
Submitted:
15 May 2024
Posted:
15 May 2024
You are already at the latest version
Abstract
Thrombotic microangiopathies (TMA) can be attributed to a wide variety of causes and the diagnostic process is often challenging, especially when the clinical scenario is full of other confounding factors. Hemolysis can be due to complement-mediated thrombotic microangiopathies and drug-induced hemolytic anemia in the appropriate clinical context. In this case presentation, two patients with TMA including a patient diagnosed with complement-mediated TMA after a through screen of other differential diagnoses, and a multiple myeloma patient on chemotherapy with primaquine-induced hemolytic anemia in the background of treatment for possible pneumocystis jirovecii pneumonia. A thorough review of the drug history of patients presenting with TMA should be conducted, along with a heightened index of suspicion for complement-mediated disorders such as complement-mediated TMA, which should be entertained after ruling out a wide variety of differential diagnoses.
Keywords:
hemolytic anemia
; complement
; thrombotic microangiopathy
; primaquine
1. Introduction
Thrombotic microangiopathies (TMA) represent a clinically heterogeneous group of hematological disorders characterized by dysfunctions of the arteriolar and capillary walls leading to microvascular thrombosis and substantial thrombocytopenia clinically [1]. The present case presentation highlights the wide clinical spectrum of presentations of TMA and the importance of maintaining a high index of suspicion for its atypical and unusual presentations and prompt institution of proper treatment, including drug-induced thrombotic microangiopathy and complement-mediated thrombotic microangiopathies.
2. Case Presentation
2.1. Case 1
A 63-year-old Chinese male was admitted for multi-level compression fractures noted on a radiograph. Investigations showed anemia with hemoglobin of 9 g/dL, hypercalcemia of 2.88 mmol/L, erythrocyte sedimentation rate >140 mm/hr, acute kidney injury, and multiple thoracic compression fractures on imaging. As multiple myeloma was suspected, a bone marrow examination was performed which showed sheets of monotypic CD138+ plasma cells involving 75-80% of the bone marrow aspirate. Further investigations confirmed the diagnosis of IgG kappa standard risk t(11:14) multiple myeloma. The patient was started on bortezomib, daratumumab, and dexamethasone (DVD) immunochemotherapy.
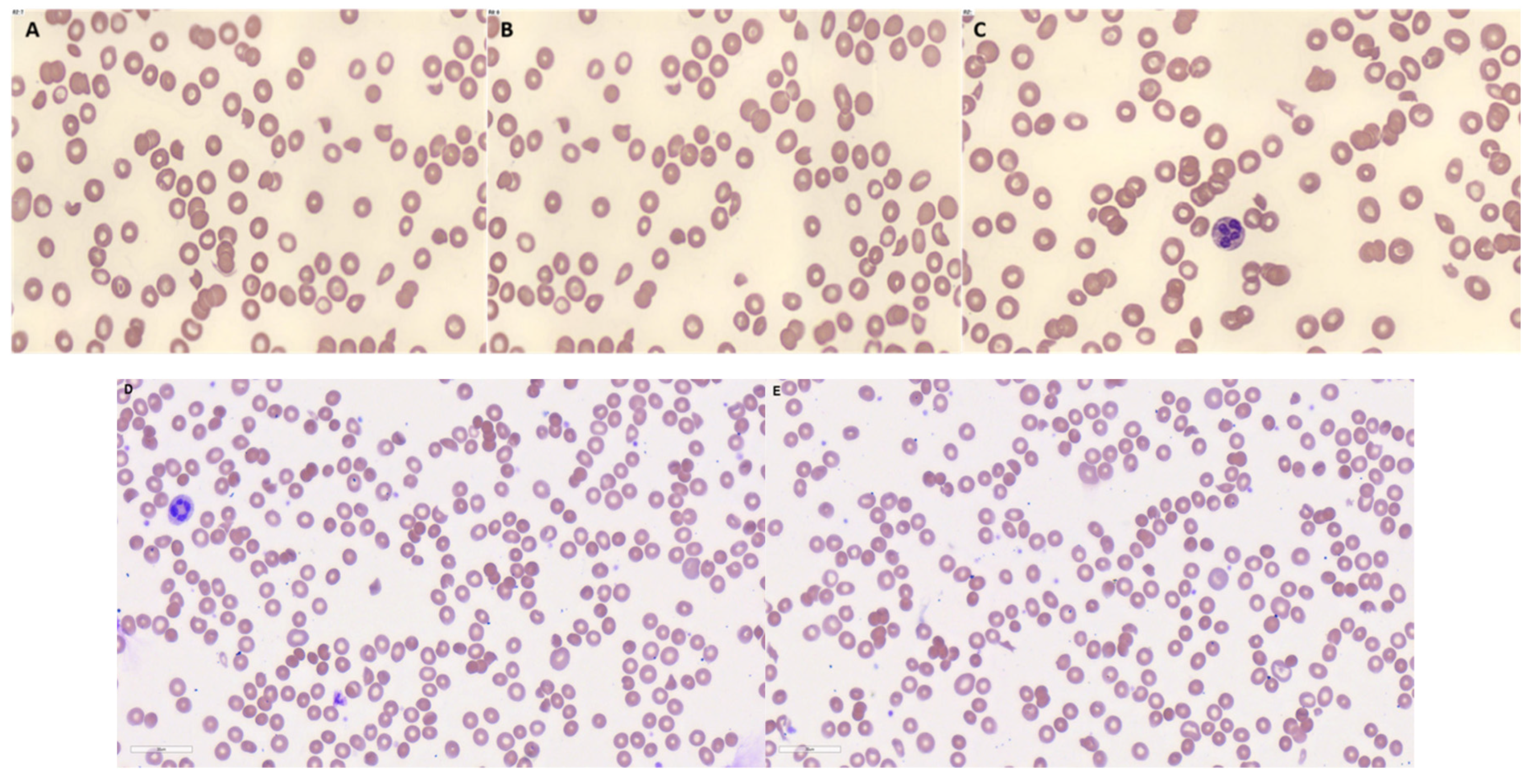
On the 12th day of his treatment, the patient was transferred to the medical intensive care unit given worsening hypoxia. Chest radiograph revealed bilateral lung infiltrates leading to concerns for Pneumocystis jirovecii pneumonia (PCP). He was subsequently intubated and empirically treated for PCP with intravenous clindamycin, hydrocortisone, and oral primaquine at 30mg per day for 5 days. 4 days after initiation of primaquine, his hemoglobin dropped to 6.8 g/dL and platelet counts to 28 x 109/L. Peripheral blood film showed cell fragmentation at about 5-8 schistocytes per high-powered field, as shown in Figure 1. Haptoglobin was <30 mg/dL, bilirubin was 16 µmol/L, lactate dehydrogenase was 2137 U/L and reticulocytes were 79.4 x 109/L. a DCT showed weakly positive IgG, and CRP was 31.7 mg/dL. These findings supported the diagnosis of TMA, thus an ADAMTS-13 was performed which was 0.32 IU/mL. While intubated, a polymerase chain reaction test for PCP was performed which was negative. Primaquine was subsequently stopped. Interestingly, the patient’s haemoglobin and platelets recovered and the fragmentation observed on the peripheral blood films resolved. His DVD chemotherapy was subsequently restarted and there was no recurrence of the TMA.
2.2. Case 2
A 53-year-old Malay female was admitted for left flank pain of 1 day, fever and dysuria, urinary frequency, and oliguria. Physical examination was unremarkable. On admission, she was noted to have acute kidney injury of creatinine 326 µmol/L, creatinine clearance 13 ml/min, urea 12 mmol/L, potassium 4.8 mmol/L, sodium 135 mmol/L, hemoglobin 11.7 g/dL, white blood cell count 9.0 x 109/L, platelet count 186 x 109/L, c-reactive protein (CRP) 33.3 mg/L. Peripheral blood film demonstrated fragmented red cells of 4 cells per high power field in keeping with thrombotic microangiopathies. Other biochemical markers showed a raised lactate dehydrogenase and bilirubin, with low haptoglobin and schistocytes in the peripheral blood film as shown in Figure 1. A direct Coombs test (DCT) was sent which was positive, with C3B/C3D 3+, and IgG 1+. Other tests were done which excluded thrombotic thrombocytopenic purpura with an ADAMTS-13 activity of 74% and disseminated intravascular coagulation with prothrombin time of 15.1 seconds, activated partial thromboplastin time of 27.3 seconds, fibrinogen 4.1 g/L, D-dimer > 4 mg/L. Other autoimmune workups were negative, including negative antiphospholipid antibodies, negative anti-nuclear antibodies, anti-double stranded DNA antibodies, anti-neutrophil cytoplasmic antibodies, and negative cryoglobulins. Anti-glomerular basement membrane antibody was positive but quantitative levels < 1.5 U/ml were suggestive of a false positive. Serologies for chronic viral infection were also negative for hepatitis B, hepatitis C and HIV. Subsequent genetic testing for the patient showed a variant of uncertain significance in the INF2 gene with a heterozygous mutation (c2288G>Ap.Arg763Lys).
Given the acute kidney injury, a renal biopsy was performed which showed glomeruli with increased cellularity, fibrinoid material including intravascular fibrin thrombin, arterial sclerosis, and acute tubular injury. The patient was urgently started on dialysis due to anuria, and Immunosuppressive therapy with plasma exchange to treat the underlying complement-mediated thrombotic microangiopathy. Immunosuppression consisted of weekly rituximab for a total of 4 cycles, IV hydrocortisone starting at approximately prednisolone 2 mg/kg) for 1 week then oral prednisolone at 1mg/kg for 2 weeks followed by a tapering steroids dose until reaching a maintenance dose of 5mg. Plasma exchange for the patient was daily for 2 weeks, which was weaned down to every other day plasma exchange for 4 days.
The patient started on regular hemodialysis but was subsequently noted to have evidence of renal recovery with good diuretic response to intravenous furosemide which was then switched to oral furosemide and eventually weaned off diuretics on discharge.
3. Discussion
Complement-mediated TMA and drug-induced TMA are the rare but important causes of microangiopathic hemolytic anemia. Complement-mediated TMA, previously referred to as atypical hemolytic uremic syndrome (aHUS), referred to HUS not caused by Shiga toxin-producing Escherichia coli infection, is associated with congenital or acquired abnormal complement activation and dysregulation, and it can present with renal and neurological dysfunction, and hypertension with a high chance of progressive renal failure if not properly recognized and treated in over one-third of the patients [2,3]. Its presentation should be carefully evaluated after a variety of alternative causes, such as thrombotic thrombocytopenic purpura, are properly excluded. A variety of genetic mutations are found to be associated with complement dysregulation and the subsequent development of complement-mediated TMA, which includes loss of function mutations in genes encoding for factor H, factor H-related proteins, membrane cofactor protein, and factor I. Additional gain of function mutations include complement proteins factor B and complement C3. The genetic mutation detected in our patient, INF2, also called inverted formin-2, is a non-complement related genetic mutation in complement-mediated TMA often found via the use of genetic testing [4]. The use of genetic testing is increasingly critical due to their elucidation of the underlying pathophysiological mechanisms behind the development of complement-mediated TMA and allows personalized and targeted therapy to be applied to patients [5]. Therapeutic plasma exchange and eculizumab, a monoclonal antibody that blocks terminal complement activation in complement-mediated thrombotic microangiopathy are the mainstay treatment options [6,7].
Suspicion for drug-induced thrombotic microangiopathy (DITMA) is based on the observation of the timing of DITMA occurrence, the pattern of disease, and the exclusion of a better explanation through investigation [8]. DITMA suspicion is amplified by DITMA resolution when the drug is withdrawn or there is a recurrent endothelial injury during re-exposure to the drug [8]. All of these criteria fit with our patient. Two major mechanisms have been postulated to cause (DITMA). One is by forming drug-dependent antibodies (immune) and the other is due to direct tissue toxicity forming platelet-rich thrombi in arterioles or capillaries (non-immune) [9]. According to published data about 78 drugs have been suspected of causing DITMA, of which the top three are quinine, cyclosporine, and tacrolimus [9]. Currently, the management of DITMA involves the discontinuation of the offending agent. Plasma exchange and immunosuppression may be reasonable if the diagnosis is uncertain. Eculizumab may be considered in in patients with non-immune DITMA, particularly those deteriorating despite drug discontinuation, and at risk of kidney failure [10]. In the case of primaquine-induced hemolytic anemia, cytoskeleton protein damage underlies the process of removal of erythrocytes when exposed to a phenolic metabolite of primaquine, namely 5-hydroxyprimaquine [11].
4. Conclusions
This case presentation highlights the importance of maintaining an index of suspicion of DITMA and complement-mediated TMA in patients with TMA and considering drug-induced etiologies and complement-mediated causes in the evaluation of patients.
Author Contributions
S.S.Y.W., C.G., K.G.E.L, C.D., collected clinical data. S.S.Y.W. and H.T. interpreted the data and wrote the draft of the manuscript. B.E.F. and P.K. reviewed the manuscript and provided feedback.
Funding
This study was not supported by any sponsor or funder.
Statement of Ethics
This case report was conducted ethically with adherence to the World Medical Association Declaration of Helsinki. This case report has obtained specific written consent from the patients for the publication of data and clinical images. The case report did not require ethic approval from the national guidelines.
Data Availability Statement
Additional data are available upon reasonable request from the corresponding author.
Conflicts of Interest
The authors declared no conflict of interest.
References
- Osti, N., et al., Case Report: Microangiopathic Hemolytic Anemia With Normal ADAMTS13 Activity. Front Med (Lausanne), 2021. 8: p. 589423. [CrossRef]
- Rafiq, A., et al., Atypical hemolytic-uremic syndrome: a case report and literature review. Am J Case Rep, 2015. 16: p. 109-14. [CrossRef]
- Catarci, S., et al., A case report of an atypical haemolytic uremic syndrome in pregnancy: something wicked this way comes. BMC Anesthesiol, 2023. 23(1): p. 94. [CrossRef]
- Feitz, W.J.C., et al., The genetics of atypical hemolytic uremic syndrome. Med Genet, 2018. 30(4): p. 400-409. [CrossRef]
- Loirat, C., et al., An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol, 2016. 31(1): p. 15-39. [CrossRef]
- Basnayake, B., et al., Atypical hemolytic uremic syndrome: a case report. J Med Case Rep, 2020. 14(1): p. 11. [CrossRef]
- Gavriilaki, E. and R.A. Brodsky, Complementopathies and precision medicine. J Clin Invest, 2020. 130(5): p. 2152-2163. [CrossRef]
- Chatzikonstantinou, T., et al., An Update in Drug-Induced Thrombotic Microangiopathy. Front Med (Lausanne), 2020. 7: p. 212. [CrossRef]
- Al-Nouri, Z.L., et al., Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood, 2015. 125(4): p. 616-8. [CrossRef]
- Efe, O., et al., Treatment of Gemcitabine-Induced Thrombotic Microangiopathy Followed by Gemcitabine Rechallenge With Eculizumab. Kidney Int Rep, 2021. 6(5): p. 1464-1468. [CrossRef]
- Bowman, Z.S., et al., Primaquine-induced hemolytic anemia: role of membrane lipid peroxidation and cytoskeletal protein alterations in the hemotoxicity of 5-hydroxyprimaquine. J Pharmacol Exp Ther, 2005. 314(2): p. 838-45. [CrossRef]
Figure 1.
(A) - (C): Red cell fragmentation in the patient with primaquine-induced thrombotic microangiopathies (D) and (E): Red cell fragmentation in the patient with complement-mediated TMA.
Figure 1.
(A) - (C): Red cell fragmentation in the patient with primaquine-induced thrombotic microangiopathies (D) and (E): Red cell fragmentation in the patient with complement-mediated TMA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



