Submitted:
28 April 2024
Posted:
29 April 2024
You are already at the latest version
Abstract
In recent years, the chiral biological effects of nanomedicines have become a subject of widespread interest. The research focus has been on understanding the influence of material chirality on cellular transcription and metabolism. Stress granules are membraneless organelles formed through liquid-liquid phase separation of G3BP1 proteins and related compartments. They have been extensively studied and proven to be closely associated with cellular damage repair and metabolism. However, the role and mechanism of chiral nanomaterials in modulating stress granules remain unknown. The aim of this study was to investigate the expression and structural characteristics of stress granules under the individual influence of chiral nanomaterials and in combination with chemotherapy. To achieve this, we constructed a library of chiral ligand-modified materials and employed immunofluorescence, live-cell imaging, and fluorescence recovery after photobleaching assays. We also used proximity labeling techniques combined with proteomics analysis to identify the protein corona adsorbed on the surface of the nanomaterials and explore their relationship with nanomaterial chirality. Our results demonstrate that the assembly of stress granules is chiral-dependent and can be regulated by chiral nanomaterials. Furthermore, we achieved enhanced chemotherapy sensitivity in cancer cells and protected normal cells by exploiting the chiral-dependent modulation of material assembly in stress granules. This study provides important insights into the regulation of membraneless cellular structures based on chiral biological effects.
Keywords:
nanomedicine
; chirality
; stress granules
; liquid-liquid phase separation
; chemotherapy
Introduction
Chirality represents a fundamental characteristic of natural biological molecules. The distinct chiral structures of molecules with chiral centers exert significant influences on their metabolic patterns within cellular environments [1,2]. Notably, the therapeutic efficacy of L-dopa in Parkinson’s disease treatment is well-established, as it can bypass the blood-brain barrier and covert to dopamine through decarboxylation [3]. Conversely, the enantiomer D-dopa lacks therapeutic effects and, instead, accumulates in the body, leading to adverse reactions [4]. Recent investigations have unveiled that the regulatory role of chirality in metabolic behavior extends beyond small molecules to include nanomaterials [5,6], giving rise to the emerging field of chiral nanomedicine. In pursuit of this objective, researchers have devised two key strategies: intrinsic chiral synthesis, involving surface microstructure modifications of nanomaterials [7] through ligand-assisted synthesis, and chiral small molecule surface modification [8]. These approaches enable the construction of diverse chiral nanomedicines. Compared to intrinsic chiral synthesis, chemical modification strategies offer benefits such as operational simplicity and precise control over the chirality of product [9]. The application of these strategies has garnered increasing attention in the realm of chiral nanomedicine research [10,11].
Mounting evidence suggests that the rational modification of chiral ligands enables the fabrication of nanomedicines that elicit distinct biological responses in cancerous and normal cells post uptake. For instance, Li et al. [12] demonstrated a pronounced disparity in toxicity between L-glutathione-modified (the tripeptide, γ-L-glutamyl-L-cysteinyl-glycine, known as glutathione) and D-glutathione-modified nanomaterials, specifically CdTe quantum dots. Moreover, in another investigation [13], it was revealed that divergent chirality-related outcomes were induced in cells received chiral gold clusters. Of note, D-glutathione-modified nanomaterials exhibited enhanced cytotoxicity towards cancer cells compared to their L-glutathione counterparts [12], underscoring the intricate interplay between cell toxicity, the chirality of nanomedicines, the chiral properties of ligands, and the inherent structure of materials. Numerous research findings suggest that exposure to chiral drugs induces cellular stress, which is tightly correlated with elevated intracellular levels of free radicals. For instance, D-glutathione-modified gold clusters were found to provoke mitochondrial release of free radicals, resulting in upregulated mRNA expression associated with steroid synthesis and peroxisome proliferator-activated receptor pathways (PPAR) in gastric cancer cells [13]. Additionally, Zhang et al. [14] accomplished in vivo cleavage of pathological Aβ amyloid aggregates by utilizing ROS provoked by chiral FexCuySe complexes, leading to improved cognitive function in murine models of Alzheimer’s disease. Furthermore, the involvement of non-free radical-dependent mechanisms in the regulation of cellular stress by chiral nanomedicines remains elusive, highlighting the significance of further elucidation of this issue to advance clinical applications in the field.
In the past decade, research has shown that in addition to metabolic and transcriptional regulation, cells form an unstable structure called stress granules (SGs) to alleviate damage caused by stress stimuli [15,16]. Stress granules are regulated by proteins such as G3BP1, which recruit specific proteins to form transient membraneless condense to protect certain mRNA and translation components from degradation or destruction when cells face stress conditions (e.g., unfolded protein response or oxidative stress) [17,18,19]. Recent studies have also shown that SGs can reduce stress damage by recruiting and imprisoning cell death execution proteins (such as Caspase 3) [20], further confirming the important role of emergency granules in repairing cellular stress damage. Meanwhile, it is yet unclear whether and how SGs affect the metabolism of chiral nanotherapeutics. In this study, we used a set of chiral ligands with different molecular structures to functionalize gold nanoparticles and construct a library of chiral nanomaterials. First, we compared the ability of different chiral nanomaterials to stimulate SGs in cancer cells and endothelial cells using immunofluorescence analysis. We then analyzed the differences in SG condensation content in different types of cells under combined stimulation of chiral nanomaterials and chemotherapeutic drugs, and the results confirmed that chiral materials have completely opposite regulatory effects on the phase separation behavior of G3BP1 in cancer cells and endothelial cells. In addition, through fluorescence recovery after photobleaching experiments (FRAP) [21], we explored the fluidity of SGs under the action of different chiral nanomaterials and found that the gelation of droplets may be a key factor leading to the weakening of the protective effect of SGs on cancer cells by chiral nanotherapeutics. To better understand the regulatory mechanism of chiral nanomaterials on SG assembly and disassembly, we used proximity labeling technology [22] combined with proteomic analysis to identify and analyze the types and functions of proteins bound by different types of materials in cells. Our research results first confirmed that the chirality of ligands can affect the adsorption of chaperons and ubiquitinated proteins on cell surfaces, thereby affecting SG assembly. Our study shows that by simply changing the chirality of nanodrugs, it is possible to selectively inhibit the liquid-liquid phase separation of G3BP1 in cancer cells, thereby increasing chemotherapy sensitivity, and reduce chemotherapy damage by enhancing SG assembly in endothelial cells. Our study confirms the potential of chiral drugs as chemotherapy sensitizers or chemotherapy damage blockers, whose effectiveness is closely related to the ability of material chirality to regulate SG assembly. This finding provides important reference significance for the development of targeted membrane-free cell structure new nanodrugs in the future.
Materials and Methods
Materials
Gold(Ⅲ) chloride trihydrate (HAuCl4·3H2O) (>99.9%), ascorbic acid (AA), cetyltrimethylammonium chloride (CTAC, 25% wt/vol), cetyltrimethylammonium bromide (CTAB, ≥98%) were all purchased from Sigma-Aldrich (United States). Sodium borohydride and ethanol were obtained from the SinoPharm (P. R. China). Cy5-PEG-SH (2 kDa), D-Histidine (≥99%), L-Histidine (≥99%), L-Cysteine (≥99%), dimethyl sulfoxide (DMSO, anhydrous) and 4-[3-(Trifluoromethyl)-3H-diazirin-3-yl] benzylamine Hydrochloride (>98%) were purchased from Aladdin (P. R. China). D-Cysteine (>98%), L-penicillamine (>98%), D-penicillamine (>99%), cis-Diammineplatinum dichloride (>65%), puromycin dihydrochloride (>98%) were purchased from Macklin (P. R. China). All chemicals were used as received without further purification. Deionized (DI) water with a resistivity of 18.2 MΩ·cm was used in all experiments, which was prepared using a Sartorius ultrapure water system (Arium mini, Germany).
Preparation of Chiral Nanomaterials
Au nanospheres were synthesized by using seed-mediated growth by following a published protocol [23]. In detail, Au seeds were prepared by mixing HAuCl4 aqueous solution (5 mL, 0.25 mM) sequentially with CTAB solution (5 mL, 100 mM) and an ice-cold aqueous NaBH4 solution (0.6 mL, 10 mM). Then, the brownish mixture was left undisturbed at 37 oC for 3 h to ensure the complete decomposition of excess NaBH4. Next, HAuCl4 solution (2 mL, 0.5 mM) were sequentially mixed with CTAC solution (2 mL, 200 mM) and aqueous AA solution (1.5 mL, 100 mM), followed by the introduction of freshly prepared seeds (10 μL) that give rise to 30-nm Au nanospheres. The unmodified Au nanospheres were collected by centrifugation at 15,000 rpm for 30 min, washed once and redispersed in DI water.
For the chiral modification, chiral ligands (20 mg/mL, including L-Histidine, D-Histidine, L-Cysteine, D-Cysteine, L-penicillamine, D-penicillamine) were added into the suspension of Au nanospheres in proportion, followed by gently shaking in the dark for 4 hours to prepare different functionalized nanomaterials. To fluorescently label Au nanomaterials for in vitro analyses, Cy5-PEG-SH (2 kDa, 20 mg/mL) was added into the reaction system during this step. Final products were collected via centrifugation at 15,000 rpm for 30 min, followed by repeated washing with DI water to remove unbound molecules.
The morphology of the obtained nanomaterials was characterized by transmission electron microscope (JEM-1400, JEOL, Japan). UV-vis-NIR spectrophotometer (1800PC, JingHua, P.R. China) was performed to obtain spectral properties of nanoparticles.
Immunofluorescence
Cell samples were fixed with 4 % paraformaldehyde (Phygene, P. R. China) for 10 min and rinsed three times with PBS (KeyGEN Bio TECH, P. R. China). Next, they were permeabilized with blocking buffer (Beyotime, P. R. China) and then incubated with primary antibodies at 4℃ overnight: G3BP1 (CST, #61559, dilution: 1:400), Cleaved Caspase-3 (CST, #9664, 1: 1000), Alexa Fluor 555-conjugated Anti-Sodium Potassium ATPase (Abcam, ab274883, 1: 100), Alexa Fluor 647-conjugated Anti-gamma H2A.X (Abcam, ab195189, 1: 200). The samples were washed three times with PBS and further incubated with secondary antibodies: Alexa Fluor-488-Goat-anti-Rabbit IgG H&L (Invitrogen, A-11008, 1: 500), Alexa Fluor 555-Goat-anti-rabbit lgG H&L (CST, #4413, 1:1000). DAPI (Invitrogen, S33025) were used to counterstain the cell nucleus.
Fluorescence Imaging and Graph Processing
Fluorescent images were sampled by total internal reflection fluorescence microscope (Thunder Imager, Leica, Germany) equipped with 405-, 488-, 561-, and 638-nm lasers, using a plan-Apochromat 100× (Numerical aperture: 1.47) and 20× objectives. The captured images were first processed by LAS X software (version 3.2.1) for computational clearing, followed by deconvolution. Imaris software (version 9.8, Oxford Instruments) was used to quantitatively analyze 3D micrographs and track the motions of SGs. Parameters including intensity sum, volume, shortest distance to recognized surface, percentage of colocalization were calculated by combining different modules provided (Surface, Spot).
Treatments
Chiral nanomaterials were dissolved in culture media and incubate with cells for 4 h (1012 nanoparticles/mL). Sodium arsenite dissolved in DMSO was used to stress cells (0.3 mM, 30 min). Cisplatin was dissolved in culture media (50 μM, 125μM, 250 μM, 375 μM, and 500 μM) and prewarmed before introducing to the cell culture medium.
Time-Lapse Study and FRAP
G3BP1-EGFP HeLa cells were seeded into an 18-well plate (ibidi, Germany) at a concentration of 5 × 103 cells/well, cultured overnight at 37 ℃ in 5% CO2. D-/L-Pen-functionalized nanomaterials (4 h) and Cisplatin (250 μM, 2 h) were co incubated with cells sequentially or separately. After washed with prewarmed PBS, these cells were next moved into a stage-top live imaging chamber (ibidi). Fluorescence recovery after photobleaching was performed using an inverted Leica laser scanning confocal microscope with a 100× oil immersion objective with numerical aperture (NA) equal to 1.47 (Apo oil immersion, Vizla, Germany). For each experiment, three droplets with 2.5–3.5 μm diameter were selected. A circular region of interest (2.5 μm diameter) was bleached once (~10 ms) with a bleaching power equal of 30 μW, time-lapses graphs were acquired every 5 s over the course. Data was analyzed using Leica LAS X software.
Photo-Proximity Labeling and Co-IP Experiments
For chemo-etching-assisted photo-proximity labeling, HeLa cells cultured in a 10-cm cell culture dish were treated with chiral nanospheres functionalized with diazirines (1012 nanoparticles/mL, 4 h). After being washed three times with PBS, cells were irradiated (20 min, 360 nm, 15 mW/cm2) in situ to covalently capture the protein from nanoparticles. Afterwards, a 2 mL of RIPA lysis buffer (Beyotime, #P0013D, 50 mM Tris-HCl, pH = 7.4, 150 mM NaCl, 0.25% (wt/vol) sodium deoxycholate) was added to cells (R.T, 10 min). The cell lysate was collected and centrifuged (18,000g for 10 min at 4 °C) to enrich the nanoparticle−protein complexes. Subsequently, the sediment was etched by I3- solution (10 mM, 2 h, 4℃) to release the protein from nanoparticles and purified by using Amicon Ultra (cutoff: 3 kDa, Millipore, United States). Identity of proteins were characterized by lable-free LC-MS analysis as described below.
For Co-IP experiment, 2 mL RIPA lysis buffer (Beyotime, P0013D) containing protease inhibitor was added to the cells after treatment, followed by the quantification of protein concentration by using BCA assay (#23225, Thermo Fisher Scientific). G3BP1 and the proteins that interact with it were enriched using commercially available Kit (#26146, Thermo Fisher Scientific) in combination with G3BP1 antibody (CST, #61559, dilution 1:50).
Proteomics Study
The proteomic analysis was conducted by Verygenome Technology Co. Ltd. (P. R. China). Samples were first reduced, alkylated, and digested with trypsin for 20 h at 37 ℃ to liberate peptides. The resulting mixtures were separated and analyzed by an Easy-nLC 1000 coupled with a Q-Exactive HF hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific). Mass spectra were analyzed using the Proteome Discoverer software. A false discovery rate (FDR) of 0.1 % was set for peptide and protein identification. Gene Ontology (https://geneontology.org) and String tools (https://string-db.org/) were harnessed to comprehensively describe the biological functions, physical and chemical properties of proteins bound to nanomaterials, including the protein class, cellular component, biological process, and molecular function. For pathway analysis, the KEGG database was harnessed.
Statistical Analysis
For all experiments, data was collected at least in triplicate. All data shown as Mean ± standard deviation (SD) or min to max (Box or Violin Plot). One-way analysis of variance (ANOVA) was used to examine the difference between three or more groups when the residues conform to a Gaussian distribution, and the correction for multiple comparisons was tested by Tukey.
Results and Discussion
Stress Conditions Induced by Chiral Nanomaterials
We used three sets of chiral ligands (D/L-cysteine, D/L-histidine, D/L-penicillamine) to functionalize gold nanoparticles. As shown in Figure 1a, the ligand modification had a negligible effect on the morphology of the gold nanoparticles. By comparing the UV/Vis spectra of the ligand-modified materials, we found that ligand binding did not result in peak broadening (Figure 1b), indicating that ligand modification did not cause nanomaterial aggregation. To investigate whether the chirality of the materials is involved in regulating SGs formation, we co-cultured HeLa cells with nanoparticles surface-modified with different optical isomers of ligands whose expression of G3BP1 was immunostained to analyze SGs expression by comparing the volume-normalized fluorescence intensity and aggregation state of G3BP1. The expression of SGs in cancer cells under different treatments is shown in Figure 1c. We observed that the chemical structure and chirality of the ligands significantly affected the uptake of the materials (Figure 1d), which may be related to differences in protein adsorption on the material surface [24,25]. The chirality of the ligands promoted the adsorption of different types of proteins on the material surface (known as the protein corona effect), ultimately changing the pathway and uptake of the materials by the cells [26,27]. Specifically, compared with L-Cys-modified materials, D-Cys-modified nanomaterials showed an 186% increase in cellular uptake, which is the largest uptake difference related to chirality among the three sets of ligands tested. In contrast, the difference in uptake between D-His and L-His-modified nanomaterials was only 9.8%. At the same time, we found that as different nanomaterials were taken up by cells, the average fluorescence intensity and aggregation state of G3BP1 protein in the cells also changed to some extent, indicating that chiral nanoparticles might have the ability to trigger cell stress whose content related tightly with the chirality of nanomaterial. We first analyzed the relationship between nanoparticle uptake and the average fluorescence intensity of G3BP1 puncta. As shown in Figure 1d,e, D-Cys-modified materials showed the highest uptake, but had the lowest expression level of G3BP1 protein compared with L- or racemic Cys-modified nanomaterial groups. Similarly, His-modified materials induced significant stress with extremely low uptake. In contrast to the above results, L-Pen-modified materials induced moderate stress in cells compared to the racemic one, which showed a significantly higher content of stress. This suggests that the chirality of the penicillamine ligand may regulate the way nanomaterial uptake by cells, thereby promoting its cell environment-dependent SGs-inducing ability.
Furthermore, we analyzed the aggregation level of G3BP1 protein before and after stimulation. Using super-resolution imaging, we analyzed the average distance between identified G3BP1 proteins (~120 nm resolution) and their five closest neighboring proteins (Figure 1f). The results showed that protein clustering was the most sensitive to chiral penicillamine. Based on the aforementioned results, we found that the chiral implication of the penicillamine ligand in regulating SGs was higher than that of cysteine and histidine groups, as indicated by the changes in G3BP1 fluorescence intensity and protein clustering analysis. As such, we focused on penicillamine ligand in the subsequent studies.
We further investigate the ability of chiral nanoparticles to induce stress granules in several other cell lines. As shown in Figure 1g,h, interestingly, normal cells exhibited lower uptake efficiency for D-Pen-modified materials compared to L-Pen-modified nanomaterials. This trend was opposite to what we observed in tumor cells and consistent with the reported selectivity of chiral ligands in regulating the uptake of nanoparticles [28]. Importantly, these findings suggest that nanoparticles modified with different chiral penicillamine ligands can effectively regulate the selectivity of drugs towards cancerous and normal endothelial cells, potentially achieving selective stress induction. Additionally, we found that the size of the stress granules induced by the nanomaterials, regardless of choices of molecular composition and chirality, were significantly smaller than that induced by sodium arsenite (Figure 1i), a classic stress granule inducer that has been extensively used for SGs induction, as well as that emerged during heat-shock and osmotic treatment (Figure S1), suggesting a lower stress level caused by nanomaterials. This confirms that the stress response solely dependent on chiral nanoparticles is insufficient to achieve significant modulation of cellular function, especially the induction of SGs.
Modulation of Stress Granule with Chiral Nanomedicines
In the present study, it has been demonstrated that modification of doxorubicin (Dox) with D- and L- Pen can induce mild SGs assembly in cancer cells, which is closely associated with chemotherapy sensitivity and cell survival [29]. Therefore, modulation of SGs has emerged as a potential therapeutic strategy for cancer treatment [30]. However, the influence of chiral materials on SG assembly and chemotherapy sensitivity in cancer cells remains unknown. In this study, we investigated the changes in chemotherapy efficacy by systematically altering the timing and concentration of cisplatin treatment after cellular uptake of chiral nanomaterials. We found that low concentrations of cisplatin (50-125 μM, 1 h, Figure 2a) were insufficient to induce SG formation within the cells. However, when the cisplatin concentration was increased to 250 μM, oxidative stress caused by chemotherapy was able to induce liquid-liquid phase separation of G3BP1, a key component of SGs [31]. The aggregation of G3BP1 reached its peak when the cisplatin concentration was raised to 375 μM (Figure 2b), but further increasing the drug concentration to 500 μM inhibited this process. This inhibition may be attributed to severe cellular damage, resulting in insufficient ATP levels to maintain the stability of membraneless structures, [32] which is consistent with previous observations. Additionally, we observed that the overall expression level of G3BP1 in SGs induced by cisplatin decreased over time (Figure 2c), while the degree of condensation showed no significant difference after a 1 hour, suggesting rapid degradation of G3BP1 (Figure 2d). Considering that SGs formed after stimulation are gradually disassembled through a series of reactions, including ubiquitination, [33] we decided to fix the cisplatin stimulation time at 1 hour in the subsequent experiments, when stress granule aggregation peaked. Furthermore, we tested the susceptibility of cells to cisplatin after the uptake of chiral Pen-modified nanomaterials (Figure 2e–g). The results showed that at cisplatin concentrations below 125 μM, there was no significant difference in SGs between the L-Pen and the control groups. However, when the cisplatin concentration was increased to 250 μM, the L-Pen-group exhibited a significantly higher efficiency of SGs assembly compared to groups received either drug simulation at the same concentration or D-Pen-modified nanomaterial. This suggests that although L-Pen-modified nanomaterials have a weaker ability to directly provoke SGs, their presence can enhance the efficiency of chemotherapy-induced SGs assembly by lowering the energy barrier for G3BP1 liquid-liquid phase separation. Interestingly, we also found that D-Pen-modified materials exerted an entirely opposite effect. Even when cells were exposed to cisplatin at 375 μM or higher concentrations, the level of SGs condensation in the cytoplasm was comparable to the effect of stimulation with lower concentration drug alone, confirming that D-Pen-functionalized nanomaterials reduced the efficiency of SGs assembly under chemotherapy stimulation (Figure 2h).
Synergy Action on Stress Granule Formation
Currently, it is not clear how stress granules formed under cisplatin stimulation regulate the efficacy of chemotherapy drugs. On one hand, reports suggest that chemotherapy drugs have the potential to enrich in membraneless organelle structures, increasing their local concentration to exert cytotoxic effects more effectively [34]. However, research also indicates that membraneless structures, particularly the formation of stress granules, may reduce the sensitivity of cancer cells to certain chemotherapy drugs [35]. Given cisplatin’s ability to specifically disrupt DNA double-strand structures [36,37], we analyzed the expression of DNA damage markers (γ-H2AX) and apoptosis-related proteins (cleaved Caspase 3) in cells before and after chemotherapy drug stimulation (Figure 3a). Importantly, we observed a significant negative correlation between the expression level of stress granules and the extent of damage within the cells (Figure 3b–e), suggesting a protective role of stress granule formation in cells.
Our previous experience indicated that D-Pen-modified materials may inhibit the assembly of stress granules SGs within cancer cells (unpublished). Therefore, we hypothesized that such an action might underly the remodeling in cellular sensitivity to cisplatin treatment. Subsequently, we analyzed the damage caused by cisplatin in cancer cells following uptake of chiral nanomaterials modified with D-Pen, with results shown in Figure 3d–e. We found that the uptake of D-Pen-modified nanomaterials led to more severe DNA damage and apoptosis induced by chemotherapeutics, in contrast to the results of L-Pen group. Furthermore, we proceeded to validate the degree of G3BP1 aggregation in U-2 OS, SGC-7901, HCT-116, and HUVEC cell lines. We observed that G3BP1 aggregation induced by L-Pen was greater than that induced by D-Pen in cancer cell lines consistently, whereas we noticed an opposite outcome in normal cells (Figure 3f).
Extensive evidence suggests that stress granules can help cells resist chemotherapy drug damage, which is closely related to their protective effect on translational elements and their ability to sequester chemotherapy drugs to reduce their effective concentration. However, the nature of these roles and mechanisms remains inconclusive, any potential involvement of SGs in the regulation of tumor cell metabolism should be further studied to clarify and enrich our understanding. In summary, we discovered that D- and L-Pen-modified nanomaterials, by inhibiting and promoting SGs formation, respectively, altered the sensitivity of cancer cells to cisplatin. Combining the selective chemosensitivity of D/L-Pen-modified materials to tumor cells, there is potential in the future to achieve precise sensitization of cancer cells towards chemotherapy while reducing damage to adjacent normal tissues.
Characterization of Multiple Treatments-Induced Stress Granules
The structure of membraneless organelles, represented by SGs, maintains its morphology through liquid-liquid phase separation, enabling the exchange of components between the dense phase (in the droplets) and the dilute phase in the environment [39,41]. This liquid-like behavior confers the ability to regulate the composition of cytoplasmic proteins within membraneless structures [41]. Furthermore, maintaining a liquid state is beneficial for organelles as it allows timely disassembly by chaperons or degradation through the ubiquitin system, ultimately restoring cellular metabolic homeostasis [41]. Facing the structural mutations or abnormal post-translational modifications, membraneless organelles may undergo transition from a liquid state to a gel or even solid state [42,43]. This conversion has been linked to the onset and progression of diseases such as cancer, cardiovascular disorders, and notably neurodegenerative conditions [44].
Nanomaterials, existing in a state between soft condensation (between solid and liquid), have increasingly been shown to alter the condensation state of proteins in the cytoplasm through adsorption [45]. However, their impact on the physical properties of membraneless organelles remains unknown. In this study, we used fluorescence recovery after photobleaching (FRAP) to investigate the mobility of SGs before and after exposure to chiral materials (Figure 4a–c). We observed that in HeLa cells, stress granules received D-Pen-modified nanomaterials showed minimal fluorescence recovery, indicating a gel-like or solid-like behavior of SGs formed under D-Pen stimulation (Figure 4b). Interestingly, although L-Pen protects tumor cells against chemotherapy, observations of SGs fluidity under L-Pen stimulation also revealed a gel-like state (Figure 4c), contradicting the conclusion of L-Pen’s protective role in cancer cells.
Considering that the functions of SGs may vary depending on their protein composition [46], we further analyzed the protein composition of stress granules stimulated by D-Pen and L-Pen using co-immunoprecipitation (co-IP). The results showed that there were 133 common proteins in both groups of SGs, with 151 and 47 types of unique proteins in D- and L-Pen groups, respecctively (Figure 4d). Apart from the difference in protein quantity, the proteins present in these two types of SGs also participate in different functions. Through gene ontology (GO) analysis of the unique proteins in the two types of SGs, we found that the proteins in D-Pen-stimulated SGs are closely associated with mRNA splicing and regulation (Figure 4d). When these proteins aggregate within low-mobility gel-like SGs, cellular protein synthesis and metabolism are significantly inhibited, leading to cell damage [47,48]. Conversely, the proteins in L-Pen-stimulated SGs are mainly involved in processes such as DNA double-strand breakage and lipid metabolism. We speculate that this functional difference may be the reason why gel-like SGs induced by L-Pen stimulation can still exert a protective effect on tumor cells.
Mechanism Study of Chiral Nanomaterials-Modulated Stress Granules Formation
In the aforementioned study, we have for the first time confirmed a correlation between the chirality of nanomaterials and the sensitivity of SGs to chemotherapy. When subjected to two different chiral nanomaterials, SGs exhibited a transition from a liquid phase to a gel phase, indicating a potential undiscovered relationship between the chirality of materials and the function of stress granules, beyond just physical property changes. Recent research has shown that both endogenous and exogenous nanomaterial surfaces are enveloped by a dynamically changing protein layer, known as the protein corona effect [49,50]. The chirality differences on the surface of nanomaterials significantly impact the composition and function of the protein corona, which in turn is closely related to various chiral-related biological effects of nanomedicines.
Based on this, we further hypothesized that the chirality differences of gold nanomaterials may regulate the assembly of SGs by recruiting different types of cytoplasmic proteins. To confirm this hypothesis, we employed a chemically etching-assisted proximity labeling strategy to separate and identify cytoplasmic proteins bound to the surface of nanomaterials. Specifically, we synthesized gold nanomaterials co-functionalized with diazirine and D-/L-Pen. Upon cellular uptake, under excitation by ultraviolet light (365 nm), the diazirine was tranformed into a highly active carbene structure, which rapidly binds to the proteins adsorbed on the material surface, converting unstable molecular interactions into stable covalent bonding [51] (illustrated in Figure 5a). Through centrifugation and mass spectrometry analysis, we identified the proteins adsorbed on chiral nanomaterials in the cytoplasm (Figure 5b,c), and from the Venn diagrams, we identified 96 and 111 common adsorbed proteins in HeLa and HUVEC cells, respectively, which were independent of the nanomaterial’s chirality. Importantly, in HeLa cells, the diversity (213 species) of proteins uniquely adsorbed to the L-Pen modified material surface was significantly greater than that of the D-Pen group (27 species). Furthermore, in HUVEC cells, the diversity of proteins adsorbed to different chiral nanomaterials showed a completely opposite trend compared to HeLa cells (44 versus 214 for L- and D-Pen groups, respectively, for a full list of components, please refer to Table S1), indicating a potentially key reason for the distinct biological behaviors exhibited by D-/L-Pen modified nanomaterials in cancer cells and endothelial cells.
By conducting GO analysis on the unique proteins adsorbed to both chiral materials in the two different cell types, we found that these adsorbed proteins participate in regulating various stress pathways, such as oxidative stress, chemical stress, and biotin stress, among others (Figure 5b–d,f). When these proteins involved in handling cellular stress are adsorbed onto the surface of nanomaterials, their accessibility is affected, leading to varying degrees of inhibition in the downstream biological functions associated with these proteins, stimulating cellular repair mechanisms. One of the key pathways through which this occurs is the translation inhibition executed by SGs.
In further analysis, we investigated the association between protein corona composition and stress response in four scenarios (two types of nanomaterials in two cell lines). The results revealed that D-Pen adsorbed 15 SG-related proteins in HeLa cells, while this number increased to 55 in HUVEC cells. Conversely, L-Pen adsorbed a total of 46 stress-regulating proteins in HeLa cells, but this number significantly decreased to 18 in HUVEC cells (please refer to Figure S2 for detailed information). These findings strongly support the notion that the functional differences of D-Pen and L-Pen in HeLa and HUVEC cells are closely related to the quantity of stress-regulating proteins adsorbed on their surfaces. Importantly, these results further confirm the chiral and cellular microenvironment-dependent nature of cytoplasmic protein corona formation.
Furthermore, upon comparison, we observed a high degree of similarity in the stress-related protein types adsorbed by L-Pen in HeLa cells and D-Pen in HUVEC cells (Figure 5e). The same phenomenon was also noted between the D-Pen group internalized by HeLa cells and the L-Pen-treated HUVEC group. Through in-depth analysis of the adsorbed protein functions, it was found that L-Pen in HeLa cells adsorbs proteins functioning as inhibitory factors of SGs (UBA1, HSP7C, HS90B, and HS90A), whereas D-Pen in HUVEC cells adsorbs various components involved in inhibiting stress granule formation or promoting their disassembly (VCP, UBA1, HSP104, HSP7C, HS90B, and HS90A). Adsorption of these inhibitory proteins by nanomaterials reduces the energy barrier for G3BP1 to assemble SGs through liquid-liquid phase separation, ultimately promoting stress granule formation and enhancing the overall stability of membrane-less organelles [54,55,56]. Conversely, L-Pen in HUVEC cells selectively adsorbs proteins that promote stress granule aggregation (UBAP2, TIAR, and TIA1), which shares functional and compositional similarities with the components adsorbed by D-Pen in HeLa cells (UBAP2, CAPRIN1) [57,58,59]. A key consequence of this interaction is the reduced ability of cells to assemble stress granules, leading to decreased diversity in stress granule composition. Enhanced interactions between structurally similar proteins may act as a driving force behind stress granule gelation.
Conclusion
Stress granules are key membrane-less organelles involved in cellular stress responses [60]. Current research on SGs primarily focuses on exploring their composition and functional differences under chemical stimuli or microenvironmental changes [29,46,61], with limited knowledge of their role in the metabolism of nanomaterials, particularly the relationship between chiral elements and SGs assembly. In this study, we demonstrate that chiral materials, both D- and L-enantiomers, do not directly induce stress granule assembly but can modulate protein adsorption behavior in the cytoplasm, ultimately affecting the dynamics of SGs assembly under stress conditions. In cancer cells, D- and L-penicillamine-modified nanoparticles exhibited high affinity towards scaffolding proteins and chaperone proteins, respectively, leading to enhanced and reduced G3BP1 liquid-liquid phase separation abilities. By combining with chemotherapy drugs, we confirmed that D-penicillamine-modified nanoparticles suppressed SGs assembly, significantly increasing cancer cell sensitivity to chemotherapy drugs, providing a new approach for adjuvant chemotherapy using chiral nanomedicines. By combining with chemotherapy drugs, we confirmed that D-penicillamine-modified nanoparticles suppressed SGs assembly, significantly increasing cancer cell sensitivity to chemotherapy drugs, providing a new approach for adjuvant chemotherapy using chiral nanomedicines. On the other hand, in normal cells, D-penicillamine-modified nanoparticles selectively adsorbed SGs disassembly-related components. A popular explanation is that certain nanomaterials can effectively reduce the energy barrier for G3BP1 liquid-liquid phase separation to take place and improve stress granule assembly efficiency, however, additional research is needed to reveal the underlying mechanisms. Although the specific mechanism by which certain nanoparticles help normal cells enhance resistance to chemotherapy drugs is so far unknown, the current findings are encouraging enough as an effective strategy to mitigate chemotherapy-induced vascular damage. Our study reveals the efficacy of chiral nanomedicines in modulating SGs, further deepening our understanding of the biological effects of chiral nanomaterials and providing important insights for the development of novel nanomedicines targeting membraneless cellular structures.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.Org.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 32371447)
Data Availability Statement
The data that support the findings of this study are presented in the study, and the raw information can be acquired from the corresponding author upon reasonable request.
Conflicts of Interest
The authors report no conflicts of interest in this work.
References
- Milton FP, Govan J, Mukhina MV, Gun’ko YK. The chiral nano-world: chiroptically active quantum nanostructures. Nanoscale Horiz. 2016;18;1(1):14-26. [CrossRef]
- Bird GH, Mazzola E, Opoku-Nsiah K, et al. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat Chem Biol. 2016;12(10):845-52. [CrossRef]
- Speranza L, di Porzio U, Viggiano D, de Donato A, Volpicelli F. Dopamine: The neuromodulator of long-term synaptic plasticity, reward and movement control. Cells. 2021;26;10(4):735. [CrossRef]
- Yanagisawa N, Ikeda S, Hashimoto T, et al. Effects of L-threo-Dops on orthostatic hypotension in Parkinson’s disease. No To Shinkei. 1998;50(2):157-63. Japanese.
- Sun M, Wang X, Guo X, et al. Chirality at nanoscale for bioscience. Chem Sci. 2022;8;13(11):3069-3081. [CrossRef]
- Ma Y, Shi L, Yue H, Gao X. Recognition at chiral interfaces: from molecules to cells. Colloids Surf B Biointerfaces. 2020;195:111268. [CrossRef]
- John M, Kalvala PR, Misra M, Menezes PL. Peening techniques for surface modification: processes, properties, and applications. Materials (Basel). 2021;9;14(14):3841. [CrossRef]
- hao Y, Yang G, Lin J, et al. Shining light on chiral inorganic nanomaterials for biological issues. Theranostics. 2021;7;11(19):9262-9295. [CrossRef]
- Zhang H, Li S, Qu A, et al. Engineering of chiral nanomaterials for biomimetic catalysis. Chem Sci. 2020;21;11(48):12937-12954. [CrossRef]
- Tee YH, Goh WJ, Yong X, et al. Actin polymerisation and crosslinking drive left-right asymmetry in single cell and cell collectives. Nat Commun. 2023;11;14(1):776. [CrossRef]
- Purcell-Milton F, Visheratina AK, Kuznetsova VA, et al. Impact of shell thickness on photoluminescence and optical activity in chiral CdSe/CdS core/shell quantum dots. ACS Nano. 2017;26;11(9):9207-9214. [CrossRef]
- Li Y, Zhou Y, Wang HY, et al. Chirality of glutathione surface coating affects the cytotoxicity of quantum dots. Angew Chem Int Ed Engl. 2011;20;50(26):5860-4. [CrossRef]
- Zhang C, Zhou Z, Zhi X, et al. Insights into the distinguishing stress-induced cytotoxicity of chiral gold nanoclusters and the relationship with GSTP1. Theranostics. 2015;1;5(2):134-49. [CrossRef]
- Zhang H, Hao C, Qu A, et al. Light-induced chiral iron copper selenide nanoparticles prevent β-amyloidopathy in vivo. Angew Chem Int Ed Engl. 2020;27;59(18):7131-7138. [CrossRef]
- Anderson P, Kedersha N. Stressful initiations. J Cell Sci. 2002;15;115(Pt 16):3227-34. [CrossRef]
- Anderson P, Kedersha N, Ivanov P. Stress granules, P-bodies and cancer. Biochim Biophys Acta. 2015;1849(7):861-70. [CrossRef]
- Kang W, Wang Y, Yang W, Zhang J, Zheng H, Li D. Research progress on the structure and function of G3BP. Front Immunol. 2021;30;12:718548. [CrossRef]
- Yang P, Mathieu C, Kolaitis RM, et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell. 2020;16;181(2):325-345.e28. [CrossRef]
- Ge Y, Jin J, Li J, Ye M, Jin X. The roles of G3BP1 in human diseases (review). Gene. 2022;5;821:146294. [CrossRef]
- Fujikawa D, Nakamura T, Yoshioka D, et al. Stress granule formation inhibits stress-induced apoptosis by selectively sequestering executioner caspases. Curr Biol. 2023;22;33(10):1967-1981.e8. [CrossRef]
- Reits EA, Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol. 2001;3(6):E145-7. [CrossRef]
- Bersuker K, Peterson CWH, To M, et al. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev Cell. 2018;8;44(1):97-112.e7. [CrossRef]
- Zheng Y, Zhong X, Li Z, et al. Successive, seed-mediated growth for the synthesis of single-crystal gold nanospheres with uniform diameters controlled in the range of 5–150 nm. Particle & Particle Systems Characterization. 2014;31(2):266-273. [CrossRef]
- Zhu M, Nie G, Meng H, Xia T, Nel A, Zhao Y. Physicochemical properties determine nanomaterial cellular uptake, transport, and fate. Acc Chem Res. 2013;19;46(3):622-31. [CrossRef]
- Zhao B, Yang S, Deng J, Pan K. Chiral graphene hybrid materials: structures, properties, and chiral applications. Adv Sci (Weinh). 2021;12;8(7):2003681. [CrossRef]
- Nguyen VH, Lee BJ. Protein corona: a new approach for nanomedicine design. Int J Nanomedicine. 2017;18;12:3137-3151. [CrossRef]
- Mahmoudi M, Landry MP, Moore A, Coreas R. The protein corona from nanomedicine to environmental science. Nat Rev Mater. 2023;24:1-17. [CrossRef]
- Yeom J, Guimaraes PPG, Ahn HM, et al. Chiral supraparticles for controllable nanomedicine. Adv Mater. 2020;32(1):e1903878. [CrossRef]
- Lee JI, Namkoong S. Stress granules dynamics: benefits in cancer. BMB Rep. 2022;55(12):577-586. [CrossRef]
- Song MS, Grabocka E. Stress granules in cancer. Rev Physiol Biochem Pharmacol. 2023;185:25-52. [CrossRef]
- Guillén-Boixet J, Kopach A, Holehouse AS, et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell. 2020;16;181(2):346-361.e17. [CrossRef]
- Begovich K, Wilhelm JE. An in vitro assembly system identifies roles for RNA nucleation and ATP in yeast stress granule formation. Mol Cell. 2020;17;79(6):991-1007.e4. [CrossRef]
- Gwon Y, Maxwell BA, Kolaitis RM, Zhang P, Kim HJ, Taylor JP. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science. 2021;25;372(6549):eabf6548. [CrossRef]
- Klein IA, Boija A, Afeyan LK, et al. Partitioning of cancer therapeutics in nuclear condensates. Science. 2020;19;368(6497):1386-1392. [CrossRef]
- Wang X, Chen T, Li C, et al. CircRNA-CREIT inhibits stress granule assembly and overcomes doxorubicin resistance in TNBC by destabilizing PKR. J Hematol Oncol. 2022;29;15(1):122. [CrossRef]
- Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;5;740:364-78. [CrossRef]
- Zhang J, Ye ZW, Tew KD, Townsend DM. Cisplatin chemotherapy and renal function. Adv Cancer Res. 2021;152:305-327. [CrossRef]
- Kedersha N, Panas MD, Achorn CA, et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol. 2016;28;212(7):845-60. [CrossRef]
- Peng PH, Hsu KW, Wu KJ. Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am J Cancer Res. 2021;15;11(8):3766-3776.
- Tong X, Tang R, Xu J, et al. Liquid-liquid phase separation in tumor biology. Signal Transduct Target Ther. 2022;8;7(1):221. [CrossRef]
- Díaz-Moreno I, De la Rosa MA. Membraneless organelles: a smart design for metabolic control. FEBS Open Bio. 2021;11(9):2388-2389. [CrossRef]
- Koppers M, Özkan N, Farías GG. Complex interactions between membrane-bound organelles, biomolecular condensates and the cytoskeleton. Front Cell Dev Biol. 2020;21;8:618733. [CrossRef]
- Li J, Zhang M, Ma W, et al. Post-translational modifications in liquid-liquid phase separation: a comprehensive review. Mol Biomed. 2022;11;3(1):13. [CrossRef]
- Wang B, Zhang L, Dai T, et al. Liquid-liquid phase separation in human health and diseases. Signal Transduct Target Ther. 2021;2;6(1):290. [CrossRef]
- Feng Q, Wilhelm J, Gao J. Transistor-like ultra-pH-sensitive polymeric nanoparticles. Acc Chem Res. 2019;18;52(6):1485-1495. [CrossRef]
- Hofmann S, Kedersha N, Anderson P, Ivanov P. Molecular mechanisms of stress granule assembly and disassembly. Biochim Biophys Acta Mol Cell Res. 2021;1868(1):118876. [CrossRef]
- Ivanov P, Kedersha N, Anderson P. Stress granules and processing bodies in translational control. Cold Spring Harb Perspect Biol. 2019;1;11(5):a032813. [CrossRef]
- Riggs CL, Kedersha N, Ivanov P, Anderson P. Mammalian stress granules and P bodies at a glance. J Cell Sci. 20201;133(16):jcs242487. [CrossRef]
- Kopac T. Protein corona, understanding the nanoparticle-protein interactions and future perspectives: A critical review. Int J Biol Macromol. 2021;1;169:290-301. [CrossRef]
- Park SJ. Protein-nanoparticle interaction: corona formation and conformational changes in proteins on nanoparticles. Int J Nanomedicine. 2020;6;15:5783-5802. [CrossRef]
- Porter NJ, Danelius E, Gonen T, Arnold FH. Biocatalytic carbene transfer using diazirines. J Am Chem Soc. 2022;25;144(20):8892-8896. [CrossRef]
- Protter DSW, Parker R. Principles and properties of stress granules. Trends Cell Biol. 2016;26(9):668-679. [CrossRef]
- Jain S, Wheeler JR, Walters RW, et al. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 201628;164(3):487-98. [CrossRef]
- Pare JM, Tahbaz N, López-Orozco J, et al. Hsp90 regulates the function of argonaute 2 and its recruitment to stress granules and P-bodies. Mol Biol Cell. 2009;20(14):3273-84. [CrossRef]
- Tolay N, Buchberger A. Role of the ubiquitin system in stress granule metabolism. Int J Mol Sci. 2022;26;23(7):3624. [CrossRef]
- Zheng Y, Zhu G, Tang Y, et al. HDAC6, A novel cargo for autophagic clearance of stress granules, mediates the repression of the type I interferon response during coxsackievirus A16 infection. Front Microbiol. 2020;31;11:78. [CrossRef]
- Peng G, Gu A, Niu H, et al. Amyotrophic lateral sclerosis (ALS) linked mutation in Ubiquilin 2 affects stress granule assembly via TIA-1. CNS Neurosci Ther. 2022;28(1):105-115. [CrossRef]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;27;147(7):1431-42. [CrossRef]
- Song D, Kuang L, Yang L, et al. Yin and yang regulation of stress granules by Caprin-1. Proc Natl Acad Sci U S A. 2022;119(44):e2207975119. [CrossRef]
- Fefilova AS, Fonin AV, Vishnyakov IE, Kuznetsova IM, Turoverov KK. Stress-induced membraneless organelles in eukaryotes and prokaryotes: bird’s-eye View. Int J Mol Sci. 2022;30;23(9):5010. [CrossRef]
- Millar SR, Huang JQ, Schreiber KJ, et al. A new phase of networking: the molecular composition and regulatory dynamics of mammalian stress granules. Chem Rev. 2023;26;123(14):9036-9064. [CrossRef]
Figure 1.
(a) TEM micrographs of differently functionalized Au nanospheres employed in this study. For the definition of various color codes, please refer to the legends in b. Scale bar: 30 nm. (b) UV-vis spectra results of Au nanospheres featuring distinct surface ligands. (c) Immunofluorescence graphs showing the condensation of stress granules in HeLa cells due to exposure to nanomaterials. A schematic is provided (middle in the upper panel) to help appreciate the presentation of graphs. The nucleus and membrane of cells were counter-stained to visulize the cell body. Here, Cys, His, and Pen stands for Cysteine, Histidine, and Penicilamine in their meso-forms, respectively. (d) Normalized uptake of fluorescently labeled nanomaterials by HeLa cells. (e) Condesation of stress granules as provoked by nanomaterials. The fluorescence intensity of G3BP1 was calculated and normalized by the volume of membraneless droplets in order to reflect the condensation content of each stress granules (n>200 droplets). (f) Shortest distance of stress granule to their closest counterparts. For median value, please refer to the violin plot in each panel (in the middle), whereas the distribution of all data points can be found underneath each plot. (g) Immunofluorescence graphs showing the condensation of stress granules in HUVEC cells received nanomaterials enclosed by D-/L-Pen molecules. For the organization of graphs and their definition, we follow the same order as described in b. (h) Difference in uptake behaviors of handed Pen-functionalized nanomaterials and condensation of stress granules between HeLa and HUVEC cells. (i) Comparison of stress granules condensation content in between cells received nanomaterials alone and that treated with sodium arsenite (NaAsO2). **, and **** indicate P < 0.01, and P < 0.0001, respectively. NS stand for non-significant statistical differences. .
Figure 1.
(a) TEM micrographs of differently functionalized Au nanospheres employed in this study. For the definition of various color codes, please refer to the legends in b. Scale bar: 30 nm. (b) UV-vis spectra results of Au nanospheres featuring distinct surface ligands. (c) Immunofluorescence graphs showing the condensation of stress granules in HeLa cells due to exposure to nanomaterials. A schematic is provided (middle in the upper panel) to help appreciate the presentation of graphs. The nucleus and membrane of cells were counter-stained to visulize the cell body. Here, Cys, His, and Pen stands for Cysteine, Histidine, and Penicilamine in their meso-forms, respectively. (d) Normalized uptake of fluorescently labeled nanomaterials by HeLa cells. (e) Condesation of stress granules as provoked by nanomaterials. The fluorescence intensity of G3BP1 was calculated and normalized by the volume of membraneless droplets in order to reflect the condensation content of each stress granules (n>200 droplets). (f) Shortest distance of stress granule to their closest counterparts. For median value, please refer to the violin plot in each panel (in the middle), whereas the distribution of all data points can be found underneath each plot. (g) Immunofluorescence graphs showing the condensation of stress granules in HUVEC cells received nanomaterials enclosed by D-/L-Pen molecules. For the organization of graphs and their definition, we follow the same order as described in b. (h) Difference in uptake behaviors of handed Pen-functionalized nanomaterials and condensation of stress granules between HeLa and HUVEC cells. (i) Comparison of stress granules condensation content in between cells received nanomaterials alone and that treated with sodium arsenite (NaAsO2). **, and **** indicate P < 0.01, and P < 0.0001, respectively. NS stand for non-significant statistical differences. .

Figure 2.
(a) Immunofluorescence graphs of cells received different treatments (cisplatin alone versus exposure in combination with chiral nanomaterials). Cell membrane and the nucleus were counter-stained with antibody of Na-K-ATPase and DAPI, respectively. (b) Condensation content of stress granules in HeLa cells received different concentrations of cisplatin (CisPt, 1 h). (c) Fluorescence of G3BP1 proteins in HeLa cells received CisPt (250 μM) as a function of time post treatment. (d) Disassemble of stress granule as a function of time post treatment with CisPt. (e,f) Condensation of stress granules in HeLa cells received combinational treatment with CisPt and D-(e) and L- (f) Pen-functionalized nanomaterials, respectively. (g) Difference in condensation content of stress granules in HeLa cells received either various concentrations of CisPt alone or combinational treatments. (h) A schematic illustrating the hand-related implication in chemotherapeutic-provoked liquid-liquid phase separation (LLPS). **, and **** indicate P < 0.01, and P < 0.0001, respectively. NS stand for non-significant statistical differences.
Figure 2.
(a) Immunofluorescence graphs of cells received different treatments (cisplatin alone versus exposure in combination with chiral nanomaterials). Cell membrane and the nucleus were counter-stained with antibody of Na-K-ATPase and DAPI, respectively. (b) Condensation content of stress granules in HeLa cells received different concentrations of cisplatin (CisPt, 1 h). (c) Fluorescence of G3BP1 proteins in HeLa cells received CisPt (250 μM) as a function of time post treatment. (d) Disassemble of stress granule as a function of time post treatment with CisPt. (e,f) Condensation of stress granules in HeLa cells received combinational treatment with CisPt and D-(e) and L- (f) Pen-functionalized nanomaterials, respectively. (g) Difference in condensation content of stress granules in HeLa cells received either various concentrations of CisPt alone or combinational treatments. (h) A schematic illustrating the hand-related implication in chemotherapeutic-provoked liquid-liquid phase separation (LLPS). **, and **** indicate P < 0.01, and P < 0.0001, respectively. NS stand for non-significant statistical differences.
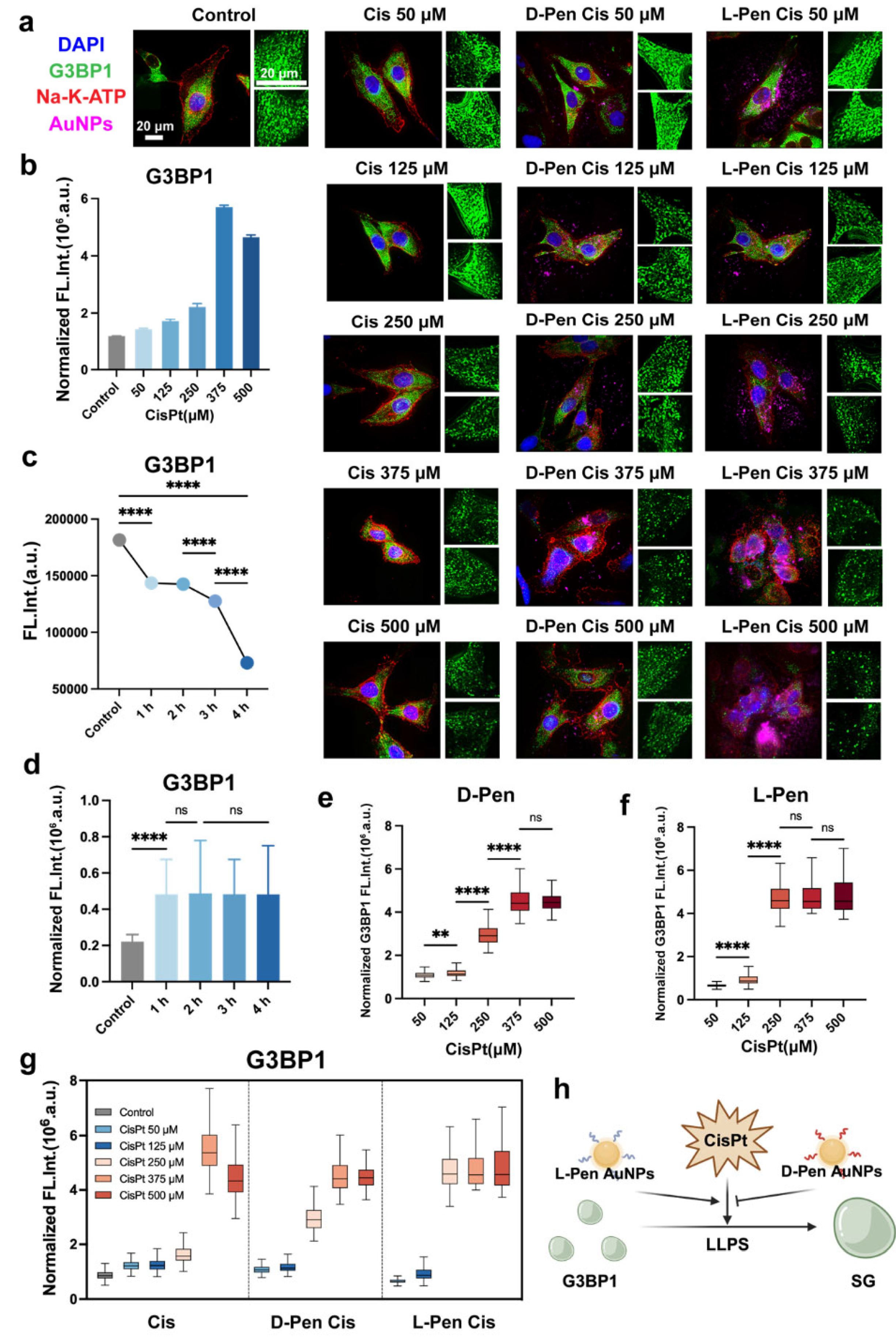
Figure 3.
(a) Immunofluorescence graphs of HeLa cells received cisplatin alone or its combination with chiral nanomaterials. Expression level of γ-H2AX (DNA damage marker) and cleaved Caspase 3 (marker of programmed apoptosis) are shown in (d,e) as box plots, respectively. (b,c) Size (b) and condensation contents (c) of stress granules in cells. For cells received neither cisplatin nor chiral nanomaterials, they serve the role as control for comparison. (f) Immunofluorescence graphs of cancerous (U2-OS, SGC-7901, and HCT-116) and normal cells (HUVEC) received chemotherapy in combination with handed Pen-functionalized nanomaterials. **, and **** indicate P < 0.01, and P < 0.0001, respectively.
Figure 3.
(a) Immunofluorescence graphs of HeLa cells received cisplatin alone or its combination with chiral nanomaterials. Expression level of γ-H2AX (DNA damage marker) and cleaved Caspase 3 (marker of programmed apoptosis) are shown in (d,e) as box plots, respectively. (b,c) Size (b) and condensation contents (c) of stress granules in cells. For cells received neither cisplatin nor chiral nanomaterials, they serve the role as control for comparison. (f) Immunofluorescence graphs of cancerous (U2-OS, SGC-7901, and HCT-116) and normal cells (HUVEC) received chemotherapy in combination with handed Pen-functionalized nanomaterials. **, and **** indicate P < 0.01, and P < 0.0001, respectively.
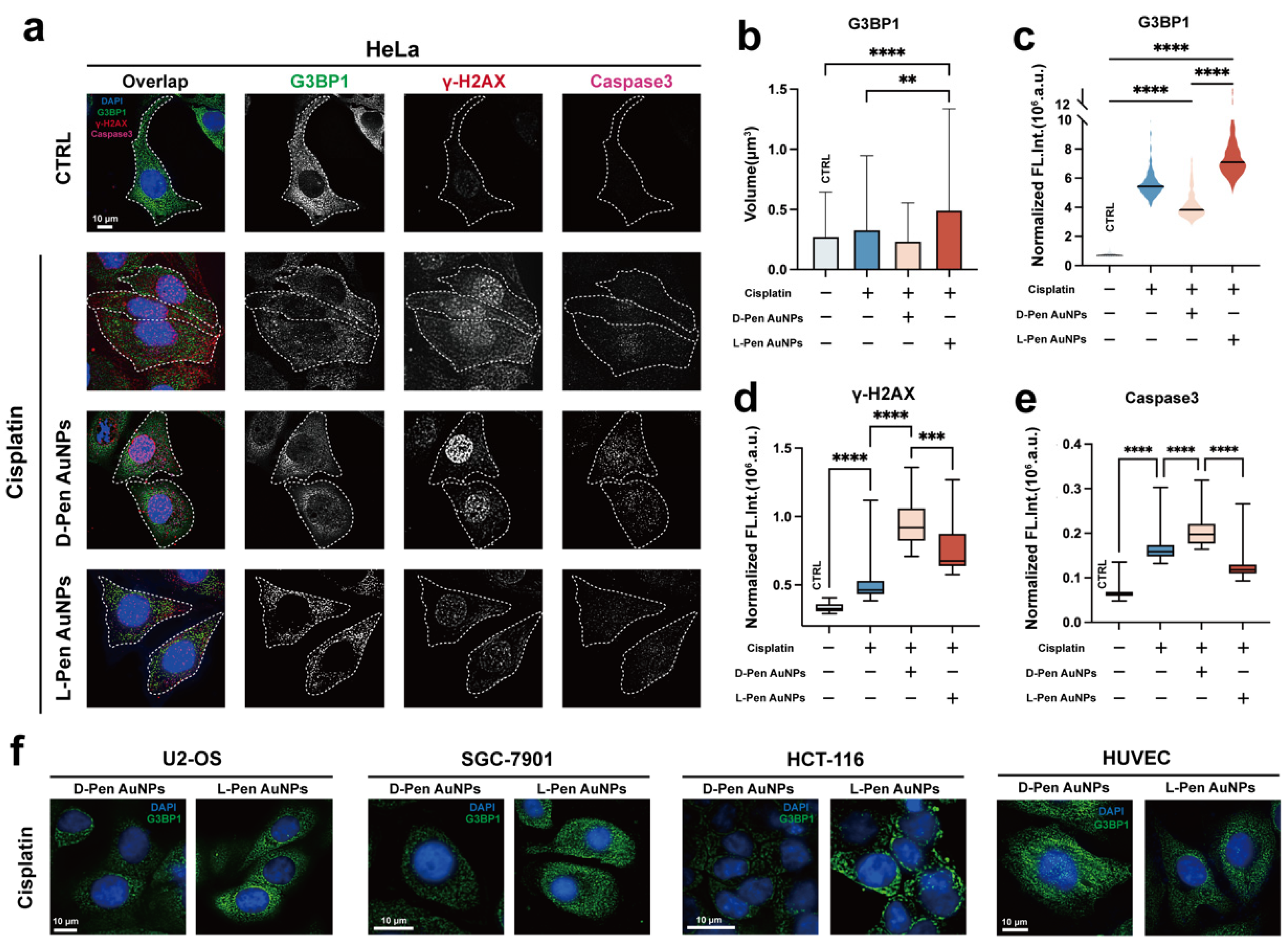
Figure 4.
(a–c) Fluorescence recovery as a function of time post photo bleaching. Relative fluorescence intensity of illuminated region was analyzed. The fluorescence recovery information of at least ten randomly depicted regions were recorded and the mean value was calculated for plotting (mean value: dashed lines). To ease the visualization, error bar was presented as shadow. Inset: representative graphs recorded at certain time points that reveal the recovery of G3BP1 fluorescence in photo-bleached stress granule. For the subcellular distribution information of illuminated region, please refer to the fluorescence graphs at bottom with lower magnification. (d) Venn diagram of proteins absorbed on D-/L-Pen-functionalized nanomaterials. In the middle of the diagram shows the overlapped components, whereas those exhibiting chirality-dependent absorption can be found on the left (for D-Pen) and right sides (for L-Pen) with corresponding GO information shown at the bottom. .
Figure 4.
(a–c) Fluorescence recovery as a function of time post photo bleaching. Relative fluorescence intensity of illuminated region was analyzed. The fluorescence recovery information of at least ten randomly depicted regions were recorded and the mean value was calculated for plotting (mean value: dashed lines). To ease the visualization, error bar was presented as shadow. Inset: representative graphs recorded at certain time points that reveal the recovery of G3BP1 fluorescence in photo-bleached stress granule. For the subcellular distribution information of illuminated region, please refer to the fluorescence graphs at bottom with lower magnification. (d) Venn diagram of proteins absorbed on D-/L-Pen-functionalized nanomaterials. In the middle of the diagram shows the overlapped components, whereas those exhibiting chirality-dependent absorption can be found on the left (for D-Pen) and right sides (for L-Pen) with corresponding GO information shown at the bottom. .
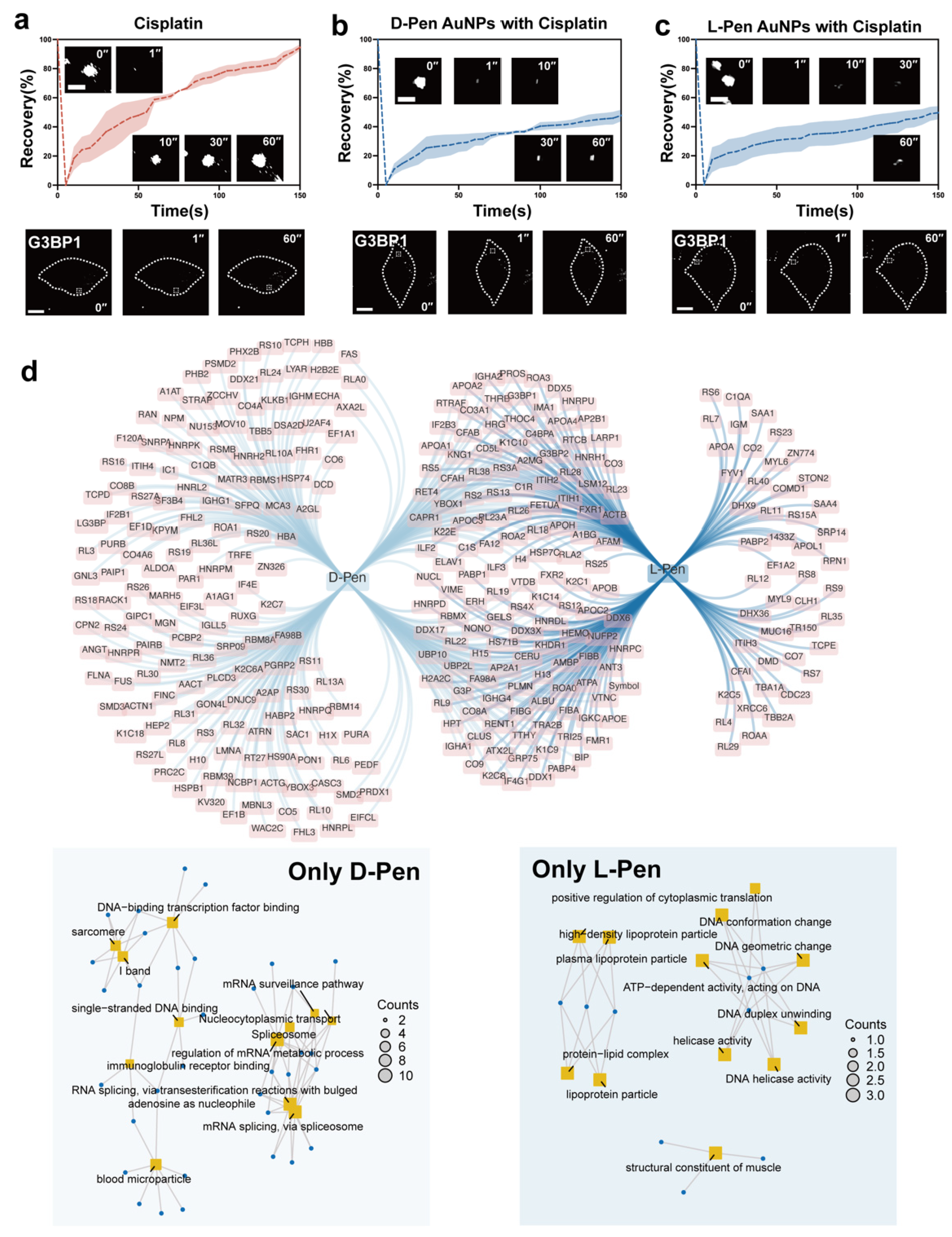
Figure 5.
(a) A schematic illustrating the pipeline of chemo-etching-assisted photo-proximity labeling and the subsequent assay to identify the intracellular protein corona components. GO information of proteins whose absorption on nanomaterials are chirality (in)dependent in HeLa ((b,d), chirality-dependent and independent, respectively) and HUVEC cells ((c,f), chirality-dependent and independent, respectively). (e) Overlapped proteins that appeared in the protein corona of L-Pen group in HeLa cells and D-Pen group in HUVEC cells and corresponding GO information (bottom).
Figure 5.
(a) A schematic illustrating the pipeline of chemo-etching-assisted photo-proximity labeling and the subsequent assay to identify the intracellular protein corona components. GO information of proteins whose absorption on nanomaterials are chirality (in)dependent in HeLa ((b,d), chirality-dependent and independent, respectively) and HUVEC cells ((c,f), chirality-dependent and independent, respectively). (e) Overlapped proteins that appeared in the protein corona of L-Pen group in HeLa cells and D-Pen group in HUVEC cells and corresponding GO information (bottom).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



