Z.; Yasuo, N.; Sekijima, M. Screening for inhibitors of main protease in sars-cov-2: In silico and in vitro approach avoiding peptidyl secondary amides. J. Chem. Inf. Model. 2022, 62, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zeng, X.; Duan, Y.; Yang, Z.; Ma, Y.; Yang, H.; Yang, X.; Liu, X. Molecular mechanism of ensitrelvir inhibiting SARS-CoV-2 main protease and its variants. Commun. Biol. 2023, 6, 694. [Google Scholar] [CrossRef] [PubMed]
- Di Chio, C.; Previti, S.; Amendola, G.; Ravichandran, R.; Wagner, A.; Cosconati, S.; Hellmich, U.A.; Schirmeister, T.; Zappalà, M.; Ettari, R. Development of novel dipeptide nitriles as inhibitors of rhodesain of Trypanosoma brucei rhodesiense. Eur. J. Med. Chem. 2022, 236, 114328. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Malla, T.R.; Owen, C.D.; Tumber, A.; Brewitz, L.; McDonough, M.A.; Salah, E.; Terasaka, N.; Katoh, T.; Lukacik, P. In vitro selection of macrocyclic peptide inhibitors containing cyclic γ2, 4-amino acids targeting the SARS-CoV-2 main protease. Nat. Chem. 2023, 1–8. [Google Scholar]
- Tyndall, J.D. S-217622, a 3CL protease inhibitor and clinical candidate for SARS-CoV-2. J. Med. Chem. 2022, 65, 6496–6498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2022, 13, 689–693. [Google Scholar] [CrossRef]
- Ahmad, B.; Batool, M.; Ain, Q.u.; Kim, M.S.; Choi, S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int. J. Mol. Sci. 2021, 22, 9124. [Google Scholar] [CrossRef]
- Zaidi, A.K.; Dehgani-Mobaraki, P. The mechanisms of action of ivermectin against SARS-CoV-2—an extensive review. J. Antibiot. 2022, 75, 60–71. [Google Scholar] [CrossRef]
- Hill, A.; Garratt, A.; Levi, J.; Falconer, J.; Ellis, L.; McCann, K.; Pilkington, V.; Qavi, A.; Wang, J.; Wentzel, H. Retracted: meta-analysis of randomized trials of ivermectin to treat SARS-CoV-2 infection. In Open Forum Infectious Diseases; Oxford University Press: Oxford, MI, USA, 2021; Volume 8, p. ofab358. [Google Scholar]
- Sharun, K.; Dhama, K.; Patel, S.K.; Pathak, M.; Tiwari, R.; Singh, B.R.; Sah, R.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J.; Leblebicioglu, H. Ivermectin, a new candidate therapeutic against SARS-CoV-2/COVID-19, 2020.
- Cobos-Campos, R.; Apiñaniz, A.; Parraza, N.; Cordero, J.; García, S.; Orruño, E. Potential use of ivermectin for the treatment and prophylaxis of SARS-CoV-2 infection. Curr. Res. Transl. Med. 2021, 69, 103309. [Google Scholar] [CrossRef] [PubMed]
- Buonfrate, D.; Chesini, F.; Martini, D.; Roncaglioni, M.C.; Fernandez, M.L.O.; Alvisi, M.F.; De Simone, I.; Rulli, E.; Nobili, A.; Casalini, G. High-dose ivermectin for early treatment of COVID-19 (COVER study): a randomised, double-blind, multicentre, phase II, dose-finding, proof-of-concept clinical trial. Int. J. Antimicrob. Agents 2022, 59, 106516. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.e.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16, 2016.
- Stewart, J.J. Application of the PM6 method to modeling proteins. J. Mol. Model. 2009, 15, 765–805. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G. Density functional theory. Annual Review of Physical Chemistry 1983, 34, 631–656. [Google Scholar] [CrossRef]
- Sham, L.J.; Schlüter, M. Density-functional theory of the energy gap. Phys. Rev. Lett. 1983, 51, 1888. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Forli, S. Using AutoDock 4 and AutoDock vina with AutoDockTools: a tutorial. Scripps Res. Inst. Mol. Graph. Lab. 2012, 10550, 1000. [Google Scholar]
- Phillips, J.C.; Zheng, G.; Kumar, S.; Kalé, L.V. NAMD: Biomolecular simulation on thousands of processors. SC’02: Proceedings of the 2002 ACM/IEEE Conference on Supercomputing. IEEE, 2002, pp. 36–36. [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell Jr, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Yamashita, Y.; Mashimo, T.; Nakamura, H. Prediction of protein- compound binding energies from known activity data: docking-score-based method and its applications. Mol. Inform. 2018, 37, 1700120. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Analysis of the frontier molecular orbitals (FMOs) of Ivermectin and Nirmatrelvir, depicting their LUMO, HOMO, and the energy gap (eV) between them.
Figure 1.
Analysis of the frontier molecular orbitals (FMOs) of Ivermectin and Nirmatrelvir, depicting their LUMO, HOMO, and the energy gap (eV) between them.
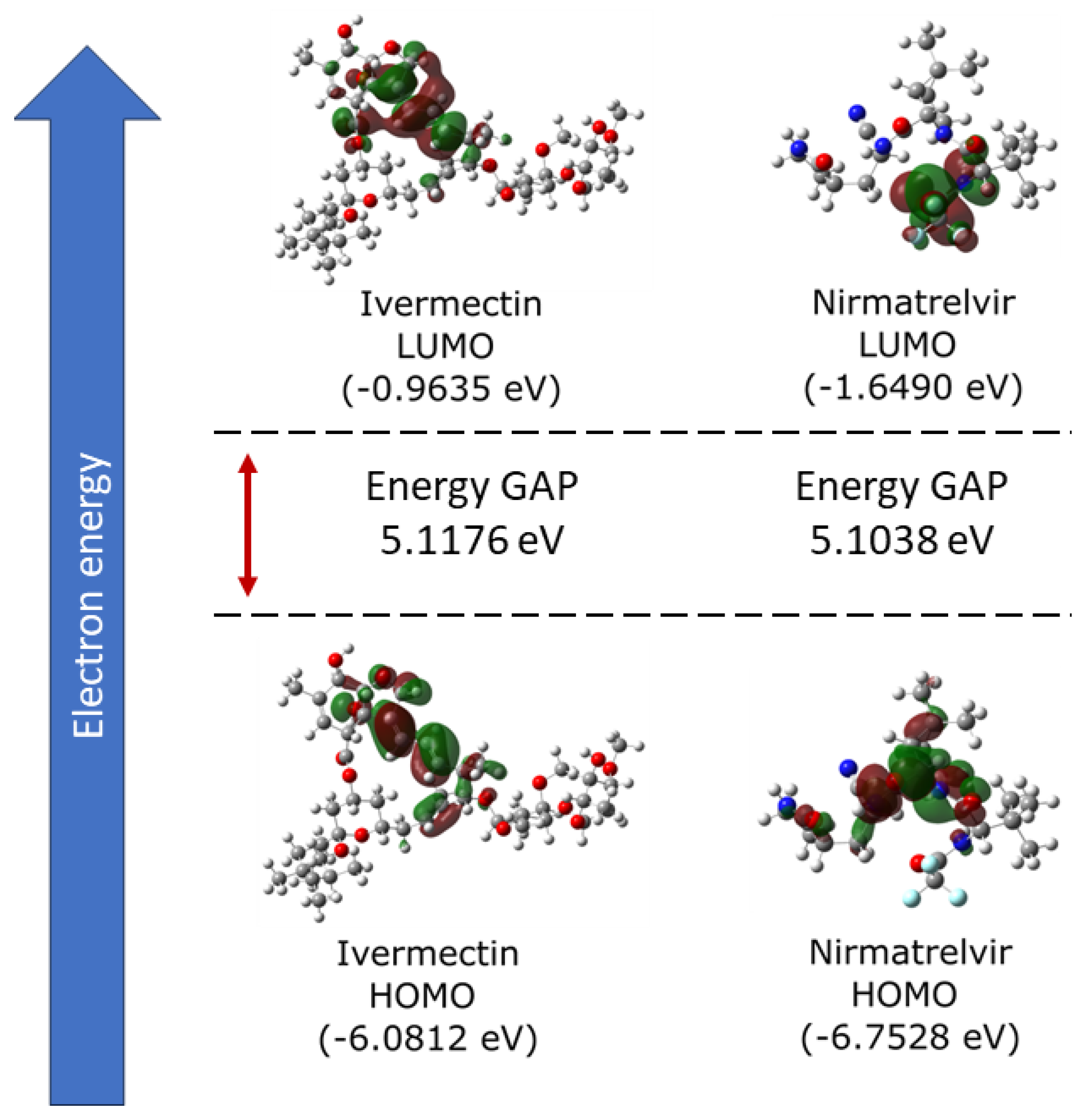
Figure 2.
Crystal Sctruture of Nirmatrelvir and from RCSB PDB Protein Data Bank (up) and Sctruture of Ivermectin and from molecular docking (down). The blue to red-color band represents the orientation from the N-terminus to the C-terminus of .
Figure 2.
Crystal Sctruture of Nirmatrelvir and from RCSB PDB Protein Data Bank (up) and Sctruture of Ivermectin and from molecular docking (down). The blue to red-color band represents the orientation from the N-terminus to the C-terminus of .
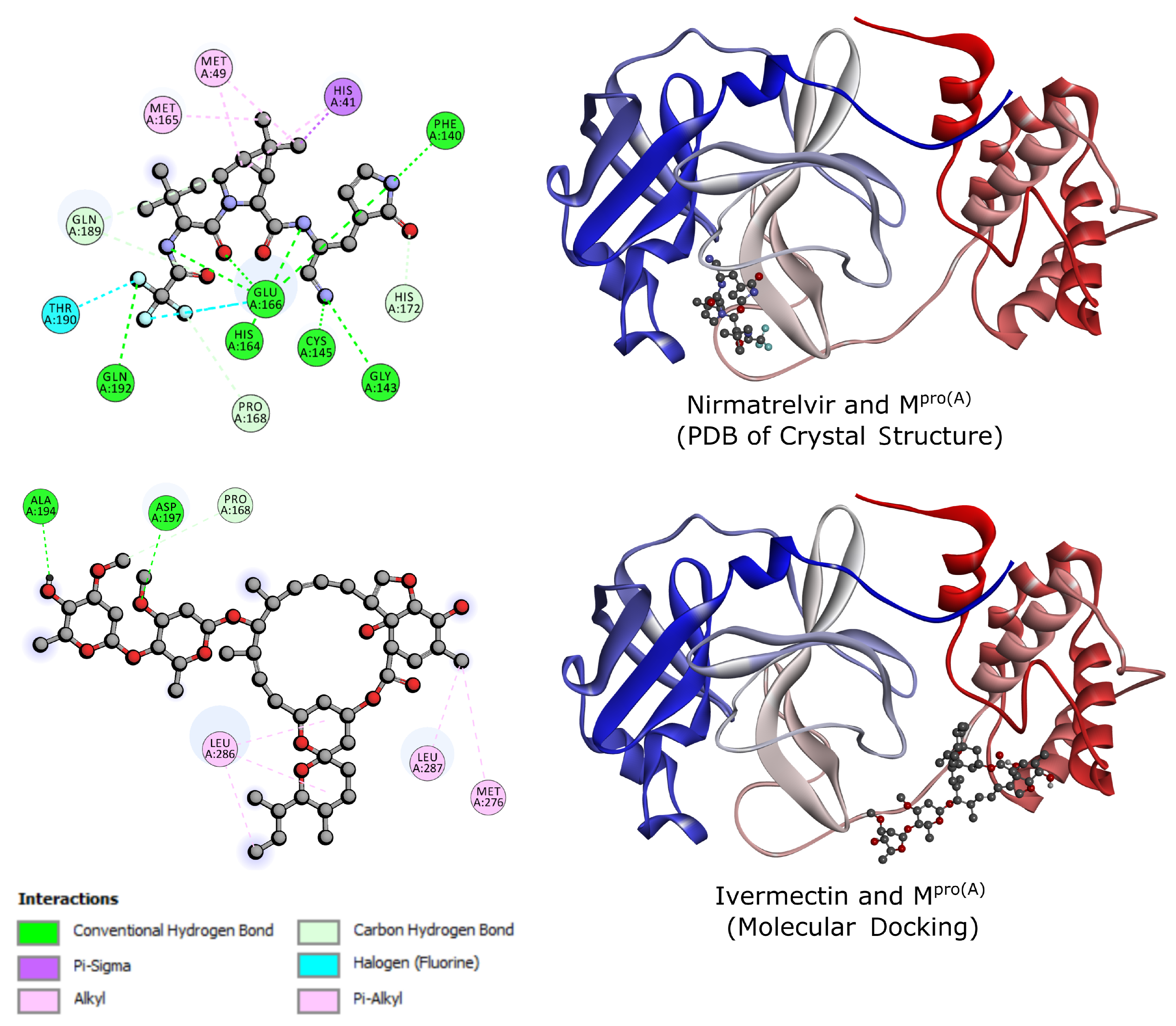
Figure 3.
Root Mean Square Deviation (upper right), Root Mean Square Fluctuation (upper left), and Number of Hydrogen Bonds (down) of in complex with Nirmatrelvir and Ivermectin throughout the simulation.
Figure 3.
Root Mean Square Deviation (upper right), Root Mean Square Fluctuation (upper left), and Number of Hydrogen Bonds (down) of in complex with Nirmatrelvir and Ivermectin throughout the simulation.
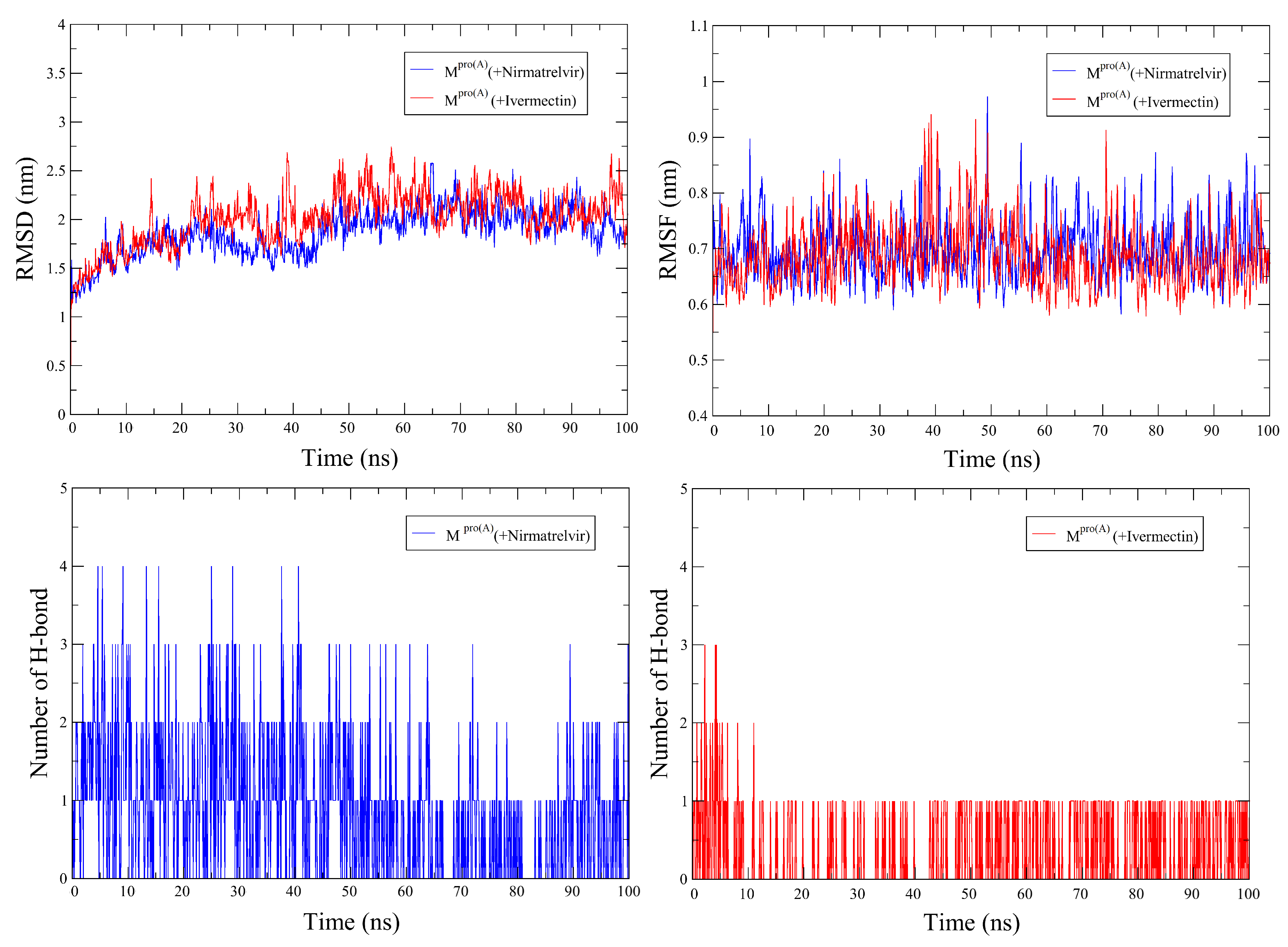
Figure 4.
By employing the optimal docking pose (see Figure 2), we examined snapshots at 50 and 100 ns intervals. These depictions reveal a significant disparity in conformational flexibility: while both ligands consistently maintain their binding to throughout the simulations, Nirmatrelvir exhibits a markedly more pronounced conformational variation compared to Ivermectin.
Figure 4.
By employing the optimal docking pose (see Figure 2), we examined snapshots at 50 and 100 ns intervals. These depictions reveal a significant disparity in conformational flexibility: while both ligands consistently maintain their binding to throughout the simulations, Nirmatrelvir exhibits a markedly more pronounced conformational variation compared to Ivermectin.

Figure 5.
Interaction of Nirmatrelvir and Ivermectin with at 50 ns and 100 ns.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



