
Submitted:
27 April 2024
Posted:
29 April 2024
You are already at the latest version
Abstract
RNA virus polymerases carry out multiple functions necessary for successful genome replication and transcription. A key tool for molecular studies of viral RNA-dependent RNA polymerases (RdRps) is a ‘minigenome’ or ‘minireplicon’ assay, in which viral RdRps are reconstituted in cells in the absence of full virus infection. Typically, plasmids expressing the viral polymerase protein(s) and other co-factors are co-transfected along with a plasmid expressing an RNA encoding a fluorescent or luminescent reporter gene flanked by viral untranslated regions containing cis-acting elements required for viral RdRp recognition. This reconstitutes the viral transcription/replication machinery and allows viral RdRp activity to be measured as a correlate of reporter protein signal. Here we report the first development of a plasmid-based minigenome assay for species A rotavirus, using a firefly luciferase reporter gene.
Keywords:
rotavirus
; minigenome
; RNA-dependent RNA polymerase
; reporter assay
Introduction
Rotaviruses (RVs) are segmented, double-stranded RNA (dsRNA) viruses that cause acute gastroenteritis in infants and young children worldwide [1]. Currently there are 11 distinct RV species (A–L, no E) with species A being the most predominant, accounting for over 90% of infections in humans and animals [2,3,4,5,6].
The RV virion is a non-enveloped triple-layered particle (TLP) containing 11 segments of dsRNA as its genome [7,8]. The core shell is formed by 60 asymmetric dimers of the viral protein 2 (VP2) and is surrounded by an intermediate layer of VP6 forming the transcriptionally active, non-infectious double-layered particle (DLP) [8,9]. The polymerase complex, composed of the RNA-dependent RNA polymerase (RdRp) (VP1) and the RNA capping enzyme (VP3), is anchored at the fivefold axes through simultaneous interactions with multiple subdomains of VP2 [10,11]. The dsRNA segments are thought to be organised within the core in a way that each genome segment interacts with one specific polymerase complex [9,12].
The RdRp has a cage-like structure with four tunnels leading to a catalytic core (residues 333–778) enclosed between the N-terminal (residues 1–332) and C-terminal domains (residues 779–1088) [13]. During transcription, the RdRp synthesises capped, non-polyadenylated, positive-sense RNA ((+)RNA) transcripts from the minus strand of the genomic dsRNA, that are extruded out of the DLP into the cytoplasm [14]. The (+)RNA functions as mRNA for viral protein translation and as templates for synthesis of new dsRNA genomes [15]. The cap-binding site of the N-terminal domain of VP1 splits the dsRNA genome through its interaction with the 5’ conserved m7GpppGGC residue of (+)RNA present in all RV segments [16,17]. After a short part of the helix is unwound, the unpaired negative sense RNA ((-)RNA) traverses towards the active site of RdRp and immediately pairs with complementary NTPs within the core that form a backbone of the nascent RNA [18]. The dsRNA genome is pushed along by the newly synthesised nascent RNA backbone until it reaches the C-terminal domain of VP1 where the coding strand reanneals with the template and reforms the dsRNA genome [13]. The presence of distinct exit tunnels ensures that the nascent RNA is released into the cytoplasm while the (-)RNA is reused in subsequent rounds of (+)RNA synthesis [13,19].
In RVs, highly conserved cis-acting elements that enhance (-)RNA synthesis were also shown to be present at the 5’-end of (+)RNA which sometimes extended into the coding region [17,20,21,22,23]. Previous studies using in vitro replication systems showed that complementary base pairing of 5’ and 3’ regions of each segment are predicted to facilitate RNA circularisation by forming panhandle structures where the 3’-GACC conserved terminal sequence extends as a single-stranded tail [17,24,25]. The RdRp specifically recognises the conserved consensus sequences at the 3’-end of (+)RNA to initiate (-)RNA synthesis during genome replication [16]. This interaction is catalytically inactive and requires the N-terminal domain of VP2 which leads to conformational changes in the priming loop within the catalytic core of VP1 stabilising the initiating nucleotide in the priming site of RdRp [26,27,28,29]. This correct alignment results in the formation of the first phosphodiester bond of the (-)RNA product [30]. Simultaneously, the priming loop retracts, allowing elongation of the dsRNA product out of the polymerase [13]. The ratio of VP1:VP2 required to achieve this replicase activity was shown to be 1:10, the same ratio that forms the vertices of the core [17,31]. Studies showed that assembly of VP2 into cores was required for RNA replication and encapsidation of VP1 and VP3 demonstrating its direct role in core assembly and packaging of newly made dsRNA products [16,29].
The above understanding is derived from in vitro biochemistry experiments, which typically require laborious protein purification and which cannot easily interrogate host interactions or rapidly test mutant viral polypeptides. While reverse genetics systems for RVs now exist [32,33,34,35], they don't easily allow separate interrogation of viral transcription. For other Baltimore groups, the study of RdRp function of plus strand viruses is readily accessible due to mini-genome assays developed in the 1990s [36] with development of assays for the study of minus strand virus RdRps following shortly thereafter [37,38,39]. Thus, the lack of a minigenome system for dsRNA viruses is a major roadblock to the study of RdRps of dsRNA viruses.
In this study, we aimed to establish a plasmid based mini-genome assay to recapitulate viral transcription and replication. We found that RV RdRp activity could be recapitulated by expressing a subset of viral structural proteins by varying ratios of VP1:VP2 constructs. Mutations of conserved residues in the catalytic core of the RdRp diminished but did not extinguish reporter activity, suggesting tolerability at these sites. This new plasmid based mini-genome system represents a more direct model for measuring the polymerase activity in vitro.
Materials and Methods
Reporter segment construction. The constructs were designed to encode the firefly luciferase gene in either a positive or negative orientation, flanked by 5’- and 3’-UTRs under a bacteriophage T7 RNA promoter (T7P), containing sequences for antigenomic hepatitis delta virus (HDV) ribozyme and T7 transcription terminator (T7T) sequences at the 3’ end. These constructs were synthesised by Invitrogen GeneArt on pMA (ampicillin resistance) vectors. Plasmids were amplified by transformation into chemically competent E. coli DH5α and purified using QIAGEN® Plasmid Midi Kit (QIAGEN) according to the manufacturer’s protocol. The inserts in each plasmid were verified by Sanger sequencing (GATC Biotech or Genewiz, Germany) using primers listed in Error! Reference source not found. Sequence results were analysed in SSE v1.4 software [40].
Cell lines. BSR-T7 cells, a derivative of baby hamster kidney fibroblasts (BHK-21 cells), constitutively expressing T7 RNA polymerase, were cultured in complete cell culture medium consisting of Glasgow’s Minimal Essential Medium (GMEM) (Gibco) supplemented with 1% tryptose phosphate broth (TPB) (Gibco), heat inactivated 10% foetal bovine serum (FBS) (Gibco) and 1% penicillin-streptomycin (Gibco). The cells were a kind gift from the laboratory of Prof. Massimo Palmarini (MRC-University of Glasgow Centre for Virus Research, UK). Cells were passaged twice weekly and maintained at 37°C, 5% CO2. At every fifth passage, the G-418 selection drug (1 mg/mL) (Scientific Laboratory Supplies) was added to cell media.

Table 1.
Sequences of primers used in this study.
| Target gene | Sequence (5' to 3') | Use |
|---|---|---|
| Fluc in pMA plasmid | TAATACGACTCACTATAGGG TCGTCCACTCGGATGGCTA |
Sequence 5’- and 3’- plasmids containing Fluc gene |
| VP1 plasmid | GGAAGGAGAGATGTACCAGGA | Sequence mutations of GDD motif in VP1 plasmid |
Site-directed mutagenesis. Site-directed mutagenesis was performed on the RF VP1 plasmid using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions but using half volume reactions. Thermal cycling parameters were: 2 min denaturing at 95°C, followed by 18 cycles of 30 sec denaturing at 95°C, 1 min primer annealing at 55°C, 6 min elongation at 68°C, with final 10 min elongation at 68°C. PCR products were digested using 1 µL of DpnI restriction enzyme to remove parental methylated DNA before transformation into competent E.coli cells. Products were visualised using gel electrophoresis. Successful mutagenesis was confirmed by Sanger sequencing.
Virus rescue. RV RF strain viruses and derivatives thereof were recovered using our previously described protocol [32]. Summarily, BSR-T7 cells in 6-well plates were co-transfected with 11 plasmids corresponding to each RV genome segment (2.5 µg for plasmids encoding NSP2 and NSP5; 0.8 µg for the remaining plasmids) using 16 µL Lipofectamine 2000 (Invitrogen) per transfection. After 24 hr incubation, MA104 cells (1 x 105 cells/well) were added to transfected BSR-T7 cells and co-cultured for 4 days in FBS-free Dulbecco’s Modified Eagle Medium (DMEM) (Sigma-Aldrich) supplemented with 0.5 µg/mL porcine pancreatic trypsin type IX (Sigma-Aldrich). Co-cultured cells were then lysed three times by freeze/thaw and lysates were incubated with trypsin at a final concentration of 10 µg/mL for 30 min to activate the virus. Lysates were then transferred to fresh MA104 cells in T25 flasks with 0.5 µg/mL porcine pancreatic trypsin type IX for up to 7 days and viruses were harvested. Mock preparations with the mutated segment omitted were generated for use as negative controls throughout. All rescue experiments were performed three times for each virus. Viruses were titred by plaque assays, and the presence of mutations in the VP1 gene segment was confirmed by Sanger sequencing (GATC Biotech or Genewiz, Germany).
Plaque assay. Plaque assays for RVs were performed using adapted methods [41,42]. Confluent monolayers of MA104 cells in 6-well plates were washed with FBS-free DMEM and infected with 800 μL of ten-fold serially diluted virus for 1 hr at 37°C 5% CO2. Following virus adsorption, 2 mL/well overlay medium was added (1:1 ratio of 2.4% Avicel (FMC Biopolymer) and FBS-free DMEM supplemented with 0.5 μg/mL porcine pancreatic trypsin type IX) and incubated for 4 days. Cells were then fixed for 1 hr with 1 mL/well of 10% neutral buffered formalin (CellPath) and stained for 1 hr with 0.1% Toluidine blue (Sigma-Aldrich) dissolved in H2O.
Luciferase assay. At 70-80% confluency, BSR-T7 cells in a 24-well plate were transfected with plasmid DNA using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol. Plasmid DNA and Lipofectamine reagent (1 µL of Lipofectamine for 1 µg of DNA) were separately diluted in 50 µL Opti-MEM. After a 5 min incubation at room temperature, diluted mixes were combined and incubated for a further 25 min. During this time, complete cell culture medium was changed to Opti-MEM (200 µL/well) and transfection mix was added to cells dropwise which were then incubated for 48 hr at 37°C, 5% CO2. Following incubation, supernatant was removed and cells were lysed in 150 µL of Active Lysis Buffer (Promega). Luciferase activity was analysed using Luciferase Assay Reagent (Promega) and signal was read on a Cytation 3 plate reader. A positive control plasmid expressing the Fluc gene driven by the cytomegalovirus (CMV) immediate early promoter, pVR1255, was used as a positive control throughout and is referred to as ‘+ve’.
Statistical analysis. GraphPad Prism v9 was used for all statistical analyses. Data are presented as mean and standard error of the mean from at least three independent experiments with technical duplicates unless otherwise stated. P values were determined by ratio paired t-test and were considered statistically significant at <0.05.
Results
Minigenome reporter construct design. The highly conserved cis-acting signals in the 5’- and 3’-UTRs of all RV segments are recognised by the viral RdRp for (+)RNA synthesis and dsRNA replication [16,17]. To set up a plasmid based mini-genome assay, initially synthetic DNA constructs were designed to express the firefly luciferase (Fluc) gene in either positive or negative orientation flanked by the 5’ and 3’ UTRs from the RF strain NSP1 gene (Figure 1A; positive and negative sense reporters were named 5’-reporter and 3’-reporter respectively). Thus, if the synthetic RV segment was recognised and transcribed and/or replicated by the viral polymerase, the luciferase levels in transfected cells would represent a measure of the transcriptional activity of the polymerase. NSP1 was selected by analogy with the influenza A virus minigenome assay, in which the open reading frame of NS1, also a broadly acting interferon antagonist, is replaced by a reporter gene [43]. The viral UTRs were flanked by T7P and HDV ribozyme sequences, as used successfully in the development of the RV reverse genetics system [33]. Thus, transcription of the resulting vector would generate full length viral (+) single stranded RNA transcripts containing native viral 5’ and 3’ termini [44].
To determine the quantity of the reporter plasmids required that could be transfected while generating only minimal background signal, increasing amounts were transfected into BSR-T7 cells (Figure 1B). The positive control (‘+ve’) luciferase-expressing plasmid produced a strong luciferase signal that increased with plasmid dose. In contrast, the RV reporter genes gave very low levels of signal, not significantly above the background of no luciferase gene at doses of 100 ng and lower (Figure 1B). Both reporters gave levels of signal that were above background at 200 ng, but this was still around 30 RLU or lower, so this was chosen as the amount of reporter construct to take forward for further assay development.
Reporter expression by RV polymerase. During RV reverse genetics, 11 plasmids (one per genome segment) are co-transfected into BSR-T7 cells for successful virus rescue, meaning that viral polymerase is functional and is able to copy both transcript polarities, thereby generating the complete viral genome. We therefore predicted that co-transfection of either of our luciferase reporters with the full complement of reverse genetics plasmids would result in an amplification of the luciferase signal above background. To test this, a dose-dependent titration was performed for all 11 plasmids in the presence of 200 ng of the 5’- and 3’-reporters to determine the minimal amount of RV plasmids needed to generate the strongest luciferase signal (Figure 2A; as in reverse genetics [32,34], NSP2 and NSP5 plasmids were used at 3.125X amounts relative to the other nine plasmids). The pVR1255 positive control plasmid produced a strong luciferase signal (Figure 2B,C). Throughout, both 5’ and 3’ reporter plasmids expressed alone gave similar levels of background to those seen in Figure 1B (data not shown), and so the cognate reporter signals were subtracted from all readings from samples transfected with RV plasmids. The 5’ reporter yielded a 2-3 log10 increase in luminescence above background between 25 – 200 ng of the 11 RG plasmids, with apparent saturation at 200 ng and a decrease at 400 ng (Figure 2B). The same trend was observed for the 3’ reporter, but overall luminescence signals were around one log10 lower than for the 5’ reporter. The 5’ reporter was therefore considered to be more suitable than the 3’-reporter for the minigenome assay, with 100-200 ng (312.5-625 ng for NSP2 and NSP5) of each reverse genetics plasmid being the optimal amount.
Generation of an inactive RdRp. The increase in luciferase signal in the presence of all RV polypeptides was suggestive but not conclusive evidence for viral polymerase activity. Therefore for further assay validation, we sought to generate a viral RdRp VP1 mutant lacking polymerase activity. The RV RdRp resembles a ‘right-handed’ architecture made up of the N-terminal domain, the core and the C-terminal domain, where the core is further split into the “palm, finger and thumb” subdomains [19,27]. Ogden et al., (2012) showed that the conserved aspartate residues within the ‘GDD’ motif in the palm subdomain (Figure 3A) were critical for RNA synthesis [26]. These conserved aspartate residues, D631 and D632, were mutated to alanine by site-directed mutagenesis, creating mutants D631A and D632A respectively (Figure 3B). To test for successful inhibition of viral RdRp activity, rescues of the VP1 mutants were attempted using reverse genetics, and as expected, no virus was recovered in the presence of the mutated polymerase (Figure 3C). This confirmed that the mutations rendered the virus replication incompetent. The same VP1 mutants were therefore tested in the minigenome assay, with 11 RV gene segments (100 ng each except 312.5 ng for NSP2 and NSP5), co-transfected into BSR-T7 with 200 ng of the 5’-reporter. As before, a pVR1255 positive control produced a strong luciferase signal (Figure 3D). Unexpectedly however, only a modest decrease in signal was observed for the D631A mutant and only the D632A mutant yielding a significant reduction, suggesting some remaining transcriptional activity of the polymerase. Thus, while the entire GDD motif appears to be essential for viral rescue, single amino acid mutations were tolerated in the minigenome assay.
Exploring the minimal requirements for RV minigenome assay. Although 11 plasmids are required for viral rescue, we considered the possibility that not all 11 segments may be needed for the minigenome assay. During RV replication, upon cell entry the loss of the outer protein layer of VP4 and VP7 triggers a conformational switch that induces polymerase activity in the now double-layered virus particle (DLP) [45]. The DLP contains a core comprising VP1, VP2 and VP3, surrounded by a shell of VP6. We therefore considered the possibility that reconstitution of the DLP alone may be sufficient for polymerase activity and whether transfection of only these four proteins with the 5’-reporter was sufficient to yield luciferase signal. However, transfecting equimolar amounts of VP1-13 and VP6 plasmids resulted in a dramatic loss of signal relative to the 11-plasmid system that was barely above background (Figure 4). However, Patton et al., (1997) showed that a molar ratio of 1 VP1 to 11 VP2, similar to that found in virion cores, produced the highest level of dsRNA synthesis in the cell-free system [46]. Therefore, we also tested whether adjusting the VP1:VP2 ratio to 1:11 would improve the signal. Indeed, when the amount of VP2 plasmid was increased 11-fold, the signal increased significantly, and was only slightly lower than that of the 11-plasmid system. This demonstrates that RV RdRp is active when only components of the DLP are expressed.
Discussion
Minigenome assays have been used to study the in-cell polymerase activity for a range of RNA viruses with single stranded genomes including influenza A virus [47], respiratory syncytial virus [48] and poliovirus [49]. As far as we are aware, in-cell reconstitution of viral polymerase activity has not been achieved previously for a virus with a double stranded RNA genome, though polymerase activity has been assayed in a cell-free system for bluetongue virus [50]. Minigenome assays have yielded significant breakthroughs in understanding of viral polymerase functions and domains, and this development represents an opportunity for such research questions to be applied to RVs.
Through this work we unexpectedly found that mutating the highly conserved catalytic GDD motif of RV VP1 only modestly impaired polymerase activity. This minor reduction in polymerase activity is likely insufficient to explain why the same mutations abrogated virus production. Possibly, this domain of VP1 has multiple functions required to complete a virus lifecycle beyond its well characterised role in dsRNA replication, such as genome packaging [19,26].
We have demonstrated that the polymerase is active when only components of the DLP are expressed, with no absolute requirement for non-structural proteins. NSP2 and NSP5 are together necessary and sufficient for the formation of viroplasms (or viroplasm-like structures), which form a sequestered environment for the accumulation of viral proteins and DLPs [45]. NSP3 replaces poly-A binding protein in ribosomal complexes to bind viral non-polyadenylated transcripts, thereby enhancing their translation (and also reducing translation of cellular polyadenylated transcripts) [51]; nevertheless, in its absence, viral polymerase activity was apparent, demonstrating that translation of viral proteins occurs readily in NSP3’s absence. To improve the dynamic range of this assay, co-expression of this subset of non-structural proteins could be explored. Co-expression of capping enzymes such as that of African swine fever virus, shown to be efficient in RV reverse genetics [52], may also augment the efficiency of this minigenome assay.
Further improvements to this system might include the co-expression of multiple viral gene segments from the same plasmid. This would reduce the number of plasmids being co-transfected, and so theoretically increase the number of cells receiving the full complement of viral proteins required for viral polymerase activity to occur. There may be a trade-off due to the possibility of larger plasmids transfecting with poorer efficiency, but we have found that analogous multi-segment plasmids improve minigenome assay efficiency in the influenza A virus system (unpublished data).
The polymerase assay system reported here was established using reverse genetics plasmids, and so all viral gene segments were under a T7 promoter, necessitating the use of BSR-T7 cells. It is likely that higher transfection efficiencies would be achieved in HEK293T cells (infectable with RV in our hands; data not shown), which are used for other minigenome assays including that of influenza A virus. Testing this would require cloning of the constructs used into a different backbone, so that viral genes are expressed under a mammalian promoter such as CMV. Alternatively, the T7 polymerase would need to be stably expressed in HEK293T cells.
Here we have established a minigenome assay for the RF strain, which is a widely used and well characterised lab strain of RV. The generalisability of this approach to other RV strains should be tested in future, using the analogous approach of generating a reporter construct encoded by flanking NSP1 UTRs of the cognate strain, analogous to strategies for maximising influenza A virus gene expression [53]. It is possible that the use of UTRs from other viral segments would yield a higher translational efficiency, which was not examined here. Reporter constructs comprising the UTRs from heterologous RV strains should also be explored as this would negate the requirement for strain-specific reporter constructs.
Author Contributions
Conceptualization, PD and EG; methodology, OD, PD and EG; validation, OD and EG; formal analysis, OD and EG; investigation, OD, CPS, BT, HWC, and EG; resources, PD and EG; data curation, OD and EG; writing – original draft preparation, OD and EG; writing – reviewing and editing, ED, HWC, PD and EG; vizualisation, OD, PD and EG; supervision, CS, PD and EG; project administration, EG; funding acquisition, PD and EG. All authors have read and agreed to the published version of the manuscript. We want to thank lab members, central support unit (CSU) and technical staff for their support and assistance with this project. We are grateful to Dr Valeria Lulla and Dr Ulrich Desselberger (both University of Cambridge) for helpful discussions.
Funding
PD: EG and CPS are supported by a BBSRC Institute Strategic Programme grant (BBS/E/RL/230002C). EG, CPS and BT are also supported by a Wellcome Trust/ Royal Society Sir Henry Dale Fellowship (211222_Z_18_Z). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. For the purpose of open access, the authors have applied a CC 775 BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Data Availability Statement
All data presented in this manuscript are available at the discretion of the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatrics 2018, 172, 958–965. [Google Scholar] [CrossRef]
- Bányai, K.; Kemenesi, G.; Budinski, I.; Földes, F.; Zana, B.; Marton, S.; Varga-Kugler, R.; Oldal, M.; Kurucz, K.; Jakab, F. Candidate new rotavirus species in Schreiber's bats, Serbia. Infection, Genetics and Evolution 2017, 48, 19–26. [Google Scholar] [CrossRef]
- Johne, R.; Schilling-Loeffler, K.; Ulrich, R.G.; Tausch, S.H. Whole Genome Sequence Analysis of a Prototype Strain of the Novel Putative Rotavirus Species L. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Tausch, S.H.; Ulrich, R.G.; Schilling-Loeffler, K. Genome analysis of the novel putative rotavirus species K. Virus Res 2023, 334, 199171. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Archives of Virology 2012, 157, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Mihalov-Kovács, E.; Gellért, Á.; Marton, S.; Farkas, S.L.; Fehér, E.; Oldal, M.; Jakab, F.; Martella, V.; Bányai, K. Candidate new rotavirus species in sheltered dogs, Hungary. Emerg Infect Dis 2015, 21, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Baker, M.L.; Jiang, W.; Estes, M.K.; Prasad, B.V. Rotavirus architecture at subnanometer resolution. J Virol 2009, 83, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Settembre, E.C.; Chen, J.Z.; Dormitzer, P.R.; Grigorieff, N.; Harrison, S.C. Atomic model of an infectious rotavirus particle. Embo j 2011, 30, 408–416. [Google Scholar] [CrossRef] [PubMed]
- McClain, B.; Settembre, E.; Temple, B.R.; Bellamy, A.R.; Harrison, S.C. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 A resolution. J Mol Biol 2010, 397, 587–599. [Google Scholar] [CrossRef]
- Estrozi, L.F.; Settembre, E.C.; Goret, G.; McClain, B.; Zhang, X.; Chen, J.Z.; Grigorieff, N.; Harrison, S.C. Location of the dsRNA-dependent polymerase, VP1, in rotavirus particles. J Mol Biol 2013, 425, 124–132. [Google Scholar] [CrossRef]
- Prasad, B.V.V.; Rothnagel, R.; Zeng, C.Q.Y.; Jakana, J.; Lawton, J.A.; Chiu, W.; Estes, M.K. Visualization of ordered genomic RNA and localization of transcriptional complexes in rotavirus. Nature 1996, 382, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, KM.; McDonald, SM.; Patton, JT. Mechanism of intraparticle synthesis of the rotavirus double-stranded RNA genome. The Journal of biological chemistry 2010, 285, 18123–18128. [Google Scholar] [CrossRef] [PubMed]
- Jenni, S.; Salgado, E.N.; Herrmann, T.; Li, Z.; Grant, T.; Grigorieff, N.; Trapani, S.; Estrozi, L.F.; Harrison, S.C. In situ Structure of Rotavirus VP1 RNA-Dependent RNA Polymerase. J Mol Biol 2019, 431, 3124–3138. [Google Scholar] [CrossRef] [PubMed]
- Crawford S.E.; Ding S.D.; Greenberg H.B.; M.K., E. Rotaviruses; Howley PM., Ed. 2023.
- Periz, J.; Celma, C.; Jing, B.; Pinkney, J.N.; Roy, P.; Kapanidis, A.N. Rotavirus mRNAS are released by transcript-specific channels in the double-layered viral capsid. Proc Natl Acad Sci U S A 2013, 110, 12042–12047. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Broering, T.J.; Nibert, M.L.; Patton, J.T. Template recognition and formation of initiation complexes by the replicase of a segmented double-stranded RNA virus. The Journal of biological chemistry 2003, 278, 32673–32682. [Google Scholar] [CrossRef] [PubMed]
- Tortorici M., A.; Shapiro B., A.; Patton J., T. A base-specific recognition signal in the 5' consensus sequence of rotavirus plus-strand RNAs promotes replication of the double-stranded RNA genome segments. RNA (New York, N.Y.) 2006, 12, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Celma, C.C.; Zhang, X.; Chang, T.; Shen, W.; Atanasov, I.; Roy, P.; Zhou, Z.H. In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nature Communications 2019, 10, 2216. [Google Scholar] [CrossRef]
- Lu, X.; McDonald, S.M.; Tortorici, M.A.; Tao, Y.J.; Vasquez-Del Carpio, R.; Nibert, M.L.; Patton, J.T.; Harrison, S.C. Mechanism for coordinated RNA packaging and genome replication by rotavirus polymerase VP1. Structure (London, England : 1993) 2008, 16, 1678–1688. [Google Scholar] [CrossRef]
- Barro, M.; Mandiola, P.; Chen, D.; Patton, J.T.; Spencer, E. Identification of sequences in rotavirus mRNAs important for minus strand synthesis using antisense oligonucleotides. Virology 2001, 288, 71–80. [Google Scholar] [CrossRef]
- Chen, D.; Barros, M.; Spencer, E.; Patton, J.T. Features of the 3'-consensus sequence of rotavirus mRNAs critical to minus strand synthesis. Virology 2001, 282, 221–229. [Google Scholar] [CrossRef]
- Navarro, A.; Trask S., D.; Patton J., T. Generation of genetically stable recombinant rotaviruses containing novel genome rearrangements and heterologous sequences by reverse genetics. J Virol 2013, 87, 6211–6220. [Google Scholar] [CrossRef] [PubMed]
- Patton J., T.; Chnaiderman, J.; Spencer, E. Open reading frame in rotavirus mRNA specifically promotes synthesis of double-stranded RNA: template size also affects replication efficiency. Virology 1999, 264, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Patton J., T. Rotavirus RNA replication requires a single-stranded 3' end for efficient minus-strand synthesis. J Virol 1998, 72, 7387–7396. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Manktelow, E.; von Kirchbach, J.C.; Gog, J.R.; Desselberger, U.; Lever, A.M. Genomic analysis of codon, sequence and structural conservation with selective biochemical-structure mapping reveals highly conserved and dynamic structures in rotavirus RNAs with potential cis-acting functions. Nucleic Acids Res 2010, 38, 7718–7735. [Google Scholar] [CrossRef] [PubMed]
- Ogden, K.M.; Ramanathan, H.N.; Patton, J.T. Mutational analysis of residues involved in nucleotide and divalent cation stabilization in the rotavirus RNA-dependent RNA polymerase catalytic pocket. Virology 2012, 431, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Steger, C.; Brown, M.; Sullivan, O.; Boudreaux, C.; Cohen, C.; LaConte, L.; McDonald, S. In Vitro Double-Stranded RNA Synthesis by Rotavirus Polymerase Mutants with Lesions at Core Shell Contact Sites. Journal of Virology 2019, 93. [Google Scholar] [CrossRef]
- Tao, Y.; Farsetta, D.L.; Nibert, M.L.; Harrison, S.C. RNA synthesis in a cage--structural studies of reovirus polymerase lambda3. Cell 2002, 111, 733–745. [Google Scholar] [CrossRef]
- Zeng, C.Q.; Estes, M.K.; Charpilienne, A.; Cohen, J. The N terminus of rotavirus VP2 is necessary for encapsidation of VP1 and VP3. J Virol 1998, 72, 201–208. [Google Scholar] [CrossRef]
- Gridley C., L.; Patton J., T. Regulation of rotavirus polymerase activity by inner capsid proteins. Current opinion in virology 2014, 9, 31–38. [Google Scholar] [CrossRef]
- Patton, J.T.; Jones, M.T.; Kalbach, A.N.; He, Y.W.; Xiaobo, J. Rotavirus RNA polymerase requires the core shell protein to synthesize the double-stranded RNA genome. J Virol 1997, 71, 9618–9626. [Google Scholar] [CrossRef]
- Diebold, O.; Gonzalez, V.; Venditti, L.; Sharp, C.; Blake, R.A.; Tan, W.S.; Stevens, J.; Caddy, S.; Digard, P.; Borodavka, A.; et al. Using Species a Rotavirus Reverse Genetics to Engineer Chimeric Viruses Expressing SARS-CoV-2 Spike Epitopes. J Virol 2022, 96, e0048822. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Komoto, S.; Kawagishi, T.; Nouda, R.; Nagasawa, N.; Onishi, M.; Matsuura, Y.; Taniguchi, K.; Kobayashi, T. Entirely plasmid-based reverse genetics system for rotaviruses. Proceedings of the National Academy of Sciences 2017, 114, 2349. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Fukuda, S.; Ide, T.; Ito, N.; Sugiyama, M.; Yoshikawa, T.; Murata, T.; Taniguchi, K. Generation of Recombinant Rotaviruses Expressing Fluorescent Proteins by Using an Optimized Reverse Genetics System. Journal of Virology 2018, 92, e00588-00518. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Kanai, Y.; Fukuda, S.; Kugita, M.; Kawagishi, T.; Ito, N.; Sugiyama, M.; Matsuura, Y.; Kobayashi, T.; Taniguchi, K. Reverse Genetics System Demonstrates that Rotavirus Nonstructural Protein NSP6 Is Not Essential for Viral Replication in Cell Culture. Journal of Virology 2017, 91, e00695-00617. [Google Scholar] [CrossRef] [PubMed]
- Percy, N.; Barclay, W.S.; Sullivan, M.; Almond, J.W. A poliovirus replicon containing the chloramphenicol acetyltransferase gene can be used to study the replication and encapsidation of poliovirus RNA. Journal of Virology 1992, 66, 5040–5046. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, M.S.; et al. Rescue of Synthetic Measles Virus Minireplicons: Measles Genomic Termini Direct Efficient Expression and Propagation of a Reporter Gene. Virology 1995, 208, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Groseth, A.; et al. RNA Polymerase I-Driven Minigenome System for Ebola Viruses. Journal of Virology 2005, 79, 4425–4433. [Google Scholar] [CrossRef]
- Lutz, A.; et al. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. Journal of Virological Methods 2005, 126, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. SSE: a nucleotide and amino acid sequence analysis platform. BMC Research Notes 2012, 5, 50. [Google Scholar] [CrossRef]
- Matrosovich, M.; Matrosovich, T.; Garten, W.; Klenk, H.-D. New low-viscosity overlay medium for viral plaque assays. Virology Journal 2006, 3, 63. [Google Scholar] [CrossRef]
- Arnold M.; Patton J.T.; McDonald S.M. Culturing, storage, and quantification of rotaviruses; 2009; Vol. Chapter 15.
- Wise, HM.; Foeglein, A.; Sun, J.; Dalton Rosa, M.; Patel, S.; Howard, W.; Anderson E., C.; Barclay W., S.; Digard, P. A Complicated Message: Identification of a Novel PB1-Related Protein Translated from Influenza A Virus Segment 2 mRNA. Journal of Virology 2009, 83, 8021–8031. [Google Scholar] [CrossRef] [PubMed]
- Roner M., R.; Joklik W., K. Reovirus reverse genetics: Incorporation of the CAT gene into the reovirus genome. Proc Natl Acad Sci U S A 2001, 98, 8036–8041. [Google Scholar] [CrossRef]
- Desselberger, U. Rotaviruses. Virus Res 2014, 190, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.T.; Jones, M.T.; Kalbach, A.N.; He, Y.W.; Xiaobo, J. Rotavirus RNA polymerase requires the core shell protein to synthesize the double-stranded RNA genome. 1997, 71, 9618–9626. [Google Scholar] [CrossRef] [PubMed]
- te Velthuis A; Long J; Barclay W. Assays to Measure the Activity of Influenza Virus Polymerase. 2018; Vol. 1836, pp. 343–374.
- Noton, S.L.; Cowton, V.M.; Zack, C.R.; McGivern, D.R.; Fearns, R. Evidence that the polymerase of respiratory syncytial virus initiates RNA replication in a nontemplated fashion. Proc Natl Acad Sci U S A 2010, 107, 10226–10231. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; Rieder, E.; Kim, D.W.; Boom, J.H.v.; Wimmer, E. Identification of an RNA Hairpin in Poliovirus RNA That Serves as the Primary Template in the In Vitro Uridylylation of VPg. 2000, 74, 10359–10370. [Google Scholar] [CrossRef] [PubMed]
- Wehrfritz, J.M.; Boyce, M.; Mirza, S.; Roy, P. Reconstitution of bluetongue virus polymerase activity from isolated domains based on a three-dimensional structural model. 2007, 86, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Vende, P.; Piron, M.; Castagné, N.; Poncet, D. Efficient Translation of Rotavirus mRNA Requires Simultaneous Interaction of NSP3 with the Eukaryotic Translation Initiation Factor eIF4G and the mRNA 3′ End. Journal of Virology 2000, 74, 7064–7071. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tacuba, L.; Feng, N.; Meade N., J.; Mellits K., H.; Jaïs P., H.; Yasukawa L., L.; Resch T., K.; Jiang, B.; López, S.; Ding, S.; et al. An Optimized Reverse Genetics System Suitable for Efficient Recovery of Simian, Human, and Murine-Like Rotaviruses. J Virol 2020, 94. [Google Scholar] [CrossRef]
- Zheng, H.; Palese, P.; García-Sastre, A. Nonconserved nucleotides at the 3' and 5' ends of an influenza A virus RNA play an important role in viral RNA replication. Virology 1996, 217, 242–251. [Google Scholar] [CrossRef]
Figure 1.
Minigenome reporter gene construct design. (A) Schematic of reporter gene construct for mini-genome assay. Plasmids under a T7 promoter (T7P) encoding the Fluc gene in either positive (5’-reporter) or negative sense (3’-reporter) flanked by the 5’- and 3’-UTRs. The plasmids included HDV ribozyme and T7 terminator sequences (T7T). (B) Dose-dependent titration of the 5’- and 3’-reporter plasmids. The pVR1255 plasmid expressing FLuc gene was used as a positive control (denoted as ‘+ve’). Mock sample contained transfection reagent only. Data are mean ± SEM from four independent experiments.
Figure 1.
Minigenome reporter gene construct design. (A) Schematic of reporter gene construct for mini-genome assay. Plasmids under a T7 promoter (T7P) encoding the Fluc gene in either positive (5’-reporter) or negative sense (3’-reporter) flanked by the 5’- and 3’-UTRs. The plasmids included HDV ribozyme and T7 terminator sequences (T7T). (B) Dose-dependent titration of the 5’- and 3’-reporter plasmids. The pVR1255 plasmid expressing FLuc gene was used as a positive control (denoted as ‘+ve’). Mock sample contained transfection reagent only. Data are mean ± SEM from four independent experiments.
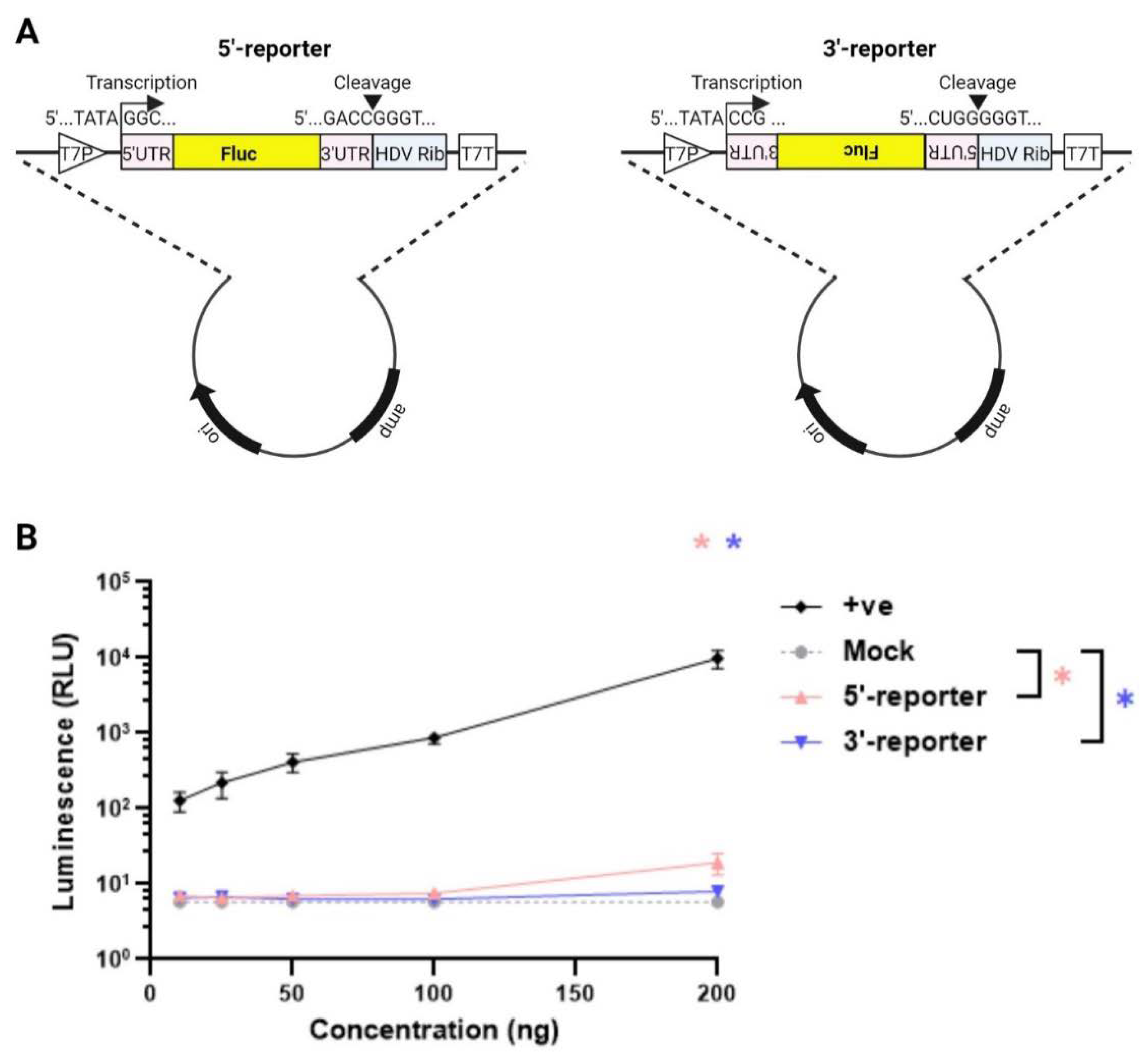
Figure 2.
Reporter expression by rotavirus polymerase. (A) Schematic of proposed mini-genome assay. RV plasmids encoding each bovine RF stain gene were co-transfected with T7 reporter plasmids expressing the Fluc gene in either positive (5’-reporter) or negative sense (3’-reporter). Luciferase activity was measured after 48 hr post transfection. Dose-dependent titration of 11 RV plasmids with 200 ng of the 5’-reporter in (B) and with 200 ng of the 3’-reporter in (C). As in reverse genetics, the amount of plasmids expressing NSP2 and NSP5 genes was increased to scale.
Figure 2.
Reporter expression by rotavirus polymerase. (A) Schematic of proposed mini-genome assay. RV plasmids encoding each bovine RF stain gene were co-transfected with T7 reporter plasmids expressing the Fluc gene in either positive (5’-reporter) or negative sense (3’-reporter). Luciferase activity was measured after 48 hr post transfection. Dose-dependent titration of 11 RV plasmids with 200 ng of the 5’-reporter in (B) and with 200 ng of the 3’-reporter in (C). As in reverse genetics, the amount of plasmids expressing NSP2 and NSP5 genes was increased to scale.
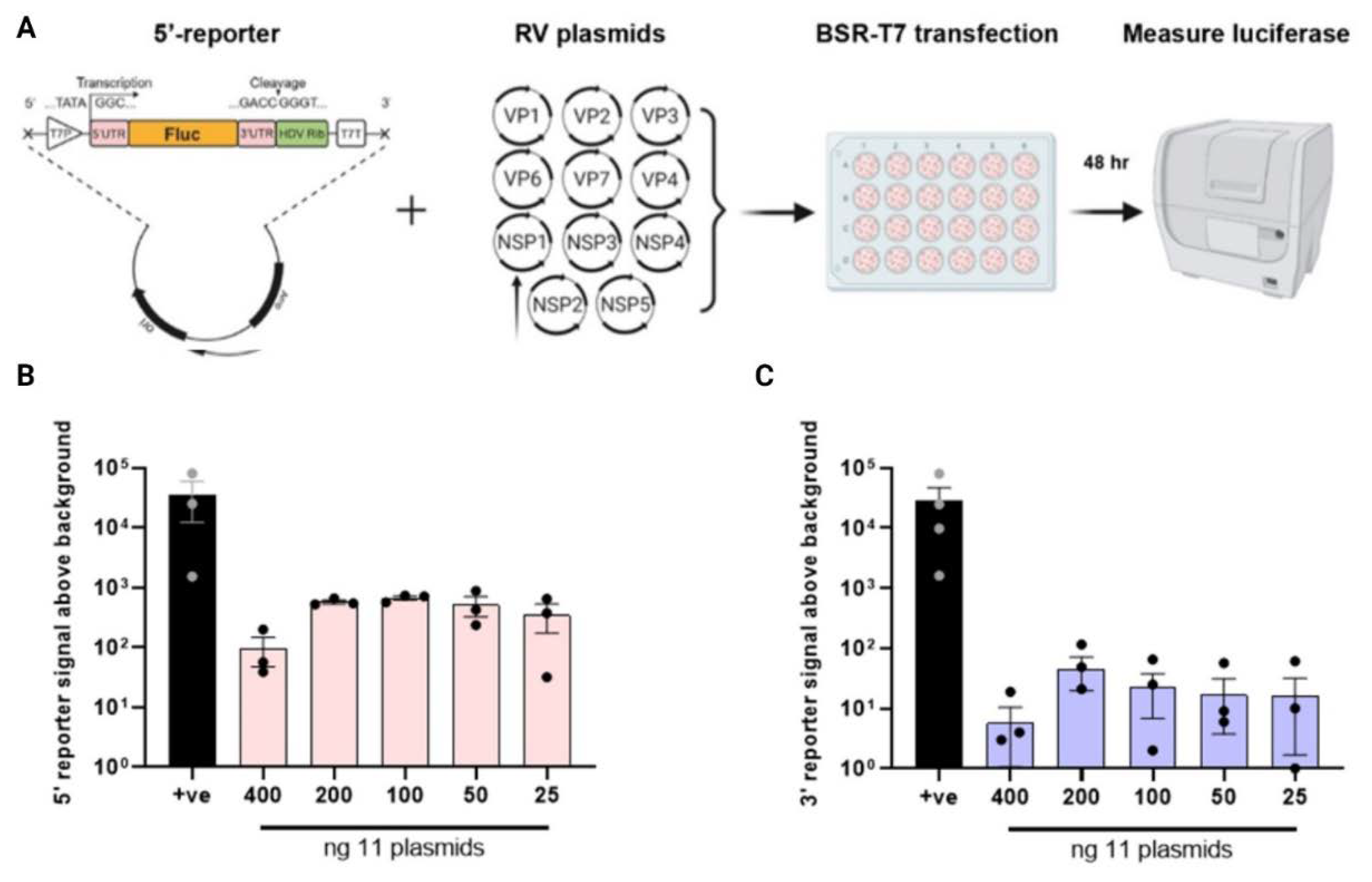
Figure 3.
Generation of inactive polymerase. (A) VP1 shown as a linear schematic and coloured according to the domain organisation with amino acid numbers labelled above. Adapted from [27], with alignments for RVH-RVL updated/added based on their reference sequences (KT962027.1 , NC_026825.2, NC_055268.1, OQ934016.1, OM101015.1). The N-terminal and C-terminal domains (deep blue) flank the core domain containing fingers (pale blue), palm (mid blue) and thumb (orange). Sequence-based alignment of RdRps across RV species and the related reovirus showing the conserved catalytic ‘GDD’ site. Dots indicate amino acid conservation. Highlighted in yellow are the two conserved aspartic acid residues targeted for mutagenesis. (B) Mutagenesis strategy for evolutionarily conserved aspartic acid residues in the VP1 catalytic domain. (C) Viral titres WT RF and of VP1 mutants. (D) Minigenome assay for VP1 mutants. In all cases, all 11 RG plasmids were transfected (with 3.125X amounts of NSP2 and NSP5 plasmids) along with 200 ng 5’-reporter. pVR1255 plasmid expressing FLuc gene was used as a positive control (denoted as ‘+ve’).
Figure 3.
Generation of inactive polymerase. (A) VP1 shown as a linear schematic and coloured according to the domain organisation with amino acid numbers labelled above. Adapted from [27], with alignments for RVH-RVL updated/added based on their reference sequences (KT962027.1 , NC_026825.2, NC_055268.1, OQ934016.1, OM101015.1). The N-terminal and C-terminal domains (deep blue) flank the core domain containing fingers (pale blue), palm (mid blue) and thumb (orange). Sequence-based alignment of RdRps across RV species and the related reovirus showing the conserved catalytic ‘GDD’ site. Dots indicate amino acid conservation. Highlighted in yellow are the two conserved aspartic acid residues targeted for mutagenesis. (B) Mutagenesis strategy for evolutionarily conserved aspartic acid residues in the VP1 catalytic domain. (C) Viral titres WT RF and of VP1 mutants. (D) Minigenome assay for VP1 mutants. In all cases, all 11 RG plasmids were transfected (with 3.125X amounts of NSP2 and NSP5 plasmids) along with 200 ng 5’-reporter. pVR1255 plasmid expressing FLuc gene was used as a positive control (denoted as ‘+ve’).
Figure 4.
Measuring RdRp activity. Luciferase activity following co-transfection of RV plasmids with 200 ng of 5’-reporter. WT RF denotes co-transfection of all 11 RG plasmids (with 3.125X amounts of NSP2 and NSP5 plasmids) with reporter. To test whether the number of RG plasmids expressed could be reduced, 4 plasmids corresponding to VP1, VP2, VP3 and VP6 were co-transfected with the 5’-reporter (‘4 plasmids + 5’ rep). To test whether the 4-plasmid system could be improved upon, the amount of the VP2 plasmid was increased 11-fold (‘VP1:VP2 ratio’). pVR1255 plasmid expressing FLuc gene was used as a positive control (denoted as ‘+ve’). Data are normalised to the 5’-reporter only sample.
Figure 4.
Measuring RdRp activity. Luciferase activity following co-transfection of RV plasmids with 200 ng of 5’-reporter. WT RF denotes co-transfection of all 11 RG plasmids (with 3.125X amounts of NSP2 and NSP5 plasmids) with reporter. To test whether the number of RG plasmids expressed could be reduced, 4 plasmids corresponding to VP1, VP2, VP3 and VP6 were co-transfected with the 5’-reporter (‘4 plasmids + 5’ rep). To test whether the 4-plasmid system could be improved upon, the amount of the VP2 plasmid was increased 11-fold (‘VP1:VP2 ratio’). pVR1255 plasmid expressing FLuc gene was used as a positive control (denoted as ‘+ve’). Data are normalised to the 5’-reporter only sample.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



