
Submitted:
29 April 2024
Posted:
01 May 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Network centrality analyses have proven successful in identifying important nodes in diverse host-pathogen interactomes. The current study presents a comprehensive investigation of the human interactome and SARS-CoV-2 host targets. We first constructed a comprehensive human interactome by compiling experimentally validated protein-protein interactions (PPIs) from eight distinct sources. Additionally, we compiled a comprehensive list of 1,449 SARS-CoV-2 host proteins and analyzed their interactions within the human interactome, which identified enriched biological processes and pathways. Seven diverse topological features were employed to reveal the enrichment of SARS-CoV-2 targets in the human interactome, with Load centrality emerging as the most effective metric. Furthermore, a novel approach called CentralityCosDist was employed to predict SARS-CoV-2 targets, which proved effective in expanding the pool of predicted targets. Pathway enrichment analyses further elucidated the functional roles and potential mechanisms associated with predicted targets. Overall, this study provides valuable insights into the complex interplay between SARS-CoV-2 and the host cellular machinery, contributing to a deeper understanding of viral infection and immune response modulation.
Keywords:
comprehensive human interactome
; SARS-CoV-2 targets
; network metrices
; centrality analyses
1. Introduction
Networks consist of nodes, which represent the systems components within the network, and edges, which represent the connections or relationships between these entities [1,2,3]. In biological systems, especially in the context of molecular biology and systems biology, network often focuses on direct and indirect interactions among genes and their products. For instance, In Protein-protein interaction (PPI) networks, every node symbolizes a protein, while edges denote the physical interactions occurring between them. Cataloging PPIs on a proteome-wide scale is commonly referred to as interactomes [1,2,3]. Interactomes exemplify the intricate web of static and dynamic interactions within living organisms, occurring both under normal steady-state conditions and in response to internal cues or external stressors. Such protein complexes play crucial functions in diverse signaling cascades, distinct cellular pathways, and a wide array of biological processes [4,5]. Specialized pathogens, including viruses, bacteria, and eukaryotic parasites, deploy a range of pathogenic molecules including virulent proteins. These proteins have evolved to interact with crucial targets within the host’s interactomes, leading to significant rewiring of information flow [2,3,6,7,8,9,10,11,12,13,14,15,16,17,18]. This manipulation of host interactomes plays a pivotal role in causing disease. Therefore, analyzing the network architecture and structural properties of host-pathogen interactomes may unveil novel components in microbial pathogenicity.
In cellular networks, this network pattern emerges from universal principles that govern network organization, indicating consistency across various network characteristics. For instance, scale-free topology describes a structure where connections among nodes exhibit a power law distribution, characterized by a few highly connected nodes and many with few connections. The “small number of nodes possessing increased connectivity” suggests that in these networks, a handful of nodes serve as hubs, connecting a significant portion of the network together. These hubs play a crucial role in maintaining efficient communication within the network, as they facilitate the flow of information between different regions or clusters of nodes. Besides network connectivity, node position is another crucial concept. Node betweenness centrality is a measure of how often a node serves as a bridge along the shortest paths between pairs of other nodes in the network. Nodes with high betweenness centrality (bottlenecks) are positioned strategically in the network, acting as critical players in a biological network. Indeed, hubs and bottlenecks are key targets for various pathogens, including viruses, bacteria, and eukaryotic pathogens, across plant and animal interactomes. For instance, following the construction of a comprehensive human interactome, analysis of network centrality unveiled that pan-viral targets predominantly consist of hubs positioned at the network’s core and enriched in fundamental biological processes [19]. A meta-analysis of host-virus interactions across 17 different viruses indicated that viruses tend to target bottlenecks and hubs. This demonstrates that viruses have evolved to disrupt the scale-free human interactome by targeting on hubs and proteins that serve as crucial communication nodes [20]. Therefore, the connectivity and position of specific nodes potentially allow us to understand how viral pathogens exploit vulnerable host cellular networks, facilitating infection, replication, or immune evasion. In addition, deciphering the viral pathogenesis can better prepare us for future pandemics, similar to the recent global pandemic of COVID-19 caused by the highly contagious and pathogenic SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2).
Here, we have constructed a comprehensive human interactome by integrating PPIs from eight large-scale experimental studies and databases. Additionally, we have compiled a comprehensive set of host targets of SARS-CoV-2. We conducted network topology analysis that involved computing seven diverse centrality measures, which were then applied to the human interactome. Subsequently, we overlaid those centrality-based prioritized nodes with SARS-CoV-2 targets. Our findings indicate that Load Centrality is the most effective in predicting viral targets, followed by PageRank, hubs, and bottlenecks. We also explored how combining diverse topological features can enhance predictive power. Furthermore, we utilized our recently developed algorithm, CentralityCosDist [21], to predict viral targets even in the presence of a small fraction of known viral targets. Overall, this integrative systems biology framework can significantly contribute to our understanding of viral pathogenesis, the identification of crucial host biological pathways, and the discovery of potential therapeutic targets.
2. Materials and Methods
2.1. Construction of a Comprehensive Human Protein Interaction Network
In this study, we constructed a comprehensive human protein-protein interaction (PPI) network by integrating data from several large-scale experimental studies and public databases. Our network included PPIs from BioPlex (“BioPlex_HCT116_55K_Dec_2019” and “BioPlex_293T_10K_Dec_2019”) [18,22,23], CoFrac [24], CUBIC [25], HuRI (“HIR2_EXP”, “HIR2_PRED”, “HI_union”) [26], and STRING v12 [27] (Figure 1B and Table S1). We also compiled a set of 1449 SARS-CoV-2 host proteins from three recent studies [28,29,30].
To ensure consistency, we mapped all protein identifiers to UniProt accession numbers using the UniProt API when necessary. Merging the PPI datasets resulted in a large network with 26,028 nodes (proteins) and 825,682 edges (interactions) (Figure 1B and Table S1). Interestingly, 1445 of the 1449 SARS-CoV-2 host interactor proteins were present in this merged network (Figure 1E).
2.2. Building a SARS-CoV-2 Host Proteins Specific Human Protein Interaction Network
From this comprehensive network, we extracted a sub-network we call the “Open network” that focuses specifically on the 1445 SARS-CoV-2 host proteins and their 19,255 direct interactions. This sub-network contains 20,700 nodes and 260,231 edges (Figure 1E).
Our integrative approach allowed us to construct a high-quality human PPI network that can serve as a valuable resource for studying viral pathogenesis and identifying potential therapeutic targets. By combining multiple large-scale datasets, we were able to expand the coverage of known human PPIs and pinpoint those most relevant to SARS-CoV-2 infection.
2.3. Network Centrality Analysis
In our investigation of the “Open network,” we employed a range of seven distinct centrality measures to pinpoint the most crucial nodes within the network. These includes, Degree centrality, Betweenness centrality, Eigenvector centrality, Closeness centrality, Load centrality, and PageRank, each offer a unique perspective on a node’s importance (Figure S1). By calculating each centrality measure for every node, we were able to generate a ranked list, with the most central nodes appearing at the top (Figure 3A). Interestingly, upon examining this ranked list, we discovered that several of the human proteins known to interact with SARS-CoV-2 were positioned prominently near the top. This finding suggests that these highly central proteins may play a vital role in the virus’s interaction with human cells.
To further refine our analysis and identify significant nodes within the network, we performed Wk-Shell analysis. We utilized the “wk-shell-decomposition” Cytoscape App, a software tool specifically designed for network analysis. Through wk-Shell analysis, we were able to peel back the layers of the network, uncovering progressively more central regions (Figure 3B–D).
2.4. Ranking Nodes with Respect to the SARS-CoV-2 Host Proteins
To identify novel human proteins that SARS-CoV-2 might potentially hijack, we employed a recently developed method called CentralityCosDist on the “Open network” (Figure S4). This method goes beyond standard network analysis by incorporating multiple centrality measures. In essence, centrality measures evaluate a node’s importance within the network. Here, we utilized eight such measures: Degree centrality, Betweenness centrality, Eigenvector centrality, Closeness centrality, Clustering coefficient, Load centrality, and PageRank. Furthermore, to enhance the effectiveness of CentralityCosDist, we strategically selected “seed nodes.” These seed nodes act as reference points within the network, guiding the algorithm’s search for new potential viral targets. We implemented a two-pronged approach for seed node selection:
- SARS-CoV-2 Host Protein Based Seeds: We utilized two variations. In the first, we selected 10% of the 1445 known SARS-CoV-2 host proteins. In the second variation, we included all 1445 known host proteins. This approach prioritizes nodes with established connections to the virus.
- Centrality Based Seeds: We identified the top 10% of nodes based on three centrality measures: Degree centrality, Betweenness centrality, and Closeness centrality. These nodes are inherently very interconnected within the network, making them prime candidates for further investigation.
By combining these seed node strategies with CentralityCosDist, we aimed to cast a wide net and uncover a diverse range of potential SARS-CoV-2 targets within the human protein network.
To preprocess our protein-protein interaction (PPI) data and perform network analyses, we utilized the Python programming language (version 3.11.4) and the NetworkX package (version 3.1). NetworkX is a widely used, open-source Python library for creating, manipulating, and studying the structure and dynamics of complex networks. To visualize our networks and generate publication-quality figures, we used the Cytoscape [31] software (version 3.10.2). To gain biological insights from our network analysis, we performed functional enrichment analysis using the Metascape web application [32].
3. Results and Discussion
3.1. A comprehensive Human Protein–Protein Interactome and SARS-CoV-2 Host Targets
Table S2 host targets, our initial step involved the assembly of a comprehensive human interactome. This encompassed the compilation of experimentally validated PPIs from eight distinct sources. Among these varied resources, the STRING database [33] emerged as pivotal, containing
A vast repository of 561,769 experimentally validated interactions involving approximately 18,000 proteins (Figure 1A,B and Table S1). Additionally, we curated PPI data from additional proteome-scale interactome studies, namely Human Interactome I and II, BioPlex, QUBIC, and CoFrac (as reviewed in [34]), alongside contributions from the Human Interactome Resource (HIR) [35] (Figure 1A,B and Table S1). Following the integration of these diverse PPI datasets, our resultant integrated human interactome featured 825,682 interactions and 26,000 nodes. Our previous study resulted in an experimentally validated high-quality interactome comprising 18,906 nodes and 444,633 edges [36]. In the current study, we have nearly doubled the number of interactions, creating a significantly expanded dataset. This enhanced interactome serves as a valuable resource for future unrelated studies, providing a more comprehensive understanding of network dynamics and facilitating further exploration into complex biological processes. Subsequently, we compiled an exhaustive compendium of SARS-CoV-2 host targets, which comprises of 1,449 host proteins (see methods section for a detail; Table S1). Upon querying these host targets within the human interactome framework, we discovered the presence of 1,445 host targets (Table S1). Intriguingly, among these, 20 host targets were found as singletons, while the remaining 1,429 were found to interact amongst themselves, forming a “closed network” encompassing 31,000 interactions (Figure 1C, Table S1). Pathway enrichment analyses conducted using Metascape revealed a plethora of enriched biological processes. These included “intracellular protein transport”, “membrane organization”, “viral infection pathways”, “RNA metabolism”, “ER protein processing and Golgi vesicle trafficking”, “RhoGTPase signaling”, “influenza infection”, “cell cycle regulation”, “neutrophil degradation”, among several other pathways (Figure 1D). Akin to this study, another proteome-scale mapping of SARS-CoV-2 targets identified 739 high-confidence binary and co-complex interactions. These interactions were found to be enriched in pathways including protein translation, mRNA splicing, Golgi transportation, neutrophil-mediated immunity, and glucose metabolism [29]. Another recent study that relied solely on the STRING database and focused on 1432 distinct proteins targeted by SARS-CoV-2 [37], our approach presents a significantly more comprehensive analysis. Their study constructed a SARS-CoV-2 relevant human interactome comprising 1111 nodes and 7043 edges, identifying enriched biological processes and functional categories such as neutrophil-mediated immunity (GO:0002446), neutrophil activation involved in immune response (GO:0002283), and viral process (GO:0016032) from the biological process category. Additionally, functional categories including dolichyl-diphosphooligosaccharide-protein glycotransferase activity (GO:0004579), GDP binding (GO:0019003), cadherin binding (GO:0045296), ATPase activity (GO:0016887), and focal adhesion (GO:0005925) were also highlighted [37]. By contrast, our methodology involved integrating data from diverse sources, resulting in a more extensive human interactome. This broader dataset facilitated a deeper exploration of network properties and pathway analyses, allowing us to uncover a wider range of biological insights. These findings offer valuable insights into the complex interplay between SARS-CoV-2 and the host cellular machinery. By shedding light on the molecular pathways involved in viral infection and immune response modulation, our study contributes to a deeper understanding of the pathogenesis of SARS-CoV-2 infection and may inform the development of novel therapeutic strategies. To comprehensively understand SARS-CoV-2 targets and their interactions within a broader context, we extended our analysis to encompass the first neighbors of these interactions within the human interactome (Figure 1E). This resulted in the establishment of a comprehensive Human-SARS-CoV-2 interactome, characterized by 20,700 nodes and 260,231 interactions (Figure 1E, Table S1). By doing so, we aimed to understand the connectivity and positioning of SARS-CoV-2 targets within the broader network, providing insights into their relationships and functional roles within the entire human interactome.
3.2. Diverse Centrality Measures to Reveal the Enrichment of SARS-CoV-2 Targets in Human Interactome
Centrality measures are crucial in network analysis, evaluating nodes’ significance by examining their connections [38]. Leveraging centrality measures enriches our understanding of network behavior, facilitating targeted interventions and revealing underlying dynamics across various biological systems. In network topology, structural centralities can generally be classified into three groups [39,40,41]: (i) Neighborhood-based centralities, such as degree centrality, coreness, and LocalRank, assess the influence of nodes by considering their relationships with neighboring nodes. (ii) Path-based centralities, including shortest path length, betweenness centrality, information centrality, closeness centrality, and Katz centrality, gauge node influence based on the distances between them within the network. (iii) Iterative refinement centralities, such as eigenvector centrality, PageRank, and LeaderRank, evaluate node influence by taking into account both the mutual interactions of node neighbors and their overall impact within the network [39,40]. We selected seven diverse centrality measures, representing the three distinct groups discussed above (Supplementary Figure 1), to discern the enrichment patterns of SARS-CoV-2 host targets. We employed a top 10% threshold to categorize nodes with high centrality across the seven topology measures. Load centrality emerged as the most effective metric for identifying SARS-CoV-2 host targets, exhibiting a prediction power exceeding 92%. Following closely behind were PageRank, bottlenecks, and hubs, with prediction powers of 90.3%, 89.9%, and 86.4%, respectively (Figure 2A, Table S2). Load centrality (Newman’s betweenness centrality) is a form of betweenness centrality. Betweenness and Load centrality differ in their approach to assessing node importance within a network. While standard betweenness centrality considers only shortest paths, Newman’s betweenness
Centrality incorporates all paths by employing a random walk method. In this method, weights are assigned based on path lengths, providing a more comprehensive evaluation of node centrality [42]. In a previous study, four different centrality measures (degree, betweenness, closeness and eigenvector) were employed on 332 SARS-CoV-2 host targets [43]. That study concluded that all centrality measures play a significant role in identifying crucial nodes within a network. As a result, median ranking scores were calculated, and the top 20 candidates were selected for functional similarity analysis. This analysis informed the construction of a drug-protein interaction network [43]. Similarly, in our previous study, network centrality analyses utilizing seven different centrality metrics identified 28 high-value SARS-CoV-2 targets. These targets are likely involved in crucial processes such as viral entry, proliferation, and survival, contributing to the establishment of infection and the progression of the disease [36]. In the present study, we identified 745 nodes that are common across seven different centrality measures. Additionally, we found 129 and 361 nodes that are common across six and five centrality measures.
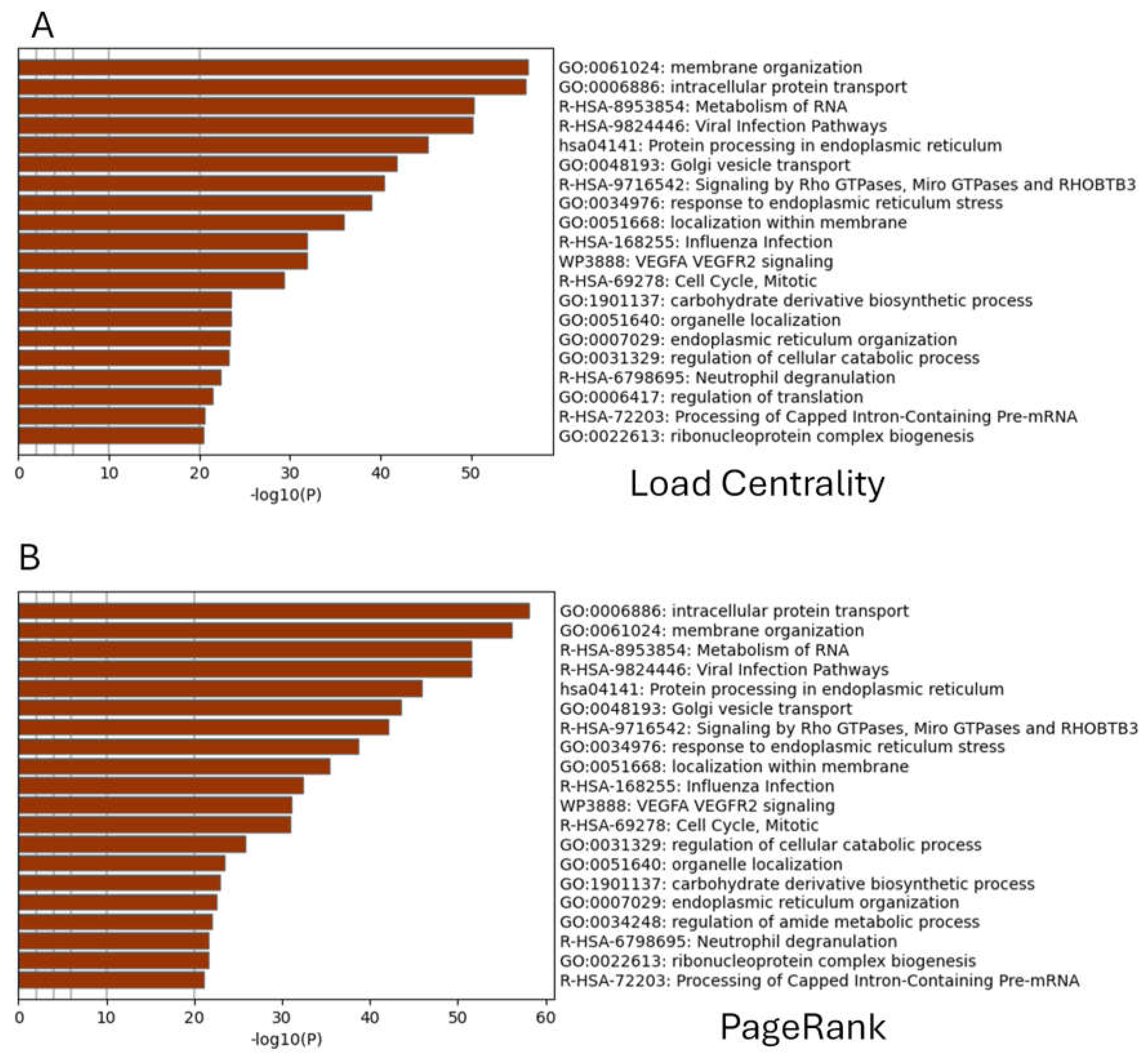
In a less densely connected network such as the Arabidopsis interactome, hubs and betweenness exhibited moderate prediction power for identifying pathogen targets, reaching up to 6.5% [15,16,17]. However, the application of weighted k-shell decomposition analyses significantly enhanced prediction power, expanding it to 40% [44,45]. Interestingly, when applying weighted k-shell decomposition to the denser human interactome, the prediction of SARS-CoV-2 targets did not exceed 40% (Figure 2B,C, Table S3). Membrane organization, protein transport, RNA metabolism, viral infection pathways, response to ER stress, cell cycle regulation, neutrophil degradation, and carbohydrate derivative biosynthesis pathways emerge as prominent biological pathways enriched across centrality measures such as Load centrality, PageRank, bottlenecks, and hubs (Figure 3A,B, Supplementary Figures S2 and S3). Taken together, we discovered that centrality measures, particularly Load centrality is an effective metric to identify SARS-CoV-2 host targets.
Figure 3.
Pathway analyses for host targets that overlaps with Load centrality (A) or PageRank (B). respectively. This indicates a robust convergence of nodes across multiple centrality metrics, highlighting their potential significance in the network and potential therapeutic targets.
Figure 3.
Pathway analyses for host targets that overlaps with Load centrality (A) or PageRank (B). respectively. This indicates a robust convergence of nodes across multiple centrality metrics, highlighting their potential significance in the network and potential therapeutic targets.
3.3. CentralityCosDist to Predict SARS-CoV-2 Targets
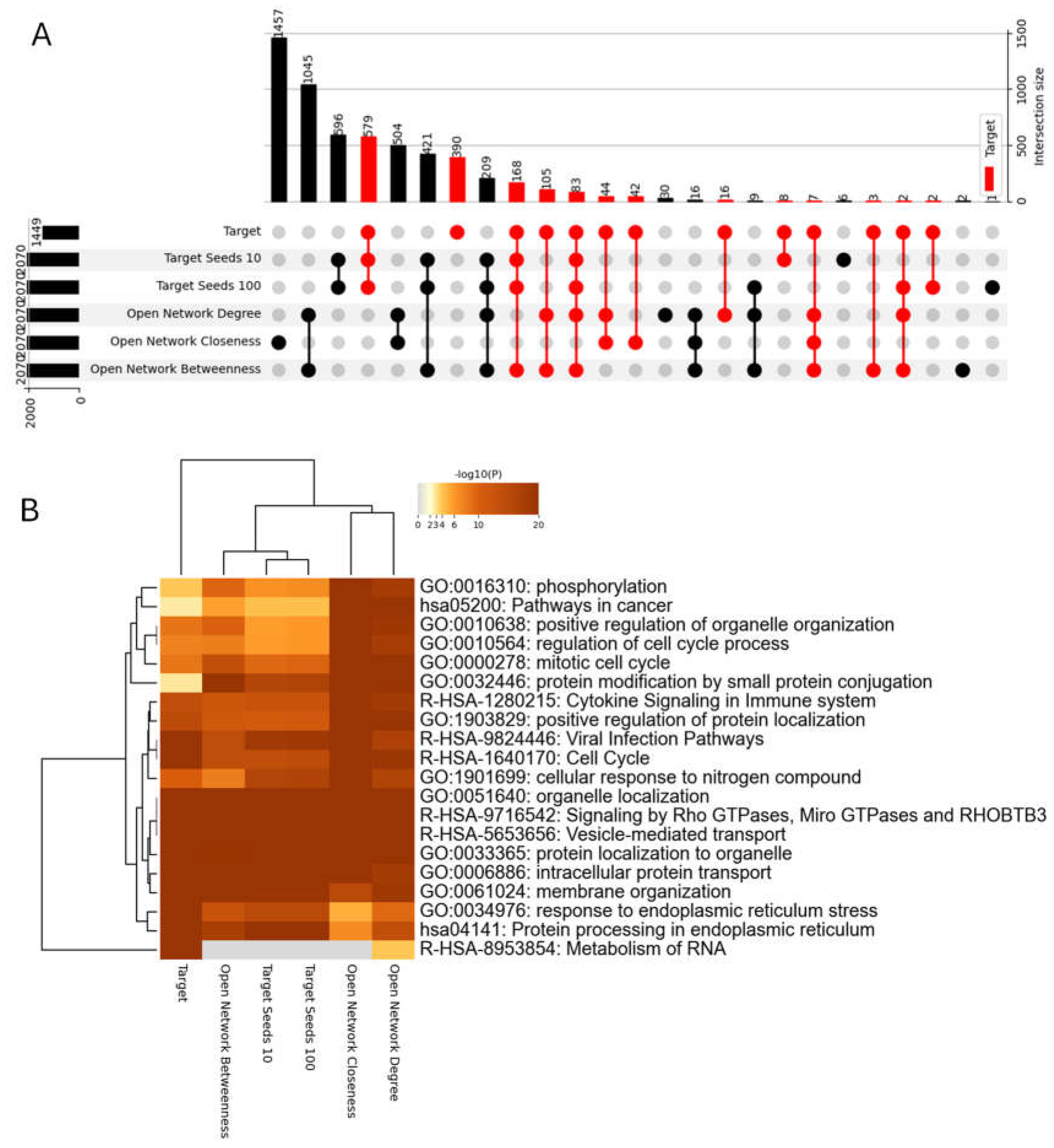
In our study, we addressed another question of how to expand the limited number of available SARS-CoV-2 targets, aiming to predict additional targets by leveraging network-based gene prioritization methods. The existing methods can generally be categorized into four groups: guilt by association, centrality measures, network propagation/random walk, and network clustering/communality analysis [38]. While each method has its strengths, they also come with their own limitations. To address these limitations, we introduced a novel approach called CentralityCosDist [38]. Briefly, this method starts with a set of seed nodes. In a given network, each node is represented as a 9-dimensional vector using 9 diverse centrality metrices. The cosine distances between the seed nodes are computed using these centrality vectors. By averaging the distances between the seed nodes and each of the other nodes, all nodes are ranked (Figure S4). This approach allow prioritize and rank the nodes based on their similarity to the seed nodes in the 9-dimensional vector space (Figure S4). We applied CentralityCosDist to a randomly selected a set of 144 SARS-CoV-2 targets (target seed 10 representing 10% of all known host targets). This was compared with target seed 100 representing what if we use all known host targets. Remarkably, this led us to predict a total of 838 targets spanning 2070 nodes, with a prediction power exceeding 57% (Figure 4A). This highlights the effectiveness of our approach in expanding the pool of predicted targets when a limited number of host targets are known. This offers valuable insights for further research into SARS-CoV-2 biology and potential therapeutic interventions. Pathway enrichment analyses for “target seed 10” revealed enrichment in several biological pathways. These pathways include organelle localization, Rho GTPase signaling, vesicle-mediated transport, response to ER stress, protein processing, and RNA metabolism, among others (Figure 4B). This comprehensive analysis provides valuable insights into the functional roles and potential mechanisms associated with “target seed 10,” shedding light on its involvement in various cellular processes.
4. Conclusions
The current study led to the construction of a comprehensive of the human protein-protein interactome and SARS-CoV-2 host targets. This expanded dataset serves as a valuable resource for future studies on other viruses and other biological questions. Additionally, we curated a comprehensive list of 1,449 SARS-CoV-2 host proteins and analyzed their interactions within the human interactome. Moreover, we employed seven diverse centrality measures in identifying crucial nodes within the network, revealing Load centrality as the most effective metric for predicting SARS-CoV-2 host targets. Our analysis highlighted the convergence of nodes across multiple centrality metrics, indicating their potential significance in the pathobiology of SARS-CoV-2. Additionally, we introduced a novel approach called CentralityCosDist to predict SARS-CoV-2 targets, which proved effective in expanding the pool of predicted targets. Pathway enrichment analyses further elucidated the functional roles and potential mechanisms associated with these predicted targets, providing valuable insights into their involvement in various cellular processes. Overall, our study offers a comprehensive understanding of SARS-CoV-2 host targets and their interactions within the human interactome. This may pave the way for future research into the pathogenesis of SARS-CoV-2 infection and the development of novel therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Centrality Metrics in Network Analysis; Figure S2: Pathway analyses for host targets that overlaps with Degree, Betweenness or Closeness Centrality. Figure S3: Pathway analyses for host targets that overlaps with Eigenvector, or Information Centrality. Figure S4: Schematics of CentralityCosDist pipeline. Table S1: Human PPIs and Seed sets. Table S2: Human PPIs (Open) and Seed sets. Table S3: Wk-Shell decomposition and CentratlityCosDist analysis results.
Author Contributions
N.K. and S.M. originally devised and refined the protocol. N.K. performed the initial analysis and data curation. S.M. and N.K. conceived the project and wrote the manuscript. S.M. supervised the analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NSF award IOS-2038872 to M.S.M.
Data Availability Statement
PPIs and SARS-CoV-2 host targets referenced in this study are available in Supplementary tables. Custom code used in this study is available from the corresponding author upon reasonable request.
Acknowledgments
The authors gratefully acknowledge the resources provided by the University of Alabama at Birmingham IT-Research Computing group for high-performance computing (HPC) support and CPU time on the Cheaha compute cluster.
Conflicts of Interest
“The authors declare no conflicts of interest.”.
References
- McCormack, M.E.; Lopez, J.A.; Crocker, T.H.; Mukhtar, M.S. Making the right connections: Network biology and plant immune system dynamics. Current Plant Biology 2016, 5, 2–12. [Google Scholar] [CrossRef]
- Pan, A.; Lahiri, C.; Rajendiran, A.; Shanmugham, B. Computational analysis of protein interaction networks for infectious diseases. Brief Bioinform 2016, 17, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, C.C.; Bangalore, P.V.; Kannar, P.; Mukhtar, M.S. Getting to the edge: Protein dynamical networks as a new frontier in plant-microbe interactions. Front Plant Sci 2014, 5, 312. [Google Scholar] [CrossRef]
- Majeed, A.; Mukhtar, S. Protein-Protein Interaction Network Exploration Using Cytoscape. Methods Mol Biol 2023, 2690, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Schopf, J.; Kogl, M.; Friedel, C.C.; Muller, M.A.; Carbajo-Lozoya, J.; Stellberger, T.; von Dall’Armi, E.; Herzog, P.; Kallies, S.; et al. The SARS-coronavirus-host interactome: Identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 2011, 7, e1002331. [Google Scholar] [CrossRef] [PubMed]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Gulbahce, N.; Yan, H.; Dricot, A.; Padi, M.; Byrdsong, D.; Franchi, R.; Lee, D.S.; Rozenblatt-Rosen, O.; Mar, J.C.; Calderwood, M.A.; et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput Biol 2012, 8, e1002531. [Google Scholar] [CrossRef]
- Abreu, A.L.; Souza, R.P.; Gimenes, F.; Consolaro, M.E. A review of methods for detect human Papillomavirus infection. Virol J 2012, 9, 262. [Google Scholar] [CrossRef]
- Calderwood, M.A.; Venkatesan, K.; Xing, L.; Chase, M.R.; Vazquez, A.; Holthaus, A.M.; Ewence, A.E.; Li, N.; Hirozane-Kishikawa, T.; Hill, D.E.; et al. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A 2007, 104, 7606–7611. [Google Scholar] [CrossRef]
- de Chassey, B.; Navratil, V.; Tafforeau, L.; Hiet, M.S.; Aublin-Gex, A.; Agaugue, S.; Meiffren, G.; Pradezynski, F.; Faria, B.F.; Chantier, T.; et al. Hepatitis C virus infection protein network. Mol Syst Biol 2008, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Roohvand, F.; Maillard, P.; Lavergne, J.P.; Boulant, S.; Walic, M.; Andreo, U.; Goueslain, L.; Helle, F.; Mallet, A.; McLauchlan, J.; et al. Initiation of hepatitis C virus infection requires the dynamic microtubule network: Role of the viral nucleocapsid protein. J Biol Chem 2009, 284, 13778–13791. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef]
- Simonis, N.; Rual, J.F.; Lemmens, I.; Boxus, M.; Hirozane-Kishikawa, T.; Gatot, J.S.; Dricot, A.; Hao, T.; Vertommen, D.; Legros, S.; et al. Host-pathogen interactome mapping for HTLV-1 and -2 retroviruses. Retrovirology 2012, 9, 26. [Google Scholar] [CrossRef]
- Arabidopsis Interactome Mapping, C. Evidence for network evolution in an Arabidopsis interactome map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, M.S.; Carvunis, A.R.; Dreze, M.; Epple, P.; Steinbrenner, J.; Moore, J.; Tasan, M.; Galli, M.; Hao, T.; Nishimura, M.T.; et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 2011, 333, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Wessling, R.; Epple, P.; Altmann, S.; He, Y.; Yang, L.; Henz, S.R.; McDonald, N.; Wiley, K.; Bader, K.C.; Glasser, C.; et al. Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 2014, 16, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Tang, K.; Tang, J.; Zeng, J.; Shen, W.; Zou, M.; Zhang, C.; Sun, Q.; Ye, X.; Li, C.; Sun, C.; et al. A network view of human immune system and virus-human interaction. Front Immunol 2022, 13, 997851. [Google Scholar] [CrossRef]
- Bosl, K.; Ianevski, A.; Than, T.T.; Andersen, P.I.; Kuivanen, S.; Teppor, M.; Zusinaite, E.; Dumpis, U.; Vitkauskiene, A.; Cox, R.J.; et al. Common Nodes of Virus-Host Interaction Revealed Through an Integrated Network Analysis. Front Immunol 2019, 10, 2186. [Google Scholar] [CrossRef]
- Kumar, N.; Mukhtar, M.S. Ranking Plant Network Nodes Based on Their Centrality Measures. Entropy 2023, 25, 676. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tang, Z.; Fan, W.; Wang, X.; Huang, L.; Jia, Y.; Wang, M.; Hu, Z.; Zhou, Y. Atlas of interactions between SARS-CoV-2 macromolecules and host proteins. Cell Insight 2023, 2, 100068. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Y.; Gupta, S.; Paramo, M.I.; Hou, Y.; Mao, C.; Luo, Y.; Judd, J.; Wierbowski, S.; Bertolotti, M.; et al. A comprehensive SARS-CoV-2-human protein-protein interactome reveals COVID-19 pathobiology and potential host therapeutic targets. Nat Biotechnol 2023, 41, 128–139. [Google Scholar] [CrossRef]
- Hu, Z.; Snitkin, E.S.; DeLisi, C. VisANT: An integrative framework for networks in systems biology. Brief Bioinform 2008, 9, 317–325. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature communications 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Luck, K.; Sheynkman, G.M.; Zhang, I.; Vidal, M. Proteome-Scale Human Interactomics. Trends Biochem Sci 2017, 42, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.P.; Ding, X.B.; Jin, J.; Zhang, H.B.; Yang, Q.L.; Chen, P.C.; Yao, H.; Ruan, L.I.; Tao, Y.T.; Chen, X. HIR V2: A human interactome resource for the biological interpretation of differentially expressed genes via gene set linkage analysis. Database (Oxford) 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mishra, B.; Mehmood, A.; Mohammad, A.; Mukhtar, M.S. Integrative Network Biology Framework Elucidates Molecular Mechanisms of SARS-CoV-2 Pathogenesis. iScience 2020, 23, 101526. [Google Scholar] [CrossRef]
- Das, J.K.; Roy, S.; Guzzi, P.H. Analyzing host-viral interactome of SARS-CoV-2 for identifying vulnerable host proteins during COVID-19 pathogenesis. Infect Genet Evol 2021, 93, 104921. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mukhtar, M.S. Ranking Plant Network Nodes Based on Their Centrality Measures. Entropy 2023, 25, 676. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Systems Biology and Machine Learning in Plant-Pathogen Interactions. Mol Plant Microbe Interact 2019, 32, 45–55. [Google Scholar] [CrossRef]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Network biology to uncover functional and structural properties of the plant immune system. Curr Opin Plant Biol 2021, 62, 102057. [Google Scholar] [CrossRef]
- Mishra, B.; Kumar, N.; Shahid Mukhtar, M. A rice protein interaction network reveals high centrality nodes and candidate pathogen effector targets. Comput Struct Biotechnol J 2022, 20, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E.J. A measure of betweenness centrality based on random walks. arXiv 2003. [Google Scholar] [CrossRef]
- Barman, R.K.; Mukhopadhyay, A.; Maulik, U.; Das, S. A network biology approach to identify crucial host targets for COVID-19. Methods 2022, 203, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Howton, T.C.; Sun, Y.; Weinberger, N.; Belkhadir, Y.; Mukhtar, M.S. Network biology discovers pathogen contact points in host protein-protein interactomes. Nature communications 2018, 9, 2312. [Google Scholar] [CrossRef] [PubMed]
- Smakowska-Luzan, E.; Mott, G.A.; Parys, K.; Stegmann, M.; Howton, T.C.; Layeghifard, M.; Neuhold, J.; Lehner, A.; Kong, J.; Grunwald, K.; et al. An extracellular network of Arabidopsis leucine-rich repeat receptor kinases. Nature 2018, 553, 342–346. [Google Scholar] [CrossRef]
Figure 1.
(A) Integration of Multiple Protein Interaction Networks to Identify a Consensus Target Cluster. The figure illustrates a funnel-like integration strategy where distinct protein interaction networks from various databases are combined. Each colored cluster represents a unique network. These networks converge to identify a consensus ‘Target’ set, denoted in red. (B) Details of number of nodes and interactions for particular network. BioPlex 55k, HuRI, STRING, HIR2 Ex, CUBIC, CoFrac, BioPlex 10k, and HIR2 P. (C) Schematic to display SARS-CoV-2 open-reading frames (ORFs) targeting Human Interactome. (D) Integrated or merged Human PPI (blue) network and Target proteins (red).
Figure 1.
(A) Integration of Multiple Protein Interaction Networks to Identify a Consensus Target Cluster. The figure illustrates a funnel-like integration strategy where distinct protein interaction networks from various databases are combined. Each colored cluster represents a unique network. These networks converge to identify a consensus ‘Target’ set, denoted in red. (B) Details of number of nodes and interactions for particular network. BioPlex 55k, HuRI, STRING, HIR2 Ex, CUBIC, CoFrac, BioPlex 10k, and HIR2 P. (C) Schematic to display SARS-CoV-2 open-reading frames (ORFs) targeting Human Interactome. (D) Integrated or merged Human PPI (blue) network and Target proteins (red).
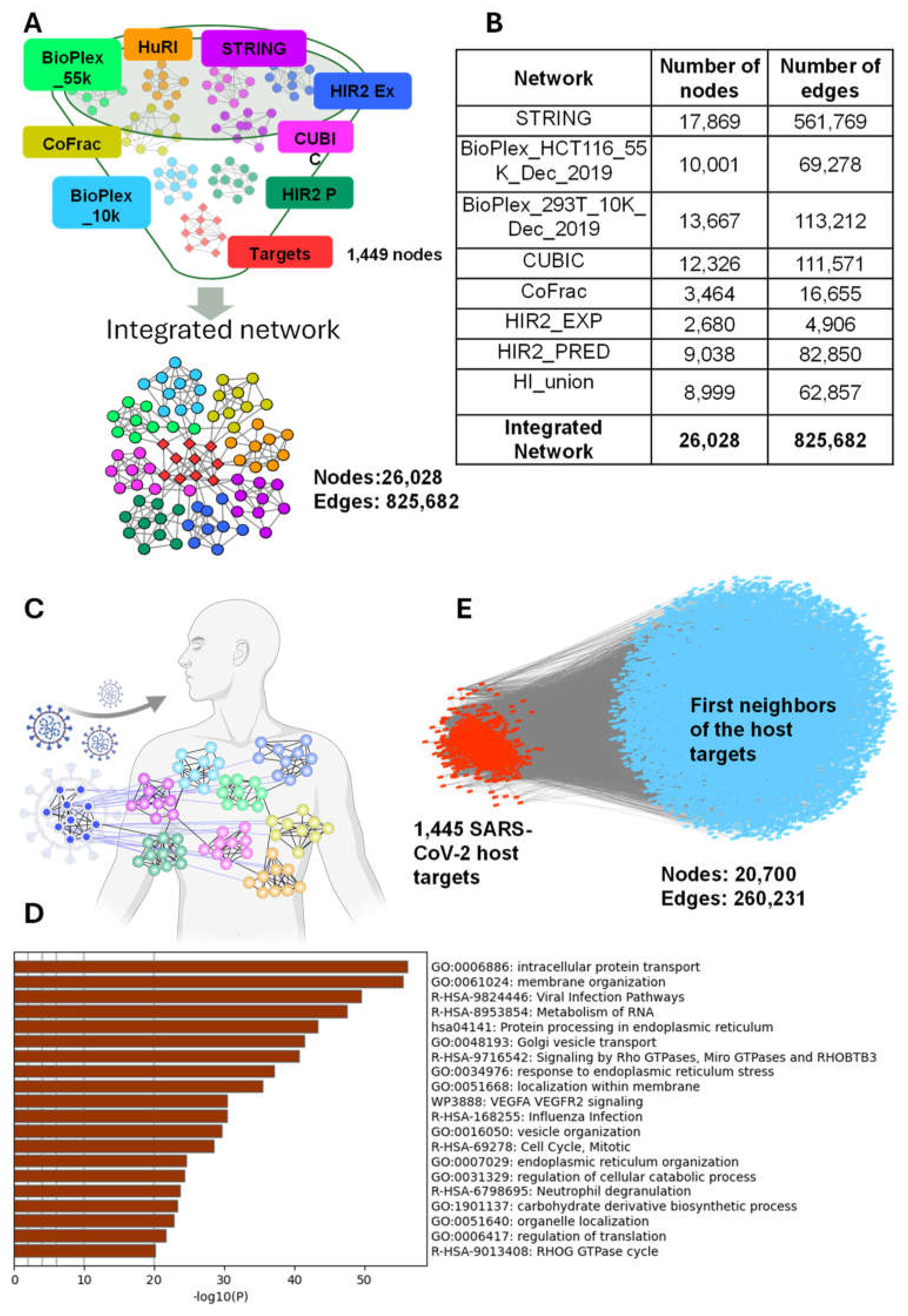
Figure 2.
(A): Centrality Measure and target protein Overlap in the Human-SARS-CoV-2 interactome. This heatmap visualizes the overlap of target protein nodes based on different centrality measures within an open network. The x-axis lists various centrality metrics such as Degree, Betweenness, Eigenvector, Closeness, Local, and PageRank Centrality. The y-axis represents the percentage of nodes within the network, ranging from the top 1% to 100%. Each cell contains a numerical value indicating the number of nodes that are target proteins across the different centrality measures, with the corresponding color gradient reflecting the degree of overlap—ranging from low (blue) to high (yellow). The high degree of overlap is indicated by the consistent appearance of higher values (indicated by yellower shades) at the top percent, particularly in the top 10%. (B) UpSet Plot of 10% Open Network Target Centrality Overlap. Overlap of top 10% (centrality ranked nodes) target protein overlapping nodes. (C) Weighted k-shell Wk-shell decomposition analysis of Network. The diagram illustrates a simple network composed of 18 nodes, each represented by a numbered circle or shell number (K1, K2, K3, and K4). The nodes are organized into a series of nested, concentric layers indicating different levels of modularity within the network. The innermost layer, highlighted in red and labeled as ‘K1’, comprises the central nodes (0-4) which form the core of the network, characterized by dense interconnectivity. The second layer, marked by a light violet shade and designated as ‘K2’, encapsulates nodes (5-7) that are closely associated with the core. The third layer, ‘K3’, shown in lighter blue, extends to include nodes (8-12 and 14), illustrating a further level of the network’s hierarchy. The outermost layer, ‘K4’, encompasses the peripheral nodes (13, 15, 16, 17), depicted in yellow, which are connected to the network but maintain fewer connections, signifying the outer tier of the modular structure. This stratified layout underscores the multi-level modularity in the network, suggesting potential functional or structural distinctions among the different groups of nodes. (D) UpSet Plot of Node Set Intersections in the Wk-Shell One thrid and one third nodes. The figure is an UpSet plot that visualizes the intersections of node sets within a specific network Wk-Shell and the target protein nodes. The plot illustrates the number of nodes shared among different subsets categorized as ‘Target,’ ‘One-third,’ and ‘Two third.’.
Figure 2.
(A): Centrality Measure and target protein Overlap in the Human-SARS-CoV-2 interactome. This heatmap visualizes the overlap of target protein nodes based on different centrality measures within an open network. The x-axis lists various centrality metrics such as Degree, Betweenness, Eigenvector, Closeness, Local, and PageRank Centrality. The y-axis represents the percentage of nodes within the network, ranging from the top 1% to 100%. Each cell contains a numerical value indicating the number of nodes that are target proteins across the different centrality measures, with the corresponding color gradient reflecting the degree of overlap—ranging from low (blue) to high (yellow). The high degree of overlap is indicated by the consistent appearance of higher values (indicated by yellower shades) at the top percent, particularly in the top 10%. (B) UpSet Plot of 10% Open Network Target Centrality Overlap. Overlap of top 10% (centrality ranked nodes) target protein overlapping nodes. (C) Weighted k-shell Wk-shell decomposition analysis of Network. The diagram illustrates a simple network composed of 18 nodes, each represented by a numbered circle or shell number (K1, K2, K3, and K4). The nodes are organized into a series of nested, concentric layers indicating different levels of modularity within the network. The innermost layer, highlighted in red and labeled as ‘K1’, comprises the central nodes (0-4) which form the core of the network, characterized by dense interconnectivity. The second layer, marked by a light violet shade and designated as ‘K2’, encapsulates nodes (5-7) that are closely associated with the core. The third layer, ‘K3’, shown in lighter blue, extends to include nodes (8-12 and 14), illustrating a further level of the network’s hierarchy. The outermost layer, ‘K4’, encompasses the peripheral nodes (13, 15, 16, 17), depicted in yellow, which are connected to the network but maintain fewer connections, signifying the outer tier of the modular structure. This stratified layout underscores the multi-level modularity in the network, suggesting potential functional or structural distinctions among the different groups of nodes. (D) UpSet Plot of Node Set Intersections in the Wk-Shell One thrid and one third nodes. The figure is an UpSet plot that visualizes the intersections of node sets within a specific network Wk-Shell and the target protein nodes. The plot illustrates the number of nodes shared among different subsets categorized as ‘Target,’ ‘One-third,’ and ‘Two third.’.
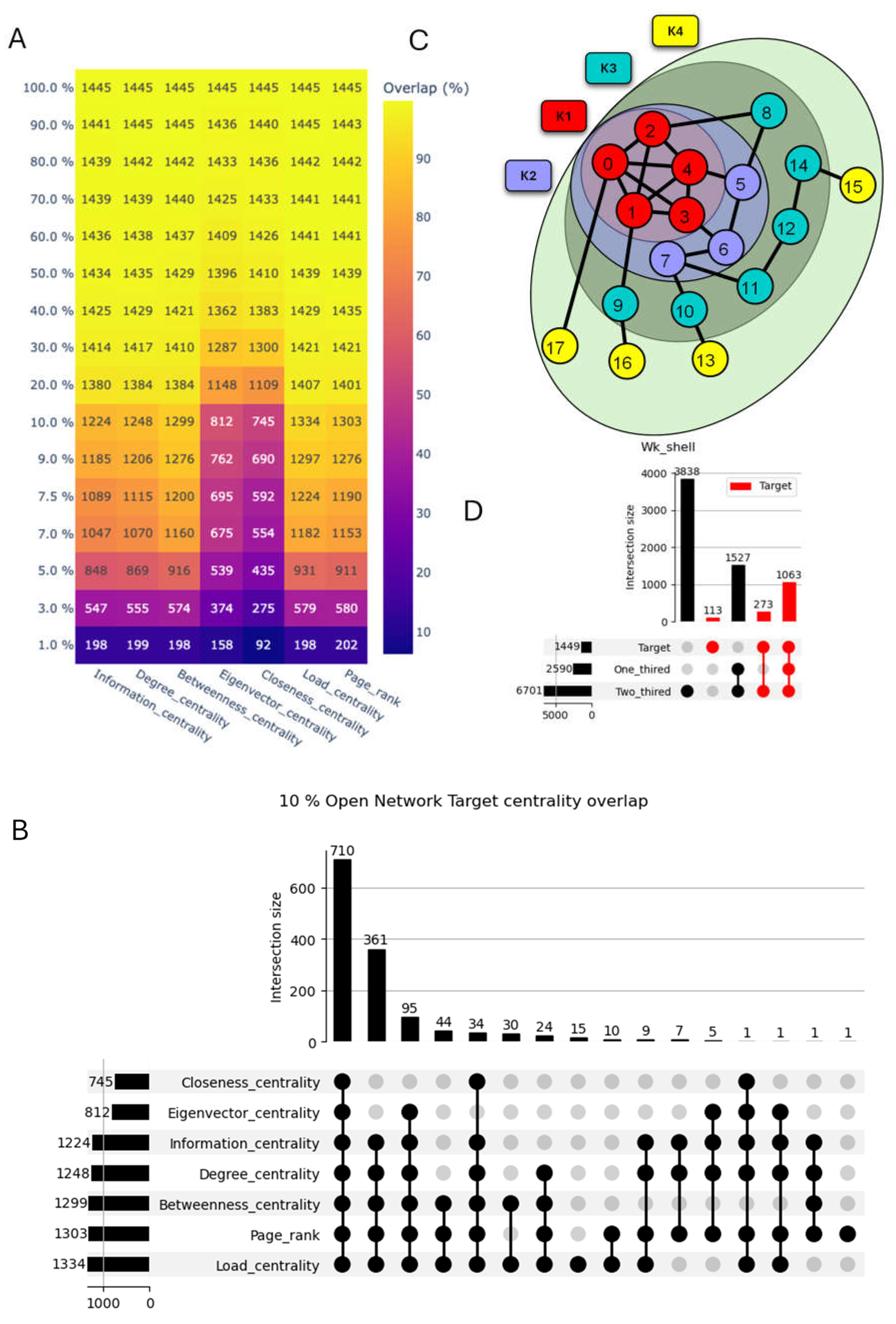
Figure 4.
(A) Comparative Analysis of CentalityCosDist Outputs with Various Seed sets. This figure presents an UpSet plot illustrating the intersections of top nodes identified by the CentalityCosDist tool across different sets of seed networks. The seeds for CentalityCosDist analysis vary, including ‘Open Network Betweenness,’ ‘Open Network Closeness,’ and ‘Open Network Degree,’ along with ‘Target Seeds 10,’ which represent the top 10% of seeds chosen at random, and ‘Target Seeds 100,’ denoting the full set of seeds amounting to 1449 nodes. The horizontal bars across the top of the plot indicate the number of top nodes (2070) identified in each CentalityCosDist run, consistent across all seed sets. The filled circles and connecting lines in the matrix reveal the overlapping results between different seed sets and the ‘Target,’ with red circles emphasizing where the ‘Target’ overlaps with other sets. Vertical bars on the right side illustrate the intersection sizes. The largest intersection is observed with the full set of seeds (‘Target Seeds 100’), while the intersections with ‘Target Seeds 10’ and centrality-based seed sets show a graded distribution of smaller sizes. This analysis demonstrates the consistency of the CentalityCosDist tool in identifying top nodes across various seed sets and particularly highlights the influence of different seed selection strategies on the outcome of the centrality distribution analysis. It offers a nuanced perspective on how the choice of seeds can affect the identification of key targets within the network. (B) Pathway analyses for host targets that are identified through CentralityCosDis.
Figure 4.
(A) Comparative Analysis of CentalityCosDist Outputs with Various Seed sets. This figure presents an UpSet plot illustrating the intersections of top nodes identified by the CentalityCosDist tool across different sets of seed networks. The seeds for CentalityCosDist analysis vary, including ‘Open Network Betweenness,’ ‘Open Network Closeness,’ and ‘Open Network Degree,’ along with ‘Target Seeds 10,’ which represent the top 10% of seeds chosen at random, and ‘Target Seeds 100,’ denoting the full set of seeds amounting to 1449 nodes. The horizontal bars across the top of the plot indicate the number of top nodes (2070) identified in each CentalityCosDist run, consistent across all seed sets. The filled circles and connecting lines in the matrix reveal the overlapping results between different seed sets and the ‘Target,’ with red circles emphasizing where the ‘Target’ overlaps with other sets. Vertical bars on the right side illustrate the intersection sizes. The largest intersection is observed with the full set of seeds (‘Target Seeds 100’), while the intersections with ‘Target Seeds 10’ and centrality-based seed sets show a graded distribution of smaller sizes. This analysis demonstrates the consistency of the CentalityCosDist tool in identifying top nodes across various seed sets and particularly highlights the influence of different seed selection strategies on the outcome of the centrality distribution analysis. It offers a nuanced perspective on how the choice of seeds can affect the identification of key targets within the network. (B) Pathway analyses for host targets that are identified through CentralityCosDis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



