
Submitted:
06 May 2024
Posted:
07 May 2024
You are already at the latest version
Abstract
Advanced glycation end-products (AGEs) form through non-enzymatic glycation of various proteins. Optic nerve degeneration is a frequent complication of diabetes, and retinal AGEs accumulation is strongly linked to diabetic retinopathy development. Type 2 diabetes mellitus (T2DM) is a major risk factor for Alzheimer's disease (AD), with patients often exhibiting optic axon degeneration in the nerve fiber layer. Notably, a gap exists in our understanding of how AGEs contribute to neuronal degeneration in the optic nerve within the context of both diabetes and AD. Our previous work demonstrated that glyceraldehyde (GA)-derived toxic advanced glycation end-products (TAGE) disrupt neurite outgrowth through TAGE-β-tubulin aggregation and tau phosphorylation in neural cultures. In this study, we further illustrated GA-induced suppression of optic nerve axonal elongation via abnormal β-tubulin aggregation in mouse retinas. Elucidating this optic nerve degeneration mechanism holds promise for bridging the knowledge gap regarding vision loss associated with DM and AD.
Keywords:
toxic advanced glycation-end products
; glyceraldehyde
; axonal elongation
; β-tubulin
; tau
1. Introduction
Axonal damage represents an early-stage neurodegenerative disorder within the central nervous system (CNS). The retina and optic nerve, being accessible regions of the CNS, offer unique substrates for investigating the impact of retinal ganglion cell (RGC) injury on optic nerve axons. The injury to RGCs, coupled with the antero-/retro-grade loss of RGC axons, manifests as a characteristic pathological alteration. Hence, comprehending the molecular mechanisms underlying axonal degeneration holds significant scientific import. Advanced glycation end-products (AGEs) form under hyperglycemic conditions via a non-enzymatic Maillard reaction between proteins and/or amino acids and reducing sugars [1]. The formation and subsequent accumulation of AGEs in various tissues progress during normal aging, with a notably accelerated pace in type 2 diabetes mellitus (T2DM). Considering that the incidence of Alzheimer's disease (AD) surpasses 2-5 times in patients with T2DM [2], numerous studies have explored whether T2DM serves as a clinical risk factor for the onset and progression of AD [3,4]. Several investigations have delved into the mechanisms underlying AGEs-induced neurotoxicity to elucidate the pathological mechanisms of T2DM-related neurodegeneration. In diabetic retinopathy, AGEs impede normal cellular functions such as axonal transport and intracellular protein trafficking [5,6]. Conversely, optic neuropathy, encompassing optic nerve degeneration and RGC loss, has been documented in AD retinas [7,8]. Optic neuropathy in AD correlates with AGEs-dependent cell death [9]. Nonetheless, no reports of AGEs-dependent axonal degeneration are associated with AD or DM.
In our previous work, we demonstrated that glyceraldehyde (GA), a metabolic intermediate of glucose (Glu) and fructose, elicits AD-like changes, including axonal degeneration, and elevates total tau and phosphorylated tau protein levels in an AGEs-dependent manner [10]. We illustrated that GA-derived AGEs exhibit robust neurotoxicity [11] and that GA-AGEs display greater neurotoxic effects compared to Glu-AGEs in neuronal cultures, thus designating them as toxic AGEs (TAGE) [1]. TAGE has been detected in axons and intracellular neuronal cells within the hippocampus and parahippocampal gyrus of patients with AD [12]. Recently, through proteomics analysis, we identified β-tubulin as one of the proteins targeted by TAGE [13]. Microtubules consist of repeating units of heterodimers between α-tubulin and β-tubulin, and their assembly is a crucial event implicated in axon outgrowth in vitro models such as SH-SY5Y, a human neuroblastoma cell line. As tau phosphorylation diminishes its binding to microtubules, GA-induced tau phosphorylation leads to axonal degeneration via TAGE-mediated abnormal aggregation of β-tubulin in SH-SY5Y cells [13]. However, the impact of TAGE-β-tubulin on axonal elongation in the adult mouse optic nerve, particularly in a model utilizing intraocular injection, remains unexplored. Hence, we utilized zymosan, known to induce axonal regeneration in adults following optic nerve injury [14,15], to investigate the effects of GA on zymosan-induced axonal elongation in adult mice after optic nerve injury. These findings hold promise for developing novel therapeutic strategies against AGEs-related axonal degeneration disorders within the CNS.
2. Results
2.1. Detection of TAGE Proteins and Identification TAGE-β-Tubulin in Retina after GA Treatment
Our prior work demonstrated that 1 mM GA induced TAGE formation in SH-SY5Y cells [13]. To investigate this further, we examined if intraocular injection of 1 mM GA could trigger TAGE formation. GA treatment significantly increased TAGE levels starting from 3 days post-injection (Figure 1A). Interestingly, we observed a significant decrease in the levels of 55 kDa monomeric β-tubulin protein from day 3 onwards following GA treatment (Figure 1B,E). Conversely, the intensity of aggregated β-tubulin bands increased in a time-dependent manner (Figure 1B,D: Lower band, C: Upper band).
2.2. Colocalization of TAGE and β-Tubulin in Retina after GA Treatment
We employed immunohistochemistry to investigate the colocalization of TAGE and β-tubulin within the retina (Figure 2). β-tubulin immunostaining at day 0 revealed positive staining primarily localized in the ganglion cell layer (GCL) and nerve fiber layer (NFL) (Figure 2A). Notably, neither the levels nor the localization of β-tubulin appeared to be significantly altered following intraocular GA treatment at 1 day (Figure 2D) or 3 days (Figure 2G).
Consistent with the β-tubulin distribution, weak TAGE signals were observed in the GCL and NFL of untreated retinas on day 0 (Figure 2A-C). However, strong TAGE immunoreactivity was evident from day 1 (Figure 2E) to day 3 (Figure 2H) following GA treatment, compared to day 0 (Figure 2B). Notably, the increased TAGE signal (Figure 1 B,E,H) in the GCL co-localized with the β-tubulin layer (Figure 2A,D,G). Merged images further confirmed the colocalization of TAGE and β-tubulin within the GCL and NFL (Figure 2C,F,I).
2.3. Pyridoxamine (PM) Dose-Dependently Inhibited TAGE Formation and TAGE-β-Tubulin Aggregation in Retina after GA Treatment
We investigated the potential of PM, an AGEs inhibitor, to counteract GA-induced TAGE formation and β-tubulin aggregation. Notably, PM at concentrations of 250-500 μM alone did not affect TAGE levels or β-tubulin aggregation compared to the vehicle control (data not shown). Slot blot analyses revealed a significant increase in TAGE formation upon GA treatment. This effect was dose-dependently attenuated by co-treatment with PM (Figure 3A). Similarly, Western blot analysis (Figure 3B) showed that GA decreased the levels of the 55 kDa monomeric β-tubulin band (Figure 3B,E) while increasing the intensity of the lower (Figure 3B,D) and upper (Figure 3B,C) bands indicative of aggregation. PM treatment significantly attenuated these GA-induced changes in a dose-dependent manner (Figure 3B-E).
2.4. PM Dose-Dependently Inhibited TAGE Formation in GCL and NFL after GA Treatment
PM showed dose-dependent suppression of TAGE levels in GCL and NFL. Intraocular injection of 1 mM GA at day 3 dramatically increased TAGE levels in both the GCL and NFL compared to the vehicle control (Figure 4A vs. Figure 4B). Notably, PM treatment at concentrations of 250 μM (Figure 4C) and 500 μM (Figure 4D) strongly attenuated TAGE levels in the GCL and NFL in a dose-dependent manner at day 3.
2.5. TAGE Inhibited Zymosan-Induced Axonal Elongation after Injury
We further investigated the in vivo effects of GA on zymosan-induced axonal elongation following nerve injury (Figure 5A-E). Intraocular zymosan injection triggered axonal elongation as revealed by GAP43 staining, compared to the vehicle control (Figure 5B,E vs. Figure 5A,E). PM alone did not affect axonal elongation in the vehicle group (data not shown). Interestingly, GA treatment significantly inhibited zymosan-induced axonal elongation compared to the vehicle control (Figure 5C,E). Furthermore, PM co-treatment significantly reversed the inhibitory effects of GA on axonal elongation (Figure 5D,E). These findings suggest a protective role for PM against GA-mediated suppression of axonal outgrowth.
2.6. GA-Increased Total Tau and Tau Phosphorylation in AGEs Dependent Manner
Previous studies have shown increased intracellular tau phosphorylation in the cortex of patients with AD [16], and we previously reported similar findings in GA-treated SH-SY5Y cells [10]. To investigate whether PM could modulate GA-induced tau phosphorylation, retinas were pre-treated with PM for 30 min followed by GA treatment for 3 days. Western blot analysis revealed significant increases in both total tau (Figure 6A,C) and tau phosphorylation (Figure 6A,B) levels upon GA treatment. PM co-treatment significantly attenuated these GA-induced increases in tau phosphorylation levels (Figure 6A-C), suggesting a protective role for PM against abnormal tau modifications.
3. Discussion
The alterations observed in the retina of patients with AD are linked to neuronal degeneration and loss, optic nerve degeneration, accumulation of amyloid-beta (Aβ) in the optic disk, and visual functional impairment [7,17,18]. Similar mechanisms of neurodegeneration observed in the brain have been demonstrated in retinal neurons, including RGCs and the optic nerve, in the eyes of patients with AD [19]. In contrast, normal visual function relies on the proper functioning of retinal neurons; thus, the loss of neuronal function must underlie vision impairment in diabetes [20]. Recent studies suggest that retinal degeneration in diabetes may result not only from vasculopathy but also from neuropathy, encompassing both neuronal and axonal loss [21]. AGEs play a pivotal role in the progression of diabetic retinopathy, contributing to dysfunction and various losses of retinal neurons [22]. Moreover, the extent of AGEs immunolabeling was higher in older donor eyes than that in younger ones [23]. Elevated levels of AGEs in RGCs and the optic nerve head signify axonal degeneration and visual loss, even in eyes affected by glaucoma [23].
Bikbova et al. reported that various AGEs, including Glu-AGEs and glyceraldehyde-AGEs (GA-AGEs), can induce apoptosis in retinal neurons and reduce the number of regenerating neurites in a retinal explant culture system [24]. Accumulating evidence suggests that cell death in retinal neurons and neurite abnormalities are linked to the early development of diabetic retinopathy [25]. However, the underlying mechanism remains elusive. This study aimed to explore the association between AD-related and AGEs-dependent axonal degeneration in the optic nerve in diabetes mellitus (DM). While most research on AD has centered on Glu-AGEs, we previously reported the critical involvement of α-hydroxyaldehydes, such as GA, a metabolic intermediate of Glu and fructose, and glycolaldehyde, including glyoxal, methylglyoxal, and 3-deoxyglucosone, in protein glycation. GA-derived AGEs exhibit higher cytotoxicity than other sugar-dependent AGEs, including Glu-AGEs [26]. Hence, GA-AGEs are also referred to as TAGE. TAGE have been detected in the axons and cytosol of neuronal cells in the hippocampus and parahippocampal gyrus of the brain tissues of patients with AD, but not in senile plaques (SP) or glial cells [27]. In contrast, Glu-AGEs were found in SP amyloid cores and astrocytes. Additionally, TAGE were exclusively detected intracellularly, while Glu-AGEs were found both intra- and extracellularly. Based on this evidence, TAGE may represent a promising candidate for treating neurodegeneration in patients with AD.
In our previous study, GA was found to reduce Aβ42 levels in culture media [10]. Additionally, GA elevated the intracellular levels of total tau and phosphorylated tau in the human neuroblastoma cell line SH-SY5Y [10]. Through proteomics analysis using matrix-assisted laser desorption ionization-time of flight mass spectrometry, we identified β-tubulin as a target protein of TAGE. Microtubules, composed of α- and β-tubulin heterodimers, play a crucial role in neurite outgrowth. In our SH-SY5Y cell model, GA facilitated the formation of TAGE-β-tubulin and abnormal β-tubulin aggregation, resulting in the inhibition of axonal elongation [13]. Interestingly, β-tubulin can also undergo glycation by Glu in a DM experimental model [28]. However, under our experimental conditions, Glu did not induce abnormal β-tubulin aggregation or inhibit neurite outgrowth [13]. GA is derived from two distinct pathways: the glycolytic and fructose metabolic pathways [29]. In hyperglycemic conditions, increased intracellular Glu stimulates the polyol pathway to generate fructose in insulin-independent nerve tissue [30]. Fructose is further metabolized to dihydroxyacetone phosphate and GA by aldolase [31]. Consequently, GA production is promoted. Aldolase proteins are expressed in RGCs and other retinal cell layers [32]. However, a high-fructose diet has been shown to induce retinopathy by suppressing synaptic plasticity [33]. Elevated levels of the fructose transporter have been detected in diabetic retinopathy [34]. These findings suggest that both GA and TAGE are produced in the retinas of patients with diabetes.
Previous studies have demonstrated that PM, a natural form of vitamin B6, acts as an inhibitor of AGEs [35]. PM suppresses the formation of TAGE-β-tubulin and alleviate the GA-induced inhibition of axonal elongation. Vitamin B6 encompasses three subtypes: PM, pyridoxal, and pyridoxine. PM can trap aldehyde groups via its amino group, thereby preventing the formation of AGEs under physiological conditions. PM can effectively suppress AGEs formation in various proteins both in vitro and in vivo, thereby preventing the development of diabetic complications such as hemoglobin glycation and lipoxidation reactions [36,37]. These compounds, including PM, serve as potent AGEs inhibitors, attenuating diabetes-related nephropathy, neuropathy, and retinopathy [38]. Moreover, a deficiency in vitamin B6 is associated with abnormal nerve growth, contributing to conditions such as schizophrenia, depression, and central neuropathy [39]. Additionally, PM inhibits the early development of retinopathy in experimental diabetic models [40].
In this study, intraocular injection of GA increased the TAGE levels in the GCL and NFL. Furthermore, GA induced β-tubulin aggregation and inhibited zymosan-induced axonal elongation even in vivo. Benowitz et al. reported that zymosan induces macrophage invasion and appears to release trophic factors such as oncomodulin, stromal cell-derived factor 1, and CCL5 chemokine that can promote axonal elongation [41,42,43].
Tau proteins, integral components of paired helical filaments, exhibit distinct characteristics such as high aggregation propensity and hyperphosphorylation [44,45]. In neurodegenerative conditions such as AD, tau dissociates from microtubules within neurofibrillary tangles and aggregates in the cytosol, facilitating self-aggregation and phosphorylation [45]. Consequently, abnormal β-tubulin aggregation may increase the detachment of tau proteins from microtubules. While the precise mechanism remains unknown, the elevation in total tau levels is believed to stem from axonal loss [46]. Recent studies have highlighted GA ability to induce AD-like alterations in vitro [47,48]. Piccirillo et al. documented GA-induced phosphorylation of tau at T212 and T214, while another study illustrated the impact of GA on tau phosphorylation at S199 and S396, alongside reduced axonal outgrowth [49,50]. Consequently, tau proteins become hyperphosphorylated at various sites, aggregating into neurofibrillary tangles within AD patient brains. Notably, tau phosphorylation at T181, T217, and T231 in CSF is a promising biomarker for AD diagnosis [51]. T217 and T231 are indicative of postsynaptic pathology, while T181 marks axonal abnormalities [52]. In our recent study, TAGE-β-tubulin accelerated abnormal aggregation and hindered neurite outgrowth, accompanied by T181 phosphorylation [10,13]. Similar outcomes were observed in the retinal optic nerve model. Further investigations are warranted since the detailed mechanisms underlying GA-induced β-tubulin aggregation and tau phosphorylation remain elusive.
While our study provides valuable insights into the pathogenic mechanisms underlying visual dysfunction in DM and AD, several limitations warrant consideration. First, the use of animal models may not fully capture the complexity of human disease progression, potentially limiting the translational relevance of our findings. Additionally, our study focused on a single time point analysis, precluding a comprehensive assessment of the long-term effects of GA-induced β-tubulin aggregation and neurite outgrowth inhibition. Furthermore, while we elucidated the involvement of AGEs in mediating neurodegeneration, the specific molecular mechanisms remain incompletely understood. Addressing these limitations in future research endeavors could provide a more comprehensive understanding of visual dysfunction in these debilitating conditions.
4. Materials and Methods
4.1. Ethics Statement
All animal care and handling procedures were approved by the Animal Care and Use Committee of Suzuka University of Medical Science (No. 78). All surgeries were performed under sodium pentobarbital anesthesia, and efforts were made to minimize suffering.
4.2. Chemicals
Zymosan, an axonal elongation inducer, was purchased from Sigma-Aldrich (Japan). The AGEs inhibitor pyridoxamine was purchased from Sigma-Aldrich (Japan).
4.3. Preparation of Anti-TAGE Antibody
Immunoaffinity-purified anti-TAGE antibodies were prepared according to previously established methods [53]. Despite their specificity, these antibodies did not recognize well-characterized AGEs structures, including triosidines, GA-derived pyridinium compounds, GA-derived pyrrolopyridinium lysine dimers, methylglyoxal-derived hydroimidazolone-1, and argpyrimidine. Moreover, the antibodies failed to detect AGEs with unknown structures, such as Glu-AGEs and fructose-derived AGEs. However, they exhibited specific recognition of unique dimeric/trimeric TAGE structures [54].
4.4. Animals and Surgery
Male C57BL6 mice aged 8–9 weeks were utilized for this study. The mice were administered sodium pentobarbital intraperitoneally at a dosage of 30–40 mg/kg and xylazine at 5 mg/kg. Intraperitoneal injections of various reagents were administered into the sclera and retina using a 30 G needle positioned 1–2 mm superior to the optic nerve head to prevent lens injury [55]. Optic nerve injury was induced 2 mm behind the eye using angle jeweler forceps (Dummont #5) for a duration of 10 sec [55]. The mice were housed in clear plastic cages and maintained under a 12-hour light/dark cycle at 23°C. PM and/or zymosan were administered intraocularly 30 min prior to GA injection.
4.5. Immunohistochemistry
Tissue fixation and cryosectioning were performed as previously described [56]. Briefly, the eyes were enucleated and fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH7.4), and cryoprotected in 20% sucrose. The eyes and optic nerves were then embedded in optimal cutting temperature compound (TissueTek; Miles, Eikhart, IN) and cryosectioned at a thickness of 12 μm (retina) and 16 μm (optic nerve). The frozen sections were mounted onto silane-coated glass slides. After washing and blocking with Blocking One (NakalaiTesque, Kyoto), sections were incubated with various antibodies, such as β-tubulin (Cell Signaling Technology, Japan), TAGE antibody, and GAP43 at 4°C overnight. The sections were then incubated with fluorescent anti-IgG (Thermo Fisher Scientific, Japan) at 23°C.
4.6. Slot Blot Analysis
To assess TAGE levels, we performed a slot blot analysis. Retinal samples were isolated and homogenized on each day of GA injection. Equal amounts of protein (30 μg) were applied to nitrocellulose membranes by vacuum filtration. The membranes were blocked with Blocking One for 1 h at 23°C and then incubated with anti-TAGE antibody. The positive bands were detected using a BCIP/NBT kit (Sera Care, Milford, MA, USA) and analyzed densitometrically as previously described [57]. All experiments were repeated at least thrice.
4.7. Western Blot Analysis
Retinal tissue proteins were extracted, and sample aliquots (30 μg) were subjected to polyacrylamide electrophoresis using a 5-20% gradient gel, as previously described [58]. The separated proteins were transferred to a nitrocellulose membrane and incubated with Blocking One and then the primary antibodies (all at 1:500): anti-β-tubulin, and anti-total tau, or anti-phosphorylated tau (Cell Signaling Technology, Japan). The secondary anti body was horseradish peroxidase-labeled anti-IgG (1:1000; Proteintech, Japan). The chemiluminescent HRP substrate (Millipore, Burlington, MA, USA). was used to detect the protein bands. The bands were densitometrically analyzed using Scion Image Software (Scion Corp., Frederick, MD, USA). All experiments were performed at least in triplicate.
4.8. Quantitation of Axonal Elongation of Optic Nerve in Vivo
The mice were sacrificed 10 days after optic nerve injury and perfused with 4% paraformaldehyde. Optic nerves were impregnated with 5% and then 20% sucrose, embedded in Optimal Cutting Temperature Compound (Sakura Fine Technical, Tokyo, Japan), and cut into longitudinal sections of 10 μm thickness. Aonal elongation was visualized by staining with a sheep anti-GAP43 antibody (1:250, the antibody was kindly provided by Dr. Benowitz, Children’s Hospital, Harvard Medical School, Boston), followed by a fluorescently labeled secondary antibody, and captured by fluorescence microscopy (BZ-9000, Keyence, Osaka, Japan). Axons were counted manually in at least eight sections per condition (six mice per treatment) at prespecified distances (250 μm) from the injury site. The numbers of axonal elongation were counted as described by groups of Dr. Benowitz [59].
4.9. Statistics
All results were presented as mean ± standard error of the mean (SEM) for 3-5 experiments. Differences between groups were analyzed using one-way ANOVA followed by Dunnett’s multiple comparison test, performed using PASW Software (SPSS Inc., Chicago, IL, USA). Statistical significance was considered at p < 0.05.
5. Conclusions
The present study demonstrates that GA induces the aggregation of β-tubulin through an AGEs-dependent mechanism, resulting in the inhibition of neurite outgrowth even in vivo. Therefore, TAGE-β-tubulin emerges as a promising target for understanding the pathogenic mechanisms contributing to visual dysfunction in both DM and AD. Our findings support the idea that therapies aimed at reducing AGEs formation, such as supplementation with preventive medications such as AGEs inhibitors, may suppress neurodegeneration in the visual systems affected by both DM and AD.
Author Contributions
Conceptualization, Y.K., H.O., and M. T.; methodology, H.O., A.F., Y.K.; formal analysis and investigation, Y.K., H.O., and A.F. Writing-original draft preparation: H.O., A.F., and Y.K.; project administration and funding acquisition, Y.K. and M.T. All authors have read and agreed to the final version of the manuscript.
Funding
This work was supported by JSPS KAKENHI, Grant Number JP 21H04865, to M.T..
Institutional Review Board Statement
Animal experiments were approved by the Suzuka University of Medical Science Animal Experiment Ethics Committee (approval number 78).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting the findings of this study are available upon request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, JI.; Koriyama, Y. Effects of Toxic AGEs (TAGE) on Human Health. Cells. 2022, 11, 2178; [Google Scholar] [CrossRef]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic advanced glycation end products (TAGE) theory in Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2006, 21, 197–208; [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A. 1994, 91, 5710–5714; [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P.; et al. Non-enzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med. 1995, 1, 693–699; [Google Scholar] [CrossRef]
- Oshitari, T. Advanced Glycation End-Products and Diabetic Neuropathy of the Retina. Int J Mol Sci. 2023, 24, 2927; [Google Scholar] [CrossRef]
- Oshitari, T. ; Roy, S: Diabetes: A potential enhancer of retinal injury in rat retinas. Neurosci letter. 2005, 390, 25–30; [Google Scholar] [CrossRef]
- Hinton, D.R.; Sadun, A.A.; Blanks, J.C.; Miller, C.A. Optic-nerve degeneration in Alzheimer's disease. N Engl J Med. 1986, 315, 485–487; [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Bassi, C.J. Optic nerve damage in Alzheimer's disease. Ophthalmology. 1990, 97, 9–17; [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Ross-Cisneros, F.N.; Aggarwal, D.; Liang, C.Y.; Sadun, A.A. Receptor for advanced glycation end products is upregulated in optic neuropathy of Alzheimer's disease. Acta Neuropathol. 2009, 118, 381–389; [Google Scholar] [CrossRef]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer's disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci Rep. 2015, 5, 13313; [Google Scholar] [CrossRef]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J Neuropathol Exp Neurol. 2000, 59, 1094–1105; [Google Scholar] [CrossRef] [PubMed]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer's disease. Acta Neuropathol. 2004, 108, 189–193; [Google Scholar] [CrossRef] [PubMed]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The Effect of Glyceraldehyde-Derived Advanced Glycation End Products on β-Tubulin-Inhibited Neurite Outgrowth in SH-SY5Y Human Neuroblastoma Cells. Nutrients. 2020, 12, 2958; [Google Scholar] [CrossRef]
- Leon, S.; Yin, Y.; Nguyen, J.; Irwin, N.; Benowitz, L.I. Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci. 2000, 20, 4615–4626; [Google Scholar] [CrossRef] [PubMed]
- de, Lima, S. ; Koriyama, Y.; Kurimoto, T.; Oliveira, J.T.; Yin, Y.; Li, Y.; Gilbert, H.Y.; Fagiolini, M.; Martinez, A.M.; Benowitz, L. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc Natl Acad Sci U S A. 2012, 109, 9149–9154; [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.W.; Li, X.; Bjorkdahl, C.; Sjogren, M.J.; Alafuzoff, I.; Soininen, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Pei, J.J. Assessments of the accumulation severities of amyloid beta-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer's brains. Neurobiol Dis. 2006, 22, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.C.; McCoubrie, P.; McDonald, B.; Jobst, K.A. Myelinated axon number in the optic nerve is unaffected by Alzheimer's disease. Br J Ophthalmol. 1995, 79, 596–600; [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.S.; Ritch, R.; Schwartz, B.; Lee, S.S.; Miller, N.R.; Chi, T.; Hsieh, F.Y. Optic nerve head and nerve fiber layer in Alzheimer's disease. Arch Ophthalmol. 1991, 109, 199–204; [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Duggan, J.; Cordeiro, M.F. Alzheimer's disease and retinal neurodegeneration. Curr Alzheimer Res. 2010, 7, 3–14; [Google Scholar] [CrossRef]
- Bao, Y.K.; Yan, Y.; Gordon, M.; McGill, J.B.; Kass, M.; Rajagopal, R. Visual Field Loss in Patients with Diabetes in the Absence of Clinically-Detectable Vascular Retinopathy in a Nationally Representative Survey. Invest Ophthalmol Vis Sci. 2019, 60, 4711–4716; [Google Scholar] [CrossRef]
- Garcia-Martin, E.; Cipres, M.; Melchor, I.; Gil-Arribas, L.; Vilades, E.; Polo, V.; Rodrigo, M.J.; Satue, M. Neurodegeneration in Patients with Type 2 Diabetes Mellitus without Diabetic Retinopathy. J Ophthalmol. 2019, 2019, 1825819; [Google Scholar] [CrossRef]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-induced pro-inflammatory responses by retinal Müller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia. 2010, 53, 2656–2666; [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Luo, C.; Yang, X. Accelerated aging in glaucoma: immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Invest Ophthalmol Vis Sci. 2007, 48, 1201–1211; [Google Scholar] [CrossRef] [PubMed]
- Bikbova, G.; Oshitari, T.; Baba, T.; Yamamoto, S. Altered Expression of NF- κ B and SP1 after Exposure to Advanced Glycation End-Products and Effects of Neurotrophic Factors in AGEs Exposed Rat Retinas. J Diabetes Res. 2015, 2015, 543818; [Google Scholar] [CrossRef]
- Bikbova, G.; Oshitari, T.; Yamamoto, S. Neuronal cell death and regeneration in diseases associated with advanced glycation end-products accumulation. Neural Regen Res. 2014, 9, 701–702; [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer's disease. J Alzheimers Dis. 2009, 16, 845–858; [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Inoue, H.; Kikuchi, S.; Takeuchi, M. Serum or cerebrospinal fluid levels of glyceraldehyde-derived advanced glycation end products (AGEs) may be a promising biomarker for early detection of Alzheimer's disease. Med Hypotheses. 2005, 64, 1205–1207; [Google Scholar] [CrossRef]
- Cullum, N.A.; Mahon, J.; Stringer, K.; McLean, W.G. Glycation of rat sciatic nerve tubulin in experimental diabetes mellitus. Diabetologia. 1991, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer's disease. Curr Pharm Des. 2008, 14, 973–978; [Google Scholar] [CrossRef]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol. 2002, 50, 325–392; [Google Scholar] [CrossRef]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660; [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Aoki, H.; Ajioka, I.; Yamazaki, M.; Abe, M.; Oh-Nishi, A.; Sakimura, K.; Sugihara, I. Detailed expression pattern of aldolase C (Aldoc) in the cerebellum, retina and other areas of the CNS studied in Aldoc-Venus knock-in mice. PLoS ONE. 2014, 9, e86679; [Google Scholar] [CrossRef] [PubMed]
- Huang, H.M.; Wu, C.W.; Chen, I.C.; Lee, Y.C.; Huang, Y.S.; Hung, C.Y.; Wu, K.L.H. Maternal high-fructose diet induced early-onset retinopathy via the suppression of synaptic plasticity mediated by mitochondrial dysfunction. Am J Physiol Endocrinol Metab. 2021, 320, E1173–E1182; [Google Scholar] [CrossRef] [PubMed]
- Trotta, M.C.; Gesualdo, C.; Petrillo, F.; Cavasso, G.; Corte, A.D.; D'Amico, G.; Hermenean, A.; Simonelli, F.; Rossi, S. ; Serum Iba-1, GLUT5, and TSPO in Patients With Diabetic Retinopathy: New Biomarkers for Early Retinal Neurovascular Alterations? A Pilot Study. Transl Vis Sci Technol. 2022, 11, 16; [Google Scholar] [CrossRef]
- Khalifah, R.G.; Baynes, J.W.; Hudson, B.G. Amadorins: Novel post-Amadori inhibitors of advanced glycation reactions. Biochem Biophys Res Commun. 1999, 257, 251–258; [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950; [Google Scholar] [CrossRef]
- Metz, T.O.; Alderson, N.L.; Chachich, M.E.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine traps intermediates in lipid peroxidation reactions in vivo: evidence on the role of lipids in chemical modification of protein and development of diabetic complications. J Biol Chem. 2003, 278, 42012–42019; [Google Scholar] [CrossRef] [PubMed]
- Metz, T.O.; Alderson, N.L.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine, an inhibitor of advanced glycation and lipoxidation reactions: a novel therapy for treatment of diabetic complications. Arch Biochem Biophys. 2003, 419, 41–49; [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.M.; Folkers, K.; Minadeo, M.; VanBuskirk, R.; Xia, L.J.; Tamagawa, H. A deficiency of vitamin B6 is a plausible molecular basis of the retinopathy of patients with diabetes mellitus. Biochem Biophys Res Commun. 1991, 179, 615–619; [Google Scholar] [CrossRef]
- Stitt, A.; Gardiner, T.A.; Alderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; Thorpe, S.R. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes. 2002, 51, 2826–2832; [Google Scholar] [CrossRef]
- Kurimoto, T.; Yin, Y.; Omura, K.; Gilbert, H.Y.; Kim, D.; Cen, L.P.; Moko, L.; Kügler, S.; Benowitz, L.I. Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci. 2010, 30, 15654–15663; [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Cui, Q.; Li, Y.; Irwin, N.; Fischer, D.; Harvey, A.R.; Benowitz, L.I. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003, 23, 2284–2293; [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wong, K.A.; Benowitz, L.I. Full-length optic nerve regeneration in the absence of genetic manipulations. JCI Insight, 1: DOI, 1645. [Google Scholar]
- Takahashi, M.; Tsujioka, Y.; Yamada, T.; Tsuboi, Y.; Okada, H.; Yamamoto, T.; Liposits, Z. Glycosylation of microtubule-associated protein tau in Alzheimer's disease brain. Acta Neuropathol. 1999, 97, 635–641; [Google Scholar] [CrossRef] [PubMed]
- Ledesma, M.D.; Bonay, P.; Avila, J. ; Tau protein from Alzheimer's disease patients is glycated at its tubulin-binding domain. J Neurochem. 1995, 65, 1658–64. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Uras, G.; Zhang, P.; Xu, S.; Yin, Y.; Liu, J.; Qin, S.; Li, X.; Allen, S.; Bai, R.; Gong, Q.; Zhang, H.; Zhu, Z.; Xu, J. Discovery of Novel Tacrine-Pyrimidone Hybrids as Potent Dual AChE/GSK-3 Inhibitors for the Treatment of Alzheimer's Disease. J Med Chem. 2021, 64, 7483–7506; [Google Scholar] [CrossRef] [PubMed]
- Bos, I.; Vos, S.; Verhey, F.; Scheltens, P.; Teunissen, C.; Engelborghs, S.; Sleegers, K.; Frisoni, G.; Blin, O.; Richardson, J.C.; Bordet, R.; Tsolaki, M.; Popp, J.; Peyratout, G.; Martinez-Lage, P.; Tainta, M.; Lleó, A.; Johannsen, P.; Freund-Levi, Y.; Frölich, L.; Vandenberghe, R.; Westwood, S.; Dobricic, V.; Barkhof, F.; Legido-Quigley, C.; Bertram, L.; Lovestone, S.; Streffer, J.; Andreasson, U.; Blennow, K.; Zetterberg, H.; Visser, P.J. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer's disease spectrum. Alzheimers Dement. 2019, 15, 644–654; [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Preziuso, A.; Piccirillo, S.; Giampieri, F.; Cianciosi, D.; Orciani, M.; Amoroso, S. The Neuroprotective Effect of L-Carnitine against Glyceraldehyde-Induced Metabolic Impairment: Possible Implications in Alzheimer's Disease. Cells. 2021, 10, 2109; [Google Scholar] [CrossRef]
- Piccirillo, S.; Preziuso, A.; Amoroso, S.; Serfilippi, T.; Miceli, F.; Magi, S.; Lariccia, V. A new K+channel-independent mechanism is involved in the antioxidant effect of XE-991 in an in vitro model of glucose metabolism impairment: Implications for Alzheimer's disease. Cell Death Discov. 2022, 8, 391; [Google Scholar] [CrossRef]
- Uras, G.; Li, X.; Manca, A.; Pantaleo, A.; Bo, M.; Xu, J.; Allen, S.; Zhu, Z. Development of p-Tau Differentiated Cell Model of Alzheimer's Disease to Screen Novel Acetylcholinesterase Inhibitors. Int J Mol Sci. 2022, 23, 14794; [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Karikari, T.K.; Ashton, N.J.; Lantero, Rodríguez, J.; Milà-Alomà, M.; Gispert, J.D.; Salvadó, G.; Minguillon, C.; Fauria, K.; Shekari, M.; Grau-Rivera, O.; Arenaza-Urquijo, E.M.; Sala-Vila, A.; Sánchez-Benavides, G.; González-de-Echávarri, J.M.; Kollmorgen, G.; Stoops, E.; Vanmechelen, E.; Zetterberg, H.; Blennow, K.; Molinuevo, J.L. ALFA Study. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer's continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med. 2020, 12, e12921; [Google Scholar] [CrossRef]
- Hirota, Y.; Sakakibara, Y.; Ibaraki, K.; Takei, K.; Iijima, K.M.; Sekiya, M. Distinct brain pathologies associated with Alzheimer's disease biomarker-related phospho-tau 181 and phospho-tau 217 in App knock-in mouse models of amyloid-β amyloidosis. Brain Commun. 2022, 4, fcac286; [Google Scholar] [CrossRef] [PubMed]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; Hayabuchi, N.; Matsui, T.; Imaizumi, T. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [(18)F]fluorodeoxyglucose positron emission tomography. Diabetes Care. 2012, 35, 2618–2625; [Google Scholar] [CrossRef]
- Sakai-Sakasai, A.; Takeda, K.; Suzuki, H.; Takeuchi, M. Structures of Toxic Advanced Glycation End-Products Derived from Glyceraldehyde, A Sugar Metabolite. Biomolecules. 2024, 14, 202; [Google Scholar] [CrossRef]
- Koriyama, Y.; Takagi, Y.; Chiba, K.; Yamazaki, M.; Sugitani, K.; Arai, K.; Suzuki, H.; Kato, S. Requirement of retinoic acid receptor β for genipin derivative-induced optic nerve regeneration in adult rat retina. PLoS ONE. 2013, 8, e71252; [Google Scholar] [CrossRef] [PubMed]
- Koriyama, Y.; Sugitani, K.; Ogai, K.; Kato, S. ; Heat shock protein 70 induction by valproic acid delays photoreceptor cell death by N-methyl-N-nitrosourea in mice. J Neurochem. 2014, 130, 707–719; [Google Scholar] [CrossRef] [PubMed]
- Ooi, H.; Nasu, R.; Furukawa, A.; Takeuchi, M.; Koriyama, Y. Pyridoxamine and Aminoguanidine Attenuate the Abnormal Aggregation of β-Tubulin and Suppression of Neurite Outgrowth by Glyceraldehyde-Derived Toxic Advanced Glycation End-Products. Front Pharmacol. 2022, 13, 921611; [Google Scholar] [CrossRef]
- Koriyama, Y.; Takagi, Y.; Chiba, K.; Yamazaki, M.; Arai, K.; Matsukawa, T.; Suzuki, H.; Sugitani, K.; Kagechika, H.; Kato, S. Neuritogenic activity of a genipin derivative in retinal ganglion cells is mediated by retinoic acid receptor β expression through nitric oxide/S-nitrosylation signaling. J Neurochem. 2011, 119, 1232–1242; [Google Scholar] [CrossRef]
- Yin, Y.; Henzl, M.T.; Lorber, B.; Nakazawa, T.; Thomas, T.T.; Jiang, F.; Langer, R.; Benowitz, L.I. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006, 9, 843–852; [Google Scholar] [CrossRef]
Figure 1.
GA increased TAGE-β-tubulin and β-tubulin aggregation in retina in a time-dependent manner. (A) TAGE were measured using slot blot analyses with anti-TAGE antibody. Graphical representation of TAGE bands in a slot blot. **p < 0.01, *p < 0.05 vs 0 days (n=3). (B–E) The levels of β-tubulin aggregation were detected using anti-β-tubulin antibody. (B) Western blot data obtained using anti-β-tubulin antibody. U: Upper band, L: Lower band, M: Monomer band. (C) The levels of the upper bands of the GA-treated β-tubulin band. (D) The levels of the lower bands of the GA-treated β-tubulin band (E) The levels of the monomer β-tubulin bands of GA-treated. **p < 0.05 vs. 0 day (n = 3).
Figure 1.
GA increased TAGE-β-tubulin and β-tubulin aggregation in retina in a time-dependent manner. (A) TAGE were measured using slot blot analyses with anti-TAGE antibody. Graphical representation of TAGE bands in a slot blot. **p < 0.01, *p < 0.05 vs 0 days (n=3). (B–E) The levels of β-tubulin aggregation were detected using anti-β-tubulin antibody. (B) Western blot data obtained using anti-β-tubulin antibody. U: Upper band, L: Lower band, M: Monomer band. (C) The levels of the upper bands of the GA-treated β-tubulin band. (D) The levels of the lower bands of the GA-treated β-tubulin band (E) The levels of the monomer β-tubulin bands of GA-treated. **p < 0.05 vs. 0 day (n = 3).
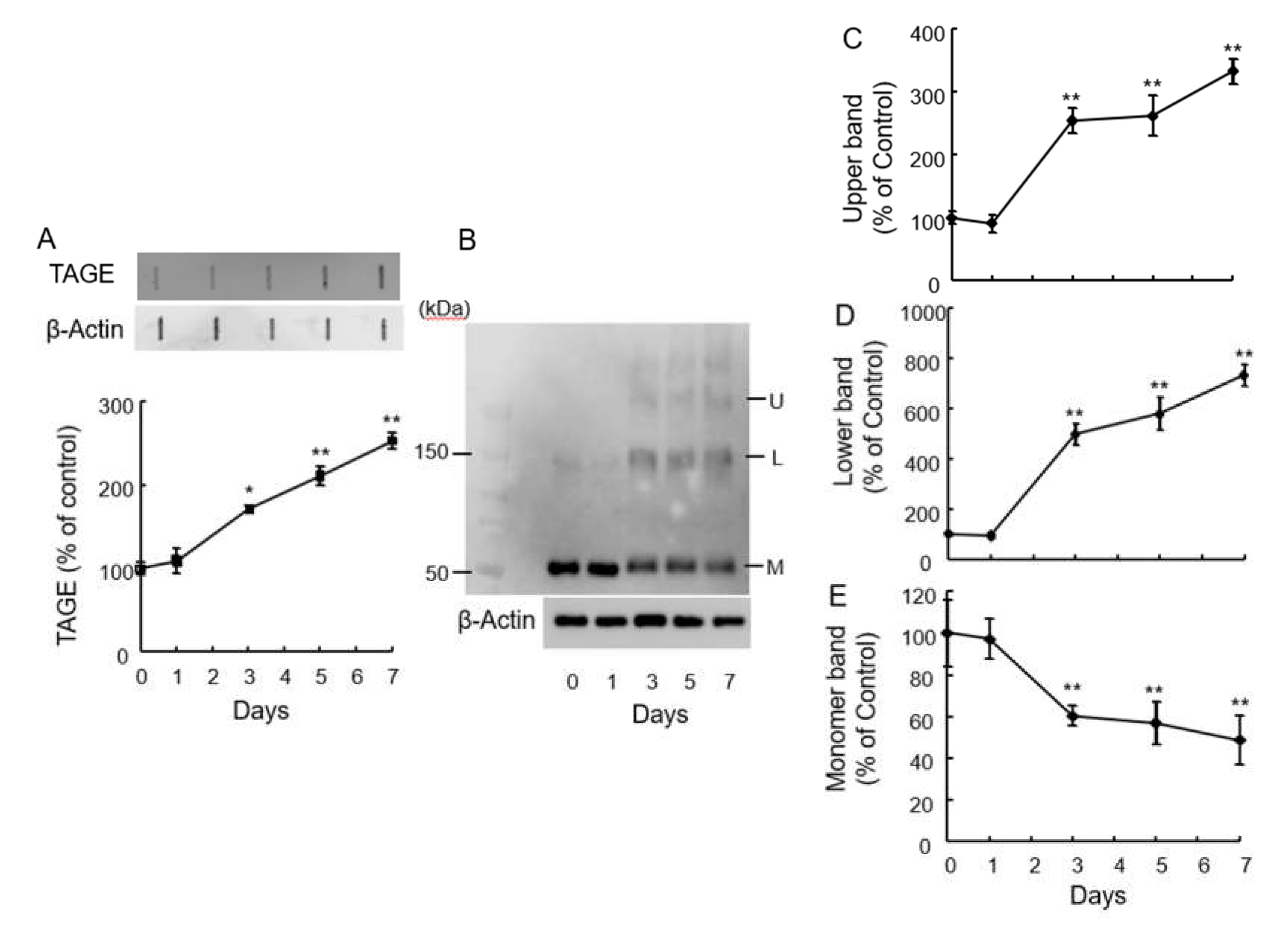
Figure 2.
TAGE immunoreactivity was colocalized with β-tubulin in retina by GA intraocular injection. (A,D,G) Immunoreactivity of TAGE was increased in GCL and NFL at 1–3 days after intraocular injection of GA (A) 0 day, (D) 1 day, (G) 3 days. (B,E,H) Immunoreactivity of β-tubulin, (B) 0 day, (E) 1 day, (H) 3 days. (C,F,I) Merged images. Scale =100 μm.
Figure 2.
TAGE immunoreactivity was colocalized with β-tubulin in retina by GA intraocular injection. (A,D,G) Immunoreactivity of TAGE was increased in GCL and NFL at 1–3 days after intraocular injection of GA (A) 0 day, (D) 1 day, (G) 3 days. (B,E,H) Immunoreactivity of β-tubulin, (B) 0 day, (E) 1 day, (H) 3 days. (C,F,I) Merged images. Scale =100 μm.
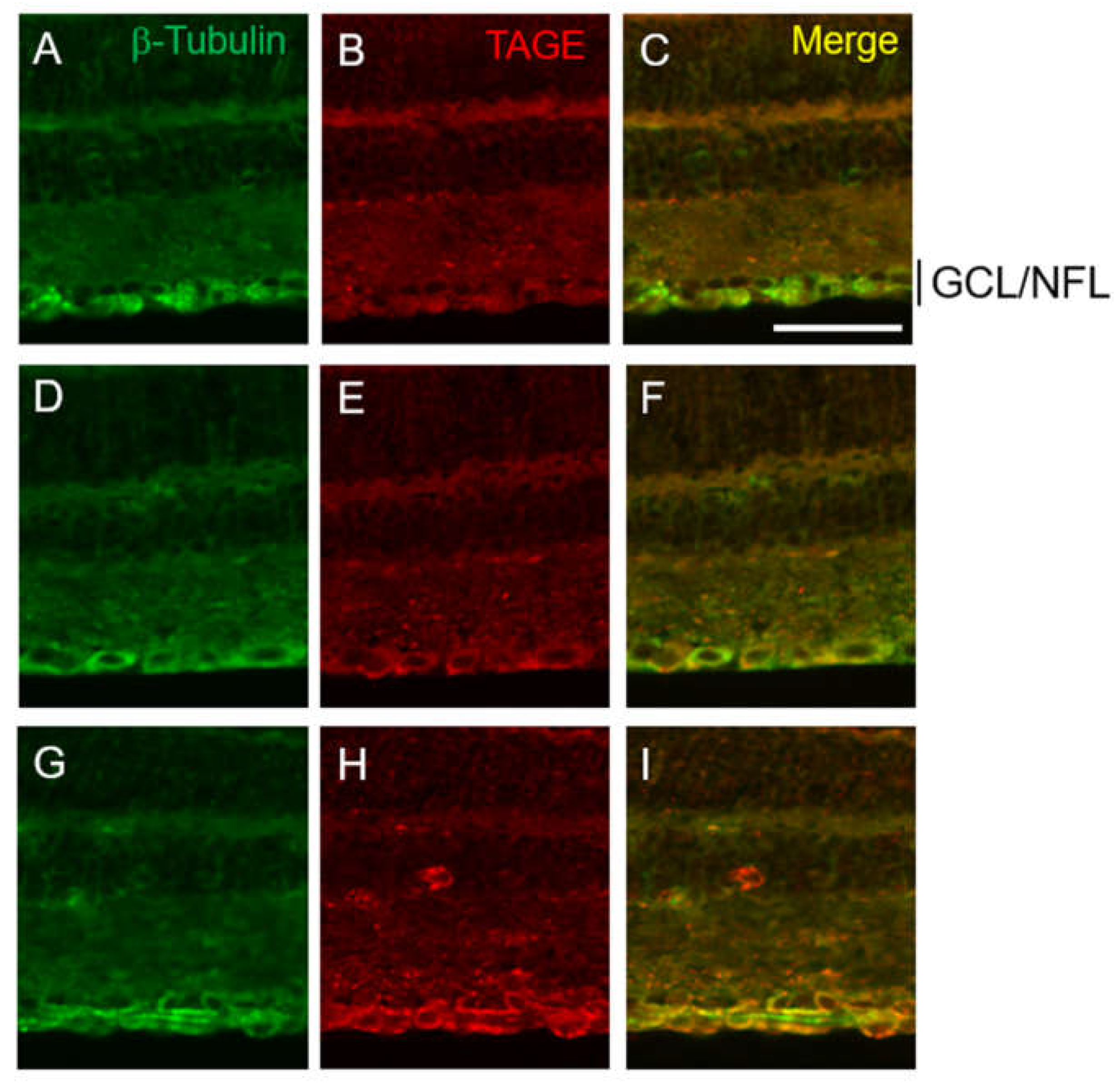
Figure 3.
PM inhibited TAGE formation and β-tubulin aggregation by GA in retina. (A) TAGE were measured using slot blot analyses with anti-TAGE antibody. Graphical representation of TAGE bands in a slot blot. ** p < 0.01, * p < 0.05 vs. vehicle control. + p < 0.01 vs. GA alone (n=3). (B) The levels of β-tubulin were detected using anti-β-tubulin antibody. U: Upper band, L: Lower band, M: Monomer band. (C-E) The levels of upper (C), lower (D) and monomer (E) bands of the GA-treated β-tubulin. **p < 0.05 vs. vehicle control, +p < 0.01 vs. GA alone (n=3).
Figure 3.
PM inhibited TAGE formation and β-tubulin aggregation by GA in retina. (A) TAGE were measured using slot blot analyses with anti-TAGE antibody. Graphical representation of TAGE bands in a slot blot. ** p < 0.01, * p < 0.05 vs. vehicle control. + p < 0.01 vs. GA alone (n=3). (B) The levels of β-tubulin were detected using anti-β-tubulin antibody. U: Upper band, L: Lower band, M: Monomer band. (C-E) The levels of upper (C), lower (D) and monomer (E) bands of the GA-treated β-tubulin. **p < 0.05 vs. vehicle control, +p < 0.01 vs. GA alone (n=3).
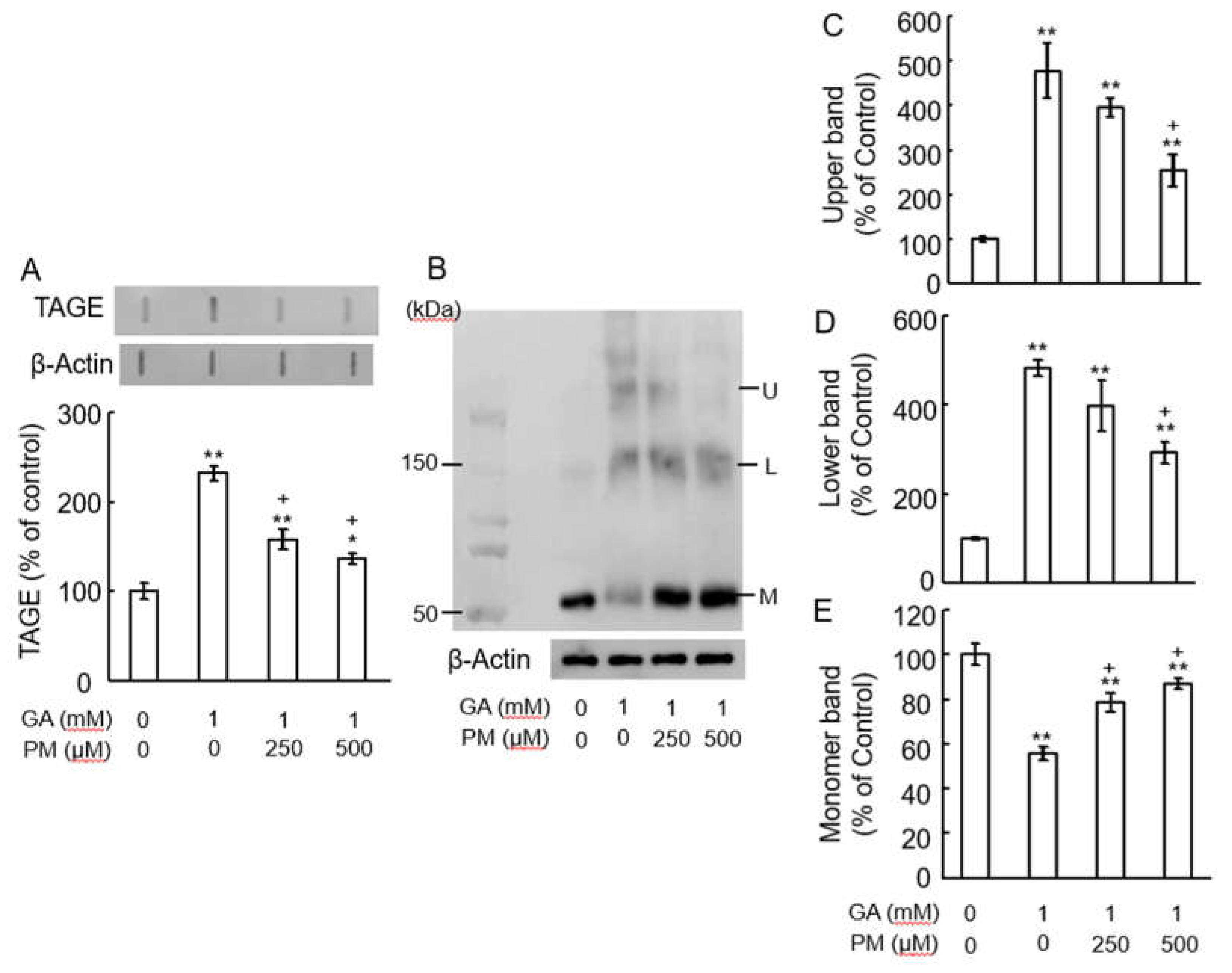
Figure 4.
PM dose-dependently suppressed TAGE- formation in GCL and NFL. (A-D) PM dose-dependently suppressed TAGE formation in GCL and NFL in retina. (A) 0 day, (B) GA, (C) GA plus 250 μM PM, (D) GA plus 500 μM PM. Scale=100 μm.
Figure 4.
PM dose-dependently suppressed TAGE- formation in GCL and NFL. (A-D) PM dose-dependently suppressed TAGE formation in GCL and NFL in retina. (A) 0 day, (B) GA, (C) GA plus 250 μM PM, (D) GA plus 500 μM PM. Scale=100 μm.
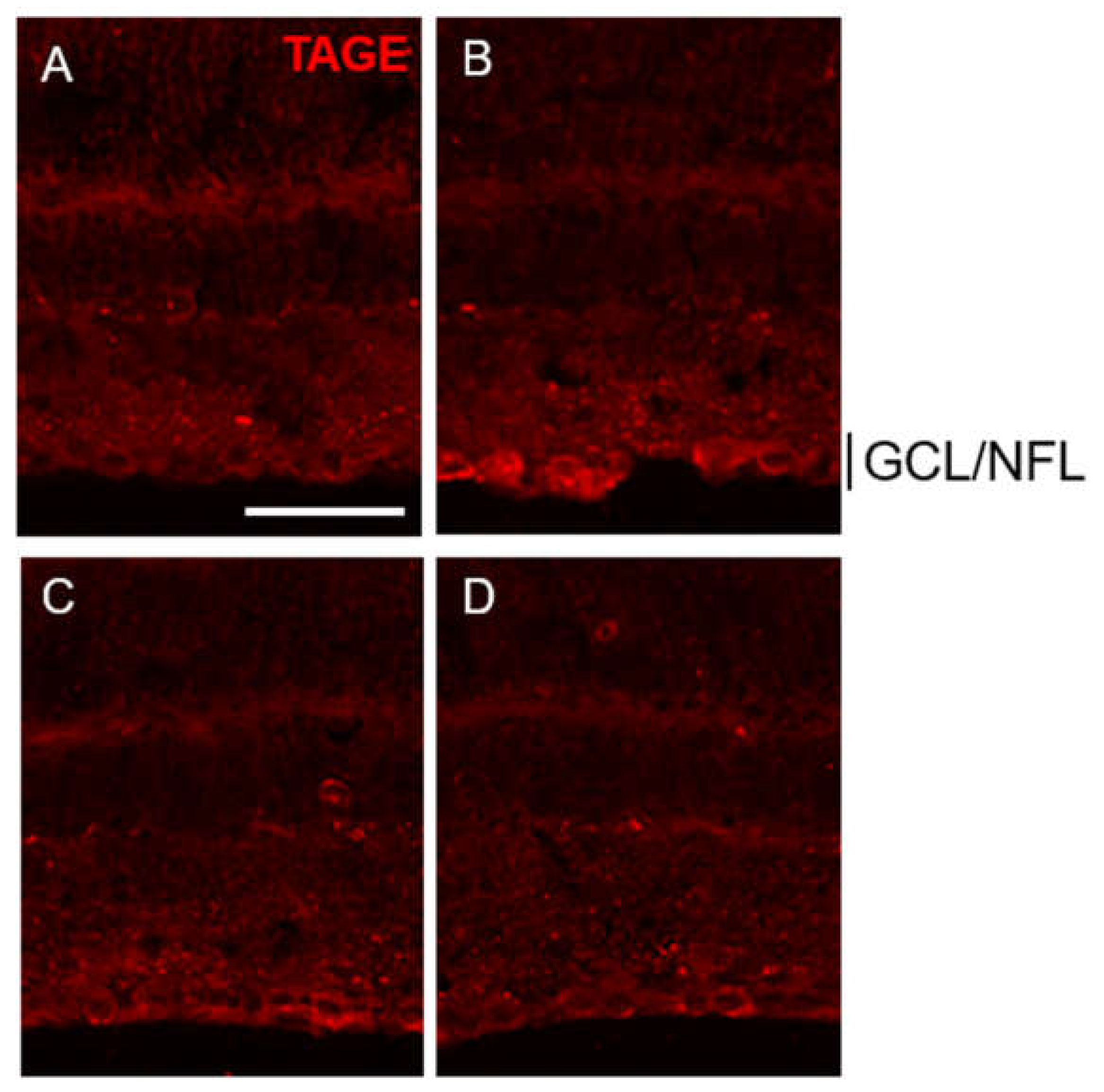
Figure 5.
Axonal elongation induced by GA was dependent on TAGE. (A–D) Longitudi nal sections of the adult mouse optic nerve showing GAP-43 positive axons extending over the injury site (asterisks) after 10 days optic nerve injury. Scale =100 μm. (A) vehicle control. (B) Zymosan, (C) Zymosan plus GA, (D) Zymosan plus GA plus PM. (E) Quantification of axonal elongation at 250 μm distal point from the injury site. **p < 0.05 vs. vehicle control. +p < 0.05 vs zymosan alone. #p < 0.01 vs. zymosan plus GA (n =8, 6 mice per each group).
Figure 5.
Axonal elongation induced by GA was dependent on TAGE. (A–D) Longitudi nal sections of the adult mouse optic nerve showing GAP-43 positive axons extending over the injury site (asterisks) after 10 days optic nerve injury. Scale =100 μm. (A) vehicle control. (B) Zymosan, (C) Zymosan plus GA, (D) Zymosan plus GA plus PM. (E) Quantification of axonal elongation at 250 μm distal point from the injury site. **p < 0.05 vs. vehicle control. +p < 0.05 vs zymosan alone. #p < 0.01 vs. zymosan plus GA (n =8, 6 mice per each group).
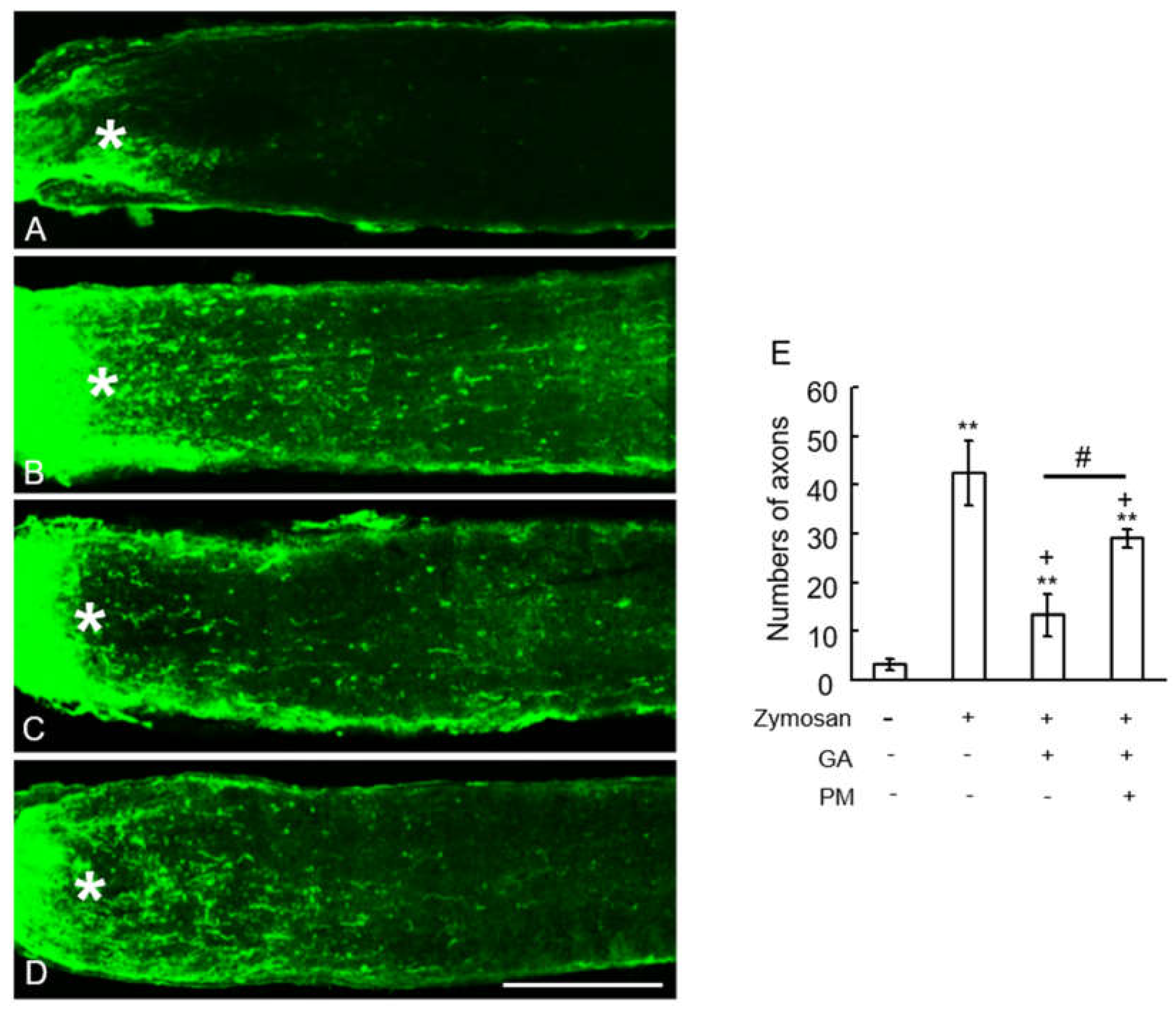
Figure 6.
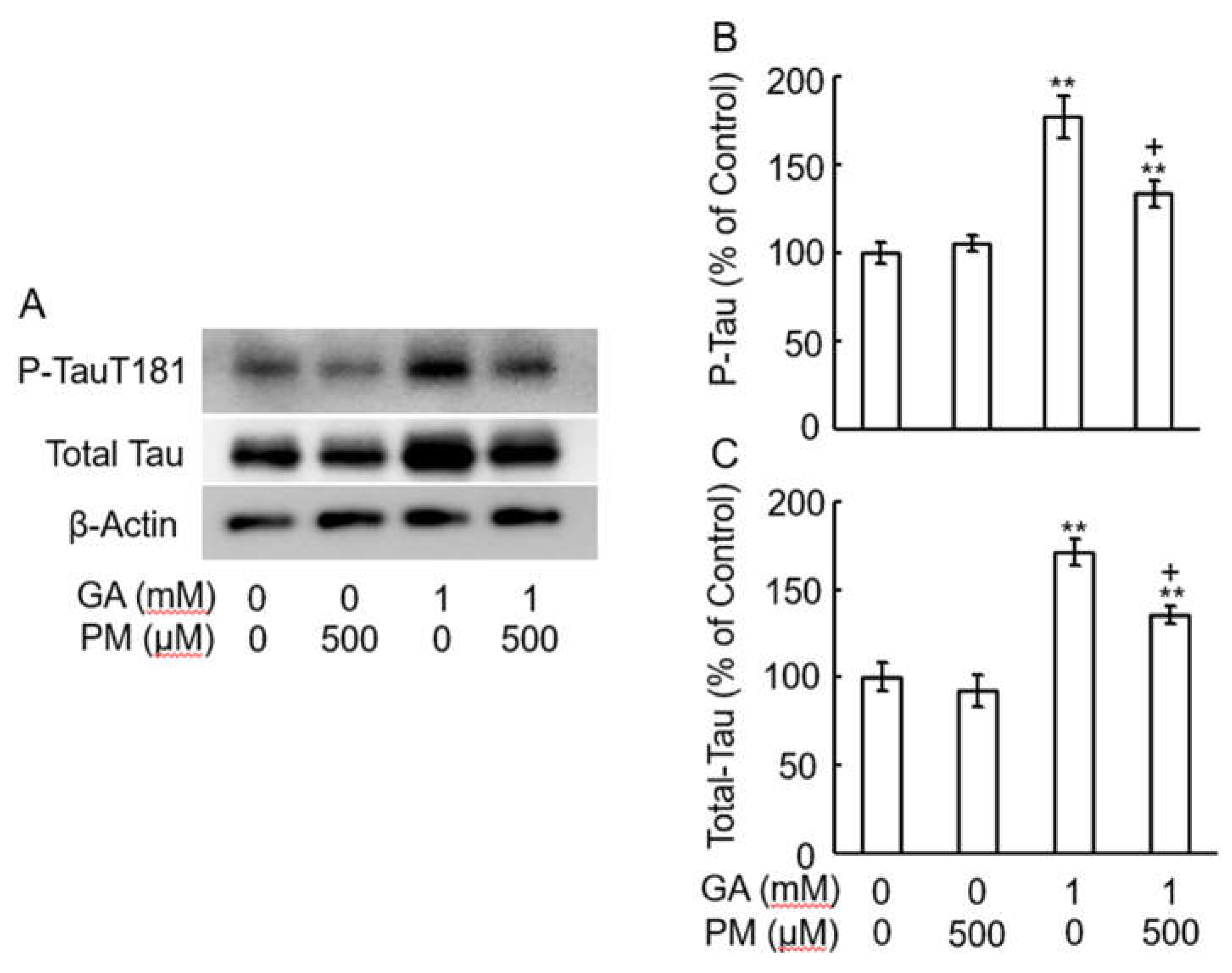
PM decreased the levels of tau-phosphorylation by GA. (A) Western blot bands. (B) Graphical representation of T-tau and (C) P-tau levels in the western blot. **p < 0.01 vs. vehicle control, +p < 0.01 vs. GA alone (n=3).
Figure 6.
PM decreased the levels of tau-phosphorylation by GA. (A) Western blot bands. (B) Graphical representation of T-tau and (C) P-tau levels in the western blot. **p < 0.01 vs. vehicle control, +p < 0.01 vs. GA alone (n=3).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



