
Submitted:
04 June 2024
Posted:
05 June 2024
You are already at the latest version
Abstract
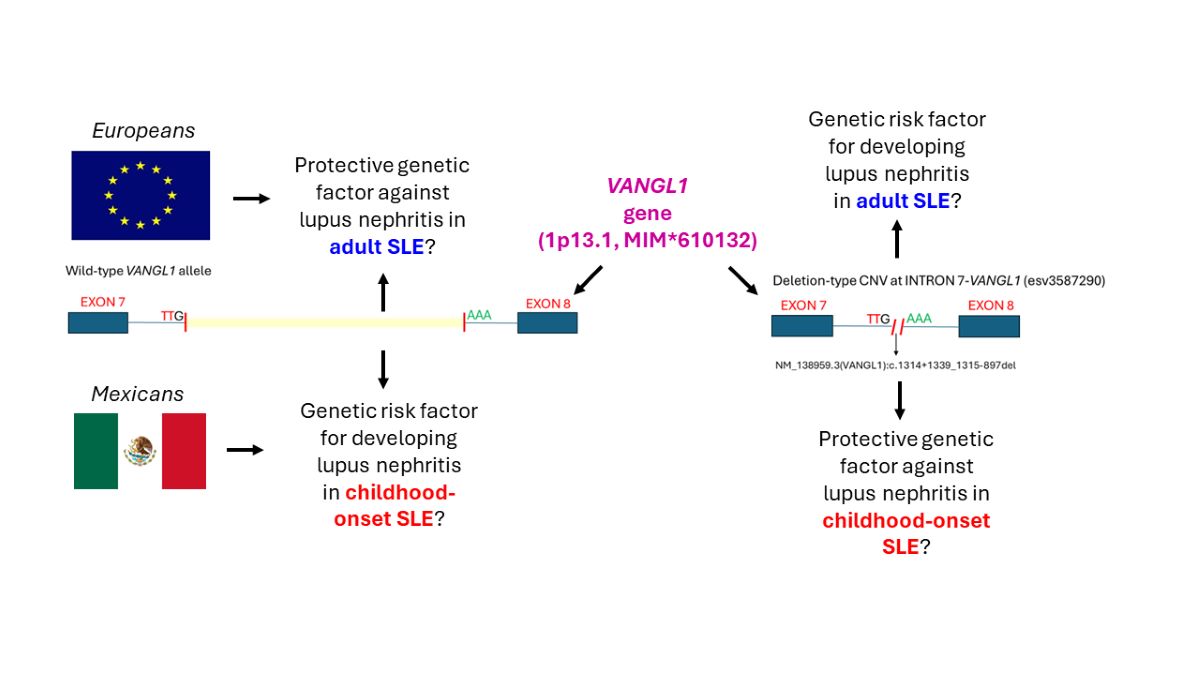
A ~3-kb deletion-type DNA copy-number variation (CNV, esv3587290) located at intron 7 of the VANGL1 gene (1p13.1, MIM*610132) has been proposed as a genetic factor for developing lupus nephritis (LN) in adult systemic lupus erythematosus (SLE) patients across European-descent populations, but its replication in other ethnicities has been inconsistent and its association with LN in childhood-onset SLE (cSLE) remains unknown. Here, we performed an exploratory association study in a sample of 66 unrelated cSLE Mexican patients (11 males, 55 females; ages 7.8 to 18.6 years). Two stratified groups were compared: cSLE patients with (N=39) or without (N=27) LN, as diagnosed by renal biopsy (N=17), proteinuria (N=33), urinary protein:creatinine ratio >0.2 (N=34), and erythrocyturia and/or granular casts in urinary sediment (N=16). For esv3587290 CNV genotyping, we performed an end-point PCR assay with breakpoint confirmation by Sanger sequencing. We also determined the allelic frequencies of the esv3587290 CNV in 181 deidentified ethnically-matched individuals (reference group). The obtained genotypes were tested for Hardy-Weinberg equilibrium (HWE) by c2 test. Associations between LN and esv3587290 CNV were tested by calculating the Odds Ratio (OR) and using Pearson's c2 tests with a 95% confidence interval and P≤0.05. The esv3587290 CNV allele (OR 0.108, 95% CI 0.034-0.33, P=0.00003) and the heterozygous genotype (OR 0.04, 95% CI 0.119-0.9811, P=0.002) showed a significant protective effect against LN development. Finally, we characterized the precise breakpoint of the esv3587290 CNV as NG_016548.1(NM_138959.3):c.1314+1339_1315-897del (https://databases.lovd.nl/shared/variants/0000918418#00025811) in our population. This report supports the notion that a broad genetic heterogeneity underlies the susceptibility for developing LN.
Keywords:
childhood-onset systemic lupus erythematosus
; lupus nephritis
; VANGL1 gene
; DNA copy-number variation
; Mexican population
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic, multisystem, autoimmune, and inflammatory disease that is most often of unknown cause. It is characterized by the production of multiple autoantibodies, especially against nuclear components (e.g., double-stranded DNA, dsDNA); this can generate inflammatory-mediated effects in any organ and/or system. Childhood-onset forms (cSLE, onset before age 18) represent 20% of all cases [1,2]. Lupus nephritis (LN) is generally estimated to occur 10-30% more frequently in cSLE (40-70%) than in adult forms of SLE [2,3,4]. This complication is among those with the highest impacts on the life quality and survival of SLE patients since it increases the risk of developing end-stage kidney disease (ESKD) [5]. LN shows an earlier onset and more severe clinical course in cSLE compared to adult SLE forms; besides it has been reported as a negative predictor of survival [4].
Identifying biomarkers for diagnosing, prognosis, and non-invasive evaluation of renal disease activity in SLE is a rapidly evolving field [6]. However, in the clinical setting, diagnosis of LN and evaluation of renal flares still rely on the assessment of traditional serum (e.g., serum creatinine (SCr), erythrocyte sedimentation rate, complement components C3 and C4, glomerular filtration rate, anti-C1q or anti-dsDNA autoantibody titers, etc.), and urinary (urine sediment, proteinuria, albumin:creatinine ratio, etc.) biomarkers, or by the histological evaluation of renal biopsies [6,7,8].
To date, genome-wide association studies (GWAS) have identified around 100 susceptibility genes for developing SLE [6,9]. The relevant genetic changes have been mainly related to single-nucleotide variations in immune-response genes (e.g., HLA-DR3 in patients of European ancestry) [9,10], along with some gene copy-number variations (CNV) in non-immune-related genes, such as VANGL1 (1p13.1, MIM*610132) [11]. VANGL1 is an essential gene in the establishment of planar cell polarity (PCP), and heterozygous variants have been associated with neural tube defects (MIM#182940) and caudal regression syndrome (MIM#600145), but not with human kidney disease. A GWAS study performed in 55 patients with SLE, 11 with Sjogren’s syndrome, and 11 healthy controls, identified three SLE patients homozygous for a VANGL1 deletion-type CNV of ~3.17 kb (esv3587290) located at intron 7. Of these patients, two had LN. These authors furthermore evaluated the esv3587290 by TaqMan® assay in 177 SLE patients of mainly European descent. The results revealed that the deletion was significantly associated with LN (χ2=27.06, 2 d.f., P<0.0001) and demonstrated a gene-dosage effect. Moreover, the esv3587290 CNV seems to be a highly prevalent allele in the Australian Aboriginal Tiwi Islander population, which presents high rates of kidney disease [11]. However, this association was not replicated in a third SLE cohort of mainly Spanish-descent patients (N=281, χ2=2.1, 1 d.f., P=0.14) [11]. Indeed, Vangl1-/+ model mice showed spontaneous deposition of IgA and IgG, but not IgM or complement, in the mesangium [11]. This led researchers to hypothesize that a deficiency of Vangl1 protein in heterozygous mice could alter the permeability of the glomerular endothelium for monomeric immunoglobulins. Despite the association of esv3587290 CNV with LN, no convincing evidence for a deleterious effect of this CNV on VANGL1 function was achieved in this study; however, the experimental murine models also supported that deficiency of Vangl1 is associated with the development of nephritis in only those Vangl1-/+ mice injected with autoreactive serum, which further support an altered glomerular endothelial permeability to autoreactive immunoglobulins. Whether this mechanism underlies the association of esv3587290 CNV with LN development in humans remains unknown [11].
African American, Hispanic, and Asian SLE patients are at greater risk for developing and presenting more severe forms of LN compared to those from European-descent populations [10]. To our knowledge, no GWAS study has yet identified any LN susceptibility locus linked to VANGL1 or the 1p13.1 region, even in populations of European ancestry [9,10,12,13]. Furthermore, Jiang et al. failed to replicate the association of LN with the esv3587290 CNV in a cohort of predominantly Spanish-descent patients [11]. Thus, additional work is needed to test whether this genetic variation could be a risk factor in other ethnicities or clinical presentations of SLE, such as the childhood-onset form. Finally, Jiang et al. employing whole-genome sequencing (WGS) described that the esv3587290 deletion-type CNV varied in size among SLE patients [11], although the authors did not report precise nucleotide-resolution breakpoints using the Human Genome Variation Society (HGVS) nomenclature [14]. This size variability could suggest that distinct mutational events may generate the CNV. Meanwhile, given its high minor allelic frequencies in different populations (0.17-0.43) [11], we cannot discard the possibility of a one-time emergence of a single common allele.
Here, we sought to determine if the VANGL1 esv3587290 deletion-type CNV is associated with LN in a sample of Mexican cSLE patients, and to characterize the precise breakpoints of this rearrangement in our cSLE patients and a reference group of ethnically-matched individuals.
2. Materials and Methods
2.1. Patient Selection
We enrolled a total of 66 unrelated Mexican children (11 males, 55 females; aged 7.8 to 18.6 years) who were born in the central region of Mexico and from Mexican parents. These patients were diagnosed with cSLE between the years 2008 to 2022 with an average age of 11.19 ± 3.31 years, as collected from the pediatric immunology service at Instituto Nacional de Pediatría, México. For inclusion, subjects were required to fulfill the criteria of the 2012 Systemic Lupus International Collaborating Clinics [15] or the 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus [16]. Patients with suspected (disease onset <5 years, parental consanguinity, or familial history of autoimmune disease in a first-degree relative) or confirmed diagnosis of a monogenic form of cSLE were not included. Diagnosis of LN was based on the presence of proteinuria (>0.5 g/dL) or a urinary protein:creatinine (UPr:Cr) ratio >0.2, erythrocyturia, the presence of granular casts in urinary sediment, and/or histopathological confirmation on renal biopsy.
To estimate the allelic esv3587290 CNV frequencies in our population and determine the necessary sample size, we included 181 deidentified genomic DNA samples from unrelated Mexican newborns. The DNA was obtained from residual dried blood spot (DBS) samples analyzed for newborn screening (reference group).
2.2. Genotyping of the esv3587290 CNV
Buccal cell swabs obtained from cSLE patients and DBS samples from the reference group were processed by the standard salting-out method to obtain genomic DNA. We initially performed a PCR assay using forward (5’-AGGGGAGGTGATGGACCCTA-3´) and reverse (5´-CTCAGACTGTAAGCGAAGGACA-3´) primers located inside exons 7 and 8 of VANGL1, respectively, to identify the esv3587290 CNV. This assay generated ~6 kb and ~3 kb PCR fragments from the wild-type and esv3587290 CNV alleles, respectively. This strategy was initially applied to 16 genomic DNA samples from the reference group, and the ~3 kb PCR fragments were identified in three individuals. These ~3 kb PCR fragments were gel excised, purified, and subjected to Sanger sequencing using a “primer-walking” strategy (primers are available upon request). As the entire ~3 kb sequence revealed a single identical breakpoint in these amplicons, we designed a new set of primers (VANGL1-INT7-CNVdelB-F: 5´-TGGCTGTTTCTTGTAATATCCC-3´ and VANGL1-INT7-CNVdelB-R: 5´-CCGACATGGTAAGCAAGC-3´) to amplify a shorter fragment (521 bp) encompassing the breakpoint boundaries of the esv3587290 CNV. To detect the non-deleted VANGL1 allele, we designed a set of primers (VANGL1-INT7A-F: 5´-ACTGATTGTCTGTTGATGCACATTT-3´ and VANGL1-INT7A-R: 5´-CACCCCCTAGGAGGGCAAT-3´) to amplify an internal region of intron 7 (357 bp) that is absent from the esv3587290 CNV sequence. These two mutually exclusive amplicons were generated by two separate monoplex end-point PCR assays (PCR conditions are available upon request) and resolved by agarose gel electrophoresis. Allelic and genotypic frequencies were obtained by direct counting in cSLE patients and reference group samples as follows: wild-type homozygous, 357-bp fragment; heterozygous, 521 and 357-bp fragments, and esv3587290 CNV homozygous, 521-bp fragment. All the 521-bp esv3587290 CNV-derived amplicons from cSLE patients and reference group individuals were subjected to direct automated Sanger sequencing and further aligned (Program Chromas Pro Version 2.1.10, Technelysium Pty Ltd., South Brisbane, Queensland, Australia) with the gene (NG_016548.1) and the Vang-like protein 1 isoform 1 (NM_138959.3) reference sequences to determine the precise breakpoints. The rearrangement was reported according to HGVS nomenclature (https://hgvs-nomenclature.org/stable/) [14].
2.3. Statistical Analysis
The sample size was calculated by applying a formula to find differences between two proportions, using reference-group allelic frequencies for wild-type (0.765) and esv3587290 CNV (0.235) VANGL1 (Table 1), using a confidence level of 99% and a power of 95%, and assuming a 10% loss to follow-up. From this, 33 individuals must be included for each cSLE group (with and without LN).
The VANGL1 genotypes obtained from the reference samples and cSLE patients were tested for Hardy-Weinberg equilibrium (HWE) using the χ2 test. Associations between the presence of LN and the presence of the esv3587290 CNV were examined by Odds Ratio (OR) calculations and Pearson’s χ2 test, using a confidence interval of 95% and a significance threshold of P≤0.05. These calculations were performed using the IBM® SPSS® Statistics software Version 25.0.
This study protocol was revised and approved by the Institutional Review Research, Biosecurity, and Ethics Committees of the National Institute of Pediatrics, Mexico (Registry 2022/030; approval date June 20, 2022). We conducted this study according to the guidelines of the Declaration of Helsinki.
3. Results
LN was present in 39 of 66 (59.1%) patients at the moment of their inclusion in this study. Seventeen patients had undergone renal biopsy and histology was available in all of them (5 with class II, 2 with class III, 8 with class IV, and 2 with class V of LN stratification), while 33 patients had 24-h proteinuria >0.5 g/dL, 34 had UPr:Cr ratio >0.2, and 16 had erythrocyturia and/or granular casts in the urinary sediment. Of the patients without LN, most of them (N=20/27) had a medical follow-up for more than 2 years, while the seven remaining had the last evaluation by immunologists between 19 and 23 months after cSLE diagnosis.
Allelic and genotypic frequencies of the esv3587290 CNV in cSLE patients and reference samples are shown in Table 1. The proportions for genotypes in both groups were consistent with HWE, as assessed by χ2 test. The esv3587290 CNV alleles and genotypes of cSLE patients showed significant associations with the presence of LN, as assessed by χ2 test. More specifically, we observed a protective effect of the esv3587290 CNV (OR 0.108, 95% CI 0.034-0.33, P=0.0003), and the heterozygous genotype (OR 0.04, 95% CI 0.119-0.9811, P=0.002) for developing LN. The relatively low number of cSLE patients in the groups with and without LN carrying a homozygous esv3587290 CNV genotype precluding χ2 testing for this comparison (Table 2).
The Sanger sequencing results of all 521-bp PCR fragments derived from the esv3587290 CNV identified in reference and cSLE samples showed an identical breakpoint, wherein deletion of 3,357 bp eliminates nearly 60% of the intron 7 sequence (5,591 bp) of VANGL1 (Figure 1). We therefore designated this common deletion as NG_016548.1(NM_138959.3):c.1314+1339_1315-897del, NC_000001.10:g.116229487_116232843del (GRCh37), or NC_000001.11:g.115686866_115690222del (GRCh38) according to HGVS nomenclature. This variant was submitted to the Leiden Open Variation Database (LOVD) of VANGL1 gene (https://databases.lovd.nl/shared/variants/0000918418#00025811).
4. Discussion
Replication studies are mainly intended to confirm genetic associations discovered through GWAS, such as that between esv3587290 CNV and SLE-related LN [11]. These studies are needed to accumulate convincing statistical evidence supporting the association and rule out spurious findings due to uncontrolled biases [17]. To the best of our knowledge, this is the first work to explore the possible association of the esv3587290 CNV with LN in a non-European population, as previously recommended [11]. Here, we assessed an SLE population of Mexican descent, which is among the ethnicities considered to have a high risk for developing LN [18]. We further focused on a clinical form of SLE different than that previously studied in this context, namely childhood-SLE, for which LN is considered more prevalent and severe than in the adult form of SLE [4].
A few genetic markers have been associated with LN in a Mexican population, including SPP1 (MIM*166490) in adult SLE [allele T of rs1126616 OR 2.0 (95% CI 1.26-3.16), P=0.003 and TT genotype under the recessive model OR 2.76 (95% CI 1.31-5.82), P=0.011] [19] and NFE2L2 (formerly NRF2; MIM*600492) in cSLE [heterozygous A/G rs35652124 genotype OR=1.81 (95% CI 1.04–3.12), P=0.032) [20]. Meanwhile, other LN-susceptibility markers previously identified in European-descent populations (e.g., PDCD1, MIM*600244) [21] or murine models (CNVs of TLR7, MIM*300365) [22], failed to show any significant association with LN in cSLE patients from a Mexican population [23,24]. These previous observations together with our present finding that esv3587290 CNV appears to protect against the development of LN in this group of Mexican patients with cSLE (Table 2), could support the idea that LN exhibits broad genetic and phenotypic heterogeneity. Our results could also agree with the lack of any evident association between LN and VANGL1, the 1p13.1 region, or PCP pathways in a GWAS performed in Hispanic, European, African American, and Asian patients [13]. We further believe that our findings and the inability of Jiang et al. to replicate their association in the third cohort suggest that, in contrast to the previous proposal [11], esv3587290 CNV genotyping is not a viable strategy for LN risk stratification, at least for non-European SLE populations and cSLE patients.
In our study population, we further characterized the precise breakpoint of the esv3587290 CNV. Whereas Jiang et al. found that this deletion varied in size when assessed using a WGS strategy [11], our Sanger sequencing approach revealed an identical deletion event in all individuals carrying one or two esv3587290 CNV copies (Figure 1). We speculate that these discrepancies may reflect different origins of the esv3587290 CNV among diverse populations, or they be related to methodological issues. The former could imply that there is a “hot-spot” for mutational events leading to distinct rearrangements, as occur for some monogenic traits (i.e., gross deletions in the DMD gene). Alternatively, some single mutational events may occur and even be associated with a founder effect [25], as appears to be the case for the esv3587290 CNV in our Mexican population. Further haplotypic analysis is warranted to support the latter hypothesis. Regarding the potential impact of methodologic issues, we note that the short-read WGS strategy is generally intended to approximately localize the breakpoints of a gross genomic structural rearrangement (e.g., a deletion-type CNV); to reach the nucleotide level of resolution, it could be necessary to apply long-range PCR and Sanger sequencing [26,27], as performed herein. Given this, we propose that it would be desirable to determine the precise nucleotide-level rearrangements of the esv3587290 CNV in other populations. In the PCR-based Sanger sequencing strategy, we used to amplify the esv3587290 CNV, the forward and reverse primers designed to anneal ~250 bp away from the breakpoints estimated by WGS [11]. There remains some possibility that allelic drop-out may have occurred due to the non-amplification of alleles bearing different breakpoints. However, we believe that this is unlikely due to the lack of departure from HWE in both study groups, along with the similarity of our allelic frequencies (Table 1) with those previously reported for esv3587290 CNV in Latino populations (~0.3) [11].
Although mis-spliced mRNA species of VANGL1 lacking exon 2 were identified in peripheral blood mononuclear cells from two homozygous esv3587290 CNV SLE patients, it remains to be determined if the esv3587290 CNV has any effect on VANGL1 function at the kidney [11]. Such a finding would support the idea that this CNV plays a genetic susceptibility role for the development of LN in humans. Interestingly, a predicted enhancer element is located inside exon 8 (encoding the 3´UTR) of VANGL1 (https://www.ncbi.nlm.nih.gov/gene/81839; chr1:116238833-116239332; GRCh37/hg19 assembly coordinates). It would be interesting to determine if the neighboring esv3587290 CNV exerts some effect on the functionality of this predicted enhancer element in human kidney tissues.
4.1. Limitations of the Study
Nonetheless, our study had several limitations: It is possible that our study sample was under-representative or phenotypically heterogeneous, given the lack of renal biopsy-proven LN in most of our patients (N=17/39 LN cSLE patients, 43.6%), which is still considered the diagnostic gold standard [7]. Obtaining such information could allow us to perform an association analysis stratified by histopathological classes, as previously recommended [11]. Also, we did not perform any ancestry analysis to determine if the reference group and cSLE patients were admixed, thus a possibility of bias by population stratification cannot be excluded. The low number of male cSLE patients (9 with LN and 2 without LN) did not allow us to perform stratification analysis by gender, so this aspect should be addressed in the future. It is important to note that the above-described aspects also were not considered by Jiang et al. [11]. Genotyping errors could have biased our analysis, as we did not use a second molecular technique to validate our PCR-based genotyping assay. However, this seems unlikely given the lack of departure from HWE for esv3587290 CNV in the reference group and cSLE patients, along with the similarity between the observed allelic frequencies (Table 1) and those previously reported in Latino populations [11]. Finally, the cross-sectional nature of this study precluded us from determining whether the included cSLE patients without LN could later develop nephritis, adding another potential confounding factor, particularly in those seven cSLE patients with normal kidney function who do not have completed the two years of follow-up. The estimated risk of developing LN in these patients is expected to be 7% [28].
5. Conclusions
The esv3587290 CNV of the VANGL1 gene was not associated with the development of LN in a sample of Mexican cSLE patients; rather, this CNV seems to be a genetic protective factor. Further replication studies on the esv3587290 CNV in other ethnicities and clinical forms of SLE are warranted to define its role as a genetic factor in the development of LN. The esv3587290 CNV in our Mexican population seems to be a unique 3,357-bp deletion that may have originated from a single mutational event.
Author Contributions
Conceptualization, M.A.A.-O.; methodology, M.A.A.-O., L.D.-G., and B.E.-O.; software, M.A.A.-O. and L.D.-G.; validation, M.A.A.-O., A.L.R.-L., L.D.-G. and A.G.-d.A.; formal analysis, M.A.A.-O., A.L.R.-L., L.D.-G., R.G.N.-V., and A.G.-d.A.; investigation, M.A.A.-O., B.E.-O., A.L.R.-L., F.E.R.-L., R.G.N.-V., and A.G.-d.A.; resources, M.A.A.-O., A.L.R.-L., F.E.R.-L., and A.G.-d.A.; data curation, M.A.A.-O., A.L.R.-L., B.E.-O., and A.G.-d.A.; writing—original draft preparation, M.A.A.-O.; writing—review and editing, M.A.A.-O., B.E.-O., A.L.R.-L., L.D.-G., F.E.R.-L., R.G.N.-V., and A.G.-d.A.; visualization, M.A.A.-O.; supervision, M.A.A.-O.; project administration, M.A.A.-O.; funding acquisition, M.A.A.-O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Instituto Nacional de Pediatría, Secretaría de Salud (Recursos Fiscales 2022–2024, Programa E022 Investigación y Desarrollo Tecnológico en Salud, Ciudad de México, México).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Research, Biosecurity, and Ethics Committees of the National Institute of Pediatrics, Mexico (Registry 2022/030). The approval date was June 20, 2022.
Informed Consent Statement
Written informed consent to participate in this study and to publish this paper was obtained either from the patients involved in the study or their parents.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found in the LOVD v.3.0 - Leiden Open Variation Database: https://www.lovd.nl/, accessed March 11, 2024; OMIM: https://www.omim.org/, accessed March 11, 2024; NCBI: https://www.ncbi.nlm.nih.gov/gene, accessed April 2, 2024. The data presented in this study are available on reasonable request from the corresponding author. All the herein reported clinically relevant genetic variants along with the available deidentified phenotypic data were submitted to the publicly available LOVD v.3.0 - Leiden Open Variation Database (https://databases.lovd.nl/shared/variants/0000918418#00025811, accessed March 11, 2024).
Acknowledgments
The authors gratefully acknowledge the patients and parents for their commitment. They also thank Roberto Edher Demetrio-Ríos and Karen Jimena Guerrero González for their assistance with the recruitment and patient sampling processes, as well as data collection and technical support.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Barsalou J, Levy DM, Silverman ED. An update on childhood-onset systemic lupus erythematosus. Curr Opin Rheumatol. 2013; 25:616–22. [CrossRef]
- Brunner HI, Holland MJ, Beresford MW, et al. American College of Rheumatology Provisional Criteria for Clinically Relevant Improvement in Children and Adolescents With Childhood-Onset Systemic Lupus Erythematosus. Arthritis Care Res (Hoboken). 2019;71(5):579-590. [CrossRef]
- Ambrose N, Morgan TA, Galloway J, Ionnoau Y, Beresford MW, Isenberg DA. Differences in disease phenotype and severity in SLE across age groups. Lupus 2016;25(14):1542–50. Epub 2016 May 4. PMID: 27147622; PMCID: PMC5089221. [CrossRef]
- Fatemi A, Matinfar M, Smiley A. Childhood versus adult-onset systemic lupus erythematosus: long-term outcome and predictors of mortality. Clin Rheumatol. 2017;36(2):343-350. [CrossRef]
- Borchers AT, Leibushor N, Naguwa SM et al. Lupus nephritis: a critical review. Autoimmun Rev 2012; 12: 174–194. Epub 2012 Sep 8. PMID: 22982174. [CrossRef]
- Alduraibi FK, Tsokos GC. Lupus Nephritis Biomarkers: A Critical Review. Int J Mol Sci. 2024 Jan 9;25(2):805. PMID: 38255879; PMCID: PMC10815779. [CrossRef]
- Abulaban KM, Brunner HI. Biomarkers for childhood-onset systemic lupus erythematosus. Curr Rheumatol Rep. 2015 Jan;17(1):471. PMID: 25475594; PMCID: PMC4980820. [CrossRef]
- Picard C, Lega JC, Ranchin B, Cochat P, Cabrera N, Fabien N, Belot A. Anti-C1q autoantibodies as markers of renal involvement in childhood-onset systemic lupus erythematosus. Pediatr Nephrol. 2017 Sep;32(9):1537-1545. Epub 2017 Mar 25. PMID: 28343355. [CrossRef]
- Vinuesa CG, Shen N, Ware T. Genetics of SLE: mechanistic insights from monogenic disease and disease-associated variants. Nat Rev Nephrol. 2023 Sep;19(9):558-572. Epub 2023 Jul 12. PMID: 37438615. [CrossRef]
- Iwamoto T, Niewold TB. Genetics of human lupus nephritis. Clin Immunol. 2017 Dec;185:32-39. Epub 2016 Sep 28. PMID: 27693588; PMCID: PMC5373939. [CrossRef]
- Jiang SH, Mercan S, Papa I, Moldovan M, Walters GD, Koina M, Fadia M, Stanley M, Lea-Henry T, Cook A, Ellyard J, McMorran B, Sundaram M, Thomson R, Canete PF, Hoy W, Hutton H, Srivastava M, McKeon K, de la Rúa Figueroa I, Cervera R, Faria R, D’Alfonso S, Gatto M, Athanasopoulos V, Field M, Mathews J, Cho E, Andrews TD, Kitching AR, Cook MC, Riquelme MA, Bahlo M, Vinuesa CG. Deletions in VANGL1 are a risk factor for antibody-mediated kidney disease. Cell Rep Med. 2021 Dec 21;2(12):100475. PMID: 35028616; PMCID: PMC8714939. [CrossRef]
- Chung SA, Brown EE, Williams AH, Ramos PS, Berthier CC, Bhangale T, Alarcon-Riquelme ME, Behrens TW, Criswell LA, Graham DC, Demirci FY, Edberg JC, Gaffney PM, Harley JB, Jacob CO, Kamboh MI, Kelly JA, Manzi S, Moser-Sivils KL, Russell LP, Petri M, Tsao BP, Vyse TJ, Zidovetzki R, Kretzler M, Kimberly RP, Freedman BI, Graham RR, Langefeld CD; International Consortium for Systemic Lupus Erythematosus Genetics. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J Am Soc Nephrol. 2014 Dec;25(12):2859-70. Epub 2014 Jun 12. PMID: 24925725; PMCID: PMC4243339. [CrossRef]
- Lanata CM, Nititham J, Taylor KE, Chung SA, Torgerson DG, Seldin MF, Pons-Estel BA, Tusié-Luna T, Tsao BP, Morand EF, Alarcón-Riquelme ME, Criswell LA. Genetic contributions to lupus nephritis in a multi-ethnic cohort of systemic lupus erythematous patients. PLoS One. 2018 Jun 28;13(6):e0199003. PMID: 29953444; PMCID: PMC6023154. [CrossRef]
- den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016 Jun;37(6):564-9. Epub 2016 Mar 25. PMID: 26931183. [CrossRef]
- Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O, Sturfelt G, Ramsey-Goldman R, Bae SC, Hanly JG, Sánchez-Guerrero J, Clarke A, Aranow C, Manzi S, Urowitz M, Gladman D, Kalunian K, Costner M, Werth VP, Zoma A, Bernatsky S, Ruiz-Irastorza G, Khamashta MA, Jacobsen S, Buyon JP, Maddison P, Dooley MA, van Vollenhoven RF, Ginzler E, Stoll T, Peschken C, Jorizzo JL, Callen JP, Lim SS, Fessler BJ, Inanc M, Kamen DL, Rahman A, Steinsson K, Franks AG Jr, Sigler L, Hameed S, Fang H, Pham N, Brey R, Weisman MH, McGwin G Jr, Magder LS. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012 Aug;64(8):2677-86. PMID: 22553077; PMCID: PMC3409311. [CrossRef]
- Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL, Jayne D, Cervera R, Costedoat-Chalumeau N, Diamond B, Gladman DD, Hahn B, Hiepe F, Jacobsen S, Khanna D, Lerstrøm K, Massarotti E, McCune J, Ruiz-Irastorza G, Sanchez-Guerrero J, Schneider M, Urowitz M, Bertsias G, Hoyer BF, Leuchten N, Tani C, Tedeschi SK, Touma Z, Schmajuk G, Anic B, Assan F, Chan TM, Clarke AE, Crow MK, Czirják L, Doria A, Graninger W, Halda-Kiss B, Hasni S, Izmirly PM, Jung M, Kumánovics G, Mariette X, Padjen I, Pego-Reigosa JM, Romero-Diaz J, Rúa-Figueroa Fernández Í, Seror R, Stummvoll GH, Tanaka Y, Tektonidou MG, Vasconcelos C, Vital EM, Wallace DJ, Yavuz S, Meroni PL, Fritzler MJ, Naden R, Dörner T, Johnson SR. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019 Sep;71(9):1400-1412. Epub 2019 Aug 6. PMID: 31385462; PMCID: PMC6827566. [CrossRef]
- Kraft P, Zeggini E, Ioannidis JP. Replication in genome-wide association studies. Stat Sci. 2009 Nov 1;24(4):561-573. PMID: 20454541; PMCID: PMC2865141. [CrossRef]
- Sánchez E, Rasmussen A, Riba L, Acevedo-Vasquez E, Kelly JA, Langefeld CD, Williams AH, Ziegler JT, Comeau ME, Marion MC, García-De La Torre I, Maradiaga-Ceceña MA, Cardiel MH, Esquivel-Valerio JA, Rodriguez-Amado J, Moctezuma JF, Miranda P, Perandones CE, Castel C, Laborde HA, Alba P, Musuruana JL, Goecke IA, Anaya JM, Kaufman KM, Adler A, Glenn SB, Brown EE, Alarcón GS, Kimberly RP, Edberg JC, Vilá LM, Criswell LA, Gilkeson GS, Niewold TB, Martín J, Vyse TJ, Boackle SA, Ramsey-Goldman R, Scofield RH, Petri M, Merrill JT, Reveille JD, Tsao BP, Orozco L, Baca V, Moser KL, Gaffney PM, James JA, Harley JB, Tusié-Luna T, Pons-Estel BA, Jacob CO, Alarcón-Riquelme ME. Impact of genetic ancestry and sociodemographic status on the clinical expression of systemic lupus erythematosus in American Indian-European populations. Arthritis Rheum. 2012 Nov;64(11):3687-94. PMID: 22886787; PMCID: PMC3485439. [CrossRef]
- Rivera-Cameras A, Gallegos-Arreola MP, Morán-Moguel MC, Salazar-Páramo M, Alcaraz-López MF, Echeverría-González G, Topete-Reyes JF, Franco-Chávez SA, Dávalos-Rodríguez IP. Association of the rs1126616 and rs9138 Variants in the SPP1 Gene among Mexican Patients with Systemic Lupus Erythematosus and Lupus Nephritis. Int J Mol Sci. 2024 Jan 13;25(2):1000. PMID: 38256074; PMCID: PMC10816335. [CrossRef]
- Córdova EJ, Velázquez-Cruz R, Centeno F, Baca V, Orozco L. The NRF2 gene variant, -653G/A, is associated with nephritis in childhood-onset systemic lupus erythematosus. Lupus. 2010 Sep;19(10):1237-42. Epub 2010 May 27. PMID: 20507872. [CrossRef]
- Johansson M, Arlestig L, Möller B, Rantapää-Dahlqvist S. Association of a PDCD1 polymorphism with renal manifestations in systemic lupus erythematosus. Arthritis Rheum. 2005 Jun;52(6):1665-9. PMID: 15934088. [CrossRef]
- Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006 Jun 27;103(26):9970-5. Epub 2006 Jun 15. PMID: 16777955; PMCID: PMC1502563. [CrossRef]
- Velázquez-Cruz R, Orozco L, Espinosa-Rosales F, Carreño-Manjarrez R, Solís-Vallejo E, López-Lara ND, Ruiz-López IK, Rodríguez-Lozano AL, Estrada-Gil JK, Jiménez-Sánchez G, Baca V. Association of PDCD1 polymorphisms with childhood-onset systemic lupus erythematosus. Eur J Hum Genet. 2007 Mar;15(3):336-41. Epub 2007 Jan 17. PMID: 17228327. [CrossRef]
- García-Ortiz H, Velázquez-Cruz R, Espinosa-Rosales F, Jiménez-Morales S, Baca V, Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis. 2010 Oct;69(10):1861-5. Epub 2010 Jun 4. PMID: 20525845. [CrossRef]
- Shotelersuk V, Larson D, Anikster Y, McDowell G, Lemons R, Bernardini I, Guo J, Thoene J, Gahl WA. CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet. 1998 Nov;63(5):1352-62. PMID: 9792862; PMCID: PMC1377545. [CrossRef]
- Ma S, Zhang Z, Fu Y, Zhang M, Niu Y, Li R, Guo Q, He Z, Zhao Q, Song Z, Wang X, Sun R. Identification of the first Alu-mediated gross deletion involving the BCKDHA gene in a compound heterozygous patient with maple syrup urine disease. Clin Chim Acta. 2021 Jun;517:23-30. Epub 2021 Feb 16. PMID: 33607070. [CrossRef]
- Cuenca-Guardiola J, de la Morena-Barrio B, García JL, Sanchis-Juan A, Corral J, Fernández-Breis JT. Improvement of large copy number variant detection by whole genome nanopore sequencing. J Adv Res. 2023 Aug;50:145-158. Epub 2022 Oct 30. PMID: 36323370; PMCID: PMC10403694. [CrossRef]
- Pennesi M, Benvenuto S. Lupus Nephritis in Children: Novel Perspectives. Medicina (Kaunas). 2023 Oct 16;59(10):1841. PMID: 37893559; PMCID: PMC10607957. [CrossRef]
Figure 1.
Schematic of the esv3587290 CNV deletion interval in intron 7 of VANGL1 and partial forward sequence alignments of the 521-bp PCR fragments revealing the identical breakpoint of the esv3587290 CNV deletion in selected cSLE patients (N=10) and reference samples (DBS, N=10). The same breakpoint was observed for the 38 and 85 esv3587290 CNV alleles (data not shown, Table 1) identified in the cSLE patients and the reference samples, respectively, leading us to establish this gene rearrangement as NG_016548.1(NM_138959.3):c.1314+1339_1315-897del.
Figure 1.
Schematic of the esv3587290 CNV deletion interval in intron 7 of VANGL1 and partial forward sequence alignments of the 521-bp PCR fragments revealing the identical breakpoint of the esv3587290 CNV deletion in selected cSLE patients (N=10) and reference samples (DBS, N=10). The same breakpoint was observed for the 38 and 85 esv3587290 CNV alleles (data not shown, Table 1) identified in the cSLE patients and the reference samples, respectively, leading us to establish this gene rearrangement as NG_016548.1(NM_138959.3):c.1314+1339_1315-897del.
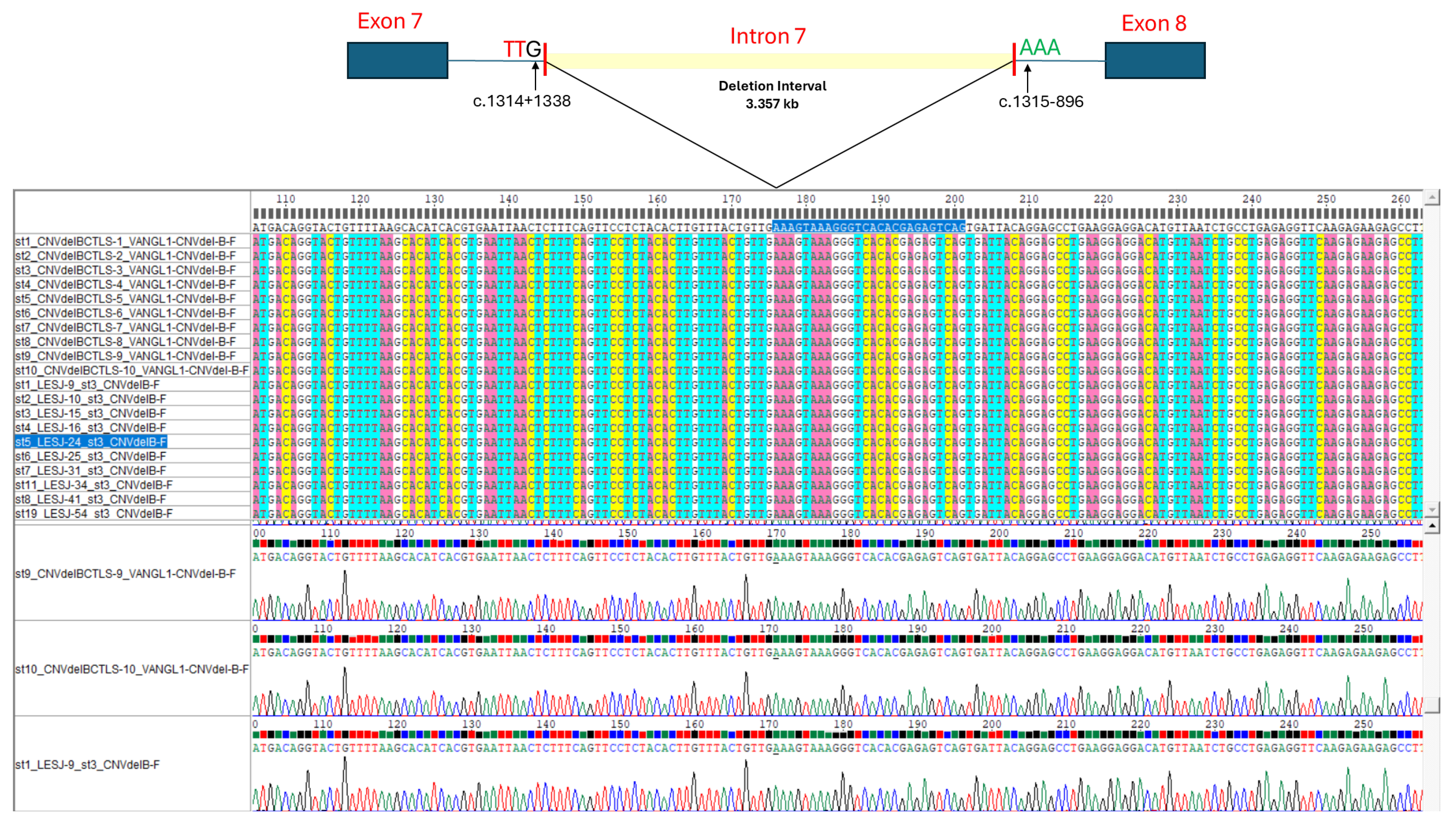

Table 1.
Allelic and genotypic frequencies of esv3587290 CNV in cSLE patients and reference samples.
Table 1.
Allelic and genotypic frequencies of esv3587290 CNV in cSLE patients and reference samples.
|
VANGL1 alleles |
Total cSLE patients (N=66, 132 alleles) |
Reference group (DBS samples) (N=181, 362 alleles) |
| Wild-type allele | 0.712 | 0.765 |
| esv3587290 CNV- deletion-type allele |
0.288 | 0.235 |
|
VANGL1 genotype |
Total cSLE patients (N=66 genotypes) |
Reference group (DBS samples) (N=181 genotypes) |
| Wild-type homozygous | 48.5% (N=32) | 58% (N=105) |
| Heterozygous | 45.5% (N=30) | 37% (N=67) |
| Homozygous esv3587290 | 6% (N=4) | 5% (N=9) |
Abbreviations: cSLE: childhood-onset systemic lupus erythematosus; CNV: copy-number variation; DBS: residual anonymized dried blood spots from Mexican unrelated neonates subjected to newborn screening program.
Table 2.
Association results and VANGL1 allelic and genotypic frequencies among cSLE patients with or without LN.
Table 2.
Association results and VANGL1 allelic and genotypic frequencies among cSLE patients with or without LN.
|
VANGL1 alleles |
Allelic frequencies cSLE patients with LN (N=39, 78 alleles) |
Allelic frequencies cSLE patients without LN (N=27, 54 alleles) |
χ2 OR (95% CI)1 |
P-Value |
| Wild-type allele | 0.769 |
0.630 |
χ2=16.41 OR=0.108 (0.034-0.33) |
P=0.000025 P=0.0003 |
| esv3587290 CNV deletion-type allele |
0.231 |
0.370 |
||
|
VANGL1 genotypes |
Genotypic frequencies cSLE patients with LN (N=39 genotypes) |
Genotypic frequencies cSLE patients without LN (N=27 genotypes) |
χ2 / OR (95% CI) |
P-Value |
| Wild-type homozygous |
59% (N=23) | 33.3% (N=9) | ||
|
Heterozygous esv3587290 |
35.9% (N=14) |
59.3% (N=16) |
χ2=4.0892 OR=0.042 (0.119-0.9811) |
P=0.02 P=0.002 |
| Homozygous esv3587290 | 5.1% (N=2) |
7.4% (N=2) | χ2= not calculable3 OR=not calculable3 |
P=not calculable P=not calculable |
1The presence of LN, esv3587290 alleles, and heterozygous genotypes were taken as the reference categories for the OR calculations. 2Wild-type homozygous vs. heterozygous for esv3587290; 3Wild-type homozygous vs. homozygous for esv3587290; Abbreviations: CI: confidence interval; cSLE: childhood-onset systemic lupus erythematosus; CNV: copy-number variation; OR: odds ratio.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



