
Submitted:
02 May 2024
Posted:
03 May 2024
You are already at the latest version
Abstract
With the rapid development of gene therapy technology in recent years, its abuse as a method of sports doping in athletic has become a concern. However, there is still room for improvement in gene doping testing methods, and a robust animal model needs to be developed. Therefore, the purposes of this study were to establish a model of gene doping using recombinant adeno-associated virus vector-9 including the human erythropoietin gene (rAAV9-hEPO), and to establish a relevant testing method. First, it was attempted to establish the model using rAAV9-hEPO on mice. The results showed a significant increase in erythrocyte volume accompanied by an increase in spleen weight, confirming the validity of the model. Next, we attempted to detect proof of gene doping by targeting DNA and RNA. Direct proof of gene doping was detected using a TaqMan-qPCR assay with certain primers/probes. In addition, some indirect proof was identified in RNAs through the combination of a TB Green qPCR assay with RNA sequencing. Taken together, these results could provide the foundation for an effective test for gene doping in human athletes in the future.
Keywords:
gene doping
; erythropoietin
; mouse model
; RNA-seq
; TaqMan-qPCR assay
 |
| Graphical abstract |
1. Introduction
Doping is the act of using prohibited substances and/or methods in sports to enhance athletic performance and success by improving physical performance [1]. The World Anti-Doping Agency (WADA), which was established in 1999, is involved in scientific research on doping, anti-doping education, the development of anti-doping strategies, and the monitoring of the World Anti-Doping Code to ensure soundness and fairness in sports worldwide [2]. However, despite WADA’s substantial efforts, doping has not been eradicated from competitive sports.
In fact, at the Tokyo 2020 Olympics in 2021, a track and field athlete was arrested for blood doping [3]. Advances in medicine and science are known to make doping more sophisticated. Therefore, we must continue to work toward establishing testing methods for any forms of doping in order to protect the fairness and health of athletes.
In recent years, gene therapy technology has been developing rapidly. In particular, the vaccine against the coronavirus disease 2019 (COVID-19) virus that shook the world is an application of this gene therapy technology. In addition, around the world, gene therapies using various vectors have been approved in patients who have muscular dystrophy, spinal muscular atrophy (SMA), hemophilia, and various other diseases [4,5]. These diseases used to be incurable, but technological innovations in gene therapy have saved patients’ lives, and it is no exaggeration to say that this is the most advanced form of medicine this century.
Gene therapy technology is evolving at this very moment, but there remains a possibility that it may be abused in the form of gene doping, and therefore, there is an urgent need to develop a method of testing for its presence. In fact, WADA, which oversees the world’s anti-doping testing, has been also wary of gene doping. The International Standard Prohibited List (2024) [6] stipulated in the World Anti-Doping Code 2021 [7], which is published with annual revisions by WADA, describes various formulations and methods used in doping. This list includes “gene doping” as a form of abuse of gene therapy technology. On this basis, our research group has been conducting research for more than seven years to establish a detection method for gene doping [8,9,10,11,12].
Erythropoietin (EPO) has a long history of use in doping [13,14,15,16,17], and is the substance causing the most alarm. In addition, recombinant adeno-associated viral (rAAV) vectors have been confirmed to be safe in gene therapy [18], and are frequently used in gene therapy clinical trials [19]. Therefore, the combination of erythropoietin andrAAV is considered to be at high risk of abuse.
In addition, as a means of indirect proof, we have recommended that the monitoring of fluctuations in RNA expression in whole-blood samples could be included in the parameters of the Athlete Biological Passport (ABP) [8]. The fundamental principle underlying the ABP involves monitoring selected biological variables that indirectly reveal the effects of doping over time rather than attempting to detect the doping substance or method itself [20]. Therefore, the regular observation of RNA expression changes in whole blood could be added as a parameter for the ABP in the future. In fact, our previous studies with a gene doping model have shown the advantage of quantifying the variability of RNA expression in whole blood [8].
Furthermore, WADA explains that the ABP can be used to identify athletes requiring further attention through intelligent, timely interpretation of passport data and can notably be used as a complement to analytical methods to further refine and strengthen overall anti-doping strategies [20]. Therefore, in this study, total RNA sequencing (RNA-seq) using whole-blood RNA was also performed to identify novel indirect proof based on the concept of the ABP.
In summary, the objectives of this study were to establish a model of gene doping using adeno-associated virus vector-9 including the human erythropoietin gene (rAAV9-hEPO), and to establish a method for detecting direct or indirect evidence of genetic doping using blood. As a result, we were able to successfully create the mouse model and prove that it is possible to accurately detect gene doping.
2. Materials and Methods
2.1. Creation of the rAAV9-hEPO Vector
To establish a robust gene-doping mouse model injected with rAAV9-hEPO, a viral vector was created via the following procedures using an outsourcing company (VectorBuilder Inc., Science City, Guangzhou, China). The pAAV expression vector containing 5’ ITRs (inverted terminal repeats) and 3’ ITRs was created with the VectorBuilder web tool. The plasmid was loaded with elements of CMVp (Human cytomegalovirus immediate early enhancer/promoter), hEPO, and WPRE (Woodchuck hepatitis virus posttranscriptional regulatory element). After that, we requested VectorBuilder to create rAAV9 using the plasmid and to obtain the virus particles by an amplification and purification method using the ultra-centrifugal method. The titers of the virus particles obtained were 2.76x10^13 vg (viral genome)/mL.
2.2. Animal Experiments
All animal experiments in this study were approved by the Animal Care Committee, University of Tsukuba (approval number: 22-125). Six-week-old male ICR mice were purchased from CREA Japan (Meguro, Tokyo, Japan) and then subjected to a 1-week acclimation period. The mice were bred and maintained in an air-conditioned animal house under specific pathogen-free conditions and subjected to a 12/12 h light/dark cycle. The mice were fed standard mice pellets and water ad libitum. At the start of the experiments, the age of the mice was 7 weeks and weights were 35.6 g±1.7 (average±SD). The animal experiments were broadly divided into short- and long-term experiments.
The short-term experiments were conducted to establish a gene-doping mouse model with the rAAV9-hEPO vectored and to develop methods to detect direct and indirect proof of gene doping. An overview of these experiments is shown in Figure 1A. After 1 week of acclimatization, mice were randomly assigned to the control (n = 8; named Con.) or rAAV9-hEPO (n = 8; named AAV-hEPO) groups. The mice in the AAV-hEPO group received injections of the rAAV9-hEPO vector (10^11 vg/100 µL/mouse) into the orbital sinus under systemic isoflurane inhalation anesthesia. The mice of the Con. group (n = 8) received injections of the 10% glycerol/PBS (100 µL/mouse) buffer used to suspend the rAAV9-hEPO vector. Ten days after the injection, whole blood was obtained with EDTA-2Na as an anticoagulant from the inferior vena cava under systemic isoflurane inhalation anesthesia, after which the mice were euthanized. The collected whole-blood samples were subjected to preprocessing about plasma separation, aliquoting, etc. for further analysis. Samples of liver and spleen were also harvested and flash-frozen in liquid nitrogen until further analysis. Other samples of spleen and liver tissues were immersed into 10% formalin neutral buffer solution overnight to obtain paraffin block specimens.
Long-term experiments including repeated sampling were conducted to investigate how long direct and indirect proof of gene doping could be positively detected from approximately one drop of whole blood. An overview of these experiments is shown in Figure 1B. After 1 week of acclimatization, small whole-blood samples (approximately 100 µL; 2 drops) from mice (n = 11) were collected with EDTA-2Na using cuts made approximately 1 mm from the tail tip (“Pre“ time point). Then, the AAV9-hEPO vector was injected using the methods described above. Subsequently, using the same method described above, blood samples were continually collected every 5 days for 30 days after the injection, as shown in Figure 1B. In order to eliminate the adverse effects of continuous blood collection on mice (for example, anemia, hematopoietic stimulation, etc.) as much as possible, we selected a blood-collection method that only obtained small amounts (approximately 100 µL) from the tail tip. The collected blood samples were divided into two samples of approximately 50 µL (equivalent to about one drop) each, and DNA and RNA were extracted from these samples and subjected to further analyses.
The animal experimental methods described so far are identical in many respects to those of our previous study [8]. In addition, the description of this later experimental method is likewise similar in many respects to the experimental methods of our previous study [8]. Therefore, the method descriptions that follow may cite reference number 8.
2.3. Measurements of General Hematopoietic Markers
Hematological indicators of red blood cells (RBCs) count, hemoglobin (HGB) level, and hematocrit (HCT) value from obtained the whole blood in the short-term experiments were measured on an automatic blood analyzer (Celltac α MEK6458; NIHON KODEN, Shinjuku, Tokyo, Japan) using 50 µL of whole blood [8]. Whole-blood volumes were also measured using 5mL syringe, during the whole-blood collection.
2.4. Enzyme-Linked Immunosorbent Assay
An enzyme-linked immunosorbent assay (ELISA) was performed to confirm the secretion of hEPO into the blood from the liver in AAV-hEPO mice in the short-term experiment. Anti-hEPO (Cat# 500-P318; PeproTech, Cranbury, NJ, USA) was diluted to 0.5 µg/mL in PBS as a capture antibody, and 100 µL of the antibody solution was then applied into ELISA plates to obtain triplicate measurements. The plate was incubated at 4 °C overnight and then washed with PBS containing 0.05% Tween 20 (PBS-T) four times; then, 100 µL of the plasma samples diluted 20-fold with PBS were added to the wells and incubated for 1 h at room temperature. Solutions of hEPO recombinant protein (Cat# 100-64; PeproTech) in PBS including 5% BSA were also added to generate a standard hEPO curve between 100 ng/mL and 195 pg/mL. After incubation, the plate was washed in the same way as described previously, and a 100 µL solution of a detection antibody (biotinylated anti-hEPO; Cat# 500-P318BT; PeproTech) diluted with PBS-T was applied to the wells at a concentration of 0.25 ng/µL, followed by incubation for 1 h at room temperature. After incubation, the plate was washed in the same way, and a 100 µL solution of HRP-conjugated streptavidin (Cat# SA00001-0; Proteintech, Rosemont, IL, USA) was diluted 5000-fold with PBS-T and was applied to the wells. The plate was incubated at room temperature for 30 min and then washed the same way. After washing, a 100 µL mixture of coloring reagent and substrate (ELISA POD Substrate A.B.T.S Kit, Cat# 14351-80; Nacalai Tesque, Nakagyo, Kyoto, Japan) was applied and incubated for 15 min, followed by application of 100 µL of stop reagent for the coloring reaction. Finally, the absorbance at 405 nm with a reference at 600 nm was measured using a microplate reader. Using the absorbance data, a standard curve as a 4-parameter logistic regression of the Rodbard equation was created in ImageJ Fiji (Life-Line version, v1.53q), and the concentration of hEPO in the plasma was calculated in duplicate measurements based on the standard curve (R2 = 0.99) [8].
2.5. TB Green qPCR Assay for Tissue
To confirm the expression of hematopoietic marker genes in the liver and spleen in the short-term experiment, a TB Green qPCR assay was performed as an intercalation method. RNA extraction from the liver and spleen was performed using RNAiso Plus (Cat# 9180; Takara Bio, Kusatsu, Shiga, Japan) according to the manufacturer’s instructions. The extracted RNA solution in Milli-Q Water (Merck Millipore, Burlington, MA, USA) was diluted and adjusted to a concentration of 100 ng/µL. Then, 500 ng of RNA was used to prepare cDNAs using PrimeScript RT Master Mix (Cat# RR036A; Takara Bio) according to the manufacturer’s instructions. The cDNAs were diluted 10× using Milli-Q Water and subjected to a quantitative real-time PCR (qPCR) assay based on the intercalator-fluorescence dye. The qPCR assay was performed to quantify the gene expression of hematopoietic markers in the liver and spleen by using TB Green Premix Ex Taq II (Cat# RR820; Takara Bio) with primers from QuantStudio 5 Real-Time PCR Systems (Thermo Fisher Scientific, Waltham, MA, USA) as duplicate measurements. The targeted gene list and primer sequences are shown in Supplementary Table S1 The template volume and primer concentrations were 2 µL and 100 nM, respectively, for a total reaction volume of 10 µL per well. Negative control wells were also established using Milli-Q water instead of the template. The conditions for thermal cycling were 95 °C for 5 min, followed by 40 cycles of 95 °C for 2 s and 60 °C for 20 s and a melt curve stage. Subsequently, the ΔΔCt method was used to calculate the relative gene expression values with reference to 18S rRNA [8].
2.6. Histology
The paraffin blocks of each tissue obtained were sectioned into 3 μm thick slices, and H&E (hematoxylin and eosin) staining was performed. After the staining, the morphology was observed under a microscope.
2.7. DNA Extraction from Whole-Blood Samples
A Maxwell RSC Blood DNA Kit (Cat# AS1400; Promega, Madison, WI, USA) was used to extract total DNA from 100 µL (short-term experiment) or 50 µL (long-term experiment) of whole blood, using a Maxwell RSC Instrument (Promega), according to the manufacturer’s instructions. The DNAs were dissolved in 50 µL of Milli-Q Water, and the solutions were subjected to the TaqMan-qPCR assays.
2.8. TaqMan qPCR Assay
A TaqMan qPCR assay was performed on the whole-blood DNA. For the detection of direct proof of gene doping in whole-blood DNA in the short- and long-term experiments, the primers and TaqMan probes were designed to target the EPO gene (2 types), WPRE (Woodchuck hepatitis virus posttranscriptional regulatory element; named Pr-WPRE), and CMVp (cytomegalovirus promoter; named Pr-CMVp) to ensure specific amplification of the rAAV9-hEPO genome using the Primer-BLAST web tool [21]. The primers and TaqMan probes for the two types of hEPO genes were designed with exon–exon junctions (exons 2–4 and 4–5; primer names are Pr-hEPO-1 and Pr-hEPO-2), which are considered non-amplifying structures in the human genome. This strategy of designing primers/probes for the hEPO gene to detect gene doping follows WADA’s guidelines [22]. The primers and probes were also checked for specificity with in silico PCR using Primer-BLAST; these evaluations confirmed the absence of amplification in the human and mouse genomes. The sequences of the primers and TaqMan probes are shown in Supplementary Table S1. The primers and probes in a double quencher system were systemized by Integrated DNA Technologies (IDT; Coralville, IA, USA). Next, the TaqMan qPCR assay was performed in duplicate to detect direct proof in whole-blood DNA by absolute quantification using PrimeTime Gene Expression Master Mix (Cat# 1055771; IDT) with the primers and TaqMan probes on QuantStudio 5 Real-Time PCR Systems (Thermo Fisher Scientific, Waltham, MA, USA). The template DNA volume was 2 µL, and the primer and probe concentrations were 200 nM and 200 nM, respectively, for a total reaction volume of 10 µL per well. Negative control wells were also established using Milli-Q water instead of a template. DNA of 6 human cell lines (fibroblast, HK2cell, Caco-2 cell, HEK293 cell, mesenchymal stem cell, and HepG2 cell) were used as the negative control. pAAV expression vectors including the hEPO gene, WPRE, and CMVp (pAAV-hEPO) were used at 10 pg/μL to prepare a standard curve for absolute quantification, and the range of the standard curves was set to 0.95 to 1.86x10^6 copy/μL for the pAAV. The conditions of thermal cycling for all primer/probe pairs were 95 °C for 5 min, followed by 40 cycles of 95 °C for 2 s and 60 °C for 20 s. All standard curves had R2 > 0.99 [8].
2.9. Sanger Sequencing
After the TaqMan qPCR, solutions including the amplicon were pooled in a 1.5 mL microtube and then subjected to electrophoresis using a 2% agarose gel. The bands of the DNA amplicons were visualized using ethidium bromide solution. Subsequently, the PCR amplicons were purified using a NucleoSpin Gel and PCR Clean-up kit (Cat# 740609; Takara Bio, Kusatsu, Shiga, Japan). Next, to check the sequence of the DNA amplicons, 5 ng of the purified DNA was subjected to Sanger sequencing via outsourcing to an external company (GENEWIZ, Shinagawa, Tokyo, Japan). The Sanger sequencing data were analyzed with BioEdit ver. 7.2.5 (developer: Tom Hall, Carlsbad, CA, USA) [8]. Alignment analysis for 42 nucleotides obtained from the Sanger sequence was performed referring to the DNA sequence of the rAAV9-hEPO.
2.10. Total RNA-Seq
Total RNA-seq was performed to identify RNA as novel indirect proof based on the concept of the ABP [20]. The total RNA of the mice (Con., N=6; AAV-hEPO, N=6) in the short-term experiments was extracted from 100 µL of whole blood using RNAiso Blood (Cat# 9112; Takara Bio) according to the manufacturer’s instructions. The RNA pellets were dissolved in 30 µL of Milli-Q Water, and the RNA solutions of 12 samples (Con.: n = 6; AAV-hEPO: n = 6) were checked for integrity using Agilent RNA 600 Nano Kit (Cat# 5067-1511; Agilent Technologies, Santa Clara, CA, USA) on a Bioanalyzer (Agilent Technologies). The RNA Integrity Number (RIN) of all samples was 9.5 or higher; thus, the RNAs of all eight samples could be subjected to library preparations for total RNA-seq. Using 500 ng of the RNAs from each sample, libraries were created using the NEBNext Ultra II RNA Library Prep Kit for Illumina and the NEBNext rRNA Depletion Kit v2 (Cat# E7770S and E7400L; New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions, and the final PCR cycle was 15. Concentrations and size distributions of the libraries were measured using an Agilent DNA 7500 kit (Cat#5067-1506; Agilent Technologies, Santa Clara, CA, USA) with Bioanalyzer. All samples were passed for analyses on NGS equipment. The libraries were pooled, and the concentrations were adjusted to 1 nM. The pooled libraries were subjected to denaturation and neutralization. Subsequently, the libraries were diluted to 1.8 pM and then applied for an NGS run using NextSeq500/550 v2.5 (75 Cycles) Kits (Cat#20024906; Illumina, San Diego, CA, USA) in the NextSeq 500 System (Illumina). The sequencing was performed with paired-end reads of 36 bases. After the sequencing run, FASTQ files were exported, and the basic information of the NGS run data were checked via the CLC Genomics Workbench 24.0 software package (QIAGEN, Hilden, Germany). In the quality assessment of the reads, a PHRED score over 20 was confirmed for 99.65% of all reads, indicating the success of the run. The read number was approximately 11.4 to 16.1 million per sample as paired-end reads [8].
2.11. Bioinformatics Analysis
The following analysis was performed to identify RNA markers (genes) as novel indirect proof based on the concept of the ABP using NGS run data. FASTQ files were mapped to the mouse genome (GRCm39) using the CLC software (QIAGEN). A statistical differential expression test was performed using the Differential Expression in Two Groups tool in the software package. A principal component analysis (PCA) plot was created using the CLC software. Transcripts Per Kilobase Million (TPM) or CPM (Counts per Million mapped reads) values were used as an expression value for figure visualizations. A cluster dendrogram was created using R (ver. 4.1.1). A tool for enrichment analysis was performed using Gene Set Enrichment Analysis (GSEA; version 4.3.3) with Mouse MSigDB (a database for enrichment analysis; mouse-ortholog hallmark gene sets; v2022.1.Mm) [23,24]. The Venny ver.2.1.0 [25] web tool was used to identify RNAs with dramatically fluctuating expression. Supplementary Table S2 shows the quantitative expression values, fold changes, and p-values of all genes obtained by analysis using the CLC software.
2.12. Measurements of mtDNA Copy Numbers in the Whole Blood
The results from the GSEA analysis of the above bioinformatics analysis showed that the gene set related to mitochondrial activity was significantly hit, so we checked the consistency of the results. The number of mitochondrial DNA (mtDNA) was measured using the whole-blood DNAs that were obtained from mice in the short-term experiment. The TB Green qPCR Assay was employed as described in Section 2.5. Primer pairs were designed to target nuclear DNA (nDNA) and mtDNA (Supplementary Table S1). The number of mtDNA per nDNA was quantified using CT values obtained from the qPCR assay.
2.13. TB Green qPCR Assay for Whole-Blood RNA
To confirm whether the genes identified as being indirect proof of gene doping showed reproducibility for the total RNA-seq results, a TB Green qPCR assay was performed for all samples (Con.: n = 8; AAV9-hEPO: n= 8) in the short-term experiment. RNA extraction from 100 µL whole-blood samples was performed using the same methods mentioned in the “Total RNA-seq” section. Subsequently, the TB Green qPCR assay for assessing whether the identified genes were indirect proof was performed using the same method described in the “TB Green qPCR assay for tissue RNAs” section. The primer sequences used in this section are shown in Supplementary Table S1. In addition, to investigate the period for which the identified indirect proof could be positively detected, the same qPCR assay was also performed using 50 µL whole-blood samples (n = 11) from the long-term experiment. Subsequently, the expression values of each targeted gene were normalized by B2m (Beta 2-microglobulin) gene expression [8].
2.14. Statistical Analysis
All data except the total RNA-seq data were statistically analyzed using GraphPad Prism version 10.2.0 (GraphPad, San Diego, CA, USA). All experimental data were first evaluated with the Shapiro–Wilk normality test to check the normality of the distributions. Subsequently, nonparametric tests were used for all data. Comparisons of three or more groups were performed with Kruskal–Wallis H tests (one-way ANOVA of ranks) followed by a two-stage Benjamini, Krieger, and Yekutieli FDR procedure as a post hoc test. A p-value less than 0.05 was considered to indicate statistical significance. All graphs without data from the bioinformatics analysis of the total RNA-seq are shown as individual plots and medians with interquartile ranges [8].
3. Results
3.1. The rAAV9-hEPO Vector Worked toward Establishing a Mouse Model of Gene Doping
Compared to the control group, the AAV-hEPO group showed a significant enlargement of the spleen and a slightly enlarged liver (Figure 2A–C), even though there was no change in body weight. In addition, blood tests showed significant increasing in blood volume, RBCs, HGB, and HCT in the AAV-hEPO group, compared to control group (Figure 2D–G).
ELISA demonstrated positive reactions (green coloration in the reaction plate; triplicate measurements; Figure 2H) in all mice of the AAV-hEPO group, with quantitative values ranging from 124 to 422 ng/mL (Figure 2I). Gene expression analysis by TB Green qPCR assay measured three hematopoietic marker genes (Gata1, Gypa, and Cd71) and hEPO genes in the liver and spleen tissue. The results showed that the all-gene expressions were significantly increased in the AAV-hEPO group compared to the control group (Figure 2J). Pathology specimens showed an increase in nuclear size in both liver and spleen in the AAV-hEPO group compared to the control group (Figure 2K). These results suggested that the rAAV9-hEPO vector worked well and was taken up by the mouse liver, where hEPO was secreted to the blood, and hematopoiesis was actively occurring in the spleen. In other words, mice that mimic gene doping with the rAAV9-hEPO vector could be established.
3.2. The Four Primer/Probe Pairs Could Detect Direct Proof of Gene Doping
The strategies to create four primers/probes are shown in Figure 3A. All primers/probes designed in this study worked and were highly sensitive with a lower limit of 0.95 copies (viral genome)/μl. In addition, the viral DNAs were not detected in whole-blood DNAs from control mice or in human cell line DNAs. In contrast, the AAV-hEPO group showed positive PCR amplification with a copy number of 34-104 copies (viral genome)/μl (Figure 3B). The amplified products (AAV-hEPO mouse and positive control only) after the qPCR assay were confirmed by electrophoresis, and all lanes showed amplified products of the predicted size (Figure 3C). The amplified products (AAV9-hEPO mice only) after its qPCR were confirmed by Sanger sequencing, and the waveform data was correctly depicted and the obtained sequence matched the target sequence (Figure 3D). These results suggest that these primer/probe pairs and the qPCR assay worked and were able to accurately detect proof of gene doping.
3.3. The Total RNA-seq Revealed Drastic Changes in RNA Expression and Identified Several Genes That Provide Indirect Proof of Gene Doping
The results of the total RNA-seq using whole blood showed that the expression of many RNAs was drastically changed. In the 3D PCA plot and cluster dendrogram, clear distance and separation by clusters between Con. and AAV-hEPO groups suggested that the two groups were in different states of gene expression (Figure 4A,B). In the statistical comparison between the Con. and AAV-hEPO groups, a total of 1144 genes were found to have significant variation when filtering for an FDR-p value < 0.001, 2-fold change, and max group mean>1 as thresholds. Of those 1144 genes, the expression of 906 genes was increased in the AAV-hEPO group, and that of the remaining 238 genes was decreased, as shown in the heatmap (Figure 4C). The results of the GSEA analysis showed that the gene set of the “heme metabolism”, “oxidative phosphorylation”, and “fatty acid metabolism” were significantly hit due to upregulation in the AAV-hEPO group (Figure 4D). The analysis to this point has shown that gene-doped mice injected with the rAAV-hEPO vector showed substantial fluctuations in RNA expression in whole blood, reflecting characteristic enrichment to some gene sets. Further bioinformatics analysis was performed to identify single gene expression with dramatic variation. Using Venn diagram analysis, strict thresholds were set, namely an FDR p-value < 0.001, a 20-fold change, and a max group mean > 20, and then 12 promising genes were identified (Figure 5A). The expressions of these 12 genes were increased in the AAV-hEPO group compared to the Con. group, as shown in the heatmap (Figure 5B). Moreover, these genes were analyzed and visualized as a bar graph (average value of the TPM; Figure 5C), scatterplot (Figure 5D), and volcano plot (Figure 5E). Consistent results were then obtained. In particular, Asns (asparagine synthetase), Shmt2 (serine hydroxymethyltransferase 2), and Mthfd2 (methylenetetrahydrofolate dehydrogenase 2), a top three gene, showed a more than 100-fold fluctuation (Figure 5C). TB Green qPCR assays were performed on these three genes to confirm the reproducibility against the total RNA-seq in all mice samples. The results were generally consistent (Figure 5F). As per the result of the GSEA analysis, mitochondrial activation of the whole blood was suspected in the AAV-hEPO group. Moreover, Shmt2 and Mthfd2 are known to be associated with mitochondrial function. To confirm their consistency, mtDNA copy numbers were quantified, and then it was found that the copy numbers were significantly increased in the AAV-hEPO group compared to the Con. group. Taken together, these results suggest that inducing the hEPO transgene in the rAAV9 vector alters the biological significance of whole-blood RNA and causes some genes to be dramatically upregulated with increasing mitochondrial numbers, which has the possibility of becoming an ABP parameter.
3.4. Proof of Gene Doping Was Detectable in about a Drop of Whole Blood for 30 Days
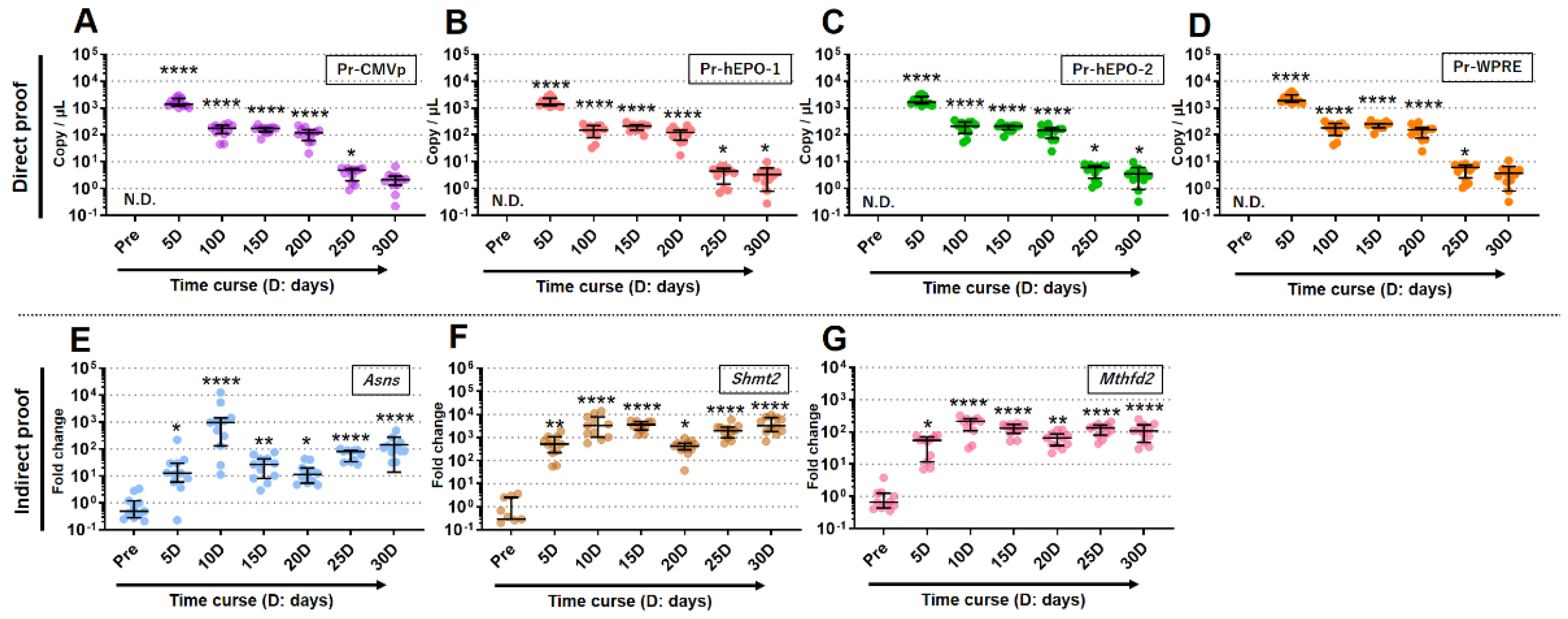
The qPCR assays were performed to detect direct or indirect proof of gene doping, using micro blood samples (about 50ul; one drop) from the tail taken repeatedly over a period of 30 days. The statistically significant detection of direct proof of gene doping with each of the primers/probes was possible up to day 25 for Pr-CMVp and Pr-WPRE and up to day 30 for Pr-hEPO-1 and Pr-hEPO-2 (Figure 6A–D). Moreover, the copy numbers of the rAAV-hEPO genome as direct proof peaked at day 5 for all primers/probes, and then unidirectionally decreased until day 30 (Figure 6A–D).
The expression values of three genes that could be indirect proof of gene doping, thus leading to the ABP concept, were significantly elevated in all periods until day 30 (Figure 6E–G). On the other hand, unlike the dynamics of direct proof, the expression level remained generally constant between day 5 and day 30. These results indicated that direct or indirect proof of gene doping could be detected using a single drop of whole blood.
4. Discussion
The first objective of this study was to create a mouse model that mimics gene doping with confirmed hematopoietic effects using rAAV9 carrying the hEPO gene. rAAV9, when injected into a mouse vein, accumulates in the liver and induces long-term expression of the target gene [26]. It has also been confirmed that the period of continuous expression is more than 9 months [26]. Therefore, in this study, it can be assumed that the injected rAAV9 reached the mice liver and induced hEPO gene expression in the hepatocytes over a long-term period. In support of this speculation, gene expression of hEPO was detected in the liver. The hEPO gene contains a secreted signal peptide, so the translated protein is also expected to circulate throughout the whole body as a hormone via blood. In fact, an ELISA confirmed the presence of human erythropoietin in the AAV-hEPO group, confirming that it circulates throughout the body. In addition, the main hematopoietic organ in adult mice is the spleen [27]. In this animal experiment, the spleen of mice was enlarged, and the expression of hematopoietic marker genes was significantly increased. Moreover, RBCs, HGB, HCT, and blood volume, which directly reflect hematopoietic effects, were also significantly increased in the AAV-hEPO group. Taken together, these results suggest that the rAAV9-hEPO vector induces the expression of hEPO in the liver after intravenous injection, and that hEPO hormone reaches the spleen through the bloodstream, thereby upregulating hematopoiesis. Therefore, the first objective in this study has been achieved, meaning that the establishment of a biological model of gene doping using AAV and hEPO genes has been accomplished for the first time. In previous studies on gene doping tests targeting hEPO, detection methods were developed by artificially mixing plasmids with blood as a spike-in test [28,29,30,31]. On the other hand, our model, however, uses an actual living organism and can consider the metabolic state of the vector, so it has the potential to be developed into a more practical model. Therefore, in the future, we plan to provide various technical support so that any researchers around the world can use our models with good reproducibility.
In this study, four specific primer/probes were generated to target DNA sequences in rAAV9-hEPO as direct proof of gene doping. For the hEPO gene, we designed the probe to span the exon–exon junction, in accordance with WADA guidelines [21]. Although the WADA guidelines do not mention the use of multiple primers/probes, the use of this method may increase the scientific validity and accuracy of the test. In addition, the use of multiple primers/probes would allow for an estimation of the type of vector used. However, in this study, one qPCR assay was performed on one target. In the future, it would be desirable to establish a multiplex assay and develop a kit with a panel comprising each vector and gene. This would reduce the test time and cost. In addition, the four primer probes used in this study detected positive reactions only in mice induced with the rAAV9-hEPO vector, with no nonspecific amplification from human DNAs or an NTC (non-template control). An analysis of the standard curve showed that the detection limit was approximately 1 copy/ul, indicating the high detection sensitivity of these primers/probes. Therefore, in the future, it may be possible for these four primers/probes to be directly used for testing of gene doping testing using blood DNA samples from athletes.
Based on the ABP concept proposed by WADA [9], we hypothesized and tested the hypothesis that RNA (gene expression) in whole blood may be targeted as a new ABP marker. As a result, in the total RNA-seq, we found that mice induced with the rAAV9-hEPO vector showed significant changes in the expression of many RNAs in whole blood, and heme metabolism, oxidative phosphorylation, and fatty acid metabolism were significantly hit in terms of their biological significance, and pathways in terms of their gene sets. Further bioinformatics analysis identified three dramatically upregulated RNAs (genes), which remained highly expressed after 30 days. These results suggest that pathway analysis using a specific set of genes or analysis for single RNA markers can be very useful as a measure of ABP. However, the total RNA-seq performed in this study is expensive in terms of reagents and analysis costs, and the equipment required is also expensive. Although the application of RNA-seq technology to ABP testing is promising, the cost and difficulty of the technology may be prohibitive. Therefore, cost reductions and technological innovations are eagerly awaited.
If this were translated to humans, it would be possible to detect genetic doping with only a trace amount of blood (one drop of blood) drawn from a fingertip after a competition. If the amount of specimen is only one drop, the development of small testing devices may also enable rapid testing for genetic doping in the field. The closest social implementation would be a PCR test for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection, also known as the COVID-19 pandemic, which has raged in recent years. With the current proliferation of various simplified test reagents and small qPCR devices, rapid PCR testing has been implemented in the field around the world. These technologies are probably very compatible with on-site rapid tests for genetic doping. Therefore, a future may not be far off in which primary on-site screening for genetic doping will be available for many athletes.
It is important to mention there are several limitations in this study. Although this study was the first in the world to establish a mouse model of gene doping using the rAAV9 vector including the hEPO gene, and the first to develop a detection method, it is only a mouse model and cannot yet be said to be applicable to actual human athletes. In future studies, the most accurate findings could be obtained by using blood samples from actual gene-doped athletes, but this is not possible due to legal or ethical aspects. As an alternative, it would be most reasonable to validate the results of this study using surplus blood samples from patients undergoing gene therapy with rAAV. As indicated in the database, many clinical trials using rAAV are being conducted around the world [19]. In addition, several Food and Drug Administration (FDA)-approved rAAV therapies [32,33,34,35] have become widely available in recent times. Research and development to prevent gene doping would require collaboration with patients receiving rAAV medications and their health care providers, as well as clinical trials. In doing so, the findings of this study would be helpful in creating safe experimental protocols and setting conditions for detection methods.
5. Conclusions
In this study, we were able to establish gene-doping model mice using the rAAV9-hEPO vector. Furthermore, not only were we able to detect direct proof of gene doping in whole blood in these mice, but we also revealed that RNA expression in whole blood fluctuates significantly, and we discovered that whole-blood RNA (gene) expression could become a new ABP parameter. The findings of this study will be beneficial for future research and development in this field.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Table S1: primers/probes; Table S2: Expression browser.
Author Contributions
Conceptualization, T.S.; methodology, T.S., Y.T. (Y. Takeuchi), and Y.K.; validation, T.S. and A.H.; formal analysis, T.S. and A.H.; investigation, T.S., N.O., and A.H.; writing—original draft preparation, T.S., N.O., K.N., and A.H.; writing—review and editing, T.S., A.H., N.O., Y.K., K.N., T.T., K.W., Y.T. (Y. Takeuchi), N.Y., and Y.T. (Y. Takahashi); visualization, T.S. and A.H.; supervision, T.S. and Y.T. (Y. Takahashi); project administration, T.S.; funding acquisition, T.S. and K.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grant of the JAPAN Anti-Doping Agency in 2021, and by the research funding for anti-doping within University of Tsukuba, Japan (academic year 2022). This research was also partially supported by the Organization for Open Facility Initiatives, University of Tsukuba, Japan (academic year 2023).
Institutional Review Board Statement
The study was conducted in accordance with the guidelines (corporation regulation; no. 50, 21 July 2005) of the animal experiments by the Animal Ethics Committee of the University of Tsukuba. The approval number was 22-125.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not appliable.
Acknowledgments
Not appliable.
Conflicts of Interest
Not appliable.
References
- A Japan Anti-Doping Agency (JADA). What Is Anti-Doping? Available online: https://www.playtruejapan.org/about/ (accessed on 5 February 2024).
- The World Anti-Doping Agency (WADA). Who We Are? Available online: https://www.wada-ama.org/en/who-we-are (accessed on 5 February 2024).
- Reuters. Olympic = Bahrain track and field athlete provisionally suspended for blood doping violation (2021, August 9). Available online: https://jp.reuters.com/article/idUSKBN2FA03U/ (accessed on 5 February 2024).
- Division of Molecular Target and Gene Therapy Products, National Institute of Health Sciences. Approved Gene Therapy Products (Updated January 23, 2024). Available online: https://www.nihs.go.jp/mtgt/pdf/section1-1.pdf (accessed on 5 February 2024).
- Li, X.; Le, Y.; Zhang, Z.; Nian, X.; Liu, B.; Yang, X. Viral Vector-Based Gene Therapy. Int. J. Mol. Sci. 2023, 24, 7736. [Google Scholar] [CrossRef] [PubMed]
- The World Anti-Doping Agency (WADA). The World Anti-Doping Code International Standard Prohibited List 2024; WADA: Montreal, QC, Canada, 2024. [Google Scholar]
- The World Anti-Doping Agency (WADA). World Anti-Doping Code 2021; WADA: Montreal, QC, Canada, 2021. [Google Scholar]
- Sugasawa, T.; Nakano, T.; Fujita, S.-I.; Matsumoto, Y.; Ishihara, G.; Aoki, K.; Yanazawa, K.; Ono, S.; Tamai, S.; Manevich, L.; et al. Proof of Gene Doping in a Mouse Model with a Human Erythropoietin Gene Transferred Using an Adenoviral Vector. Genes (Basel) 2021, 12, 1249. [Google Scholar] [CrossRef] [PubMed]
- Yanazawa, K.; Sugasawa, T.; Aoki, K.; Nakano, T.; Kawakami, Y.; Takekoshi, K. Development of a Gene Doping Detection Method to Detect Overexpressed Human Follistatin Using an Adenovirus Vector in Mice. PeerJ 2021, 9, e12285. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, T.; Aoki, K.; Yanazawa, K.; Takekoshi, K. Detection of Multiple Transgene Fragments in a Mouse Model of Gene Doping Based on Plasmid Vector Using TaqMan-qPCR Assay. Genes (Basel) 2020, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Sugasawa, T.; Yanazawa, K.; Watanabe, K.; Takemasa, T.; Takeuchi, Y.; Aita, Y.; Yahagi, N.; Yoshida, Y.; Kuji, T.; et al. The Detection of Trans Gene Fragments of hEPO in Gene Doping Model Mice by Taqman qPCR Assay. PeerJ 2020, 8, e8595. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, T.; Aoki, K.; Watanabe, K.; Yanazawa, K.; Natsume, T.; Takemasa, T.; Yamaguchi, K.; Takeuchi, Y.; Aita, Y.; Yahagi, N.; et al. Detection of Transgenes in Gene Delivery Model Mice by Adenoviral Vector Using ddPCR. Genes (Basel) 2019, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.A.A.C.; Cohen Tervaert, J.M.; Schepers, F.M.L.; Vliegenthart, A.D.B.; Rotmans, J.I.; Daniels, J.M.A.; Burggraaf, J.; Cohen, A.F. Erythropoietin doping in cycling: Lack of evidence for efficacy and a negative risk-benefit: Erythropoietin doping in cycling. Br. J. Clin. Pharmacol. 2013, 75, 1406–1421. [Google Scholar] [CrossRef] [PubMed]
- DiMeo, P. Why Lance Armstrong? Historical context and key turning points in the ‘Cleaning Up’ of professional cycling. Int. J. Hist. Sport 2014, 31, 951–968. [Google Scholar] [CrossRef]
- Wadler, G.I. The status of doping and drug use and the implications for boxing. Clin. Sports Med. 2009, 28, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Fitch, K. Proscribed drugs at the Olympic Games: Permitted use and misuse (doping) by athletes. Clin. Med. 2012, 12, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R. Analytical study of doping cases of banned substances during Olympics games from 1968 to 2012. Int. J. Phys. Educ. Sports Health 2016, 3, 3–37. [Google Scholar]
- Maurya, S.; Sarangi, P.; Jayandharan, G.R. Safety of Adeno-Associated Virus-Based Vector-Mediated Gene Therapy-Impact of Vector Dose. Cancer Gene Ther. 2022, 29, 1305–1306. [Google Scholar] [CrossRef] [PubMed]
- Gene Therapy Clinical Trials Worldwide. Vectors. Available online: https://a873679.fmphost.com/fmi/webd/GTCT (accessed on 9 February 2024).
- The World Anti-Doping Agency (WADA). Athlete Biological Passport (ABP) Operating Guidelines; WADA: Montreal, QC, Canada, 2021. [Google Scholar]
- Primer Designing Tool. Available online: https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 16 April 2024).
- The World Anti-Doping Agency (WADA). Gene Doping Detection Based on Polymerase Chain Reaction (PCR), Version 1.0; WADA: Montreal, QC, Canada, 2021. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Castanza, A.S.; Recla, J.M.; Eby, D.; Thorvaldsdóttir, H.; Bult, C.J.; Mesirov, J.P. Extending Support for Mouse Data in the Molecular Signatures Database (MSigDB). Nat. Methods 2023, 20, 1619–1620. [Google Scholar] [CrossRef] [PubMed]
- Venny ver.2.1.0. Available online: https://csbg.cnb.csic.es/BioinfoGP/venny.html (accessed on 11 April 2024).
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV Serotypes 1-9 Mediated Gene Expression and Tropism in Mice after Systemic Injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Iseki, A.; Morita, Y.; Nakauchi, H.; Ema, H. Hematopoietic Stem Cells in the Mouse Spleen. Blood 2008, 112, 2421. [Google Scholar] [CrossRef]
- Baoutina, A.; Coldham, T.; Bains, G.S.; Emslie, K.R. Gene Doping Detection: Evaluation of Approach for Direct Detection of Gene Transfer Using Erythropoietin as a Model System. Gene Ther. 2010, 17, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Gene Doping Detection by next Generation Sequencing. Available online: https://www.wada-ama.org/en/resources/scientific-research/gene-doping-detection-next-generation-sequencing (accessed on 16 April 2024).
- Marchand, A.; Roulland, I.; Semence, F.; Ericsson, M. EPO Transgene Detection in Dried Blood Spots for Antidoping Application. Drug Test. Anal. 2021, 13, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- de Boer, E.N.; van der Wouden, P.E.; Johansson, L.F.; van Diemen, C.C.; Haisma, H.J. A Next-Generation Sequencing Method for Gene Doping Detection That Distinguishes Low Levels of Plasmid DNA against a Background of Genomic DNA. Gene Ther. 2019, 26, 338–346. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Gene Therapy for Treatment of Certain Patients with Duchenne Muscular Dystrophy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-certain-patients-duchenne-muscular-dystrophy (accessed on 16 April 2024).
- FDA Approves First Gene Therapy for Adults with Severe Hemophilia A. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-adults-severe-hemophilia (accessed on 16 April 2024).
- Office of the Commissioner FDA Approves Innovative Gene Therapy to Treat Pediatric Patients with Spinal Muscular Atrophy, a Rare Disease and Leading Genetic Cause of Infant Mortality. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (accessed on 16 April 2024).
- FDA Approves Novel Gene Therapy to Treat Patients with a Rare Form of Inherited Vision Loss. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (accessed on 16 April 2024).
Figure 1.
An overview of animal experiments. A: short-term experiments; B: long-term experiments including repeated sampling.
Figure 1.
An overview of animal experiments. A: short-term experiments; B: long-term experiments including repeated sampling.
Figure 2.
Establishment of a mouse model of gene doping with the rAAV9-hEPO vector. A: Body weight; B: tissue weight of liver and spleen; C: external view of tissue of representative samples; D: blood volume; E: RBCs; F: HGB; G: HCT; H: ELIZA reaction plates (N = 8 per group; measured in triplicate); I: quantification of plasma hEPO by ELISA; J: gene expression analysis of hematopoietic marker genes in each tissue; K: pathological specimen images of liver and spleen (HE staining). N.D.: not detected. * p<0.05, ** p<0.01, *** p<0.001.
Figure 2.
Establishment of a mouse model of gene doping with the rAAV9-hEPO vector. A: Body weight; B: tissue weight of liver and spleen; C: external view of tissue of representative samples; D: blood volume; E: RBCs; F: HGB; G: HCT; H: ELIZA reaction plates (N = 8 per group; measured in triplicate); I: quantification of plasma hEPO by ELISA; J: gene expression analysis of hematopoietic marker genes in each tissue; K: pathological specimen images of liver and spleen (HE staining). N.D.: not detected. * p<0.05, ** p<0.01, *** p<0.001.
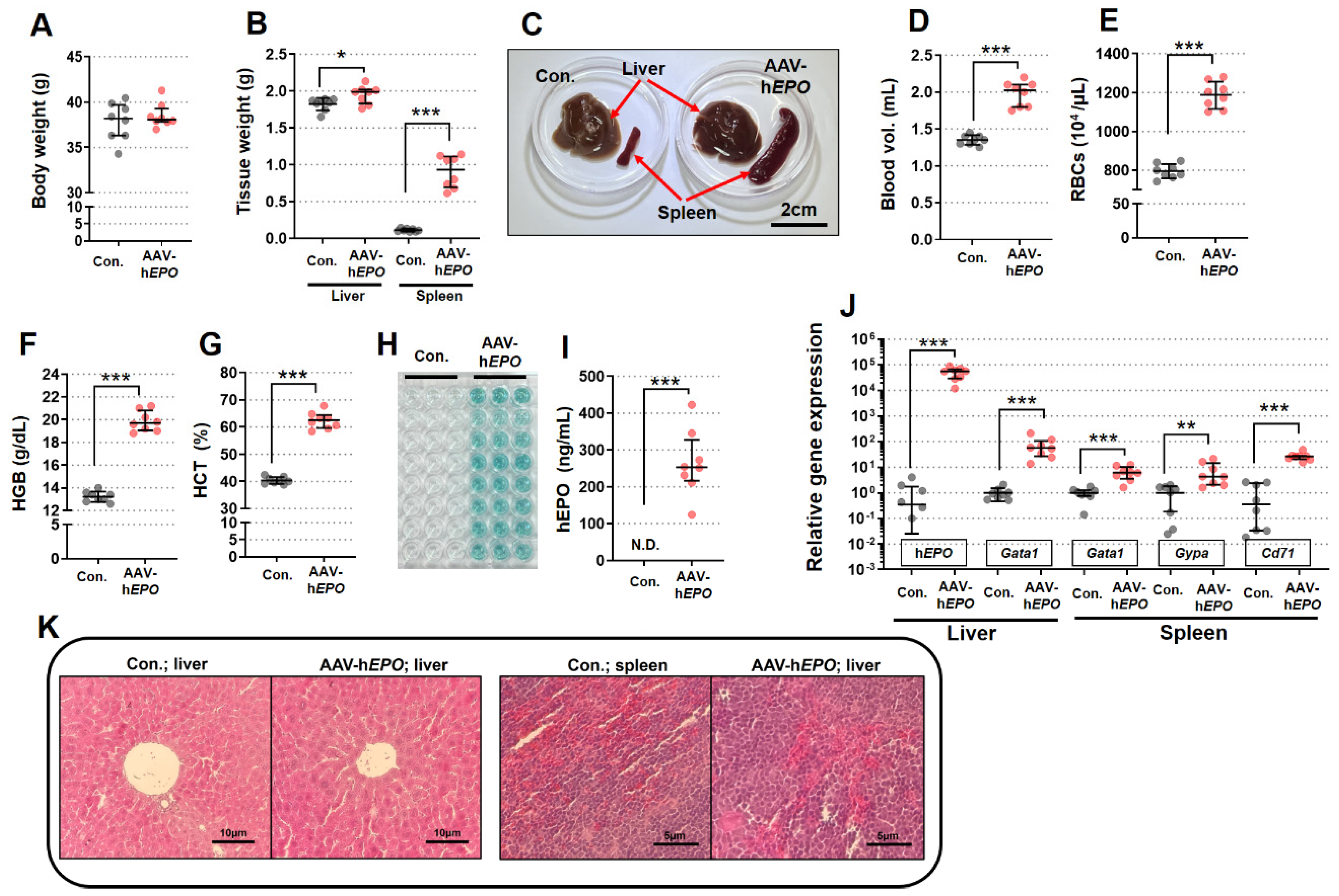
Figure 3.
Detection of direct proof of gene doping in whole-blood DNA. A: Designing strategy of primer/probe pairs for the rAAV9-hEPO vector; B: detection of the direct proof using four primer/probe pairs; C: gel electrogram of the amplicons; D: waveforms and sequences of Sanger sequences using the amplicons as templates. N.D.: not detected; P.C.: positive control; S.: samples; R.S.: reference sequence. *** p<0.001 vs. Con. or human cells.
Figure 3.
Detection of direct proof of gene doping in whole-blood DNA. A: Designing strategy of primer/probe pairs for the rAAV9-hEPO vector; B: detection of the direct proof using four primer/probe pairs; C: gel electrogram of the amplicons; D: waveforms and sequences of Sanger sequences using the amplicons as templates. N.D.: not detected; P.C.: positive control; S.: samples; R.S.: reference sequence. *** p<0.001 vs. Con. or human cells.
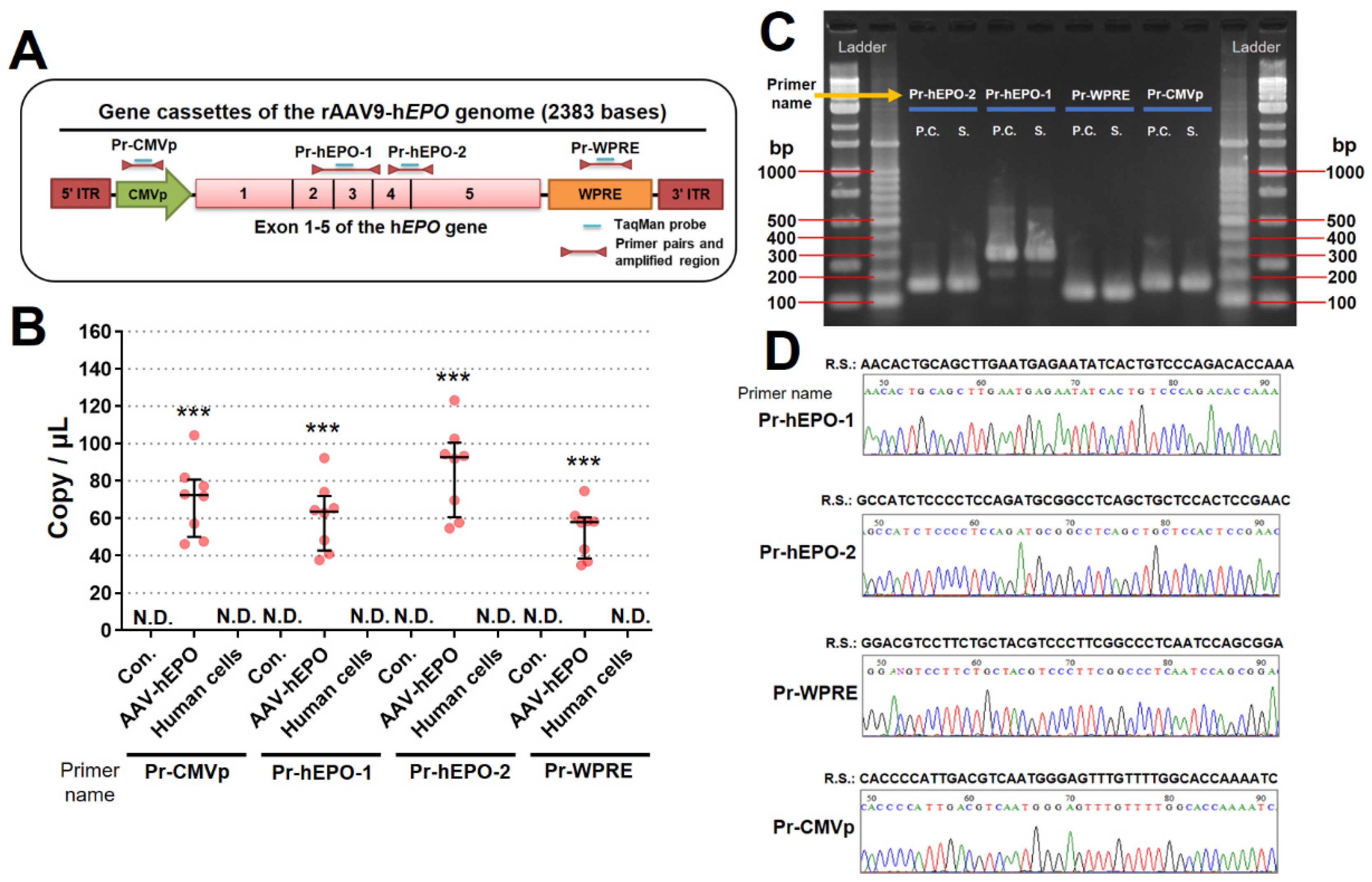
Figure 4.
Understanding the results and overall gene expression of total RNA-seq and bioinformatics analysis. A: PCA plot; B: cluster dendrogram; C: heatmap for the 1144 genes; D: results of top 3 gene sets on the GSEA analysis. NES: normalized enriched score.
Figure 4.
Understanding the results and overall gene expression of total RNA-seq and bioinformatics analysis. A: PCA plot; B: cluster dendrogram; C: heatmap for the 1144 genes; D: results of top 3 gene sets on the GSEA analysis. NES: normalized enriched score.
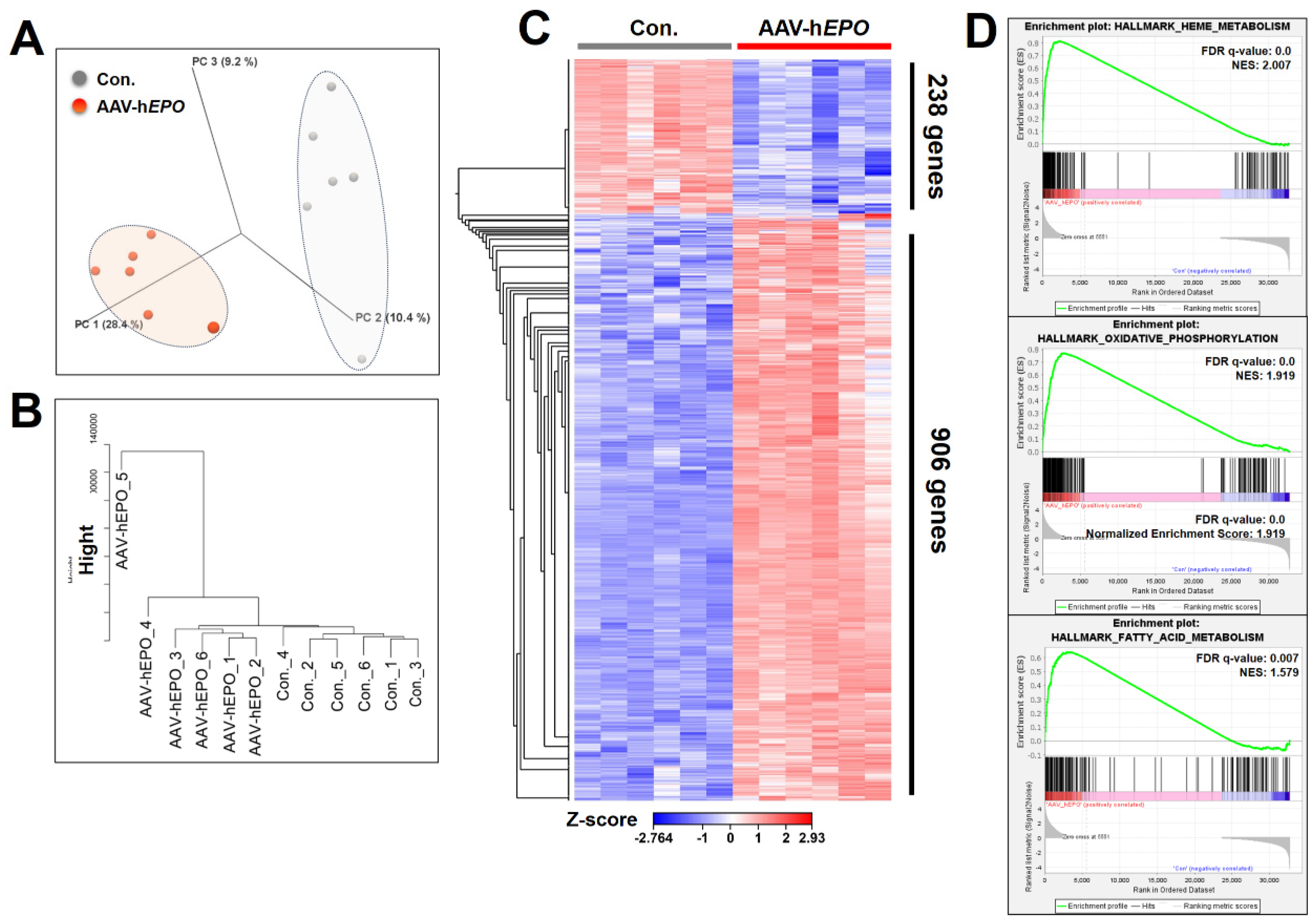
Figure 5.
Identified 12 promising genes with drastic change in the expression. A: Venn diagram analysis to identify the 12 genes; B: heatmap of the 12 genes; C: bar graph of the relative expression value (average of TPM value) of 12 genes; D: scatter plot collared the 12 genes; E: volcano plot collared the 12 genes; F: TB Green qPCR assay for all mice (N=8, respectively) of the top 3 genes. G: mtDNA copy numbers. *** p<0.001.
Figure 5.
Identified 12 promising genes with drastic change in the expression. A: Venn diagram analysis to identify the 12 genes; B: heatmap of the 12 genes; C: bar graph of the relative expression value (average of TPM value) of 12 genes; D: scatter plot collared the 12 genes; E: volcano plot collared the 12 genes; F: TB Green qPCR assay for all mice (N=8, respectively) of the top 3 genes. G: mtDNA copy numbers. *** p<0.001.
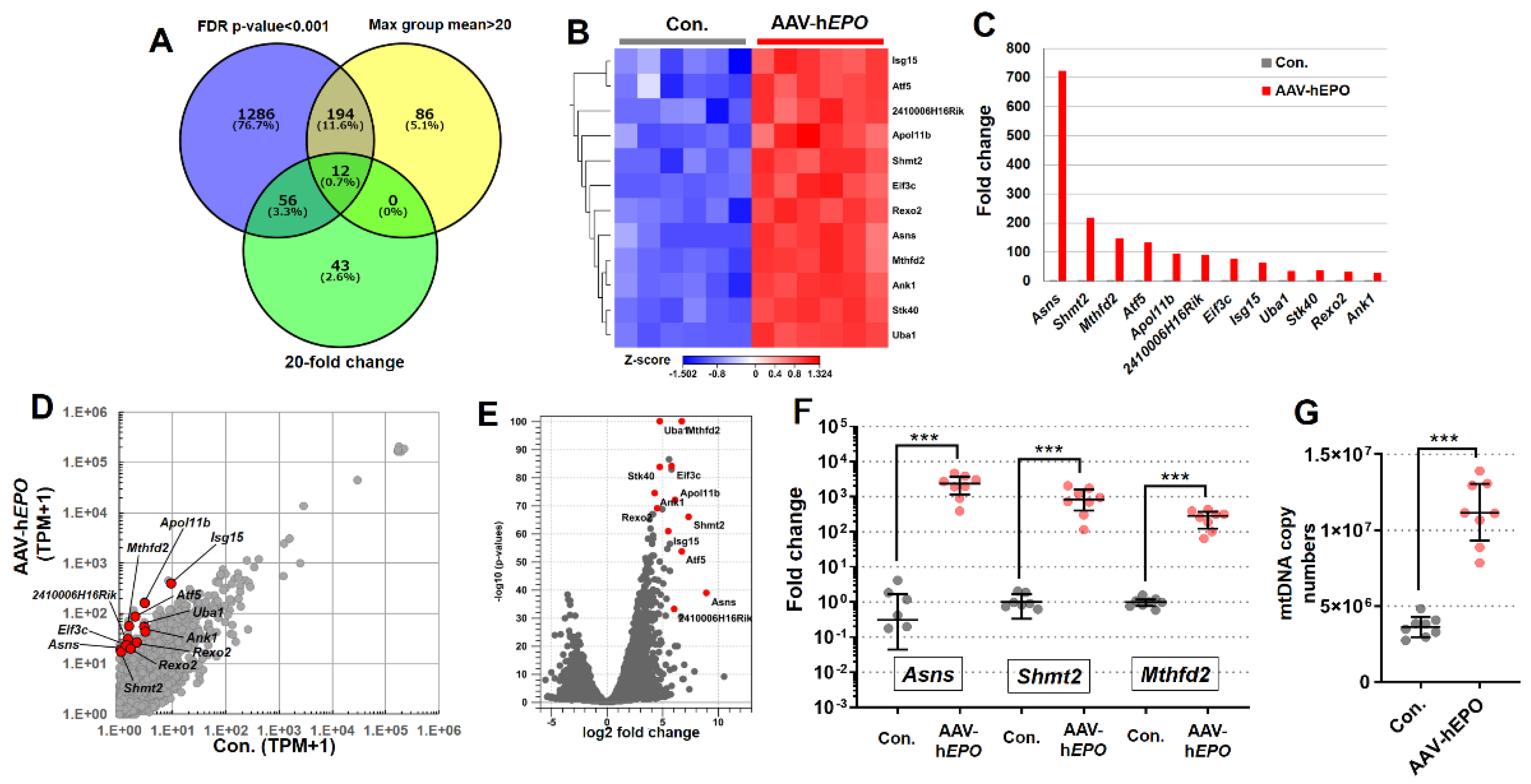
Figure 6.
Detection of direct or indirect proof of gene doping for 30 days. A to D: Detection of direct proof using each primers-probe; E to G: detection of indirect proof as 3 gene expressions leading to the concept of ABP. * p<0.05, ** p<0.01, **** p<0.0001 vs. Pre, respectively.
Figure 6.
Detection of direct or indirect proof of gene doping for 30 days. A to D: Detection of direct proof using each primers-probe; E to G: detection of indirect proof as 3 gene expressions leading to the concept of ABP. * p<0.05, ** p<0.01, **** p<0.0001 vs. Pre, respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



