
Submitted:
01 May 2024
Posted:
03 May 2024
You are already at the latest version
Abstract
Monodisperse and semi-facetted ultra-small templated mesoporous silica nanoparticles (US-MSN) of 20-25 nm were synthesized using a short-time hydrolysis of tetraethoxysilane (TEOS) at room temperature followed by a dilution for nucleation quenching. According to dynamic light scattering (DLS), a two-step pH adjustment was necessary for growth termination and colloidal stabilization. The pore size was controlled by cetyltrimethylammonium bromide (CTAB) and the tiny amount of neutral surfactant F127 was added for minimizing the coalescence between US-MSN and favoring the transition towards internal ordering. Flocculation eventually occured allowing to harvest a powder by centrifugation ( 60% silica yield after one month). The scanning transmission electron microscopy (STEM) and 3D high-resolution transmission electron microscopy (3D-HR-TEM) images revealed that the US-MSNs are partially ordered. The 2D FT transmorm images evidences the coexistance of four, five and sixfold patterns characterizing an “on-the-edge” crystallization step between amorphous raspberry and hexagonal pore array morphologies, typical of a protocrystalline state. Calcination preserved this state and yields a powder characterized by a packing developping a hierarchical porosity centered at 3.9 ± 0.2 (internal pores) and 68 ± 7 nm (packing voids) of high potential as support for separation and catalysis.
Keywords:
mesoporous silica
; nanoparticles
; cristalinity
; synthesis
; monodispersion
; porosity
1. Introduction
The quest for size control of porous particles in the nanometric scale range below 100 nm is a very active research field. As stable colloidal suspensions, they provide an alternative to liquid crystalline lipid or polymeric nano carriers for magnetic or optical labeling and vectorization useful for imaging and drug delivery in medical diagnosis and therapy.[1,2,3,4,5]These nanosized objects can be also applied as building units to create hierarchically structured porosity so as to minimize internal molecular diffusion for the design of adsorbents and catalysts. This is useful in many different fields such as industrial production of chemicals and pollution control. In both cases, well-defined physical properties such as monodispersion and size calibration are key points for a better diffusion control in living systems and for an optimal pharmacological use. Sizes smaller than 100 nm are required to avoid capillary plugging in living systems (embolism). Most of the inorganic nanocargos developed up to now using inorganic matrices are mainly designed from colloidal suspensions of non-porous gold nanoparticles functionalized on their external surface; they are already in the ultra small size range below 20 nm.[6,7] Silica-based nanocargos are also developed owing to their well-defined internal porosity in the micropore (< 2 nm, zeolite) or in the mesopore (2 to 50 nm, micelle templated material) ranges. Despite their interest for molecular retention and controlled diffusivity, there are very scarce examples combining fine tunings of both porosity and grain size at such a very small scale of length.[1,8,9] The first article on mesoporous nanoparticles was published in 2001 reporting polydispersed mesostructured silicas (20 to 100 nm), with regular array of channels observed only for particles larger than 50-60 nm.[10] The synthesis was inspired from a recipe of bulk periodic mesoporous silica (PMS) of MCM-41 or MCM-48 types using a cationic surfactant cetyltrimethylammonium as pore structuring agent. [10,11,12] Up to now, mesoporous silica nanoparticles with well defined cubic or hexagonal mesophases are reported for sizes larger than 70 nm.[13] These systems exhibit attractive properties such as high pore volume, large specific surface area and surfaces easy to functionalize from their silanol groups.[14,15] To keep the size as small as possible, the first parameter to take into account is the supersaturation that determines the number of nuclei from which growth takes place. Accordingly, nucleation quenching using dilution was the first approached adopted.[10,11,12]Chemical quenching using functionalized organosilanes was also applied since it blocks the polycondensation and also depending on the organic function (thiol or amine), controls the surface charges stabilizing small sizes and yielding different morphologies.[11,16,17,18,19] Furthermore, stabilization of nanoparticles in the range of 25 - 200 nm using ammonia, ethanol and triethanolamine (TEA) additives are also reported.[16,20,21] In particular, TEA is claimed to act both as a base and as a chelating agent for the oligomeric silica precursors yielding nanosized particles (20 - 40 nm) that spontaneously merge one with another into hierarchical porous silica materials.[16] Using the same additive, Bein et al. successfully stabilized colloidal solutions of well controlled size and shape nanoparticles in high yield and, notably without the need for dilution.[21] However, these spherical nanoparticles exhibit little ordering and weak X-ray diffraction (XRD) diffraction.[16,22,23] This is due to a radial array of channels, which appears suitable for drug delivery since it allows size control even in the presence of organosilanes.[16,17,18,19,24] The resistance of such radial structured mesoporous nanoparticles towards dilution in water or physiological media remains limited to few days, which may present an advantage in some medical applications like diagnosis by imaging or short time drug delivery control.[25] Fine pore structure tuning from radial arrays to raspberry and worm-like shapes is obtained using polyolamine at the appropriate concentrations, though no neat facetting nor periodic order could be observed.[26] In contrast, Imai et al. showed that the addition of the neutral template F127 to cetyltrimethylammonium bromide (CTAB) leads to well-faceted nanoparticles which exhibit a somewhat large distribution of sizes and some shape diversity.[27,28] The nonionic triblock copolymer F127 is considered here as a growth suppressant. The inutility of dilution and pH controls are consistent with the chemical growth control mode of action of both additives. There is one example of monodispersion attested below 50 nm combining initial high dilution with adddition of TEA.[29] Despite a much lower quantity of TEA compared with the earlier reports (0.25 instead of 1 mol eq. vs TEOS), these particles still exhibit a very low degree of ordering as reported in all the other contributions utilizing the TEA additive.[21,22,23] Therefore, the actual size threshold for well structured mesoporous particles is still represented by the transitional ovoid particles of 50 nm depicted first by Mann et al. in 2002 and can be proposed as the upper limit for ultra-small nanosized mesoporous objects.[12] To our knowledge, scarce examples of structured ultrasmall mesoporous silica nanoparticles (US-MSN) have been observed in polydisperse systems and only one claim of monodispersion with no details of the synthetic process nor detailed investigation of their structural state.[30]
Herein we address both issues, showing that the nucleation and growth quenching sequences using dilution and pH control in the presence of a tiny amount of nonionic surfactant lead to monodisperse US-MSN quenched in “on-the-edge” crystallization state. This is documented thanks to a thorough 3D HR-TEM investigation using 2D-image FT transform analysis. Their surprinzingly high stability under the beam and calcination prompted us to investigate the porosity and surface area of interest for catalysis and separation applications.
2. Materials and Methods
2.1. Synthesis of the Nanoparticles
Synthesis of MSN-23-A: The molar ratio of the synthesis solution was 1 SiO2 : 0.5 NaOH : 0.12 CTAB : 5.2 10-5 F127 : 130 H2O. CTAB (0.2 g, 5.5 10-4 mol) and F127 (0.003 g, 2.4 10-7 mol) were dissolved in a solution of NaOH (1 M, 2.3 g) and distilled H2O (8.78 g). After a short time (40 s), TEOS (1 ml) was added. Dilution by 4, 6 or 8 times were then performed by adding 50, 75 or 100 ml of distilled water, respectively. Four minutes later, the pH was adjusted to ca. 5.5 using about 1.1 ml of 2M HCl solution. Finally, 3 hours later, NaOH was added to increase the pH up to ca. 7. The stirring was stopped after 1 h. The suspension was then kept at RT statically, and found stable for more than half a year. A solid was obtained by centrifugation, washing and re-dispersing in distilled water under ultrasonic treatment for several times. Surfactants were removed by calcination under air flow while the temperature was increased from 25 to 550 °C for 6 h and then kept at 550 °C for another 6 h. Elemental analysis. MSN-23-A (wt %): C, 47.15; N, 2.89; H, 9.30; residual mass at 1000 °C, 63.
Syntheses of MSN-100-B and MSN-100-A : MSN-100-B was synthesized as described elsewhere and is characterized as nanoparticles with different shapes and an average size of 100 nm (TEM image in Figure S8). [27,28] MSN-100-A was obtained from MSN-100-B upon treatment at pH 5.5 for 3 hours followed by neutralization according to the same procedure as the two last steps of the synthesis of MSN-23-A. Elemental analysis. MSN-100-B (wt %): C, 31.75; N, 2.43; residual mass at 1000 °C from TGA, 33.1 ; MSN-100-A (wt %): C, 26.88; N, 1.53; residual mass at 1000 °C, 44.2.
2.2. Transmission Electron Microscopy
A drop of diluted MSN colloidal solution was deposited on a carbon grid and dried at room temperature. The high-angle annular dark-field (HAADF)-STEM, also known as Z-contrast, images was taken at room temperature using a JEOL JEM-2100F field-emission microscope equipped with a JEOL ADF detector. The microscope was operated at 200 kV. The probe size and camera length are 0.45 nm and 8 cm, respectively. The regular HR-TEM images were obtained on a JEOL 2010. TEM tilting experiments were carried out, using also a JEOL 2010 microscope operated at 200 kV and equipped with a home-made sample holder designed for tilting on a large angular range comprised between -20 and +20°.[31]
2.3. Images Analysis
Filtered images and diffraction patterns were obtained from direct and inverse 2D Fourier transformed combined with ad hoc gray threshold filtering using IMAGEJ ™ free software led to presented here. Diffraction pattern by 2D FT of images needed contrast enhancement and eventually contrast inversion for a better observation. In addition, rings at ca. 0.9, 1.8 nm-1 and above in the reciprocal space (diffraction image obtained by FT) are due to optical artifacts (picture grain). Gray threshold filtering allows to remove these rings then, FT filtered pictures were obtained.
2.3. DLS, XR, TGA and Nitrogen Isotherms
DLS measurements were performed at room temperature using a Cordouan DLS135 instrument and applied on the synthetic solution either as it is, or after dilution and ultrasonication to reach the size of non-aggregated particles.9 XRD was carried out on both as-synthesized and calcined samples with a Bruker (D8 Advance) diffractometer using Cu Kα monochromatic radiation. TGA was performed on the as-synthesized sample with a Netzsch STA 409 PC/PG instrument under air flow (30 ml min-1) with a temperature increase rate of 10 °C min-1. Nitrogen sorption isotherms were measured at 77 K on calcined samples with a Belsorp-max. A pre-treatment at 350°C under vacuum was performed before sorption measurement
3. Results and Discussion
3.1. Time Controlled Synthesis Approach
The synthesis protocol of the US-MSN is schematized in Figure 1, where timing, addition of chemicals, pH values together with the reaction steps are provided. Note the additional treatment for 3 h at pH 5.5 and the presence of F127 at the beginning of the synthesis that are the novel features in comparison to Mann et al. reports [10,11,12] TEOS was added to an aqueous solution (pH~13) containing CTAB and a tiny amount of triblock copolymer Pluronic F127, i. e., 1.5 (wt) % with CTAB/F127 molar ratio of 2,300. The above reaction mixture was stirred at room temperature for 40 s. Then, a rapid aqueous dilution by 6 times led to a pH drop down to 11.5. After 4 minutes a pH adjustment down to ~ 5.5 was performed using HCl followed after 3 hours by neutralization using NaOH and equilibration at pH 7 for another hour under stirring. The final colloidal solution was stocked as such at room temperature and found stable for at least 6 months. The as-synthesized mesoporous silica nanoparticles (23 ± 3 nm, Figure 1b, vide infra) using this acidic treatment before neutralization are referred as to MSN-23-A. A white powder obtained by centrifugation (11,000 rpm) was washed 3 times with distilled water and eventually calcined under air flow at 550°C to remove the organic surfactants (see experimental section). For comparison, another set of nano particles were prepared according to the acidic route (pH 0.5, instead of basic) and dual templating technic reported by Imai et al. that uses no quenching sequence and yields average particle size of 100 nm: the sample was referred to as MSN-100-B.[27,28] In the latter case, the TEOS concentration was close to that used in the synthesis of MSN-23-A, for about five times more CTA+ and fifty times more F127.
3.2. Structure and Morphology
High resolution transmission electron microscopy (HR-TEM) and scanning electron microscopy (SEM) images showed that both as-synthesized and calcined MSN-23-A samples contained nanoparticles with narrow size distribution in the range 18 to 25 nm that were rather faceted (Figure 2 and S1, respectively). XRD diagrams exhibited a strong peak at a d spacing of 4.9 ± 0.1 nm, which was much more intense than all previous reports concerning nanoparticles in the size range 20 to 50 nm (see introduction). There was also a distinguishable shoulder on the high angle side that covered the range where secondary peaks were expected for hexagonal or cubic MCM-41 or MCM-48 types of structure (Figure 2a). Unfortunately, the peaks were too broad to be resolved due to the very small size of the ordered domains or to their faulted nature. A close examination of their shapes revealed that the angles between facets ranged from 100 to 150°. Despite a large number of US-MSN observed for each sample (> 200), rectangular or square shapes were too scarce or too difficult to be observed and the typical 2D hexagonal array of channels was not observed discarding regular 2D hexagonal or cubic arrays. Although such a structure is clearly observed for nanoparticles or fibers as small as 60 to 100 nm, [10-13,32] further investigations are necessary to clarify this point. Indeed, distorted channels with various angles of emergence out of the particles could blur the observation. Alternatively, preferential orientation of the particles on the TEM grid could complicate the observations.
To further document this point, diffraction patterns and FT filtered images of the calcined samples were analyzed thoroughly. Indeed, the channels being empty, better-contrasted images of internal structure were obtained. For instance Figure 3 depicts a cluster of 20 to 30 nm particles with periodically contrasted bodies consistent with internal pores of nearly the same sizes of about 3 nm large. The three selected areas (yellow frames) exhibit also complex diffraction patterns dominated by dots forming rings, consistent with a mixture of monodomains (blue arrows point some of the most contrasted dots). This evidences that the neighboring particles were not sharing the same direction of ordering. In the center of the cluster in Figure 3a, the ordered domains seemed more extended but still belonging to two different hexagonal crystals, the orientation of which differed one to another by ca. 10°. A grey threshold filtering on these diffraction patterns afforded a simplified picture that still kept the the most important features and led to the same conclusion, i. e., the presence of two different hexagonal domains tilted by 10° (Figure 3b). The hexagonal diffracting patterns exhibited dots at reciprocal distances of 0.258 ± 0.004 nm-1, i.e., 3.9 nm accounting for a0 = 4.5 nm. This was apparently at odd with the XRD peak at 4.9 nm, which would correspond to a channel-to-channel distance of 4.9 or 5.7 nm for a cubic array or a hexagonal one. Alternatively, when enough pores were aligned one with another, an intensity profile analysis of their TEM images exhibited periodicities of 5.3 ± 0.1 nm just in between distances calculated assuming simple cubic or hexagonal arrays. These apparent discrepancies were assigned to a mixture of symmetries and/or an absence of medium range order inside the nanoparticles. Besides, there are obvious defects in all the areas selected. In particular, the particle lying at the bottom of Figure 3 is characterized by a step defect crossing at 45° through its structure. A survey through other regions confirmed that pure hexagonal array was not the dominating feature.
To get deeper insights on the intimate ordering of the particles and eventually to address the question on preferential orientation or unusual symmetry, HR-TEM images of particles were taken at different incident angles by tilting the sample under the beam from -20 to +20° (Figure 4). At first sight, there was no obvious apparition of parallel channels while most of the particles looked roughly alike consistent with no preferential orientation. Another advantage of tilting was the change of the contrast between pore walls and pore voids. This was clearly seen on particle A, D and E. By contrast, particles B and C as well as F and G were superimposed one onto another generating a moiré pattern preventing any simple interpretation in terms of structure. The white spots in particles A, D and E corresponding to pores aligned with the direction of observation appeared with different level of contrast. Their diameters appeared also smaller than the diameter measured from the N2 adorsorption-desorption curves (1 to 2 nm instead of 3.9 nm, vide infra). Channel diameter seen smaller than it should is usually attributed channel curvature in bulky MCM41.[33,34] Indeed, short range curvature characterized on large ovoid MCM-41 nanoparticles (> 50 nm) could be the rationale though it was inconsistent with the presence of facets and an intense XRD peak.[12] A radial array of channels generated in the presence of TEA molecules was also discarded for the same reasons.[16,22,23] This suggested that the pores are connected one to another through restricted apertures.
Furthermore, Figure 4 showed that the cluster of particles at different tilted angles generated a diffraction pattern consistent with different domains ill-oriented one with another. Therefore, each particle was investigated individually by 2D FT analysis using the region of the image delimited by yellow frames on the FT filtered image. Individual diffraction patterns were obtained for each portion of image included in the frame. At the top left, particle A exhibited the most facetted aspect among US-MSN of this cluster and has a diffraction pattern depending on the tilt angle : ill defined at -20 and 0°, orthorhombic at -15 and +10°, close to hexagonal at -10°, and 1D-ordered (with two opposite diffraction points) at +20°. This is typical of a 3D ordering with some diffraction images that are much better orientated than others. On the top left of the 2D FT filtered image taken at +10°, there was a definitive cubic array with a step defect on the right side. The rationale is a mixture of faulted hexagonal and cubic symmetries in the same nanoparticle. At the bottom center particle E exhibited a diffraction pattern with an overall four fold symmetry axis at -20° tilt and a rather sixfold symmetry damonating at 0° tilts. Strickingly at -20° particle E, which had the least facetted appearance, possessed a circular array of pores on the right side and a faulted hexagonal symmetry on the left side. There was also an obvious fivefold array lying in the center of the particle. At a tilt of -15° the diffraction seemed based on a hexagonal pattern ackwardly splitted into a four leaf clover pattern and a top side with four faces of half of an octagon. Particle D on the bottom right of each image exhibited a 90° clockwise rotated muffin like shape at -20°, the base of which had the form of half of a hexagon. The corresponding diffraction pattern is definitively showing an order though complicated to assign. Both particle F and G presented more faulted structures than other particles. On particle C that emerged between particles A and B, parallel diffracting lines appeared at a tilt of +20° (center right image, Figure 4). This was a rare example that could account for a tranversal view of a 1D hexagonal array of channels expected in a MCM-41 mesophase. A large survey showed that the particles of Figure 4 were more representative by far than those of Figure 3. Therefore, particles characterized by well-defined short-range order and ill-defined medium range order were the dominant features (Figure 4) while “good looking” particles with a quasi-hexagonal close packing array represented rather rare instances (Figure 3). All these particles were prefiguring cubic or hexagonal arrays of pores but did not yet arrived to this level of ordering.
As a complement to TEM, the dynamic light scattering (DLS) study provided the hydrodynamic diameters of the particles or their aggregates moving coherently with some solvent and surfactant molecules.[9] In the synthetic solution, the hydrodynamic diameters was distributed between 24 to 41 nm accounting for both isolated and aggregated nanoparticles (Figure S3a). In fact, after dilution and ultrasonication treatment, the flocculates were dismantled giving rise to mondispersed particles characterized as a single DLS peak at 18 ± 2 nm. This confirm the very narrow distribution of sizes centered at a size slightly smaller than those characterized by HR-TEM (23 ± 3 nm) (Figure 2c). Aggregates were indeed seen in TEM images as superimposition of small primary particles. These fragile aggregates or nanoflocculates were themselves small enough to yield stable colloidal solutions at room temperature (RT) for several months. After more than half a year at pH ~ 6.5 to 7, the average size of the flocculates increased slightly yielding a distribution ranging from 37 to 70 nm (Figure S3b) though the primary particle sze was unchanged according to HR-TEM observation. This would correspond to ca. 3 to 12 or 8 to 60 US-MSNs per flocculate in fresh solution or in 3 month aged solution, respectively. This slow flocculation allowed harvesting more particles by centrifugation after longer period of time. For instance, the silica yield were close to few percents from fresh solution and ca. 60 % after one month. Note that both CTA+ and silica yields were identical as CTA+/SiO2 molar ratio is the same in US-MSN than in the synthesis solution (see experimentals).
3.3. structural and Textural Porosity
The nitrogen adsorption-desorption profile of the calcined ultra small nanoparticles MSN-23-A was compared to those of MSN-100-B and bulk MCM-41 (Figure 5). Both types of nanoparticles presented a steep step in the reduced pressure range P/P0 = 0.27 - 0.34 that is typical of the capillary condensation in a templated internal mesoporous network of diameter 3.9 nm for MSN-23-A and bulk MCM-41 and 3.5 nm for MSN-100-B according to the BdB method (DBDB).[35,36] Note that the size distribution in MSN-23-A was ± 0.2 nm and was twice larger than MSN-100-B and bulk MCM-41.[13,14,27,28,35,36,37].
The height of the adsorption plateau taken before a second hysteresis loop at P/P0 = 0.8 was similar for MSN-100-B and bulk MCM-41 and stood for internal pore volumes of 0.83 and 0.88 ± 0.02 cm3 g-1 while it was smaller for MSN-23-A, 0.63 ± 0.02 cm3 g-1. The pore wall thickness calculated from XRD and pore sizes was similar for MSN-100-B and bulk MCM-41, i. e., 0.9 and 1.0 ± 0.1 nm, respectively and much larger for MSN-23-A., 1.8 ± 0.2 nm assuming a hexagonal array. Note that TEM contrast profile analysis provide a smaller estimate of 1.4 ± 0.2 nm for the latter.
Assuming the same pore wall density as for bulk MCM-41 and considering the pore volume of the latter as reference, the internal pore volume would vary as the ratio of pore diameter over pore wall thickness, i. e., 0.49 (from XRD and adsorption) or 0.63 (wall thickness from TEM contrast analysis) and 0.88 cm3/g for MSN-23-A and MSN-100-B, respectively. Obviously, the best wall thickness estimate of 1.4 nm came from TEM. This smaller estimate was still 40% larger than in MSN-100-B and in bulk MCM-41 materials. Indeed, the pore walls of MCM-41 are known to contain asperities large enough to fit argon atoms as most likely does MSN-100-B.[38] Accordingly, MSN-23-A might possess less or smaller asperities than than the other materials.
The vertical hysteresis loops at high relative pressure seen only for nanoparticles were assigned to interparticle void (Figure 5, insert). Indeed, the absence of such a loop in bulk MCM-41 indicated that the voids between grains or fibers were too large (> 200 nm) for N2 adsorption-desorption measurements. In MSN-100-B and in MSN-23-A, their sizes and volumes calculated on the adsorption branch were 38 and 68 nm for 0.94 ± 0.01 and 1.41 ± 0.02 cm3 g-1, respectively. In the former, the voids were smaller than the particle size indicating a rather efficient packing density. In contrast, MSN-23-A exhibited larger voids sizes than the particle size consistent with a rather low packing density for an extremely large unoccupied volume. Narrow void size distribution and high volumes of about 1 cm3g-1 are also reported by some of us in packed monodispersed 90 nm zeolite.[9]
3.4. Thermal and Hydrothermal Stability
Like for bulk MCM-41, the peak intensity of the XRD diffraction pattern was twice more intense with no noticeable broadening consistent with a full retention of structure and a mere replacement of the surfactant by air and some adsorbed water coming from ambient humidity (Figure 2a).[37] Moreover, the absence of shift of the main XRD peak after calcination indicated that MSN-23-A did not undergo any shrinkage up to 550°C in air, contrary to most of the reported bulk materials.[14,15] The US-MSNs that were stable for several months in water in the presence of surfactant were also subjected to stability tests in distilled water overnight after the removal of the surfactant by calcination. MSN-23-A showed a remarkable stability towards shrinkage (no XRD peak shift) and little lost of ordering (little intensity loss for same peak width, Figure S2). By contrast, calcined MSN-100-B exhibited a significant shrinkage and a drastic lost of ordering (intensity loss and shift towards high diffraction angles). This was a strong indication that the polycondensation of the siliceous wall was terminated at room temperature and led to a highly stable structure. This corroborated the above hypothesis for thicker and/or denser pore wall.
3.5. Surfactant to Silica Balance in US-MSN
The amount of surfactant retained in the as-synthesized nanoparticles was estimated from TGA (Figure S2). Below 150 °C, the mass loss was due to water desorption (2 to 3 % of the total mass loss). Above 150 °C, the mass loss was assigned to the surfactant oxidation.[39] In the latter range of temperatures, a much larger mas loss was measured for MSN-100-B (67 ± 2 %) than that for MSN-23-A (37 ± 1 %) or for bulk MCM-41 (40 ± 1 wt%, accounting in the latter case for ca. 0.17 surfactant/Si molar ratio).[14,36] Taking bulk MCM-41 as a reference and assuming an equivalent pore filling factor by the surfactant in all three materials, the mass losses should be proportional to internal pore volumes. Accordingly from N2 physisorption data, these volumes would be ca. 35 wt% in MSN-23-A and only 42 wt% in MSN-100-B, accounting for 0.12 and 0.16 surfactant/Si mol ratio, respectively. The 25 wt% excess of mass loss in MSN-100-B might be due to the presence of the much higher loading in secondary surfactant F127 (70 wt% in MSN-100-B and 1.5 wt% in MSN-23-A).[22,27,40] However, this was at odd in both solids with the elemental analysis revealing a C/N ratio very close to 19, which mostly characterizes CTA+ cations discarding the presence of F127 in the nanoparticles after washing the powder by distilled water. Then, it would be tempting to assign the strong surfactant retention to the smaller size of the interparticle voids in MSN-100-B (38 nm) than in MSN-23-A (68 nm).[41] However, the synthesis of MSN-100-B ended in a very acidic media apparently favorable for a high cationic surfactant retention since washing with water at pH 5.5 -7 eliminated this excess of surfactant (Figure S2). Then, nanoparticles of MSN-100-B and bulk MCM-41 were alike concerning internal pore filling with surfactant while MSN-23-A contained less surfactant molecules in agreement with thicker or denser porewall.
3.6. synthesis Parameters Versus Particle Size, Morphology and Colloidal Stability
The best results were obtained with Si:CTA+:F127 molar ratio of 19200: 2300:1 used for MSN-23-A. The nanoparticles exhibited a positive zeta potential evolving with time from +24 to +32 mV at pH 5.5, which was not high enough to avoid flocculation for long time storage at such pH (Table S1). Thus, an adjustment to pH 7 was necessary and after 3 hours, during which the zeta potential increased progressively from +42 to +59 mV leading to colloidal solution stable for several months (Figure S3). Positive potentials were consistent with a double layer of cationic surfactant covering the negatively charged silica surface.[29] The increase of potential after neutralization by the addition of NaOH could be related to Na+ adsorption. However, this element was not detected by EDX. The rationale was rather a higher CTA+ coverage favored by a higher density of surface silanolate groups (SiO-) generated at higher pH.
The effect of dilution on nucleation quenching was illustrated in Figure 6. For a dilution by 4 (MSN-18-A) instead of 6, the average size was similar (~ 20 nm) but the size distribution was wider (15 to 30 nm) and the shapes were more heterogeneous (Figure 6a). The channels were also more difficult to observe in the TEM images suggesting a less ordered structure. In addition, these particles aggregated into large and compact domains. These differences in comparison with the best samples were assigned to an inefficient nucleation quenching, keeping supersaturation conditions at work for secondary nucleations to proceed, hence, broadening the distribution of sizes. For a dilution by 8 times, the nanoparticles became larger, ranging from 25 to 40 nm consistent with a lower number of nuclei (MSN-30-A , Figure 6b). Therefore, the nucleation blockage seemed likely acheived while larger distribution of sizes and shapes suggested that the nuclei were instable and underwent Oswald ripening.
The effect of varying the proportions of surfactants between them and also relative to the silicon precursors was also investigated. With twice more F127 (2,300 CTA+ to 2 F127), heterogeneous particle shapes and sizes are produced, some rather small (~ 25 nm) and others much larger (up to ~ 100 nm) with among them ellipsoidal particles of 40 to 60 nm (Figure S4a). The latter resembled those described previously in the absence of F127.[12] For twice less F127 (2,300 CTA+ : 0.5 F127), small facetted particles of about 17 nm presenting various shapes were mixed with larger ones of about 20-25 nm (Figure S4b). This might be due to a lack of secondary surfactant that covers inappropriately each particles. When CTA+ concentration was doubled instead, i.e., 4,600 CTA+: 1 F127, larger sizes were observed in the range 18 to 40 nm (Figure S4c). In addition, the particles exhibited irregular shapes, from ill-facetted to non-spheroid. This was likely due to the merging of primary particles and/or face dependent growth rate. On the contrary, decreasing two times the CTA+ concentration but not that of F127 (1,150 CTA+ : 1 F127) yields rather relatively well-structured small particles. These conditions corresponded to twice less surfactant of both kinds than in the first modification proposed above, (Figure S4d). However, more particle merging took place and particle isolation by utlracentrifugation more difficult to acheive. The rationale behind these experiments was the control of nucleation rate by CTA+ and its quenching by dilution while the neutral F127 minimizes the coalescence of particles one with another.
Indeed, higher concentration of CTA+ led to smaller particles due to higher supersaturation conditions that leads to a larger number of nuclei and therefore to smaller particles. Then, a too small dilution produced larger size distribution with ill-blocked nucleation conditions while high concentration led to merging. The latter effect seemed better controlled eventually toghether with Oswald ripening in the presence of F127, the latter being likely adsorbed on the US-MSN surface. Note that an over-concentration of F127 was detrimental to the particle size distribution, showing that F127 should be restrained to its role on the surface and should not interfere with CTA+ on both nucleation rate and internal pore structuring. Indeed in MSN-100-B, a smaller pore size than in bulk MCM-41 or in the present particles revealed the presence of both surfactants in the internal structure. The optimum CTA+/F127 ratio proposed here has been found by trial and error since it was not possible to detect its presence on the surface of the US-MSN.
The pH control was also decisive for the formation of well-structured particles. In fact, during the treatment at pH = 5.5 for 3 hours, small oily droplets of TEOS were still present and progressively disappeared. In this sequence, the system was still in a slow growth regime where particle merging or coalescence could take place without the dual control by CTA+ and F127. Indeed, TEM images showed that the particles had not yet reached neither the final internal ordering nor their final sizes (7 to 10 nm) (Figure 7). Their smooth surface and the absence of visible pores suggested that they were covered by an amorphous layer of silica precursor that might contain partly hydrolyzed TEOS and presents some analogies with the immature nanoblocks observed during the induction period of a zeolite growth.[9] Contrary to zeolitic nanoblocks, the actual US-MSN were not crystalline and cannot be considered as nuclei stricto sensu since they did not present any defined structure found in bulk MCM-41 or 48 materials. The second difference with the zeolitic growth process is a smaller synthesis temperature of 25° C instead of 80°C. The nanoblock aggregation into larger particles of ca. 90 nm was avoided here probably using higher dilution and acidic quenching. This is consistent with previous studies showing that nanoparticles are preferentially generated between pH 3 and 6 using CTA+ alone.[42]
3.7. Role of F127 Additive as Charge Density Modulator and Particle Size Stabilizer
By contrast with all previous works using large quantities of additives, here very small amounts of F127 sufficed (typically 1.5 wt%, F127/Si molar ratio of 1 to 2300). It was previously stated that the neutral surfactant acts as a growth suppressant by fully covering the particle external surface.[27,28] In the present case, there was not enough F127 to cover the external surface of the particles. The positive zeta potential showed rather that the surface was covered by CTA+ surfactant in agreement with previous observations.[43] Then F127 is likely to intervene in this CTA+ layers since it prevented the coalescence of the US-MSN.
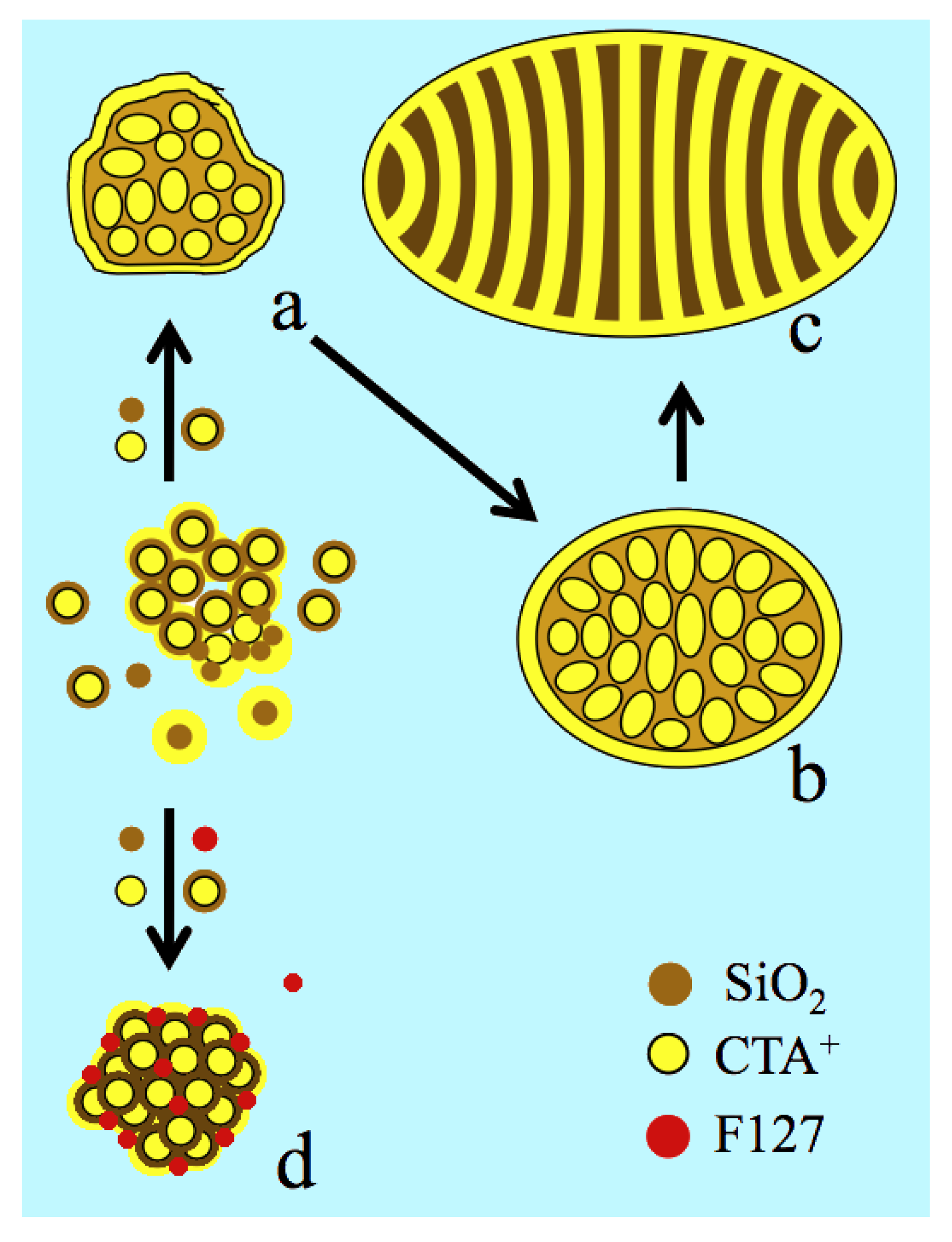
Indeed, one expects the formation of a {S+, I-} interface on the external surface like in the pores, where S+ is the cationic surfactant and I- the inorganic surface. The internal curvature that drives the control on such a type of mesophases (lamellar, hexagonal or cubic) is obtained from the surfactant to silicon ratio or from synthesis temperature that affects the condensation of silica and the silanol density.[44,45,46,47] A perfect match of charge density would yield flat surfaces while a mismatch generates convex or concave surfaces depending on which side the charge density is the highest (Figure S5).[48] Since the surfactant is also adsorbed on the external surface, it affects also the particle morphology and size.[42,48] Along this reasonning, it is stress that F127 was modulating the external charge density releasing the surface tension of the US-MSN. Such a role is reported for the surfactant counter ions X- that intervene directly in the double electrical interface represented as {S+, mX-, (1-m)I-}.[48,49,50,51] Changing the cationic head group or intercalating anionic surfactant produces also a curvature control.[52,53] Since neutral organic molecules and neutral alkyl-polyethylene block co-polymers favor the formation of MCM-48 cubic mesophase,[54,55] they likely act on the charge density matching. F127, which is an ethyleneoxide-propyleneoxide EO-PO-EO triblock co-polymer is likely to behave similarly and stabilize the nanoparticles and prevent their coalescence (Figure 8).
3.7. Protocrystalline US-MSN as Precursor State of Nuclei
The actual nanoparticles were clearly connected to the “raspberry like” particles that appears in the very beginning of the growth process, just before the formation of the channel in the ovoid particles (Figure 8).[11,12] The difference lied in the faceting appearing together with a higher degree of ordering attributed to the presence of F127. Both “raspberry like” particles and the actual protocrystalline US-MSN are likely sharing a common genesis from the aggregation of silica-coated micelles, leading to larger pore wall and smaller specific surface area than in MCM-41 or MCM-48.[12] In addition, the short range order in the packed silica-coated micelles clearly indicated that US-MSN structure solidified before any external forces like surface tension would drive any further ordering. Note that the soft matter transition avoided here was reported at much larger scale of sizes for nascent SBA-15 and SBA-16 nuclei.[18,19] Thus, the diversity of arrays within each US-MSN and among them corresponded to a mixture of nascent particles at different stages of maturity, the evolution of which was quenched in the presence of the stabilizing effect of F127. They could be considered as nano quasicrystal nuclei with respect to the presence of some characteristic non periodical pentagonal or octagonal arrays tough they do not develop their nascent structure all through their volume.[56] Paracrystallinity is another state of matter related to materials presenting short and medium range order with a lack of long range order where crystallized nuclei are embedded in an amorphous matter.[57] This describes the transitional states between amorphous and crystalline phase in vapodeposited thin film of germanium or silicon semiconductor, in silica natural deposits (petrification), in opals and diatom earth deposits.[58,59,60,61,62]. However, most of the US-MSN typically those represented in Figure 4 exhibit a lower degree of structural maturation that is better described as protocystalline. To our knowledge, this term has been introduced by Collins et al. in 1998 to describe an “on-the-edge” state before crystallization in vapodeposited thin film of amorphous hydrogenated silicon exhibiting high performance for solar cell applications.[63,64] The mixture of Si-Si and Si-H bonds creates voids and stablizes the amporphous state together with crystalline nanophase. This structural mixture is the so-called paracrystalline regime.[64,65,66] There is a parallel with the chemistry of porous silica, possessing SiOH groups at the pore interfaces, where the spatial ordering is facilitated by large particles and small pores. This is documented by a large number of reports dealing with micro-, meso- and macroporous materials. [18,19,67,68,69,70]
The actual US-MSNs represent the first isolation of the on-the-edge state of ordering in porous nanoparticles, hence considered as nano protocrystals, which are soft in solution but can be quenched upon drying and eventually strengthen under calcination or under the electron beam during TEM observation. Their colloidal solutions may be useful for the synthesis of novel materials and structures such as hierarchical porous powder materials, as shown here, or coatings on specific substrates.
4. Conclusions
In the present study, a series of monodispersed US-MSN smaller than 50 nm were synthesized in good yield at room temperature using a modified cationic templated route to which was applied a time controlled sequence of dilution and pH adjustments in the presence of a minute amount of F127 neutral surfactant. The slight acidic treatment allowed nucleation quenching for a better size control while the final neutralization step improved the colloidal stabilization for at least 6 months. F127 was assumed to play a complementary role as a charge density modulator at the external surface preventing the coalescence of particles. HR-TEM observations at different tilt angles and a thorough analysis of the images clearly demonstrate that the US-MSN particles are on the crystallization edge characterized by partial short-range order, typical of the protocrystalline state. After calcination, the powder exhibits a well-defined hierarchical dual porosity in the mesoporous range (internal pore ~ 4 nm) (interpartical voids ~ 7 nm). These structurally stable US-MSN protocrystals are good candidates not only for separation and catalysis but also for medical applications as nanocarriers in their colloidal state.
Supplementary Materials
The following are available online at www.mdpi.com/xxx/s1, Table S1: Zeta potential analysis, Figure S1: HRTEM image of MSN-23-A, Figure S2: XRD patterns and TGA, Figure S3: DLS size distribution of MSN-20-A, Figure S4: TEM images of US-MSN, Figure S5: Scheme of charge density mismatch principle, and Figure S6: TEM of MSN-100.
Author Contributions
LB was in charge of the research funding acquisition, administration and conceptualization. LB was sharing the supervision with BA who both participated with FG in writing the original draft. The reviewing process was performed by LB, BA and ME. The synthesis methodology was designed by LB in concertation with F.G who performed all the experiences and sampling. Excepted TEM, all the other physical characterization executed in the chemistry department were analyzed (data curation included) by FG and validated by L.B. and B.A., who both took care of the corresponding resources. The TEM experiments were performed in Lyon under the supervision of PP, images analysis and treatments were performed by LB while TE was providing the ressources. All authors have read and agreed to the published version of the manuscript excepted PS who died before the submission process.” Laurent Bonneviot: orcid.org/0000-0002-9092-8966; Belén Albela: orcid.org/0000-0002-2182-2375; Thierry Epicier :orcid.org/0000-0001-5552-1418 ; Mohamad El Eter : orcid.org/0009-0000-8548-0880.
Funding
The running cost of this research was supported the “ Laboratoire de chimie de l’ENS de Lyon” covered by a general grant the CNRS and the “Ministère de l’Enseignement supérieure et de la recherche” of the French government. The postdoc salary of FG was supported by a King Abdullah University of Science and Technology (KAUST) project, award No. UK-C0017.
Institutional Review Board Statement
Not applicable
Acknowledgments
The authors thank Prof. J.-L. Guth for valuable discussion and suggestion. This work is dedicated to the memory of our dear collegue and friend Pascal Perriat, who died before the publication of this study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Vivero-Escoto, J.L.; Slowing, II; Trewyn, B.G.; Lin, V.S.Y. Mesoporous Silica Nanoparticles for Intracellular Controlled Drug Delivery. Small 2010, 6, 1952–1967. [CrossRef] [PubMed]
- Vallet-Regí, M.; Colilla, M.; González, B. Medical applications of organic-inorganic hybrid materials within the field of silica-based bioceramics. Chem. Soc. Rev. 2011, 40, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Cotí, K.K.; Belowich, M.E.; Liong, M.; Ambrogio, M.W.; Lau, Y.A.; Khatib, H.A.; Zink, J.I.; Khashab, N.M.; Stoddart, J.F. Mechanised nanoparticles for drug delivery. Nanoscale 2009, 1, 16–39. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Padilla, D.; Durfee, P.N.; Brown, P.A.; Hanna, T.N.; Liu, J.W.; Phillips, B.; Carter, M.B.; et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nature Materials 2011, 10, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.; Angelov, B.; Mutafchieva, R.; Lesieur, S.; Couvreur, P. Self-Assembled Multicompartment Liquid Crystalline Lipid Carriers for Protein, Peptide, and Nucleic Acid Drug Delivery. Acc. Chem. Res. 2011, 44, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Frens, G. CONTROLLED NUCLEATION FOR REGULATION OF PARTICLE-SIZE IN MONODISPERSE GOLD SUSPENSIONS. Nature-Physical Science 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Sari, S.M.C.; Debouttière, P.J.; Lamartine, R.; Vocanson, F.; Dujardin, C.; Ledoux, G.; Roux, S.; Tillement, O.; Perriat, P. Grafting of colloidal stable gold nanoparticles with lissamine rhodamine B:: an original procedure for counting the number of dye molecules attached to the particles. J. Mater. Chem. 2004, 14, 402–407. [Google Scholar] [CrossRef]
- Lerouge, F.; Melnyk, O.; Durand, J.O.; Raehm, L.; Berthault, P.; Huber, G.; Desvaux, H.; Constantinesco, A.; Choquet, P.; Detour, J.; et al. Towards thrombosis-targeted zeolite nanoparticles for laser-polarized <SUP>129</SUP>Xe MRI. J. Mater. Chem. 2009, 19, 379–386. [Google Scholar] [CrossRef]
- Gao, F.; Sougrat, R.; Albela, B.; Bonneviot, L. Nanoblock Aggregation-Disaggregation of Zeolite Nanoparticles: Temperature Control on Crystallinity. J. Phys. Chem. C 2011, 115, 7285–7291. [Google Scholar] [CrossRef]
- Fowler, C.E.; Khushalani, D.; Lebeau, B.; Mann, S. Nanoscale Materials with Mesostructured Interiors. Adv. Mater. 2001, 13, 649–652. [Google Scholar] [CrossRef]
- Sadasivan, S.; Khushalani, D.; Mann, S. Synthesis and shape modification of organo-functionalised silica nanoparticles with ordered mesostructured interiors. J. Mater. Chem. 2003, 13, 1023–1029. [Google Scholar] [CrossRef]
- Sadasivan, S.; Fowler, C.E.; Khushalani, D.; Mann, S. Nucleation of MCM-41 nanoparticles by internal reorganization of disordered and nematic-like silica surfactant clusters. Angew. Chem. Int. Ed. 2002, 41, 2151–2153. [Google Scholar] [CrossRef]
- Kim, T.W.; Chung, P.W.; Lin, V.S.Y. Facile Synthesis of Monodisperse Spherical MCM-48 Mesoporous Silica Nanoparticles with Controlled Particle Size. Chem. Mater. 2010, 22, 5093–5104. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. ORDERED MESOPOROUS MOLECULAR-SIEVES SYNTHESIZED BY A LIQUID-CRYSTAL TEMPLATE MECHANISM. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Wan, Y.; Zhao. On the Controllable Soft-Templating Approach to Mesoporous Silicates. Chem. Rev. 2007, 107, 2821–2860. [CrossRef] [PubMed]
- El Haskouri, J.; de Zárate, D.O.; Guillem, C.; Beltrán-Porter, A.; Caldés, M.; Marcos, M.D.; Beltrán-Porter, D.; Latorre, J.; Amorós, P. Hierarchical porous nanosized organosilicas. Chem. Mater. 2002, 14, 4502–4504. [Google Scholar] [CrossRef]
- Büchel, G.; Grün, M.; Unger, K.K.; Matsumoto, A.; Tsutsumi, K. Tailored syntheses of nanostructured silicas:: Control of particle morphology, particle size and pore size. Supramol. Sci. 1998, 5, 253–259. [Google Scholar] [CrossRef]
- Mesa, M.; Sierra, L.; Patarin, J.; Guth, J.L. Morphology and porosity characteristics control of SBA-16 mesoporous silica. Effect of the triblock surfactant Pluronic F127 degradation during the synthesis. Solid State Sciences 2005, 7, 990–997. [Google Scholar] [CrossRef]
- Mesa, M.; Sierra, L.; Guth, J.L. Contribution to the study of the formation mechanism of mesoporous SBA-15 and SBA-16 type silica particles in aqueous acid solutions. Microporous Mesoporous Mater. 2008, 112, 338–350. [Google Scholar] [CrossRef]
- Qiao, Z.A.; Zhang, L.; Guo, M.Y.; Liu, Y.L.; Huo, Q.S. Synthesis of Mesoporous Silica Nanoparticles via Controlled Hydrolysis and Condensation of Silicon Alkoxide. Chem. Mater. 2009, 21, 3823–3829. [Google Scholar] [CrossRef]
- Möller, K.; Kobler, J.; Bein, T. Colloidal suspensions of mercapto-functionalized nanosized mesoporous silica. J. Mater. Chem. 2007, 17, 624–631. [Google Scholar] [CrossRef]
- Moller, K.; Kobler, J.; Bein, T. Colloidal suspensions of nanometer-sized mesoporous silica. Adv. Funct. Mater. 2007, 17, 605–612. [Google Scholar] [CrossRef]
- Kobler, J.; Möller, K.; Bein, T. Colloidal suspensions of functionalized mesoporous silica nanoparticles. ACS Nano 2008, 2, 791–799. [Google Scholar] [CrossRef]
- Cauda, V.; Argyo, C.; Schlossbauer, A.; Bein, T. Controlling the delivery kinetics from colloidal mesoporous silica nanoparticles with pH-sensitive gates. J. Mater. Chem. 2010, 20, 4305–4311. [Google Scholar] [CrossRef]
- Cauda, V.; Schlossbauer, A.; Bein, T. Bio-degradation study of colloidal mesoporous silica nanoparticles: Effect of surface functionalization with organo-silanes and poly(ethylene glycol). Microporous Mesoporous Mater. 2010, 132, 60–71. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, L.-L.; Jiang, J.-G.; Calin, N.; Lam, K.-F.; Zhang, S.-J.; Wu, H.-H.; Wu, G.-D.; Albela, B.; Bonneviot, L.; et al. Facile Large-Scale Synthesis of Monodisperse Mesoporous Silica Nanospheres with Tunable Pore Structure. Journal of the American Chemical Society 2013, 135, 2427–2430. [Google Scholar] [CrossRef]
- Ikari, K.; Suzuki, K.; Imai, H. Grain size control of mesoporous silica and formation of bimodal pore structures. Langmuir 2004, 20, 11504–11508. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ikari, K.; Imai, H. Synthesis of silica nanoparticles having a well-ordered mesostructure using a double surfactant system. Journal of the American Chemical Society 2004, 126, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Urata, C.; Aoyama, Y.; Tonegawa, A.; Yamauchi, Y.; Kuroda, K. Dialysis process for the removal of surfactants to form colloidal mesoporous silica nanoparticles. Chem. Commun. 2009, 5094–5096. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Gao, F.; Valtchev, V. Nanosized inorganic porous materials: fabrication, modification and application. Journal of Materials Chemistry A 2016, 4, 16756–16770. [Google Scholar] [CrossRef]
- Benlekbir, S.; Epicier, T.; Bausach, M.; Aouine, M.; Berhault, G. STEM HAADF electron tomography of palladium nanoparticles with complex shapes. Philos. Mag. Lett. 2009, 89, 145–153. [Google Scholar] [CrossRef]
- Zhou, W.J.; Albela, B.; Perriat, P.; He, M.Y.; Bonneviot, L. Accessibility Control on Copper(II) Complexes in Mesostructured Porous Silica Obtained by Direct Synthesis using Bidentate Organosilane Ligands. Langmuir 2010, 26, 13493–13501. [Google Scholar] [CrossRef] [PubMed]
- Edler, K.J.; Reynolds, P.A.; White, J.W.; Cookson, D. Diffuse wall structure and narrow mesopores in highly crystalline MCM-41 materials studied by X-ray diffraction. J. Chem. Soc.-Faraday Trans. 1997, 93, 199–202. [Google Scholar] [CrossRef]
- Edler, K.J.; Reynolds, P.A.; White, J.W. Small-angle neutron scattering studies on the mesoporous molecular sieve MCM-41. J. Phys. Chem. B 1998, 102, 3676–3683. [Google Scholar] [CrossRef]
- Broekhoff, J.C.P.; De Boer, J.H. Studies on pore systems in catalysts: XI. Pore distribution calculations from the adsorption branch of a nitrogen adsorption isotherm in the case of ‚Äúink-bottle‚Äù type pores. J. Catal. 1968, 10, 153–165. [Google Scholar] [CrossRef]
- Abry, S.; Lux, F.; Albela, B.; Artigas-Miquel, A.; Nicolas, S.; Jarry, B.; Perriat, P.; Lemercier, G.; Bonneviot, L. Europium(III) Complex Probing Distribution of Functions Grafted using Molecular Stencil Patterning in 2D Hexagonal Mesostructured Porous Silica. Chem. Mater. 2009, 21, 2349–2359. [Google Scholar] [CrossRef]
- Spanoudaki, A.; Albela, B.; Bonneviot, L.; Peyrard, M. The dynamics of water in nanoporous silica studied by dielectric spectroscopy. European Physical Journal E 2005, 17, 21–27. [Google Scholar] [CrossRef]
- Muroyama, N.; Yoshimura, A.; Kubota, Y.; Miyasaka, K.; Ohsuna, T.; Ryoo, R.; Ravikovitch, P.I.; Neimark, A.V.; Takata, M.; Terasaki, O. Argon adsorption on MCM-41 mesoporous crystal studied by in situ synchrotron powder X-ray diffraction. J. Phys. Chem. C 2008, 112, 10803–10813. [Google Scholar] [CrossRef]
- Kleitz, F.; Schmidt, W.; Schüth, F. Calcination behavior of different surfactant-templated mesostructured silica materials. Microporous Mesoporous Mater. 2003, 65, 1–29. [Google Scholar] [CrossRef]
- Ikari, K.; Suzuki, K.; Imai, H. Structural control of mesoporous silica nanoparticles in a binary surfactant system. Langmuir 2006, 22, 802–806. [Google Scholar] [CrossRef]
- A 100 nm particle covered by a double (tail to tail) layer of cationic surfactant ~ 4 nm thick would lead to an excess of 40 mol% of surfactant corresponding to a total loss of 50 wt% that is smaller that the measured mass loss. Note also that filling completely the inter granular space would produce a material with a total mass loss of ca. 62 wt%, which is close to the measurement for MSN-100-B.
- Muto, S.; Imai, H. Relationship between mesostructures and pH conditions for the formation of silica-cationic surfactant complexes. Microporous Mesoporous Mater. 2006, 95, 200–205. [Google Scholar] [CrossRef]
- A strong adsorption of F127 producing a 1 nm thick monolayer on the surface of a 22 nm particle will develop a volume that is 30 % that of the particle. Knowing that the internal pore volume filled by the surfactant afford 58% of the US-MSN volume for a 0.12 CTA/Si molar ratio, the external monolayer will correspond to 0.02 F127/Si for a F127 : CTA+ molar ratio of ca. 1:6. In comparison, a double layer of CTA+, which is ca. 4 nm thick would develop a volume 2.5 times larger than that of a 22 nm particle. Then, the external CTA+ would account for 4.3 times more than the internal CTA+. Since, the silica yield reaches at least 60% with a surfactant to silicon stoechiometry in solution equal to that of the particle without external CTA+, the external CTA+ double layer covers at most 16% (= 0.67/4.3 where 0.67 holdf for the ratio = 0.4/0.6 of unreacted surfactant). By comparison, in the conditions adopted by Imai et al. in refs 26 and 27 with little more than two times of CTA+ for larger particles of 100 nm, there is enough CTA+ to develop more than a double layer of CTA (ca. 1.5).
- Vartuli, J.C.; Schmitt, K.D.; Kresge, C.T.; Roth, W.J.; Leonowicz, M.E.; McCullen, S.B.; Hellring, S.D.; Beck, J.S.; Schlenker, J.L.; Olson, D.H.; et al. EFFECT OF SURFACTANT SILICA MOLAR RATIOS ON THE FORMATION OF MESOPOROUS MOLECULAR-SIEVES - INORGANIC MIMICRY OF SURFACTANT LIQUID-CRYSTAL PHASES AND MECHANISTIC IMPLICATIONS. Chem. Mater. 1994, 6, 2317–2326. [Google Scholar] [CrossRef]
- Monnier, A.; Schuth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Maxwell, R.S.; Stucky, G.D.; Krishnamurty, M.; Petroff, P.; Firouzi, A.; et al. COOPERATIVE FORMATION OF INORGANIC-ORGANIC INTERFACES IN THE SYNTHESIS OF SILICATE MESOSTRUCTURES. Science 1993, 261, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Landry, C.C.; Tolbert, S.H.; Gallis, K.W.; Monnier, A.; Stucky, G.D.; Norby, F.; Hanson, J.C. Phase transformations in mesostructured silica/surfactant composites. Mechanisms for change and applications to materials synthesist. Chem. Mater. 2001, 13, 1600–1608. [Google Scholar] [CrossRef]
- Huo, Q.; Margolese, D.I.; Ciesla, U.; Demuth, D.G.; Feng, P.; Gier, T.E.; Sieger, P.; Firouzi, A.; Chmelka, B.F. Organization of Organic Molecules with Inorganic Molecular Species into Nanocomposite Biphase Arrays. Chem. Mater. 1994, 6, 1176–1191. [Google Scholar] [CrossRef]
- Echchahed, B.; Morin, M.; Blais, S.; Badiei, A.R.; Berhault, G.; Bonneviot, L. Ion mediation and surface charge density in phase transition of micelle templated silica. Microporous Mesoporous Mater. 2001, 44, 53–63. [Google Scholar] [CrossRef]
- Oye, G.; Sjöblom, J.; Stöcker, M. Synthesis and characterization of siliceous and aluminum-containing mesoporous materials from different surfactant solutions. Microporous Mesoporous Mater. 1999, 27, 171–180. [Google Scholar] [CrossRef]
- Wang, L.Z.; Shao, Y.F.; Zhang, J.L.; Anpo, M. Synthesis of MCM-48 mesoporous molecular sieve with thermal and hydrothermal stability with the aid of promoter anions. Microporous Mesoporous Mater. 2006, 95, 17–25. [Google Scholar] [CrossRef]
- Badiei, A.R.; Cantournet, S.; Morin, M.; Bonneviot, L. Anion effect on surface density of silanolate groups in As-synthesized mesoporous silicas. Langmuir 1998, 14, 7087–7090. [Google Scholar] [CrossRef]
- Huo, Q.S.; Margolese, D.I.; Stucky, G.D. Surfactant control of phases in the synthesis of mesoporous silica-based materials. Chem. Mater. 1996, 8, 1147–1160. [Google Scholar] [CrossRef]
- Ogura, T.; Sakai, K.; Sakai, H.; Abe, M. Synthesis of highly ordered mesoporous silica with a lamellar structure using assembly of cationic and anionic surfactant mixtures as a template. J. Phys. Chem. C 2008, 112, 12184–12187. [Google Scholar] [CrossRef]
- Galarneau, A.; Cangiotti, M.; di Renzo, F.; Sartori, F.; Ottaviani, M.F. Synthesis of large-pore micelle-templated silico-aluminas at different alumina contents. J. Phys. Chem. B 2006, 110, 20202–20210. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, R.; Joo, S.H.; Kim, J.M. Energetically favored formation of MCM-48 from cationic-neutral surfactant mixtures. J. Phys. Chem. B 1999, 103, 7435–7440. [Google Scholar] [CrossRef]
- Shechtman, D.; Blech, I.; Gratias, D.; Cahn, J.W. METALLIC PHASE WITH LONG-RANGE ORIENTATIONAL ORDER AND NO TRANSLATIONAL SYMMETRY. Phys. Rev. Lett. 1984, 53, 1951–1953. [Google Scholar] [CrossRef]
- Voyles, P.M.; Zotov, N.; Nakhmanson, S.M.; Drabold, D.A.; Gibson, J.M.; Treacy, M.M.J.; Keblinski, P. Structure and physical properties of paracrystalline atomistic models of amorphous silicon. J. Appl. Phys. 2001, 90, 4437–4451. [Google Scholar] [CrossRef]
- Treacy, M.M.J.; Gibson, J.M.; Keblinski, P.J. Paracrystallites found in evaporated amorphous tetrahedral semiconductors. J. Non-Cryst. Solids 1998, 231, 99–110. [Google Scholar] [CrossRef]
- Scurfield, G.; Segnit, E.R. PETRIFACTION OF WOOD BY SILICA MINERALS. Sediment. Geol. 1984, 39, 149–167. [Google Scholar] [CrossRef]
- Gruner, J.W. Cristobalite in bentonite. Am. Mineral. 1940, 25, 587–590. [Google Scholar]
- Banerjee, A.; Wenzel, T. Black opal from Honduras. Eur. J. Mineral. 1999, 11, 401–408. [Google Scholar] [CrossRef]
- Bustillo, M.A.; Martínez-Frías, J. Green opals in hydrothermalized basalts (Tenerife Island, Spain):: alteration and aging of silica pseudoglass. J. Non-Cryst. Solids 2003, 323, 27–33. [Google Scholar] [CrossRef]
- Koh, J.H.; Lee, Y.H.; Fujiwara, H.; Wronski, C.R.; Collins, R.W. Optimization of hydrogenated amorphous silicon p-i-n solar cells with two-step i layers guided by real-time spectroscopic ellipsometry. Appl. Phys. Lett. 1998, 73, 1526–1528. [Google Scholar] [CrossRef]
- Wronski, C.R.; Collins, R.W. Phase engineering of a-Si:H solar cells for optimized performance. Sol. Energy 2004, 77, 877–885. [Google Scholar] [CrossRef]
- Morral, A.F.I.; Cabarrocas, P.R.I.; Clerc, C. Structure and hydrogen content of polymorphous silicon thin films studied by spectroscopic ellipsometry and nuclear measurements. Physical Review B 2004, 69, 10. [Google Scholar] [CrossRef]
- Mahan, A.H.; Roy, B.; Reedy, R.C.; Readey, D.W.; Ginley, D.S. Rapid thermal annealing of hot wire chemical-vapor-deposited <i>a</i>-Si:H films:: The effect of the film hydrogen content on the crystallization kinetics, surface morphology, and grain growth -: art. no. 023597. J. Appl. Phys. 2006, 99, 9. [Google Scholar] [CrossRef]
- Ng, E.P.; Chateigner, D.; Bein, T.; Valtchev, V.; Mintova, S. Capturing Ultrasmall EMT Zeolite from Template-Free Systems. Science 2012, 335, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Holland, B.T.; Blanford, C.F.; Stein, A. Synthesis of macroporous minerals with highly ordered three-dimensional arrays of spheroidal voids. Science 1998, 281, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Vaudreuil, S.; Bousmina, M.; Kaliaguine, S.; Bonneviot, L. Synthesis of macrostructured silica by sedimentation-aggregation. Adv. Mater. 2001, 13, 1310–1312. [Google Scholar] [CrossRef]
- Vaudreuil, S.; Bousmina, M.; Kaliaguine, S.; Bonneviot, L. Preparation of macrostructured metal oxides by sedimentation-aggregation. Microporous Mesoporous Mater. 2001, 44, 249–258. [Google Scholar] [CrossRef]
Figure 1.
Time dependent scheme of MSN-23-A synthesis: black dots depict oily droplets of partially hydrolyzed TEOS, black and white checkerboard filled circles are US-MSN and dash areas around circles stand for surfactant layers covering the particles.
Figure 1.
Time dependent scheme of MSN-23-A synthesis: black dots depict oily droplets of partially hydrolyzed TEOS, black and white checkerboard filled circles are US-MSN and dash areas around circles stand for surfactant layers covering the particles.
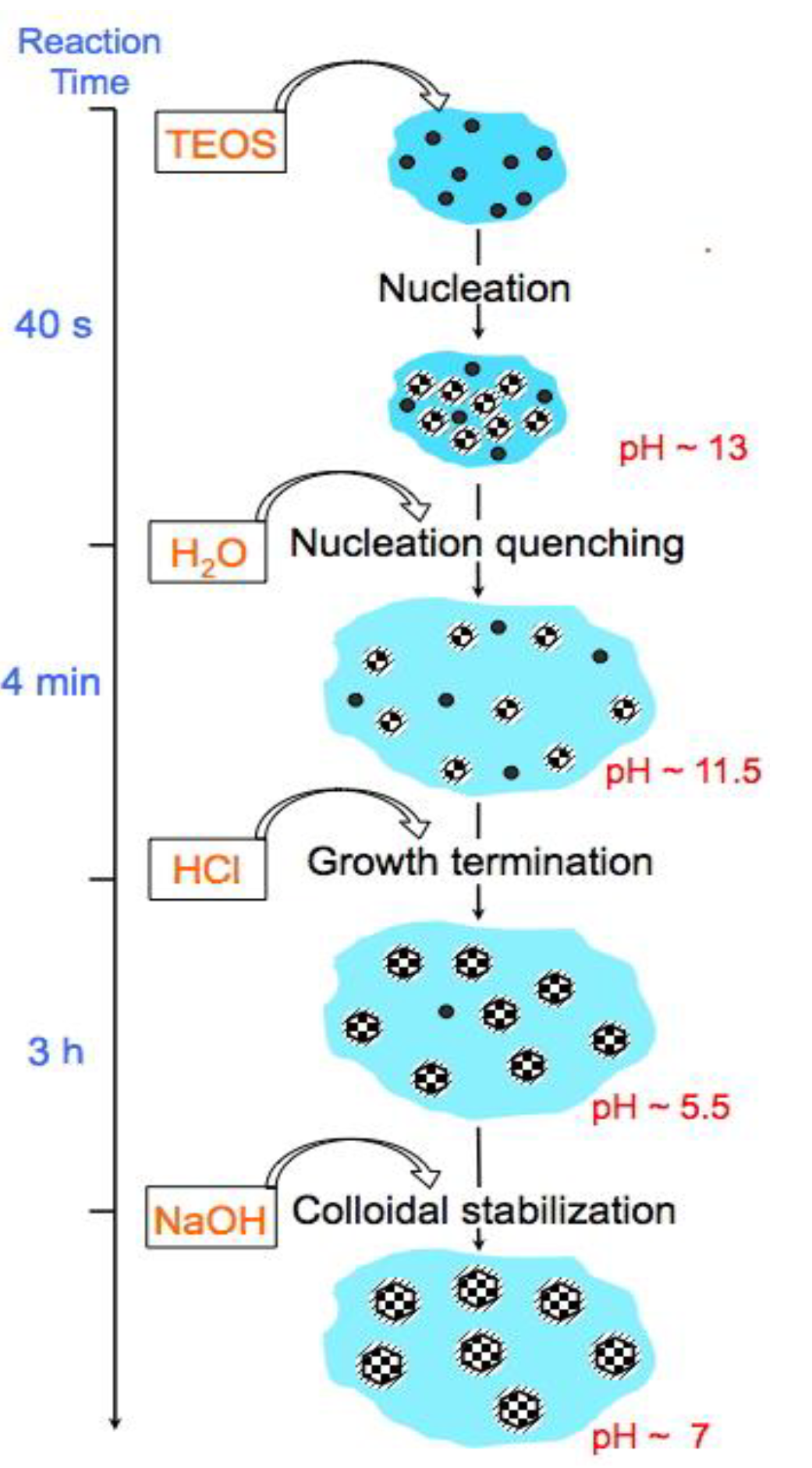
Figure 2.
(a) XRD patterns, (b) HRTEM and STEM images, and (c) DLS size distribution of ultrasonicated as-synthesized MSN-23-A nanoparticles. Composition of the synthesis solution: 1 SiO2 : 0.5 NaOH : 0.12 CTAB : 0.000052 F127 : 130 H2O, nucleation quenching using dilution by 6 times (CTA : F127 = 2300:1 molar ratio).
Figure 2.
(a) XRD patterns, (b) HRTEM and STEM images, and (c) DLS size distribution of ultrasonicated as-synthesized MSN-23-A nanoparticles. Composition of the synthesis solution: 1 SiO2 : 0.5 NaOH : 0.12 CTAB : 0.000052 F127 : 130 H2O, nucleation quenching using dilution by 6 times (CTA : F127 = 2300:1 molar ratio).
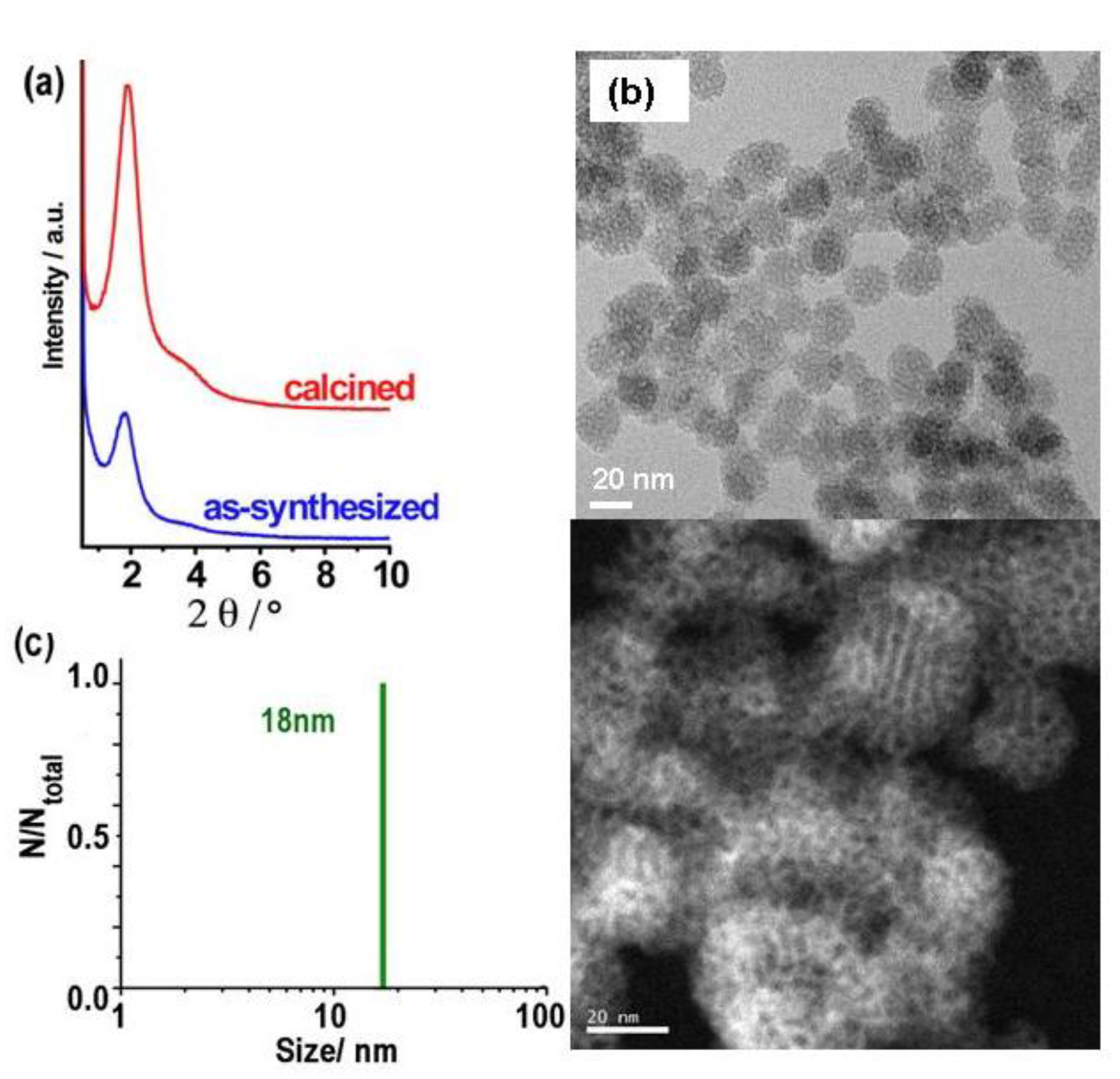
Figure 3.
HR-TEM image of calcined MSN-23-A on a typical cluster of US-MSN particles; (a) areas selected for 2D FT transform producing diffraction patterns represented in reverse contrast (yellows arrows connect the FT transformed area with the corresponding diffraction pattern) : blue arrows are provided on the pattern as a guide to the eyes, (b) same TEM image with the same area as in (a) replaced by their back FT transform (red-brown arrows connect the grey threshold filtrated diffraction pattern with the corresponding back FT transformed area; blue arrows have the same function as in (a)).
Figure 3.
HR-TEM image of calcined MSN-23-A on a typical cluster of US-MSN particles; (a) areas selected for 2D FT transform producing diffraction patterns represented in reverse contrast (yellows arrows connect the FT transformed area with the corresponding diffraction pattern) : blue arrows are provided on the pattern as a guide to the eyes, (b) same TEM image with the same area as in (a) replaced by their back FT transform (red-brown arrows connect the grey threshold filtrated diffraction pattern with the corresponding back FT transformed area; blue arrows have the same function as in (a)).
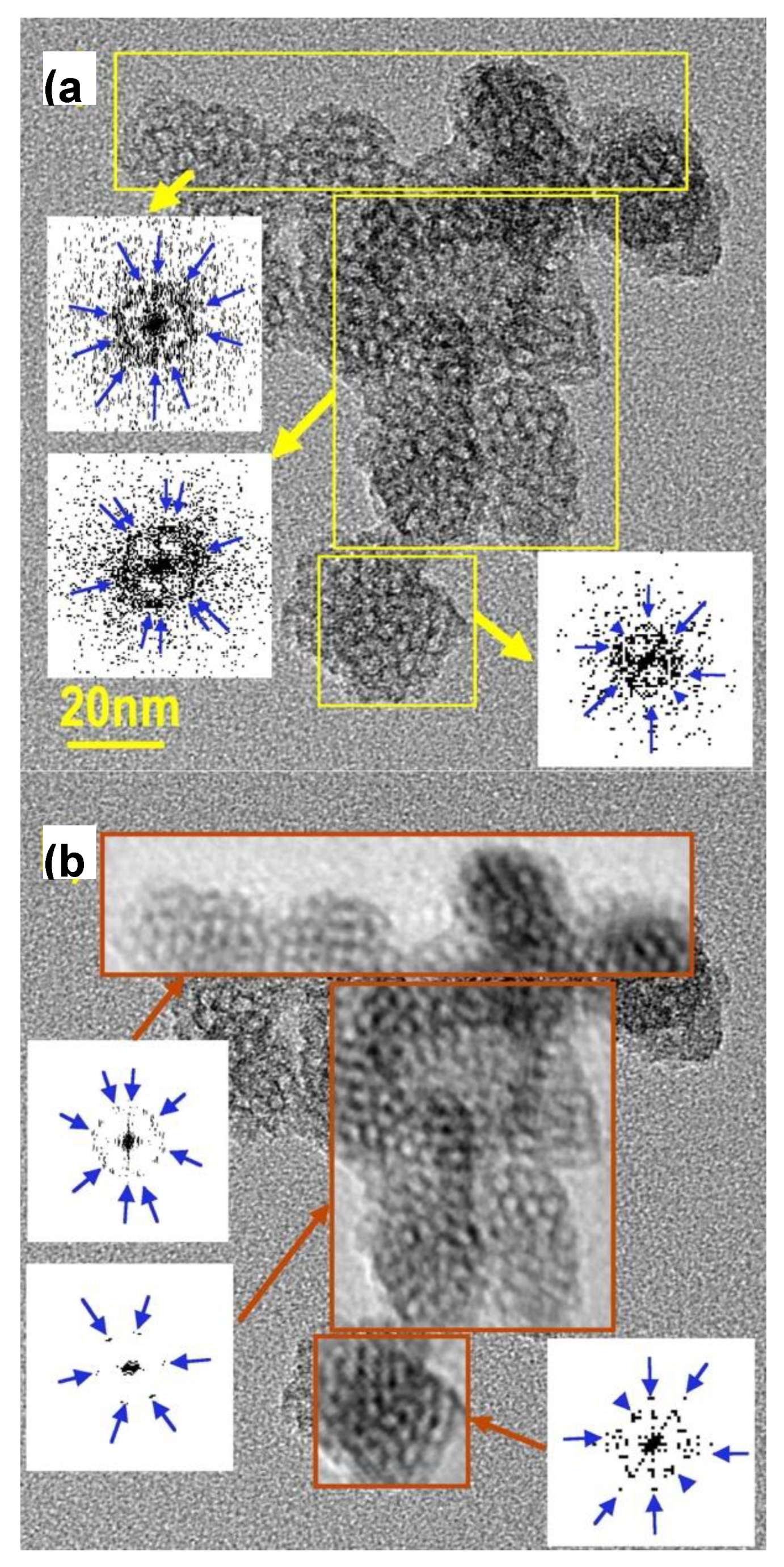
Figure 4.
HR-TEM of a typical cluster of nanoparticles of calcined MSN-23-A at different tilt angles; rough images (top), corresponding 2D FT images (bottom), tilt angles (top right of each image of the top series) with the same scale (top right image), diffraction pattern provided below each images, total FT (top series) and individual particle diffraction (bottom series) for only A, E and D on the first four images avoiding superimposition and moiré effect (see text); F and G taken together and also A and E diffraction patterns are provided in the two last images on the bottom right while diffraction pattern for D is not given because of the superimposition with particle B; note that in the last image (+20°C) particle C pointed by the longest blue arrow emerges in between particle A, B above E.
Figure 4.
HR-TEM of a typical cluster of nanoparticles of calcined MSN-23-A at different tilt angles; rough images (top), corresponding 2D FT images (bottom), tilt angles (top right of each image of the top series) with the same scale (top right image), diffraction pattern provided below each images, total FT (top series) and individual particle diffraction (bottom series) for only A, E and D on the first four images avoiding superimposition and moiré effect (see text); F and G taken together and also A and E diffraction patterns are provided in the two last images on the bottom right while diffraction pattern for D is not given because of the superimposition with particle B; note that in the last image (+20°C) particle C pointed by the longest blue arrow emerges in between particle A, B above E.
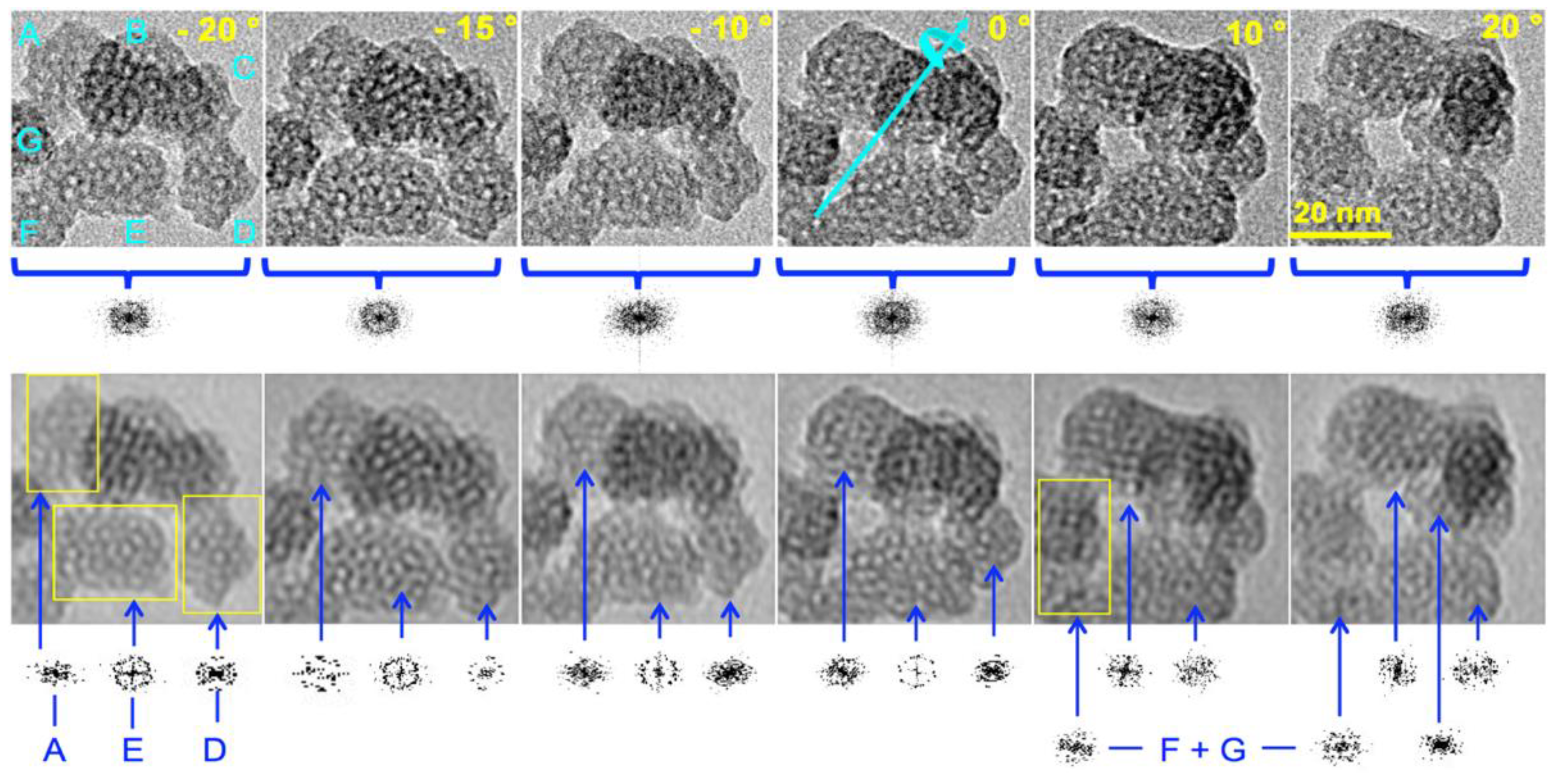
Figure 5.
N2 adsorption-desorption isotherms of MSN-23-A (red round mark), MSN-100-B (blue square mark), and bulk classical MCM-41 (black triangle mark); insert: enlarged part from 0.85 to 1.0 P/P0 ; adsorption (full symbol) and desorption (empty symbol).
Figure 5.
N2 adsorption-desorption isotherms of MSN-23-A (red round mark), MSN-100-B (blue square mark), and bulk classical MCM-41 (black triangle mark); insert: enlarged part from 0.85 to 1.0 P/P0 ; adsorption (full symbol) and desorption (empty symbol).
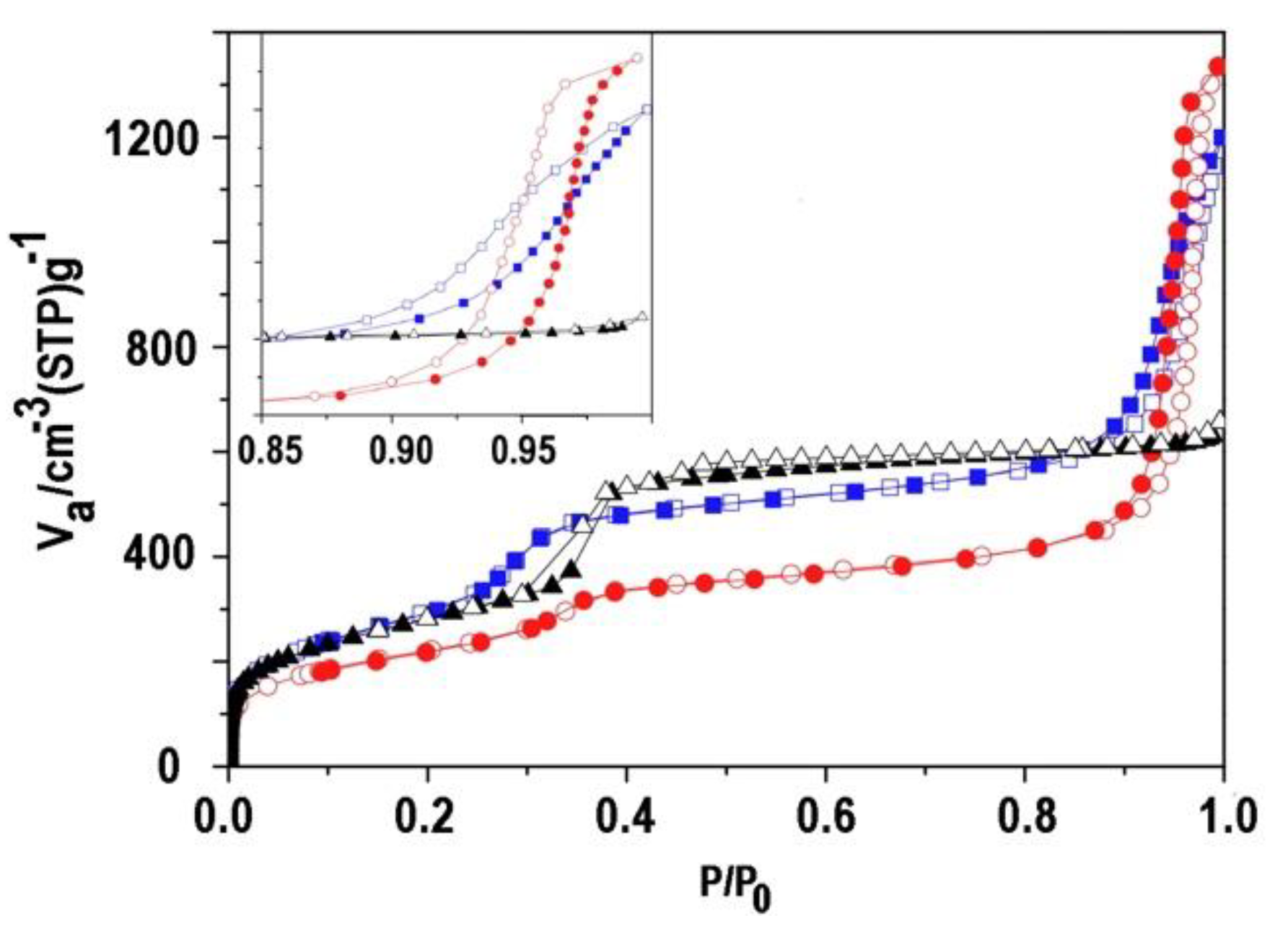
Figure 6.
TEM images of MSN-18-A and MSN-30-A obtained with the same surfactant molar ratio as in the synthesis of MSN-23-A with dilution of 4 (a) and 8 (b) instead of 6 in Figure 1.
Figure 6.
TEM images of MSN-18-A and MSN-30-A obtained with the same surfactant molar ratio as in the synthesis of MSN-23-A with dilution of 4 (a) and 8 (b) instead of 6 in Figure 1.

Figure 7.
TEM image of MSN-23-A at pH 5.5, before the neutralization step.

Figure 8.
Nanoparticle morphologies produced from silica-micelles hybrids among them silica coated micelles according to the F127 free route (a, b and c, ref. [12]) and in the presence of F127 including aggregation, ordering and stabilization of the silica coated packing (d); CTA+ in yellow color, circles for spherical micelles or strips for surfactant double layers around the micelles and F127 in red; light and dark brown colors for silicate oligomer precursors and polymerized silica, respectively: (a) ill-ordered strawberry-like particle, (b) intermediate state of nanoparticles implying elongation of micelles and pre-ordering, (c) ovoid particles (~ 50, see Figure S4a), (d) US-MSN produced from 3D array of nano hollow spheres in the presence of F127.
Figure 8.
Nanoparticle morphologies produced from silica-micelles hybrids among them silica coated micelles according to the F127 free route (a, b and c, ref. [12]) and in the presence of F127 including aggregation, ordering and stabilization of the silica coated packing (d); CTA+ in yellow color, circles for spherical micelles or strips for surfactant double layers around the micelles and F127 in red; light and dark brown colors for silicate oligomer precursors and polymerized silica, respectively: (a) ill-ordered strawberry-like particle, (b) intermediate state of nanoparticles implying elongation of micelles and pre-ordering, (c) ovoid particles (~ 50, see Figure S4a), (d) US-MSN produced from 3D array of nano hollow spheres in the presence of F127.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



