
Submitted:
13 May 2024
Posted:
14 May 2024
You are already at the latest version
Abstract
Alternative splicing is an essential regulatory mechanism for gene expression in mammalian cells contributing to protein, cellular, and species diversity. Isoform switching, the process where cells alter the expression of various splice forms (isoforms) of a gene, plays a critical role in cellular function and adaptation. Our study provides a comprehensive analysis of splice forms prevailing across ten of the most studied human cell lines using public RNAseq data. We analyzed the occurrence of splice forms and found substantial justification for revising the canonical sequences for 56 genes, suggesting that these alternative splice forms should be recognized as canonical. Additionally, distinct profiles of splice form prevalence across these cell lines offer new insights into their specific biological roles. Our findings reinforce and expand upon previous insights into the link between isoform expression and oncological processes. Notably, we identified three genes exhibiting statistically significant changes between cancerous and normal tissues. Moreover, a comparative analysis of prevailing isoforms across diverse cell lines with prior research unveiled 140 common splice variants associated with cancer, highlighting their importance for prognostication and understanding oncogenesis. We revealed unique patterns and variations in isoform switching of these lines that provide new insights into the importance of selecting specific ones for research.
Keywords:
alternative splicing
; splice form
; isoform switching
1. Introduction
Alternative splicing (AS) is a fundamental mechanism that significantly enhances biological diversity at both the RNA and protein levels within eukaryotic organisms. This diversified repertoire plays a pivotal role in “fine tuning” of numerous biological processes, encompassing organismal development, homeostasis maintenance, and programmed cell death, underscoring the indispensability of gene splice variants (isoforms) in these intricate pathways [1,2] Moreover, about 50% of expressed genes have tissue-specific transcripts that can modulate the functionality, subcellular localization, and stability of their corresponding RNA or protein molecules [3]. The current level of development compels us to delve deeper into gene analysis. Therefore, the generalized value of gene expression is no longer adequate even for fundamental tasks on calibrating the quantity of PCR using housekeeping genes [4].
Cumulative evidence from large-scale inquiries has delineated numerous splice variants correlating with the pathogenesis of oncological conditions [2,5,6]. The complexity of biological processes is maintained by the variability of expressed genes, as well as by the full or partial duplication of molecular pathways, which prevents any definitive claims about the oncogenicity of a particular splice form. Variations in the amino acid sequences of alternatively spliced proteoforms originating from the same gene may result in substantial or subtle differences in their biological functions. For instance, the apoptosis regulator BCL-X gives rise to two protein isoforms: the long splice variant (BCL-XL), which confers protection against apoptosis, and the short splice variant (BCL-XS), which promotes it [7].
Transcriptome and proteome profiling methods facilitate the characterization of splice variants representation. RNA sequencing technology (RNA-seq) emerges as a validated quantitative tool for assessing cellular expression, enabling the discovery and identification of novel transcripts arising from AS. The high sensitivity of nucleic acid sequencing, in contrast to proteomic approaches (such as mass spectrometry and enzyme-linked immunosorbent assay), affords a broader spectrum of detectable transcripts reflecting AS events.
Human cell lines, due to their availability and comprehensive molecular description, serve as invaluable tools in a range of scientific research, ranging from cytotoxicity testing to elucidating metabolic pathways and gene functions [8]. Scrutinizing differences in splice form expression between different cell lines can reveal novel insights into their biology that may evade conventional gene-level analyses. To dissect the functions of splice forms, it is important not only to choose a cell line predominantly expressing the target splice form but also to comprehend the entire spectrum of splice variants within the sample and incorporate this distribution into research endeavors. This approach facilitates the investigation of the specific functions and impact of individual splice form on cellular processes.
With the bioinformatics advancements, accurate quantification of gene splice variants using RNA sequencing has become a reality, empowering nuanced investigations into cellular processes [9]. As part of our study, we analyzed the isoform switching dynamics across ten of the most prevalent human cell lines using publicly available datasets.
2. Results
2.1. Data Description
Our study aims to examine the prevalence and expression of splice variants in human cell lines. Utilizing publicly available data, we focused on the ten most extensively studied cell lines as determined by their prevalence in the NCBI SRA database (Figure 1). Among these, nine cell lines were of cancerous origin, including two breast cancer models (MCF-7 and MDA-MB-231), along with cervical (HeLa), liver (HepG2), lung (A549), colorectal (HCT116), prostate (LNCaP) cancer cells, as well as leukemic lines K562 and THP-1. Additionally, we included one non-cancerous cell line, HEK293T, renowned for its robust growth and transfection efficiency. To ensure the integrity of our investigation, we exclusively selected only control samples in datasets, representing each cell line in its untreated state. This approach was adopted to accurately capture the natural baseline from which isoform switching could be assessed, free from external influences that might potentially confound the results.
To mitigate batch effects across samples, we employed the Combat algorithm [10]. Samples displaying a Pearson correlation coefficient of less than 0.7 compared to other samples at transcript expression levels were excluded from subsequent analysis. All choosing cell lines have more than 10 transcriptomic profiling datasets.
On average, each cell line under study exhibited the expression of 17,717 genes and 38,936 transcripts. HEK293T displayed the highest diversity (18,548 genes, 40,856 transcripts), while HepG2 showed the lowest (with 17,178 genes and 37,894 transcripts). Further details regarding the average data quality for each cell line is presented in Supplementary Table S1.
2.2. Algorithm Description
The definition of isoform switching lacks universal consensus as different studies introduce different methodologies tailored to answering specific biological inquiries [5]. Generally, the concept of "isoform switching" entails a comparison of transcriptomic profiles between normal and abnormal types of the same tissue to discern differential transcript usage and expression patterns. Such an approach is not directly applicable to the assessment of AS profiles across multiple cell lines. To elucidate the functional significance of the splice form in the implementation of molecular processes, priority should be given to proteoforms exhibiting predominant expression relative to other splice forms encoded by the same gene. This consideration is imperative when interpreting results of the experiments on gene knockout or knockdown.
Detecting switching isoforms necessitates analyzing a substantial number of samples due to inherent heterogeneity even within identical tissue types. While cell lines offer molecular stability, the correlation coefficient between samples from the same cell line in transcript expression analyses is quite moderate, despite similar gene expression profiles [5]. This circumstance obliges the analysis of a larger number of samples of the same cell line. Hence, we focused on those cases where predominant splice forms were observed in more than half of the samples.
Based on the distribution of expression levels across the samples (Supplementary File 2), we formulated criteria for defining prevailing splice forms: 1) the presence of expression of two or more splice forms (>0.1 TPM, transcripts per million) encoded by one gene; 2) log2 Fold Change (log2FC) of at least 2 between the expression of two splice forms; and 3) and the expression of at least one of these splice forms exceeding 1 TPM.
The difficulty we encountered was the variability of the same splice form between samples. We noted considerable variability in transcript expression levels across samples, with an average coefficient of variation of 130%. Remarkably, this parameter remained relatively stable even with increasing expression levels, plateauing at approximately 50% for expression levels exceeding 50 TPM (Supplementary file 3). To ensure data robustness, we identified cases of splice form predominance within each sample and subsequently calculated their occurrence across samples.
The difficulty we encountered was the variability of the same splice form between samples. We noted considerable variability in transcript expression levels across samples, with an average coefficient of variation of 130%. Remarkably, this parameter remained relatively stable even with increasing expression levels, plateauing at approximately 50% for expression levels exceeding 50 TPM. To ensure data robustness, we identified cases of splice form predominance within each sample and subsequently calculated their occurrence across samples.
2.3. Splicing Diversity within Cell Lines
For each cell line, an average of 3,700 prevailing splice forms were identified, disregarding their occurrence in individual samples (either in one or all, Figure 2). The highest number of such instances was observed in the HEK293T (4,592) and HCT116 (4,188) cultures, whereas the lowest count was recorded in K562 (3,003). Furthermore, approximately 31% of splice form predominance events were observed in at least half of the samples across all cell lines. Based on this assessment, A549 and LNCaP cells exhibit relatively low heterogeneity, with approximately 41.5% and 43.9%, respectively, of their prevailing splice forms detected in the majority of samples. In contrast, HEK293T (19.4%) and HCT116 (26.8%) display the highest levels of heterogeneity.
Remarkably, within cell lines, splice form predominance varies across samples: with the same splice form being major in some samples and minor - in others. HEK293T cells exhibit the highest diversity in such cases of internal isoform switching, with 333 genes displaying varying expression transcripts, while LNCaP and A549 cultures demonstrate the lowest, with only 38 and 36 genes, respectively, demonstrating such variability.
When analyzing cases where splice forms of one gene are prevalent in more than 50% of the samples, we observed slight variations across different cell lines. In HEK293T cells, the majority of such cases are retained in HEK293T cells, where 217 genes account for approximately 65.2% of such cases. LNCaP cells exhibit the smallest number of splice forms - with only 22 genes, representing 58% of genes encoding transcripts with different expression levels. MCF-7 cells show an even lower relative prevalence at 52.3%. For splice forms prevailing in over 90% of the samples under study, specific genes are retained in HCT116, MDA-MB-231, THP-1 and HeLa cultures. Notably, we identified 15 genes in HCT116 cells, 14 in MDA-MB-231, two in THP-1 and one in HeLa (Supplementary Table S4). Across most cell lines, one splice form is expressed in the vast majority of samples (60-80%) for differentially expressed transcripts. Among these cases, only 20-40% are defined as canonical variants (Figure 3). A canonical variant is the most representative gene product form, typically characterized as the longest sequence available. It is widely expressed and encoded by conserved exons found in orthologous sequences. A notable exception is the A549 cell line, where all prevailing isoforms were canonical. Additionally, more than 50% of canonical splice forms were observed in LNCaP and HeLa cultures. It is worth mentioning that canonical variants tend to be present across different samples, rather than being specific to a distinct sample.
The initial assumption regarding genes featuring switching isoforms suggested a universal presence across different cell lines, with the diversity of splice forms serving as a distinguishing characteristic. However, our analysis revealed that out of a total gene of 1038, only 270 were shared among various cell lines (Supplementary Table S4), underscoring that 74% of genes possess uniqueness for each of the cell lines.
Functionally, the subset of 270 genes common to multiple cell lines is involved in fundamental processes, such as signal transduction (adj. p-value = 0.001), cell cycle (adj. p-value = 0.001) and post-translational protein modification (adj. p-value = 0.02), as well as oncogenesis (Supplementary Table S4). Among oncogenesis pathways, the pivotal role of apoptosis (adj. p-value = 0.01) in maintaining tissue health by orchestrating cellular self-destruction when damaged stands out. In contrast, the VEGFA-VEGFR2 (adj. p-value = 0.03) pathway emerges as a driver of angiogenesis essential for healing and development. Furthermore, cell cycle checkpoints (adj. p-value = 0.03) safeguard DNA integrity by regulating cell division to impede the replication of aberrant cells and thereby thwarting cancer progression. In particular, for the MDA-MB-231 (breast adenocarcinoma) and HeLa (cervical adenocarcinoma) cultures, the hallmark presence of the common oncogene TP53 is notable. Additionally, the RRM2 gene emerges as a common variable gene for across HeLa, HCT116 (colorectal carcinoma), K562 (kidney cancer) and HepG2 (hepatoblastoma) cell lines, with its canonical form P31350-1 prevailing in all four cases. Overexpression of this gene is associated with unfavorable prognosis in liver and colorectal cancers, actively contributing to tumor development and progression [11,12].
Our enrichment analysis targeted genes which exhibit isoform switching within individual cell lines. While key biological pathways remained elusive for HEK293T, LNCAP, HeLa and THP-1 cells, significant pathways were discerned in other instances where involved genes played essential roles in a variety of fundamental cell functions, including cell cycle regulation, apoptosis, RNA splicing, signal transduction, and metabolism. Interesting examples include the unique variable gene - CDKN2B - in HeLa cells, encoding function-independent tumor suppressor protein [13]. This gene is frequently altered in human cancers. In HeLa cells, the CDKN2B coding sequence remains intact, and there is minimal DNA methylation in the CpG islands of its promoter regions. This cell line exhibits a high degree of aneuploidy, with an average of about three copies of the CDKN2B locus [14]. Another example is the CTNND1 gene, which is differentially expressed in the MDA-MB-231 cell line. Previous research demonstrated that knocking down CTNND1 in this cell line reduced cell adhesion but enhanced migration and invasion, suggesting its potential significance as a predictor of bone metastasis in breast cancer [15].
These findings illuminate the tissue-specific nature of genes exhibiting internal isoform switching and underscore the differential expression of such genes as a possible mechanism to preserve the properties of progenitor cells.
2.4. Splicing Diversity across Cell Lines
An intriguing aspect of splice variants is their differential expression patterns in distinct cell types. This means that one splice form may predominate in one cell type, while another splice form prevails in a different cell context, despite originating from the same gene. Within our analysis of 10 diverse cell lines, we observed alterations in the prevailing splice form of 14 genes (Figure 4).
For instance, the SLK gene exhibited the highest occurrence in six cell lines. Notably, in A549, THP1, K562, HepG2, and MDA-MB-231 cells, the non-canonical splice variant Q9H2G2-2 prevailed, while in LNCAP, the canonical one - Q9H2G2-1 - was dominant. According to the Protein Atlas, SLK gene is highly expressed in the liver and is involved in cytoplasmic microtubule organization, protein autophosphorylation, and regulation of focal adhesion assembly. Similarly, the PFKFB3 gene responsible for cell cycle progression and prevention of apoptosis showcased a shift in prevailing splice forms, with Q16875-3 prevailing in four cell lines (THP1, HEK293T, HCT116, LNCAP), while the canonical form Q16875-1 was predominant in A549 culture.
2.5. Comparative Analysis of Splice Variant Prevalence among Cell Lines
In our analysis of the ten examined cell lines, we identified significant instances of one alternatively spliced transcript predominating over others in 2,829 genes, excluding the differentially prevalent splice forms discussed earlier. Across all cell lines, we observed 344 (12%) prevailing splice forms encoded by genes involved in fundamental biological processes crucial for cellular and organismal viability. These genes cover key aspects of metabolism ("Metabolism Of Proteins", adj. p-value < 0.001), cell cycle ("Cell Cycle", adj. p-value < 0.001), and cell division ("Mitotic Metaphase And Anaphase", "M Phase", adj. p-value < 0.001) (Supplementary Table S4).
Upon comparing expression profiles across cell lines, we observed a high correlation between prevailing isoforms (Supplementary Figure S5). This consistency underscores the potential functional importance of these isoforms in maintaining cellular health.
We identified 800 unique splice forms exclusively present in one cell line, twice the number of splice forms detected across all cell lines (Figure 5). The highest numbers of unique splice variants were found in THP-1 cells (144 genes) and LNCaP cells (138 genes), whereas the smallest number was found in HeLa cells—33 (Supplementary Table S4). These unique splice forms may perform culture-specific functions, as evidenced by enrichment analysis using ARCHS4 database [16]. For instance, the RAI14 gene in the A549 cell line emerges as a potential biomarker for lung adenocarcinoma, up-regulated and highly expressed in that cell line [17]. Similarly, EFHD1, actively expressed in breast cancer, represents a unique gene in the MCF7 cell line [18]. In the HepG2 cell line, the gene SERPINA1 was unique, which is robustly expressed in these cells and therefore serves as a good model for studying endogenous structure of the native 5′-UTR [19].
Within the range of two to nine cell lines, 1,685 splice variants were detected. Among these, A549 and HeLa lines share 662 prevalent splice variants, surpassing the 609 shared between A549 and MCF7. Although these three cell lines originate from different tissue types, their splicing profiles are similar. Pearson correlation coefficients of splice variant expression levels between the cell lines were also high: at 0.93 between A549 and HeLa, and between A549 and MCF7.
Among A549, HeLa, and MCF7 cell lines, 477 common prevalent splice forms are identified, the highest number among any trio of cell lines. Despite similarities in prevalent splice forms, noticeable differences in their expression exist between cells. Interestingly, genes common to all three lines play a key role in fundamental biological processes such as the Cell Cycle, Metabolism, and DNA Repair, with a minimal probability of random error (adj. p-value < 0.001). Among four cell lines, the highest number of prevalent splice forms was observed between A549, HeLa, MCF-7 and LNCaP - totaling 340. Although LNCaP is included in this group, it does not feature among the five cell lines with the highest number of splice forms. This set included A549, MCF7, HeLa, MDA-MB-253, and THP-1, which collectively exhibit 237 common variants. Similarly, the group of six cell lines (A549, THP-1, K562, MCF7, HeLa, and MDA-MB-231) demonstrates the maximum number of common splice variants among similar groups - totalling 173.
As the number of cell lines in various groups increases, not only does the number of common prevalent splice forms decrease, but the specificity of gene functions associated with oncogenesis also intensifies. Genes within these groups participate in key biological pathways such as transcriptional regulation by TP53 [20], clathrin-mediated endocytosis [21], and the RHOBTB3 ATPase cycle [22]. These pathways highlight their crucial roles in the development and progression of cancer, exemplified by RhoBTB3, which has been shown to promote cancer growth.
2.6. Selection of the Canonical Variant
Out of all 2,829 prevalent splice variants, 23% deviate from the canonical variant (https://www.uniprot.org/help/canonical_and_isoforms, accessed on 3 April 2024). Moreover, among the splice variants prevalent in all 10 cell lines, alternatively spliced variants constituted 16.2% (Supplementary Table S4). For the corresponding genes, we did not identify “exotic” biological pathways: they participate in a diverse spectrum of cellular processes including structural support, signaling pathways, regulation of gene expression, metabolism, and DNA repair. Of particular interest in the context of tumorigenesis are the genes SMAD2 and TRIM24. SMAD2 is pivotal in TGF-β signaling cascade, regulating critical processes such as cell proliferation, differentiation, and apoptosis. Several studies have demonstrated the oncogenic effect of this gene in response to TGF-β1 stimulation in cancer [23,24]. Similarly, TRIM24 interacts with numerous nuclear receptors and coactivators, modulating the transcription of target genes in various cancers [25]. TRIM24 also influences cell proliferation and apoptosis regulation, partly through modulating p53/TP53 protein levels [26].
In over half of the examined cell lines (ranging from five to nine), the prevalence of a specific splice form is documented for 842 genes, with 19% of these being alternative to the canonical variant. The proportion of prevalent splice forms diverging from the canonical sequence in each cell line ranges from 15 to 21%, with the highest observed in A549 (138 genes) and the lowest in HEK293T (86 genes). Functionally, genes featuring alternative prevalent sequences are associated with processes such as RNA binding and poly(A) binding (adj. p < 0.05) according to GeneOntology. In the HCT116 cell line, genes are involved in the cadherin binding pathway (adj. p-value = 0.008). Cadherins form catenin complexes with catenins, essential for cell-to-cell adherence, and any destabilization of these complexes can promote tumor progression. Furthermore, in the MDA-MB-231 cell line, unique pathways including ubiquitin protein ligase binding (adj. p-value = 0.02) and ubiquitin-like protein ligase binding (adj. p = 0.02) were also observed.
2.7. Relevance to Cancer Development
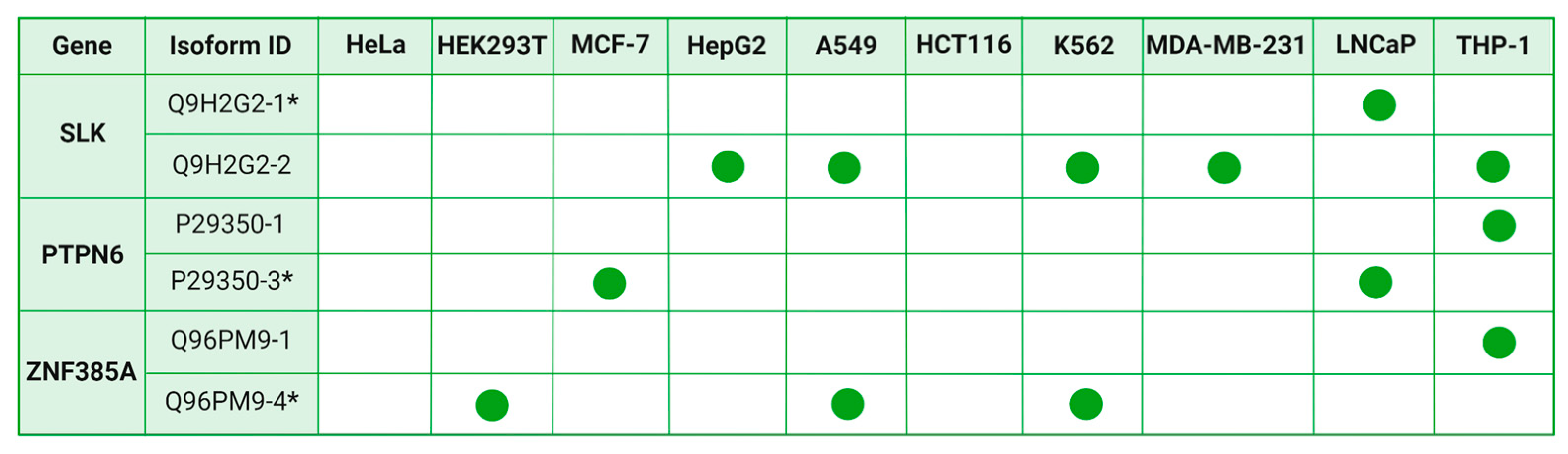
To elucidate the association between isoform and oncogenic processes, we cross-referenced our findings with results previously obtained in Dolgalev et al.’s study [5], which identified isoform switching events across 12 types of cancer compared to normal tissue. Firstly, we compared our dataset, which includes 14 genes with isoform switching between cell lines,with previously identified genes that exhibit isoform switching events when transitioning from normal to tumorous conditions. Consequently, we identified three genes where splice form changes between cancerous and normal tissue were statistically significant. For the ZNF385A gene (Figure 6), the canonical isoform Q96PM9-4 predominated in cancer tissues of patients with lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), while non-canonical variant Q96PM9-1 exhibited opposite trend. In A549, K562, HEK293T, and HCT116 cells, the canonical form was prevalent, whereas in THP1 predominantly expressed the non-canonical form. For the gene PTPN6, previous studies indicated lower expression of the non-canonical variant P29350-1 in LUSC cells compared to normal cells. In our study, we observed the prevalence of the canonical variant P29350-3 in the MCF7 and LNCaP cell lines. Conversely, the non-canonical variant was more prevalent in the THP1 cell line. Furthermore, the non-canonical variant Q9H2G2-2 of the SLK gene, as reported in the study by Dolgalev et al., was expressed at low levels in four types of cancer cells—breast cancer (BRCA), prostate adenocarcinoma (RDAD), LUAD and LUSC — compared to normal tissue. Consistent with these findings, our study revealed predominance of this non-canonical variant in five cell lines — A549, THP1, K562, HepG2, and MDA-MB-231 - while the canonical variant Q9H2G2-1 was prevalent in the LNCaP cell line.
The subsequent step involved comparing the comprehensive list of prevalent isoforms (2,829 genes) with findings from a prior publication [5]. This comparison led to identifying 140 shared splice variants associated with cancer. Among these, seven splice forms occur in all cell lines, 58 are detected in two to nine lines, and 37 are unique to just one cell culture. The list of variants observed in two to nine cell lines includes the canonical variant Q9H5V8-1 of the CDCP1 gene, which is involved in cancer survival, growth, metastasis, and therapy resistance [27]. This canonical variant is prevalent in half of the cell lines, including A549, K562, HCT116, LNCAP, and MDA-MB-231. Another canonical variant that appears in two to nine cell lines is P30281-1, encoded by the CCND3 gene. This gene is crucial for regulating the transition from the G1 to S phase in the cell cycle and is associated with poor prognosis in breast and bladder cancers [28,29]. The only cell line in which this gene's splice form is not prevalent is HepG2.
3. Discussion
The results of this work unveil a fascinating aspect of molecular biology-unique profiles of the prevalence of splice forms encoded by one gene are observed depending on the type of cells. This intricate phenomenon, even in a small number of cell lines, reveals that the proportion of prevailing splice forms common to all biological samples is surprisingly low - only 12%. These splice forms, if this trend continues in a more significant number of cells, could be considered canonical variants. In fact, 56 out of 344 of these splice forms are represented by the non-canonical sequence. The term "canonical sequence" is crucial within the framework of maintaining the "one gene - one protein" approach in the analysis and interpretation of data. In terms of functional annotation, all existing systems are based on describing the role of a protein/gene in biological processes in a single entity, summarizing the functions implemented by all proteoforms encoded by the same gene. Nevertheless, due to the fact that one of the first points in the definition of a canonical variant is its most frequent occurrence, it is likely that available information about a function will mostly relate to this particular variant. In any case, understanding the prevailing splice form within the object of study in order to describe its function is crucially important for deciphering the role of genes in cellular processes. For the ten cell lines, we defined 654 prevailing splice forms that do not match the canonical sequence.
It is necessary to make a final decision on the change in the canonical form after a thorough analysis of not only all the cell lines but also human tissue samples. Given the limited number of cell lines and relevant samples available, our results should be considered preliminary. Gene expression, as a set of transcript expressions, is a more constant value within samples of a single cell line than the expression of each transcript [30]. At the same time, for normalizing the use of housekeeping genes, it has been proposed that the focus is on stably expressed transcripts [4]. To minimize stochasticity in the results, a minimum of 10 samples were chosen, with a cutoff value of 5%, to ensure that the picture does not significantly change regarding the number of prevailing splice forms. The second parameter was chosen to be the 50% prevalence of samples [31].
By increasing the number of cell lines for analysis, we open up a world of possibilities. This expansion will not only allow us to clarify the canonical variant but also the genes for which their splice forms are likely to have different functions. The identified expression profiles of splice forms can already be the basis for a more targeted study of their functions. This progress is particularly useful for proteomics studies, such as AP-MS or knockout experiments. As part of the study of protein function, more and more attention is paid to the complete composition of the molecular profile of the sample. Considering the environment allows us to minimize false positive results, as is the case with different localizations of proteins in Y2H experiments. This will probably make it possible, in the long term, to better identify low- and high-molecular partners for specific proteoforms. This is a direct path towards a more accurate search for drug targets and design of drugs.
Within this framework, terminology also arises: how to correctly reflect a change in the prevailing splice form. The accepted concept of isoform switching refers to a significant difference in the expression of splice forms in normal and oncology tissue, with a direct transition to the dominant variant. Isoform switching is a special case of differential expression of several transcripts encoded by the same gene. Nevertheless, in the example of stable and one-dimensional objects - cell lines, a change in the prevailing transcript is observed not only between different types of cells (14 genes), but also within cell lines (270 genes). These genes involved in key processes including signal transduction, cell cycle, and protein modification, all pivotal for oncogenesis. For example, specific to MDA-MB-231 and HeLa cell lines, the common oncogene TP53 is prominent. Additionally, the RRM2 gene, prevalent in HeLa, HCT116, K562, and HepG2 cell lines, is linked to poor outcomes in liver and colorectal cancers, significantly contributing to tumor progression.
Genes that are the prevailing transcript between different types of cells (14 genes) are associated with tumor processes. For example, for the SLK gene encoding the Ste20-like protein kinase that mediates apoptosis, we observe its prevailing splice form Q9H2G2-2 in 4 cell lines and the prevalence of Q9H2G2-1 in one cell line. Non-canonical variant Q9H2G2-2, according to the results of analysis of data from the TCGA project, was low expressed in four types of tumors: breast cancer, prostate adenocarcinoma, lung adenocarcinoma, and lung squamous cell carcinoma compared to normal tissue [5]. In cancer cells, Q9H2G2-1 predominates, while in normal cells, this was observed for Q9H2G2-2.
It should be noted that the difference in the amino acid sequences of these splice forms is only 30 amino acids. However, this part of the sequence is difficult to detect for current proteomic technologies. Thus, this illustration shows the likely erroneous choice of the canonical sequence variant and the cell lines that should be used to annotate the function of each proteoform.
4. Materials and Methods
4.1. Data
The search for raw public RNAseq data was performed in the NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra, accessed on January 22, 2024). We applied criteria established in a previous study [30]: total first/second fragment count > 20,000,000; average read length > 60 base pairs; untreated cells; Illumina sequencing technology. Accession numbers for the experiments are provided in Supplementary Table S1.
4.2. Data Analysis
Quality control was performed using FastQC [32]. Gene expression and isoform analysis were performed using Salmon (mapping-based mode, version 1.6.0) [33]. The batch effect was corrected with pyComBat [10].
For genomic annotation, we used the GTF file downloaded from the Ensembl resource (GRCh38, release 111). The Uniprot database was acquired using BioMart [34] and focused our analysis on manually annotated proteins from the Swiss-Prot database. This led to a total of 24,049 isoforms corresponding to 10,369 proteins, along with 32,832 protein-coding transcripts and 10,371 genes.
Computations and visualization were carried out in the Python software environment (version 3, available at http://www.python.org, accessed on January 22, 2024).
5. Conclusions
The interaction of proteins underlies biological processes. Thus, in the discussion of protein function or gene ontology, the analysis of splice-mediated transcripts must be carried out through the lens of their translation. This study is focused on assessing the prevalence of splice forms, currently annotated as translatable, according to the UniProt resource.
Considering the 10 most popular cell lines in transcriptomic studies as objects, it was shown that based on the prevalence of splice forms for a number of genes, there are justified grounds for canonical sequence alteration. First and foremost, 56 splice forms, alternative to the current canonical sequence, should be considered.
Distinct profiles of splice form prevalence are the primary candidates for exploring specific roles of each in biological processes. The association of several splice forms with oncodenic processes, including switching, provides support for the development of biomarkers for pathological changes in cell metabolism.
Thus, our results confirm and extend previous findings regarding the association of isoform expression with oncological processes. We identified three common genes with statistically significant changes between cancerous and normal tissues. Furthermore, comparing prevailing isoforms across different cell lines with previous studies revealed 140 common splice variants associated with cancer, underscoring their potential value for prognostication and understanding oncogenesis. During the transition from cells in cell lines to organs and tissues, it is expected that the frequency of internal switching events will increase.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.V.P.; methodology, E.V.P. and V.A.A.; formal analysis, V.A.A.; writing—original draft preparation, E.V.P., and V.A.A.; writing—review and editing, E.V.P. and O.I.K.; visualization, V.A.A.; supervision, E.V.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by RSF No.21-74-10061.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ye, J.; Blelloch, R. Regulation of Pluripotency by RNA Binding Proteins. Cell Stem Cell 2014, 15, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sebestyén, E.; Zawisza, M.; Eyras, E. Detection of Recurrent Alternative Splicing Switches in Tumor Samples Reveals Novel Signatures of Cancer. Nucleic Acids Res. 2015, 43, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Huber, W. Alternative Start and Termination Sites of Transcription Drive Most Transcript Isoform Differences across Human Tissues. Nucleic Acids Res. 2018, 46, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Hounkpe, B.W.; Chenou, F.; de Lima, F.; De Paula, E.V. HRT Atlas v1.0 Database: Redefining Human and Mouse Housekeeping Genes and Candidate Reference Transcripts by Mining Massive RNA-Seq Datasets. Nucleic Acids Res. 2021, 49, D947–D955. [Google Scholar] [CrossRef] [PubMed]
- Dolgalev, G.; Poverennaya, E. Quantitative Analysis of Isoform Switching in Cancer. Int. J. Mol. Sci. 2023, 24, 10065. [Google Scholar] [CrossRef] [PubMed]
- Vitting-Seerup, K.; Sandelin, A. The Landscape of Isoform Switches in Human Cancers. Mol. Cancer Res. MCR 2017, 15, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Passacantilli, I.; Sette, C. Alternative Splicing and Cell Survival: From Tissue Homeostasis to Disease. Cell Death Differ. 2016, 23, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Arzumanian, V.A.; Kiseleva, O.I.; Poverennaya, E.V. The Curious Case of the HepG2 Cell Line: 40 Years of Expertise. Int. J. Mol. Sci. 2021, 22, 13135. [Google Scholar] [CrossRef]
- Bao, J.; Vitting-Seerup, K.; Waage, J.; Tang, C.; Ge, Y.; Porse, B.T.; Yan, W. UPF2-Dependent Nonsense-Mediated mRNA Decay Pathway Is Essential for Spermatogenesis by Selectively Eliminating Longer 3’UTR Transcripts. PLoS Genet. 2016, 12, e1005863. [Google Scholar] [CrossRef]
- Behdenna, A.; Colange, M.; Haziza, J.; Gema, A.; Appé, G.; Azencott, C.-A.; Nordor, A. pyComBat, a Python Tool for Batch Effects Correction in High-Throughput Molecular Data Using Empirical Bayes Methods. BMC Bioinform. 2023, 24, 459. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, J.; Guo, S.; Xue, X.; Wang, Y.; Qiu, S.; Cui, J.; Ma, L.; Zhang, X.; Wang, J. RRM2 Protects against Ferroptosis and Is a Tumor Biomarker for Liver Cancer. Cancer Cell Int. 2020, 20, 587. [Google Scholar] [CrossRef]
- Liu, Q.; Guo, L.; Qi, H.; Lou, M.; Wang, R.; Hai, B.; Xu, K.; Zhu, L.; Ding, Y.; Li, C.; et al. A MYBL2 Complex for RRM2 Transactivation and the Synthetic Effect of MYBL2 Knockdown with WEE1 Inhibition against Colorectal Cancer. Cell Death Dis. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Xu, L.; Bennett, L.; Hooks, J.C.; Liu, J.; Zhou, Q.; Liem, P.; Zheng, Y.; Skapek, S.X. Identification of De Novo Enhancers Activated by TGFβ to Drive Expression of CDKN2A and B in HeLa Cells. Mol. Cancer Res. MCR 2019, 17, 1854–1866. [Google Scholar] [CrossRef]
- Landry, J.J.M.; Pyl, P.T.; Rausch, T.; Zichner, T.; Tekkedil, M.M.; Stütz, A.M.; Jauch, A.; Aiyar, R.S.; Pau, G.; Delhomme, N.; et al. The Genomic and Transcriptomic Landscape of a HeLa Cell Line. G3 Bethesda Md 2013, 3, 1213–1224. [Google Scholar] [CrossRef]
- Gong, C.; Lin, Q.; Fang, X.; Jiang, W.; Li, J.; Li, J.; Shi, Y.; Luo, Q.; Liu, Z.; Cen, Y. CTNND1 to Mediate Bone Metastasis of Triple-Negative Breast Cancer via Regulating CXCR4. J. Clin. Oncol. 2021, 39, e13045–e13045. [Google Scholar] [CrossRef]
- Lachmann, A.; Torre, D.; Keenan, A.B.; Jagodnik, K.M.; Lee, H.J.; Wang, L.; Silverstein, M.C.; Ma’ayan, A. Massive Mining of Publicly Available RNA-Seq Data from Human and Mouse. Nat. Commun. 2018, 9, 1366. [Google Scholar] [CrossRef]
- Yuan, C.; Hu, H.; Kuang, M.; Chen, Z.; Tao, X.; Fang, S.; Sun, Y.; Zhang, Y.; Chen, H. Super Enhancer Associated RAI14 Is a New Potential Biomarker in Lung Adenocarcinoma. Oncotarget 2017, 8, 105251–105261. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Stavnes, H.T.; Risberg, B.; Nesland, J.M.; Wohlschlaeger, J.; Yang, Y.; Shih, I.-M.; Wang, T.-L. Gene Expression Signatures Differentiate Adenocarcinoma of Lung and Breast Origin in Effusions. Hum. Pathol. 2012, 43, 684–694. [Google Scholar] [CrossRef]
- Grayeski, P.J.; Weidmann, C.A.; Kumar, J.; Lackey, L.; Mustoe, A.M.; Busan, S.; Laederach, A.; Weeks, K.M. Global 5’-UTR RNA Structure Regulates Translation of a SERPINA1 mRNA. Nucleic Acids Res. 2022, 50, 9689–9704. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Khan, I.; Steeg, P.S. Endocytosis: A Pivotal Pathway for Regulating Metastasis. Br. J. Cancer 2021, 124, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, Y.-J. RhoBTB3 Regulates Proliferation and Invasion of Breast Cancer Cells via Col1a1. Mol. Cells 2022, 45, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Bertrand-Chapel, A.; Caligaris, C.; Fenouil, T.; Savary, C.; Aires, S.; Martel, S.; Huchedé, P.; Chassot, C.; Chauvet, V.; Cardot-Ruffino, V.; et al. SMAD2/3 Mediate Oncogenic Effects of TGF-β in the Absence of SMAD4. Commun. Biol. 2022, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Girolami, I.; Veronese, N.; Smith, L.; Caruso, M.G.; Reddavide, R.; Leandro, G.; Demurtas, J.; Nottegar, A. The Activation Status of the TGF-β Transducer Smad2 Is Associated with a Reduced Survival in Gastrointestinal Cancers: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 3831. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Li, Y.; Zhang, W.; Alvarez, A.A.; Song, L.; Tang, J.; Gao, W.-Q.; Hu, B.; Cheng, S.-Y.; Feng, H. TRIM24 Is an Oncogenic Transcriptional Co-Activator of STAT3 in Glioblastoma. Nat. Commun. 2017, 8, 1454. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Allton, K.; Duncan, A.D.; Barton, M.C. TRIM24 Is a P53-Induced E3-Ubiquitin Ligase That Undergoes ATM-Mediated Phosphorylation and Autodegradation during DNA Damage. Mol. Cell. Biol. 2014, 34, 2695–2709. [Google Scholar] [CrossRef]
- Khan, T.; Kryza, T.; Lyons, N.J.; He, Y.; Hooper, J.D. The CDCP1 Signaling Hub: A Target for Cancer Detection and Therapeutic Intervention. Cancer Res. 2021, 81, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-Y.; Li, R.; Zhang, Q.-J.; Wang, Y.; Jiang, Y.; Meng, Q.-Y.; Xi, Q.-L.; Wu, G.-H. Prognostic Role of Cyclin D2/D3 in Multiple Human Malignant Neoplasms: A Systematic Review and Meta-Analysis. Cancer Med. 2019, 8, 2717–2729. [Google Scholar] [CrossRef] [PubMed]
- Blanca, A.; Requena, M.J.; Alvarez, J.; Cheng, L.; Montironi, R.; Raspollini, M.R.; Reymundo, C.; Lopez-Beltran, A. FGFR3 and Cyclin D3 as Urine Biomarkers of Bladder Cancer Recurrence. Biomark. Med. 2016, 10, 243–253. [Google Scholar] [CrossRef]
- Arzumanian, V.; Pyatnitskiy, M.; Poverennaya, E. Comparative Transcriptomic Analysis of Three Common Liver Cell Lines. Int. J. Mol. Sci. 2023, 24, 8791. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Krasnov, G.S.; Zaretsky, A.R.; Zhavoronkov, A.; Nyushko, K.M.; Moskalev, A.A.; Karpova, I.Y.; Afremova, A.I.; Lipatova, A.V.; Kochetkov, D.V.; et al. Differential Expression of Alternatively Spliced Transcripts Related to Energy Metabolism in Colorectal Cancer. BMC Genomics 2016, 17, 1011. [Google Scholar] [CrossRef]
- Trivedi, U.H.; Cezard, T.; Bridgett, S.; Montazam, A.; Nichols, J.; Blaxter, M.; Gharbi, K. Quality Control of Next-Generation Sequencing Data without a Reference. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart--Biological Queries Made Easy. BMC Genomics 2009, 10, 22. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Number of datasets containing transcriptomic experiment results for specific cell lines (as of February 14, 2024). The histogram is built using the query {cell_line}[All Fields] AND control[All Fields]) AND (biomol rna[Properties] AND platform illumina[Properties]).
Figure 1.
Number of datasets containing transcriptomic experiment results for specific cell lines (as of February 14, 2024). The histogram is built using the query {cell_line}[All Fields] AND control[All Fields]) AND (biomol rna[Properties] AND platform illumina[Properties]).
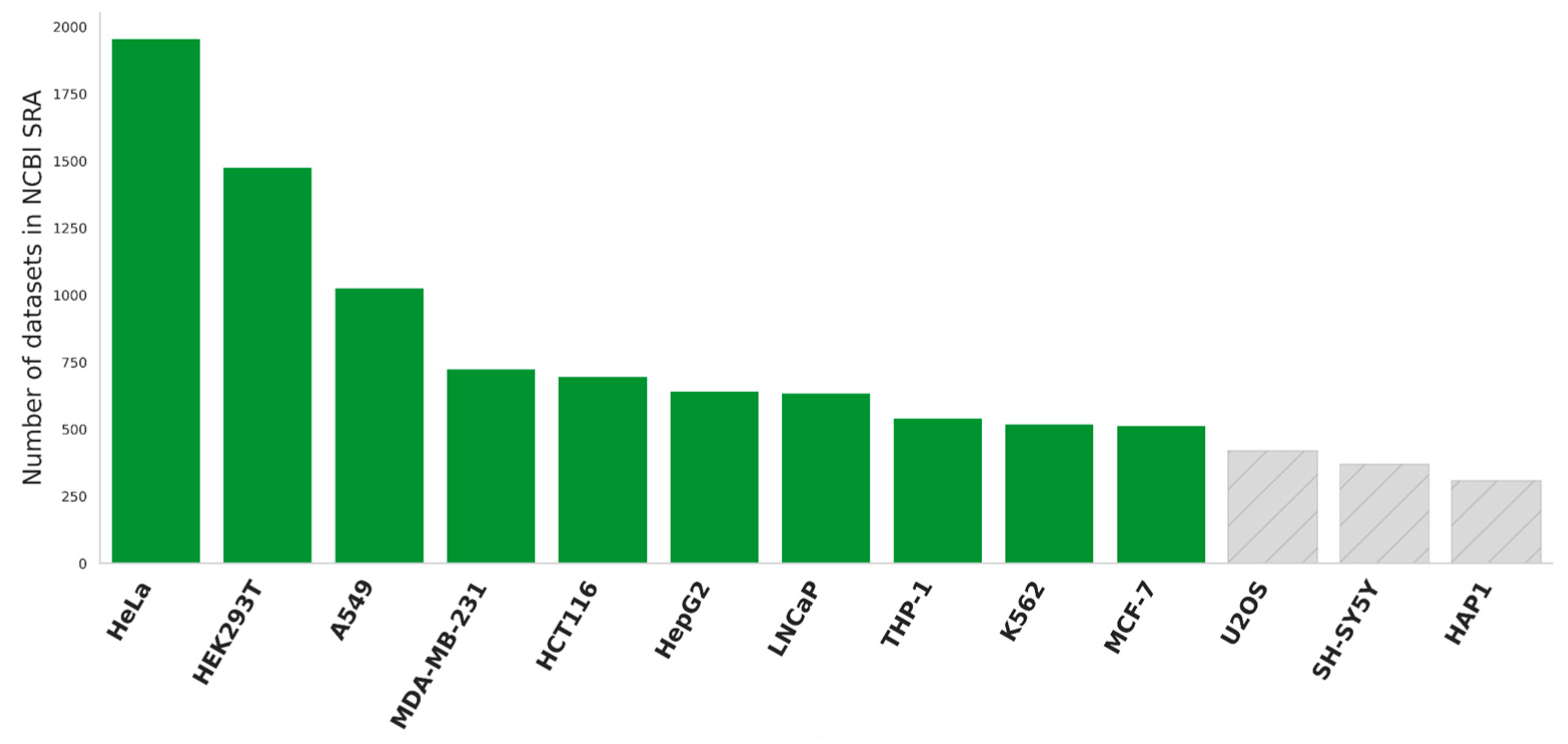
Figure 2.
The number of cases of prevalent splice forms for 10 cell lines. Green bars indicate the number of prevalent splice variants that occur in more than half of the samples within the same cell culture. Yellow bars indicate the number of prevalent splice forms that occur in less than half of the samples.
Figure 2.
The number of cases of prevalent splice forms for 10 cell lines. Green bars indicate the number of prevalent splice variants that occur in more than half of the samples within the same cell culture. Yellow bars indicate the number of prevalent splice forms that occur in less than half of the samples.
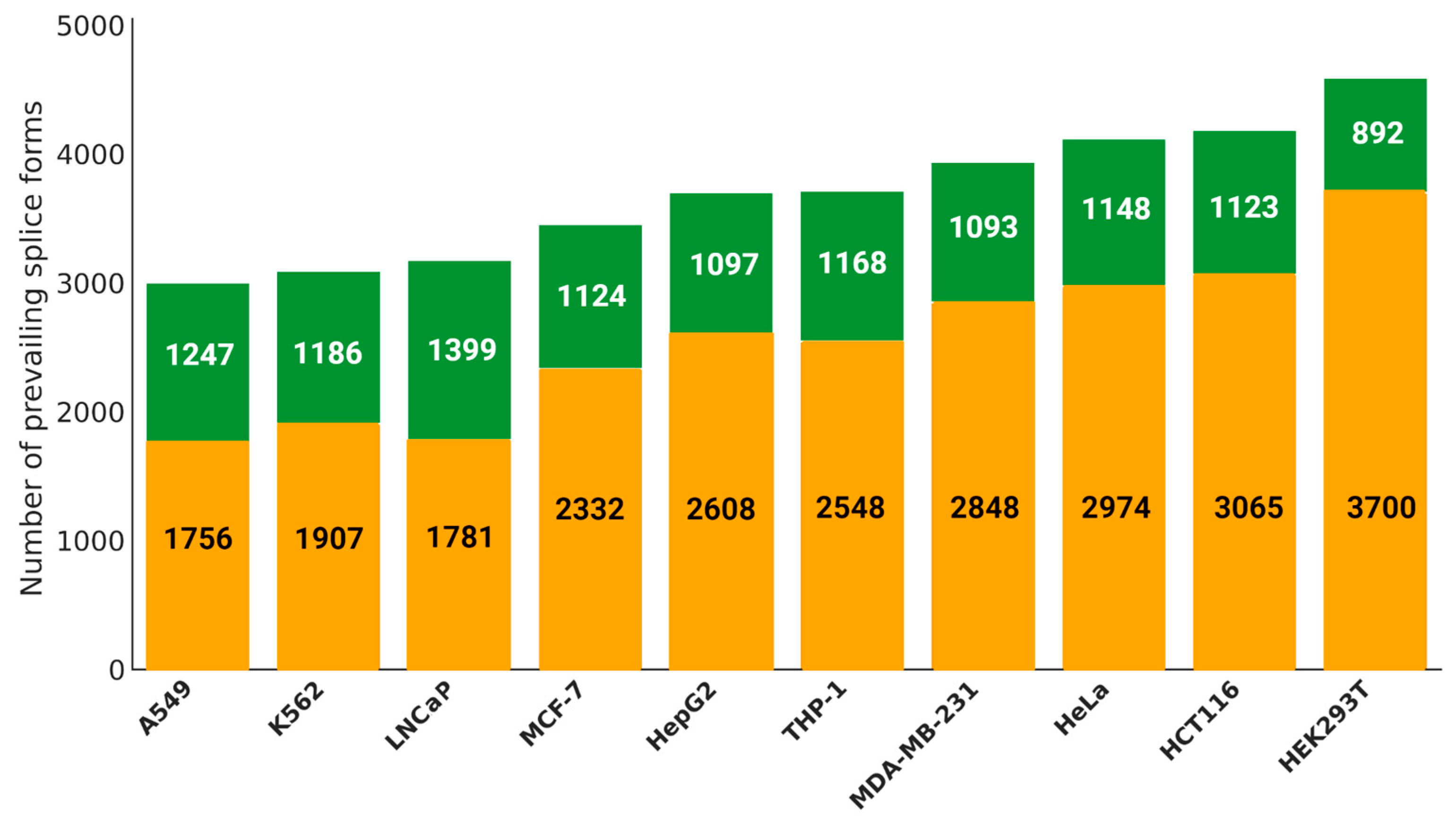
Figure 3.
Distribution of canonical (green bars) and non-canonical (yellow bars) splice forms that are expressed in more than 80% of samples. A canonical variant (marked with asterisk) is the most representative gene product, typically characterized as the longest sequence available. It is widely expressed and encoded by conserved exons found in orthologous sequences.
Figure 3.
Distribution of canonical (green bars) and non-canonical (yellow bars) splice forms that are expressed in more than 80% of samples. A canonical variant (marked with asterisk) is the most representative gene product, typically characterized as the longest sequence available. It is widely expressed and encoded by conserved exons found in orthologous sequences.
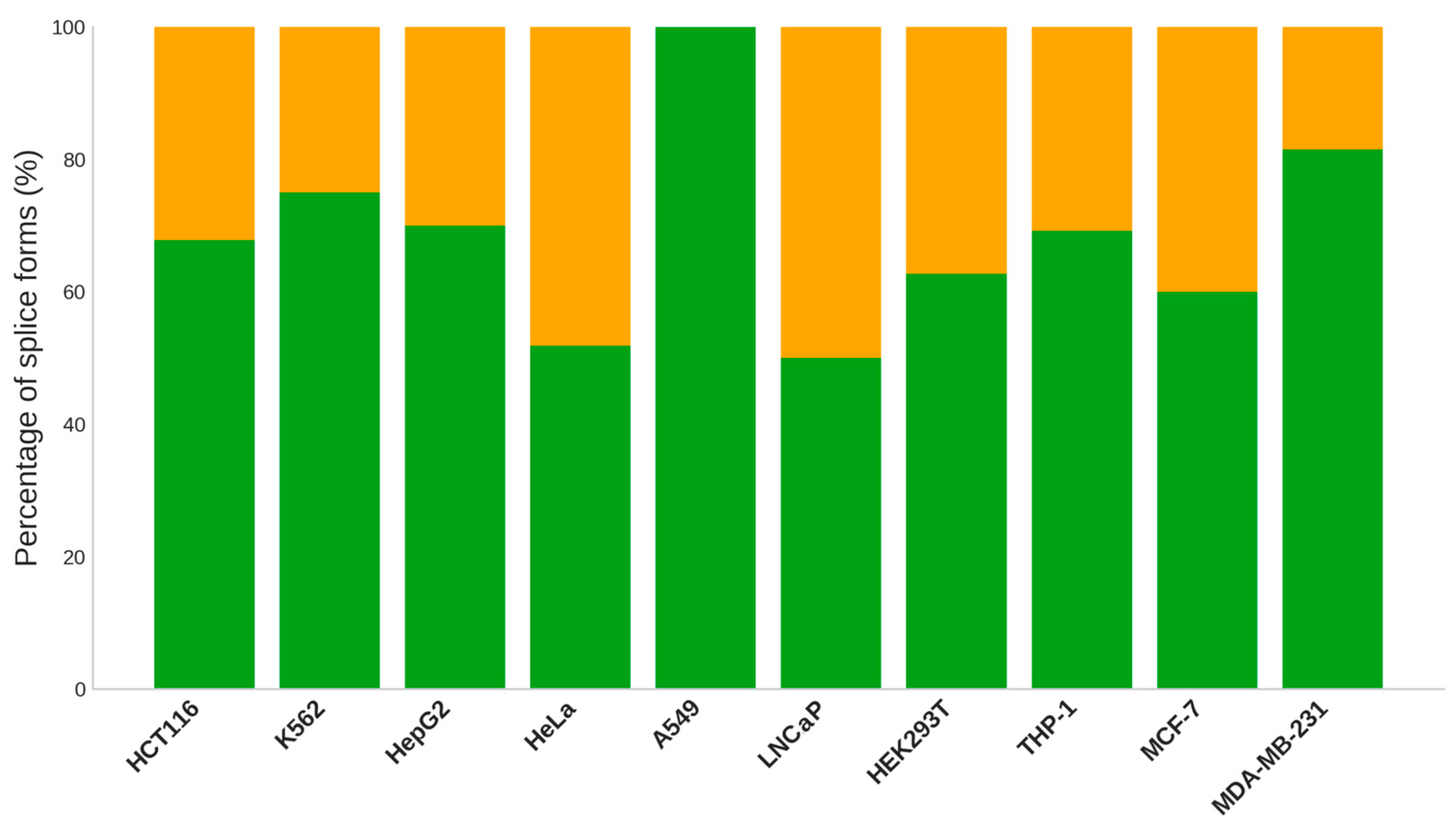
Figure 4.
Occurrence of switching splice variants among ten cell lines. Y axis reflects the number of cell lines where this isoform variant is expressed. Shaded bars indicate canonical splice forms.
Figure 4.
Occurrence of switching splice variants among ten cell lines. Y axis reflects the number of cell lines where this isoform variant is expressed. Shaded bars indicate canonical splice forms.
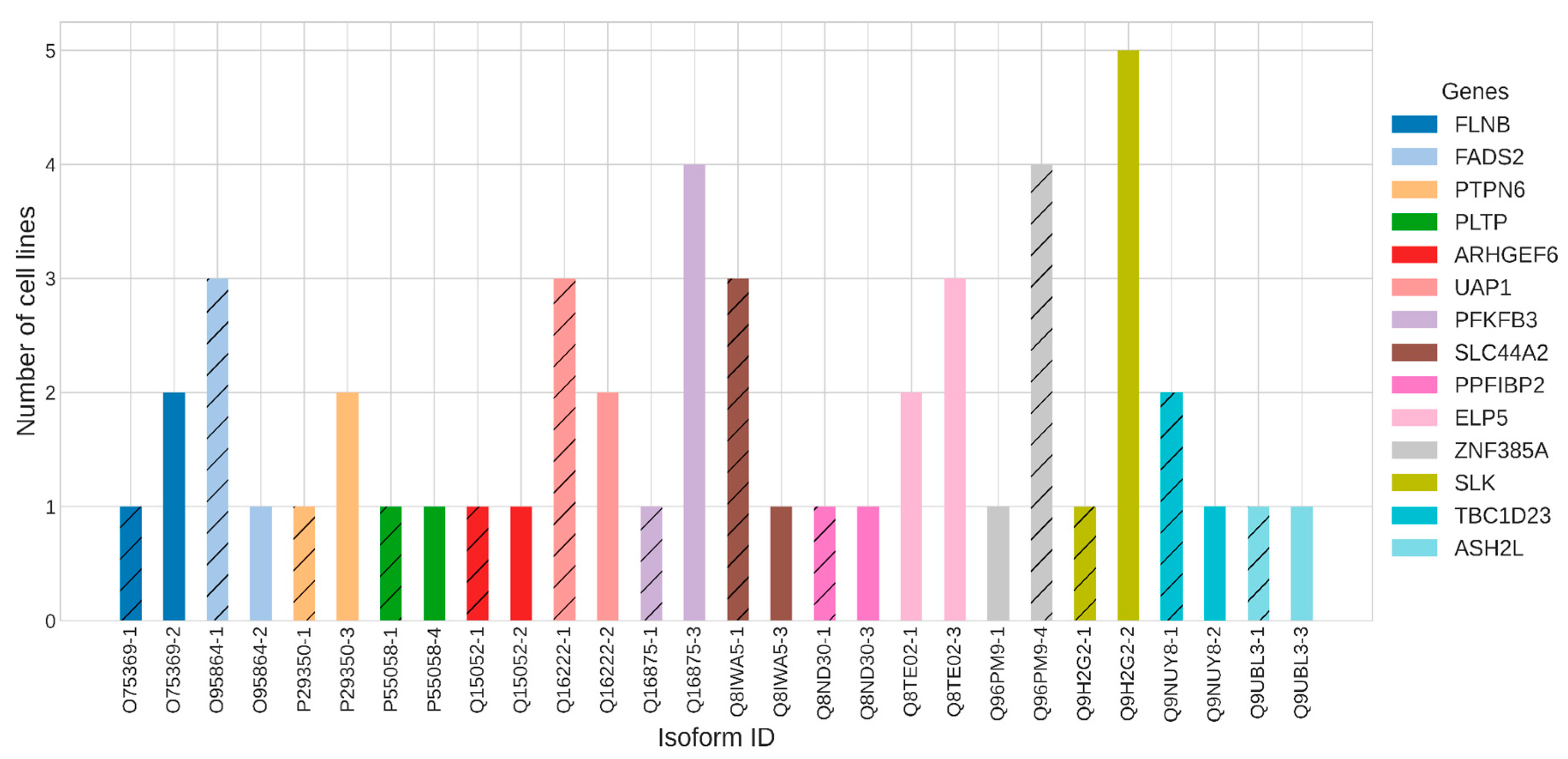
Figure 5.
Occurrence of prevalent splice forms in cell lines under study.
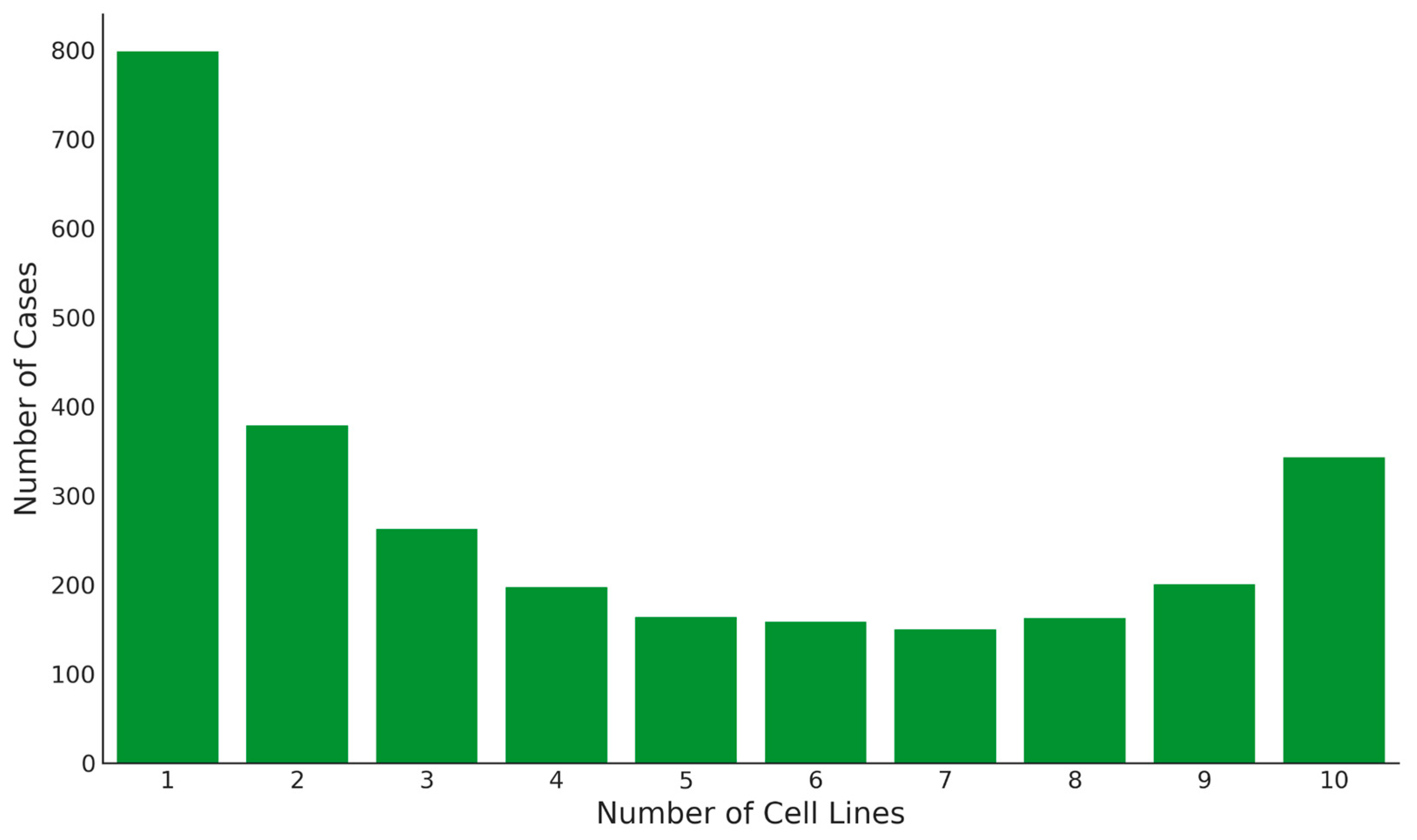
Figure 6.
Matrix of occurrence of cancer-associated splice variants in cell lines. Canonical splice forms are marked with an asterisk.
Figure 6.
Matrix of occurrence of cancer-associated splice variants in cell lines. Canonical splice forms are marked with an asterisk.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



