
Submitted:
14 May 2024
Posted:
14 May 2024
You are already at the latest version
Abstract
Amitriptyline is a tricyclic antidepressant commonly used for depressive disorders and pre-scribed off-label for several neurological conditions like neuropathic pain, migraine and anxiety. Amitriptyline potentiates the monoaminergic transmission by blocking the reuptake of norepi-nephrine and serotonin. However, antidepressants also act on additional targets that contribute to either therapeutic or adverse effects and may suggest new indications for their repurposing. Furthermore, since depression is often a co-morbidity in patients with chronic conditions (i.e. cancer, neurodegenerative diseases) the use of antidepressants interfering with pathways in-volved in cell homeostasis may affect the progression and outcome of these pathologies.
Here we investigate the effects of amitriptyline on proliferation and autophagy in human SH-SY5Y neuroblastoma cells. Dose and time-dependent upregulation of the autophagy markers LC3II and autophagy receptor p62, with accumulation of LAMP1 positive compartments, were observed in SH-SY5Y cells exposed to amitriptyline. Alteration of autophagy was accompanied by reduced cell viability and decreased clonogenic capacity, without a significant induction of apoptosis. Decrease viability and clonogenic activity were still observed in autophagy deficient Atg5 -/- MEF and following pre-treatment of SH-SY5Y culture with the autophagy inhibitor chloroquine suggesting that the amitriptyline’s effects on cell proliferation were not caused by the modulation of autophagy.
Our findings demonstrate that amitriptyline acts on pathways that are crucial for cell and tissue homeostasis and pose the basis for further studies on the potential application of these effects and the consequences during long-term antidepressant treatment.
Keywords:
amitriptyline
; antidepressant
; autophagy
; cell proliferation
; neuroblastoma
; SH-SY5Y
1. Introduction
Affecting 121 million people worldwide, depressive disorders are the second cause of Years Lived with Disability (DALY) in the age category 15-44 and the fourth leading contributor to the global burden of disease in developing countries [1]. Depression is often a common co-morbidity in patients with several chronic medical diseases including neurodegenerative diseases, stroke, epilepsy, multiple sclerosis, autoimmunity [2,3,4,5], cancer [6,7] and it is a post-partum complication [8,9,10,11].
Antidepressants are the first-line medications for moderate and severe depression and among the most widely prescribed drugs [12]. Their consumption has significantly raised in the last two decades with a five-fold increase in the United States and a three-fold increase in European countries [13]. A further rise is linked to the recent coronavirus 2019 (COVID-19) pandemic which contributed to worsening mental health crisis, especially among adolescents and young adults [14,15]. Aside from their use in managing depressive symptoms, antidepressant drugs are used to treat people suffering from other mental health issues and physical conditions, including anxiety, psychotic disorders, attention deficit, insomnia, migraine, neuropathic pain, premenstrual dysphoric disorder, gastrointestinal and genitourinary pathologies [16,17,18,19,20,21,22,23,24]. Moreover, following the concept of drug repurposing, antidepressant drugs are now being explored for their usefulness in diseases beyond their therapeutic indication, for example in cancer treatment [25,26].
Indeed, although the primary action of antidepressants is the regulation of monoamine concentration in the synaptic space, several studies suggested that they possess anticancer properties [25,26,27], which encompass apoptosis induction [28], restriction of cellular energy metabolism [29], antioxidant activities [30], inhibition of angiogenesis [31] and modulation of immune response [32].
Tricyclic antidepressants (TCAs), introduced in the market in 1959 for the treatment of major depressive disorders (MDD), act by inhibiting the reuptake of norepinephrine and serotonin and elevating the concentrations of these neurotransmitters within the synaptic cleft [33,34]. Amitriptyline belongs to the TCAs class of antidepressants and it has been FDA approved to treat MDD in adults [35] and used off-label to treat anxiety, post-traumatic stress disorder, insomnia, chronic pain (diabetic neuropathy, fibromyalgia), irritable bowel syndrome, interstitial cystitis (bladder pain syndrome), migraine prophylaxis, postherpetic neuralgia, sialorrhea [36,37,38] and for post-COVID headaches [39].
Recently, emerging evidence has suggested that several antidepressant drugs are able to interfere with autophagy, a highly conserved catabolic pathway that delivers cytoplasmic components to lysosomes for degradation [40], and this may be involved in their ability to suppress tumour growth [41,42,43,44,45].
Here we explored the effects of the TCA antidepressant amitriptyline on neuroblastoma cells focusing on the modulation of autophagy and its role in cell proliferation.
2. Materials and Methods
Reagents
Amitriptyline hydrochloride (A8404), dimethyl sulfoxide (DMSO), chloroquine (CQ; C6628) and bafilomycin A1 (BafA1; B1793) were purchased from Sigma-Aldrich (Milan, Italy).
Cell Cultures and Treatments
Adherent human SH-SY5Y neuroblastoma cells, obtained from ICLC-IST (Genoa, Italy), were grown in RPMI 1640 medium (Gibco, Life Technologies, Paisley, UK) supplemented with: 10% heat-inactivated foetal bovine serum (FBS; Gibco, Life Technologies, Paisley, UK), 1 mM sodium pyruvate and 2 mM glutamine (Gibco, Life Technologies, Paisley, UK), 100 IU/ml penicillin and 100 μg/ml streptomycin (Gibco, Life Technologies, Paisley, UK). Wild type (Atg5 +/+) and Atg5-deficient (Atg5 -/-) mouse embryonic fibroblasts (MEF) were purchased from RIKEN Bio-Resource Cell Bank (Japan) and grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Life Technologies, Paisley, UK) supplemented with 10% FBS, 100 IU/ml penicillin (Gibco, Life Technologies, Paisley, UK) and 100 μg/ml streptomycin (Gibco, Life Technologies, Paisley, UK). Cell cultures were maintained at 37°C in a humidified atmosphere with 5% CO2. Cells, cultured in 75 cm2 flasks, were seeded onto 96, 24 or 6-well plates; 24h after plating, the medium was replaced with fresh medium (control), medium supplemented with vehicle or the indicated compound. A stock solution of amitriptyline (5 mM) was prepared in water and diluted in culture medium to obtain the final concentrations of 5-120 μM. Amitriptyline concentrations were chosen based on previously published in vitro studies [46,47].
CQ, dissolved in water (50 mM) and diluted in culture medium, was applied to SH-SY5Y cells at the final concentration of 50 μM [48] 2h before the addition of the antidepressant, and maintained throughout the period of treatment.
Stock solution of BafA1 (1 mM) was prepared in DMSO and further diluted in culture medium to the final concentration of 100 μM [49]; cells were treated with BafA1 for the last 4h of exposure to amitriptyline. DMSO (0.01%) was added to the medium of vehicle-treated cultures.
Cell Viability Study
Cell viability was assessed by trypan blue dye exclusion test (0.4% w/v) and cell death reported as the percentage of stained (non-viable) vs total cells counted [50].
Cell metabolic activity, as indirect measure of cell viability, was evaluated by the quantification of the intracellular reduction of the yellow dye 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT; Sigma-Aldrich, Milan, Italy) to purple formazan. Cells, seeded and treated onto 96-well plates, were incubated with 100 μl of MTT (0.5 mg/ml) for 1h in a humidified 5% CO2 incubator at 37°C. At the indicated time points (24, 48 or 72h) medium was removed, and formazan crystals were solubilized by the addition of 100 μl of DMSO. Absorbance was measured at 540/690 nm by a microplate spectrophotometer (Synergy H1 plate reader, BioTek Instruments, Inc., Winooski, VT, USA). For each experimental conditions the absorbance of 4-8 wells was averaged. Data were expressed as percentage of cell survival vs. control cultures (set to 100%). Each experiment was performed in triplicate.
Immunocytochemistry
For immunocytochemical staining, SH-SY5Y cells were plated onto poly-L-lysine (Sigma-Aldrich, Milan, Italy) coated coverslips and cultured for 24h before being exposed to amitriptyline 5-60 μM. At the indicated time, culture medium was removed and cells were washed with phosphate-buffered saline (PBS, pH 7.4), fixed with formalin solution (containing 4% paraformaldehyde; Sigma-Aldrich, Milan, Italy) for 15 minutes at room temperature (RT) and washed three times in PBS. Cells were permeabilized with a solution of Triton 0.1% for 10 minutes at RT, washed three times with PBS and blocked with 10% donkey serum (DS; Sigma-Aldrich, Milan, Italy) for 30 min. Coverslips were incubated with primary antibodies against rabbit microtubule-associated protein 1 light chain 3 (LC3; code PD036, MBL International Corporation, Nagoya, Japan; 1∶500 dilution), mouse lysosomal-associated membrane protein 1 (LAMP1; Developmental Studies Hybridoma Bank, Iowa City, IA, USA; 1∶200 dilution), rabbit sequestosome 1 (p62/SQSTM1; code sc-25575, Santa-Cruz; 1∶50 dilution), in 5% DS/PBS overnight at 4°C. After three washes with PBS, coverslips were incubated with Alexa Fluor secondary antibodies (Alexa Fluor 488 donkey anti-rabbit (1:500 dilution) and Alexa Fluor 594 donkey anti-mouse (1:1000 dilution) (Molecular Probes, Life Technologies Paisley, UK) for 1h at RT. Control experiments were prepared in the absence of primary antibody to exclude secondary antibody non-specific staining. Coverslips were mounted with Vectashield solution containing 4′,6-diamidino-2-phenylindole (DAPI, AB104139, Abcam, Cambridge, UK) to visualize nuclei. Images were acquired using a confocal microscope (FV3000, Olympus Corporation, Tokyo, Japan).
Phase-Contrast Microscopy, Measurement of Cells Confluence and Neurites Count
SH-SY5Y cells were observed under an inverted phase contrast microscope (CKX53, Olympus Corporation, Tokyo, Japan) with a 10X objective and 4 images for each condition were taken and analyzed with ImageJ software. The percentage value of the white area (cells) divided by the total area of the image corresponded to the value of confluence indicated as percentage of area covered by cells [51].
The number of neurite for each cell was quantified using the ImageJ software package (NIH, Bethesda, MD, USA) [52]. Thirty cells for each experimental condition were analyzed. Data were expressed as the average of the neurite number/cell. Each experiment was performed in triplicate.
Live-Cell Labeling of Acidic Compartments
For lysosomal staining, SH-SY5Y cells were grown on glass-bottom dish and treated with amitriptyline (5-60 μM) for 24h. Cells were washed with media and loaded with a lysosomal probe (LysoTracker Red DND-99, Thermo Fisher Scientific, Waltham, MA, USA). LysoTracker (60 nM) was used according to the manufacturer’s instruction. Images were acquired by a confocal microscope (FV3000; Olympus Corporation, Tokyo, Japan).
TUNEL Assay
Apoptosis was detected using the DeadEnd™ Fluorometric TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labelling) System (cod. G3250, Promega Madison, Wisconsin, USA) according to the manufacturer's instructions. Briefly, SH-SY5Y cells, cultured on coverslips, were fixed with 10% PFA, rinsed three times with PBS, and equilibrated with 100 µl Equilibration Buffer at RT for 10 minutes. Fixed cells were incubated with 50 µl of Terminal Deoxynucleotidyl Transferase (TdT) solution for 1h at 37°C in a humidified environment. The reaction was stopped by incubation with 2x SSC solution for 15 minutes and coverslip were mounted with Vectashield mounting media with DAPI (Abcam, Cambridge, UK) to visualize nuclei. Images were acquired using a confocal microscope (FV3000, Olympus Corporation, Tokyo, Japan). TUNEL-positive cells were expressed as the ratio of TUNEL-positive cells / total cells.
Protein Extraction and Western Blot Analysis
Cells were lysed in ice-cold RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% IGEPAL and 0.5% sodium deoxycholate) containing protease inhibitor (code P8349; Sigma-Aldrich, Milan, Italy) and phosphatase inhibitor cocktails (code 05726; Sigma-Aldrich, Milan, Italy). Lysates were centrifuged for 15 min at 14.000 g at 4°C and supernatants were assayed for protein content with the Bio-Rad DC Protein Assay Kit (Bio-Rad Laboratories, Milan, Italy). Equal amount (10 g) of total proteins for each condition was resolved by 8, 12 or 15% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto PVDF membranes (Immobilon-P, Sigma-Aldrich, Milan, Italy). Membranes were blocked with 5% non-fat milk (Santa Cruz Biotechnology, Dallas, USA) in Tris-buffered saline (TBS) containing 0.05% Tween 20 for 1h at RT. Primary antibodies were incubated overnight at 4°C, and then with a horseradish peroxidase (HRP)-conjugated secondary antibody for 1h at RT. Protein bands were visualized with the ECL Western Blotting Detection kit (Santa Cruz Biotechnology, Dallas, USA) and the chemiluminescence signal detected using X-ray films (Santa Cruz Biotechnology, Dallas, USA). Autoradiographic films were scanned, digitalized at 600 dpi and band quantification was performed using ImageJ software (NIH, Bethesda, MD, USA).
The following primary antibodies and dilutions were used: anti-LC3 1∶2000 (code PD036; MBL International Corporation, Nagoya, Japan; 1∶500 dilution), anti-p62/SQSTM1 1∶4000 (code P0067, Sigma-Aldrich, Milan, Italy), anti-actin 1∶1000 (clone AC-40; Sigma-Aldrich, Milan, Italy), anti-LAMP1 1:1000 (Developmental Studies Hybridoma Bank, Iowa City, IA, USA). HRP-conjugated goat anti-rabbit or anti-mouse IgG (Pierce Biotechnology, Rockford, IL, U.S.A) were used as secondary antibodies.
Clonogenic Assay
SH-SY5Y, Atg5 +/+ and Atg5 -/- MEF cells, plated into 6-well plates at a density of 500 and 300 cells/well respectively, were treated with medium containing amitriptyline (5-120 μM) for 12 days. Cells were fixed and stained for 30 minutes with a mixture of 6.0% glutaraldehyde and 0.5% crystal violet (Sigma-Aldrich, Milan, Italy). Plates were imaged using an inverted phase contrast microscope (CKX53; Olympus Corporation, Tokyo, Japan) and colonies with more than 50 cells were automatically counted using the "colony blob count tool" of NIH ImageJ software (Bethesda, MD, USA).
Cell Cycle Analysis
SH-SY5Y were plated in 6 well plates and 24h later treated with amitriptyline (5-60 M) for 24, 48 or 72h. Cells were washed twice with PBS and fixed with ice-cold 70% ethanol overnight at -20°C. After centrifugation, a solution containing 20 μg/ml propidium iodide (PI), 0.17 mg/ml RNAse-A and 0.1% Triton X-100 (Sigma Aldrich, Milan, Italy) was added to each tube and cells were stained for 20 minutes at RT in the dark and under constant agitation. DNA content was measured by flow cytometry (CytoFLEX Beckman, Beckman Coulter, Milan, Italy). Data analysis was performed using CytExpert Beckman Coulter software (Beckman Coulter, Milan, Italy).
Statistical Analysis
Data are expressed as the mean ± S.E.M. of the indicated number of independent experiments and evaluated statistically for differences by ANOVA followed by Tukey-Kramer test for multiple comparisons. Where indicated, Student's t test was used to evaluate differences between two means. A value of P<0.05 was considered statistically significant. The statistical significance was analyzed using GraphPad Prism 8.3.0 software (GraphPad Soft-ware, Inc., San Diego, CA, USA).
3. Results
3.1. Amitriptyline Induced a Concentration and Time-Dependent Reduction of Cell Viability in SH-SY5Y Cultures
Under control conditions SH-SY5Y cells grow as monolayer displaying a flattened morphology and cytoplasmic extensions (Figure 1A). However, in cultures exposed to increasing concentration of amitriptyline 15-60 μM for 24h a significant percentage of cells rounded up and a reduction of the neuritic processes (Figure 1B) was observed in cells exposed to the higher concentrations 30-60 μM suggesting that cell retraction and detachment is undergoing (Figure 1A-D).
To evaluate whether these morphological changes were associated to or preceded cell death, cell viability was evaluated in SH-SY5Y exposed to amitriptyline for 24, 48 and 72h.
Treatment of cultures with amitriptyline 15-120 μM, but not with 5 μM, induced a concentration and time-dependent reduction of cell viability measured as mitochondrial activity (Figure 1E) and altered cell membrane integrity evaluated by trypan blue exclusion test (Figure 1F).
IC50 was calculated for cell viability at each considered time points and corresponded to 81.03 +2, 59.78 +2, 43.60 +2 μM at 24, 48 and 72h respectively (Figure 1G).
Since alteration of cell adhesion is often associated with apoptosis [53,54] we performed TUNEL assay. As shown in Figure 2, exposure of SH-SY5Y cells to amitriptyline 5, 15 or 30 μM for 24h did not significantly increase the number of TUNEL-positive cells as compared to control cultures, whereas a small but significant population of apoptotic cells was detected in cultures treated with amitriptyline 60 μM (Figure 2A, B). No activation of caspase-3 was observed by western blotting analysis at any of the concentrations and time points analyzed (data not shown).
3.2. Amitriptyline Reduced SH-SY5Y Clonogenic Capacity
Analysis of the images obtained by phase-contrast microscopy showed that, at the seeded density, the surface area covered by growing cells exposed to amitriptyline 5-60 μM for 24, 48 and 72h, was significantly lower as compared to control cultures (Figure 1C, D).
This can be partially due to the cytotoxic effects of the treatment (Figure 1E, F). However, exposure to the tricyclic antidepressant may also alter the proliferation rate of neuroblastoma cells similarly to what has been reported in other cell lines [25,55]. Therefore, we tested the effects of amitriptyline on the growth and proliferation of SH-SY5Y cultures. As shown in Figure 3, amitriptyline 15-60 μM significantly reduced SH-SY5Y clonogenic capacity (i.e. the capability of a single cell to form a colony) in a concentration-dependent manner (Figure 3A, B).
To further investigate the effects of amitriptyline on cell proliferation we performed cell cycle analysis in cultures exposed to increasing concentration of amitriptyline (5-60 μM) for 24, 48 and 72h (Figure 3C). Significant changes in cell cycle phases distribution, with increase of S and G1 phases were reported only when cultures were exposed to amitriptyline 60 μM. Lower concentrations of the antidepressant did not affect the distribution of cell cycle phases at any of the analyzed time points (5-30 μM) (Figure 3C). No significant changes were reported in the value of hypodiploid SubG1 phase (data not shown).
3.3. Autophagy Is Modulated in SH-SY5Y Cell Cultures Exposed to Amitriptyline
It has been previously reported that tricyclic antidepressants affect autophagy pathway in several immortalized and primary cells [41,46,56,57,58,59]. To investigate if modulation of autophagy also occurs in SH-SY5Y cells exposed to amitriptyline, we studied the expression of the microtubule-associated protein 1 light-chain (LC3). LC3I is the soluble cytoplasmic form of the protein which, once converted in LC3II by lipidation, is stably associated with autophagosomes (i.e. the vesicular double membrane structures where the autophagic cargo is loaded) [60].
Immunofluorescence analysis showed that, following exposure to increasing concentration of amitriptyline for 24h, LC3 immunoreactivity is upregulated in SH-SY5Y cultures as compared to control cultures, with redistribution of the fluorescence signal in bright dots localized at the perinuclear region (FIGURE 4A). Most of these LC3-positive punctua colocalized with the lysosomal associated membrane protein 1 (LAMP1), indicating that fusion of autophagosomes with lysosomes occurred (Figure 4A).
Western blotting analysis confirmed that, 24h exposure to amitriptyline 15-60 μM significantly induced a concentration-dependent accumulation of LC3II (Figure 5 A, B). This effect was maintained at 48 and 72h but only when cultures were exposed to amitriptyline 30 or 60 μM (Figure 5 A, C, D). Accumulation of LC3II was absent at lower concentration of amitriptyline (5 μM) at all considered time points (Figure 5).
Upregulation of LC3II was associated, after 72h incubation, with a concentration dependent accumulation of the autophagy substrate and receptor p62/SQSTM-1, while no significant changes were reported at shorter time points (Figure 5). Nevertheless, 24h exposure to amitriptyline 15-60 μM led to a heterogeneous distribution of p62 immunoreactivity with the appearance of p62 bodies partially colocalizing with LAMP1 positive compartments (Figure 4B).
This would suggest that, at this time point, p62/SQSTM-1 is efficiently recruited into the lysosomal compartment and, together with the upregulation of LC3II and its colocalization with LAMP1, would indicate that enhancement of functional autophagy is triggered by the TCA.
However, the delayed accumulation of p62 might imply that, after 72h exposure, the efficiency of autophagosome clearance is impaired or insufficient in amitriptyline treated cells.
To confirm this hypothesis, we performed autophagic flux assay by studying the effect of amitriptyline in the presence and absence of the lysosomal inhibitor BafA1, which prevents lysosomal acidification and blocks the fusion between autophagosomes and lysosomes [61].
When lysosomal activity was inhibited by BafA1, a significant increase of LC3II was still reported in cultures exposed to amitriptyline for 24 (Figure 6A) or 72h (Figure 6B) as compared to vehicle treated cells supporting the hypothesis that the rate of autophagosomal formation was enhanced by the antidepressant.
3.4. Amitriptyline Does Not Affect Lysosomal pH but Induces Lysosomes Accumulation
Lysosomes are highly dynamic organelles serving as degradation hubs for autophagy. Based on its cationic amphiphilic properties, off target effects of amitriptyline could be linked to its accumulation within lysosomes [62,63,64].
To investigate the effects of amitriptyline on the lysosomal arm of the autophagy pathway we used the fluorescent dye Lysotracker Red (LTR), an acidotropic probe that is trapped and therefore labels cellular acidic compartments, including lysosomes and autolysosomes [65]. As expected, BafA1, which acts as a specific inhibitor of vacuolar-type H+-ATPase [66] and was here used as positive control, reduced LTR intensity compared to control cultures (Figure 7A). On the contrary, a concentration-dependent increase of LTR intensity was reported after 24h exposure to amitriptyline suggesting that no changes in acidic lysosomal pH were occurring at any of the tested antidepressant concentrations (Figure 7A).
The staining with the fluorescent dye also showed a concentration-dependent increase of the cytoplasmic area occupied by acidic compartments in cells exposed to amitriptyline 5-60 μM for 24h (Figure 7A).
This observation was supported by LAMP1 western blotting analyses. Indeed, a concentration-dependent increase of LAMP1 expression was detected after 24h of treatment with the antidepressant at the concentration of 15-60 μM (Figure 7B).
3.5. Autophagy Modulation Does Not Take Part to the Cytotoxic Effects of Amitriptyline
To investigate if modulation of autophagy was involved in the reduction of cell viability and clonogenic capacity induced by amitriptyline, cells were preincubated with chloroquine (CQ), an autophagy inhibitor that prevents autophagosome-lysosome fusion and blocks the degradative activity of lysosomes [66].
As shown in Figure 8, CQ itself significantly reduced cell viability in control cultures and further potentiated the effect of amitriptyline after 72h incubation (Figure 8B). No significant effects due to autophagy inhibition were reported in cultures exposed to amitriptyline 5-30 μM for 24h, while a further reduction of cell viability was detected at the highest concentration tested (60 μM) (Figure 8A).
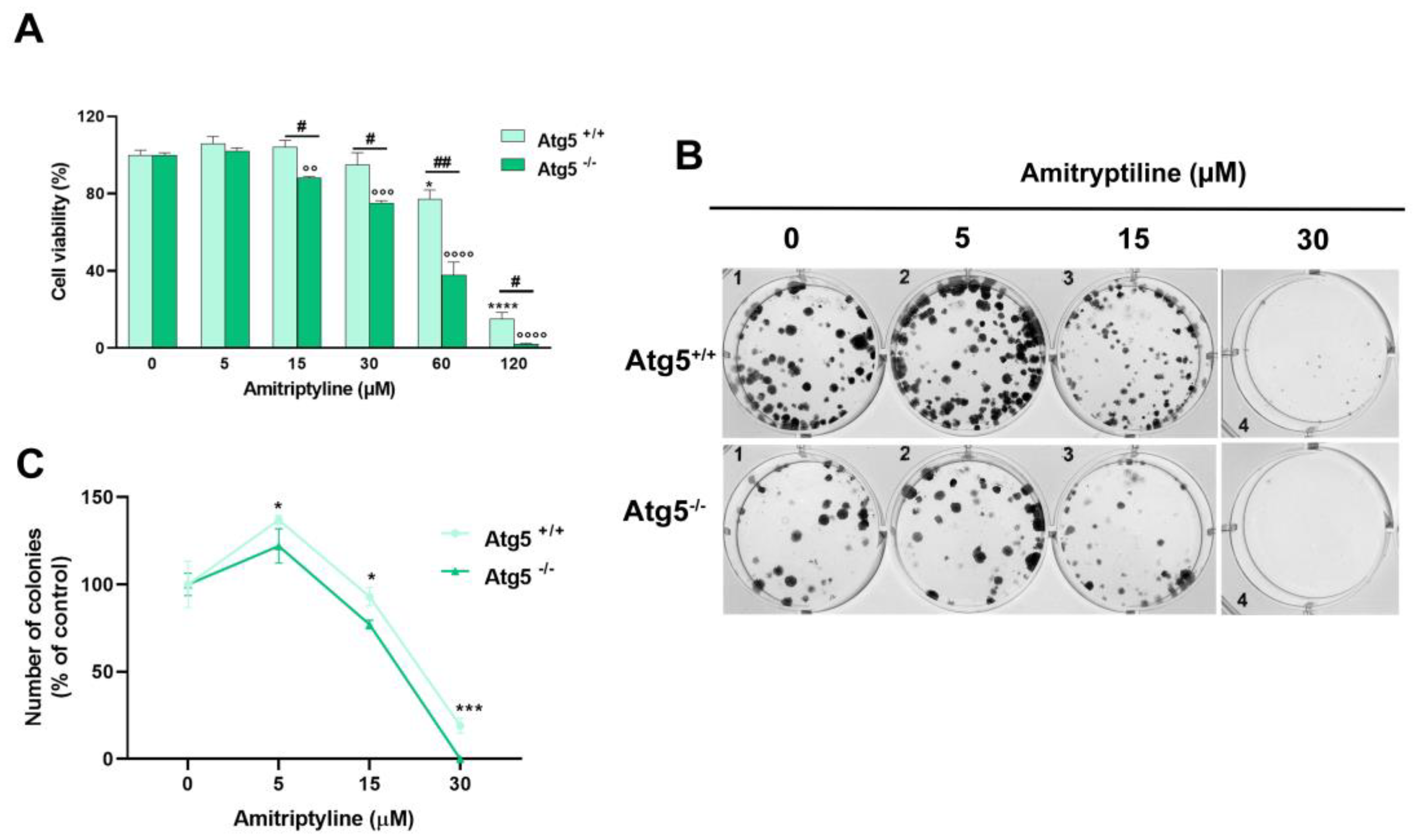
To strengthened the observation that modulation of autophagy is not the key mechanism responsible for the cytotoxic effect of amitriptyline but rather a cytoprotective pathway triggered by the exposure to the antidepressant, we evaluated the effect of amitriptyline in autophagy deficient Atg5 -/- MEF. As reported in Figure 9A, autophagy deficiency drastically reduced cell viability in cells exposed to amitriptyline 15-120 μM compared to wild type Atg5 +/+ MEF. No effect on viability was reported in both autophagy deficient and wild type MEF treated with lower concentration of amitriptyline (5 μM) (Figure 9A).
3.6. Reduced Clonogenic Capacity in Amitriptyline Treated Cells Does Not Depend on Autophagy Modulation
To investigate the relation between the modulation of autophagy and the reduced clonogenic capacity in amitriptyline treated neuroblastoma cells we used autophagy deficient MEF. However as shown in Figure 9B the deletion of Atg5 gene in Atg5 -/- MEF significantly altered the morphology and distribution of colonies as compared to Atg5 +/+ MEF making the results following the treatment not comparable (Figure 9 B, C).
Then the capacity of amitriptyline to suppress colony formation was evaluated in SH-SY5Y pretreated with the autophagy inhibitor CQ. Inhibition of autophagy significantly reduced the number of colonies formed after 12 days of exposure to amitriptyline 15 and 30 μM (Figure 8C) suggesting that autophagy does not cause, but rather buffers, the effect of amitriptyline on cell proliferation. This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
4. Discussion
Autophagy is an evolutionary conserved catabolic pathway that ensures organelle and protein homeostasis through their lysosomal degradation [40]. This process represents a critical cellular response to either physiological and pathological stimuli and its alteration, or inherited mutation on autophagy related genes (Atg), have been linked to several human diseases, including neurodegenerative diseases and cancer [67,68,69,70]. Several common drugs have been found to be able to modulate autophagy with molecular mechanisms that have been only partially identified and are often independent from their main pharmacological targets [71,72,73]. This implies the opportunity to repurpose those drugs in diseases with known alteration of the autophagy pathway and, on the other hand, rises questions on the possibility that some therapeutic treatments may affects the progression of co-existing diseases in which autophagy plays a relevant role.
In the present study we showed that, in SH-SY5Y neuroblastoma cells, amitriptyline, a TCA drug prescribed for depressive syndromes and pain, modulates autophagy in a time and dose-dependent manner with upregulation of the autophagosome-associated form of LC3, LC3II, and a delayed accumulation of the autophagy receptor/substrate p62. The effects on autophagy were not involved in the decreased cell viability and clonogenic capacity observed following exposure of neuroblastoma cultures to the TCA, since genetic and pharmacological inhibition of autophagy did not prevent, but rather increase, amitriptyline cytotoxicity.
Recent studies demonstrated that diverse antidepressant drugs have inherited anticancer activity; in particular TCAs are cytotoxic to several cancer cell lines in vitro [74,75,76] and are able to reverse multidrug resistance of tumor cells in vitro and in tumor bearing mice [56,77,78,79].
Amitriptyline has been shown to exert, through different molecular mechanisms, antitumor effects in several type of cancers including colon carcinoma, lung cancer, breast cancer, glioblastoma, multiple myeloma, melanoma and hepatocarcinoma [80,81,82,83].
Apoptotic cell death was induced by amitriptyline in human multiple myeloma cell lines and primary cells by decreasing histone deacetylases (HDACs) (HDAC-3, 6, 7, 8) expression and inhibiting HDAC activity; furthermore, reduction of cyclin D2 arrested cells in the G0/G1 phase of the cell cycle [84]. Cell viability and proliferation of uterine leiomyosarcoma cell were suppressed by amitriptyline treatment and apoptosis induction was mediated by the upregulation of the non-selective neurotrophin receptor (NTR) p75NTR [85]. In lung cancer cells amitriptyline activated TRAIL (tumor necrosis factor-related apoptosis-inducing ligand)-induced apoptosis by increasing death receptor (DR) 4 and 5 [86]. Decreased tumor cell proliferation though the reduction of Ki-67 and inhibition of -catenin [87] was reported in hepatocellular carcinoma cells treated with amitriptyline, while in glioblastoma multiforme (GBM) amitriptyline induced cell death by interfering with mitochondrial function [88].
Here we observed a dose and time-dependent cytotoxic effect of amitriptyline in human neuroblastoma cells reporting a reduction of cell viability and cell proliferation. However, under our experimental conditions we didn’t detect a direct and quantitively reasonable correlation with apoptotic hallmarks, such as DNA fragmentation, caspase-3 activation and increase of hypodiploid events. Therefore, it can be hypothesized that other forms of cell death are activated in neuroblastoma cells following the exposure to the TCA.
This is supported by the work of Lee and colleagues which reported that, in SH-SY5Y cells, amitriptyline and desipramine induced cell death, but not apoptosis [47]. Indeed, cell death, as well as mitochondrial damage and oxidative stress induced by the TCA, was attenuated by antioxidant but not by inhibitors of caspases, Parp-1, cathepsin or calpains suggesting that it was different from conventional apoptosis or programmed necrosis [47]. Accordingly, caspase-independent cell death following exposure to amitriptyline has been reported in hepatoma HepG2 cells [89]. In this same study an early activation of autophagy, with increase of LC3II, Beclin-1 and Atg12-Atg5, and upregulation of LAMP1, was reported, suggesting that autophagy activation preceded cell death that eventually occurred by necrosis or autophagy-apoptosis switch [89,90].
In our study, we observed an early increase of LC3 lipidation which occurs in the first 24h of exposure to amitriptyline. Autophagy flux experiments showed that amitriptyline is still able to increase LC3II levels even under the presence of the autophagy inhibitor BafA1 implying that treatment with the TCA is associated with autophagy induction rather than inhibition of autophagosomal degradation. This hypothesis is also supported by the increase of LC3 immunofluorescence signal in cells treated with amitriptyline and the colocalization observed between LC3/p62 and p62/LAMP1. However, the delayed accumulation of the autophagy substrate p62 would suggest that the formation of autophagosomes overcame the cell capacity to degrade their content through the lysosomal system therefore limiting autophagosomal turnover.
Basic lipophilic compounds, like amitriptyline and other TCAs, accumulates into acidic intracellular compartment, such as lysosomes, and may perturb vesicular pH therefore disturbing the autophagy process [46,91,92,93,94]. However, in our study, acidic lysosomal pH is not modified by the treatment with amitriptyline as demonstrate by the efficient loading of the acidotropic probe LTR. In agreement with this observation, the study by Kornhuber and colleagues showed that lysosomal pH is not changed by amitriptyline [95]. Nevertheless, the accumulation of amitriptyline into the lysosomal lumen, as lysosomotropic drug, may affect lysosomal membrane permeability [95] and lysosomal enzymes activity accounting for the delay of autophagic cargo degradation [96,97].
Several studies on the cytotoxic effect of TCA have shown that this is often mediated by autophagy dysregulation [55]. However, controversial findings on the influence of amitriptyline on autophagy and its role in the cytotoxicity of the antidepressant among different types of cancer have been reported. For example, in hepatocellular carcinoma cells, amitriptyline induced an early autophagy activation and pharmacological or genetic inhibition of autophagy exacerbates the toxic effects of amitriptyline increasing apoptosis [89]. Viceversa, in lung cancer cells, amitriptyline inhibited autophagy by blocking the fusion of autophagosomes with lysosomes; amitriptyline-induced autophagy blockade increased DR4 and DR5 expression enhancing TRAIL-mediated apoptotic cell death [86].
In our study, inhibition of autophagy by CQ, which prevents the process of autophagosome-lysosome fusion, did not exert significant effects on cell viability when cultures were exposed to amitriptyline for 24h, while exacerbated the cytotoxic effects of the antidepressant after longer exposure and further reduced clonogenic capacity. These data support the hypothesis that, under our experimental conditions, modulation of autophagy is activated by amitriptyline as a cytoprotective mechanism, however the newly formed autophagosomes may not be efficiently degraded through the lysosomal system, either because it is overloaded and above the degradation rate or functionally impaired. This hypothesis is also supported by previous observation showing that when autophagy is inhibited in the step of autophagy induction (i.e. 3-MA treatment) the TCA-induced cell death is not reduced neither aggravated [47] since it occurs before the overloading of the lysosomal degradation system.
In summary our study shows that amitriptyline exerts cytotoxic and antiproliferative effects on neuroblastoma cells while modulating autophagy. The induction of TCA-mediated autophagy is a protective mechanism and it is not responsible for the observed cytotoxicity. Further studies are need to investigate the molecular mechanisms underlying the reported effects and to translate these observations in vivo. Nevertheless, our study, together with the previously published amount of literature, poses the basis for further investigation on the potential exploitation of these effects for therapeutic interventions and on the consequences of long-term antidepressant treatment.
Author Contributions
Conceptualization, R.R. and A.A.; methodology, A.A., M.L.L., A.S., E.L. and R.R.; data curation: A.A. and R.R.; supervision: R.R.; visualization: G.B. and M.T.C.; formal analysis, R.R. and A.A.; writing original draft preparation, R.R. and A.A.; writing review and editing, R.R., A.A., A.S., M.L.L., E.L., G.B. and M.T.C.
Funding
This research received no external funding.
Acknowledgments
We thank Dr Francesca Giordano for skillful technical support with FACS experiments.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Friedrich, M.J. Depression Is the Leading Cause of Disability Around the World. Jama 2017, 317, 1517. [Google Scholar] [CrossRef] [PubMed]
- Wan-Fei, K.; Hassan, S.T.S.; Sann, L.M.; Ismail, S.I.F.; Raman, R.A.; Ibrahim, F. Depression, anxiety and quality of life in stroke survivors and their family caregivers: A pilot study using an actor/partner interdependence model. Electronic physician 2017, 9, 4924–4933. [Google Scholar] [CrossRef] [PubMed]
- Guekht, A. Epilepsy, Comorbidities and Treatments. Current pharmaceutical design 2017, 23, 5702–5726. [Google Scholar] [CrossRef] [PubMed]
- Brozek, P.; Brachmanska, M.; Rabiczko, K.; Bulska, W.; Ciulkowicz, M.; Krzystanek, E. Depression, sleep disturbances and anxiety in patients with relapsing-remitting multiple sclerosis: a longitudinal cohort observation. Psychiatria Danubina 2017, 29, 464–468. [Google Scholar] [PubMed]
- Baquero, M.; Martín, N. Depressive symptoms in neurodegenerative diseases. World journal of clinical cases 2015, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.J.; Chan, M.; Bhatti, H.; Halton, M.; Grassi, L.; Johansen, C.; Meader, N. Prevalence of depression, anxiety, and adjustment disorder in oncological, haematological, and palliative-care settings: a meta-analysis of 94 interview-based studies. The Lancet. Oncology 2011, 12, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, B.; Hyphantis, T.N.; Valpione, S.; Perini, G.; Maes, M.; Morris, G.; Kubera, M.; Köhler, C.A.; Fernandes, B.S.; Stubbs, B.; et al. Depression in cancer: The many biobehavioral pathways driving tumor progression. Cancer treatment reviews 2017, 52, 58–70. [Google Scholar] [CrossRef]
- Becker, M.; Weinberger, T.; Chandy, A.; Schmukler, S. Depression During Pregnancy and Postpartum. Current psychiatry reports 2016, 18, 32. [Google Scholar] [CrossRef]
- Martínez-Paredes, J.F.; Jácome-Pérez, N. Depression in Pregnancy. Revista Colombiana de psiquiatria (English ed.) 2019, 48, 58–65. [Google Scholar] [CrossRef]
- Szpunar, M.J.; Malaktaris, A.; Baca, S.A.; Hauger, R.L.; Lang, A.J. Are alterations in estradiol, cortisol, and inflammatory cytokines associated with depression during pregnancy and postpartum? An exploratory study. Brain, behavior, & immunity - health 2021, 16, 100309. [Google Scholar] [CrossRef]
- Comorbid depression in medical diseases. Nature reviews. Disease primers 2020, 6, 70. [CrossRef] [PubMed]
- Karrouri, R.; Hammani, Z.; Benjelloun, R.; Otheman, Y. Major depressive disorder: Validated treatments and future challenges. World journal of clinical cases 2021, 9, 9350–9367. [Google Scholar] [CrossRef] [PubMed]
- Cherrie, M.; Curtis, S.; Baranyi, G.; McTaggart, S.; Cunningham, N.; Licence, K.; Dibben, C.; Bambra, C.; Pearce, J. Use of sequence analysis for classifying individual antidepressant trajectories to monitor population mental health. BMC psychiatry 2020, 20, 551. [Google Scholar] [CrossRef] [PubMed]
- Kleine, R.; Galimov, A.; Hanewinkel, R.; Unger, J.; Sussman, S.; Hansen, J. Impact of the COVID-19 pandemic on young people with and without pre-existing mental health problems. Scientific reports 2023, 13, 6111. [Google Scholar] [CrossRef] [PubMed]
- Ferwana, I.; Varshney, L.R. The impact of COVID-19 lockdowns on mental health patient populations in the United States. Scientific reports 2024, 14, 5689. [Google Scholar] [CrossRef]
- Bonilla-Jaime, H.; Sánchez-Salcedo, J.A.; Estevez-Cabrera, M.M.; Molina-Jiménez, T.; Cortes-Altamirano, J.L.; Alfaro-Rodríguez, A. Depression and Pain: Use of Antidepressants. Current neuropharmacology 2022, 20, 384–402. [Google Scholar] [CrossRef]
- Feighner, J.P. Overview of antidepressants currently used to treat anxiety disorders. The Journal of clinical psychiatry 1999, 60 Suppl 22, 18–22. [Google Scholar]
- Liu, Y.; Xu, X.; Dong, M.; Jia, S.; Wei, Y. Treatment of insomnia with tricyclic antidepressants: a meta-analysis of polysomnographic randomized controlled trials. Sleep medicine 2017, 34, 126–133. [Google Scholar] [CrossRef]
- Burch, R. Antidepressants for Preventive Treatment of Migraine. Current treatment options in neurology 2019, 21, 18. [Google Scholar] [CrossRef]
- Scuteri, D.; Vulnera, M.; Piro, B.; Bossio, R.B.; Morrone, L.A.; Sandrini, G.; Tamburin, S.; Tonin, P.; Bagetta, G.; Corasaniti, M.T. Pattern of treatment of behavioural and psychological symptoms of dementia and pain: evidence on pharmacoutilization from a large real-world sample and from a centre for cognitive disturbances and dementia. European journal of clinical pharmacology 2021, 77, 241–249. [Google Scholar] [CrossRef]
- Pearlstein, T. Selective serotonin reuptake inhibitors for premenstrual dysphoric disorder: the emerging gold standard? Drugs 2002, 62, 1869–1885. [Google Scholar] [CrossRef]
- Maidment, I.D. The use of antidepressants to treat attention deficit hyperactivity disorder in adults. Journal of psychopharmacology (Oxford, England) 2003, 17, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.; Vos, R.; Tack, J. The influence of citalopram on interdigestive gastrointestinal motility in man. Alimentary pharmacology & therapeutics 2010, 32, 289–295. [Google Scholar] [CrossRef] [PubMed]
- van Kerrebroeck, P.; Abrams, P.; Lange, R.; Slack, M.; Wyndaele, J.J.; Yalcin, I.; Bump, R.C. Duloxetine versus placebo in the treatment of European and Canadian women with stress urinary incontinence. BJOG : an international journal of obstetrics and gynaecology 2004, 111, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chang, X.; Huang, Y.; He, D. The application of antidepressant drugs in cancer treatment. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2023, 157, 113985. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yang, X.; Yu, B. Repurposing antidepressants for anticancer drug discovery. Drug discovery today 2022, 27, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Bielecka, A.M.; Obuchowicz, E. Antidepressant drugs as a complementary therapeutic strategy in cancer. Experimental biology and medicine (Maywood, N.J.) 2013, 238, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.B.; Wang, T.T.; Zhu, H.L.; Wang, H.K.; Sun, W.; Zhao, L.F. Vortioxetine induces apoptosis and autophagy of gastric cancer AGS cells via the PI3K/AKT pathway. FEBS open bio 2020, 10, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Qiu, Y.; Yang, L.; Peng, L.; Xia, Z.; Hou, L.N.; Fang, C.; Qi, H.; Chen, H.Z. Desipramine induces apoptosis in rat glioma cells via endoplasmic reticulum stress-dependent CHOP pathway. Journal of neuro-oncology 2011, 101, 41–48. [Google Scholar] [CrossRef]
- Zafir, A.; Ara, A.; Banu, N. Invivo antioxidant status: a putative target of antidepressant action. Progress in neuro-psychopharmacology & biological psychiatry 2009, 33, 220–228. [Google Scholar] [CrossRef]
- Kannen, V.; Hintzsche, H.; Zanette, D.L.; Silva, W.A., Jr.; Garcia, S.B.; Waaga-Gasser, A.M.; Stopper, H. Antiproliferative effects of fluoxetine on colon cancer cells and in a colonic carcinogen mouse model. PloS one 2012, 7, e50043. [Google Scholar] [CrossRef]
- Fang, C.K.; Chen, H.W.; Chiang, I.T.; Chen, C.C.; Liao, J.F.; Su, T.P.; Tung, C.Y.; Uchitomi, Y.; Hwang, J.J. Mirtazapine inhibits tumor growth via immune response and serotonergic system. PloS one 2012, 7, e38886. [Google Scholar] [CrossRef]
- Hamon, M.; Blier, P. Monoamine neurocircuitry in depression and strategies for new treatments. Progress in neuro-psychopharmacology & biological psychiatry 2013, 45, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Thour, A.; Marwaha, R. Amitriptyline. In StatPearls; StatPearls Publishing Copyright © 2024, StatPearls Publishing LLC.: Treasure Island (FL) ineligible companies. Disclosure: Raman Marwaha declares no relevant financial relationships with ineligible companies, 2024. [Google Scholar]
- Dopheide, J.A. Recognizing and treating depression in children and adolescents. American journal of health-system pharmacy : AJHP : official journal of the American Society of Health-System Pharmacists 2006, 63, 233–243. [Google Scholar] [CrossRef]
- Radley, D.C.; Finkelstein, S.N.; Stafford, R.S. Off-label prescribing among office-based physicians. Archives of internal medicine 2006, 166, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, S.; Sloan, G.; Petrie, J.; White, D.; Bradburn, M.; Julious, S.; Rajbhandari, S.; Sharma, S.; Rayman, G.; Gouni, R.; et al. Comparison of amitriptyline supplemented with pregabalin, pregabalin supplemented with amitriptyline, and duloxetine supplemented with pregabalin for the treatment of diabetic peripheral neuropathic pain (OPTION-DM): a multicentre, double-blind, randomised crossover trial. Lancet (London, England) 2022, 400, 680–690. [Google Scholar] [CrossRef]
- Farag, H.M.; Yunusa, I.; Goswami, H.; Sultan, I.; Doucette, J.A.; Eguale, T. Comparison of Amitriptyline and US Food and Drug Administration-Approved Treatments for Fibromyalgia: A Systematic Review and Network Meta-analysis. JAMA network open 2022, 5, e2212939. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martinez, A.; Guerrero-Peral Á, L.; Arias-Rivas, S.; Silva, L.; Sierra, Á.; Gago-Veiga, A.B.; García-Azorín, D. Amitriptyline for post-COVID headache: effectiveness, tolerability, and response predictors. Journal of neurology 2022, 269, 5702–5709. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: process and function. Genes & development 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Rein, T. Is Autophagy Involved in the Diverse Effects of Antidepressants? Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhuang, X.; Zhang, L.; Qiao, T. Antidepressants Fluoxetine Mediates Endoplasmic Reticulum Stress and Autophagy of Non-Small Cell Lung Cancer Cells Through the ATF4-AKT-mTOR Signaling Pathway. Frontiers in pharmacology 2022, 13, 904701. [Google Scholar] [CrossRef] [PubMed]
- Chinnapaka, S.; Bakthavachalam, V.; Munirathinam, G. Repurposing antidepressant sertraline as a pharmacological drug to target prostate cancer stem cells: dual activation of apoptosis and autophagy signaling by deregulating redox balance. American journal of cancer research 2020, 10, 2043–2065. [Google Scholar]
- Hwang, H.Y.; Shim, J.S.; Kim, D.; Kwon, H.J. Antidepressant drug sertraline modulates AMPK-MTOR signaling-mediated autophagy via targeting mitochondrial VDAC1 protein. Autophagy 2021, 17, 2783–2799. [Google Scholar] [CrossRef]
- Cavaliere, F.; Fornarelli, A.; Bertan, F.; Russo, R.; Marsal-Cots, A.; Morrone, L.A.; Adornetto, A.; Corasaniti, M.T.; Bano, D.; Bagetta, G.; et al. The tricyclic antidepressant clomipramine inhibits neuronal autophagic flux. Scientific reports 2019, 9, 4881. [Google Scholar] [CrossRef]
- Kwon, Y.; Bang, Y.; Moon, S.H.; Kim, A.; Choi, H.J. Amitriptyline interferes with autophagy-mediated clearance of protein aggregates via inhibiting autophagosome maturation in neuronal cells. Cell death & disease 2020, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Hong, S.; Kim, N.; Shin, K.S.; Kang, S.J. Tricyclic Antidepressants Amitriptyline and Desipramine Induced Neurotoxicity Associated with Parkinson's Disease. Molecules and cells 2015, 38, 734–740. [Google Scholar] [CrossRef]
- Gentile, D.; Berliocchi, L.; Russo, R.; Bagetta, G.; Corasaniti, M.T. Effects of the autophagy modulators d-limonene and chloroquine on vimentin levels in SH-SY5Y cells. Biochemical and biophysical research communications 2020, 533, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Cassiano, M.G.; Ciociaro, A.; Adornetto, A.; Varano, G.P.; Chiappini, C.; Berliocchi, L.; Tassorelli, C.; Bagetta, G.; Corasaniti, M.T. Role of D-Limonene in autophagy induced by bergamot essential oil in SH-SY5Y neuroblastoma cells. PloS one 2014, 9, e113682. [Google Scholar] [CrossRef]
- Corasaniti, M.T.; Bilotta, A.; Strongoli, M.C.; Navarra, M.; Bagetta, G.; Di Renzo, G. HIV-1 coat protein gp120 stimulates interleukin-1beta secretion from human neuroblastoma cells: evidence for a role in the mechanism of cell death. British journal of pharmacology 2001, 134, 1344–1350. [Google Scholar] [CrossRef]
- Yordanov, Y.I. Hep G2 cell culture confluence measurement in phase-contrast micrographs - a user-friendly, open-source software-based approach. Toxicology mechanisms and methods 2020, 30, 146–152. [Google Scholar] [CrossRef]
- Pemberton, K.; Mersman, B.; Xu, F. Using ImageJ to Assess Neurite Outgrowth in Mammalian Cell Cultures: Research Data Quantification Exercises in Undergraduate Neuroscience Lab. Journal of undergraduate neuroscience education : JUNE : a publication of FUN, Faculty for Undergraduate Neuroscience 2018, 16, A186–a194. [Google Scholar] [PubMed]
- Su, J.M.; Wang, L.Y.; Liang, Y.L.; Zha, X.L. Role of cell adhesion signal molecules in hepatocellular carcinoma cell apoptosis. World journal of gastroenterology 2005, 11, 4667–4673. [Google Scholar] [CrossRef]
- Suzanne, M.; Steller, H. Letting go: modification of cell adhesion during apoptosis. Journal of biology 2009, 8, 49. [Google Scholar] [CrossRef]
- He, L.; Fu, Y.; Tian, Y.; Wang, X.; Zhou, X.; Ding, R.B.; Qi, X.; Bao, J. Antidepressants as Autophagy Modulators for Cancer Therapy. Molecules (Basel, Switzerland) 2023, 28. [Google Scholar] [CrossRef]
- Rossi, M.; Munarriz, E.R.; Bartesaghi, S.; Milanese, M.; Dinsdale, D.; Guerra-Martin, M.A.; Bampton, E.T.; Glynn, P.; Bonanno, G.; Knight, R.A.; et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. Journal of cell science 2009, 122, 3330–3339. [Google Scholar] [CrossRef]
- Gassen, N.C.; Hartmann, J.; Schmidt, M.V.; Rein, T. FKBP5/FKBP51 enhances autophagy to synergize with antidepressant action. Autophagy 2015, 11, 578–580. [Google Scholar] [CrossRef]
- Zschocke, J.; Zimmermann, N.; Berning, B.; Ganal, V.; Holsboer, F.; Rein, T. Antidepressant drugs diversely affect autophagy pathways in astrocytes and neurons--dissociation from cholesterol homeostasis. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2011, 36, 1754–1768. [Google Scholar] [CrossRef]
- Zschocke, J.; Rein, T. Antidepressants encounter autophagy in neural cells. Autophagy 2011, 7, 1247–1248. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. The international journal of biochemistry & cell biology 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Funk, R.S.; Krise, J.P. Cationic amphiphilic drugs cause a marked expansion of apparent lysosomal volume: implications for an intracellular distribution-based drug interaction. Molecular pharmaceutics 2012, 9, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, I.; Clemmensen, K.K.B.; Fogde, D.L.; Dietrich, T.N.; Giacobini, J.D.; Bilgin, M.; Jäättelä, M.; Maeda, K. Cationic amphiphilic drugs induce accumulation of cytolytic lysoglycerophospholipids in the lysosomes of cancer cells and block their recycling into common membrane glycerophospholipids. Molecular biology of the cell 2024, 35, ar25. [Google Scholar] [CrossRef] [PubMed]
- Vater, M.; Möckl, L.; Gormanns, V.; Schultz Fademrecht, C.; Mallmann, A.M.; Ziegart-Sadowska, K.; Zaba, M.; Frevert, M.L.; Bräuchle, C.; Holsboer, F.; et al. New insights into the intracellular distribution pattern of cationic amphiphilic drugs. Scientific reports 2017, 7, 44277. [Google Scholar] [CrossRef] [PubMed]
- Chazotte, B. Labeling lysosomes in live cells with LysoTracker. Cold Spring Harbor protocols 2011, 2011, pdbprot5571. [Google Scholar] [CrossRef]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. The Journal of biological chemistry 1991, 266, 17707–17712. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. The Journal of clinical investigation 2015, 125, 65–74. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. International journal of molecular sciences 2018, 19. [Google Scholar] [CrossRef]
- Adornetto, A.; Parisi, V.; Morrone, L.A.; Corasaniti, M.T.; Bagetta, G.; Tonin, P.; Russo, R. The Role of Autophagy in Glaucomatous Optic Neuropathy. Frontiers in cell and developmental biology 2020, 8, 121. [Google Scholar] [CrossRef]
- Berliocchi, L.; Maiarù, M.; Varano, G.P.; Russo, R.; Corasaniti, M.T.; Bagetta, G.; Tassorelli, C. Spinal autophagy is differently modulated in distinct mouse models of neuropathic pain. Molecular pain 2015, 11, 3. [Google Scholar] [CrossRef]
- Shi, Q.; Pei, F.; Silverman, G.A.; Pak, S.C.; Perlmutter, D.H.; Liu, B.; Bahar, I. Mechanisms of Action of Autophagy Modulators Dissected by Quantitative Systems Pharmacology Analysis. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Li, X.; Xu, H.L.; Liu, Y.X.; An, N.; Zhao, S.; Bao, J.K. Autophagy modulation as a target for anticancer drug discovery. Acta pharmacologica Sinica 2013, 34, 612–624. [Google Scholar] [CrossRef]
- Gao, L.; Jauregui, C.E.; Teng, Y. Targeting autophagy as a strategy for drug discovery and therapeutic modulation. Future medicinal chemistry 2017, 9, 335–345. [Google Scholar] [CrossRef]
- Arimochi, H.; Morita, K. Characterization of cytotoxic actions of tricyclic antidepressants on human HT29 colon carcinoma cells. European journal of pharmacology 2006, 541, 17–23. [Google Scholar] [CrossRef]
- Xia, Z.; Bergstrand, A.; DePierre, J.W.; Nässberger, L. The antidepressants imipramine, clomipramine, and citalopram induce apoptosis in human acute myeloid leukemia HL-60 cells via caspase-3 activation. Journal of biochemical and molecular toxicology 1999, 13, 338–347. [Google Scholar] [CrossRef]
- Levkovitz, Y.; Gil-Ad, I.; Zeldich, E.; Dayag, M.; Weizman, A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. Journal of molecular neuroscience : MN 2005, 27, 29–42. [Google Scholar] [CrossRef]
- Rácz, B.; Spengler, G. Repurposing Antidepressants and Phenothiazine Antipsychotics as Efflux Pump Inhibitors in Cancer and Infectious Diseases. Antibiotics (Basel, Switzerland) 2023, 12. [Google Scholar] [CrossRef]
- Varga, A.; Nugel, H.; Baehr, R.; Marx, U.; Hevér, A.; Nacsa, J.; Ocsovszky, I.; Molnar, J. Reversal of multidrug resistance by amitriptyline in vitro. Anticancer research 1996, 16, 209–211. [Google Scholar]
- O'Brien, F.E.; Dinan, T.G.; Griffin, B.T.; Cryan, J.F. Interactions between antidepressants and P-glycoprotein at the blood-brain barrier: clinical significance of in vitro and in vivo findings. British journal of pharmacology 2012, 165, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Asensi-Cantó, A.; Rodríguez-Braun, E.; Beltrán-Videla, A.; Hurtado, A.M.; Conesa-Zamora, P. Effects of imipramine on cancer patients over-expressing Fascin1; description of the HITCLIF clinical trial. Frontiers in oncology 2023, 13, 1238464. [Google Scholar] [CrossRef]
- Cordero, M.D.; Sánchez-Alcázar, J.A.; Bautista-Ferrufino, M.R.; Carmona-López, M.I.; Illanes, M.; Ríos, M.J.; Garrido-Maraver, J.; Alcudia, A.; Navas, P.; de Miguel, M. Acute oxidant damage promoted on cancer cells by amitriptyline in comparison with some common chemotherapeutic drugs. Anti-cancer drugs 2010, 21, 932–944. [Google Scholar] [CrossRef]
- Motafeghi, F.; Shahsavari, R.; Mortazavi, P.; Shokrzadeh, M. Anticancer effect of paroxetine and amitriptyline on HT29 and A549 cell lines. Toxicology in vitro : an international journal published in association with BIBRA 2023, 87, 105532. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yeh, C.T. Anticancer Effects of Antidepressants in Hepatocellular Carcinoma Cells. Anticancer research 2023, 43, 1201–1206. [Google Scholar] [CrossRef]
- Mao, X.; Hou, T.; Cao, B.; Wang, W.; Li, Z.; Chen, S.; Fei, M.; Hurren, R.; Gronda, M.; Wu, D.; et al. The tricyclic antidepressant amitriptyline inhibits D-cyclin transactivation and induces myeloma cell apoptosis by inhibiting histone deacetylases: in vitro and in silico evidence. Molecular pharmacology 2011, 79, 672–680. [Google Scholar] [CrossRef]
- Pula, G.; Pistilli, A.; Montagnoli, C.; Stabile, A.M.; Rambotti, M.G.; Rende, M. The tricyclic antidepressant amitriptyline is cytotoxic to HTB114 human leiomyosarcoma and induces p75(NTR)-dependent apoptosis. Anti-cancer drugs 2013, 24, 899–910. [Google Scholar] [CrossRef]
- Zinnah, K.M.A.; Park, S.Y. Sensitizing TRAIL-resistant A549 lung cancer cells and enhancing TRAIL-induced apoptosis with the antidepressant amitriptyline. Oncology reports 2021, 46. [Google Scholar] [CrossRef]
- Evason, K.J.; Francisco, M.T.; Juric, V.; Balakrishnan, S.; Lopez Pazmino Mdel, P.; Gordan, J.D.; Kakar, S.; Spitsbergen, J.; Goga, A.; Stainier, D.Y. Identification of Chemical Inhibitors of β-Catenin-Driven Liver Tumorigenesis in Zebrafish. PLoS genetics 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Higgins, S.C.; Pilkington, G.J. The in vitro effects of tricyclic drugs and dexamethasone on cellular respiration of malignant glioma. Anticancer research 2010, 30, 391–397. [Google Scholar]
- Villanueva-Paz, M.; Cordero, M.D.; Pavón, A.D.; Vega, B.C.; Cotán, D.; De la Mata, M.; Oropesa-Ávila, M.; Alcocer-Gomez, E.; de Lavera, I.; Garrido-Maraver, J.; et al. Amitriptyline induces mitophagy that precedes apoptosis in human HepG2 cells. Genes & cancer 2016, 7, 260–277. [Google Scholar] [CrossRef]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. International journal of biological sciences 2014, 10, 1072–1083. [Google Scholar] [CrossRef]
- Ashoor, R.; Yafawi, R.; Jessen, B.; Lu, S. The contribution of lysosomotropism to autophagy perturbation. PloS one 2013, 8, e82481. [Google Scholar] [CrossRef]
- Guan, Y.; Li, X.; Umetani, M.; Boini, K.M.; Li, P.L.; Zhang, Y. Tricyclic antidepressant amitriptyline inhibits autophagic flux and prevents tube formation in vascular endothelial cells. Basic & clinical pharmacology & toxicology 2019, 124, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug metabolism and disposition: the biological fate of chemicals 2013, 41, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Nadanaciva, S.; Lu, S.; Gebhard, D.F.; Jessen, B.A.; Pennie, W.D.; Will, Y. A high content screening assay for identifying lysosomotropic compounds. Toxicology in vitro : an international journal published in association with BIBRA 2011, 25, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Henkel, A.W.; Groemer, T.W.; Städtler, S.; Welzel, O.; Tripal, P.; Rotter, A.; Bleich, S.; Trapp, S. Lipophilic cationic drugs increase the permeability of lysosomal membranes in a cell culture system. Journal of cellular physiology 2010, 224, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell discovery 2020, 6, 6. [Google Scholar] [CrossRef]
- Xu, J.; Gu, J.; Pei, W.; Zhang, Y.; Wang, L.; Gao, J. The role of lysosomal membrane proteins in autophagy and related diseases. The FEBS journal 2023. [CrossRef]
Figure 1.
Effects of amitriptyline on cell morphology and viability of SH-SY5Y neuroblastoma cells. (A) Phase contrast microscopy images (10x objective) showing SH-SY5Y cell cultures morphology under control conditions (0) and following treatment with amitriptyline 5-60 μM for 24, 48 and 72h. (B, C, D) Histograms showing the number of neurites per cell (B) and percentage of covered area (C, D) in SH-SY5Y control cultures (0) and cultures exposed to increasing concentration of amitriptyline 5-60 μM for 24, 48 and 72h. Each bar represents the mean ± s.e.m. (standard error of the mean) of 3-4 independent experiments. (E, F) Concentration dependent cytotoxicity of amitriptyline in SH-SY5Y cell cultures. Cells were treated with amitriptyline 5-120 μM for 24, 48 and 72h and cell viability evaluated by MTT assay (E) or trypan blue assay (F). Histograms represent the mean ± s.e.m. of 3-4 independent experiments. (G) IC50 was calculated for cell viability after 24, 48 and 72h exposure to amitriptyline. (*p<0.05; **p<0.01; ****p<0.0001 vs 0 24h; #p<0.05; ##p<0.01; ###p<0.001; ####p<0.0001 vs 0 48h; °p<0.05; °°°p<0.001; °°°°p<0.0001 vs 0 72h; §§p<0.01; §§§ p<0.001).
Figure 1.
Effects of amitriptyline on cell morphology and viability of SH-SY5Y neuroblastoma cells. (A) Phase contrast microscopy images (10x objective) showing SH-SY5Y cell cultures morphology under control conditions (0) and following treatment with amitriptyline 5-60 μM for 24, 48 and 72h. (B, C, D) Histograms showing the number of neurites per cell (B) and percentage of covered area (C, D) in SH-SY5Y control cultures (0) and cultures exposed to increasing concentration of amitriptyline 5-60 μM for 24, 48 and 72h. Each bar represents the mean ± s.e.m. (standard error of the mean) of 3-4 independent experiments. (E, F) Concentration dependent cytotoxicity of amitriptyline in SH-SY5Y cell cultures. Cells were treated with amitriptyline 5-120 μM for 24, 48 and 72h and cell viability evaluated by MTT assay (E) or trypan blue assay (F). Histograms represent the mean ± s.e.m. of 3-4 independent experiments. (G) IC50 was calculated for cell viability after 24, 48 and 72h exposure to amitriptyline. (*p<0.05; **p<0.01; ****p<0.0001 vs 0 24h; #p<0.05; ##p<0.01; ###p<0.001; ####p<0.0001 vs 0 48h; °p<0.05; °°°p<0.001; °°°°p<0.0001 vs 0 72h; §§p<0.01; §§§ p<0.001).
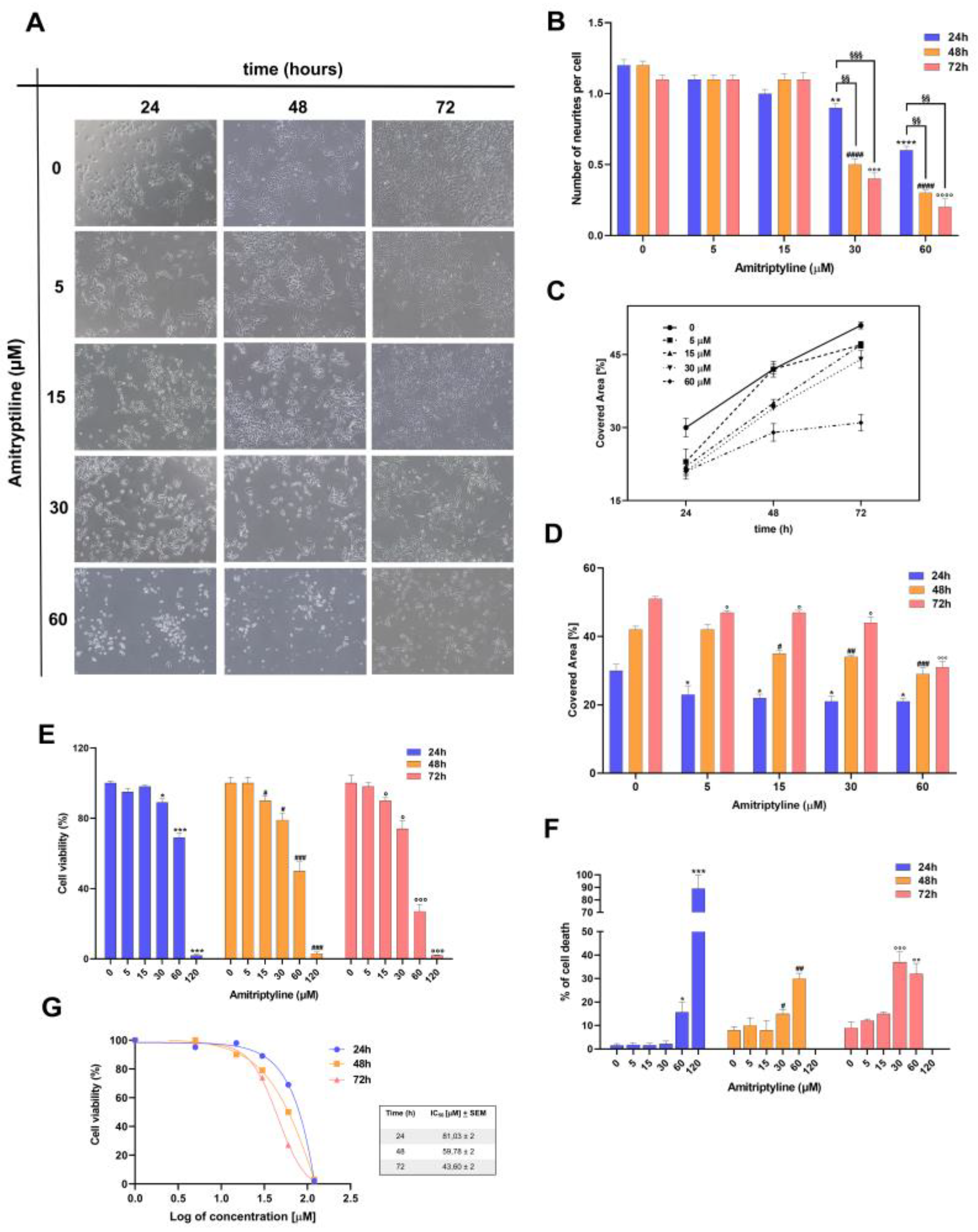
Figure 2.
Analysis of TUNEL-positive cells in SH-SY5Y cultures exposed to amitriptyline for 24h. (A) Representative microphotographs of cells treated with amitriptyline for 24h and stained (green) by TUNEL assay. Cell nuclei were counterstained with DAPI (blue). (B) Apoptotic cell index was calculated as the percentage of TUNEL-positive cells versus total cell counted per microscopic field. 10 fields were acquired for each experimental condition using a confocal microscope equipped with a 20X objective. Data were reported as mean ± s.e.m. of n=5 independent experiments. (***p<0.001 vs control 0).
Figure 2.
Analysis of TUNEL-positive cells in SH-SY5Y cultures exposed to amitriptyline for 24h. (A) Representative microphotographs of cells treated with amitriptyline for 24h and stained (green) by TUNEL assay. Cell nuclei were counterstained with DAPI (blue). (B) Apoptotic cell index was calculated as the percentage of TUNEL-positive cells versus total cell counted per microscopic field. 10 fields were acquired for each experimental condition using a confocal microscope equipped with a 20X objective. Data were reported as mean ± s.e.m. of n=5 independent experiments. (***p<0.001 vs control 0).
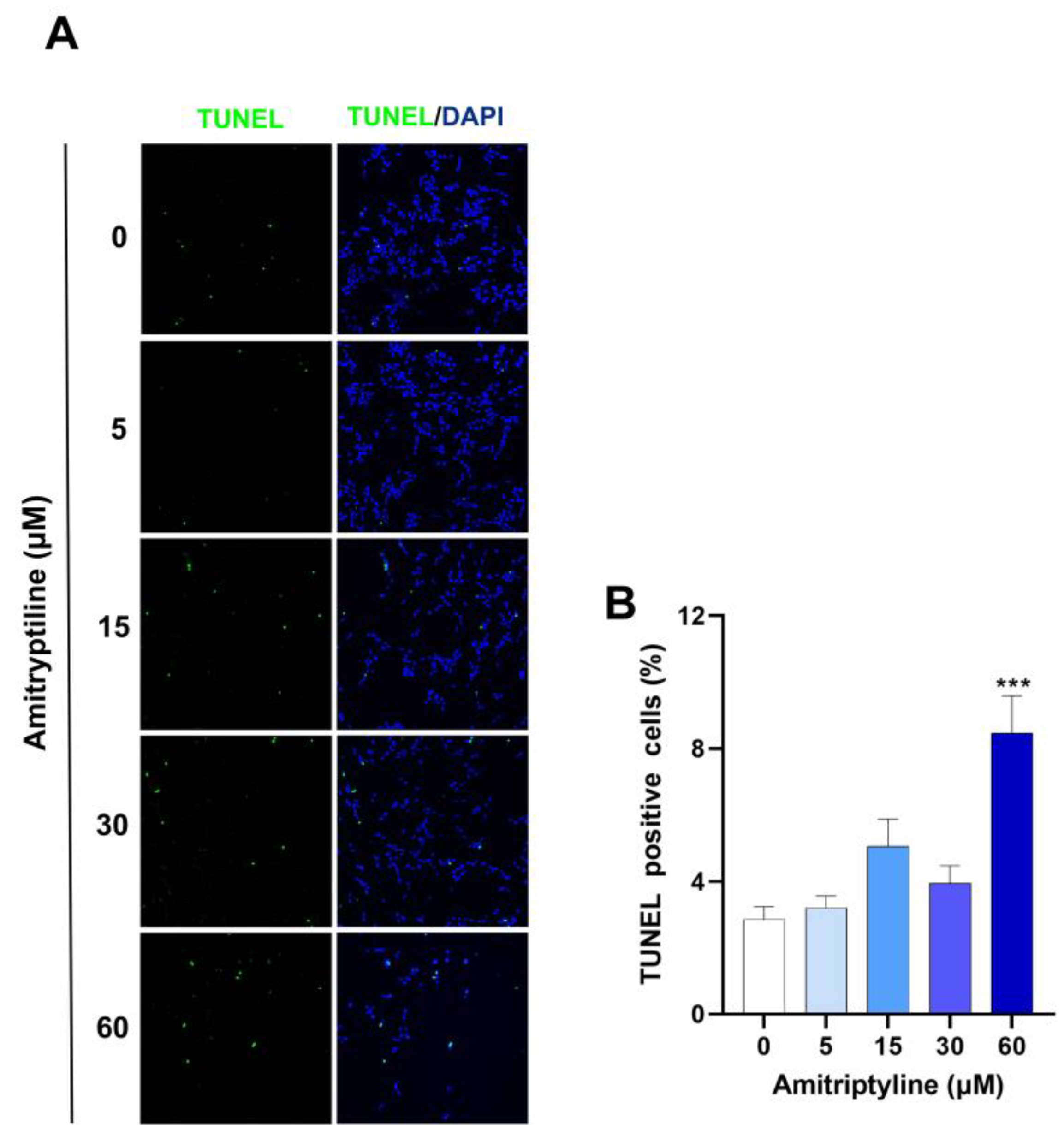
Figure 3.
Amitriptyline reduces clonogenic capacity and affects cell cycle distribution of SH-SY5Y cells. (A) Representative image of a clonogenic assay showing the concentration-dependent inhibition of colony formation induced by incubation with amitriptyline 15-60 μM for 12 days. (B) Quantification of the colonies formed expressed as percentage of the untreated control (0). Results are reported as mean ± s.e.m. of n=5 independent experiments (**p<0.01; ****p<0.0001). (C) Histograms show the distribution of cell cycle phases following exposure to amitriptyline 5-60 μM for 24, 48 and 72h. Results are expressed as mean ± s.e.m. of n=4 independent experiments (*p<0.05; **p<0.01; ***p<0.001 vs untreated control 0).
Figure 3.
Amitriptyline reduces clonogenic capacity and affects cell cycle distribution of SH-SY5Y cells. (A) Representative image of a clonogenic assay showing the concentration-dependent inhibition of colony formation induced by incubation with amitriptyline 15-60 μM for 12 days. (B) Quantification of the colonies formed expressed as percentage of the untreated control (0). Results are reported as mean ± s.e.m. of n=5 independent experiments (**p<0.01; ****p<0.0001). (C) Histograms show the distribution of cell cycle phases following exposure to amitriptyline 5-60 μM for 24, 48 and 72h. Results are expressed as mean ± s.e.m. of n=4 independent experiments (*p<0.05; **p<0.01; ***p<0.001 vs untreated control 0).
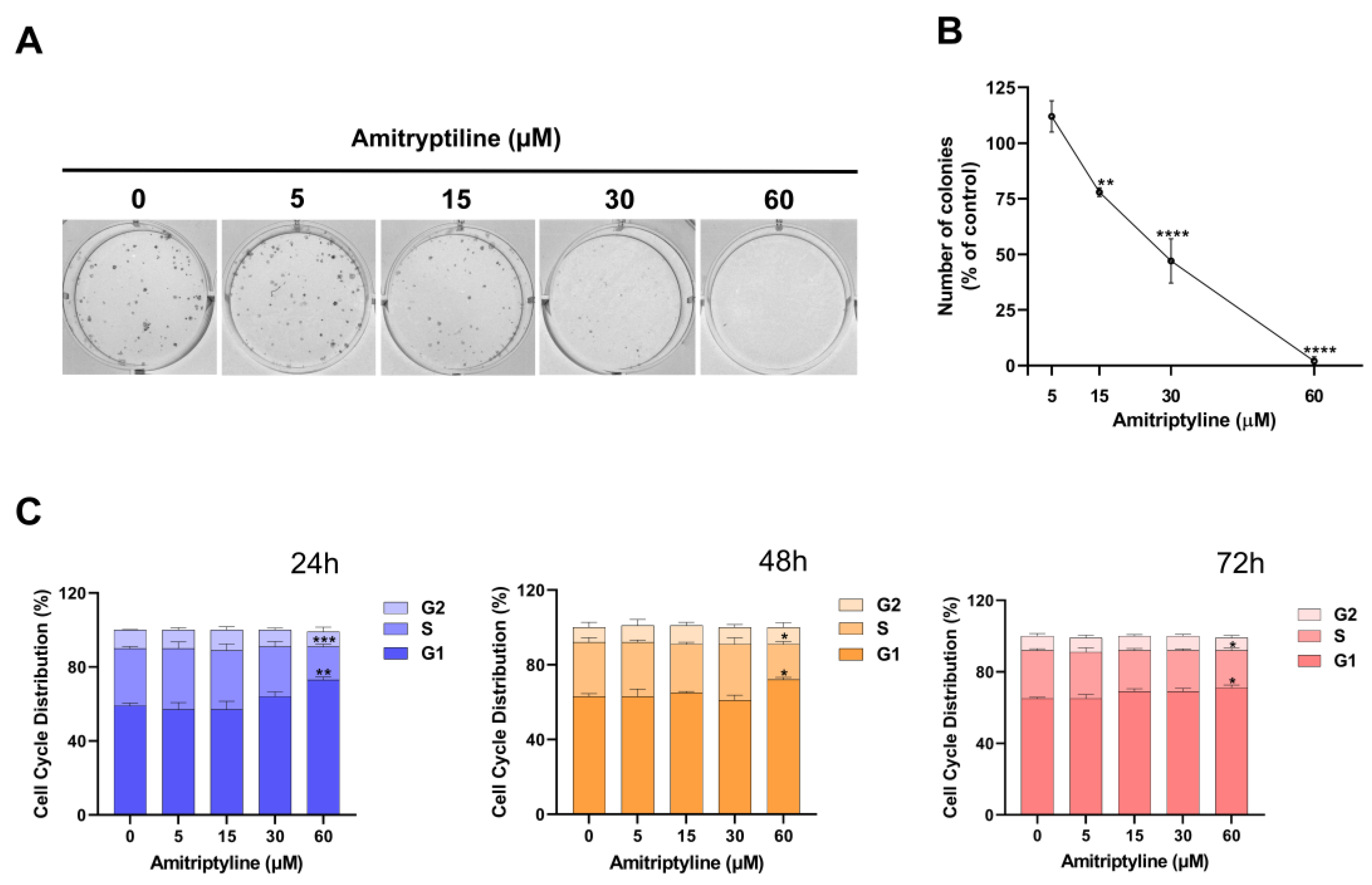
Figure 4.
LC3, p62 and LAMP1 immunoreactivity in SH-SY5Y cells exposed to amitriptyline. (A) Representative confocal images of SH-SY5Y cells labeled with anti-LC3 (green) and anti-LAMP1 (red) antibodies. Following exposure to increasing concentration of amitriptyline 15-60 μM for 24h, LC3 immunoreactivity is upregulated in SH-SY5Y cultures as compared to control cultures and partially co-localizes with LAMP1-positive structures. (B) Images showing the distribution of anti-p62 (green) and LAMP1 (red) immunoreactivity in neuroblastoma cells treated with amitriptyline 15-60 μM for 24h. Nuclei were counterstained with DAPI (blue). n=4. Images were acquired with a confocal microscope equipped with a 20x objective with 2x zoom.
Figure 4.
LC3, p62 and LAMP1 immunoreactivity in SH-SY5Y cells exposed to amitriptyline. (A) Representative confocal images of SH-SY5Y cells labeled with anti-LC3 (green) and anti-LAMP1 (red) antibodies. Following exposure to increasing concentration of amitriptyline 15-60 μM for 24h, LC3 immunoreactivity is upregulated in SH-SY5Y cultures as compared to control cultures and partially co-localizes with LAMP1-positive structures. (B) Images showing the distribution of anti-p62 (green) and LAMP1 (red) immunoreactivity in neuroblastoma cells treated with amitriptyline 15-60 μM for 24h. Nuclei were counterstained with DAPI (blue). n=4. Images were acquired with a confocal microscope equipped with a 20x objective with 2x zoom.
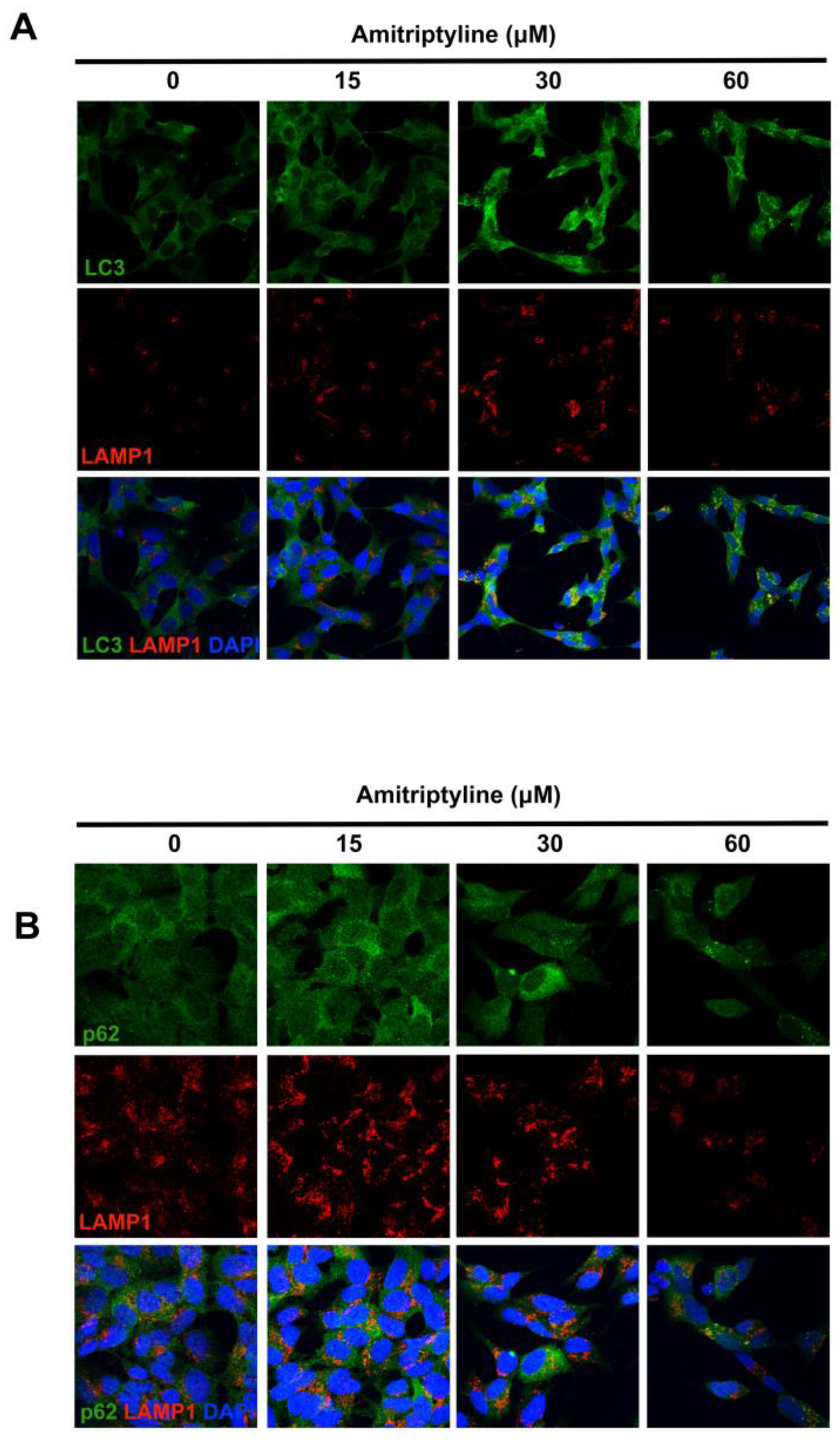
Figure 5.
Amitriptyline modulates LC3 and p62 protein expression in SH-SY5Y neuroblastoma cells. (A) Representative immunoblot showing p62 and LC3 expression in SH-SY5Y cells exposed to amitriptyline 5-60 μM for 24 (B), 48 (C) and 72h (D). Actin was used as loading control. (B, C, D) Histograms report the results of the densitometric analysis of the bands normalized to loading control following treatment with amitriptyline for 24, 48 and 72h respectively. Data are reported as mean ± s.e.m. of n=3-5 independent experiments. (Significance was determined via Student’s t test; *p<0.05, **p<0.01, ***<0.001 vs untreated control 0).
Figure 5.
Amitriptyline modulates LC3 and p62 protein expression in SH-SY5Y neuroblastoma cells. (A) Representative immunoblot showing p62 and LC3 expression in SH-SY5Y cells exposed to amitriptyline 5-60 μM for 24 (B), 48 (C) and 72h (D). Actin was used as loading control. (B, C, D) Histograms report the results of the densitometric analysis of the bands normalized to loading control following treatment with amitriptyline for 24, 48 and 72h respectively. Data are reported as mean ± s.e.m. of n=3-5 independent experiments. (Significance was determined via Student’s t test; *p<0.05, **p<0.01, ***<0.001 vs untreated control 0).
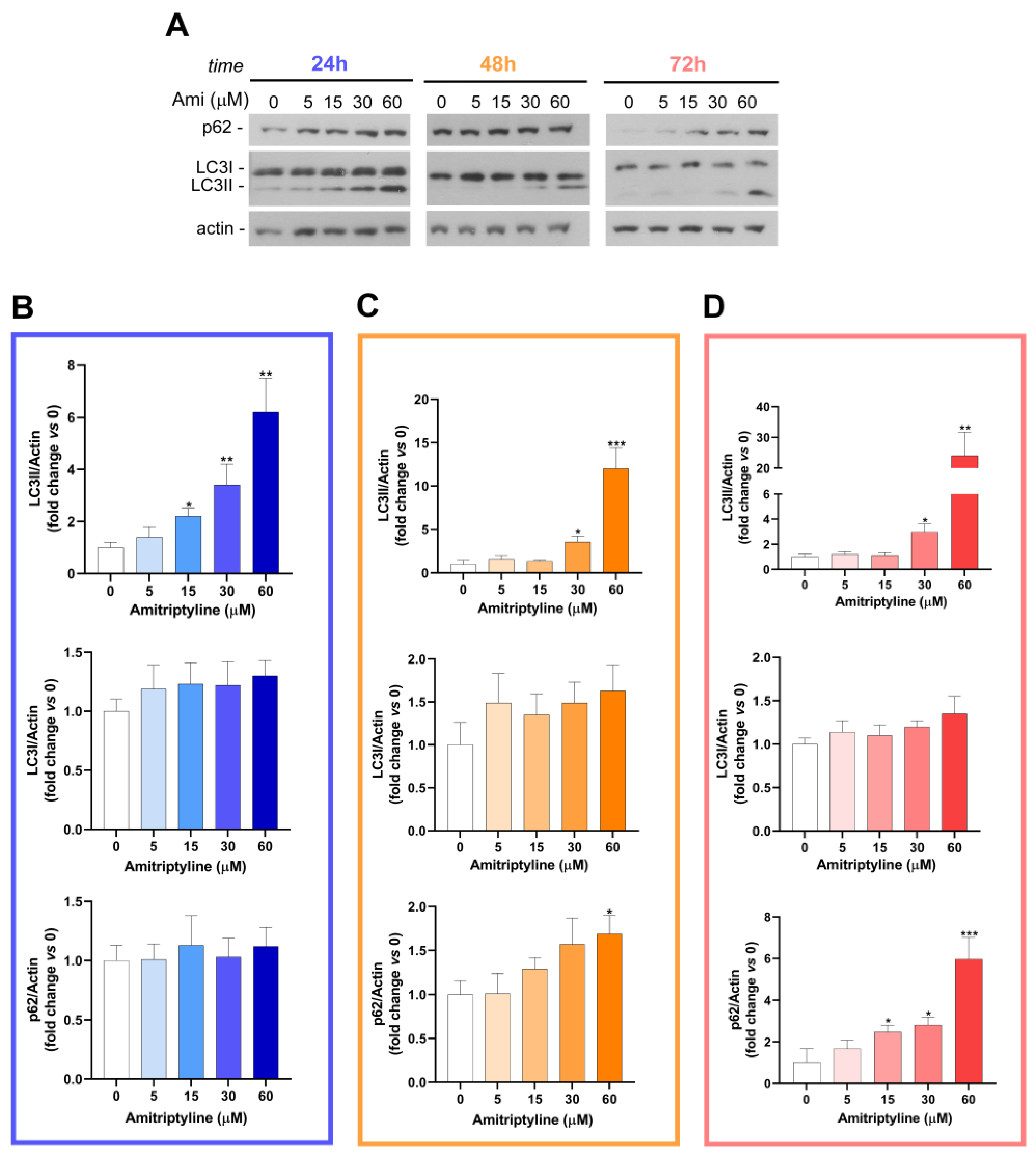
Figure 6.
Amitriptyline increases autophagy flux in SH-SY5Y cells. Representative immunoblot showing the level of LC3 proteins in SH-SY5Y treated with amitriptyline for 24 (A) or 72h (B) and incubated with the lysosomal inhibitor Bafilomycin A1 (+BafA1; 100 μM) or vehicle (-BafA1) for the last 4h of treatment. Actin was used as loading control. Histograms show the LC3II/Actin optical density ratio reported as mean ± s.e.m. of n=3-5 independent experiments. (Significance was determined via Student’s t test; *p<0.05; **p<0.01 vs 0 - BafA1; °p<0.05; °°p<0.01, °°°p<0.001 vs corresponding concentration of amitriptyline -BafA1; #p<0.05; ##p<0.01). (Significance was determined via Student’s t test; *p<0.05, **p<0.01, ***<0.001 vs untreated control 0).
Figure 6.
Amitriptyline increases autophagy flux in SH-SY5Y cells. Representative immunoblot showing the level of LC3 proteins in SH-SY5Y treated with amitriptyline for 24 (A) or 72h (B) and incubated with the lysosomal inhibitor Bafilomycin A1 (+BafA1; 100 μM) or vehicle (-BafA1) for the last 4h of treatment. Actin was used as loading control. Histograms show the LC3II/Actin optical density ratio reported as mean ± s.e.m. of n=3-5 independent experiments. (Significance was determined via Student’s t test; *p<0.05; **p<0.01 vs 0 - BafA1; °p<0.05; °°p<0.01, °°°p<0.001 vs corresponding concentration of amitriptyline -BafA1; #p<0.05; ##p<0.01). (Significance was determined via Student’s t test; *p<0.05, **p<0.01, ***<0.001 vs untreated control 0).
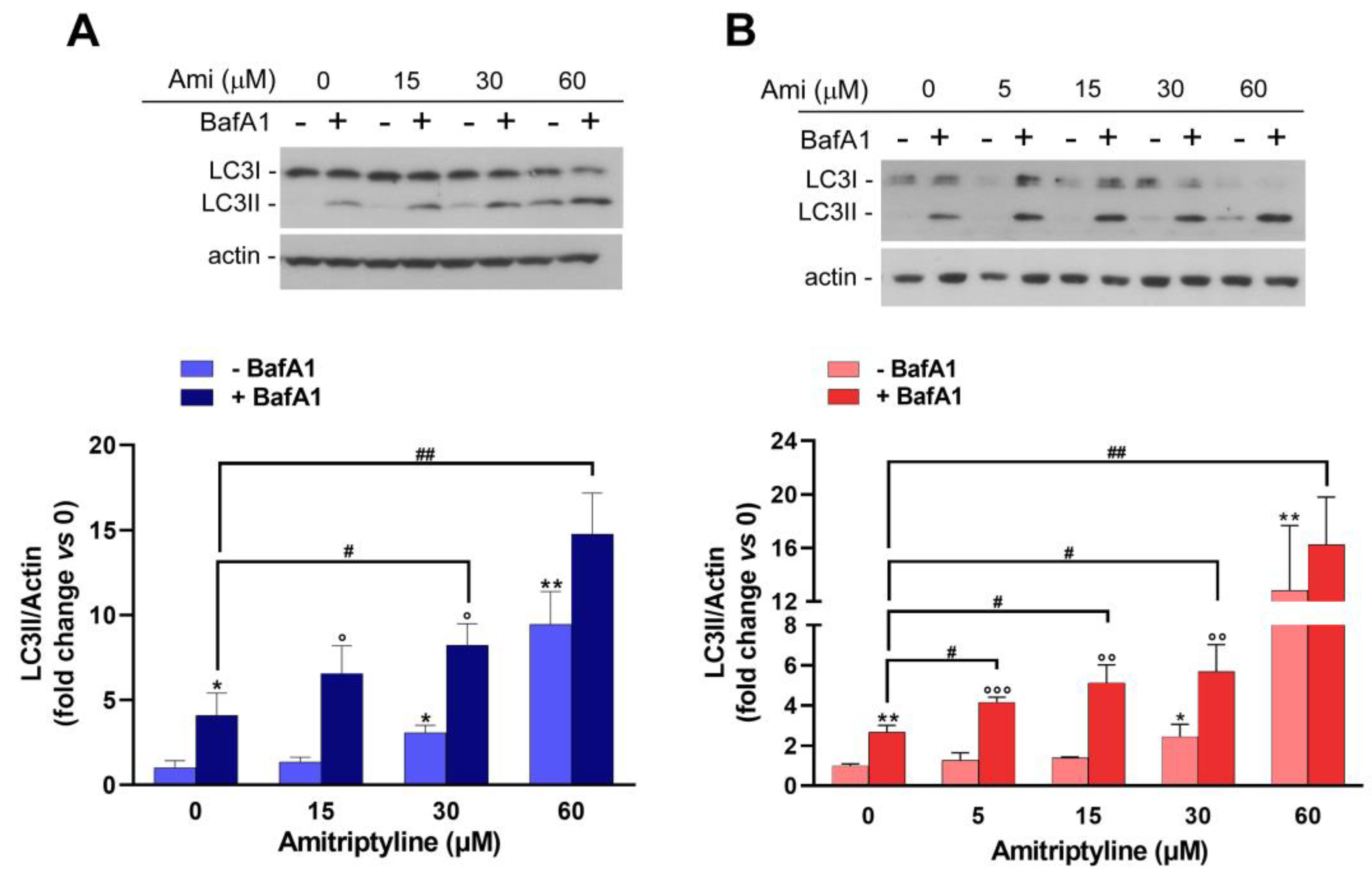
Figure 7.
Effects of amitriptyline on lysosomal pH and LAMP1 expression in SH-SY5Y cells. (A) Representative images of Lysotracker Red (LTR) staining of SH-SY5Y cells exposed to amitriptyline 5-60μM for 24h. BafA1 (100 μM) is used as positive control to reduce lysosomal pH. A concentration-dependent increase of LTR intensity is reported after 24h exposure to AMI. Images were acquired with a confocal microscope equipped with a 20x objective. (B) Western analysis showing the dose-dependent increase of LAMP1 expression in SH-SY5Y cells treated with amitriptyline 5-60 μM for 24h. Histograms show the densitometric analysis of the bands normalized to loading control (actin) and reported as mean ± s.e.m. of n=4 independent experiments. (Significance was determined via Student’s t test; *p<0.05).
Figure 7.
Effects of amitriptyline on lysosomal pH and LAMP1 expression in SH-SY5Y cells. (A) Representative images of Lysotracker Red (LTR) staining of SH-SY5Y cells exposed to amitriptyline 5-60μM for 24h. BafA1 (100 μM) is used as positive control to reduce lysosomal pH. A concentration-dependent increase of LTR intensity is reported after 24h exposure to AMI. Images were acquired with a confocal microscope equipped with a 20x objective. (B) Western analysis showing the dose-dependent increase of LAMP1 expression in SH-SY5Y cells treated with amitriptyline 5-60 μM for 24h. Histograms show the densitometric analysis of the bands normalized to loading control (actin) and reported as mean ± s.e.m. of n=4 independent experiments. (Significance was determined via Student’s t test; *p<0.05).
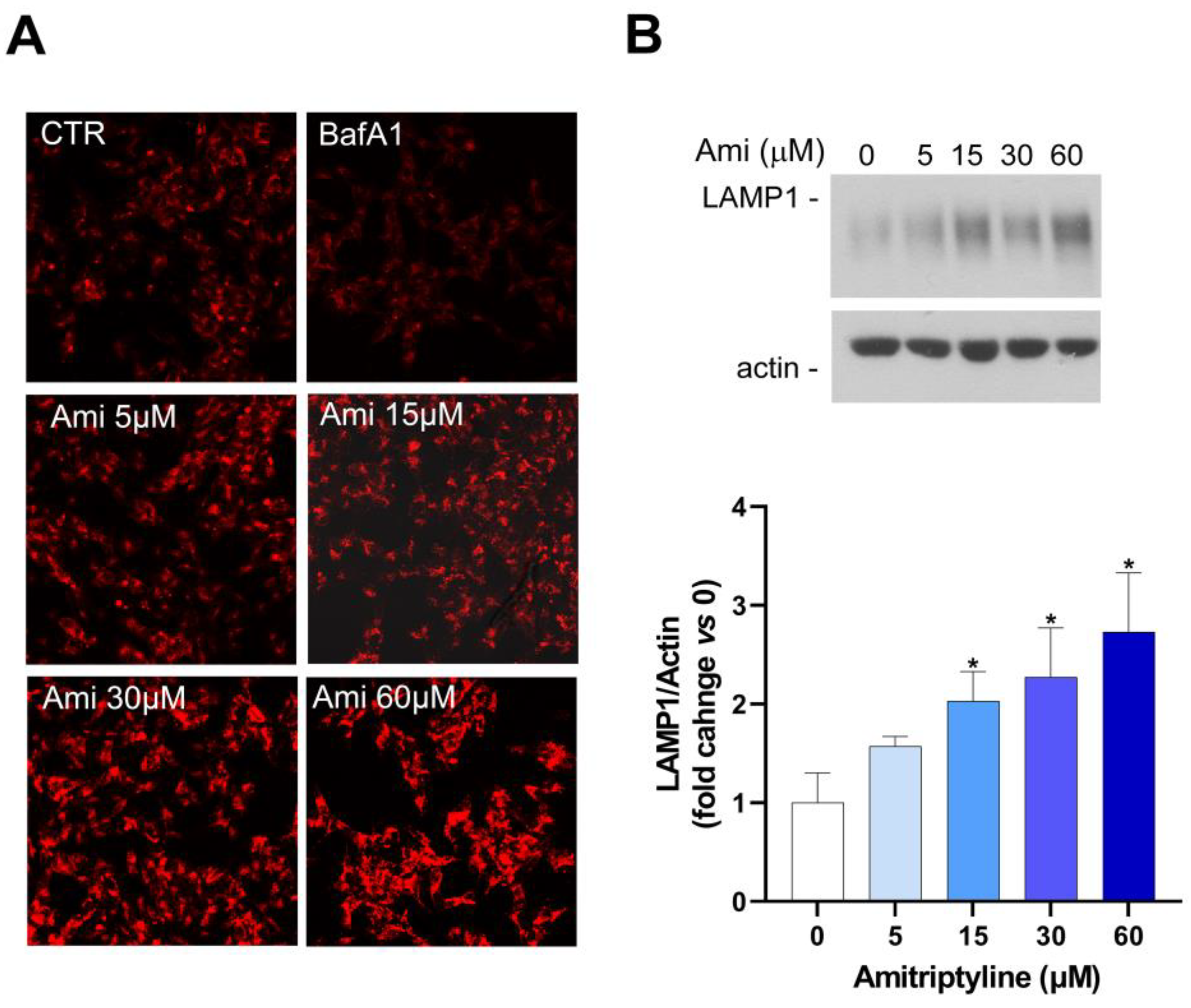
Figure 8.
Inhibition of autophagy by chloroquine does not revert the cytotoxic and antiproliferative effects of amitriptyline in SH-SY5Y. SH-SY5Y cells were preincubated with chloroquine (CQ, 20 μM) for 2h and then exposed to amitriptyline 5-60 μM for 24 (A) and 72h (B). Cell viability was evaluated by MTT assay. CQ itself significantly reduced cell viability in untreated cells and exacerbate the cytotoxic effect of amitriptyline after 72h incubation. Data (mean ± s.e.m. of n=5 independent experiments) are expressed as percentage of the untreated control (0 - CQ). (C) Representative image of clonogenic assay of SH-SY5Y pretreated with CQ and incubated with amitriptyline 5-60 μM for 12 days. The graph shows the quantification of the colonies formed in four independent experiments; the results (mean ± s.e.m.) are expressed as relative colony number compared to untreated cells (0 - CQ). (*p<0.05; ***p<0.001; ****p<0.0001 vs 0 – CQ; °p<0.05; °°p<0.01; °°°p<0.001; °°°°p<0.0001 vs corresponding concentration of amitriptyline -CQ; #p<0.05; ###p<0.001).
Figure 8.
Inhibition of autophagy by chloroquine does not revert the cytotoxic and antiproliferative effects of amitriptyline in SH-SY5Y. SH-SY5Y cells were preincubated with chloroquine (CQ, 20 μM) for 2h and then exposed to amitriptyline 5-60 μM for 24 (A) and 72h (B). Cell viability was evaluated by MTT assay. CQ itself significantly reduced cell viability in untreated cells and exacerbate the cytotoxic effect of amitriptyline after 72h incubation. Data (mean ± s.e.m. of n=5 independent experiments) are expressed as percentage of the untreated control (0 - CQ). (C) Representative image of clonogenic assay of SH-SY5Y pretreated with CQ and incubated with amitriptyline 5-60 μM for 12 days. The graph shows the quantification of the colonies formed in four independent experiments; the results (mean ± s.e.m.) are expressed as relative colony number compared to untreated cells (0 - CQ). (*p<0.05; ***p<0.001; ****p<0.0001 vs 0 – CQ; °p<0.05; °°p<0.01; °°°p<0.001; °°°°p<0.0001 vs corresponding concentration of amitriptyline -CQ; #p<0.05; ###p<0.001).
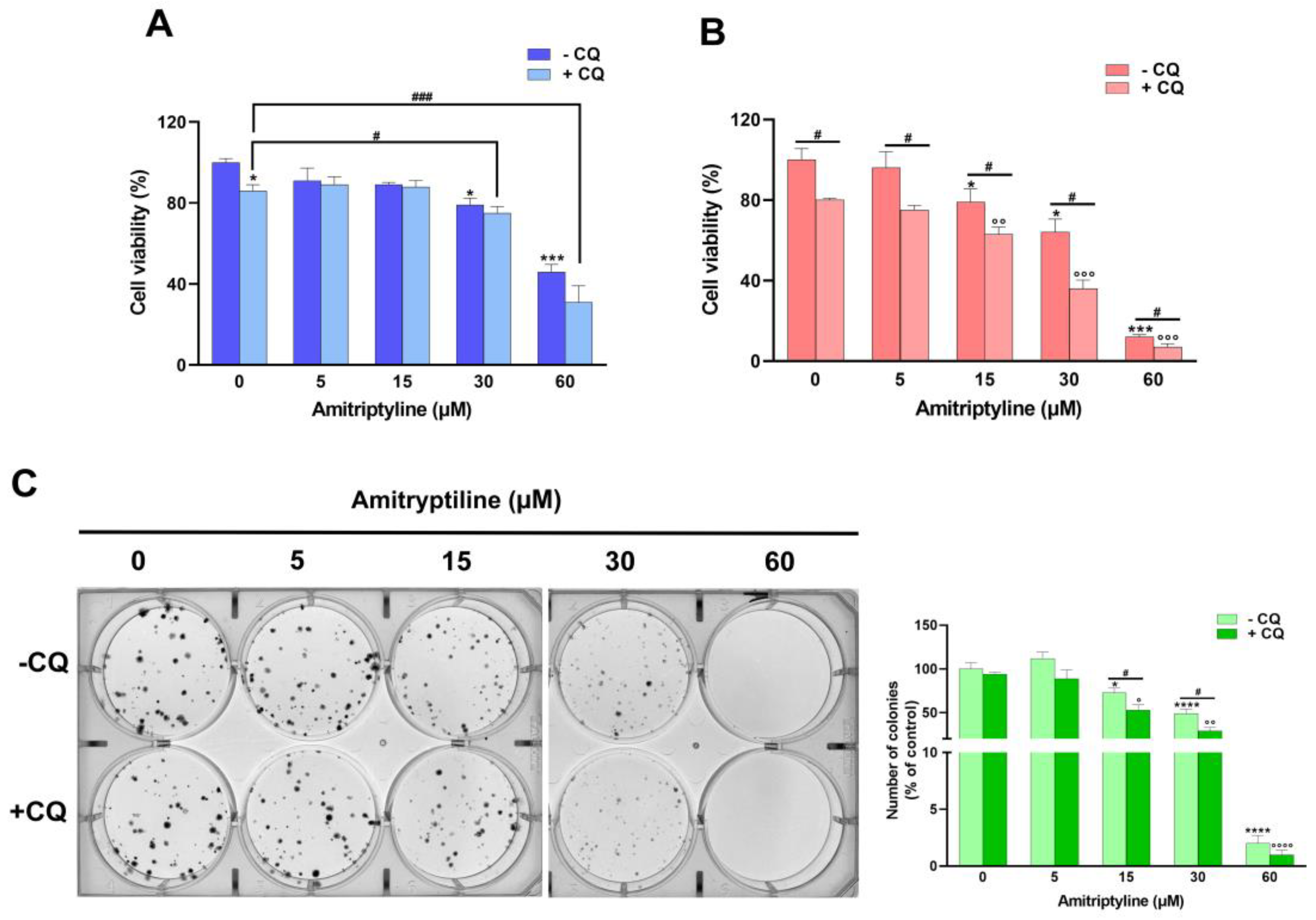
Figure 9.
Effects of amitriptyline on cell viability and clonogenic capacity in autophagy-deficient Atg5 -/- mouse embryonic fibroblast (MEF) (A) Atg5-deficient (Atg5 -/-) and wild type (Atg5 +/+) mouse embryonic fibroblasts (MEF) were treated with amitriptyline 5-120μM for 24h and cell viability was measured by MTT assay. Cell viability was significantly reduced in Atg5 -/- compared to Atg5 +/+ MEF following treatment with amitriptyline 15-120 μM. Histograms represent the mean ± s.e.m. of 3 independent experiments (*p<0.05; ****p<0.0001 vs 0 Atg5 +/+; °p<0.05; °°°p<0.001; °°°°p<0.0001 vs 0 Atg5 -/-; #p<0.05; ##p<0.01). (B) Representative images of a clonogenic assay in Atg5 -/- and Atg5 +/+ MEF cells treated with amitriptyline. (C) Quantification of the colonies expressed as percentage of the relative untreated control (0). Results are reported as mean ± s.e.m. of n=4-5 independent experiments (*p<0.05; ***p<0.001 vs relative untreated control).
Figure 9.
Effects of amitriptyline on cell viability and clonogenic capacity in autophagy-deficient Atg5 -/- mouse embryonic fibroblast (MEF) (A) Atg5-deficient (Atg5 -/-) and wild type (Atg5 +/+) mouse embryonic fibroblasts (MEF) were treated with amitriptyline 5-120μM for 24h and cell viability was measured by MTT assay. Cell viability was significantly reduced in Atg5 -/- compared to Atg5 +/+ MEF following treatment with amitriptyline 15-120 μM. Histograms represent the mean ± s.e.m. of 3 independent experiments (*p<0.05; ****p<0.0001 vs 0 Atg5 +/+; °p<0.05; °°°p<0.001; °°°°p<0.0001 vs 0 Atg5 -/-; #p<0.05; ##p<0.01). (B) Representative images of a clonogenic assay in Atg5 -/- and Atg5 +/+ MEF cells treated with amitriptyline. (C) Quantification of the colonies expressed as percentage of the relative untreated control (0). Results are reported as mean ± s.e.m. of n=4-5 independent experiments (*p<0.05; ***p<0.001 vs relative untreated control).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



