
Submitted:
13 May 2024
Posted:
14 May 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Papain-like protease PLpro, a domain within a large polyfunctional protein nsp3, plays key roles in the life cycle of SARS-CoV-2, being responsible for the first events of cleavage of a polyprotein into individual proteins (nsp1-4) as well as for the suppression of cellular immunity. Here, we developed a new genetically encoded fluorescent sensor, named PLpro-ERNuc, for detection of PLpro activity in living cells using translocation-based readout. The sensor was designed as follows. A fragment of nsp3 protein was used to direct the sensor on the cytoplasmic surface of endoplasmic reticulum (ER) membrane, thus closely mimicking the natural target of PLpro. The fluorescent part included two bright fluorescent proteins - red mScarlet I and green mNeonGreen, separated by a linker with the PLpro cleavage site. Nuclear localization signal (NLS) was attached to ensure accumulation of mNeonGreen into the nucleus upon cleavage. We tested PLpro-ERNuc in the model of recombinant PLpro expressed in HeLa cells. The sensor demonstrated expected cytoplasmic reticular network in the red and green channels in the absence of protease, and efficient translocation of the green signal into nuclei in the PLpro-expressing cells (14-fold increase of the nucleus/cytoplasm ratio). Then, we used PLpro-ERNuc in the model of Huh7.5 cells infected with SARS-CoV-2 virus, where it showed robust ER-to-nucleus translocation of the green signal in the infected cells 24 h post infection. We believe that PLpro-ERNuc represents a useful tool for screening PLpro inhibitors as well as for monitoring virus spread in a culture.
Keywords:
COVID-19
; SARS-CoV-2
; coronavirus
; protease PLpro
; live cell imaging
; genetically encoded probes
; green fluorescent protein
1. Introduction
COVID-19 (Coronavirus disease 2019) is an acute respiratory infection caused by the SARS-CoV-2 virus, which caused a pandemic in 2020 and led to enormous socio-economic consequences for humanity. Currently, the number of deaths since the beginning of the pandemic across the entire human population is more than 7 million people [1].
SARS-CoV-2 is an enveloped single-stranded (+)RNA Betacoronavirus. To enter a host cell, SARS-CoV-2 virion binds to the ACE2 receptor on the cell surface. After a 30 kb long RNA genome is released into the cell cytoplasm, the translation of a replicase gene begins. The replicase gene encodes two main open reading frames - ORF1a и ORF1b, which give rise to 16 non-structural proteins (nsp1–nsp16), necessary for viral RNA synthesis [2]. Cap-dependent translation results in two polyproteins - pp1a or pp1ab, which are further cleaved into nsp proteins by SARS-CoV-2 proteases. Coronavirus has two proteases – a 33-kDa chymotrypsin-like protease Mpro (the main protease) encoded by nsp5, and a 36-kDa papain-like protease (PLpro), which is the part of multi-domain membrane-bound protein nsp3 [3]. PLpro cleaves the viral polyprotein precursors, pp1a and pp1ab, at three sites which results in liberation of the first non-structural proteins (nsp1–nsp4) essential for viral replication process [4], while Mpro cuts polyproteins at 11 sites generating the remaining 12 nsps [3]. In addition to proteolytic activity, PLpro exhibits deubiquitinating and deISG15ylating (interferon-induced gene 15) activities that are responsible for host immune system suppression, facilitating viral proliferation and replication [5]. All of the above functions of PLpro, including the fact that it represents the earliest viral enzymatic activity which triggers all subsequent events, make PLpro an excellent target for anti-SARS-CoV-2 therapeutics.
The 250 amino acid PLpro domain is a part of nsp3, the largest protein (1945 amino acid residues) encoded by the SARS-CoV-2 genome (Figure 1). Nsp3 acts as a membrane-anchored protein with four transmembrane segments that pass through the endoplasmic reticulum (ER) membrane, while most of the protein is exposed to the cell cytosol [3].
The importance of proteases has led to extensive efforts in the development of screening systems to study their enzymatic activity, as well as to test protease inhibitors as drug candidates. To study the activity of cellular proteases genetically encoded fluorescent sensors are widely used [6]. The ability to use fluorescence microscopy in experiments with living cells makes it possible to study dynamic processes in real time. Due to high pathogenicity of SARS-CoV-2, experiments with the live virus should be conducted at biosafety level 3 (BSL-3), which seriously limits the possibility of screening in the laboratory environment. In this case, screening systems based on genetically encoded fluorescent sensors in mammalian cells are highly effective, safe and almost the only tool to perform this task. Such screening systems have two main advantages. Firstly, live cell-based experiments allow to exclude compounds that have a cytotoxic effect, as well as compounds that are impermeable to the cell membrane. Secondly, the cleavage of the substrate in the cytoplasm of the cell is physiologically similar to the process of cleavage of the viral polyprotein by protease in a virus-infected cell [7].
Recently, FlipGFP-based screening systems have been developed to test inhibitors of coronavirus proteases - PLpro [7] and Mpro [8]. Reporter system is based on the FlipGFP protein, which generates fluorescent signal only after beta-strand reorientation [9]. The linker between the crucial for reorientation 10th and 11th beta-strands of the FlipGFP contained PLpro or Mpro cleavage site. In the absence of active protease, no visible fluorescence was observed during sensor expression in mammalian cells. Protease expression led to β10-11 reorientation with formation of a functional GFP β-barrel resulting in a noticeable green fluorescent signal in the system. When inhibitors were added, a dose-dependent decrease in fluorescence was observed. This system allows the screening of antiviral chemicals at biosafety level 2 (BSL-2) [8,9].
FRET sensors are also used as a tool for monitoring protease activity in real time. The assay is typically based on a FRET-pair of fluorescent proteins separated by a protease cleavage site, the proteolysis of which results in the disappearance of FRET. A FRET-pair of cyan and yellow fluorescent proteins (CFP-YFP) separated by the Mpro recognition site was used to develop a reporter for the main protease activity [10]. When the FRET-sensor was expressed in mammalian cells, a yellow fluorescent YFP signal generated as a result of energy transfer from the excited CFP donor to the fused YFP acceptor was detected in the system. Separation of the two fluorescent proteins in case of linker proteolysis by Mpro led to FRET signal disruption. Addition of a Mpro inhibitor (GC376), blocked the cleavage of CFP-YFP linker and thus maintained the FRET signal in the system [10]. Thus, the following FRET-based assay can be considered as a platform for in vitro compound testing and primary screening of potential Mpro inhibitors.
Intracellular translocation-based sensors provide another convenient way to detect protease activity in live cells. Many of them are based on a fluorescent protein with NLS that is retained in cytoplasm but translocates into the nucleus upon cleavage by viral protease. The cleavage sites correspond to polypeptide sequences in a polyprotein of the respective virus or to proteins that are targeted by viral protease. One of the pioneer studies in this field developed a sensor based on an antiviral protein MAVS (also known as IPS-1, Cardif or VISA) that is localized on the outer mitochondrial membrane and interacts with cytosolic RNA helicase RIG-I, which senses double-stranded viral RNA [11]. As naive MAVS is targeted by the NS3 protease of hepatitis C virus, upon HCV infection fused EGFP-MAVS sensor showed redistribution of green fluorescence from mitochondrial networks to diffuse cytoplasm staining, while RFP-NLS-MAVS allowed monitoring of translocation of RFP to nuclei shortly within 10-16 h post-infection. Recently, a similar sensor was developed for detection of enterovirus 71 infection whose 2A protease also cleaves MAVS [12]. This sensor could potentially be used for a plethora of viruses, as MAVS was shown to be digested by proteases of cytomegalovirus [13], MERS [14], hepatitis A virus [14] and other infections [15]. Noteworthy, similar sensors with other proteins of antiviral response of a host cell targeted by infections were also described [15].
Notably, SARS-CoV-2 PLpro domain as well as its viral protein targets are situated near the cytoplasmic surface of ER (Figure 1). Such a compartmentalization can be functionally important bringing all the players into the same space with 2D-like characteristics of diffusion of the membrane-anchored proteins. We recently developed a translocation-based sensor of PLpro activity, which was attached to the ER membrane by the tail-anchor (TA) short sequence [16]. Here, we designed an improved version of this sensor that closely recapitulates the native substrate of PLpro and ensures a high-contrast detection of PLpro-triggered ER-to-nucleus translocation of the fluorescent signal in live cells.
2. Results
2.1. Sensor Design
Our previously developed translocation-based sensor for PLpro activity [16] consisted of a fusion of the green mNeonGreen and red mScarlet I fluorescent proteins, which are characterized by fast maturation and high brightness in mammalian cells [13,14]. PLpro recognition site was inserted between mNeonGreen and mScarlet I, whereas the whole sensor was attached to the cytoplasmic surface of ER by a short membrane localization signal (so called “tail anchor”, TA) from the tyrosine phosphatase PTP1B. As a result, the sensor localized to the ER, and cleavage by PLpro liberated mNeonGreen to be diffusely distributed throughout the cell including nucleus. Thus, nucleus-to-cytoplasm ratios in green and red channels can be used as a measure of PLpro activation [16].
Here, we present a new sensor variant called PLpro-ERNuc, which was improved in two ways. First, we used a natural target-mimicking attachment to ER to ensure efficient cleavage by the native viral PLpro. The sensor design was changed by replacing TA sequence by the fragment of original sequence from SARS-CoV-2 nsp3 molecule (amino acid residues 1341-1567) (Figure 2A). mNeonGreen and mScarlet I fluorescent proteins were separated by the linker containing PLpro cleavage site with LKGGAPTKV motif that corresponds to the nsp2–nsp3 junction from the SARS-CoV-2 polyprotein. Second, we added a nuclear localization signal (NLS) to the C-terminus of the reporter, which ensures accumulation of mNeonGreen in the nucleus after cleavage by PLpro. Thus, a clearer cytoplasm-to-nucleus translocation is expected for PLpro-ERNuc.
2.2. Testing PLpro-ERNuc Sensor in Cells Expressing Recombinant PLpro
The first series of experiments with the sensor was performed on mammalian cells transfected with PLpro. To mimic the natural localization of the viral PLpro, we generated PLpro-Plus genetic construct containing PLpro catalytic domain and adjacent nsp3 domains including ER transmembrane regions TM1-4 (Figure 2B). The blue fluorescent protein mTagBFP2 [17] separated by the “self-cleaving” T2A peptide [18] was used for selection of transfected mammalian cells. Co-expression of PLpro-Plus and PLpro-ERNuc was expected to result in a proximity of the protease domain to the target cleavage site (Figure 2C).
We generated a stable HeLa cell line expressing PLpro-ERNuc using lentiviral transduction method. This line was then transiently transfected with the PLpro-Plus vector. The next day, the PLpro-Plus expression was checked by the presence of the blue fluorescent signal, whereas the sensor signal was assessed in two channels: red (mScarlet I) and green (mNeonGreen).
In control cells expressing PLpro-ERNuc only, an expected cytoplasmic reticular network (ER) fluorescent signal was observed in both red and green channels (Figure 3A). In cells, which also expressed SARS-CoV-2 PLpro protease, the red fluorescence was still localized to the ER. At the same time, the green signal was no longer associated with the reticular network, but became fully accumulated in the cell nucleus (Figure 3A). To quantify the observed translocation, we calculated the nucleus/cytosol ratio in the green channel and normalized it to the nucleus/cytosol ratio in the red channel for the same regions of interest (ROIs) as previously suggested [16] (Figure 3B). The calculated ratio for PLpro-ERNuc increased about 14.3 times in the cells with PLpro expression compared to the rested cells (the mean ratio before translocation – 1.20, after translocation – 17.17).
We concluded that the observed changes fully correspond to the expected PLpro-catalyzed cleavage of the sensor into the nuclear-localized mNeonGreen protein and the ER-bound nsp3-mScarlet I part. Ratio calculations confirmed a robust high-contrast readout provided by the reporter.
2.3. Testing PLpro-ERNuc Sensor in the Model of SARS-CoV-2 Infection Of Cultured Cells
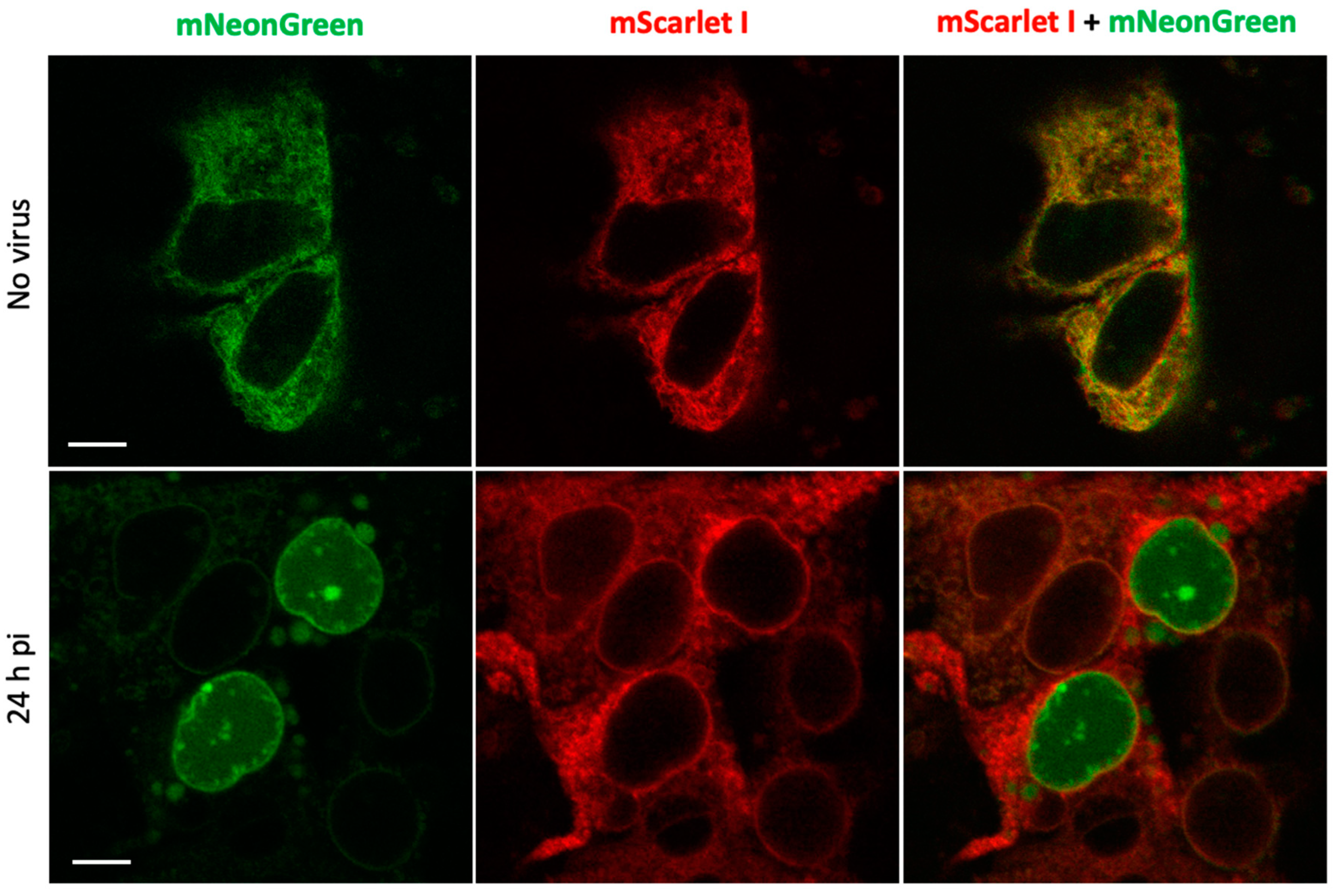
Next, we evaluated the performance of the reporter in the context of SARS-CoV-2 infection in human hepatoma Huh7.5 cell line that is permissive to SARS-CoV-2 infection [19]. To increase reproducibility of imaging experiments, we generated a stable line of Huh7.5 cells expressing PLpro-ERNuc by lentiviral transduction. The cells were infected with SARS-CoV-2 virus that was removed two hours post-infection (h.p.i.). Unlike the previous series of experiments, where the proteolytic activity was provided by a genetically encoded protease (PLpro-Plus construct), here the linker cleavage in the sensor was performed by viral protease expressed during SARS-CoV-2 replication. Confocal fluorescent imaging demonstrated that in mock-treated cells (Figure 4) as well as in virus-infected cells at early stages of infection (4-8 h.p.i., not shown) both red and green fluorescent signals were localized in the ER network. Imaging at 24 h.p.i., showed that in some cells the green reticular network mostly disappeared and green fluorescent signal accumulated in the nucleus (Figure 4).
The obtained results are consistent with the above genetically encoded protease experiments and confirm the effectiveness of the sensor in experiments with the active virus.
3. Discussion
Fully genetically encoded fluorescent sensors represent a powerful tool that enables direct detection of various activities in living cells without addition of any external chemicals or antibodies. Such sensors for viral proteases can be used in two main areas: (i) high-throughput or high-content screening of protease inhibitors (antivirals), and (ii) monitoring the spread of viral infection at the single cell level (mostly in the cell culture models).
Here we developed fluorescent sensor PLpro-ERNuc specifically designed for SARS-CoV-2 PLpro protease. Compared to previously published PLpro sensors, PLpro-ERNuc has several advantages. Firstly, the new sensor design implies intracellular localization and protein context that are very close to that of natural target of PLpro. Secondly, PLpro-ERNuc is characterized by much higher (about 10-fold) dynamic range than our previous version of the sensor. Importantly, we observed no cleavage and translocation of the sensor in the absence of PLpro. Thus, the LKGGAPTKV motif in the linker is not recognized by endogenous cellular proteases, which ensures reporter selectivity.
PLpro-ERNuc demonstrated a robust readout in both overexpressed protease and SARS-CoV-2 infection models. As PLpro-ERNuc-based screening can be performed in living cells, it is potentially suitable to simultaneously assess cytotoxicity and cell membrane permeability of the tested compounds. Sensors for visualization of infected cells are highly needed by virologists, as they allow monitoring virus spread in a culture. First, it allows measuring protease activity in the context of expression with other viral proteins and altered by the virus the intracellular membrane milieu that could affect localization of the protease and its activity. Second, this can show the kinetics of virus spread in cell culture. Moreover, for some infections it may show direct cell-to-cell transmission through tight junctions [20] leading to identification of yet unknown mechanisms of transmission and investigation of mechanisms of antiviral drug resistance. In addition, screening platforms based on native virus application possess several benefits. During a coronaviral infection, PLpro is expressed in the cell along with many other viral proteins. It means that cross-viral protein interactions can affect the potential protease inhibitor. Additionally, coronaviral infection induces significant cellular membrane rearrangements that do not happen during transient transfection of protease-encoding plasmid.
4. Materials and Methods
4.1. General Methods and Materials
DNA oligonucleotides for the PCR were commercially synthesized by Evrogen (Moscow, Russia). An Encyclo PlusPCR kit (Evrogen) was used for the routine PCR amplifications. ScreenMix-HS with HS Taq (Evrogen) was used for the screening of E. coli colonies after the transformation. The length of the PCR products was verified on 1.2% TAE agarose gels. The PCR products of interest were then cut and purified with a Cleanup Mini kit (Evrogen). The DNA plasmids were cultured overnight and then extracted using a Plasmid Miniprep kit (Evrogen). The gene sequences were verified by Sanger sequencing. Huh7.5 cells were a kind gift of C.M.Rice (Rockefeller University, NY, USA) and Apath L.L.C. (St. Louis, MO, USA). SARS-CoV-2 (passage 4) corresponding to hCoV-19/Russia/Moscow-PMVL-12/2020 strain with infectivity of 106 TCID50/ml, was from the Russian State collection of viruses at the National Research Center for Epidemiology and Microbiology.
4.2. DNA Cloning
The DNA constructs were cloned using the Golden Gate cloning system [21]. The genes of interest were cloned into Level 0 and Level 1 backbones obtained from a MoClo Toolkit (AddGene Kit #1000000044). BpiI (BbsI) and Eco31I (BsaI) restriction endonucleases (Thermo Scientific, MA, USA) and T4 DNA ligase (Evrogen) were used for the MoClo procedure.
The expression vector encoding SARS-CoV-2 nsp3 was a gift from Fritz Roth (AddGene plasmid #141257 [22]). PLpro-coding DNA fragments were generated by PCR amplification on the template of the nsp3 coding sequence and included ubiquitin-like domain 2 (Ubl2, residues 746–805), PLpro catalytic domain (residues 806–1058), nucleic acid-binding domain (NBD, residues 1059–1200), marker domain (MD, residues 1201–1340) and four transmembrane regions (TM1-4, residues 1341–1567) [23]. Transfection efficiency was visualized using mTagBFP2 protein fused to the “self-cleaving” T2A peptide GSGEGRGSLLTCGDVEENPGP [18].
DNA constructs for the PLpro-ERNuc consisted of two fluorescent proteins (mNeonGreen and mScarlet I) connected by an amino acid linker GGGST-LKGGAPTKV-GGSGS, where LKGGARTKV corresponded with the nsp2–nsp3 junction from the SARS-CoV-2 polyprotein. On the N-terminus the sensor contained four transmembrane regions (TM1-4, residues 1341–1567) from SARS-CoV-2 nsp3, which ensured ER localization of the sensor, and three Y-domains (Y1-3, residues 1568–1945), which provided natural context for the nsp2–nsp3 cleavage site. On the С-terminus of the sensor the 26 amino acid nuclear localization signal (NLS) was added (DPKKKRKVDPKKKRKVDPKKKRKSGL), which provided green signal accumulation in nucleus after proteolysis.
All elements of the translation unit including coding sequences, promoter and terminator were assembled into Level 1 expression vectors using the Golden Gate cloning system. DNA construct encoding the PLpro was put under the control of CMV promoter and possessed SV40 poly(A) sequence. DNA construct encoding the PLpro-ERNuc was put under CMV promoter and was assembled into a pLVT-like vector for further lentiviral production.
4.3. Cell culture and Transfection
Imaging experiments reported in the manuscript were performed with HeLa and Huh-7.5 cells. HEK293T cells were used for lentivirus production. HeLa and HEK293T cells were cultured in complete DMEM: Dulbecco’s Modified Eagle Medium, DMEM (PanEco, Moscow, Russia), supplemented with 10% fetal bovine serum (FBS) (BioSera, Nuaille, France), 100 U/mL penicillin, and 100 mg/mL streptomycin (PanEco) at 37 °C in 5% CO2. Huh-7.5 cells were cultured in Dulbecco’s Modified Eagle Medium (PanEco) supplemented with 10% FBS (BioSera), 2 mM L-Glutamine (Gibco, MA, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin (PanEco) at 37 °C in 5% CO2. For the live cell imaging experiments, the DMEM was replaced by imaging media: MEM (PanEco) supplemented with 10% FBS (BioSera) and 20 mM HEPES (Corning, NY, USA). Transfections were performed in Opti-MEM (Gibco) with GenJect-39 (Molecta, Moscow, Russia) transfection reagent according to manufacturers’ protocol (1:1 DNA:GenJect-39 ratio).
4.4. Lentivirus Production and Stable HeLa Cell Line Generation
Lentivirus was produced from a lentiviral pLVT-like vector transfected into HEK293T cells using a polyethyenimine (PEI) transfection protocol. HEK293T cells were seeded at 3.8×106 cells per plate in 10 cm tissue culture plates (SPL Life Sciences, Gyeonggi-do, Korea) in complete DMEM at 37 °C, 5% CO2. After 24 hours cells were transfected in Opti-MEM (Gibco) with a mixture of 3 transfection plasmids (pR8.91, pMDG, lentiviral transfer plasmid encoding PLpro-ERNuc) with PEI 25K (Polysciences, Warrington, USA) according to manufacturers’ protocol (1:3 DNA:PEI ratio). Virus was harvested at 48 hours post-transfection and was concentrated by ultracentrifugation at 100,000 g (Beckman, California, USA) at 4°C for 3 hours.
For lentiviral transduction HeLa cells were incubated with the lentivirus for 24 hours. Fluorescence-activated cell sorting (FACS) was performed using BD FACSAria III (BD Biosciences, New Jersey, USA).
4.5. SARS-CoV-2 Infection Assay
Huh7.5 cells were seeded on 10 cm dishes (SPL Life Sciences, Gyeonggi-do, Korea) at a density of 1x106 in DMEM supplemented with 10% FBS. When the cells reached 50% confluence, they were transduced with the lentivirus. Three days post-transduction the cells were harvested and seeded in glass bottom cell culture dishes with 15-mm inserts (Nest, Jiangsu, China) in the same medium.
Infection with SARS-CoV-2 was performed as described in [19]. Briefly, when the cells on confocal dishes reached 40% density, the virus was added for two hours in DMEM with 2.5% serum at a multiplicity of infection (MOI) 0.1. Then, the cells were fixed with metanol-acetone as described in [24], followed by transfer to PBS. Confocal 8-bit digital images were acquired using a Leica TCS SP5 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany) equipped with a HCX PLAPO CS 63x 1.4NA oil immersion lens. Fluorescence was excited at 488 nm and registered in the 510–535 nm range in the green channel and excited at 543 nm and registered in the 570-600 nm range in the red channel.
4.6. Live Cell Imaging
For the live cell imaging experiments HeLa stable line expressing PLpro-ERNuc sensor was used. Cells were seeded on glass-bottomed 35 × 10 mm dishes (SPL Life Sciences) and were incubated at 37 °C in 5% CO2 overnight. The next day, cells were transfected with PLpro-Plus plasmid. Imaging experiments were performed 24 hours post-transfection using a BZ-9000 inverted fluorescence microscope (Keyence, Osaka, Japan). The imaging was carried out at 37 ℃ with a 60 × PlanApo 1.40 NA oil objective (Nikon, NY, USA). TexasRed OP79302 SB filter (excitation 562/40 nm, emission 624/40), GFP-BP OP66836 BZ filter (excitation 470/40 nm, emission 535/50), and 49021-ET-EBFP2/Coumarin/Attenuated DAPI filter (excitation 405/10 nm, emission 460/25) (Chroma Technology, VT, USA) were used to visualize mScarlet I, mNeonGreen, and mTagBFP2 respectively.
4.7. Image Analysis
The images were processed using Fiji ImageJ [25]. For the calculation of the nucleus/cytoplasm ratio, the mean fluorescence was measured in the nucleus and cytoplasmic ROIs, respectively; the same ROIs in the green and red channels were used for each individual cell. For all calculations, the mean fluorescence intensity of the background out of the cells was subtracted. For experiments stating p-values, a paired Student’s t-test was performed. p-values < 0.05 were considered as significant.
5. Conclusions
Here, we used a bioinspired design to construct PLpro-ERNuc - a new genetically encoded fluorescent sensor for live-cell monitoring activity of SARS-CoV-2 PLpro protease. A fragment of nsp3 protein including four ER-associated transmembrane regions was used to place the sensor at intracellular localization and protein context highly similar to that of the native target of PLpro. The use of two fluorescent proteins, one of which remains attached to ER all the time and another one translocated from ER to the cell nucleus upon sensor cleavage with PLpro, enabled easy-to-detect high-contrasting ratiometric (and thus independent on the sensor expression level) readout. We demonstrated efficient functioning of PLpro-ERNuc in both recombinant PLpro overexpression and SARS-CoV-2 virus infection cell models. These two models correspond to the main areas of a viral protease sensor application, namely safe virus-free cell-based screening of protease inhibitors to develop novel antivirals, and monitoring virus spreading for basic studies.
Author Contributions
E.L.S., A.V.I., and K.A.L. designed the research; E.L.S., O.N.I., I.T.F., and A.V.I. acquired the data; E.L.S. analyzed the data; E.L.S., A.V.I., and K.A.L. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
Experiments with infectious virus were supported by the Russian Science Foundation (grant #21-74-10146). Imaging experiments with overexpressed recombinant PLpro were supported by grant no. 075-15-2019-1789 from the Ministry of Science and Higher Education of the Russian Federation, allocated to the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Pirogov Russian National Research Medical University, Russia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO COVID-19 Dashboard. Available online: http://data.who.int (accessed on 4 May 2024).
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The Molecular Virology of Coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural Insights into SARS-CoV-2 Proteins. J. Mol. Biol. 2021, 433, 166725. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. SARS-CoV-2 Papain-Like Protease: Structure, Function and Inhibition. Chembiochem 2022, 23, e202200327. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of Papain-like Protease from SARS-CoV-2 and Its Complexes with Non-Covalent Inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Ong, I.L.H.; Yang, K.-L. Recent Developments in Protease Activity Assays and Sensors. Analyst 2017, 142, 1867–1881. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Xia, Z.; Lambrinidis, G.; Townsend, J.A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent Sci 2021, 7, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Froggatt, H.M.; Heaton, B.E.; Heaton, N.S. Development of a Fluorescence-Based, High-Throughput SARS-CoV-2 3CLpro Reporter Assay. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Zhang, Q.; Schepis, A.; Huang, H.; Yang, J.; Ma, W.; Torra, J.; Zhang, S.-Q.; Yang, L.; Wu, H.; Nonell, S.; et al. Designing a Green Fluorogenic Protease Reporter by Flipping a Beta Strand of GFP for Imaging Apoptosis in Animals. J. Am. Chem. Soc. 2019, 141, 4526–4530. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.; Häge, S.; Herrmann, A.; Wangen, C.; Kicuntod, J.; Jungnickl, D.; Tillmanns, J.; Müller, R.; Fraedrich, K.; Überla, K.; et al. Methodological Development of a Multi-Readout Assay for the Assessment of Antiviral Drugs against SARS-CoV-2. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Jones, C.T.; Catanese, M.T.; Law, L.M.J.; Khetani, S.R.; Syder, A.J.; Ploss, A.; Oh, T.S.; Schoggins, J.W.; MacDonald, M.R.; Bhatia, S.N.; et al. Real-Time Imaging of Hepatitis C Virus Infection Using a Fluorescent Cell-Based Reporter System. Nat. Biotechnol. 2010, 28, 167–171. [Google Scholar] [CrossRef]
- Li, X.; Yang, E.; Li, X.; Fan, T.; Guo, S.; Yang, H.; Wu, B.; Wang, H. MAVS-Based Reporter Systems for Real-Time Imaging of EV71 Infection and Antiviral Testing. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human Cytomegalovirus-Encoded US9 Targets MAVS and STING Signaling to Evade Type I Interferon Immune Responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- van Huizen, M.; Vendrell, X.M.; de Gruyter, H.L.M.; Boomaars-van der Zanden, A.L.; van der Meer, Y.; Snijder, E.J.; Kikkert, M.; Myeni, S.K. The Main Protease of Middle East Respiratory Syndrome Coronavirus Induces Cleavage of Mitochondrial Antiviral Signaling Protein to Antagonize the Innate Immune Response. Viruses 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wu, L.; Yuan, Y.; Wang, H.; Yin, H.; Li, M.; Chai, L.; Liang, R.; Liu, Y.; Zhao, D.; et al. Species-Specific Cleavage of cGAS by Picornavirus Protease 3C Disrupts Mitochondria DNA-Mediated Immune Sensing. PLoS Pathog. 2023, 19, e1011641. [Google Scholar] [CrossRef] [PubMed]
- Sokolinskaya, E.L.; Putlyaeva, L.V.; Polinovskaya, V.S.; Lukyanov, K.A. Genetically Encoded Fluorescent Sensors for SARS-CoV-2 Papain-like Protease PLpro. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Subach, O.M.; Cranfill, P.J.; Davidson, M.W.; Verkhusha, V.V. An Enhanced Monomeric Blue Fluorescent Protein with the High Chemical Stability of the Chromophore. PLoS One 2011, 6, e28674. [Google Scholar] [CrossRef]
- Ibrahimi, A.; Vande Velde, G.; Reumers, V.; Toelen, J.; Thiry, I.; Vandeputte, C.; Vets, S.; Deroose, C.; Bormans, G.; Baekelandt, V.; et al. Highly Efficient Multicistronic Lentiviral Vectors with Peptide 2A Sequences. Hum. Gene Ther. 2009, 20, 845–860. [Google Scholar] [CrossRef]
- Smirnova, O.A.; Ivanova, O.N.; Fedyakina, I.T.; Yusubalieva, G.M.; Baklaushev, V.P.; Yanvarev, D.V.; Kechko, O.I.; Mitkevich, V.A.; Vorobyev, P.O.; Fedorov, V.S.; et al. SARS-CoV-2 Establishes a Productive Infection in Hepatoma and Glioblastoma Multiforme Cell Lines. Cancers 2023, 15. [Google Scholar] [CrossRef]
- Xiao, F.; Fofana, I.; Heydmann, L.; Barth, H.; Soulier, E.; Habersetzer, F.; Doffoël, M.; Bukh, J.; Patel, A.H.; Zeisel, M.B.; et al. Hepatitis C Virus Cell-Cell Transmission and Resistance to Direct-Acting Antiviral Agents. PLoS Pathog. 2014, 10, e1004128. [Google Scholar] [CrossRef]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS One 2011, 6, e16765. [Google Scholar] [CrossRef]
- Kim, D.-K.; Knapp, J.J.; Kuang, D.; Chawla, A.; Cassonnet, P.; Lee, H.; Sheykhkarimli, D.; Samavarchi-Tehrani, P.; Abdouni, H.; Rayhan, A.; et al. A Comprehensive, Flexible Collection of SARS-CoV-2 Coding Regions. G3 2020, 10, 3399–3402. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural Biology of SARS-CoV-2: Open the Door for Novel Therapies. Signal Transduct Target Ther 2022, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Tunitskaya, V.L.; Eliseeva, O.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Zakirova, N.F.; Khomich, O.A.; Kalis, M.; Latyshev, O.E.; Starodubova, E.S.; Ivanova, O.N.; et al. Prokaryotic Expression, Purification and Immunogenicity in Rabbits of the Small Antigen of Hepatitis Delta Virus. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of SARS-CoV-2 nsp3 domain organization. Ubl1 - ubiquitin-like domain 1, HVR - hypervariable region, Mac1 - macrodomain 1, SUD - SARS-unique domain, Ubl2 - ubiquitin-like domain 2, PLpro - papain-like protease, NBD - nucleic acid-binding domain, MD - marker domain, TM 1-4 - transmembrane regions 1-4, Y - the Y domain. Numbers indicate the position of the amino acid.
Figure 1.
Schematic representation of SARS-CoV-2 nsp3 domain organization. Ubl1 - ubiquitin-like domain 1, HVR - hypervariable region, Mac1 - macrodomain 1, SUD - SARS-unique domain, Ubl2 - ubiquitin-like domain 2, PLpro - papain-like protease, NBD - nucleic acid-binding domain, MD - marker domain, TM 1-4 - transmembrane regions 1-4, Y - the Y domain. Numbers indicate the position of the amino acid.
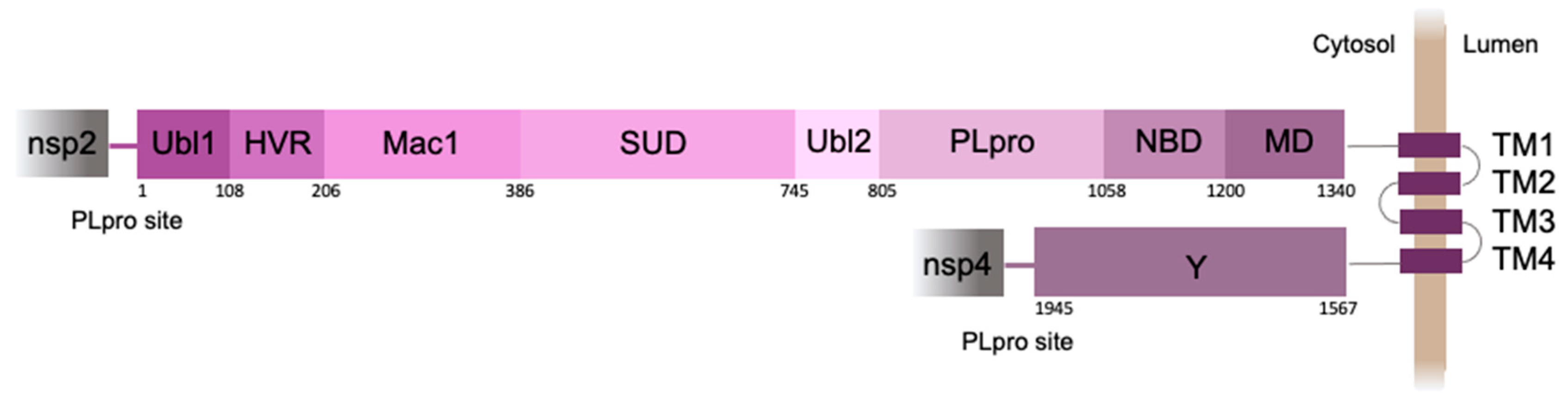
Figure 2.
Diagrams of the genetic constructs. PLpro-ERNuc (A) and PLpro-Plus (B) constructs used for imaging experiments in mammalian cells and their proposed intracellular localization (C). Indication of nsp3 domains is described in Figure 1. mScarlet I, mNeonGreen, mTagBFP2 - red, green and blue fluorescent proteins, respectively. T2A - “self-cleaving” T2A peptide, NLS - nuclear localization signal. The gray arrow indicates targeting of the PLpro cleavage site by the protease.
Figure 2.
Diagrams of the genetic constructs. PLpro-ERNuc (A) and PLpro-Plus (B) constructs used for imaging experiments in mammalian cells and their proposed intracellular localization (C). Indication of nsp3 domains is described in Figure 1. mScarlet I, mNeonGreen, mTagBFP2 - red, green and blue fluorescent proteins, respectively. T2A - “self-cleaving” T2A peptide, NLS - nuclear localization signal. The gray arrow indicates targeting of the PLpro cleavage site by the protease.
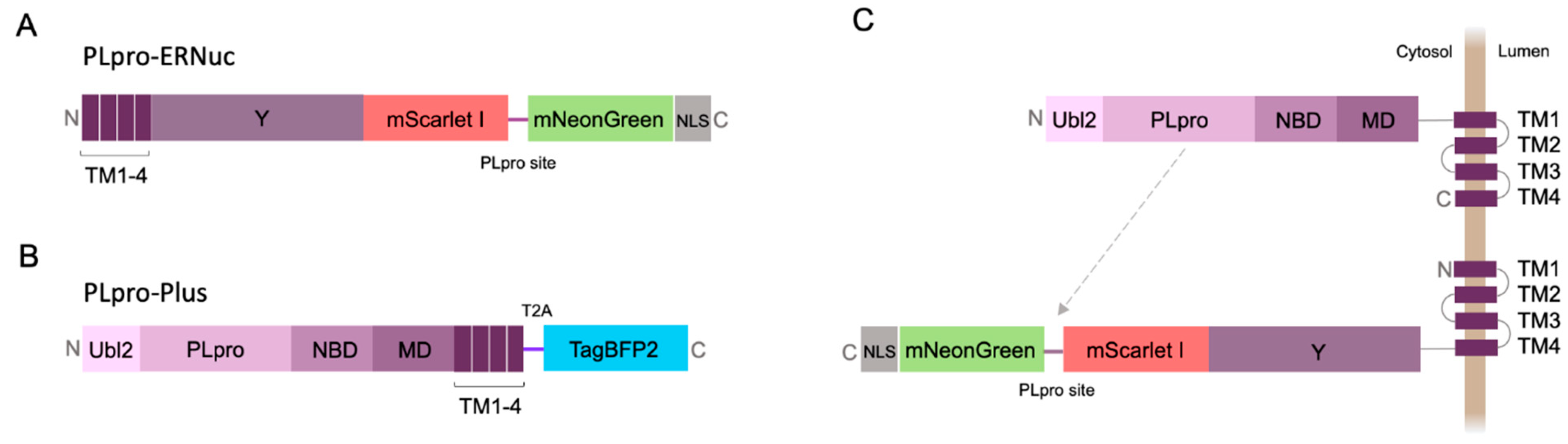
Figure 3.
PLpro-ERNuc translocation-based fluorescent sensor for SARS-CoV-2 PLpro protease activity. (A) Representative cell images of HeLa cells expressing PLpro-ERNuc. The upper panel: HeLa cells expressing PLpro-ERNuc in the absence of PLpro protease. The lower panel: HeLa cells expressing PLpro-ERNuc transiently transfected with PLpro-Plus. Scale bars, 10 μm. (B) Quantification of nucleus/cytosol ratios for cells expressing PLpro-ERNuc in the absence of PLpro (left, n = 107 cells) and in the presence of PLpro (right, n = 103 cells). In the box plot median, mean, 1st, and 3rd quartiles as well as minimum and maximum values are shown. The red (1) and green (2) boxes are the ratio of fluorescence in the nucleus to the cytoplasm in the corresponding channels, and the yellow (3) boxes are the ratio of the obtained values to each other. Statistical significance is shown in the right graph in comparison with corresponding values in the left graph (**** - p-value < 0.0001, ns - not significant).
Figure 3.
PLpro-ERNuc translocation-based fluorescent sensor for SARS-CoV-2 PLpro protease activity. (A) Representative cell images of HeLa cells expressing PLpro-ERNuc. The upper panel: HeLa cells expressing PLpro-ERNuc in the absence of PLpro protease. The lower panel: HeLa cells expressing PLpro-ERNuc transiently transfected with PLpro-Plus. Scale bars, 10 μm. (B) Quantification of nucleus/cytosol ratios for cells expressing PLpro-ERNuc in the absence of PLpro (left, n = 107 cells) and in the presence of PLpro (right, n = 103 cells). In the box plot median, mean, 1st, and 3rd quartiles as well as minimum and maximum values are shown. The red (1) and green (2) boxes are the ratio of fluorescence in the nucleus to the cytoplasm in the corresponding channels, and the yellow (3) boxes are the ratio of the obtained values to each other. Statistical significance is shown in the right graph in comparison with corresponding values in the left graph (**** - p-value < 0.0001, ns - not significant).
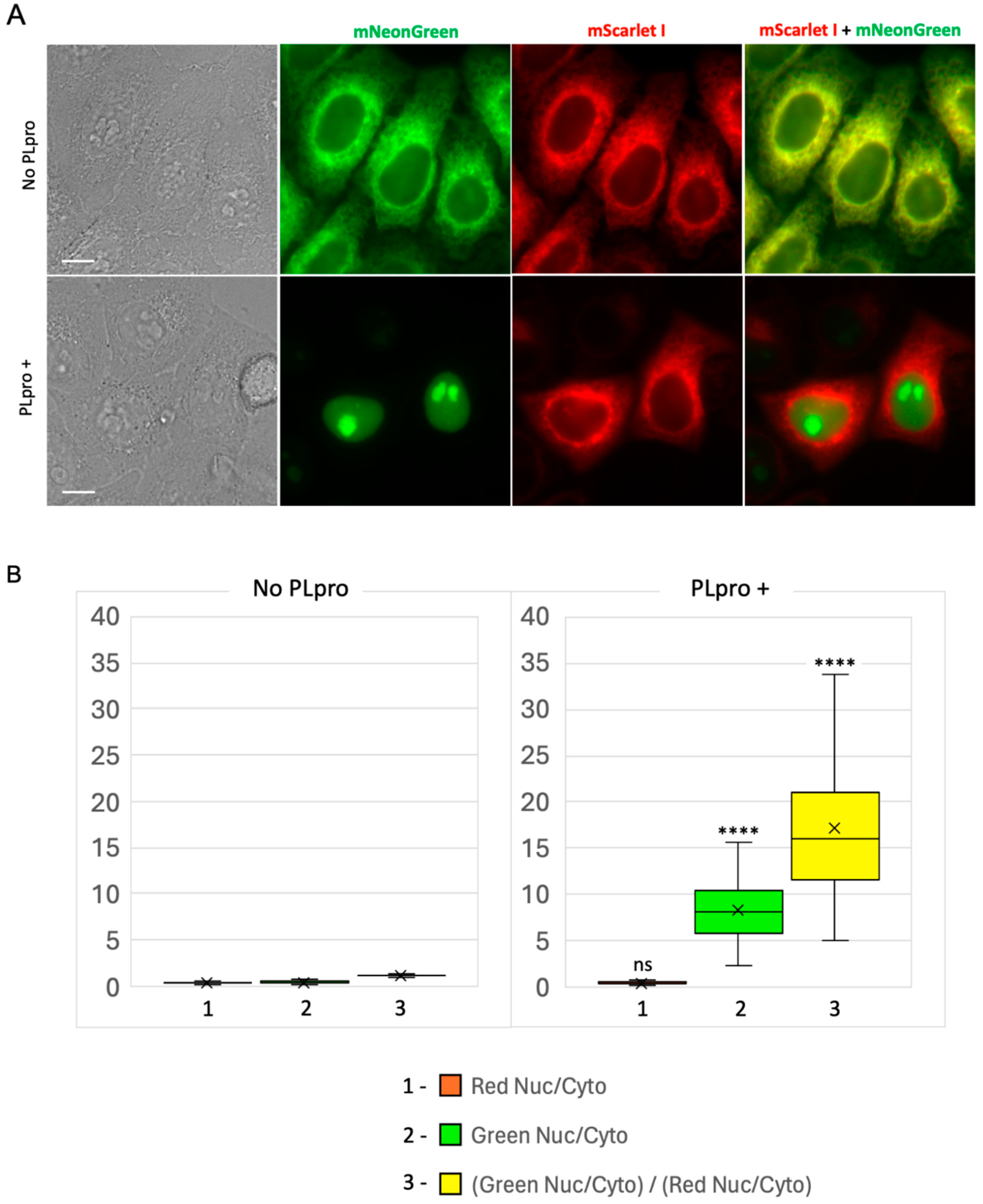
Figure 4.
Assessment of SARS-CoV-2 infection of Huh7.5 cells expressing PLpro-ERNuc. Confocal microscopy of fixed cells in the green and red channels in the absence of SARS-CoV-2 infection (upper panels) and 24 h post-infection (bottom panels). Scale bars, 10 μm.
Figure 4.
Assessment of SARS-CoV-2 infection of Huh7.5 cells expressing PLpro-ERNuc. Confocal microscopy of fixed cells in the green and red channels in the absence of SARS-CoV-2 infection (upper panels) and 24 h post-infection (bottom panels). Scale bars, 10 μm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



