Submitted:
30 April 2024
Posted:
01 May 2024
You are already at the latest version
Abstract
Duchenne muscular dystrophy (DMD) is caused by genetic mutations in the cytoskeletal-sarcolemmal anchor protein dystrophin. Repeated cycles of sarcolemmal tearing and repair lead to a variety of secondary cellular and physiological stressors that are thought to contribute to weakness, atrophy, and fibrosis. Collectively, these stressors can contribute to a pro-inflammatory milieu in locomotor, cardiac, and respiratory muscles. Given the many unwanted side effects that accompany current anti-inflammatory steroid-based approaches for treating DMD (e.g., glucocorticoids), there is a need to develop new therapies that address inflammation and other cellular dysfunctions. Adiponectin receptor (AdipoR) agonists, which stimulate AdipoR1 and R2 isoforms on various cell types, have emerged as therapeutic candidates for DMD due to their anti-inflammatory, anti-fibrotic, and pro-myogenic properties in pre-clinical human and rodent DMD models. Although these molecules represent a new direction for therapeutic intervention, the mechanisms through which they elicit their beneficial effects are not yet fully understood, and DMD-specific data is limited. The overarching goal of this review is to investigate how adiponectin signaling may ameliorate pathology associated with dystrophin deficiency through inflammatory-dependent and -independent mechanisms, and to determine if current data supports their future progression to clinical trials.
Keywords:
Duchenne muscular dystrophy
; cardiomyopathy
; inflammation
; skeletal muscle
; adiponectin
1. Overview of Muscular Dystrophy
1.1. Introduction
Muscular dystrophies (MD) are hereditary or genetic (spontaneous/non-hereditary) diseases characterized by severe progressive deterioration of locomotor, respiratory, and/or cardiac muscles. While there are many types of muscular dystrophies, X-linked dystrophinopathy defined by over 70 different mutations in the dystrophin gene are the most common and occurs predominantly in males [1,2]. Such mutations can lead to a complete absence of dystrophin (Duchenne muscular dystrophy; DMD) or a truncated transcript (Becker muscular dystrophy; BMD) [3,4,5,6]. While DMD is generally more severe than BMD, both diseases cause muscle weakness, atrophy, and fibrosis leading to reduced mobility, respiratory and cardiac dysfunction, as well as reduced lifespans. Dystrophin mutations often result in additional systemic dysfunctions including cognitive impairment, digestive abnormalities, anxiety, depression, or obesity [7,8,9].
While there is no cure, ongoing pre-clinical research seeks to restore the normal genetic sequence through emerging gene editing technologies such as CRISPR or produce truncated dystrophin transcripts with exon skipping and microdystrophin gene therapies [10,11,12]. Exon skipping therapy was recently approved by the FDA in the USA for certain mutations, but the technology must be adapted for each of the numerous mutations. Their ability to partially improve muscle dysfunction underscores the importance of maintaining glucocorticoid therapy as a major standard of care given systemic inflammation is a major contributor to muscle dysfunction [13,14,15] for virtually all persons with DMD. While the effectiveness of glucocorticoids in slowing the decline of muscle function demonstrates the value in targeting secondary contributors to these diseases, the eventual decline in muscle function and side effects (i.e. attenuated growth, obesity, mood disorders, other) [16] underscores the importance of developing additional treatments that provide benefits for most patients regardless of the specific underlying mutation.
Exogenously administered agonists of adiponectin (ApN) and its downstream cell-surface receptors have emerged as an attractive candidate for pharmacological intervention given their anti-inflammatory properties in DMD [17,18,19,20,21,22]. Although pre-clinical data from in vivo rodent and in vitro human cell-based models suggests that exogenous ApN agonism may be beneficial for attenuating some of the secondary physiological stressors associated with DMD, further insight is required to elucidate the specific mechanisms. In addition to attenuating inflammation, ApN also induces considerable metabolic reprogramming in muscle [10,23,24,25,26,27,28,29]. In this regard, the purpose of this review is to provide an overview of the pathophysiology of DMD with a perspective of translating disease mechanisms to the development of ApN agonist therapies.
1.2. Muscle Damage and Inflammation in DMD
Dystrophin is an indispensable component of cellular architecture given that it is responsible for anchoring the actin cytoskeleton to the dystrophin-associated glycoprotein complex (DGC) at the cell membrane (sarcolemma) to maintain cellular stability [30]. Dystrophin-deficient models are characterized by loss of myofiber integrity, essentially rendering muscle fibres susceptible to contraction-induced damage [31]. In DMD, muscle fibres demonstrate chronic damage that arises from contraction-relaxation cycles. However, the regenerative capacity of muscle fibres eventually exhausts, leading to impaired homeostatic muscle repair and turnover [32,33,34]. This cycle leads to muscle fibre necrosis and fibrofatty replacement of necrotic tissue, as well as atrophy, as a last-resort mechanism to uphold cyto-structure [35].
Key to the pathology of DMD is prolonged activation of the innate immune system in response to the chronic contraction-induced damage of muscle fibres [36]. The innate immune response is triggered when granulocytes, monocytes and monocyte-derived-macrophages, and dendritic cells are triggered by damage-associated molecular patterns (DAMPs) that leak from damaged muscle fibres [37,38]. DAMPs trigger the recruitment of macrophages and neutrophils to sites of damage by binding to their pathogen recognition receptors (PRRs), which include toll-like receptors (TLR2/4/7) [33,38,39,40,41]. Interestingly, data has demonstrated that deleting TLR2 or administering a TLR7/9 antagonist in C57BL/10.mdx mice (commonly utilized rodent model of DMD) reduces muscle inflammation and improves skeletal muscle function, thus supporting the notion that PRRs play a pivotal role in promoting muscle degeneration [42,43].
Following TLR activation, downstream inflammatory signaling is mediated by nuclear factor kappa B (NF-κB) [44], c-Jun NH2-terminal kinase (JNK) [45], and interferon regulatory factors (IRFs), which are activated by tumour necrosis factor alpha (TNFα) [46], interleukin (IL) 6 (IL-6) [47], and the myeloid differentiation primary response 88 (MyD88)-dependent pathways [34,39]. NF-κB activation induces the expression of pro-inflammatory genes in the nucleus [37,39] including IL-6, which promotes inflammation. IL-6 also interferes with muscle satellite cell populations and impedes muscle regeneration [48,49].
The induction of pro-inflammatory signaling events occurs in M1 classically-activated macrophages [36,50]. Since DMD, by definition, is characterized by asynchronous cycles of muscle damage and repair, M1 macrophages must be continuously recruited to sites of damage to sustain an immune response. Consequently, a high concentration of pro-inflammatory cytokines such as TNFα, IL-6, and IL-1β perpetuates a chronic inflammatory state [51]. Although many different chemo-attractive molecules can stimulate the recruitment of immune cells to dystrophic muscles, C-C motif chemokine receptor type 2 (CCR2) has demonstrated a significant role for recruiting inflammatory cells to sites of injury in C57BL/10.mdx muscle [32,36]. During early phases of inflammation, elevated pro-inflammatory cytokine concentrations can lead to the production of inducible nitric oxide synthase (iNOS), which alongside other cytoplasmic and mitochondrial oxidizing radicals [52,53], can significantly damage dystrophin deficient skeletal muscle by increasing damage to surrounding tissues and causing aberrant cell lysis [50]. While M1 pro-inflammatory macrophages generally induce damage, M2 CD206-expressing alternatively-activated anti-inflammatory macrophages release anti-inflammatory cytokines like IL-10, IL-4, and insulin-like growth factor-1 (IGF-1) which downregulate iNOS production and promote muscle repair in dystrophin deficient muscle [50]. Among the many responsibilities of M2 macrophages, they are vital for regulating skeletal muscle regeneration by ensuring the proliferation and maturation of muscle progenitor cells, which include satellite cells and collagen-secreting fibroblasts [54].
These steps culminate in two major mechanisms regulating muscle dysfunction. First, inflammation inhibits muscle satellite cells and regeneration [55]. Second, continual recruitment of M2 macrophages leads to increased release of transforming growth factor beta (TGFβ) that stimulates fibroblast activity and production of extracellular matrix (ECM) proteins including excessive collagen to create a form of ‘reactive fibrosis’ [56]. The balance between classically activated M1 populations and alternatively-activated M2 populations remains critical to consider when examining processes that maximize the reparative potential of muscle.
Neutrophils remove cellular debris that accumulates in damaged regions [57]. Studies have shown that neutrophils are recruited to sites of injury at early stages of dystrophinopathy in C57BL/10.mdx mice, and can be approximately 30% more numerous as macrophages in dystrophic muscle [50,58]. Despite the protective properties of neutrophils in healthy physiological systems, they can also impair regeneration in dystrophic muscle by stimulating the secretion of myeloperoxidase (MPO), which is predominantly involved in catalyzing the production of hypochlorous acid (HOCl) – a damaging and reactive oxidant – in the presence of hydrogen peroxide (H2O2) and chloride (Cl-), at sites of inflammation [36,59,60]. Data has shown that golden retriever muscular dystrophy (GRMD) muscle exhibits significantly higher levels of MPO compared to healthy WT muscles, thus suggesting that neutrophil-derived MPO might be contributing to muscle damage by inducing oxidative stress [59]. In addition to MPO release, proteomic analyses of C57BL/10.mdx muscle has also revealed elevated production of neutrophil elastase (NE), compared to healthy WT muscle [61]. NE can be particularly damaging in dystrophic muscle due to its propensity to impair myoblast survival and proliferation by promoting cell adhesion molecule (CAM) degradation [61].
Cytotoxic T-lymphocytes, or the CD4+ T-cells have been identified as additional mediators of the dystrophic immune response. These cells, which can be subdivided into regulatory T-cells (Tregs) and conventional T helper (Th) cells, have been associated with reductions to muscle inflammation and damage in dystrophic muscle [36]. The differentiation of Tregs from naïve T-cells is controlled by the transcription factor FoxP3, whose expression is induced by TGFβ [36]. The presence of Tregs has generally been cited as being beneficial for dystrophic muscle given that they are immunosuppressive and express the anti-inflammatory cytokine, IL-10 [36]. Two separate models, one employing rapamycin-treated C57BL/10.mdx muscle to demonstrate elevated Tregs [62] and the other depleting Tregs via antibody depletion of CD25+ cells [63], implicated Tregs in reducing muscle fibre damage, managing serum creatine kinase (CK) concentrations, and also reducing muscle inflammation and interferon γ (IFNγ) expression [36,50]. Additionally, ablation of FoxP3-expressing Tregs exacerbated mdx muscle damage and led to elevated IFNγ expression and reductions to the expression of the M2 macrophage-specific marker, CD206 [50]. Collectively, this data suggests that although Tregs exist at extraordinarily low frequency of occurrence in sites of damage, they still play a vital role in the transitionary stages of early C57BL/10.mdx pathology to later-onset regenerative stages [36].
Many of these inflammatory responses are attenuated by common glucocorticoids used as standard of care (e.g. prednisolone) for DMD. For example, a study conducted by [64] investigated the effects of prednisolone treatment in C57BL/10.mdx mice between 2 and 4 weeks of age on several immune-related markers, including pro-inflammatory macrophages (using an anti-F4/80 marker), CD4+ T cells, and CD8+ T cells in quadriceps and soleus muscle. The group determined that prednisolone treatment reduced F4/80-positive macrophages (57-59% reduction), CD4+ T cells (50-60% reduction), and CD8+ T cells (48-58% reduction) in both muscles [64], suggesting that glucocorticoids may play an essential role in modulating the DMD-induced immune response. The effects of glucocorticoids on macrophage markers and T cell activation has been well characterized in several other pathological models with robust inflammation as well [65,66]. Interestingly, work investigating the effects of glucocorticoid treatment on human peripheral lung inflammation in asthma identified significant elevations to neutrophils following treatment, accompanied by improvements to lung function [67], which may attest to the beneficial/pleotropic role of neutrophil recruitment at sites of injury and damage such as in DMD. However, the side effects of glucocorticoids warrant consideration of alternative therapies. As mentioned previously, ApN is an attractive candidate for consideration in DMD due to its anti-inflammatory properties – however, the mechanisms through which these anti-inflammatory effects are elicited still require elucidation as discussed in the next section. Understanding the role of ApN in various pathological models serves as a foundation for investigating how exogenous ApN administration might be efficacious in DMD for attenuating secondary physiological stressors.
2. Physiological Role of Adiponectin in Non-Dystrophic Models
2.1. Cellular Properties and Structural Features
ApN and its receptors (which exist as structurally-related AdipoR1 and AdipoR2 isoforms) have been implicated in a myriad of functions, ranging from regulating cellular metabolism to anti-inflammatory effects in both skeletal muscle and cardiac tissue [21,68,69]. In addition to AdipoR1 and AdipoR2, T-cadherin (T-Cad) is another cell surface molecule that has a significant affinity for high molecular weight oligomers of ApN [70]. Although T-Cad can bind to ApN, its lack of intracellular signaling domain impedes its consideration as an ApN signaling receptor [70].
ApN is a 30-kDa multimeric protein, abundantly secreted by mature adipocytes within white adipose tissue (WAT), that consists of a globular C-terminal domain and a collagen-like N-terminal domain [70,71,72]. Although the collagenous domain allows ApN to be secreted into the bloodstream as three oligomeric complexes, including a low molecular weight (LMW) trimer form (67 kDa), middle molecular weight (MMW) hexamer form (140 kDa), and high molecular weight (HMW) (300 kDa) form [73], the HMW oligomer in particular elicits insulin-sensitizing and cardioprotective properties [74]. While the trimer is formed by hydrophobic interactions in its globular heads, and is stabilized by non-covalent interactions of the collagen-like domains, the hexamer and HMW forms of ApN require intermolecular disulfide bond formation between highly conserved cysteine residues [74,75]. This is particularly important because several post-translational modifications, including hydroxylation and glycosylation on conserved lysine residues, are vital for assembly and secretion of HMW ApN [74].
While ApN is produced by WAT in its full-length (fAd) form, fAd can be proteolytically/enzymatically cleaved to the smaller globular (gAd) form by neutrophil elastase produced from monocytes or macrophages [76,77]. In addition to secretion by WAT, ApN can also be secreted by human and murine liver parenchymal cells [78], skeletal muscle myocytes [79], cardiac epithelial cells [80], endothelial cells [81], osteoblasts [71,82] and kidney tubular cells [83]. ApN can represent between 0.01-0.05% of total plasma protein (~2-20 μg/ml range) in humans, thus attesting to its abundance in circulation [70,84,85,86]. Despite being a stable protein in plasma, ApN has a relatively short half-life in circulation of only ~45-75 mins in humans, and is cleared primarily by the liver [87].
ApN is an attractive therapeutic candidate for a host of pathologies [88] given that its circulating levels have been negatively correlated with cardiovascular disease (CVD), cancer, and metabolic syndrome, while high circulating concentrations have been correlated with healthy physiological systems. In this regard, extensive research has demonstrated that ApN is an important regulator of carbohydrate and fat metabolism in multiple tissues as described below.
2.2. Multi-Organ and Inflammatory Regulation of ApN
Through interactions with hepatic AdipoR1 and AdipoR2 receptors, ApN exerts regulatory control over glucose uptake and fat metabolism by reducing hepatic gluconeogenesis [89,90], glycogenolysis [91], and lipogenesis [24,92], while enhancing hepatic fatty acid oxidation [23,93].
AdipoR1 is the predominant adiponectin receptor isoform found in skeletal muscle [24]. ApN regulates skeletal muscle glucose and fat metabolism and insulin sensitivity in part through AMPK-p38-MAPK signaling and PPAR-α induction [24,94,95]. Since ApN is able to activate AMPK by interacting with AdipoR1 and its downstream adaptor protein <adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1)> [90], it is able to stimulate fatty acid oxidation and glucose entry into muscle cells [23,93].
The cardioprotective properties of ApN can be partly attributed to its effects on cardiac metabolism, apoptosis, autophagy, and hypertrophy [88,96]. Through AdipoR1-APP1 interactions, ApN signals AMPKα2, which regulates fatty acid β-oxidation by stimulating acetyl-CoA carboxylase (ACC) phosphorylation, thus enhancing fatty acid oxidation in the heart [97].
ApN has long been recognized for its propensity to shift macrophage polarization towards an M2 anti-inflammatory phenotype (Figure 1), at least in murine peritoneal cavity and adipose tissue [98]. Of note, ApN induces production of the anti-inflammatory cytokine IL-10, while also suppressing the growth and proliferation of bone marrow-derived macrophage progenitors without affecting haematopoietic cell lines in human leukocytes [70,99,100]. While macrophages from ApN-null mice demonstrate a M1 pro-inflammatory phenotype by releasing higher levels of tumour necrosis factor (TNF-α), monocyte chemoattractant protein (MCP-1), and IL-6 compared to WT mice, the elevations to these cytokines is reversed by exogenous recombinant ApN administration [98] (Figure 1). Accordingly, while ApN is able to suppress differentiation and classical activation of M1 pro-inflammatory macrophages, it is also able to promote M2 anti-inflammatory macrophage proliferation and expression of cytokines like IL-10, Arg-1, and Mgl-1 [98,99,101,102,103] (Figure 1). To date, little is known about the role of ApN in regulating T cell and neutrophil function. Although previous studies have demonstrated that AdipoR1 receptors are expressed in murine Tregs [104], the mechanism through which this relationship confers anti-inflammatory effects requires further insight.
3. Synthetic Adiponectin Receptor Agonists
The discovery and design of AdipoR agonists that activate downstream signaling cascades have attracted great effort given that it is difficult and expensive to produce biologically active recombinant ApN with an optimized dosage and route of administration for pre-clinical or clinical use [88] given its large multimeric size and requirement for extensive posttranslational modifications [106]. Several molecules that activate AdipoR have been developed and explored in a variety of conditions including preclinical models of DMD.
3.1. AdipoRon
AdipoRon is a synthetic small-peptide AdipoR agonist that acts via both AdipoR1 and AdipoR1 to exert ApN-like effects [107,108]. It is the most studied AdipoR agonist available. Given that AdipoRon was the first orally active endogenous AdipoR agonist, many studies have been conducted to test its efficacy across different pathologies. Rodent models utilizing AdipoRon have determined that it has the ability to ameliorate insulin resistance, diabetes, and inflammation, in addition to its antiproliferative properties in various cancer models [108,109]. Subsequent studies demonstrated limited potency and specificity which prompted the development of other AdipoR agonists [107,108].
3.2. ALY688
The 10 amino acid-long peptidomimetic ADP355 was developed following the identification of the critical receptor binding domain of ApN. This compound demonstrated high specificity for both receptor isoforms, was modified to be more resistant to proteolytic degradation and demonstrated greater potency than ApN [106,110,111]. Now called ALY688, this small peptide was shown to lower inflammation in inflammatory disorders such as dry eye disease and reduce fibrosis in the liver [94,112,113]. Recent work using ALY688 has demonstrated its efficacy at increasing basal glucose uptake and enhancing insulin-stimulated glucose uptake in skeletal muscle cells, while also improving glucose handling when administered to mice on a high-fat high-sucrose diet [94]. Additionally, the same group also determined that daily subcutaneous ALY688 administration to 10-12-week-old C57BL/6 mice subjected to pressure overload attenuated cardiac hypertrophy, cardiac remodeling, fibrosis, and several cytokines consistent with inflammation, including IL-6, TLR-4, and IL-1β [114].
4. Pre-Clinical Development of Adiponectin-Receptor Agonists for DMD
Boys with DMD have lower serum ApN concentrations [115] as do male mdx mice [116]. As such, several investigations have explored the potential of AdipoR signaling to prevent inflammation, metabolic dysfunction, and fibrosis in pre-clinical models of DMD.
Using C57BL/10.mdx mice, overexpression of ApN improved whole-body measures of muscle function including grip strength, wire test, and treadmill activity while reducing muscle damage and markers of inflammation [17]. AdipoRon treatment in this same model of DMD reduced inflammatory markers and muscle damage in skeletal muscle, stimulated markers of regeneration, lowered general marks of oxidative stress, and improved similar whole-body tests of muscle function [18]. This foundational work provides novel evidence to suggest that ApN signaling can beneficially influence whole-body indices of dystrophin deficiency-induced damage. Specifically, the work demonstrates a relationship between both genetically- and pharmacologically elevated ApN in C57BL/10.mdx mice and attenuations of the dystrophic phenotype. These findings are important because they position ApN as a prospective therapeutic target and potential biomarker to aid in addressing DMD.
Two recent studies explored the potential for ALY688 to modify the disease process in mouse models of DMD. In one study, daily subcutaneous administration of ALY688 for 2 months beginning at 4 weeks of age in C57BL/10.mdx mice improved treadmill activity, wire test, and grip strength while lowering tibialis anterior necrosis and fibrosis [21]. Reductions in IL-1β and TNFα, the M1-type macrophage marker CD68+ as well as the lipid peroxidation product 4-HNE (4-Hydroxynonenal) were reduced in the limb muscle gastrocnemius. ALY688 treatment increased the expression of muscle differentiation and maturation factors and enhanced the myogenic program leading to partial increases in revertant dystrophin-expressing fibres in quadriceps. mRNA levels of Mrf4, a marker of late muscle differentiation, was halved in C57BL/10.mdx mice and partially restored with ALY688 as were other regeneration markers (Figure 2). ALY688 also lowered fibrosis in quadriceps and the fibrotic regulators TGFβ and p-SMAD2 while activating adenosine monophosphate kinase (AMPK) which has been shown to mediate AdipoR-mediated reductions in inflammation and fibrosis, and stimulate both glucose and fatty acid oxidation [19,24,117] (Figure 2). Incubation of myotubes derived from DMD patients with ALY688 lowered IL-1β and TNFα and increased the expression of utrophin, a dystrophin homologue – the latter also being increased in the treated C57BL/10.mdx tibialis anterior muscle. As discussed by the authors [21], the collective results suggest a possible role for ALY688 in lowering inflammation and fibrosis through AMPK activation given this mechanism was previously demonstrated in C57BL/10.mdx mice [118].
In a second study, ALY688 was injected daily into D2.mdx mice – a more severe model of DMD than the C57BL10.mdx model– beginning at day 7 of age up to day 28 of age in order to determine the early effects of AdipoR agonism on muscle remodeling [22]. In the diaphragm, treatment reduced fibrosis in relation to lower IL-6 mRNA but increased IL-6 and TGFβ protein contents. ALY688 lowered mitochondrial complex I-stimulated H2O2 emission (form of reactive oxygen species; ROS) without restoring pyruvate oxidation (a marker of glucose oxidation) that was shown to be lower in untreated D2.mdx mice assessed with high-resolution respirometry in vitro. Treatment lowered diaphragm force production assessed in vitro while quadriceps were not affected and remained lower compared to WT mice. Serum CK – a marker of sarcolemmal damage – were decreased by high doses of ALY688 and increased with low doses, while grip strength, cage hang time, and voluntary wheel running were unaffected with treatment. No changes in AMPK phosphorylation were observed but it was noted that future studies could consider whether the signaling effects are rapid and transient and would be captured by assessing tissues shortly after the last drug injection. The early prevention of diaphragm fibrosis and markers of muscle damage at high doses of treatment in these 4-week-old mice suggests future studies could examine longer term treatment into adulthood when complex muscle development during adolescence is complete.
Collectively, ALY688 demonstrates contrasting effects on inflammatory cascades that may be dependent on the age of assessments, the mouse model employed, the duration of treatment, or the muscles selected for analyses. While no studies of ApN/AdipoR agonists have been performed in humans, some work has assessed the effects of ApN on human DMD-derived myotubes separate from the lower IL-1β and TNFα responses to ALY688 treatments discussed above [21]. Specifically, ApN incubations reduced several pro-inflammatory cytokines including TNFα, IL-17A and CCL28 [19] while also decreasing the NLRP3 inflammasome [20] (Figure 2). ApN treatment in human DMD-derived myotubes increases IL-6 [19] similar to the findings of ALY688 treatment in young D2.mdx mice discussed above [22], but in contrast to the effects seen with longer treatments and later ages in C57BL10.mdx mice [21]. The repression of NLRP3 is notable given mdx/NLRP3-KO cross-bred mice reverse the increases of caspase-1 activity and contents of the pro-inflammatory cytokines IL-1β, IL-18, and TNFα seen in C57BL/10.mdx controls [20].
5. Conclusion and Future Directions
Pre-clinical studies have demonstrated improved skeletal muscle quality following overexpression of ApN or treatment with AdipoRon or ALY688 in mdx mouse models of DMD. The precise mechanisms are not fully elucidated, although reduced inflammation and metabolic reprogramming have been identified in some but not all of these approaches. Careful consideration of specific pre-clinical models, stage of disease, treatment duration, and muscle heterogeneity should be considered when constructing experimental designs with direct assessments of muscle force generating capacities complemented with whole body functional assessments. The divergent responses of cytokines between studies investigating ALY688 in mdx mouse models of DMD, despite a consistent response of lower fibrosis, may be consistent with prior reports that show specific cytokines can be both pro- or anti-inflammatory depending on specific contexts [119] which underscores the complexities of cytokines in remodeling dystrophic muscle during disease progression. Furthermore, resolving the apparent complexities in the inflammatory and metabolic responses to these approaches will give new insight into the fundamental mechanisms by which adiponectin receptors modify disease development in pre-clinical models in addition to guiding translational efforts towards clinical trials. In this regard, the considerable expense of synthesizing ApN due to its large size and requirement for extensive posttranslational modification [111] and limited potency and specificity of AdipoRon [108] limits their potential for further clinical development. Peptidomimetics such as ALY688 have potential for clinical development and therefore warrant further consideration for pre-clinical research throughout the disease process in DMD.
Funding
No funding was provided for the writing of this manuscript.
Competing interests
S.G., G.S., and C.G.R.P. have been previously funded by Allysta Pharmaceuticals. G.S. is a Scientific Advisor for Allysta Pharmaceuticals. The authors declare no conflicts of interest.
Author contributions
S.G., G.S., C.G.R.P. contributed to the preparation, writing, and editing of this paper.
References
- Nigro, G.; Comi, L.I.; Limongelli, F.M.; Giugliano, M.A.M.; Politano, L.; Petretta, V.; Passamano, L.; Stefanelli, S. Prospective Study of X-linked Progressive Muscular Dystrophy in Campania. Muscle Nerve 1983. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2020. [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Prim. 2021. [CrossRef] [PubMed]
- Aartsma-Rus, A.; den Dunnen, J.T. Phenotype Predictions for Exon Deletions/Duplications: A User Guide for Professionals and Clinicians Using Becker and Duchenne Muscular Dystrophy as Examples. Hum. Mutat. 2019. [CrossRef] [PubMed]
- Nicolas, A.; Raguènès-Nicol, C.; Yaou, R. Ben; Hir, S.A. Le; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.F.; et al. Becker Muscular Dystrophy Severity Is Linked to the Structure of Dystrophin. Hum. Mol. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Holtzer, C.; Meaney, F.J.; Andrews, J.; Ciafaloni, E.; Fox, D.J.; James, K.A.; Lu, Z.; Miller, L.; Pandya, S.; Ouyang, L.; et al. Disparities in the Diagnostic Process of Duchenne and Becker Muscular Dystrophy. Genet. Med. 2011. [Google Scholar] [CrossRef]
- Latimer, R.; Street, N.; Conway, K.C.; James, K.; Cunniff, C.; Oleszek, J.; Fox, D.; Ciafaloni, E.; Westfield, C.; Paramsothy, P. Secondary Conditions among Males with Duchenne or Becker Muscular Dystrophy. J. Child Neurol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mirski, K.T.; Crawford, T.O. Motor and Cognitive Delay in Duchenne Muscular Dystrophy: Implication for Early Diagnosis. J. Pediatr. 2014. [Google Scholar] [CrossRef] [PubMed]
- Shahrizaila, N.; Kinnear, W.J.M.; Wills, A.J. Respiratory Involvement in Inherited Primary Muscle Conditions. J. Neurol. Neurosurg. Psychiatry 2006. [CrossRef]
- Chey, Y.C.J.; Arudkumar, J.; Aartsma-Rus, A.; Adikusuma, F.; Thomas, P.Q. CRISPR Applications for Duchenne Muscular Dystrophy: From Animal Models to Potential Therapies. WIREs Mech. Dis. 2023. [Google Scholar] [CrossRef]
- Fatehi, S.; Marks, R.M.; Rok, M.J.; Perillat, L.; Ivakine, E.A.; Cohn, R.D. Advances in CRISPR/Cas9 Genome Editing for the Treatment of Muscular Dystrophies. Hum. Gene Ther. 2023. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. Biological and Genetic Therapies for the Treatment of Duchenne Muscular Dystrophy. Expert Opin. Biol. Ther. 2023. [CrossRef] [PubMed]
- Escolar, D.M.; Hache, L.P.; Clemens, P.R.; Cnaan, A.; McDonald, C.M.; Viswanathan, V.; Kornberg, A.J.; Bertorini, T.E.; Nevo, Y.; Lotze, T.; et al. Randomized, Blinded Trial of Weekend vs Daily Prednisone in Duchenne Muscular Dystrophy. Neurology 2011. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Zaidman, C.M.; Golumbek, P.T.; Cradock, M.M.; Flanigan, K.M.; Kuntz, N.L.; Finkel, R.S.; McDonald, C.M.; Iannaccone, S.T.; Anand, P.; et al. Twice-Weekly Glucocorticosteroids in Infants and Young Boys with Duchenne Muscular Dystrophy. Muscle and Nerve 2019. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Zelikovich, A.S.; Jiang, Z.; Peek, C.B.; Demonbreun, A.R.; Kuntz, N.L.; Barish, G.D.; Haldar, S.M.; Bass, J.; McNally, E.M. Pulsed Glucocorticoids Enhance Dystrophic Muscle Performance through Epigenetic-Metabolic Reprogramming. JCI Insight 2019. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the Treatment of Duchenne Muscular Dystrophy. Cochrane Database Syst. Rev. 2016. [CrossRef] [PubMed]
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of Adiponectin in the Pathogenesis of Dystrophinopathy. Skelet. Muscle 2015. [Google Scholar] [CrossRef] [PubMed]
- Abou-Samra, M.; Selvais, C.M.; Boursereau, R.; Lecompte, S.; Noel, L.; Brichard, S.M. AdipoRon, a New Therapeutic Prospect for Duchenne Muscular Dystrophy. J. Cachexia. Sarcopenia Muscle 2020. [Google Scholar] [CrossRef]
- Lecompte, S.; Abou-Samra, M.; Boursereau, R.; Noel, L.; Brichard, S.M. Skeletal Muscle Secretome in Duchenne Muscular Dystrophy: A Pivotal Anti-Inflammatory Role of Adiponectin. Cell. Mol. Life Sci. 2017. [Google Scholar] [CrossRef]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 Inflammasome by Adiponectin Rescues Duchenne Muscular Dystrophy. BMC Biol. 2018. [Google Scholar] [CrossRef]
- Dubuisson, N.; Versele, R.; Davis-López de Carrizosa, M.A.; Selvais, C.M.; Noel, L.; Planchon, C.; Van den Bergh, P.Y.K.; Brichard, S.M.; Abou-Samra, M. The Adiponectin Receptor Agonist, ALY688: A Promising Therapeutic for Fibrosis in the Dystrophic Muscle. Cells 2023. [Google Scholar] [CrossRef]
- Bellissimo, C.A.; Gandhi, S.; Castellani, L.N.; Murugathasan, M.; Delfinis, L.J.; Thuhan, A.; Garibotti, M.C.; Seo, Y.; Rebalka, I.A.; Hsu, H.H.; et al. The Slow-Release Adiponectin Analog ALY688-SR Modifies Early-Stage Disease Development in the D2.Mdx Mouse Model of Duchenne Muscular Dystrophy. Am. J. Physiol. - Cell Physiol. [CrossRef]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.S.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic Cleavage Product of 30-KDa Adipocyte Complement-Related Protein Increases Fatty Acid Oxidation in Muscle and Causes Weight Loss in Mice. Proc. Natl. Acad. Sci. U. S. A. 2001. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin Stimulates Glucose Utilization and Fatty-Acid Oxidation by Activating AMP-Activated Protein Kinase. Nat. Med. 2002. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B.; Somwar, R.; Maida, A.; Fang, X.; Bikopoulos, G.; Sweeney, G. Globular Adiponectin Increases GLUT4 Translocation and Glucose Uptake but Reduces Glycogen Synthesis in Rat Skeletal Muscle Cells. Diabetologia 2005. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.B.; McAinch, A.J.; Macaulay, S.L.; Castelli, L.A.; O’Brien, P.E.; Dixon, J.B.; Cameron-Smith, D.; Kemp, B.E.; Steinberg, G.R. Impaired Activation of AMP-Kinase and Fatty Acid Oxidation by Globular Adiponectin in Cultured Human Skeletal Muscle of Obese Type 2 Diabetics. J. Clin. Endocrinol. Metab. 2005. [Google Scholar] [CrossRef]
- Liu, Y.; Turdi, S.; Park, T.; Morris, N.J.; Deshaies, Y.; Xu, A.; Sweeney, G. Adiponectin Corrects High-Fat Diet-Induced Disturbances in Muscle Metabolomic Profile and Whole-Body Glucose Homeostasis. Diabetes 2013. [Google Scholar] [CrossRef]
- Liu, Y.; Sweeney, G. Adiponectin Action in Skeletal Muscle. In Proceedings of the Best Practice and Research: Clinical Endocrinology and Metabolism; 2014. [Google Scholar]
- Marette, A.; Liu, Y.; Sweeney, G. Skeletal Muscle Glucose Metabolism and Inflammation in the Development of the Metabolic Syndrome. Rev. Endocr. Metab. Disord. 2014. [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A Role for the Dystrophin-Glycoprotein Complex as a Transmembrane Linker between Laminin and Actin. J. Cell Biol. 1993. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin Protects the Sarcolemma from Stresses Developed during Muscle Contraction. Proc. Natl. Acad. Sci. U. S. A. 1993. [Google Scholar] [CrossRef]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory Monocytes Promote Progression of Duchenne Muscular Dystrophy and Can Be Therapeutically Targeted via CCR 2. EMBO Mol. Med. 2014. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-Mediated Pathology in Duchenne Muscular Dystrophy. Sci. Transl. Med. 2015. [CrossRef]
- Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021. [CrossRef] [PubMed]
- Carpenter, S.; Karpati, G. Duchenne Muscular Dystrophy: Plasma Membrane Loss Initiates Muscle Cell Necrosis Unless It Is Repaired. Brain 1979. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018.
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I - Promoting Inflammation and Immunity. Sultan Qaboos Univ. Med. J. 2015.
- Petrof, B. The Role of Innate Immunity in Dystrophic Diaphragm Pathology. FASEB J. 2020. [Google Scholar] [CrossRef]
- Herbelet, S.; Rodenbach, A.; De Paepe, B.; De Bleecker, J.L. Anti-Inflammatory and General Glucocorticoid Physiology in Skeletal Muscles Affected by Duchenne Muscular Dystrophy: Exploration of Steroid-Sparing Agents. Int. J. Mol. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.L.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-ΚB Signaling in Macrophages and Myofibers Promotes Muscle Degeneration in Duchenne Muscular Dystrophy. J. Clin. Invest. 2007. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Li, Q.; Ding, J.; Liang, F.; Gusev, E.; Lapohos, O.; Fonseca, G.J.; Kaufmann, E.; Divangahi, M.; Petrof, B.J. TLR4 Is a Regulator of Trained Immunity in a Murine Model of Duchenne Muscular Dystrophy. Nat. Commun. 2022. [Google Scholar] [CrossRef]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-Inflammatory Drugs for Duchenne Muscular Dystrophy: Focus on Skeletal Muscle-Releasing Factors. Drug Des. Devel. Ther. 2016.
- Henriques-Pons, A.; Yu, Q.; Rayavarapu, S.; Cohen, T. V.; Ampong, B.; Cha, H.J.; Jahnke, V.; Van der meulen, J.; Wang, D.; Jiang, W.; et al. Role of Toll-like Receptors in the Pathogenesis of Dystrophin-Deficient Skeletal and Heart Muscle. Hum. Mol. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; Van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a Novel Anti-Inflammatory and Membrane-Stabilizer, Improves Muscular Dystrophy without Side Effects. EMBO Mol. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, S.M.; Walsh, G.S.; Balazsi, K.; Seale, P.; Sandoz, J.; Hierlihy, A.M.; Rudnicki, M.A.; Chamberlain, J.S.; Miller, F.D.; Megeney, L.A. Activation of JNK1 Contributes to Dystrophic Muscle Pathogenesis. Curr. Biol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Kuru, S.; Inukai, A.; Kato, T.; Liang, Y.; Kimura, S.; Sobue, G. Expression of Tumor Necrosis Factor-α in Regenerating Muscle Fibers in Inflammatory and Non-Inflammatory Myopathies. Acta Neuropathol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; De Benedetti, F.; et al. Increased Levels of Interleukin-6 Exacerbate the Dystrophic Phenotype in Mdx Mice. Hum. Mol. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Adamo, S.; Berghella, L. The JAK/STAT Pathway in Skeletal Muscle Pathophysiology. Front. Physiol. 2019. [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and Morphological Improvement of Dystrophic Muscle by Interleukin 6 Receptor Blockade. EBioMedicine 2015. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ Promotes Muscle Damage in the Mdx Mouse Model of Duchenne Muscular Dystrophy by Suppressing M2 Macrophage Activation and Inhibiting Muscle Cell Proliferation. J. Immunol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidbal, J.G. Shifts in Macrophage Phenotypes and Macrophage Competition for Arginine Metabolism Affect the Severity of Muscle Pathology in Muscular Dystrophy. Hum. Mol. Genet. 2009. [Google Scholar] [CrossRef]
- Burelle, Y.; Khairallah, M.; Ascah, A.; Allen, B.G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Alterations in Mitochondrial Function as a Harbinger of Cardiomyopathy: Lessons from the Dystrophic Heart. J. Mol. Cell. Cardiol. 2010. [CrossRef]
- Hughes, M.C.; Ramos, S. V.; Turnbull, P.C.; Edgett, B.A.; Huber, J.S.; Polidovitch, N.; Schlattner, U.; Backx, P.H.; Simpson, J.A.; Perry, C.G.R. Impairments in Left Ventricular Mitochondrial Bioenergetics Precede Overt Cardiac Dysfunction and Remodelling in Duchenne Muscular Dystrophy. J. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019. [CrossRef]
- Ribeiro, A.F.; Souza, L.S.; Almeida, C.F.; Ishiba, R.; Fernandes, S.A.; Guerrieri, D.A.; Santos, A.L.F.; Onofre-Oliveira, P.C.G.; Vainzof, M. Muscle Satellite Cells and Impaired Late Stage Regeneration in Different Murine Models for Muscular Dystrophies. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the Process of Fibrosis in Duchenne Muscular Dystrophy. Biomed Res. Int. 2014. [CrossRef]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The Role of Neutrophils in Inflammation Resolution. Semin. Immunol. 2016. [CrossRef] [PubMed]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A Nitric Oxide Synthase Transgene Ameliorates Muscular Dystrophy in Mdx Mice. J. Cell Biol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Duong, M.N.; Turner, R.; Le Guiner, C.; Boyatzis, A.; Kettle, A.J.; Grounds, M.D.; Arthur, P.G. Levels of Inflammation and Oxidative Stress, and a Role for Taurine in Dystropathology of the Golden Retriever Muscular Dystrophy Dog Model for Duchenne Muscular Dystrophy. Redox Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne Muscular Dystrophy: Myonecrosis, Inflammation and Oxidative Stress. DMM Dis. Model. Mech. 2020. [CrossRef] [PubMed]
- Arecco, N.; Clarke, C.J.; Jones, F.K.; Simpson, D.M.; Mason, D.; Beynon, R.J.; Pisconti, A. Elastase Levels and Activity Are Increased in Dystrophic Muscle and Impair Myoblast Cell Survival, Proliferation and Differentiation. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Eghtesad, S.; Jhunjhunwala, S.; Little, S.R.; Clemens, P.R. Rapamycin Ameliorates Dystrophic Phenotype in Mdx Mouse Skeletal Muscle. Mol. Med. 2011. [Google Scholar] [CrossRef]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. XA Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell 2013. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Lee, J.J.; Tidball, J.G. Prednisolone Decreases Cellular Adhesion Molecules Required for Inflammatory Cell Infiltration in Dystrophin-Deficient Skeletal Muscle. Neuromuscul. Disord. 2004. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.K.K.; Cuello, C.; Bertouch, J. V.; Roberts-Thomson, P.J.; Ahern, M.J.; Smith, M.D.; Youssef, P.P. Effects of Pulse Methylprednisolone on Macrophage Chemotactic Protein-1 and Macrophage Inflammatory Protein-1α in Rheumatoid Synovium. J. Rheumatol. 2001. [Google Scholar]
- Ferdman, R.M.; Church, J.A. Immunologic and Virologic Effects of Glucocorticoids on Human Immunodeficiency Virus Infection in Children: A Preliminary Study. Pediatr. Infect. Dis. J. 1994. [Google Scholar] [CrossRef] [PubMed]
- Hauber, H.P.; Gotfried, M.; Newman, K.; Danda, R.; Servi, R.J.; Christodoulopoulos, P.; Hamid, Q. Effect of HFA-Flunisolide on Peripheral Lung Inflammation in Asthma. J. Allergy Clin. Immunol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kadowaki, T. Physiological and Pathophysiological Roles of Adiponectin and Adiponectin Receptors in the Integrated Regulation of Metabolic and Cardiovascular Diseases. Int. J. Obes. 2008. [CrossRef] [PubMed]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted Disruption of AdipoR1 and AdipoR2 Causes Abrogation of Adiponectin Binding and Metabolic Actions. Nat. Med. 2007. [Google Scholar] [CrossRef] [PubMed]
- Turer, A.T.; Scherer, P.E. Adiponectin: Mechanistic Insights and Clinical Implications. Diabetologia 2012. [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017. [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A Novel Serum Protein Similar to C1q, Produced Exclusively in Adipocytes. J. Biol. Chem. 1995. [Google Scholar] [CrossRef]
- Magkos, F.; Sidossis, L.S. Recent Advances in the Measurement of Adiponectin Isoform Distribution. Curr. Opin. Clin. Nutr. Metab. Care 2007. [CrossRef]
- Hui, X.; Lam, K.S.; Vanhoutte, P.M.; Xu, A. Adiponectin and Cardiovascular Health: An Update. Br. J. Pharmacol. 2012. [CrossRef] [PubMed]
- Tsao, T.S.; Tomas, E.; Murrey, H.E.; Hug, C.; Lee, D.H.; Ruderman, N.B.; Heuser, J.E.; Lodish, H.F. Role of Disulfide Bonds in Acrp30/Adiponectin Structure and Signaling Specificity: Different Oligomers Activate Different Signal Transduction Pathways. J. Biol. Chem. 2003. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Giannoni, E.; Taddei, M.L.; Chiarugi, P. Globular Adiponectin Activates Motility and Regenerative Traits of Muscle Satellite Cells. PLoS One 2012. [Google Scholar] [CrossRef]
- Waki, H.; Yamauchi, T.; Kamon, J.; Kita, S.; Ito, Y.; Hada, Y.; Uchida, S.; Tsuchida, A.; Takekawa, S.; Kadowaki, T. Generation of Globular Fragment of Adiponectin by Leukocyte Elastase Secreted by Monocytic Cell Line THP-1. Endocrinology 2005. [Google Scholar] [CrossRef]
- Yoda-Murakami, M.; Taniguchi, M.; Takahashi, K.; Kawamata, S.; Saito, K.; Choi-Miura, N.H.; Tomita, M. Change in Expression of GBP28/Adiponectin in Carbon Tetrachloride-Administrated Mouse Liver. Biochem. Biophys. Res. Commun. 2001. [Google Scholar] [CrossRef] [PubMed]
- Delaigle, A.M.; Jonas, J.C.; Bauche, I.B.; Cornu, O.; Brichard, S.M. Induction of Adiponectin in Skeletal Muscle by Inflammatory Cytokines: In Vivo and in Vitro Studies. Endocrinology 2004. [Google Scholar] [CrossRef]
- Patel, J. V.; Abraheem, A.; Dotsenko, O.; Creamer, J.; Gunning, M.; Hughes, E.A.; Lip, G.Y.H. Circulating Serum Adiponectin Levels in Patients with Coronary Artery Disease: Relationship to Atherosclerotic Burden and Cardiac Function. J. Intern. Med. 2008. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Galgani, M.; Carbone, F.; Rocca, C. La; Formisano, L.; Matarese, G. Role of Adipokines Signaling in the Modulation of T Cells Function. Front. Immunol. 2013. [CrossRef]
- Berner, H.S.; Lyngstadaas, S.P.; Spahr, A.; Monjo, M.; Thommesen, L.; Drevon, C.A.; Syversen, U.; Reseland, J.E. Adiponectin and Its Receptors Are Expressed in Bone-Forming Cells. Bone 2004. [Google Scholar] [CrossRef]
- Onodera, T.; Wang, M.Y.; Rutkowski, J.M.; Deja, S.; Chen, S.; Balzer, M.S.; Kim, D.S.; Sun, X.; An, Y.A.; Field, B.C.; et al. Endogenous Renal Adiponectin Drives Gluconeogenesis through Enhancing Pyruvate and Fatty Acid Utilization. Nat. Commun. 2023. [Google Scholar] [CrossRef]
- Maeda, K.; Okubo, K.; Shimomura, I.; Funahashi, T.; Matsuzawa, Y.; Matsubara, K. CDNA Cloning and Expression of a Novel Adipose Specific Collagen-like Factor, ApM1 (Adipose Most Abundant Gene Transcript 1). Biochem. Biophys. Res. Commun. 1996. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cheng, K.K.Y.; Vanhoutte, P.M.; Lam, K.S.L.; Xu, A. Vascular Effects of Adiponectin: Molecular Mechanisms and Potential Therapeutic Intervention. Clin. Sci. 2008. [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.I.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical Decrease of an Adipose-Specific Protein, Adiponectin, in Obesity. Biochem. Biophys. Res. Commun. 1999. [Google Scholar] [CrossRef]
- Halberg, N.; Schraw, T.D.; Wang, Z. V.; Kim, J.Y.; Yi, J.; Hamilton, M.P.; Luby-Phelps, K.; Scherer, P.E. Systemic Fate of the Adipocyte-Derived Factor Adiponectin. Diabetes 2009. [Google Scholar] [CrossRef]
- Liu, Y.; Vu, V.; Sweeney, G. Examining the Potential of Developing and Implementing Use of Adiponectin-Targeted Therapeutics for Metabolic and Cardiovascular Diseases. Front. Endocrinol. (Lausanne). 2019. [CrossRef]
- Park, S.; Kim, D.S.; Kwon, D.Y.; Yang, H.J. Long-Term Central Infusion of Adiponectin Improves Energy and Glucose Homeostasis by Decreasing Fat Storage and Suppressing Hepatic Gluconeogenesis without Changing Food Intake. J. Neuroendocrinol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Deepa, S.S.; Etzler, J.C.; Ryu, J.; Mao, X.; Fang, Q.; Liu, D.D.; Torres, J.M.; Jia, W.; Lechleiter, J.D.; et al. Adiponectin Activates AMP-Activated Protein Kinase in Muscle Cells via APPL1/LKB1-Dependent and Phospholipase C/Ca2+/Ca2+/Calmodulin-Dependent Protein Kinase Kinase-Dependent Pathways. J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous Glucose Production Is Inhibited by the Adipose-Derived Protein Acrp30. J. Clin. Invest. 2001. [Google Scholar] [CrossRef]
- Awazawa, M.; Ueki, K.; Inabe, K.; Yamauchi, T.; Kaneko, K.; Okazaki, Y.; Bardeesy, N.; Ohnishi, S.; Nagai, R.; Kadowaki, T. Adiponectin Suppresses Hepatic SREBP1c Expression in an AdipoR1/LKB1/AMPK Dependent Pathway. Biochem. Biophys. Res. Commun. 2009. [Google Scholar] [CrossRef]
- Khoramipour, K.; Chamari, K.; Hekmatikar, A.A.; Ziyaiyan, A.; Taherkhani, S.; Elguindy, N.M.; Bragazzi, N.L. Adiponectin: Structure, Physiological Functions, Role in Diseases, and Effects of Nutrition. Nutrients 2021. [CrossRef]
- Sung, H.K.; Mitchell, P.L.; Gross, S.; Marette, A.; Sweeney, G. ALY688 Elicits Adiponectin-Mimetic Signaling and Improves Insulin Action in Skeletal Muscle Cells. Am. J. Physiol. - Cell Physiol. [CrossRef]
- Bruce, C.R.; Mertz, V.A.; Heigenhauser, G.J.F.; Dyck, D.J. The Stimulatory Effect of Globular Adiponectin on Insulin-Stimulated Glucose Uptake and Fatty Acid Oxidation Is Impaired in Skeletal Muscle from Obese Subjects. Diabetes 2005. [Google Scholar] [CrossRef]
- Park, M.; Sweeney, G. Direct Effects of Adipokines on the Heart: Focus on Adiponectin. In Proceedings of the Heart Failure Reviews; 2013. [Google Scholar]
- Fang, X.; Palanivel, R.; Cresser, J.; Schram, K.; Ganguly, R.; Thong, F.S.L.; Tuinei, J.; Xu, A.; Abel, E.D.; Sweeney, G. An APPL1-AMPK Signaling Axis Mediates Beneficial Metabolic Effects of Adiponectin in the Heart. Am. J. Physiol. - Endocrinol. Metab. [CrossRef]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin Promotes Macrophage Polarization toward an Anti-Inflammatory Phenotype. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Enrich, B.; Tilg, H. Adiponectin Induces the Anti-Inflammatory Cytokines IL-10 and IL-1RA in Human Leukocytes. Biochem. Biophys. Res. Commun. 2004. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Oritani, K.; Takahashi, I.; Ishikawa, J.; Matsuyama, A.; Ouchi, N.; Kihara, S.; Funahashi, T.; Tenner, A.J.; Tomiyama, Y.; et al. Adiponectin, a New Member of the Family of Soluble Defense Collagens, Negatively Regulates the Growth of Myelomonocytic Progenitors and the Functions of Macrophages. Blood 2000. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Szmitko, P.E.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Chan, L.; Al-Omran, M.; Teoh, H. Adiponectin Primes Human Monocytes into Alternative Anti-Inflammatory M2 Macrophages. Am. J. Physiol. - Hear. Circ. Physiol. [CrossRef] [PubMed]
- Ajuwon, K.M.; Spurlock, M.E. Adiponectin Inhibits LPS-Induced NF-ΚB Activation and IL-6 Production and Increases PPARγ2 Expression in Adipocytes. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, M. Adiponectin: A Versatile Player of Innate Immunity. J. Mol. Cell Biol. 2016. [CrossRef]
- Ramos-Ramírez, P.; Malmhäll, C.; Tliba, O.; Rådinger, M.; Bossios, A. Adiponectin/AdipoR1 Axis Promotes IL-10 Release by Human Regulatory T Cells. Front. Immunol. 2021. [Google Scholar] [CrossRef]
- Zhao, D.; Xue, C.; Li, J.; Feng, K.; Zeng, P.; Chen, Y.; Duan, Y.; Zhang, S.; Li, X.; Han, J.; et al. Adiponectin Agonist ADP355 Ameliorates Doxorubicin-Induced Cardiotoxicity by Decreasing Cardiomyocyte Apoptosis and Oxidative Stress. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Otvos, L.; Haspinger, E.; La Russa, F.; Maspero, F.; Graziano, P.; Kovalszky, I.; Lovas, S.; Nama, K.; Hoffmann, R.; Knappe, D.; et al. Design and Development of a Peptide-Based Adiponectin Receptor Agonist for Cancer Treatment. BMC Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Li, R.; Lau, W.B.; Yuan, Y.X.; Liang, B.; Li, R.; Gao, E.H.; Koch, W.J.; Ma, X.L.; et al. AdipoRon, the First Orally Active Adiponectin Receptor Activator, Attenuates Postischemic Myocardial Apoptosis through Both AMPK-Mediated and AMPK-Independent Signalings. Am. J. Physiol. - Endocrinol. Metab. [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.I.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A Small-Molecule AdipoR Agonist for Type 2 Diabetes and Short Life in Obesity. Nature 2013. [Google Scholar] [CrossRef] [PubMed]
- Sapio, L.; Nigro, E.; Ragone, A.; Salzillo, A.; Illiano, M.; Spina, A.; Polito, R.; Daniele, A.; Naviglio, S. AdipoRon Affects Cell Cycle Progression and Inhibits Proliferation in Human Osteosarcoma Cells. J. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. Potential Adiponectin Receptor Response Modifier Therapeutics. Front. Endocrinol. (Lausanne). 2019. [CrossRef] [PubMed]
- Otvos, L.; Knappe, D.; Hoffmann, R.; Kovalszky, I.; Olah, J.; Hewitson, T.D.; Stawikowska, R.; Stawikowski, M.; Cudic, P.; Lin, F.; et al. Development of Second Generation Peptides Modulating Cellular Adiponectin Receptor Responses. Front. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Smith, T.; Rahman, K.; Thorn, N.E.; Anania, F.A. Adiponectin Agonist ADP355 Attenuates CCL4-Induced Liver Fibrosis in Mice. PLoS One 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; La Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-Derived Active Peptide ADP355 Exerts Anti-Inflammatory and Anti-Fibrotic Activities in Thioacetamide-Induced Liver Injury. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Dadson, K.; Sung, H.K.; Ayansola, O.; Mirzaesmaeili, A.; Noskovicova, N.; Zhao, Y.; Cheung, K.; Radisic, M.; Hinz, B.; et al. Cardioprotection by the Adiponectin Receptor Agonist ALY688 in a Preclinical Mouse Model of Heart Failure with Reduced Ejection Fraction (HFrEF). Biomed. Pharmacother. 2024. [Google Scholar] [CrossRef]
- Hathout, Y.; Liang, C.; Ogundele, M.; Xu, G.; Tawalbeh, S.M.; Dang, U.J.; Hoffman, E.P.; Gordish-Dressman, H.; Conklin, L.S.; van den Anker, J.N.; et al. Disease-Specific and Glucocorticoid-Responsive Serum Biomarkers for Duchenne Muscular Dystrophy. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zelikovich, A.S.; Quattrocelli, M.; Salamone, I.M.; Kuntz, N.L.; McNally, E.M. Moderate Exercise Improves Function and Increases Adiponectin in the Mdx Mouse Model of Muscular Dystrophy. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 Regulate PGC-1α and Mitochondria by Ca 2+ and AMPK/SIRT1. Nature 2010. [Google Scholar] [CrossRef] [PubMed]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; Ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK Activation Regulates LTBP4-Dependent TGF-Β1 Secretion by Pro-Inflammatory Macrophages and Controls Fibrosis in Duchenne Muscular Dystrophy. Cell Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Turpin-Nolan, S.; Febbraio, M.A. IL-6 Family Cytokines as Potential Therapeutic Strategies to Treat Metabolic Diseases. Cytokine 2021. [CrossRef] [PubMed]
Figure 1.
Adiponectin promotes monocyte differentiation towards anti-inflammatory macrophage phenotype that is responsible for attenuating dystrophin deficiency-induced impairments to cardiac and skeletal muscle. It has been demonstrated that, at least in human atherosclerotic tissue, ApN has the ability to influence monocytes to differentiate towards the M2 anti-inflammatory macrophage phenotype [101]. This is particularly important given that dystrophic skeletal muscle exhibits elevations to several pro-inflammatory cytokines including IL-6 [47] and TNF-α [46] that exacerbate the pathology associated with dystrophin deficiency. Given that the pleotropic cytokines IL-10 (elevated by ApN agonism in certain inflammatory conditions) and TGF-β (downregulated by ApN agonism in mdx skeletal muscle) are generally associated with ‘alternatively-activated’ macrophages [51,105], ApN serves as a prospective intervention for inducing this anti-inflammatory phenotype. Made using www.biorender.com .
Figure 1.
Adiponectin promotes monocyte differentiation towards anti-inflammatory macrophage phenotype that is responsible for attenuating dystrophin deficiency-induced impairments to cardiac and skeletal muscle. It has been demonstrated that, at least in human atherosclerotic tissue, ApN has the ability to influence monocytes to differentiate towards the M2 anti-inflammatory macrophage phenotype [101]. This is particularly important given that dystrophic skeletal muscle exhibits elevations to several pro-inflammatory cytokines including IL-6 [47] and TNF-α [46] that exacerbate the pathology associated with dystrophin deficiency. Given that the pleotropic cytokines IL-10 (elevated by ApN agonism in certain inflammatory conditions) and TGF-β (downregulated by ApN agonism in mdx skeletal muscle) are generally associated with ‘alternatively-activated’ macrophages [51,105], ApN serves as a prospective intervention for inducing this anti-inflammatory phenotype. Made using www.biorender.com .
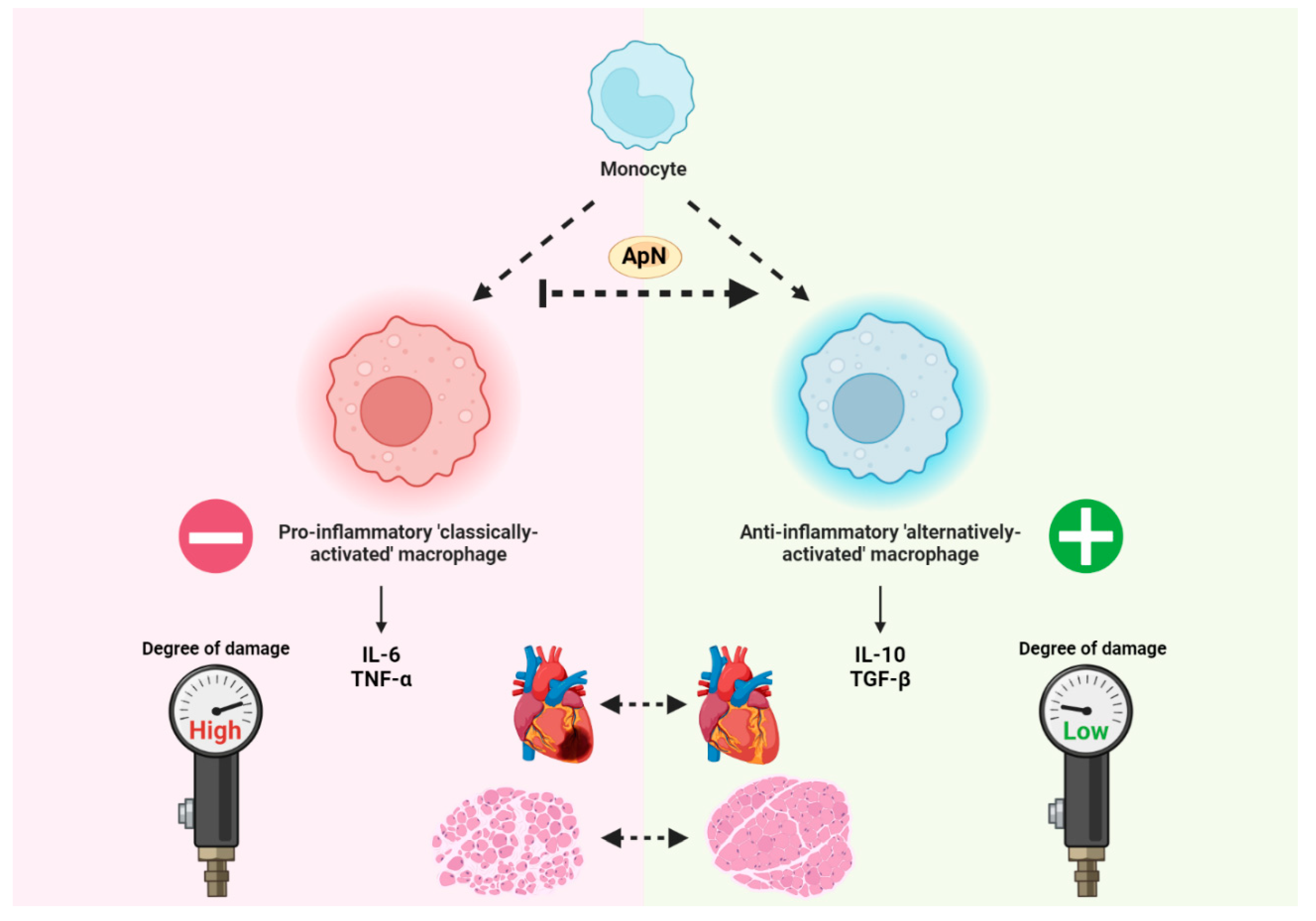
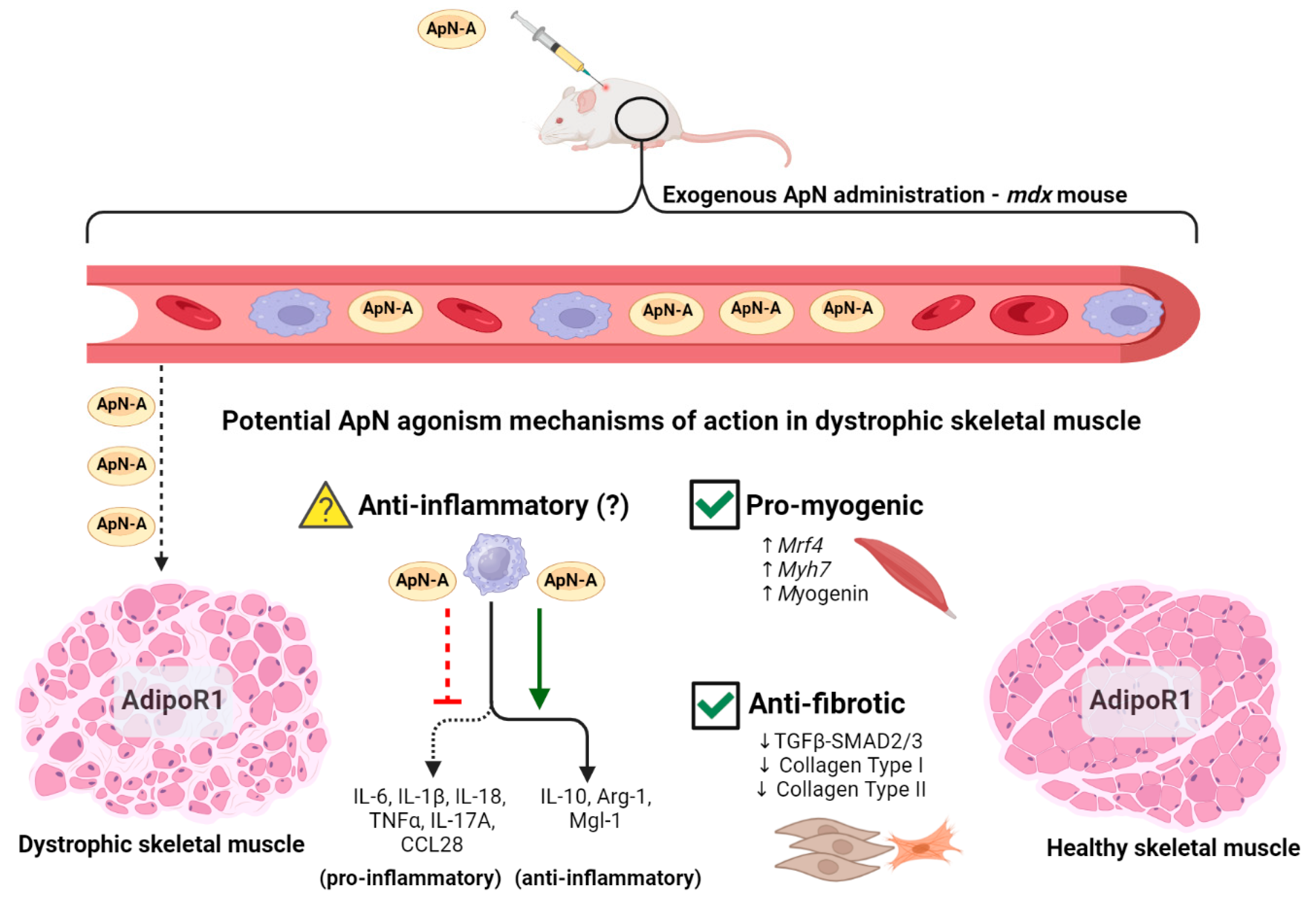
Figure 2.
Exogenous ApN agonist administration may improve the inflammatory, myogenic, and fibrotic profile of skeletal muscle in dystrophin deficient mice. Dystrophic mice exhibit low circulating ApN levels [17], and restoring these levels with exogenous ApN agonists may shift the pro-inflammatory macrophage phenotype [19] towards the anti-inflammatory phenotype, characterized by elevations to IL-10, Arg-1, and Mgl-1, although this is yet to be proven in mdx mice (denoted by question mark) [98,99,102,103]. Additionally, as previously demonstrated in mdx mice, ApN can improve the myogenic profile of the dystrophin deficient skeletal muscle by upregulating Mrf4, Myh7, and Myogenin, while simultaneously attenuating fibrosis by downregulating TGFβ-SMAD2/3, Collagen Type I, and Collagen Type II (check mark denotes mdx data) [21]. By restoring indices of perturbed inflammation (not yet entirely demonstrated in mdx), myogenesis, and fibrosis in dystrophic rodent skeletal muscle, ApN agonism has the potential to improve the detrimental phenotype associated with dystrophin deficiency. Legend: ApN-A = Adiponectin agonist. Made using www.biorender.com.
Figure 2.
Exogenous ApN agonist administration may improve the inflammatory, myogenic, and fibrotic profile of skeletal muscle in dystrophin deficient mice. Dystrophic mice exhibit low circulating ApN levels [17], and restoring these levels with exogenous ApN agonists may shift the pro-inflammatory macrophage phenotype [19] towards the anti-inflammatory phenotype, characterized by elevations to IL-10, Arg-1, and Mgl-1, although this is yet to be proven in mdx mice (denoted by question mark) [98,99,102,103]. Additionally, as previously demonstrated in mdx mice, ApN can improve the myogenic profile of the dystrophin deficient skeletal muscle by upregulating Mrf4, Myh7, and Myogenin, while simultaneously attenuating fibrosis by downregulating TGFβ-SMAD2/3, Collagen Type I, and Collagen Type II (check mark denotes mdx data) [21]. By restoring indices of perturbed inflammation (not yet entirely demonstrated in mdx), myogenesis, and fibrosis in dystrophic rodent skeletal muscle, ApN agonism has the potential to improve the detrimental phenotype associated with dystrophin deficiency. Legend: ApN-A = Adiponectin agonist. Made using www.biorender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



