
Submitted:
08 May 2024
Posted:
08 May 2024
You are already at the latest version
Abstract
Abstract
Cerebral microbleed(s) (CMBs) are increasingly being viewed not only as a marker for cerebral small vessel disease (SVD) but also as having an increased risk for the development of stroke (hemorrhagic/ischemic) and aging-related dementia. Recently, brain endothelial cell activation and dysfunction and blood-brain barrier dysfunction and/or disruption have been shown to be associated with SVD, enlarged perivascular spaces, and the development and evolution of CMBs. CMBs are a known disorder of cerebral microvessels that are visualized as 3-5mm, smooth, round or oval, and hypointense (black) lesions seen only on T2*-weighted gradient recall echo or susceptibility-weighted sequences on MRI images. CMBs (a global problem) are known to occur with high prevalence in community-dwelling older individuals. Since our current global population is the oldest recorded in history and only expected to continue to grow, we can only expect the health care burdens associated with CMBs to also grow. CMBs should raise a red flag regarding the increased risk of large symptomatic neurologic intracerebral hemorrhages. Importantly, CMBs are also currently regarded as markers of diffuse vascular and neurodegenerative brain damage. Thus, it is essential that we try to learn as much as we can about their development, evolution, and their relation to impaired cognition, dementia, and neurodegeneration.
Keywords:
Alzheimers Disease
; BBB
; Cerebral microbleeds
; Dementia
; Hypertension
; MRI
; Neurovascular unit
; cerebral small vessel disease
; Transmission electron microscopy
1. Introduction
Neuroimaging by magnetic resonance imaging (MRI) of cerebral microbleeds (CMBs) are being increasingly identified as important structural remodeling changes as hypodense small, smooth and rounded structures (2-5mm and sometimes up to 10mm) on T2*-weighted gradient recall echo (T2*-GRE) or susceptibility-weighted sequences MRI images (Figure 1) [1,2,3,4].
CMBs may now be considered as markers for small vessel disease (SVD) [1] that strongly suggest an increased risk for stroke (hemorrhagic and/or ischemic) and stroke mortality [3]. They are also currently associated with the clinical presence of hypertension (HTN), cerebral amyloid angiopathy (CAA), advancing age, cerebro-cardiovascular disease, SVD, late onset or sporadic Alzheimer’s disease (LOAD), cardiovascular disease (CVD), and chronic kidney disease [1,5,6]. Notably, the Rotterdam study revealed that within the general population, those with an increase in the number of microbleeds as demonstrated by MRI were associated with an increased risk of stroke (both hemorrhagic and ischemic) [7]. The risk differs for the subtypes of stroke depending on the location of the cerebral microbleeds (cortical-lobar CAA-related; subcortical – deep, white matter, basal ganglia (BG), thalamus, cerebellar are (HTN)-related) [8]. Notably, those with the largest microbleed burden were at highest risk of stroke and microbleeds not only mark the progression of cerebrovascular pathology but also represent a precursor of stroke [7].
SVD may be defined as the sum of all neuropathological processes, which affect small vessels of the brain including small arteries, arterioles, capillaries, venules, and small veins [9,10,11]. Importantly, SVD consists of multiple MRI identifiable, aberrant findings, which includes, recent small subcortical infarcts, lacunes, enlarged perivascular spaces (EPVS), white matter hyperintensities (WMH), CMBs, and brain atrophy of cortical neurons (Figure 2) [1,12,13].
As their name implies, CMBs are currently known to develop as a result of a small accumulation of cerebral microvessel blood components (erythrocytes-red blood cell(s) (RBCs), plasma, hemoglobin, hemosiderin or hemosiderin from surrounding macrophages) that have escaped microvessels lumens due to brain endothelial cells activation/dysfunction (BECact/dys) and subsequent or concurrent blood-brain barrier dysfunction or disruption (BBBdd) with increased microvessel permeability and leakage of RBCs, leukocytes, fluids, and solutes. Additionally, there may be a loss of the arterial vessel walls media vascular smooth muscle cell (VSMC) integrity (due to deposition of amyloid beta in CAA and hyalinosis and/or arteriolosclerosis in hypertensive vasculopathy with rupture), which allows for the escape of luminal blood and its contents to aberrantly reside within the CNS parenchymal interstitium [1,14,15]. The escaped blood from the lumen now aberrantly residing within the neuronal interstitial parenchyma is capable of instigating a brain injury with a known response to injury wound healing mechanism. This response to injury is due to the neurotoxic contents of red blood cell (RBCs) hemoglobin metabolism including hemosiderin which gives these structures a flare component as well as their hypodense, black appearance on T2*-GRE MRI. Additionally, the plasma that is extruded from these microbleeds contains neurotoxic thrombin, plasmin, and the components within the complement cascade [1,15,16]. The information regarding the possibility of the BECact/dys and subsequent or concurrent NVU BBBdd is currently gaining a great deal of support [17,18,19,20,21].
During the past two decades there has been increasing and widespread clinical and research use of MRI (~1966-2000) and our understanding of CMBs has undergone exponential growth. CMBs were originally thought to be asymptomatic markers of SVD; however, emerging data has shown an association between microbleeds and cognitive impairment - dementia, with an associated increased risk for ischemic and large symptomatic strokes [3,7,22]. CMBs are considered to be MRI-defined lesions corresponding to small deposits of blood components (due mainly to hemosiderin that accumulates in perivascular macrophages to allow MRI identification by GRE from previous episodes of small amounts of bleeding in the brain primarily in the neuronal interstitial spaces or interstitium) that are most commonly related and thought to be markers for small vessel damage, small vessel disease and concurrently raise a red flag for the increased risk of large symptomatic neurologic intracerebral hemorrhages [ICH] [1,3,7,22]. Indeed, CMBs have generated a great interest as MRI GRE markers for SVD prone to bleeding, with accumulating evidence that they are related to an increased risk of stroke (hemorrhagic and/or ischemic infarcts) and thus identify those individuals who are at greater risk for the development of larger symptomatic ICH [1,3,7,22,23,24]. Therefore, it is essential that we try to learn as much as we can about their evolutionary development and how and why they result in neuronal damage in addition to ischemic and/or hemorrhagic stroke.
In summary, knowledge has expanded greatly regarding CMBs during these past two decades [3] and are considered to be MRI-defined lesions corresponding to small deposits of blood components (due mainly to paramagnetic qualities of hemosiderin that accumulates in perivascular macrophages to allow MRI identification by T2*-GRE from previous episodes of small amounts of bleeding in the brain primarily in the neuronal interstitial spaces or interstitium) [3]. These bleeds are thought to be markers for small vessel damage, and concurrently raise a red flag for the increased risk of future large symptomatic ICH [1,3,22]. The presence of increased CMBs is related to an increased risk of stroke (ischemic and/or hemorrhagic infarcts) and thus identify those individuals who are at greater risk for the development of larger symptomatic ICH [1,3,22,23,24]. CMBs may also predict cerebral bleeding after a stroke [25] and their presence is regarded as a marker of diffuse vascular and neurodegenerative brain damage [26]. Additionally, CMBs may be considered to be amongst the most prevalent known neurologic processes and are also known to have major implications in regards to stroke, dementia and/or cognitive impairment, and aging [9,27,28]. Therefore, it is essential that we continue to expand our data base of knowledge and learn as much as we can about their important evolutionary role in development as to how and why they evolve.
2. A Possible Sequence of Events in the Development of Cerebral Microvessel Bleeds (CMBs):
Cerebral microvessels including small arteries, precapillary arterioles, true capillaries, post capillary venules, and veins are capable of becoming leaky due to increased permeability and multifactorial rupturing as a result of BECact/dys and/or BBBdd and these findings of leakage and rupture are sources for the development of CMBs. [29,30].
2.1. Brain Endothelial Cell activation and dysfunction (BECact/dys)
The monolayer of BECs, similar to systemic ECs, play an important multifunctional role in cerebrovascular homeostasis by regulating blood fluidity, fibrinolysis, vascular tone, permeability, angiogenesis, leukocyte, RBC, and platelet adhesion as well as aggregation [31]. Importantly, BECs may also be considered as the gate keepers and sentinel cells between the circulating blood and neuronal parenchyma and in their quiesent state (non-activated) protect the brain from peripheral neurotoxins in health along with its NVU BBB, pericytes, ECM and tightly adherent perivascular astrocytes.BECact/dys indicates activation of the cellular machinery that upregulate cell-surface inflammatory adhesion proteins such as vascular cellular adhesion molecule-1 (VCAM-1), intercellular cellular adhesion molecule-1 (ICAM-1), and endothelial leukocyte adhesion molecule (ELAM or E-selectin) in order to call up peripheral leukocytes for their adherence to the endothelium (activation).BEC dysfunction occurs when there is decreased bioavailable nitric oxide (NO) from any cause [32,33].
I-CAM, V-CAM, and E-Selectin become activated by peripherally-derived systemic injurious stimuli including pCC such that there is an impaired synthesis of NO by the endothelial nitric oxide synthase (eNOS) enzyme. This may occur concurrent with eNOS uncoupling due to oxidation of tetrahydrobiopterin (BH4) oxidation to BH3 or BH2.BH4 must be completely reduced in order to run the eNOS reaction to generate NO to result in decreased bioavailable NO [17,32,34]. Further, BECs may also be considered as gate keepers and sentinel cells between the circulating blood and neuronal parenchyma and the BEC in its quiesent state (non-activated) to protect the brain from peripheral neurotoxins in health along with its NVU BBB, pericytes, ECM and tightly adherent perivascular astrocytes.
There are multiple injurious species from the peripheral circulation, which are capable of instigating BECact/dys with upregulation of inflammatory signaling as well as promoting a decrease in bioavailable nitric oxide, since these two frequently present concomitantly (Figure 3) [13,16,35].
Once the NVU undergoes BECact/dys the BEC is capable of synthesizing and secreting numerous brain-derived cytokines and chemokines specifically from activated BEC (becCC or cnsCC) to contrast with pCC.These becCC include the cytokines: Interleukin-1beta (IL-1β), interleukin-6 (Il-6), interleukin-8 (IL-8), tumor necrosis alpha (TNFα).While chemokines include (MCP-1) or (CCL2), (CCL5) or (RANTES), plus others that, in turn, stimulate the brains’ reactive microglia and astrocytes (rMGCs, rACs) to produce even more cnsC/C. Additionally, the BECs, rMGCs, and rACs are also capable of actively secreting reactive oxygen species (ROS), resulting in the creation of the reactive species interactome (RSI) of reactive oxygen, nitrogen, sulfur species (RONSS), which in turn activate local matrix metalloproteinases -2, -9 (constitutive MMP-2 and inducible MMP-9 respectively) that are capable of degrading the glia limitans allowing the proinflammatory leukocytes to breech this 2nd barrier to result in neuroinflammation(Figure 4 and Figure 5) [15,36,37].
In either a step-wise fashion or concurrently BECact/dys gives rise to BBB dysfunction or disruption [17].
2.2. Blood-Brain Barrier Dysfunction and/or Disruption (BBBdd) with Increased Permeability
Wang et al., make a strong statement in that, they state: “Blood-brain barrier (BBB) dysfunction or disruption (BBBdd) is considered to be the event that initiates CMBs development.” [17]. This group believes strongly that BBBdd is responsible for CMBs as put forth earlier in the text along with multiple other authors [18,19,20,21]. Risk factors for the development of CMBs consist of advancing age, HTN, CAA, type 2 diabetes mellitus (T2DM), smoking, and previous strokes [1,3,17].
Once BECact/dys has occurred, this allows for the development of BBBdd due largely to the activation of redox-sensitive MMPs via inflammation generated by cytokine ROS production. These MMPs not only promote the dysfunction and degradation of the TJ/AJs but also allows for the degradation of the glia limitans the outermost barrier of the PVU to allow for step 2 as portrayed in Figure 6 to allow the escape of leukocytes, red blood cells, and plasma into the surrounding parenchymal interstitial spaces to result in neuroinflammation and CMBs as in Figure 4 and Figure 5 [36]. Thus, BECact/dys may be considered a promoter of BBBdd, which in turn allows for the breeching of the PVU/PVS glia limitans the outermost barrier to enter the neuropil interstitial spaces (Figure 4 and Figure 5).
Wang et al. conclude with the following paragraph as follows; “In conclusion, despite many details that still require study, considerable evidence suggests that BBB dysfunction appears to play a significant role in the development and progression of CMBs. Risk factors for CMBs can exacerbate BBB breakdown through the vulnerability of the BBB to anatomical and functional changes. To reduce the burden of CMBs, it is necessary to increase awareness of BBB alterations and perform additional research to increase our knowledge regarding their relationship with CMBs.” [17]. Indeed, BBBdd allows for increased permeability and this could lead to the extravasation of RBCs from cerebral microvessels into the neuropil interstitial space, which leads to the deterioration of the brain’s environment and further aggrevate brain degeneration due to the development of CMBs [17,18,19,20,21,38]. Additionally, the next or concurrent steps in the progression to CMBs would be one of two primary etiologies or mechanisms HTN or CAA to develop CMBs. Notably, primarily basal, deep, infratentorial or occasionally mixed CMBs develop in hypertensive vasculopathy and lobar or cortical CMBs develop almost exclusively in CAA [7,39].
2.3. Hypertensive (HTN) Vasculopathy
HTN is the 2nd leading cause for CMBs following advancing age [17] and is a major risk factor for CMBs [29,40]. CMBs pathogenesis is thought to be primarily a result of the vicious cycle of oxidative stress and inflammation as in Figure 6 and their remodeling effects on the VSMCs, which results in a loss of integrity and allows these cerebral microvessels to be prone to rupture [29]. Additionally, these hypertension-induced microbleeds are also worsened by aging [41] and amyloid pathology [42]. Cerebral microbleeds associate with worse cognitive function, and underlying mechanisms may involve not only local brain injury but also chronic inflammation [43]. HTN related CMBs are known to occur primarily in the subcortical – deep, white matter, BG, thalamus, brain stem, and cerebellar regions [3,8].
HTN affects each of the cell-types in the NVU by various mechanisms, which result in BECact/dys, BBBdd, and impaired neurovascular unit (NVU) function with impaired neurovascular responsiveness. Endothelial dysfunction results from reduced NO bioavailability, as a result of impaired eNOS function via reduced expression, mislocalization, impaired phosphorylation, and eNOS uncoupling, thus resulting in reduced NO production and decreased bioavailability and increased ROS production by perivascular macrophage(s) (PVMФs) [44]. Subsequently, HTN allows cerebral microvessels to become a net producer of damaging ROS instead of vasculoprotective NO.
Angiotensin II (AngII)) type 1 receptor (AT1R) activation results in the activation of NADPH oxidase 2 in both BECs and PVMФs, and a potential interaction with toll-like receptor 4 (TLR4) in ECs, which contribute to dysfunction and disruption of the BBB or BBBdd. Neurovascular coupling is impaired by PVMФs-derived ROS, aldosterone -induced damage of inwardly rectifying potassium channel 2.1 (KIR2.1) and endothelial hyperpolarization, as well as altered calcium signaling in astrocytic endfeet. Also, pericytes express Nox4 (NADPH oxidase 4), which is upregulated by AngII and may contribute to vascular inflammation [44,45].
HTN is associated with BECact/dys and BBBdd and once these are established, they play an important role in the further development of CMBs and their progression with increased permeability and leakage of RBCs [17,44,45]. Importantly, inflammation, pCC, becCC, cnsCC, ROS, SNS, RAAS, and specifically Ang II are now known to play important roles in the development and perpetuation of HTN [17,44,45].
2.4. Cerebral Amyloid Angiopathy (CAA)
CAA may be characterized by the deposition of primarily misfolded amyloid beta (Aβ (1-40) misfolded proteins in the media and adventitia of small and mid-sized cerebral arteries (less commonly in the capillaries and veins) and leptomeninges [46]. It occurs most often in its sporadic form; however, mutant variants are known and frequently familial.Its clinical presentation includes strokes both ischemic and hemorrhagic presentation with primary intracerebral hemorrhage being most common due to ruptured vessels with bleeding.Aβ (1-40) is known to be vasculotoxic to the media’s VSMCs and results in a loss of integrity and weakness of the arterial media with subsequent rupture. Aβ (1-40), which is a product of amyloid precursor protein (APP) cleaved by β-secretase 1 (BACE-1) and γ-secretases is the primary protein deposited in these microvessels [47,48].
Aβ aggregation and deposition due to either excess production or impaired clearance by perivascular spaces is known to be vasculotoxic to both BECs and VSMCs that result in a loss of integrity of the microvessel media, which predisposes these arterioles to rupture allowing the formation of CMBs.For example, in sporadic CAA and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), accumulation of misfolded Aβ protein deposits without evidence of increased production strongly suggest impairments in clearance [49]. As Aβ (1-40) continues to deposit over time in the media [50] it results in the loss of vascular integrity and rupture subsequently develops with the extravasation of fluids, solutes and RBCs that form CMBs, which also activate perivascular macrophages and associates with inflammation. These CMBs reside in the lobar or cortical regions in contrast to the deep infratentorial basal ganglia hypertension-derived CMBs [8]. Additionally, the extruded Aβ and inflammation will have an effect on the BECs to result in BECact/dys that may also instigate BBBdd with increased permeability to result in further CAA associated injury [51].
Also, Aβ is capable of disrupting the BEC mitochondrial metabolic pathways by inhibiting the tricarboxylic acid cycle, electron transport chain, and oxidative phosphorylation [53,54].
Interestingly, we have previously treated streptozotocin-induced diabetic mice that developed BBB dysfunction and disruption thought to be due to the glucotoxicity effect on BEC mitochondria and excess mitochondrial ROS production. Topiramate is a mitochondrial specific carbonic anhydrase inhibitor used clinically as an antiseizure medication, which serves as specific mitochondria carbonic anhydrase inhibitor (antioxidant) and topiramate treatment prevented the BBB dysfunction and disruption increased permeability as measured by 14C-sucrose measurements as well as protecting tight and adherent junction BBBs from attenuation and loss of tight and adherens junction by ultrastructure studies (Figure 7) [55].
Notably, Aβ (1-40) is capable of binding to the receptor for advanced glycation end-products (RAGE) to generate ROS, which are capable of activating MMP-2 and 9 that are capable of degrading tight and adherens junctions (TJ/AJ) to result in in BBBdd with increased permeability [56,57]. Also, Aβ (1-40) is capable of activating BEC to promote the secretion of proinflammatory becCC including Il-1β, IL-6, and MCP-1 molecules that are capable of recruiting even more peripheral immune cells into the brain [58].
In summary, CAA is a definite age-related disease. For example, in the Religious Orders Study (404 individuals) 84 % of participants had CAA [59] and this study also found that CAA frequently co-occurred with LOAD with an estimated 78–98% of individuals with LOAD also having CAA [60]; however, only ~25% of patients with LOAD also have severe CAA [61].
Since CAA and HTN are the most common clinical causes of CMBs after advanced aging [62], a table comparing CAA and HTN similarities and differences in relation as to how they are associated with the development of CMBs that are identified by (2-5mm and sometimes up to 10mm) on T2*-GRE or SWSI MRI is presented (Table 1).
Since there are considerable similarities between CAA and HTN, could there be a synergism if they co-occurred? Indeed, HTN is frequently observed in CAA individuals [63] and previous studies have suggested that HTN may accelerate CMBs in CAA [64]. Further, microscopic and immunohistochemistry studies have previously shown that HTN-related arteriolosclerosis and CAA pathological changes of AB (1-40) deposition in the media VSMC regions often coexist [65,66]. Interestingly, Zhu et al. sought to find if there was an association between HTN vasculopathy and CAA by studying MRIs of 222 individuals who presented with ICHs. They studied 222 (mean age of 59.88 ±13.56) and found a significant association between HTN vasculopathy and CAA and SVD in these individuals. They felt these findings suggested a possible synergistic effect between HTN vasculopathy. CCA and SVD in ICH; however, further studies will be required to answer this proposed question with certainty [67].
3. Transmission Electron Microscopy (TEM) Imaging of Brain Endothelial Cell Activation and Dysfunction (BECact/dys), Blood-Brain Barrier Dysfunction and Disruption (BBBdd) With Cerebral Microbleeds (CMBs)
CMBs may be present even before we can observe them on MRI by histopathology and ultrastructural TEM studies. Previous studies have determined that there is a link between T2DM and the development of LOAD (Alzheimer’s disease) [68] and that there are underlying mechanisms that have been proposed to help explain the association of T2DM and cognitive impairment, which include BECact/dys, BBBdd, inflammation, and insulin resistance [68,69,70,71,72,73]. Now according to Teng et al. recent findings, we can now add the existance of SVD, which includes CMBs that are included to underlie an additional cause of cognitive impairment along with lacunes, EPVS, and WMH [69]. Notably, these possible mechanisms are also important factors, which contribute to the pathogenesis and development of SVD, which contributes to cognitive impairment [74].
Importantly, T2DM is known to be associated with significant ultrastructure TEM brain remodeling with the development of SVD, cognitive impairment and dysfunction (CID), vascular cognitive impairment and dementia (VCID), MCI and depression with SVD including EPVS and CMBs. Notably, in the 20-week-old female obese, insulin resistance, metabolic syndrome, type 2 diabetes mellitus, preclinical diabetic db/db mouse models CMBs were found only in db/db mice and not control models or those db/db models treated with the antidiabetic medication empagliflozin [15,75,76,77,78,79,80,82,83]. In the diabetic db/db models we were able to make multiple observations of BECact/dys including BEC abrupt thickening and loss of cytoplasm electron density, BEC Basement membrane thickening, with aberrant vacuole-like bodies, leukocyte, red blood cell, and platelet adherence to the activated BECs. (Figure 8) [15,82,83]
We were also able to observe the presence of CMBs by observing the free homogeneous electron-dense regions of free plasma or RBCs that had previously extruded from adjacent small NVU capillaries. Even though these NVU imaged capillaries appeared to be intact without observable disruption, one cannot deny the escape or extrusion of their luminal electron-dense and homogeneous plasma contents and RBCs to reside freely within the interstitial spaces of the adjacent neuropil (Figure 9, Figure 10 and Figure 11) [13,15,82,83].
The NVU capillary BECs in this model had intact TJ/AJ by ultrastructure observations in these regions; however, overall, they demonstrated multiple changes of BECact/dys such as pericyte endfeet retraction, basement membrane thickening, perivascular astrocyte endfeet detachment and retraction [13,82]. Importantly, Wang et al. have noted that the findings of CMBs is dependent upon the paramagnetic properties of hemosiderin or erythrocytes that have passed through BEC that have developed BECact/dys and feels that it is worth considering CMBs without microvessel rupture as shown in the above obese diabetic db/db models (Figure 9, Figure 10 and Figure 11) to be worthy of further studies [17].
Notably, T2DM and metabolic syndrome have been found to be associated with an increased risk and an association with lobar CMBs in T2DM [4] and deep CMBs in metabolic syndrome [78]. Importantly, in our diabetic db/db models the CMBs were all located to the lobar cortical regions III; however, they were very close to the transition zones between layers III and the white matter regions.
We have now observed and learned that CMBs manifested by plasma and RBC extrusions from adjacent small NVU capillary microvessels may occur early by 20-weeks of age in the preclinical female diabetic db/db mouse models (Figure 9, Figure 10 and Figure 11).Therefore, it now becomes somewhat evident that CMBs may also be part of a spectrum disorder as are the other aberrant remodeling changes found with SVD such as lacunes, EPVS, and WMHs [13]. Also, the previous spectrum figure describing these spectrum changes regarding EPVS may also be pertinent to the development and evolution of CMBs (Figure 12) [13].
4. Atrial Fibrillation (AF) Association with Cerebral Microbleeds and Stroke
AF is the most common cause of cardioembolism and stroke and long-term oral anticoagulation is the mainstay for therapy [84]. Also, it is known that stroke (hemorrhagic and ischemic) risk is increased by three to five-fold in those individuals with chronic AF [85]. Unfortunately, studies suggest that individuals in general with CMBs both with and without AF are at an elevated risk for future stroke and in particular ICH [86,87,88]. Importantly, the prevalence of CMBs is significantly higher in individuals with chronic AF than those who do not [89].
AF is one of the most common arrhythmias and is known to increase with aging similar to CMBs and its prevalence is increasing within our global aging population [90]. Further, AF results in cerebrovascular dysfunction with impaired cerebral blood flow (CBF) [91]. Additionally, Junejo et al., have provided solid evidence for diminished cerebral blood flow, cerebral autoregulation, neurovascular coupling, and cerebrovascular carbon dioxide reactivity, which supports diminished cerebrovascular vasodilatory reserve in AF patients when compared to control participants in sinus rhythm.It was recently found (March 2023) that in those individuals who have AF that are on antithrombotic therapy for secondary prevention after ischemic stroke or transient ischemic attack, the presence of CMBs was associated with increased risk of both subsequent intracerebral hemorrhage and ischemic stroke with a greater association for increased intracerebral hemorrhage [92].
An algorithm has been recently proposed by Fisher. which incorporates MRI screening to the anticoagulation decision-making protocols [93]. Their algorithm includes individuals with chronic non-valvular AF based on ≥ age 60 should have MRI screening prior to beginning oral anticoagulation. Fisher has recommend starting oral anticoagulation in those who have none or less than five subcortical CMBs. However, in those with any or ≥ five CMBs, he recommends neurologic consultation and avoiding warfarin in preference to using non-oral anticoagulants if indeed anticoagulation is still thought to outweigh the possible side effects of ICH. Also, he recommends repeating MRIs for comparison who develop neurological deficits and discontinue anticoagulation if there is a progression of CMBs. While this approach may not be accepted by all it does importantly incorporate the detrimental role of increased CMBs and a role for personalized medicine [93].
Stroke (increased up to five-fold) is the leading complication of chronic non-valvular AF [85,94] and 80% of strokes are caused by arterial occlusion of cerebral arteries, whereas the remaining 20% are caused by intracerebral hemorrhages [95]. Unfortunately, this presents somewhat of a conundrum, in that, antithrombotic therapy can act like a double-edged sword with both beneficial and adverse effects and therefore the benefit risk ratio must be considered at all points in therapy [96]. Yet, oral antithrombotic therapy (OAT) remains the best treatment option to prevent cardioembolism in AF [97]. Because of the above associations and the everchanging landscape regarding anticoagulation in association with CMBs, it is recommended that the most up-to-date recommendations regarding anticoagulation to be followed.
5. Apolipoprotein E Association with CMBs
Apolipoprotein E is known to be important in lipid metabolism, lipid transport, and membrane biosynthesis in sprouting and synaptic remodeling [98]. The presence of the APOE-ε4 allele increases the amount of A accumulation in the brain (vascular and parenchymal) and when the amount ofAPOE-ε4 was increased, this also paralleled the prevalence of CMBs [99]. Ingala et al. also found that CMBs occurred in lobar regions and co-localized with white matter hyperintensities. Interestingly, they found that APOE-ε2 allele did not protect from developing CMBs, whereas the allele APOE-ε3 was neuroprotective [99]. Additionally, this group found that those individuals homozygous for the APOE- ε4 genotype had more fragile microvessels in lobar locations and co-occurred with WMH, which suggested increased vascular vulnerability for the development of CMBs [99].
6. Conclusions
This narrative review suggests that BECact/dys and subsequent or concurrent BBBdd play a key and central role in the pathophysiology of SVD, and specifically CMBs in the increased risk for ICH.However, their occurance and increased risk for ICH may still need further confirmation in larger longitudinal studies in human individuals.
The finding of CMBs in numbers ≥ 10 are indeed a ‘red flag’ for clinicians and researchers and have become an important and independent predictor for the increased risk of intracerebral hemorrhage (ICH) [73]. Further, in the author’s opinion this risk of ICH is strengthened if the CMBs occur in mixed regions of the brain, i.e., both lobar and deep - infratentorial regions that may indicate the presence of both CAA and HTN vasculopathy and a recent publication pointed to the importance of the role of simultaneous multiple intracerebral hemorrhages (SMICH) that are characterized by symptomatic intracerebral hemorrhages within different arterial territories and some had both CAA and hypertensive vasculopathy. While this study did not answer author’s opinion regarding synergism, it nevertheless pointed to the co-occurance and possible importance of CMBs occurring simultaneously in lobar and deep (BG) regions [100].
The presence of CMBs in numbers ≥ 10 suggest that there is a significant degree of microvessel disease with increased SVD presence with loss of microvessel integrity and increased vulnerability to undergo extravasation of microvessel blood luminal contents to undergo diapedesis and/or rhexis (rupture).The subsequent increase in CMBs, may increase the risk for both ischemic and hemorrhagic intracerebral stroke.However, based on the recent literature and current evidence, the presence of CMBs should not be a contraindication to intravenous thrombolysis for the treatment of acute cerebral infarction [101]. Additionally, Chacon-Portillo et al. have stated that CMBs should not dictate treatment of acute stroke [102].
There are considerable studies that suggest that aging, APOE-ε4, HTN, and peripheral systemic inflammation, and neuroinflammation correlate with the increased risk for the development of CMBs as well as the discussion in section 2 of this text (Figure 13) [17,43,103,104,105,106,107].
Currently, it is becoming more and more obvious considering the clinical significance of CMBs that it is necessary to place a greater emphasis on studying the development and progression of CMBs.Many details regarding the development and evolution of CMBs still require further study and even though there is considerable evidence that BECact/dys and BBBdd appear to play a very important key role in the development in the development of CMBs and stroke (both ischemic and intracerebral hemorrhage), more studies are necessary.
Future Directions
Late-onset Alzheimer’s disease (LOAD) is responsible for 95% verses genetic or familial early-onset AD (EOAD) for 5% of the extracellular neuritic Aβ plaques and intracellular hyperphosphorylated neurofibrils that form neurofibrillary tangles and contribute to neurodegeneration (ND).CAA amyloid (Aβ1-40) occurs primarily in the VSMC media and adventitial layers of small arteries, arterioles, and leptomeninges regions and result in vascular dysfunction due to loss of VSMC (apoptosis) and thinning of the media with microvessel dysfunction, loss of integrity with leakage of luminal blood contents due to extravasation via rhexis or rupture and/or diapedesis.Additionally, CAA is an age-related disease that contributes to neurodegeneration.VaD is caused primarily by HTN with associated SVD that includes CMBs, arteriolosclerosis or hyalinosis/lipohyalinosis and even intracerebral atherosclerosis as a microvascular hypertensive vasculopathy with damage to the VSMC media with extravasation due to loss of VSMC with thinning and leakage of luminal blood contents due to extravasation via rhexis or rupture and or leakage.There exists considerable variability to the estimated prevalence of VaD.However, Kling et al. suggest that its prevalence is between 11-18% [108]. Jellinger states that its prevalence is between 8-10% [109]. Each of these three (LOAD, CAA, and VaD) contributing clinical risk for the development of neurodegeneration and also, share a common factor of being age-related [110].
Since LOAD, CAA, and VaD share many overlaps and co-exist in age-related neurodegeneration (ND) and associate with CMBs, should we begin thinking about ND with a new perspective or begin thinking about new paradigm shifts in regards to the role of vascular diseases and neurodegeneration?Kling et al. have recently discussed this in greater depth and provided some suggestions [108]. There is considerable overlap in risk factors and findings at autopsies that are found in LOAD, CAA, and VaD (Alzheimer’s disease and Vascular dementia) [68. 108]. As a result of the above discussions, we should consider a paradigm shift in which the focus of research is shifted to also include the multiple shared and overlapping risk factors, autopsy findings, and the associated interacting mechanisms [68,108]. Maybe, we should be thinking more and more about utilizing the terms mixed dementias (LOAD and VaD) and co-occurring dementias more often [68].
In Summary, this narrative review has strongly suggested, outlined, and discussed in sections 2.1. and 2.2. that BECact/dys and BBBdd play important roles in the development of CMBs and strokes (either ischemic or hemorrhagic). However, the current discussion regarding these important roles may need further conformation in larger longitudinal studies in human individuals by MRI with stronger magnification strength, such as 7Tesla to better identify CMBs number that are occurring in either lobar or deep BG – infratentorial regions.
Funding
Author did not receive any grants or other funding.
Institutional Review Board Statement
The tissues provided for the representative electron microscopic images utilized in this manuscript were all approved in advance by the University of Missouri Institutional Animal Care and Use Committee (No. 190). The animals were cared for in accordance with National Institutes of Health guidelines and by the Institutional Animal Care and Use Committees at the Harry S. Truman Memorial Veterans Hospital and the University of Missouri, Columbia, MO, USA, which conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data and materials can be provided upon reasonable request.
Acknowledgments
The author would like to acknowledge Tatyana Shulyatnikova for the contribution of many artistic illustrations and editing of this manuscript. The author would also like to acknowledge DeAna Grant Research Specialist of the Electron Microscopy Core Facility at the Roy Blunt NextGen Precision Health Research Center, University of Missouri, Columbia, Missouri. The author also acknowledges the kind support of the William A. Banks Lab at the VA Medical Center, Seattle, Washington.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
AGE/RAGE, advanced glycation end products/receptor for advanced glycation end products; BBB, blood–brain barrier; BEC(s), brain endothelial cell(s); BECact/dys, brain endothelial cell activation/dysfunction; BBB, blood-brain barrier; BBBdd, blood-brain barrier dysfunction/disruption; BG. Basal ganglia; BH4, tetrahydrobiopterin; BM, basement membrane; CAA, cerebral amyloid angiopathy; CMBs, cerebral microbleeds; CL, capillary lumen; CVD, cerebral vascular disease; eNOS, endothelial nitric oxide synthase; EPVS, enlarged perivascular spaces; GRE, gradient recall echo;HTN, hypertension; ICH, intracerebral hemorrhages; ISF, interstitial fluid; ISS, interstitial space; LOAD, late-onset Alzheimer’s disease;; MGCs, microglia cells; MMP-2,-9, matrix metalloproteinase-2,-9; MRI, magnetic resonance image; NVU, neurovascular unit; NO, nitric oxide; PVS, perivascular spaces; RBCs, red blood cells; SVD, cerebral small vessel disease; T2DM, type 2 diabetes mellitus; TEM, transmission electron microscopy; TI/AJs, tight and adherens junctions; T2*-GRE, T2*-weighted gradient recall echo;; VaD, vascular dementia; VSMCs, vascular smooth muscle cells; WMH, white matter hyperintensities.
References
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neuro. 2013;12(8):822-38. [CrossRef]
- Greenberg SM, Vernooij MW, Cordonnier C; et al., Microbleed Study Group. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. [CrossRef]
- Lee J, Sohn EH, Oh E. Lee AY. Characteristics of Cerebral Microbleeds. Dement Neurocogn Disord. 2018;17(3):73–82. [CrossRef]
- Shao P, Xu H, Sheng X, Quin R, Ma J, Luo Y; et al. Lobar Cerebral Microbleeds Are Associated With Cognitive Decline in Patients With Type 2 Diabetes Mellitus. Front Neurol. 2022;13:84320. [CrossRef]
- Yakushiji Y, Hara H. Cerebral microbleeds: Clinical features and management. Rinsho Shinkeigaku. 2012;52(11):1106-9. [CrossRef]
- Elmståhl S, Ellström K, Siennicki-Lantz A. Abul-Kasim K. Association between cerebral microbleeds and hypertension in the Swedish general population “Good Aging in Skåne” study. J Clin Hypertens (Greenwich). 2019;21(8): 1099–1107. [CrossRef]
- Akoudad S, Portegies MLP, Koustall PJ, Hofman A, van der Lugt A, Ikram MA, Vernooij MW. Cerebral Microbleeds Are Associated With an Increased Risk of Stroke. The Rotterdam Study. Circulation. 2015;132(6):509-516. [CrossRef]
- Yakushiji Y, Werring DJ. Cerebrovascular disease: Lobar cerebral microbleeds signal early cognitive impairment. Nat Rev Neurol. 2016;12(12):680-682. [CrossRef]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689-701. [CrossRef]
- Marini S, Anderson CD, Rosand J. Genetics of Cerebral Small Vessel Disease. Stroke. 2020;51(1):12-20. [CrossRef]
- Gao Y, Li D, Lin J, Thomas AM, Miao J, Chen D, Li S, Chu C. Cerebral small vessel disease: Pathological mechanisms and potential therapeutic targets. Front. Aging Neurosci. 2022; 14:961661. [CrossRef]
- Zhao L, Lee A, Fan YH, Mok VCT, Shi L. Magnetic resonance imaging manifestations of cerebral small vessel disease: Automated quantification and clinical application. Chin Med J (Engl). 2021;134(2):151–160. [CrossRef]
- Shulyatnikova T, Hayden MR. Why Are Perivascular Spaces Important?
- Medicina (Kaunas). 2023 ;59(5):917. 10 May. [CrossRef]
- Stokum JA, Cannarsa GJ, Wessell AP, Shea P, Wenger N, Simard JM. When the Blood Hits Your Brain: The Neurotoxicity of Extravasated Blood. Int J Mol Sci. 2021; 22(10): 5132. [CrossRef]
- Hayden, MR. A Closer Look at the Perivascular Unit in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Biomedicines. 2024;12(1):96. [CrossRef]
- Hayden, MR. Brain Injury: Response to Injury Wound-Healing Mechanisms and Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Medicina. 2023, 59, 1337. [Google Scholar] [CrossRef] [PubMed]
- Wang H, Zhang Cl, Qiu Ym, Chen Aq, Li Yn, Hu B. Dysfunction of the Blood-brain Barrier in Cerebral Microbleeds: From Bedside to Bench. Aging Dis. 2021;12(8):1898-1919. [CrossRef]
- Cheng Z, Dai L, Wu Y, Cao Y, Chai X, Wang P, Liu C, Ni M, Gao F, Wang Q and Lv X (2023) Correlation of blood–brain barrier leakage with cerebral small vessel disease including cerebral microbleeds in Alzheimer’s disease. Front. Neurol. 2023;14:1077860. [CrossRef]
- Li Y, Li M, Zuo L, Li X, Hou Y, Hu W. Cerebral Microbleeds Are Associated with Widespread Blood-Brain Barrier Leakage. Eur Neuro. 2023;86(6):395-403. [CrossRef]
- Lee JM, Zhai G, Liu Q, Gonzales ER, Yin K, Yan P, Hsu CY, Vo KD, Lin W: Vascular permeability precedes spontaneous intracerebral hemorrhage in stroke-prone spontaneously hypertensive rats. Stroke. 2007;38(12):3289-3291. [CrossRef]
- Freeze WM, Jacobs HIL, Schreuder FHBM, Oostenbrugge RJV, Backes WH, Verhey FR, Klijn CJ. Blood-Brain Barrier Dysfunction in Small Vessel Disease Related Intracerebral Hemorrhage. Front. Neuro. 2018; 9:926. [CrossRef]
- Wilson D, Ambler G, Lee KJ, Lim JS, Shiozawa M, Koga M; et al. Cerebral microbleeds and stroke risk after ischaemic stroke or transient ischaemic attack: A pooled analysis of individual patient data from cohort studies. Lancet Neurol. 2019;18(7):653-665. [CrossRef]
- Nishikawa T, Ueba T, Kajiwara M, Fujisawa I, Miyamatsu N, Yamashita K. Cerebral microbleeds predict first-ever symptomatic cerebrovascular events. Clin Neuro Neurosurg. 2009;111(10):825-8. [CrossRef]
- Jeon S-B, Kang D-W, Cho A-H, Lee E-M, Choi CG, Kwon SU; et al. Initial microbleeds at MR imaging can predict recurrent intracerebral hemorrhage. J Neurol. 2007;254(4):508–512. [CrossRef]
- Senior, K. Microbleeds may predict cerebral bleeding after stroke. Lancet. 2002;359(9308):769. [CrossRef]
- Akoudad S, Wolters FJ, Viswanathan A, de Bruijn RF, der Lugt AV, Hofman A, Koudstaal PJ, Ikram MA, Vernooij MW. Association of Cerebral Microbleeds With Cognitive Decline and Dementia. JAMA Neurol. 2016;73(8):934-943. [CrossRef]
- Thompson CS, Hakim AM. Living beyond our physiological means: Small vessel disease of the brain is an expression of a systemic failure in arteriolar function: A unifying hypothesis. Stroke. 2009;40(5):e322-e330. [CrossRef]
- Charidimou, A. Werring DJ. Cerebral microbleeds: Detection, mechanisms and clinical challenges. Future Neurol. 2011;6(5): 587–611. [CrossRef]
- Ungvari Z, Tarantini S, Kirkpatrick AC, Csiszar A, Prodan CI. Cerebral microhemorrhages: Mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol. 2017;312(6):H1128-H1143. [CrossRef]
- Fulop GA, Tarantini S, Yabluchanskiy A, Molnar A, Prodan CI, Kiss T, Csipo T, Lipecz A, Balasubramanian P, Farkas E; et al. Role of age-related alterations of the cerebral venous circulation in the pathogenesis of vascular cognitive impairment. Am. J. Physiol. Heart Circ. Physiol. 2019; 316, H1124–H1140. [CrossRef]
- Stolarz AJ, Mu S, Zhang H, Fouda AY, Rusch NJ, Ding Z. Opinion: Endothelial Cells - Macrophage-Like Gatekeepers? Front Immunol. 2022;13:902945. [CrossRef]
- Liao, JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013;123(2):540-541. [CrossRef]
- Pober, JS. Cytokine-mediated activation of vascular endothelium. Am J Pathology. 1988;133(3):426-433.
- Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009; 297(5): H1829–H1836. [CrossRef]
- Hayden MR, Tyagi SC. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc Diabetol. 2002;1:3. [CrossRef]
- Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008 Dec;67(12):1113-21. [CrossRef]
- Hayden MR The Brain Endothelial Cell Glycocalyx Plays a Crucial Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Life (Basel). 2023;13(10):1955. [CrossRef]
- Noguchi-Shinohara M, Komatsu J, Samuraki M, Matsunari I, Ikeda T, Sakai K; et al. Cerebral amyloid angiopathy-related microbleeds and cerebrospinal fluid biomarkers in Alzheimer’s disease. J Alzheimers Dis. 2017;55:905-913. D oi 10.3233/JAD-160651.
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Salman RAS, Warch S; et al. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neuro. 2009;8(2):165-174. [CrossRef]
- Petrea RE, O’Donnell A, Beiser AS, Habes M, Aparicio H, DeCarli C, Seshadri S, Romero JR. Mid to late life hypertension trends and cerebral small vessel disease in the framingham heart study. Hypertension. 2020; 76:707–714. [CrossRef]
- Toth P, Tarantini S, Springo Z, Tucsek Z, Gautam T, Giles CB, Wren JD, Koller A, Sonntag WE, Csiszar A; et al. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: Role of resveratrol treatment in vasoprotection. Aging Cell. 2015; 14:400–408. [CrossRef]
- Nyúl-Tóth A, Tarantini S, Kiss T, Toth P, Galvan V, Tarantini A, Yabluchanskiy A, Csiszar A, Ungvari Z. Increases in hypertension-induced cerebral microhemorrhages exacerbate gait dysfunction in a mouse model of alzheimer’s disease. Geroscience. 2020; 42:1685–1698. [CrossRef]
- Poels MM, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, Krestin GP, Breteler MMB, Vernooij MW. Cerebral microbleeds are associated with worse cognitive function: The rotterdam scan study. Neurology. 2012; 78:326–333. [CrossRef]
- Baggeroer CE, Cambronero FE, Savan NA, Jefferson AL, Santisteban MM. Basic Mechanisms of Brain Injury and Cognitive Decline in Hypertension. Hypertension. 2024;81(1):34-44. [CrossRef]
- Katsi V, Marketou M, Maragkoudakis S, Didagelos M, Charalambous G, Parthenakis F, Tsioufis C, Tousoulis DJ. Blood–brain barrier dysfunction: The undervalued frontier of hypertension. J Hum Hypertens. 2020;34(10):682–691. [CrossRef]
- Pezzini A, Del Zotto E, Volonghi I, Giossi A, Costa P, Padovani A. Cerebral amyloid angiopathy: A common cause of cerebral hemorrhage. Curr Med Chem. 2009;16(20):2498-2513. [CrossRef]
- Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA. 1993 Nov 15;90(22):10836-40. [CrossRef]
- Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010;12(1):1-12. [CrossRef]
- Mestre H, Kostrikov S, Mehta RI, Nedergaard M. Perivascular Spaces, Glymphatic Dysfunction, and Small Vessel Disease. Clin Sci (Lond). 2017;131(17): 2257–2274. [CrossRef]
- Keable A,Fenna K,Yuen HM, Johnston DA, Smyth NR, Smith C; et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta. 2016;1862(5):1037-1046. [CrossRef]
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30-42. [CrossRef]
- Hsu MJ, Hsu CY, Chen BC, Chen MC, Ou G, Lin CH. Apoptosis signal-regulating kinase 1 in amyloid beta peptide-induced cerebral endothelial cell apoptosis. J Neurosci. 2007;27(21):5719-5729. [CrossRef]
- Parodi-Rullán R, Sone JY, Fossati S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer's Disease. J Alzheimers Dis. 2019;72(4):1019-1039. [CrossRef]
- Teng T, Ridgley DM, Tsoy A, Sun GY, Askarova S, Lee JC. Azelnidipine Attenuates the Oxidative and NFκB Pathways in Amyloid-β-Stimulated Cerebral Endothelial Cells. ACS Chem Neurosci. 2019; 10:209-215. 2019;10(1):209-215. [CrossRef]
- Salameh TS, Shah GN, Price TO, Hayden MR, Banks WA. Blood–Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J Pharmacol Exp Ther. 2016;359(3):452-459. [CrossRef]
- Du H, Li P, Wang J, Qing X, Li W. The interaction of amyloid β and the receptor for advanced glycation endproducts induces matrix metalloproteinase-2 expression in brain endothelial cells. Cell Mol Neurobiol. 2012;32(1):141-147. [CrossRef]
- Lee JM, Yin KJ, Hsin I, Chen S, Fryer JD, Holtzman DM; et al. Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann Neurol. 2003;54(3):379-382. [CrossRef]
- Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO; et al. (2009). Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer's brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis. 2009;34(1):95-106. [CrossRef]
- Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA (2011). Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011; 69(2): 320–327. [CrossRef]
- Jellinger, KA. Alzheimer disease and cerebrovascular pathology: An update. J Neural Transm Vienna Austria 1996. 2002;109:813–836. [CrossRef]
- 61 Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology. 1996; 46:1592–1596.
- Reddy ST, Savitz SI. Hypertension-Related Cerebral Microbleeds. Case Rep Neurol. 2020;12(3): 266-269. . https://doi.org/. [CrossRef]
- Zhang S, Wang Z, Zheng A, Yuan R, Shu Y, Zhang S; et al. Blood pressure and outcomes in patients with different etiologies of intracerebral hemorrhage: A multicenter cohort study. J Am Heart Assoc. 2020;9:e016766. [CrossRef]
- Stanisavljevic A, Schrader JM, Zhu X, Mattar JM, Hanks A, Xu F; et al. Impact of non-pharmacological chronic hypertension on a transgenic rat model of cerebral amyloid Angiopathy. Front Neurosci. 2022;16:811371. [CrossRef]
- Jolink WMT, van Veluw SJ, Zwanenburg JJM, Rozemuller AJM, van Hecke W, Frosch MP; et al. Histopathology of cerebral microinfarcts and microbleeds in spontaneous intracerebral hemorrhage. Transl Stroke Res. 2022;14:174–184. [CrossRef]
- Fazekas F, Kleinert R, Roob G, Kleinert G, Kapeller P, Schmidt R; et al. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: Evidence of microangiopathy-related microbleeds. AJNR Am J Neuroradiol. 1999;20:637–642.
- Zhu Y, Liu L, Zhong L, Cheng Y, Zhang S, Wu B, Wang D, Xu M. The association between hypertensive angiopathy and cerebral amyloid angiopathy in primary intracerebral hemorrhage. Front Neurol. 2023;14:1257896. [CrossRef]
- Hayden, MR. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019;9(10):262. [CrossRef]
- Teng Z, Feng J, Liu R, Dong Y, Chen H, Xu J, Jiang X, Li R, Lv P. Cerebral Small Vessel Disease is Associated with Mild Cognitive Impairment in Type 2 Diabetes Mellitus. Diabetes Metab Syndr Obes. 2022;15:1985–1994. [CrossRef]
- Srikanth V, Sinclair AJ, Hill-Briggs F, Moran C, Biessels GJ. Type 2 diabetes and cognitive dysfunction-towards effective management of both comorbidities. Lancet Diabetes Endocrinol. 2020;8(6):535–545. [CrossRef]
- Biessels GJ, Despa F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat Rev Endocrinol. 2018;14(10):591–604. [CrossRef]
- Kullmann S, Kleinridders A, Small DM, Fritsche A, Häring HU. Preissl H, Henri M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020;8(6):524–534. [CrossRef]
- Chen YC, Lu BZ, Shu YC, Sun YT. Spatiotemporal dynamics of cerebral vascular permeability in Type 2 diabetes-related cerebral microangiopathy. Front Endocrinol. 2021;12:805637. [CrossRef]
- Hamilton O, Backhouse EV, Janssen E; et al. Cognitive impairment in sporadic cerebral small vessel disease: A systematic review and meta-analysis. Alzheimers Dement. 2021;17(4):665–685. [CrossRef]
- Jiang Q, Zhang L, Ding G, Davoodi-Bojd E, Li Q, Li L; et al. Impairment of the glymphatic system after diabetes. J. Cereb. Blood Flow Metab. 2017;37:1326–1337. [CrossRef]
- Munia, OB. Association of Type 2 Diabetes Mellitus With Perivascular Spaces and Cerebral Amyloid Angiopathy in Alzheimer’s Disease: Insights From MRI Imaging. Dement Neurocogn Disord. 2023;22(3):87–99. [CrossRef]
- Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes—Systematic overview of prospective observational studies. Diabetologia. 2005;48:2460–2469. [CrossRef]
- 79. Mitaki S, Takayoshi H, Nakagawa T, Nagai A, Oguro H, Yamaguchi S.Metabolic syndrome is associated with incidence of deep cerebral microbleeds. PLoS ONE, 0194. [CrossRef]
- Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, Waberski B; et al. Long-Term Effect of Diabetes and Its Treatment on Cognitive Function. N. Engl. J. Med. 2007;356:1842–1852. [CrossRef]
- Van Stolten TT, Sedaghat S, Carnethon MR, Launer LL, Stehouwer CDA. Cerebral microvascular complications of type 2 diabetes: Stroke, cognitive dysfunction, and depression. The Lancet Diabetes and Endocrinology. 2020;8(4):325-336. [CrossRef]
- McCrimmon RJ, Ryan CM, Frier BM. Diabetes and cognitive dysfunction. Lancet. 2012;379: 2291–2299. [CrossRef]
- Hayden MR, Grant DG, Aroor AR, DeMarco VG. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia. 2018;1: 220–244. [CrossRef]
- Hayden MR, Grant DG, Aroor AR, DeMarco VG. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019;9(3):57. [CrossRef]
- Selim M, Diener HC. Atrial Fibrillation and Microbleeds. Stroke. 2017;48(10):2660-2664. [CrossRef]
- Wolf PA, Dawber TR, Thomas HE, Kannel WB. Epidemiologic assessment of chronic atrial fibrillation and risk of stroke: The Framingham study. Neurology. 1978;28:073-977. doi. 10.1212/wnl.28.10.973.
- Bokura H, Saika R, Yamaguchi T, Nagai A, Oguro H, Kobayashi S; et al. Microbleeds are associated with subsequent hemorrhagic and ischemic stroke in healthy elderly individuals. Stroke. 2011; 42:1967-1871. [CrossRef]
- Jeon SB, Kang DW, Cho AH, Lee EM, Choi CG, Kwon SU; et al. Initial microbleeds at MR imaging can predict recurrent intracerebral hemorrhage. J Neurol. 2007;254:508-512. [CrossRef]
- Thijs V, Lemmens R, Schoofs C, Görner A, Van Damme P, Schrooten M; et al. Microbleeds and the risk of recurrent stroke. Stroke. 2010;41:20005-2009. [CrossRef]
- Hirata Y, Kato N, Muraga K, ShindonA, Nakamura N, Matsuur K; et al. Cerebral Microbleeds With Atrial Fibrillation After Ablation Therapy. Front Cell Neurosci. 2022;16: 818288. [CrossRef]
- Bunch, TJ. Atrial fibrillation and dementia. Circulation. 2020;142(7):618-620. [CrossRef]
- Junejo RT, Lip GYH, Fisher JP. Cerebrovascular Dysfunction in Atrial Fibrillation. Front Physiol. 2020;11:1066. [CrossRef]
- Soo Y, Zietz A, Mok VCT, Yie B, Polymeris AA, Seiffge D, Ambler G; et al.Impact of Cerebral Microbleeds in Stroke Patients with Atrial Fibrillation. Annal of Neuro. 2023;94(1):61-74. [CrossRef]
- Fisher, M. MRI screening for chronic anticoagulation in atrial fibrillation. Fron Neuro. 2013;4:137. [CrossRef]
- Lip GYH. Atrial fibrillation in 2011: Stroke prevention in AF. Natl Rev Cardiol. 2011;9(2):71-73. [CrossRef]
- VIP: De Meyer SF, Stoll G, Wagner DD, Kleinschnitz C. von Willebrand factor: An emerging target in stroke therapy. Stroke. 2012;43 (2):599–606. [CrossRef]
- Stirbys, P. Review And Insights Into The Bleeding Mechanism Incited By Antithrombotic Therapy: Mechanistic Nuances Of Dual Pro-Hemorrhagic Substrate Incorporating Drug-Induced Microvascular Leakage. J Atr Fibrillation. 2015;8(2):1254. [CrossRef]
- Raffaele D, Steen H, Lars W, Felicita A, Harald A, Fedor B; et al. General mechanisms of coagulation and targets of anticoagulants (Section I). Position Paper of the ESC Working Group on Thrombosis--Task Force on Anticoagulants in Heart Disease. Thromb.Haemost. 2013;109 (4):569–579. [CrossRef]
- Huang Y, Mahley RW. Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol Dis. 2014;72PA:3-12. [CrossRef]
- Ingala S, Mazzai L, Sudre CH, Salvadó G, Brugulat-Serrat A, Wottschel V; et al. The relation between APOE genotype and cerebral microbleeds in cognitively unimpaired middle- and old-aged individuals. Neurobiology of Aging. 2020;95:104-114. [CrossRef]
- Li J, Shen D, Zhou Y, Jin Y, Jin L, Ye X, Tong L, Gao F. Underlying microangiopathy and functional outcome of simultaneous multiple intracerebral hemorrhage. Front Aging Neurosci.22022;14: 1000573. [CrossRef]
- Wang S, Lv Y, Zheng X, Qiu J, Chen HS. The impact of cerebral microbleeds on intracerebral hemorrhage and poor functional outcome of acute ischemic stroke patients treated with intravenous thrombolysis: A systematic review and meta-analysis. J Neurol. 2017;264(7):1309-1319. [CrossRef]
- Chacon-Portillo, MA, Llinas RH, Marsh EB. Cerebral microbleeds shouldn’t dictate treatment of acute stroke: A retrospective cohort study evaluating risk of intracerebral hemorrhage. BMC Neurol. 2018;18:33. [CrossRef]
- Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: A matter of blood-brain barrier dysfunction? J Exp Med. 2017;214(11):3151-3169. [CrossRef]
- Poel MM, Vernooij MW, Ikram MA, Hofman A, Krestin GP, van der Lugt A, Breteler MM. Prevalence and risk factors of cerebral microbleeds: An update of the Rotterdam scan study. Stroke. 2010;41(10 Suppl): S103-106. [CrossRef]
- Nahirney PC, Reeson P, Brown CE. Ultrastructural analysis of blood-brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J Cereb Blood Flow Metab.
- 2016;36(2):413-25. [CrossRef]
- Bai T, Yu S, Feng J. Advances in the Role of Endothelial Cells in Cerebral Small Vessel Disease. Front. Neurol. 2022;13, 2022. [CrossRef]
- Wang H, Zhang Cl, Qiu Ym, Chen Aq, Li Yn, Hu B. Dysfunction of the Blood-brain Barrier in Cerebral Microbleeds: From Bedside to Bench. Aging Dis. 2021;12(8):1898-1919. [CrossRef]
- Kling MA, Trojanowski JQ, Wolk DA, Lee VMY, Arnold SE Vascular Disease and Dementias: Paradigm Shifts to Drive Research in New Directions. Alzheimers Dement. 2013; 9(1): 76–92. [CrossRef]
- Jellinger, KA. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol. 2007;113(4):349-388. [CrossRef]
- Attems J,Jellinger KA. The overlap between vascular disease and Alzheimer’s disease - lessons from pathology. BMC Med. 2014; 12: 206. [CrossRef]
Figure 1.
Cerebral microbleeds (CMBs) pathologic lesions with varying sizes (usually 2-5 mm and less than 10mm) and different locations (lobar (red asterisks) verses deep, infratentorial, white matter basal ganglia (BG) (red arrows) in T*2-weighted gradient recall echo (GRE)/susceptibility weighted images (SWI) MRI images. This revised image provided with permission by CC 4.0 [4].
Figure 1.
Cerebral microbleeds (CMBs) pathologic lesions with varying sizes (usually 2-5 mm and less than 10mm) and different locations (lobar (red asterisks) verses deep, infratentorial, white matter basal ganglia (BG) (red arrows) in T*2-weighted gradient recall echo (GRE)/susceptibility weighted images (SWI) MRI images. This revised image provided with permission by CC 4.0 [4].
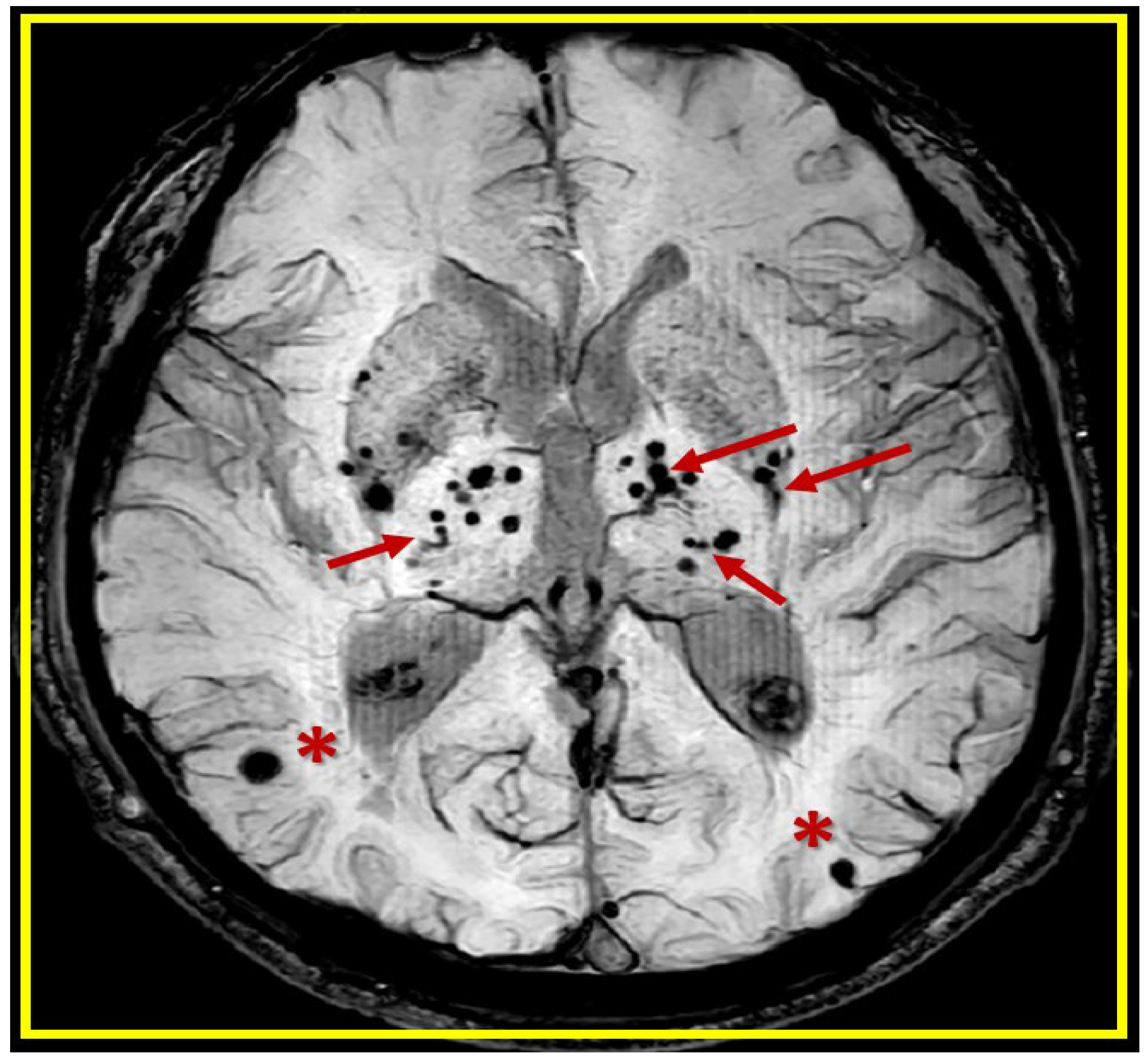
Figure 2.
Comparing similarities and differences between the 5 identifiable structures of cerebral small vessel disease (SVD) in a table-like figure. 1. Lacunes (a footprint of stroke); 2. Enlarged perivascular spaces (EPVS) (biomarker of glymphatic (GLY) system pathway dysfunction (dys); 3. White matter hyperintensities (WMH) (footprint of ischemia); 4. Cerebral microbleeds (CMBs) (biomarkers of SVD/stroke with hemorrhage or infarct; 5. recent small subcortical infarcts (historical or MRI findings of recent infarction similar to lacunes parameters but with greater flair suggesting recent occurance, not presented in this table-like figure). Location of CMBs has further clinical importance, in that lobar/cortical CMBs are CAA-related and deep, basal, infratentorial CMBs are hypertension-related. Revised table-like figure image provided with permission by CC 4.0 [13]. CAA, cerebral amyloid angiopathy; mm, millimeter.
Figure 2.
Comparing similarities and differences between the 5 identifiable structures of cerebral small vessel disease (SVD) in a table-like figure. 1. Lacunes (a footprint of stroke); 2. Enlarged perivascular spaces (EPVS) (biomarker of glymphatic (GLY) system pathway dysfunction (dys); 3. White matter hyperintensities (WMH) (footprint of ischemia); 4. Cerebral microbleeds (CMBs) (biomarkers of SVD/stroke with hemorrhage or infarct; 5. recent small subcortical infarcts (historical or MRI findings of recent infarction similar to lacunes parameters but with greater flair suggesting recent occurance, not presented in this table-like figure). Location of CMBs has further clinical importance, in that lobar/cortical CMBs are CAA-related and deep, basal, infratentorial CMBs are hypertension-related. Revised table-like figure image provided with permission by CC 4.0 [13]. CAA, cerebral amyloid angiopathy; mm, millimeter.
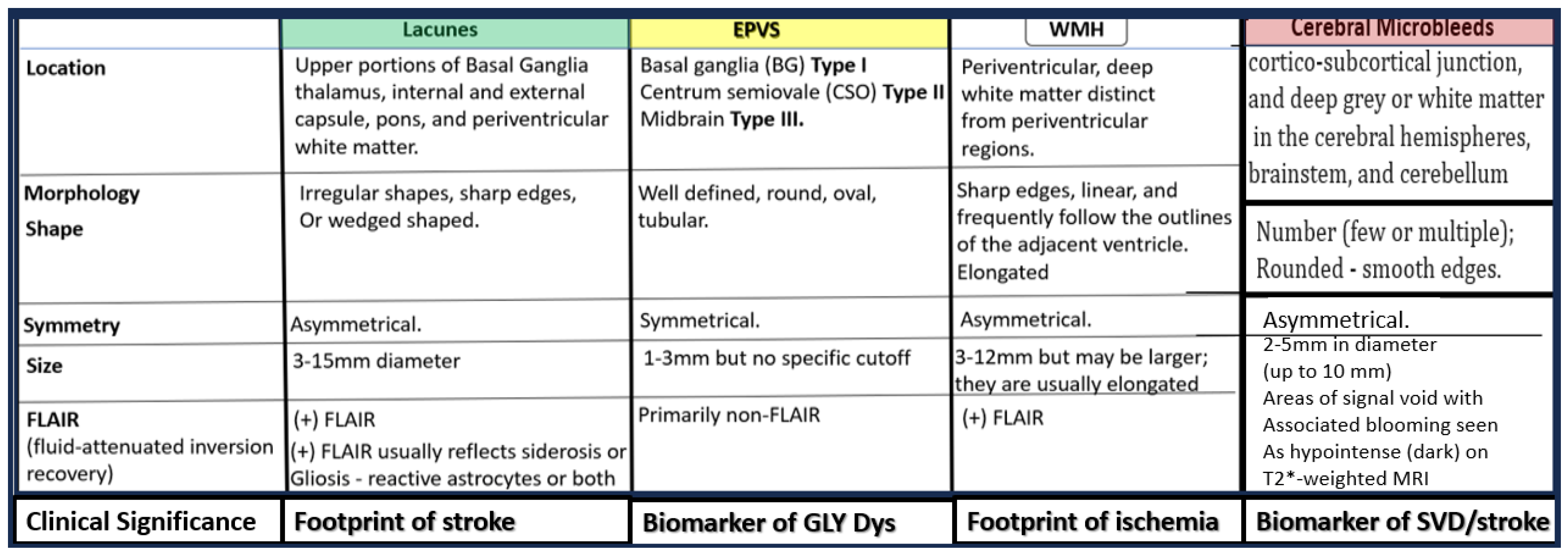
Figure 3.
Brain endothelial cell activation and dysfunction (BECact/dys) instigates neurovascular unit blood-brain barrier dysfunction/disruption (NVU BBBdd) and is responsible for the development of markers for small vessel disease (SVD) including cerebral microbleeds (CMBs). Initially note the red-dashed line at the top of this image since it demarks the location of the multiple injurious species MIS that are responsible for the initial brain endothelial cell injury in multiple clinical diseases and structural abnormalities, which importantly included SVD and CMBs. Also, note that below the BEC abluminal surface the multiple molecular consequences of the luminal multiple injurious stimuli that may be directly or indirectly related to the development of CM Bs as well as other structural changes of SVD found on MRI. Please note that pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) are not shown; however peripheral cytokines and chemokines. BH4 uncoupling not shown. This highly modified image provided with permission by CC 4.0 [16]. Ang II, angiotensin two; BBB, blood-brain barrier; BEC, brain endothelial cell; BECact/dys, brain endothelial cell activation/dysfunction; BH4, tetrahydrobiopterin; CCL2, chemokine (C-C motif) ligand 2; ecGCx, endothelial glycocalyx; intercellular cellular adhesion molecule-1, ICAM-1, IL-1β, interleukin-1β; IL-6, interleukin-6; cellular adhesion molecule; JAMs, junctional adhesion molecules; LDL, low density lipoprotein cholesterol; LPa, lipoprotein little a; MCP-1, monocyte chemotactic protein-1; NO, nitric oxide; ONOO, peroxinitrite; pnsCC, peripheral nervous system cytokines and chemokines; peroxinitrite; NVU, neurovascular unit; peroxinitrite; RBC, red blood cell; RONSS, reactive oxygen, nitrogen, sulfur species; ROS, reactive oxygen species; RSI, reactive species interactome; T, transcytosis; TNFα, tumor necrosis factor alpha; VCAM-1, vascular cellular adhesion molecule-1; WBC, white blood cell.
Figure 3.
Brain endothelial cell activation and dysfunction (BECact/dys) instigates neurovascular unit blood-brain barrier dysfunction/disruption (NVU BBBdd) and is responsible for the development of markers for small vessel disease (SVD) including cerebral microbleeds (CMBs). Initially note the red-dashed line at the top of this image since it demarks the location of the multiple injurious species MIS that are responsible for the initial brain endothelial cell injury in multiple clinical diseases and structural abnormalities, which importantly included SVD and CMBs. Also, note that below the BEC abluminal surface the multiple molecular consequences of the luminal multiple injurious stimuli that may be directly or indirectly related to the development of CM Bs as well as other structural changes of SVD found on MRI. Please note that pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) are not shown; however peripheral cytokines and chemokines. BH4 uncoupling not shown. This highly modified image provided with permission by CC 4.0 [16]. Ang II, angiotensin two; BBB, blood-brain barrier; BEC, brain endothelial cell; BECact/dys, brain endothelial cell activation/dysfunction; BH4, tetrahydrobiopterin; CCL2, chemokine (C-C motif) ligand 2; ecGCx, endothelial glycocalyx; intercellular cellular adhesion molecule-1, ICAM-1, IL-1β, interleukin-1β; IL-6, interleukin-6; cellular adhesion molecule; JAMs, junctional adhesion molecules; LDL, low density lipoprotein cholesterol; LPa, lipoprotein little a; MCP-1, monocyte chemotactic protein-1; NO, nitric oxide; ONOO, peroxinitrite; pnsCC, peripheral nervous system cytokines and chemokines; peroxinitrite; NVU, neurovascular unit; peroxinitrite; RBC, red blood cell; RONSS, reactive oxygen, nitrogen, sulfur species; ROS, reactive oxygen species; RSI, reactive species interactome; T, transcytosis; TNFα, tumor necrosis factor alpha; VCAM-1, vascular cellular adhesion molecule-1; WBC, white blood cell.
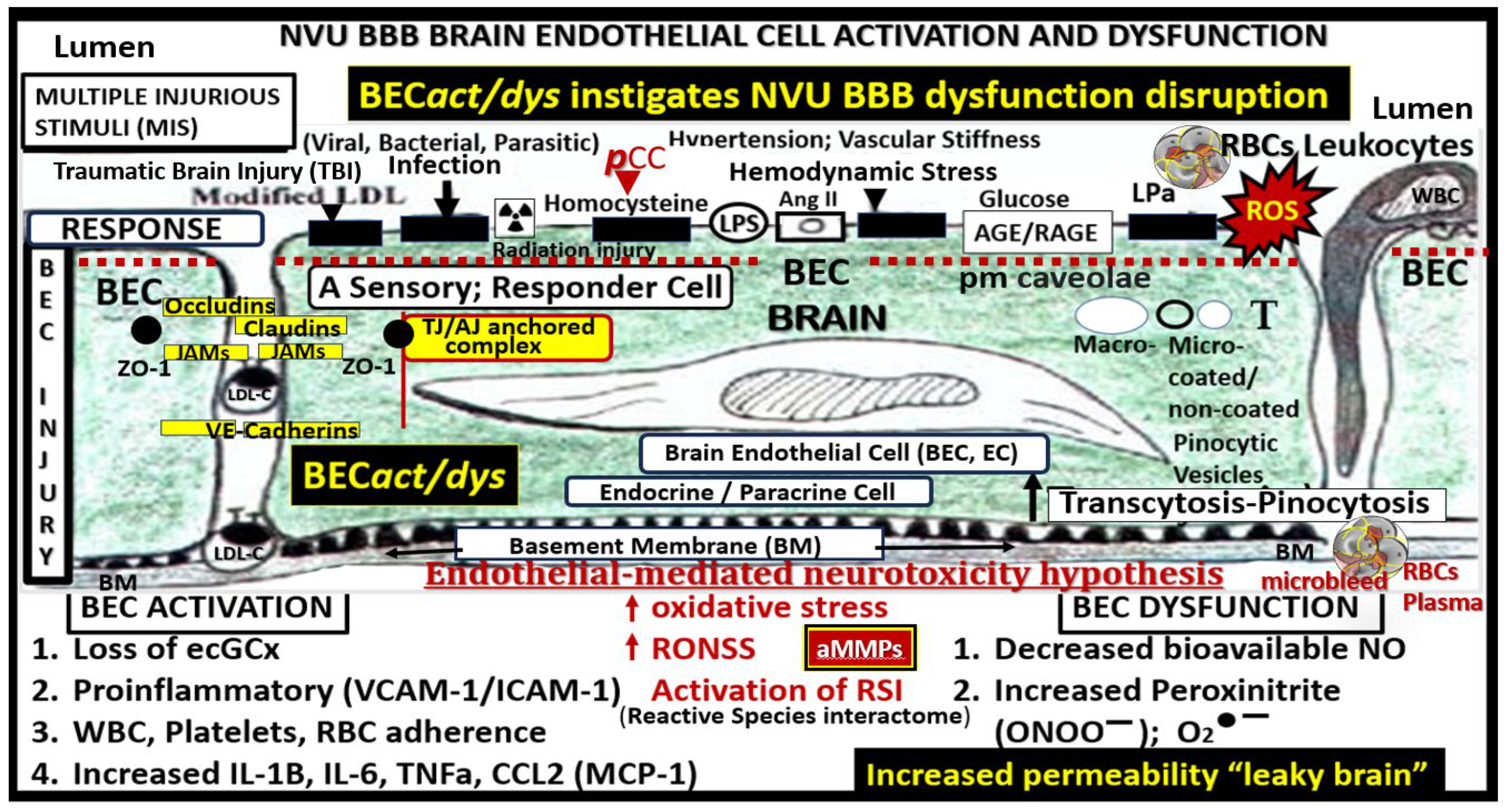
Figure 4.
Comparison of the true capillary neurovascular unit (NVU) to the postcapillary venule perivascular unit (PVU). The NVU protoplasmic perivascular astrocyte endfeet (pvACef) (pseudo-colored blue) within the true capillary illustration (A) are the connecting and creating cells that allow remodeling of the normal perivascular unit (PVU (B)) perivascular spaces (PVS) to transform and remodel into the pathologic enlarged perivascular space (EPVS, which measure 1–3 mm on magnetic resonance imaging). (A) Hand-drawn and pseudo-colored control true capillary neurovascular unit (NVU)). Note that when the brain endothelial cells (BECs) become activated and NVU BBB disruption develops, due to BEC activation and dysfunction (BECact/dys) (from multiple causes), there develops an increased permeability of fluids, peripheral cytokines and chemokines, and peripheral immune cells with a neutrophile (N) depicted herein penetrating the tight and adherens junctions (TJ/AJs) paracellular spaces to enter the postcapillary venule along with monocytes (M) and lymphocytes (L) into the postcapillary venule PVS of the PVU (B) this image illustrates step one of the two-step process of neuroinflammation. The postcapillary venule contains the PVU, which includes the normal PVS that has the capability to remodel to the pathological EPVS. Note how the proinflammatory leukocytes enter the PVS along with fluids, solutes, and peripheral and endothelial cell-derived cytokines/chemokines from an activated, disrupted, and leaky NVU in (A). Note how the pvACef (pseudo-colored blue) and its glia limitans (pseudo-colored brown in the control NVU in (A) to the cyan color with exaggerated thickness for illustrative purposes in (B)) that faces and adheres to the NVU BM extracellular matrix and faces the PVS PVU lumen, since this has detached and separated and allowed the creation of a perivascular space that transforms to an EPVS in (B). Also, note how the glia limitans becomes pseudo-colored red, once the EPVS have developed and then becomes breeched due to activation of matrix metalloproteinases and degradation of the proteins in the glia limitans, which allow neurotoxins and proinflammatory cells to leak into the interstitial spaces of the neuropil and mix with the ISF and result in neuroinflammation (step two) of the two-step process of neuroinflammation [36]. Importantly step 2 also allows for the free extravasation of blood components including neurotoxic red blood cells (hemoglobin metabolic byproducts including hemosiderin, which allows for the hypodensities on GRE MRI images and plasma that contains neurotoxic thrombin, fibrin, plasmin, hemoglobin metabolic byproducts such as hemosiderin and free iron to instigate further neuroinflammation, oxidative stress and activation of local MMPs that contribute to ongoing degradation of the PVS outer barrier (glia limitans) in a vicious cycle once it is instigated. Note that the dysfunctional pvACef AQP4 water channel is associated with the dysfunctional bidirectional signaling between the neurons (N) and the dysfunctional pvACef AQP4 water channel. Modified image provided with permission by CC 4.0 [15]. AQP4, aquaporin 4; Asterisk, tight and adherens junction; BBB, blood–brain barrier; BM, both inner (i) and outer (o) basement membrane; dACef and dpvACef, dysfunctional astrocyte endfeet; EC, brain endothelial cell; ecGCx, endothelial glycocalyx; EVPS, enlarged perivascular space; fAQP4, functional aquaporin 4; GL, glia limitans; H2O,water; L, lymphocyte; M, monocyte; N, neutrophile and neuron; Pc, pericyte; PVS, perivascular space; PVU, perivascular unit; RBC(s), red blood cells; rPVMΦ, resident perivascular macrophage; TJ/AJ. tight and adherens junctions.
Figure 4.
Comparison of the true capillary neurovascular unit (NVU) to the postcapillary venule perivascular unit (PVU). The NVU protoplasmic perivascular astrocyte endfeet (pvACef) (pseudo-colored blue) within the true capillary illustration (A) are the connecting and creating cells that allow remodeling of the normal perivascular unit (PVU (B)) perivascular spaces (PVS) to transform and remodel into the pathologic enlarged perivascular space (EPVS, which measure 1–3 mm on magnetic resonance imaging). (A) Hand-drawn and pseudo-colored control true capillary neurovascular unit (NVU)). Note that when the brain endothelial cells (BECs) become activated and NVU BBB disruption develops, due to BEC activation and dysfunction (BECact/dys) (from multiple causes), there develops an increased permeability of fluids, peripheral cytokines and chemokines, and peripheral immune cells with a neutrophile (N) depicted herein penetrating the tight and adherens junctions (TJ/AJs) paracellular spaces to enter the postcapillary venule along with monocytes (M) and lymphocytes (L) into the postcapillary venule PVS of the PVU (B) this image illustrates step one of the two-step process of neuroinflammation. The postcapillary venule contains the PVU, which includes the normal PVS that has the capability to remodel to the pathological EPVS. Note how the proinflammatory leukocytes enter the PVS along with fluids, solutes, and peripheral and endothelial cell-derived cytokines/chemokines from an activated, disrupted, and leaky NVU in (A). Note how the pvACef (pseudo-colored blue) and its glia limitans (pseudo-colored brown in the control NVU in (A) to the cyan color with exaggerated thickness for illustrative purposes in (B)) that faces and adheres to the NVU BM extracellular matrix and faces the PVS PVU lumen, since this has detached and separated and allowed the creation of a perivascular space that transforms to an EPVS in (B). Also, note how the glia limitans becomes pseudo-colored red, once the EPVS have developed and then becomes breeched due to activation of matrix metalloproteinases and degradation of the proteins in the glia limitans, which allow neurotoxins and proinflammatory cells to leak into the interstitial spaces of the neuropil and mix with the ISF and result in neuroinflammation (step two) of the two-step process of neuroinflammation [36]. Importantly step 2 also allows for the free extravasation of blood components including neurotoxic red blood cells (hemoglobin metabolic byproducts including hemosiderin, which allows for the hypodensities on GRE MRI images and plasma that contains neurotoxic thrombin, fibrin, plasmin, hemoglobin metabolic byproducts such as hemosiderin and free iron to instigate further neuroinflammation, oxidative stress and activation of local MMPs that contribute to ongoing degradation of the PVS outer barrier (glia limitans) in a vicious cycle once it is instigated. Note that the dysfunctional pvACef AQP4 water channel is associated with the dysfunctional bidirectional signaling between the neurons (N) and the dysfunctional pvACef AQP4 water channel. Modified image provided with permission by CC 4.0 [15]. AQP4, aquaporin 4; Asterisk, tight and adherens junction; BBB, blood–brain barrier; BM, both inner (i) and outer (o) basement membrane; dACef and dpvACef, dysfunctional astrocyte endfeet; EC, brain endothelial cell; ecGCx, endothelial glycocalyx; EVPS, enlarged perivascular space; fAQP4, functional aquaporin 4; GL, glia limitans; H2O,water; L, lymphocyte; M, monocyte; N, neutrophile and neuron; Pc, pericyte; PVS, perivascular space; PVU, perivascular unit; RBC(s), red blood cells; rPVMΦ, resident perivascular macrophage; TJ/AJ. tight and adherens junctions.
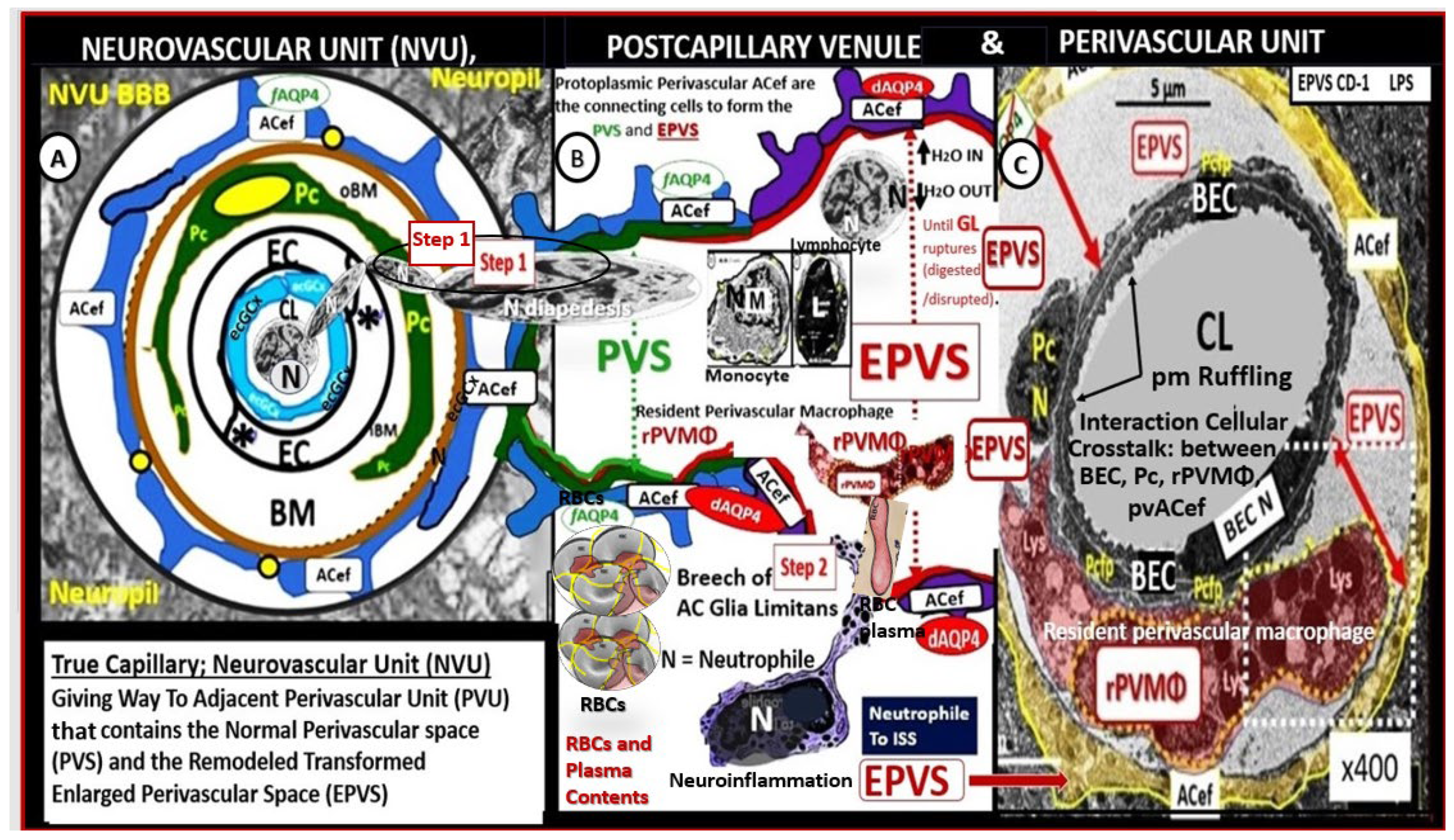
Figure 5.
Perivascular astrocyte endfeet (ACef), neurovascular unit (NVU), perivascular unit (PVU), perivascular space PVS, and enlarged perivascular space (EPVS). The NVU is located centrally; note the absence of the endothelial glycocalyx (ecGCx) surface layer, which occurs in many neurovascular and neurodegenerative diseases with impaired cognition that also include obesity, metabolic syndrome (MetS), and type 2 diabetes mellitus (T2DM). Increased NVU permeability via BECact/dys BBB disruption due to multiple clinical neurovascular and neurodegenerative diseases allows the entry of proinflammatory leukocytes into the PVU PVS in postcapillary venules. The accumulation of proinflammatory cells’ oxidative stress with increased ROS will activate local and regional matrix metalloproteinase (MMP)—a proteolytic enzyme capable of degrading the glia limitans of the pvACef to allow the breeching of the postcapillary perivascular space and the entry of proinflammatory leukocytes, red blood cells (RBCs), solutes, and neurotoxins into the interstitial spaces (ISSs) to result in CMBs, neuroinflammation and increased CNSC/C, impaired cognition, and neurodegeneration via synaptic and neuronal loss with neural atrophy. Note the isolated synapse (uncradled) in the upper left-hand side of illustration. Image reproduced with permission by CC by 4.0 [37]. AC, astrocyte; ACef, perivascular astrocyte endfeet; AQP4, aquaporin-4; BEC, brain endothelial cell; N, nucleus; n, = nucleolus; Pc, pericyte; PVU, perivascular unit; pvMGC, perivascular microglia cell; rMGC, reactive microglia cell; rPVMΦ, resident perivascular macrophage; ROS, reactive oxygen species. Modified image provided with permission by CC 4.0 [36,37].
Figure 5.
Perivascular astrocyte endfeet (ACef), neurovascular unit (NVU), perivascular unit (PVU), perivascular space PVS, and enlarged perivascular space (EPVS). The NVU is located centrally; note the absence of the endothelial glycocalyx (ecGCx) surface layer, which occurs in many neurovascular and neurodegenerative diseases with impaired cognition that also include obesity, metabolic syndrome (MetS), and type 2 diabetes mellitus (T2DM). Increased NVU permeability via BECact/dys BBB disruption due to multiple clinical neurovascular and neurodegenerative diseases allows the entry of proinflammatory leukocytes into the PVU PVS in postcapillary venules. The accumulation of proinflammatory cells’ oxidative stress with increased ROS will activate local and regional matrix metalloproteinase (MMP)—a proteolytic enzyme capable of degrading the glia limitans of the pvACef to allow the breeching of the postcapillary perivascular space and the entry of proinflammatory leukocytes, red blood cells (RBCs), solutes, and neurotoxins into the interstitial spaces (ISSs) to result in CMBs, neuroinflammation and increased CNSC/C, impaired cognition, and neurodegeneration via synaptic and neuronal loss with neural atrophy. Note the isolated synapse (uncradled) in the upper left-hand side of illustration. Image reproduced with permission by CC by 4.0 [37]. AC, astrocyte; ACef, perivascular astrocyte endfeet; AQP4, aquaporin-4; BEC, brain endothelial cell; N, nucleus; n, = nucleolus; Pc, pericyte; PVU, perivascular unit; pvMGC, perivascular microglia cell; rMGC, reactive microglia cell; rPVMΦ, resident perivascular macrophage; ROS, reactive oxygen species. Modified image provided with permission by CC 4.0 [36,37].
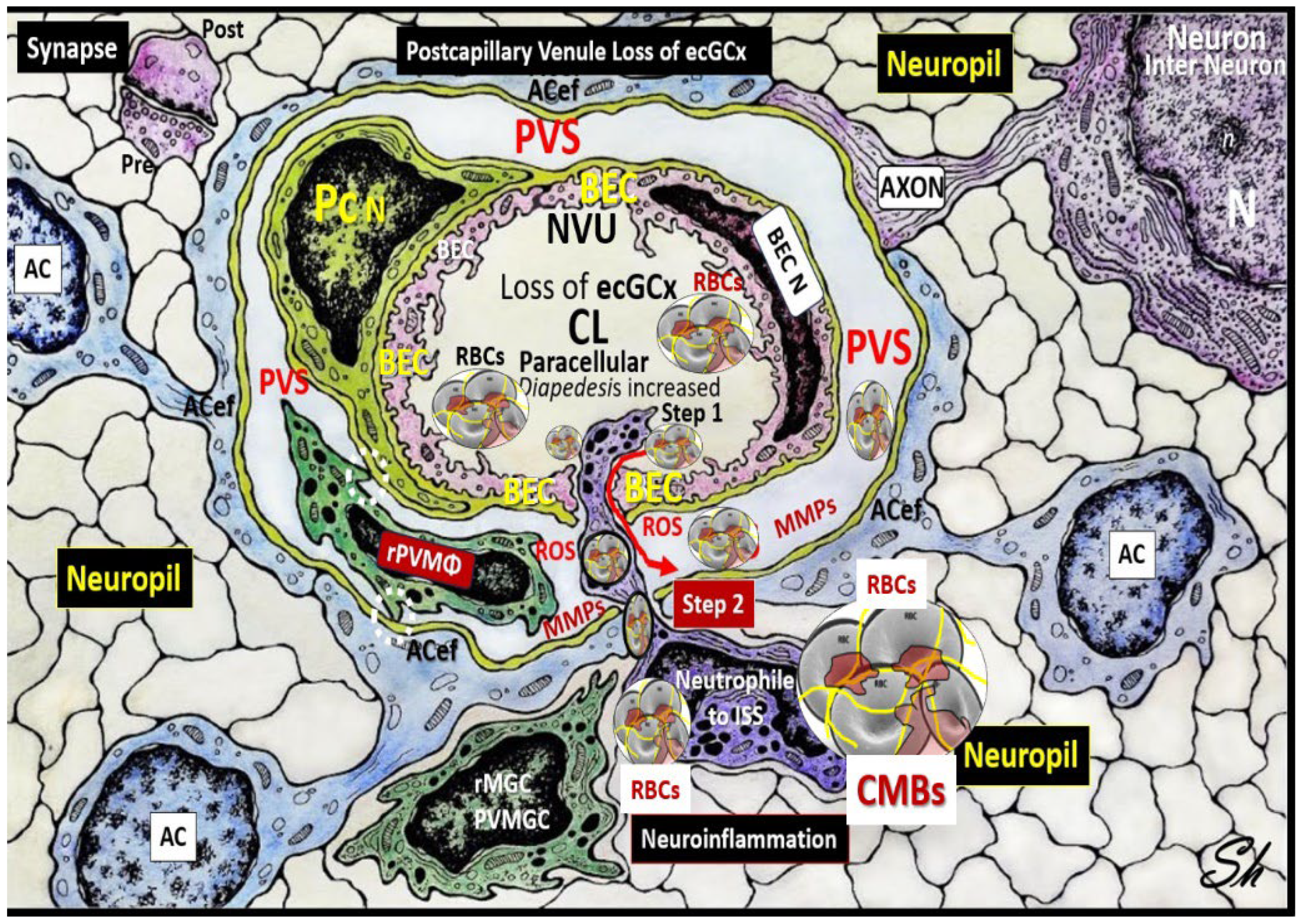
Figure 6.
Possible reoccurring sequence of events resulting in a vicious cycle with increased cerebral microbleeds and leaky brains that lead to impaired cognition and neurodegeneration. Arrowheads, lead to; BECact/dys, brain endothelial cell activation/dysfunction; BBBdd, blood-brain barrier dysfunction, disruption; CMBs, cerebral microbleeds; EPVS, enlarged perivascular spaces; MMP, matrix metalloproteinases; NFkappaB, nuclear factor kappa B; pCC, peripheral cytokines/chemokines; ROS, reactive oxygen species; RONSS, reactive oxygen, nitrogen, sulfur, species; RSI, reactive species interactome; SVD, small vessel disease; TJ/AJ, tight and adherens junctions; VE-Cadherins, vascular endothelial cadherins; WMH, white matter hyperintensities.
Figure 6.
Possible reoccurring sequence of events resulting in a vicious cycle with increased cerebral microbleeds and leaky brains that lead to impaired cognition and neurodegeneration. Arrowheads, lead to; BECact/dys, brain endothelial cell activation/dysfunction; BBBdd, blood-brain barrier dysfunction, disruption; CMBs, cerebral microbleeds; EPVS, enlarged perivascular spaces; MMP, matrix metalloproteinases; NFkappaB, nuclear factor kappa B; pCC, peripheral cytokines/chemokines; ROS, reactive oxygen species; RONSS, reactive oxygen, nitrogen, sulfur, species; RSI, reactive species interactome; SVD, small vessel disease; TJ/AJ, tight and adherens junctions; VE-Cadherins, vascular endothelial cadherins; WMH, white matter hyperintensities.
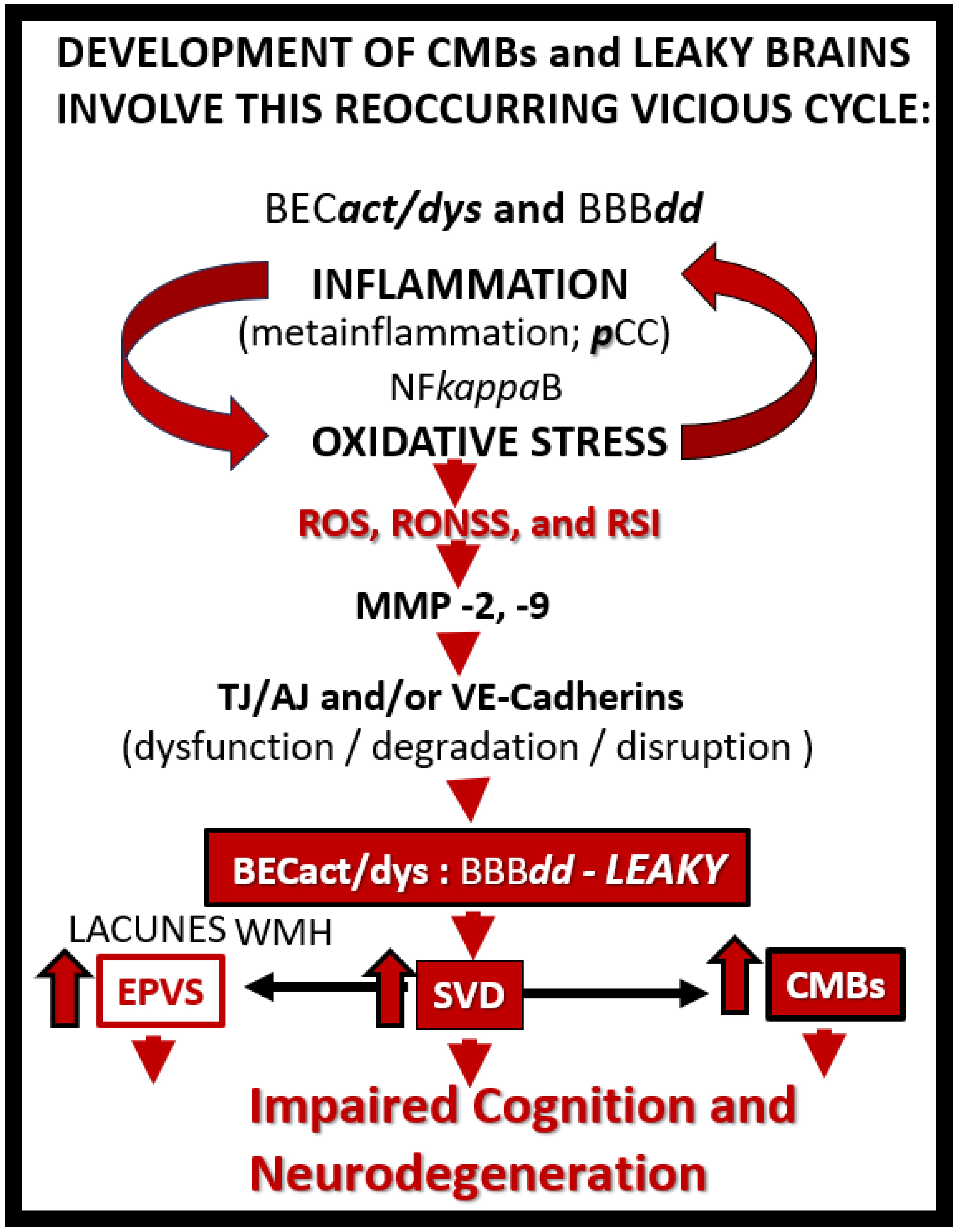
Figure 7.
Attenuation and/or loss of tight and adherence junction(s) (TJ/AJs) paracellular blood-brain barrier (BBB) in male CD-1 streptozotocin-induced (STZ) diabetic preclinical mice models resulting in blood-brain barrier dysfunction and disruption (BBBdd) protected by the carbonic anhydrase inhibitor (Topiramate a mitochondria antioxidant) in the mid brain as compared to the cerebellum. The STZ-induced type I diabetic mice revealed disruption of the BBB by 14C-sucrose measurements. Panel A demonstrates three prominent elongated electron dense TJ/AJ (white and black arrows). Panel B depicts a discontinuous and disrupted TJ/AJ (black arrows) into three distinct segments in the midbrain. Note how the TJ/AJ tend to form at EC-EC overlap junctions. Panel C demonstrates that treatment with Topiramate prevented this disruption in the brain endothelial cell BBB (yellow and black arrows and yellow dashed line below the intact BBB TJ/AJ) in the midbrain. Revised figure images provided with permission by CC 4.0 [55]. Magnification x3,000; scale bar = 0.5μm (A); x10,000; scale bar = 0.2μm in (B) and (C).
Figure 7.
Attenuation and/or loss of tight and adherence junction(s) (TJ/AJs) paracellular blood-brain barrier (BBB) in male CD-1 streptozotocin-induced (STZ) diabetic preclinical mice models resulting in blood-brain barrier dysfunction and disruption (BBBdd) protected by the carbonic anhydrase inhibitor (Topiramate a mitochondria antioxidant) in the mid brain as compared to the cerebellum. The STZ-induced type I diabetic mice revealed disruption of the BBB by 14C-sucrose measurements. Panel A demonstrates three prominent elongated electron dense TJ/AJ (white and black arrows). Panel B depicts a discontinuous and disrupted TJ/AJ (black arrows) into three distinct segments in the midbrain. Note how the TJ/AJ tend to form at EC-EC overlap junctions. Panel C demonstrates that treatment with Topiramate prevented this disruption in the brain endothelial cell BBB (yellow and black arrows and yellow dashed line below the intact BBB TJ/AJ) in the midbrain. Revised figure images provided with permission by CC 4.0 [55]. Magnification x3,000; scale bar = 0.5μm (A); x10,000; scale bar = 0.2μm in (B) and (C).
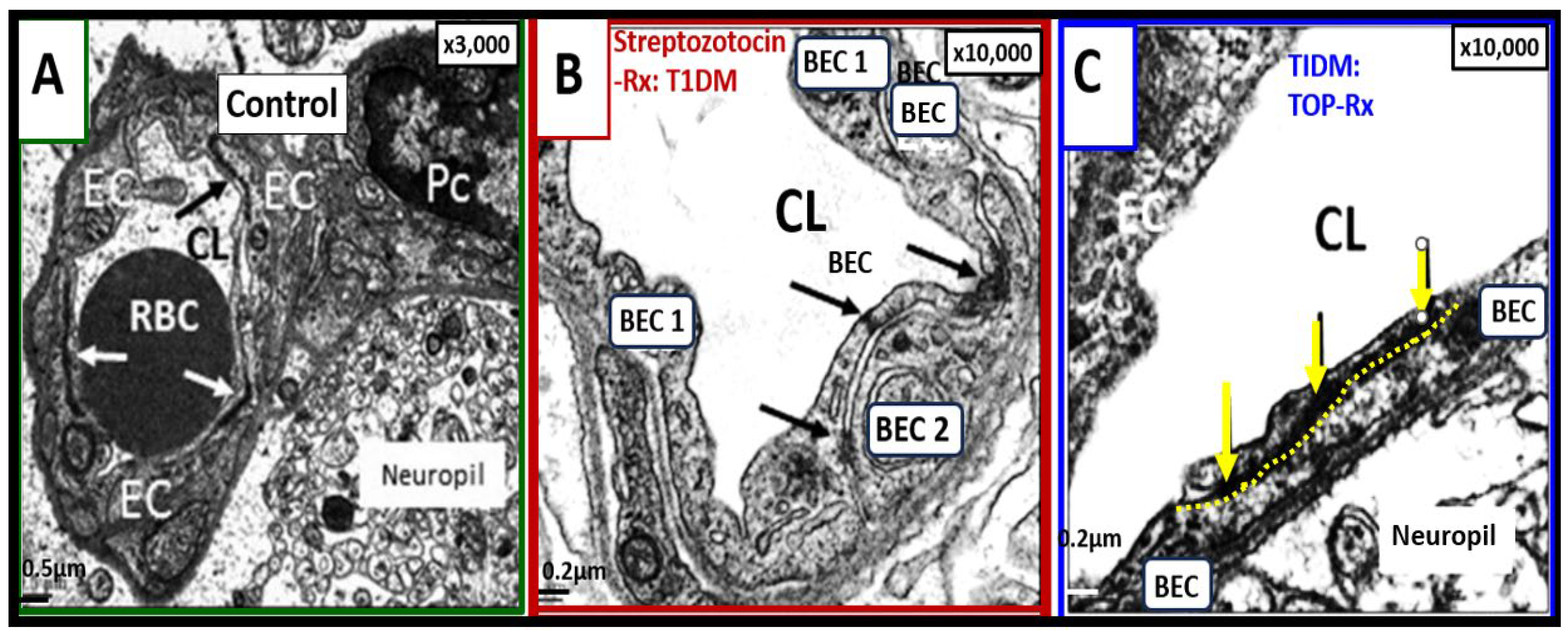
Figure 8.
Ultrastructural images of brain endothelial cell activation/dysfunction (BECact/dys) in 20-week- old obese diabetic db/db female models. (A) demonstrates the normal neurovascular unit (NVU) blood-brain barrier (BBB) capillary and note the thinness of the moderate electron dense cytoplasm. (B) depicts regions of abrupt thickened electron lucent (red arrows) with vacuolization of the basement membrane (BM) in obese diabetic female db/db models with BECact/dys as compared to control panel A. (C) depicts BM thickening with increased vacuole-like bodies (V). (D), (E), (F), and (G) depict monocyte (D), lymphocyte (E) leukocytes, platelet (F), and red blood cell (G) adherence to the plasma membrane of brain endothelial cells in BECact/dys db/db models. Images reproduced with permission by CC 4.0 [13,82]. Original magnification = ×2000; scale bar = 1 μm. ACfp, astrocyte foot processes; Cl, capillary lumen; EC, brain endothelial cells; MP, microparticle of the platelet.
Figure 8.
Ultrastructural images of brain endothelial cell activation/dysfunction (BECact/dys) in 20-week- old obese diabetic db/db female models. (A) demonstrates the normal neurovascular unit (NVU) blood-brain barrier (BBB) capillary and note the thinness of the moderate electron dense cytoplasm. (B) depicts regions of abrupt thickened electron lucent (red arrows) with vacuolization of the basement membrane (BM) in obese diabetic female db/db models with BECact/dys as compared to control panel A. (C) depicts BM thickening with increased vacuole-like bodies (V). (D), (E), (F), and (G) depict monocyte (D), lymphocyte (E) leukocytes, platelet (F), and red blood cell (G) adherence to the plasma membrane of brain endothelial cells in BECact/dys db/db models. Images reproduced with permission by CC 4.0 [13,82]. Original magnification = ×2000; scale bar = 1 μm. ACfp, astrocyte foot processes; Cl, capillary lumen; EC, brain endothelial cells; MP, microparticle of the platelet.
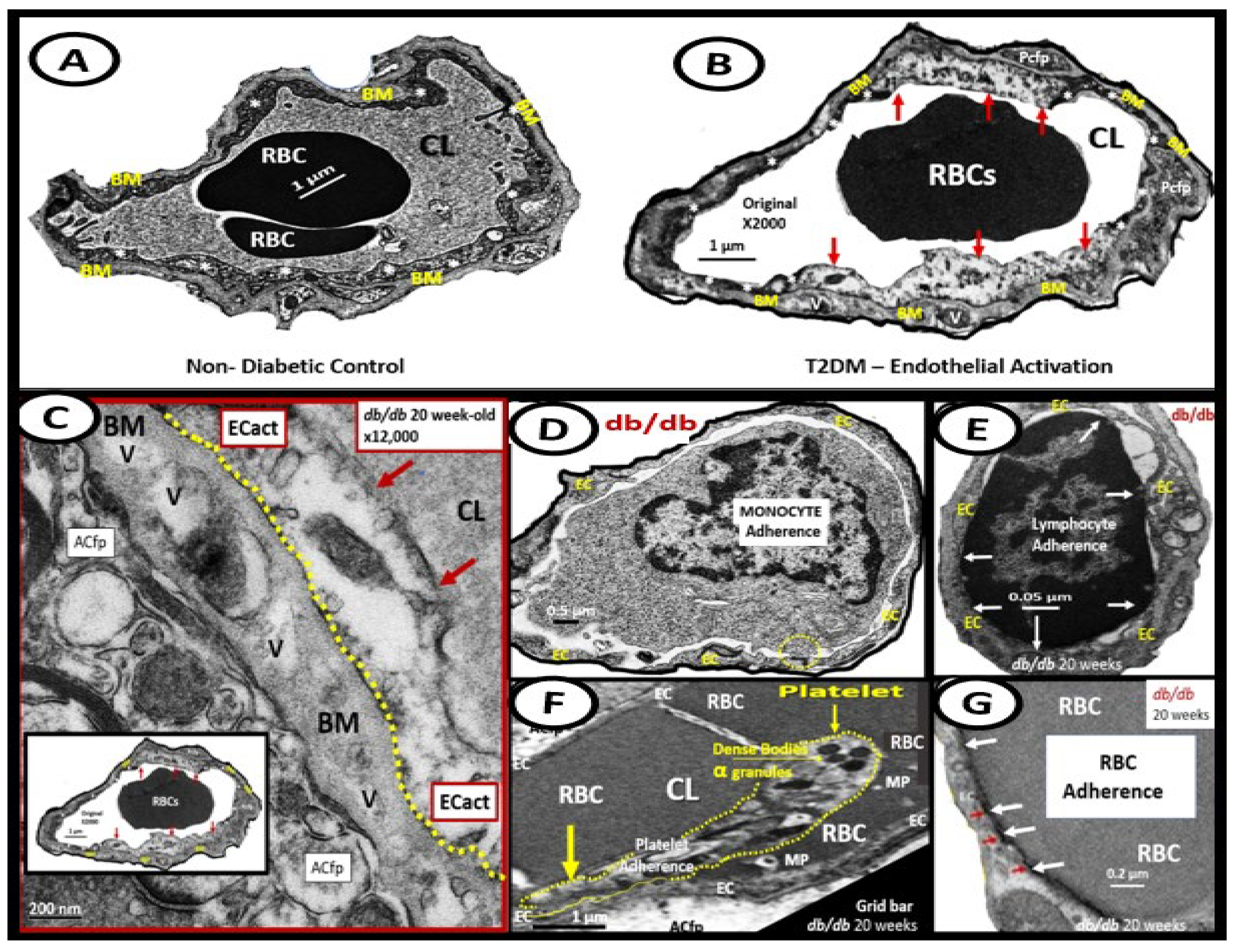
Figure 9.
Six panel image depicting cerebral microbleeds-hemorrhages in preclinical female obese metabolic syndrome, and type 2 diabetes mellitus db/db genetic mice models from frontal cortical layer III.Each of these six panels except for panel D depict a cerebral microbleed identified by a large white X. Note in panels B and C the homogeneous electron-dense staining could also represent free extruded plasma. Also, importantly note that in panel E the homogenous electron-dense staining may represent a free RBC within the neuropil due to the presence of the multiple small electron lucent vacuoles (white in color) that aid in the identification of this being a RBC. Note in panel D the aberrant mitochondria (pseudo-colored yellow outlined in red) in the brain endothelial cell and also in the adjacent aMGC, which suggest activation of brain endothelial cell. Images provided by CC 4.0 [15,82]. aMGC, reactive microglia cell; hashtags, microbleeds most likely plasma; N, nucleus; numbers (1), (2), adjacent capillary neurovascular units; RBC, red blood cell; X, microbleeds. Magnifications and scale bars vary from image to image and are present in each panel.
Figure 9.
Six panel image depicting cerebral microbleeds-hemorrhages in preclinical female obese metabolic syndrome, and type 2 diabetes mellitus db/db genetic mice models from frontal cortical layer III.Each of these six panels except for panel D depict a cerebral microbleed identified by a large white X. Note in panels B and C the homogeneous electron-dense staining could also represent free extruded plasma. Also, importantly note that in panel E the homogenous electron-dense staining may represent a free RBC within the neuropil due to the presence of the multiple small electron lucent vacuoles (white in color) that aid in the identification of this being a RBC. Note in panel D the aberrant mitochondria (pseudo-colored yellow outlined in red) in the brain endothelial cell and also in the adjacent aMGC, which suggest activation of brain endothelial cell. Images provided by CC 4.0 [15,82]. aMGC, reactive microglia cell; hashtags, microbleeds most likely plasma; N, nucleus; numbers (1), (2), adjacent capillary neurovascular units; RBC, red blood cell; X, microbleeds. Magnifications and scale bars vary from image to image and are present in each panel.
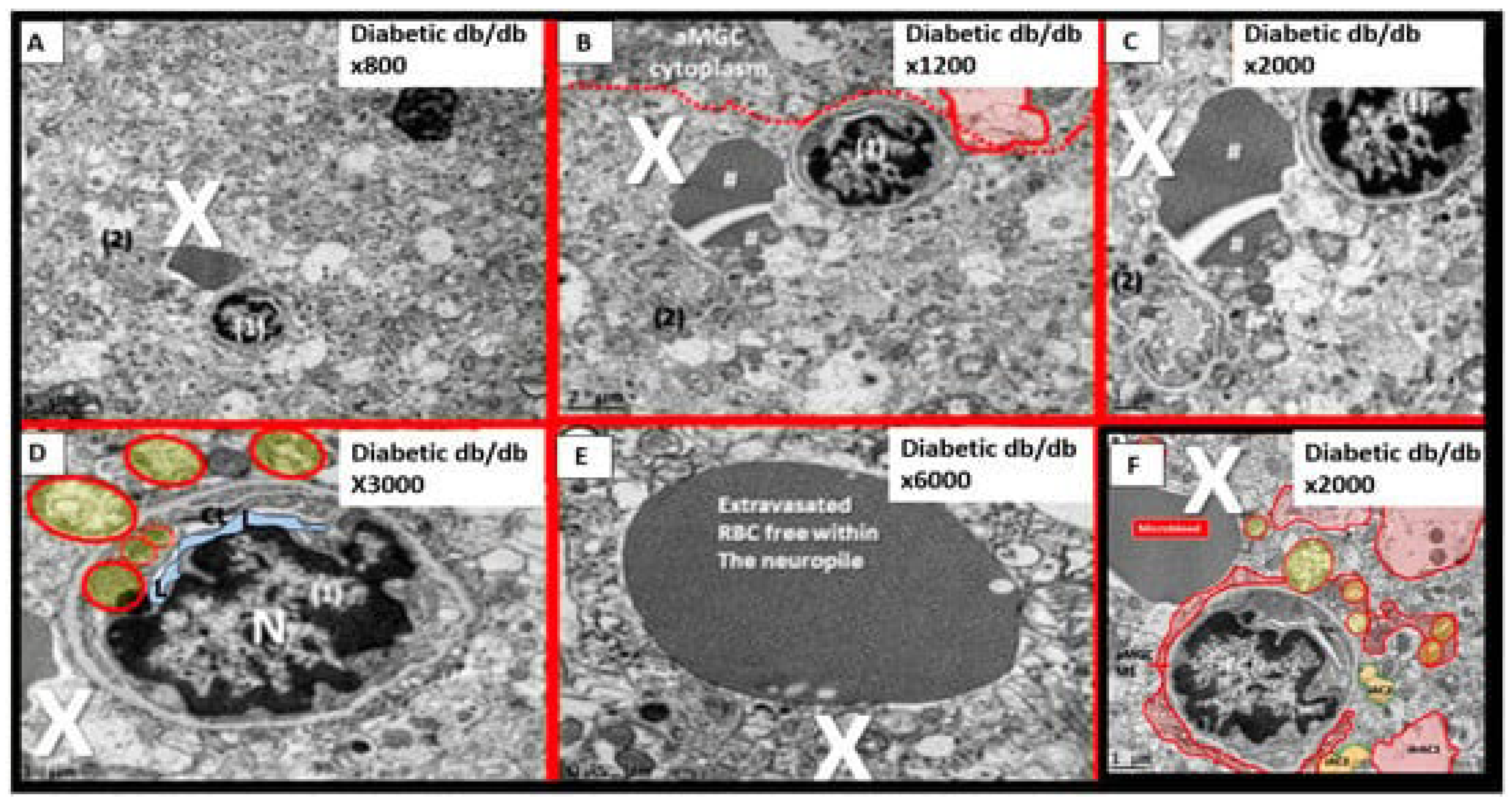
Figure 10.
Extrusion of a free RBC into the neuropil from an adjacent capillary neurovascular unit in the cortical layer III from the diabetic db/db model at low and high magnification. Note the labeled microbleed associated with the nearby small NVU capillary, the encroaching reactive microglia cell M1-like proinflammatory cell (aMGC M1 - like) cytoplasmic process enveloping the aberrant small neurovascular unit capillary, and the detached and retracted astrocytes pseudo-colored red (drAC1) and a couple of intact attached astrocytes (pseudo-colored yellow) iAC2). Images provided by CC 4.0 [82] Magnification x800; scale bar = 1μm (panel A) and Magnification x2000; scale bar = 1 μm (panel B). EC N, brain endothelial cell nucleus; aMGC, reactive microglia cell.
Figure 10.
Extrusion of a free RBC into the neuropil from an adjacent capillary neurovascular unit in the cortical layer III from the diabetic db/db model at low and high magnification. Note the labeled microbleed associated with the nearby small NVU capillary, the encroaching reactive microglia cell M1-like proinflammatory cell (aMGC M1 - like) cytoplasmic process enveloping the aberrant small neurovascular unit capillary, and the detached and retracted astrocytes pseudo-colored red (drAC1) and a couple of intact attached astrocytes (pseudo-colored yellow) iAC2). Images provided by CC 4.0 [82] Magnification x800; scale bar = 1μm (panel A) and Magnification x2000; scale bar = 1 μm (panel B). EC N, brain endothelial cell nucleus; aMGC, reactive microglia cell.

Figure 11.
A microbleed plasma extrusion (~5μm) immediately adjacent to a contracted lumen microvessel (~5μm). Note how the lumen of this microvessel (pseudo-colored light blue) is nearly collapsed and that the brain endothelial cell (BEC) nucleus is contracted with extremely prominent chromatin condensation instead of being heterogenous suggesting BEC activation and dysfunction. These similar morphological contracted BEC remodeling changes and nuclear remodeling changes were observed in the aortic endothelium of activated endothelial cells in female Western diet fed mice at 20-weeks of age.Also, note that the reactive microglia (pseudo-colored red) encircle this microvessel that it contains multiple aberrant mitochondria (aMt), which provide excessive mitochondria-derived reactive oxygen species that provide BEC injury for the response to injury wound healing mechanisms at the level of this microvessel to result in BEC activation and dysfunction. Importantly, note reactive astrocyte detachment and separation of reactive perivascular astrocytes. These remodeling changes allow for microvessel disruption and microbleeds. Image provided by CC 4.0 [82,83]. AC, astrocyte; CMB, cerebral microbleed; EC N, brain endothelial nucleus; iAC, intact attached astrocyte; rMGC, reactive microglia cell; Pc, pericyte; X, microbleed-microhemorrhage.
Figure 11.
A microbleed plasma extrusion (~5μm) immediately adjacent to a contracted lumen microvessel (~5μm). Note how the lumen of this microvessel (pseudo-colored light blue) is nearly collapsed and that the brain endothelial cell (BEC) nucleus is contracted with extremely prominent chromatin condensation instead of being heterogenous suggesting BEC activation and dysfunction. These similar morphological contracted BEC remodeling changes and nuclear remodeling changes were observed in the aortic endothelium of activated endothelial cells in female Western diet fed mice at 20-weeks of age.Also, note that the reactive microglia (pseudo-colored red) encircle this microvessel that it contains multiple aberrant mitochondria (aMt), which provide excessive mitochondria-derived reactive oxygen species that provide BEC injury for the response to injury wound healing mechanisms at the level of this microvessel to result in BEC activation and dysfunction. Importantly, note reactive astrocyte detachment and separation of reactive perivascular astrocytes. These remodeling changes allow for microvessel disruption and microbleeds. Image provided by CC 4.0 [82,83]. AC, astrocyte; CMB, cerebral microbleed; EC N, brain endothelial nucleus; iAC, intact attached astrocyte; rMGC, reactive microglia cell; Pc, pericyte; X, microbleed-microhemorrhage.
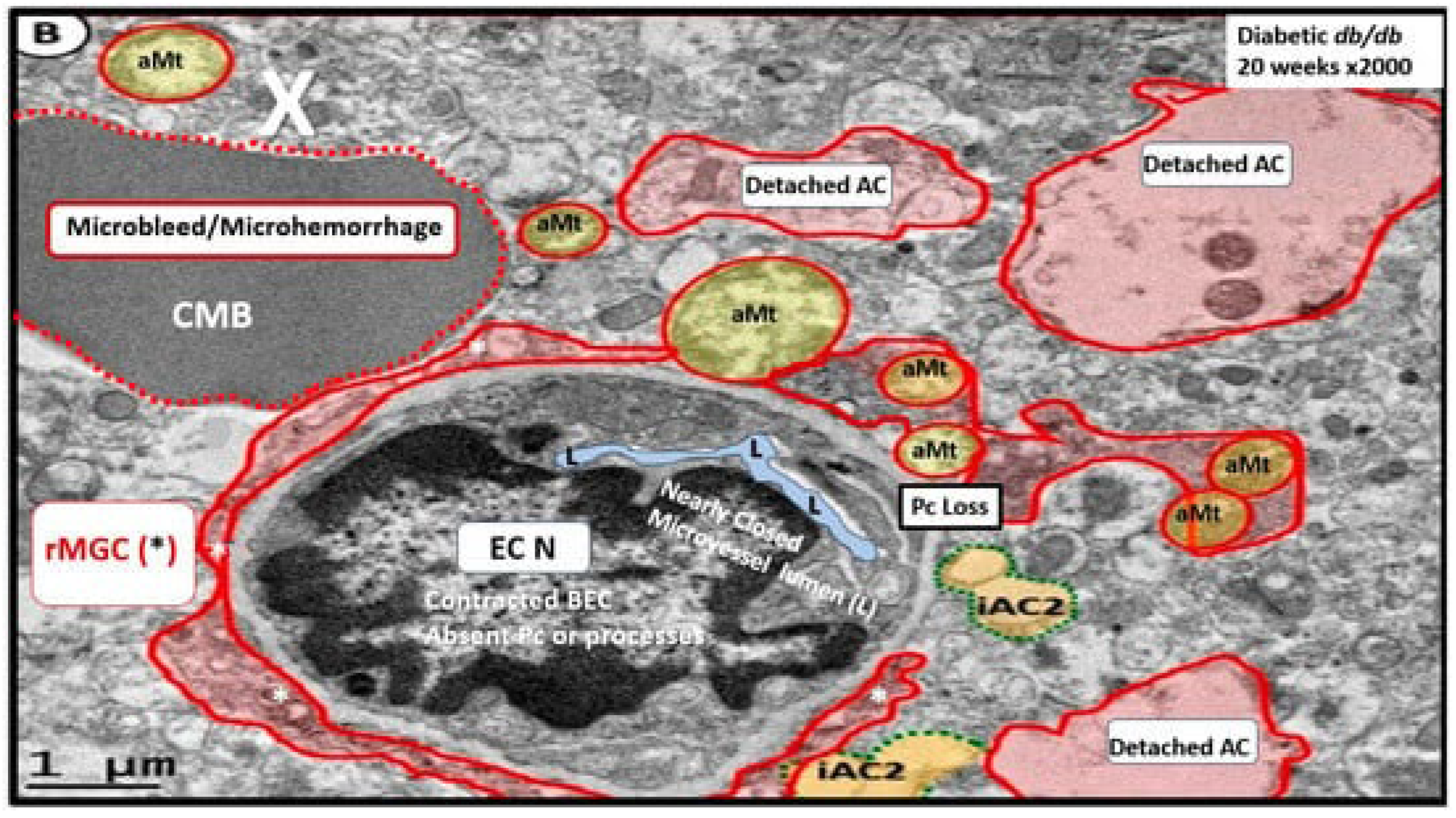
Figure 12.
Small vessel disease (SVD) comparisons and EPVS and CMBs spectrum disorders. Upper panel A depicts the comparisons between the 4 major components of SVD: lacunes, enlarged perivascular spaces (EPVS), white matter hyperintensities (WMH), and cerebral microbleeds (CMBs). Note that panel A is a reproduction of previous figure 2.Lower panel B depicts the spectrum of SVD with emphasis on EPVS and CMBs. Note the importance of EPVS and CMBs are each now considered to be biomarkers for microvessel SVD and that time plays an important role in the development of SVD with emphasis on EPVS and CMBs. Note the asterisks denote the emphasis on EPVS and CMBs that are two of the 4 components of SVD biomarkers.aBEC, activated brain endothelial cells; ACfps, astrocyte foot processes; AQ4, aquaporin 4; CSF, cerebral spinal fluid; ISF, interstitial fluid; LOAD, late-onset Alzheimer’s disease; MRI, magnetic resonance image or imaging; PVS. perivascular spaces; SAS, subarachnoid space.
Figure 12.
Small vessel disease (SVD) comparisons and EPVS and CMBs spectrum disorders. Upper panel A depicts the comparisons between the 4 major components of SVD: lacunes, enlarged perivascular spaces (EPVS), white matter hyperintensities (WMH), and cerebral microbleeds (CMBs). Note that panel A is a reproduction of previous figure 2.Lower panel B depicts the spectrum of SVD with emphasis on EPVS and CMBs. Note the importance of EPVS and CMBs are each now considered to be biomarkers for microvessel SVD and that time plays an important role in the development of SVD with emphasis on EPVS and CMBs. Note the asterisks denote the emphasis on EPVS and CMBs that are two of the 4 components of SVD biomarkers.aBEC, activated brain endothelial cells; ACfps, astrocyte foot processes; AQ4, aquaporin 4; CSF, cerebral spinal fluid; ISF, interstitial fluid; LOAD, late-onset Alzheimer’s disease; MRI, magnetic resonance image or imaging; PVS. perivascular spaces; SAS, subarachnoid space.

Figure 13.
Schematic for aging, APOE-ε4 genotype, T2DM, HTN, and inflammation/neuroinflammation in the development of cerebral microbleeds (CMBs). Note that each of the major color-coded components AGING yellow, APOE-ε4 (blue), Hypertension (orange), Inflammation/Neuroinflammation (red), and their respective components may all contribute to the development and evolution of the centrally located CMBs.Also, note the importance of BECact/dys and BBBdd as a possible final common pathway for the development of CMBs. Note that ROS and MMPs are placed in a colored dark-red box with yellow lettering since they are possibly the final common signaling pathway for the development of CMBs due the degradation of the glia limitans of the perivascular units’ perivascular spaces outermost limiting barrier membrane. APOE-ε4, apolipoprotein epsilon 4; BBB, blood-brain barrier; BBBdd, blood-brain barrier dysfunction and/or disruption; BECact/dys, brain endothelial cell activation and dysfunction; CAA, cerebral amyloid angiopathy; MMPs, matrix metalloproteinases; NO, nitric oxide; PKC, protein kinase C; ROS, reactive oxygen species, upward arrow, increase; downward arrow, decrease.
Figure 13.
Schematic for aging, APOE-ε4 genotype, T2DM, HTN, and inflammation/neuroinflammation in the development of cerebral microbleeds (CMBs). Note that each of the major color-coded components AGING yellow, APOE-ε4 (blue), Hypertension (orange), Inflammation/Neuroinflammation (red), and their respective components may all contribute to the development and evolution of the centrally located CMBs.Also, note the importance of BECact/dys and BBBdd as a possible final common pathway for the development of CMBs. Note that ROS and MMPs are placed in a colored dark-red box with yellow lettering since they are possibly the final common signaling pathway for the development of CMBs due the degradation of the glia limitans of the perivascular units’ perivascular spaces outermost limiting barrier membrane. APOE-ε4, apolipoprotein epsilon 4; BBB, blood-brain barrier; BBBdd, blood-brain barrier dysfunction and/or disruption; BECact/dys, brain endothelial cell activation and dysfunction; CAA, cerebral amyloid angiopathy; MMPs, matrix metalloproteinases; NO, nitric oxide; PKC, protein kinase C; ROS, reactive oxygen species, upward arrow, increase; downward arrow, decrease.
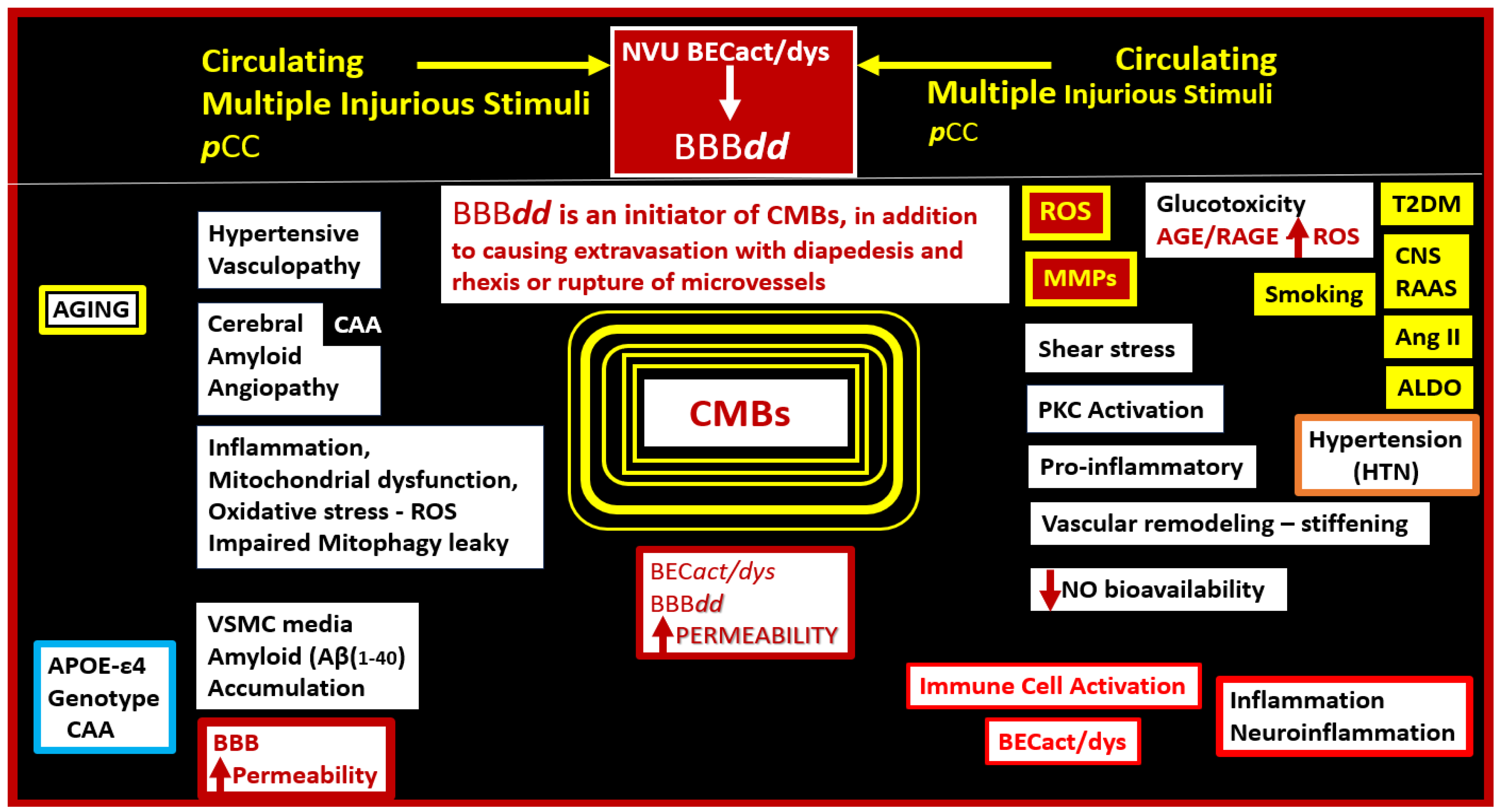

Table 1.
Similarities and differences between cerebral amyloid angiopathy (CAA) and hypertension (HTN) as related to the formation of cerebral microbleeds (CMBs). Aβ (1-40), amyloid beta 1 through 40; ALDO, aldosterone; Ang II, angiotensin two; ApoE-ε4, apolipoprotein E epsilon 4 genotype; BBBdd, blood-brain barrier dysfunction and/or disruption; BEC(s), brain endothelial cells; BECact/dys, brain endothelial cell activation and dysfunction; CNS, central nervous system; ECM, extracellular matrix; RAAS, brain renin-angiotensin-aldosterone system; SNS, sympathetic nervous system; VSMCs, vascular smooth muscle cells.
Table 1.
Similarities and differences between cerebral amyloid angiopathy (CAA) and hypertension (HTN) as related to the formation of cerebral microbleeds (CMBs). Aβ (1-40), amyloid beta 1 through 40; ALDO, aldosterone; Ang II, angiotensin two; ApoE-ε4, apolipoprotein E epsilon 4 genotype; BBBdd, blood-brain barrier dysfunction and/or disruption; BEC(s), brain endothelial cells; BECact/dys, brain endothelial cell activation and dysfunction; CNS, central nervous system; ECM, extracellular matrix; RAAS, brain renin-angiotensin-aldosterone system; SNS, sympathetic nervous system; VSMCs, vascular smooth muscle cells.
| FACTOR | CEREBAL AMYLOID ARTERIOPATHY(CAA) | HYPERTENSION (HTN) |
|---|---|---|
| Definition | CAA is a condition where amyloid beta Aβ (1-40) protein builds up in the walls of arteries and arterioles primarily to the media VSMCs, ECM and adventitia in the CNS, increasing the risk of cerebral microbleeds (CMBs). | Hypertension refers to high blood pressure, which can lead to changes in small blood vessels (microvessels) in the brain, potentially causing microbleeds (CMBs). |
| Molecular | Cerebral amyloid beta (Aβ1-40) accumulation in microvessels resulting in loss of integrity to the vessel wall with extrusion of blood contents by rhexis (rupture) and/or diapedesis. | Peripheral and CNS RAAS activation, SNS, increased AngII and ALDO. Multifactorial genetic and environmental causes dysfunction and damage to intimal BECs, media VSMCs with loss of integrity and extrusion of luminal blood contents by rhexis (rupture) and/or diapedesis. |
| Location | Lobar (cortex, gray-white matter junction, subcortical white matter and leptomeninges) location. | Deep (infratentorial, basal ganglia, lacunal, internal and external capsule, thalamus, and brainstem. |
| Mechanisms - Pathogenesis | Vascular rupture due to endothelial and VSMC remodeling with loss of vascular integrity and rupture due to Aβ (1-40) direct effect on VSMC and ECM.BECact/dys and BBBdd. | Vascular stiffness, increased pulse pressure, vascular hyalinosis, atherosclerosis, arteriolosclerosis, oxidative stress, and inflammation. BECact/dys and BBBdd. |
| Associated conditions | Often seen in conjunction with Alzheimer’s disease and older age ≥ 60. | Age of onset may vary but usually increases with aging and can lead to various cardiovascular conditions such as heart disease and stroke. |
| Risk factors | Aging, genetics (e.g., presence of ApoE-ε4 allele genotype), and history of Alzheimer's disease | Obesity, sedentary lifestyle, smoking, excessive alcohol consumption, and family history of hypertension. |
| Clinical presentation | Most often asymptomatic but can manifest as cognitive decline, seizures, or intracerebral hemorrhage with neurologic deficits. | Most often asymptomatic but can lead to symptoms of stroke, cognitive impairment, or neurological deficits if severe. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



