
Submitted:
13 August 2024
Posted:
14 August 2024
You are already at the latest version
Abstract
Vaginitis is a widespread issue for women worldwide, yet current diagnostic tools are lacking. Bacterial vaginosis (BV) is the most prevalent type of vaginitis, found in 10-50% of reproductive aged women. Current diagnostic methods for BV rely on clinical criteria, microscopy, or detection of a few microbes by qPCR. However, many vaginal infections lack a single etiological agent and are characterized by changes in the vaginal microbiome community structure (e.g. BV is defined as a loss of protective lactobacilli resulting in an overgrowth of anaerobic bacteria). Shotgun metagenomic sequencing provides a comprehensive view of all the organisms present in the vaginal microbiome (VMB) allowing for a better understanding of all potential etiologies. Here, we describe a robust VMB metagenomics sequencing test with a sensitivity of 93.1%, specificity of 90%, a negative predictive value of 93.4%, and a positive predictive value of 89.6% certified by Clinical Laboratory Improvement Amendments (CLIA), the College of American Pathologist (CAP) and the Clinical Laboratory Evaluation Program (CLEP). We sequenced over 7,000 human vaginal samples with this pipeline and described general findings and comparison to US census data.
Keywords:
vaginal microbiome
; shotgun metagenomic sequencing
; vaginitis
1. Introduction
The vaginal microbiome (VMB) is a complex ecosystem of microorganisms that live inside the vagina. A Lactobacillus-dominant microbiome provides protection against infection, while a highly diverse microbiome is often associated with symptoms and vaginal infections [1].
The most common vaginal infection is bacterial vaginosis (BV), a condition that affects up to 29% of women annually [2]. Other conditions of the VMB include vulvovaginal candidiasis (VVC), which will affect 30-50% of women at least once in their lifetime [3], and trichomoniasis, which will affect about 2% of women in the US annually [4].
It has long been understood that vaginal lactobacilli protect against pathogen colonization by producing lactic acid, hydrogen peroxide and antimicrobial peptides [5]. BV is defined as a loss of lactobacilli and an overgrowth of anaerobic organisms [1]; however, it can also occur alongside infections with yeast or aerobes [6]. The first diagnostic methods for BV relied heavily on symptoms and detection of lactobacilli by microscopy (i.e. Amsel criteria and Nugent score [7,8]). However, symptom-based diagnosis is often inaccurate because different conditions may cause similar symptoms [9] and co-infections can be common [6]. Additionally, microscopic methods fail to identify the specific organisms present, instead relying on a ratio of rod-shaped bacteria seen with microscopy.
More recently, multiple qPCR panels have been developed for BV diagnosis [10], and while PCR has a very low limit of detection, it can only detect the specific organisms it was designed to detect. Therefore, qPCR misses other potential pathogens that may be contributing to symptoms as well as the relative amount of protective microbes in the VMB.
Recent research into the VMB has relied on 16s rRNA sequencing due to its relatively low cost and semi-comprehensive assessment of the microbiome [11,12]. Clinical applications of 16S sequencing for bacteria, and ITS for fungi, are also growing in application.
The next advancement in sequencing technology following targeted region amplicon sequencing is shotgun metagenomic sequencing. This method that can provide strain-level resolution of all DNA-based microorganisms including bacteria and yeast from the same assay [13]. Additionally, shotgun sequencing allows for in-depth study of the function and potential interactions between species as it can identify the genomic differences and pathogenic or biochemical pathways.
Here we describe the Evvy test, a novel shotgun metagenomics-based test that is approved by the Clinical Laboratory Improvement Amendments (CLIA), the College of American Pathologist (CAP) and the Clinical Laboratory Evaluation Program (CLEP) for VMB profiling that can be used to identify all bacteria and fungi present in a vaginal sample. Additionally, we provide an overview of findings from seven thousand samples, including demographic representation compared to United States (US) census data.
2. Materials and Methods
2.1. Evvy Test Workflow
An overview of the workflow for Evvy’s VMB laboratory developed test (LDT) is shown in Figure 1. A medical provider submits a lab order for an Evvy vaginal health test. A sample collection kit is shipped directly to the patient. The patient self-collected a vaginal swab and ships the sample at ambient temperature to a CLIA, CAP, and CLEP certified lab (CLIA 45D1086390, CAP 7214171, PFI 9433Microgen DX, TX, US). Samples are processed, which includes a chemical and mechanical lysis, host depletion, and DNA extraction using an automated extraction handling instrument. NGS libraries are prepared, multiplexed, quality checked and sequenced on the Illumina NovaSeq (Illumina, San Diego, CA). Sequencing data is processed through Evvy’s pipeline, which is specifically designed to characterize the vaginal microbiome. Samples that pass detection thresholds are reported to the provider and patient.
2.2. Isolate Sequencing
A total of 168 isolates were included in the sensitivity and specificity study. Isolates were ordered from ATCC (Table S1), and spiked into collection tubes containing DNA preservatives. All samples were processed using the shotgun metagenomic sample prep described below. Sensitivity, specificity, positive and negative predictive values were calculated as previously described [14]. To be considered a positive, samples must have at least 20,000 reads, >50% relative abundance, and >90% identity to the input ATCC. False positives include ATCC strains that passed thresholding but had inaccurate species identifications (i.e. not matching ATCC isolates input). The test set also included true negatives, including ATCC strains that were not represented in the classification database, as well as molecular grade water controls.
2.3. Limit of Detection and Precision Studies
The limit of detection and precision studies were carried out with a Gram-negative organism (Klebsialle oxytoca), a Gram-positive organism (Lactobacillus gasseri), and a fungal species (Candida albicans) available through ATCC. On each of 3 days, 8 ten-fold dilutions were prepared and run through the protocol as three technical replicates. The limit of detection (LOD) was determined as the concentration of cells that could be detected in ≥95% of samples tested with a coefficient of variation ≤5%.
Inter-assay and Intra-assay reproducibility (precision) were demonstrated by comparing the average relative abundance (the reported value) within and between runs over three days of testing by two different technicians. To be considered reproducible the coefficient of variation of the average relative abundance must be less than 5% for each day of testing (intra-assay reproducibility) and less than 5% over 3 days of testing (inter-assay reproducibility) at the established LOD.
2.4. Mock Community Analysis
Whole cell vaginal mock communities were purchased from ATCC (MSA-2007) that contains equal amounts of Gardnerella vaginalis, Lactobacillus gasseri, Mycoplasma hominis, Prevotella bivia, Streptococcus agalactiae, and Lactobacillus jensenii. Prior to DNA extraction, Hep2 cells (CCL-23) were spiked in to mimic the host DNA seen within a vaginal sample. The mock communities plus host were serially diluted (1:10), then processed through the normal Evvy pipeline. This experiment was done in duplicate, and one representative is show in in Figure 2. The undiluted samples went through an in-silico dilution process, where the undiluted sample was subsampled to 1-4 log fewer sequence reads (from 1.6 million reads to 160 reads).
2.5. Study Participants, Ethics, and Sample Collection and Transportation
Clinical samples were obtained from participants on the Evvy platform in the US. All study participants consented to participate in the study, and all study procedures were approved by a federally accredited Institutional Review Board (IRB# 20220118.evvy). Participants represented varying ages and ethnic groups.
Patients self-collected vaginal swabs and place them into Copan eNAT collection tube (Copan, Murrieta, CA), samples were shipped to the laboratory (Microgen DX, TX, US) and processed through a CLIA/CAP/CLEP certified shotgun metagenomics pipeline within 5 days. Each participant completed a questionnaire which includes symptoms, related diagnoses, and demographic information.
2.6. Shotgun Metagenomics Analysis of Vaginal Samples
For the shotgun metagenomics analysis, a validated pipeline was used. Vaginal samples undergo host depletion with PMA [15], chemical lysis using metapolyzyme (Sigma), followed by bead beating and DNA extraction on an automated extraction instrument (KingFisher FLEX, Thermo Scientific). Each extraction batch includes, a negative control (NC, water), and a mock community positive control (PC - ATCC MSA-2007).
Evvy’s bioinformatics platform includes quality control, host depletion, and taxonomic profiling. Trimmomatic [16] was used to trim and filter raw reads to remove low quality bases. Following quality filtering, remaining data are further refined by eliminating human DNA sequences by mapping to the latest human reference (GRCh38). Remaining reads are mapped to a proprietary database of precomputed genomic signatures for species level classification. Only classifications with >0.75% relative abundance are included in reports.
The unique signatures comprising the database are computed from microbial whole genomes isolated from the urogenital tract and contains more than 4000 microbial genomes, representing 700 bacterial species. Genomes were collected from publicly available repositories from any isolate documented to be collected from “urogenital” or “vaginal” sources. To accurately represent the phylogenetic diversity of underrepresented taxa, some genomes were included from other human microbiome sites. Public repositories are incomplete in their representation of the vaginal microbiome, for this reason we also included metagenome assembled genomes generated from Evvy test data.
3. Results
3.1. Sensitivity and Specificity
The sensitivity and specificity study included a total of 162 ATCC strains (Table S1) plus 50 16S negative patient vaginal samples spiked with molecular grade water (total of 202 samples). Samples must have had at least 20 thousandr reads. Any sample that had greater than 50% relative abundance and greater than 90% identity were considered positive. Any sample that did not meet those thresholds was considered a negative. True positives are samples with accurate species classification, while a false positive was a sample that passed all thresholds but had inaccurate species classification. We note that eight of eleven (72%) of the false positives were accurate at the genus level (Sup table 1). True negatives were samples that did not meet the thresholds because they were not included in the database, therefore it is expected that they do not produce accurately identified reads. False negatives failed threshold requirements even though they are included in the database. We note that two of the seven false negatives had accurate overall classification and four of the remaining five were accurate at the genus level. Therefore 85% (6/7) false negatives had semi-accurate IDs even though they did not meet thresholding requirements (Sup Table 1
Ninety-five samples were true positives with data from ATCC and the NovaSeq6000 matching. Forty-nine isolates were not included in the reference database and therefore produced true negative results, plus the 50 negative patient samples spiked with water. The test therefore had a measured sensitivity of 93.1%, specificity of 90%, a negative predictive value (NPV) of 93.4%, and a positive predictive value (PPV) of 89.6% (Table 1).
3.2. Limit of Detection and Precision
The bacterial LOD was determined to be between 1,640 - 3,000 colony forming units (CFU)/mL and the fungal LOD was determined to be 2,400 CFU/mL. The fungal species could be detected at 240 CFU/mL; however, the inter-assay reproducibility was unacceptable at this concentration (CV%=17.5) (Table S2).
All species included in the inter- and intra-assay reproducibility study were reproducible within runs at the assay limit of detection over the three testing days of the study (Table S2).

Table 2.
Average coefficient of variance (CV) for inter- and intra-assay reproducability.
| Average Inter-assay CV | Average Intra-assay CV | |
|---|---|---|
| K. oxytoca | 1.57 | 4.30 |
| L. gasseri | 1.60 | 2.03 |
| C. albicans | 2.56 | 1.49 |
3.3. Mock Community Analysis
Wet lab serial dilutions of mock community plus host cells show a stepwise decrease in the detection of mock community as the amount of DNA decreases (Figure 2A). An expected corresponding amount of “kit-ome” is detected [17], listed here as “other”. All components of the mock community could be detected at the 10^3 dilution, which reached the sequencing limit of detection of twenty thousand reads.
An in-silico subsampling of the initial mock community sample shows that our algorithm is accurate at detecting all components of the mock community at read levels ranging from 1.6 million reads (no dilution) to 160 reads (4 log decrease) (Figure 2B)
3.4.1. Findings from Evvy’s Vaginal Metagenomics Test
Using the Evvy’s sampling and taxonomy classification pipeline, a total of 7,158 samples have been sequenced in 4.5 months. Of those samples, 178 microbial species have been identified above the detection threshold. The top 10 species most commonly detected include all 4 named Gardnerellas (G. vaginalis, G. swidinskii, G. piotii, G. leopoldii), Lactobacillus iners, Lactobacillus crispatus, Fannyhessae (previously Atopobium) vaginae, Bifidobacterium animalis, Prevotella bivia, and Prevotella timonensis (Table 2). The top 10 species with the highest average abundance include Lactobacillus and Bifidobacterium species, two Gardnerella species, and Alloscardovia omnicolens (Table 2). To illustrate the community structure in samples containing these commonly found organisms, we pulled five random samples for each of the most abundant species (Figure 3). We see that some samples are not diverse and consist primarily of a single taxa (i.e. L. crispatus, L. gasseri, or Gardnerella dominant), while other samples are more diverse (i.e. L. helveticus, Prevotella and Gardnerella non-dominant samples (Figure 3). This is consistent with previously published vaginal microbiome literature [18,19].
Table 2.
The top ten species and genera identified in 7,158 human vaginal samples, based on their prevalence.
Table 2.
The top ten species and genera identified in 7,158 human vaginal samples, based on their prevalence.
| Top 10 species by frequency of detection | Percentage of tests detected | Top 10 Species by relative abundance | Average Abundance | Percent of tests detected |
|---|---|---|---|---|
| Gardnerella vaginalis | 99% | Lactobacillus crispatus | 33% | 93% |
| Gardnerella swidinskii | 99% | Lactobacillus helveticus | 23% | 0.1% |
| Lactobacillus iners | 95% | Bifidobacterium dentium | 19% | 0.9% |
| Gardnerella piotii | 95% | Lactobacillus iners | 18% | 95% |
| Lactobacillus crispatus | 93% | Lactobacillus gasseri | 15% | 14% |
| Gardnerella leopoldii | 78% | Bifidobacterium longum | 13% | 2% |
| Prevotella bivia | 46% | Bifidobacterium bifidum | 13% | 0.7% |
| Fannyhessae vaginae | 33% | Gardnerella swidinskii | 12% | 99% |
| Bifidobacterium animalis | 31% | Alloscardovia omnicolens | 12% | 2% |
| Prevotella timonensis | 27% | Gardnerella vaginalis | 11% | 99% |
3.4.2. Co-occurrence of Pathogens Detected
Shotgun metagenomics can detect a variety of bacteria and yeast, allowing for accurate identification of all potential bacterial pathogens. Using the Evvy test, multiple potential pathogens can be identified within each sample. Across 7,158 samples, Candida species (C. albicans, C. glabrata, and C. parapsilosis) rarely co-occurred, but were commonly found alongside both anaerobic, BV-associated organisms, like Prevotella, Mobiluncus, Megasphaera, and Gardnerella (Table S3). Aerobic organisms (specifically, E. coli, Klebsiella, E. faecalis, and Group B Strep) also commonly co-occurred with BV-associated organisms (Table S3).
3.4.3. Comparison to US Census Data
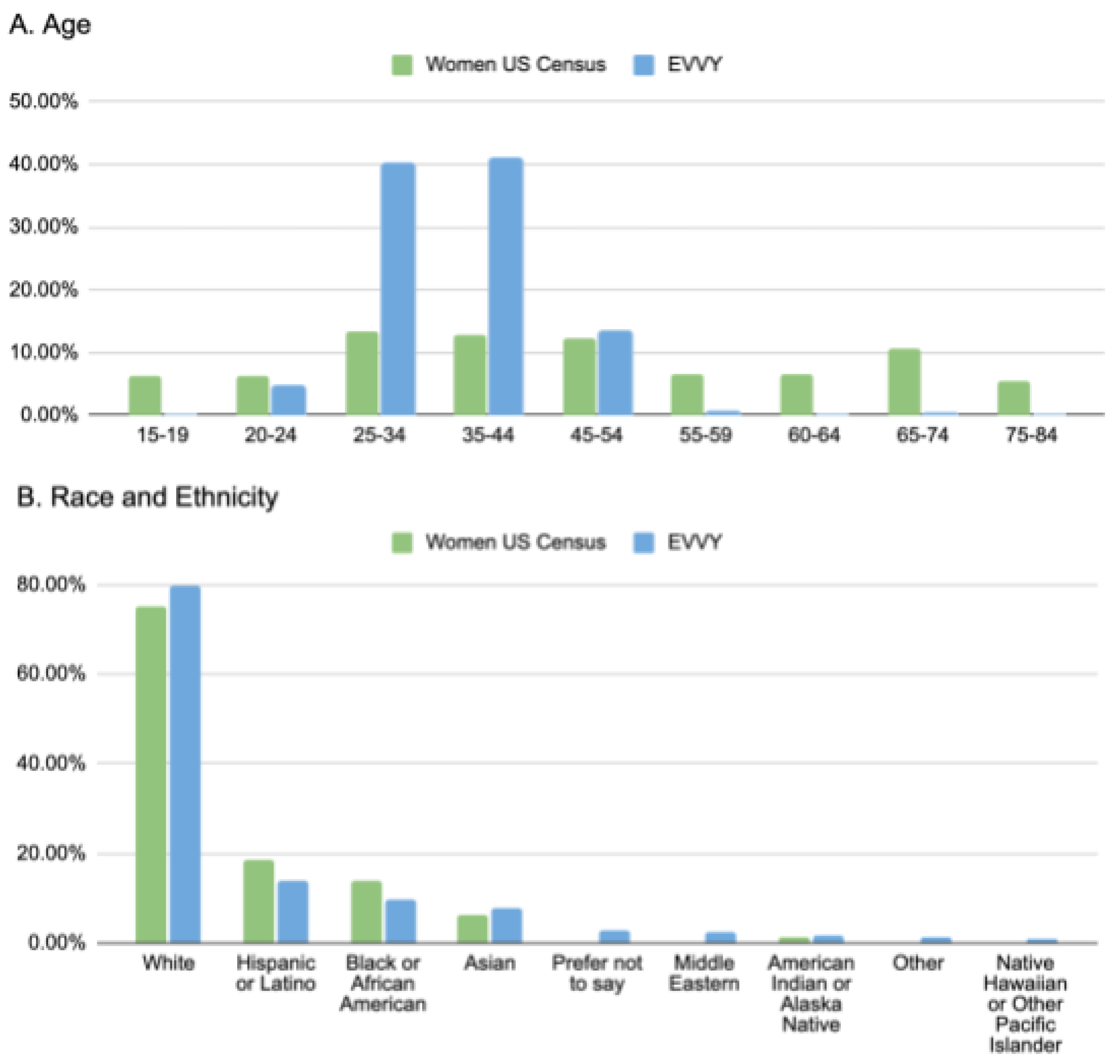
A comparison of the 7,158 samples to the US census data shows that Evvy users tend to be younger (aged 20-44) with fewer Evvy users are above age 55 than compared to the US population as a whole (Figure 3A). Evvy samples closely match the distributions of race and ethnicity across the US, with similar proportions of Hispanic or Latino, Black or African American, Asian, and Native American or Alaskan Natives (Figure 3B).
Figure 4.
Comparison of Evvy’s user data to the 2020 US census data. Percent of samples stratified by (a) age, or (b) self-reported race and ethnicity compared to reported US census data.
Figure 4.
Comparison of Evvy’s user data to the 2020 US census data. Percent of samples stratified by (a) age, or (b) self-reported race and ethnicity compared to reported US census data.
4. Discussion
In this report, we describe the validation of the first CLIA/CAP/CLEP certified shotgun metagenomic test for vaginal microbiome (VMB) samples. The Evvy test includes patient-collected vaginal swabs, complete sample lysis, DNA extraction, sequencing library preparation, NovaSeq sequencing, and a bioinformatic analysis platform that includes taxonomic classification to the species level.
This metagenomic test provides a comprehensive view of the microbiome that can be used for patient diagnoses, increasing access to care through self-sampling at home, and future research into the VMB.
Vaginitis affects 29% of women annually [2], and is one of the primary reasons women seek medical care [20]. Yet the average wait time for an OB-GYN appointment is 31 days [21]. The Evvy test can expand access to care by providing the ability to self-test, receive lab results, and physician-lead care from home.
Studies show that vaginitis patients are more likely to be misdiagnosed than correctly diagnosed by a doctor [9]. The Evvy pipeline demonstrates accurate detection of vaginal isolates with a sensitivity of 93.1%, specificity of 90%, a negative predictive value (NPV) of 93.4%, and a positive predictive value (PPV) of 89.6%. These numbers are above the acceptable range for a clinical metagenomics test [22,23] and allow for accurate characterization of the vaginal microbiome.
Vaginitis remains poorly understood [24] and symptoms can often be the result of mixed infections [6]. A co-occurrence analysis showed that different Candida species were found to rarely co-occur together. But Candida did co-occur with BV organisms as well as a variety of aerobic organisms (E. coli, Klebsiella, E. faecalis, and Group B Strep). Previous work from Evvy’s group also identified that when aerobic organisms are present, symptoms tend to be more severe [25]. PCR tests only detect a small panel of organisms, for example no currently available PCR panel detects anaerobic organisms or all Candida species. This illustrates how the Evvy test allows for a broader characterization of the microorganisms present in vaginal samples, enabling a provider to evaluate multiple etiologies with one test.
One limitation of the Evvy test is the turnaround time from sampling to metagenomic sequencing results (about 12 days). Therefore, the Evvy test may not be the appropriate test for every patient. PCR may be a more appropriate choice when physicians are looking for the presence of specific low abundance pathogens. While shotgun metagenomics allows for breadth of organism detection, PCR still has a much lower limit of detection than any current next generation sequencing (NGS) technology available. However, for patients with negative PCR tests, a history of recurrent infections, or suspected co-infections, shotgun metagenomic sequencing of the VMB can be useful for informing diagnosis and treatment. Providers looking for a more comprehensive view of the VMB may also leverage this type of test. Moreover, recent research has highlighted the relationship between VMB dysbiosis and a wide array of gynecological and obstetric conditions [12,26,27,28,29,30,31,32].
Through this testing platform, Evvy has generated the largest mNGS vaginal microbiome and health profile dataset ever collected. Evvy’s platform includes patients from all fifty states and very closely matches 2020 US census data for race and ethnicity. To our knowledge, no other dataset has included both the large sample sizes as well as breadth of racial and demographic representation. Its wide representation means that the Evvy’s multi-dimensional dataset can be used to better understand how the VMB changes in response to treatments, life events (pregnancy, menopause, etc), and in association with different gynecological diseases. These analyses will be described in upcoming publications.
In conclusion, the Evvy test represents a significant advancement in the diagnosis and understanding of vaginitis. Its capability to provide a more comprehensive view of the VMB opens new avenues for personalized medicine and enhances our understanding of the microbiome's role in female health.
5. Conclusions
Here we report a new CLIA/CAP/CLEP certified laboratory developed test that comprehensively and accurately identifies organisms in the vaginal microbiome. The test has sensitivity of 93.1%, specificity of 90%, a negative predictive value (NPV) of 93.4%, and a positive predictive value (PPV) of 89.6%. An analysis of seven thousand results of the Evvy test show a high co-occurrence of potential pathogenic organisms, and racial representation similar to the US population. This test provides a more comprehensive characterization of the composition of the whole vaginal microbiome with one swab.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, K.T-W. and P.N.; methodology, K.T-W. and E.H.H..; validation, C.P. and C.J.; formal analysis, K.T-W., M.G., G.O., and C.J.; resources, C.P. and P.N..; data curation, K.T-W., G.O., NS, JW; writing—original draft preparation, K.T-W..; writing—review and editing, E.H.H., C.P., G.O., M.G., J.W., N.S., P.N.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Evvy.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of Viome Institutional Review Board (IRB# 20220118.evvy 06/07/2023).
Informed Consent Statement
All study participants consented to de-identified research.
Data Availability Statement
This research was sponsored by Evvy and the authors of the paper who have access to the data are employees or scientific collaborators of Evvy who have signed contracts with Evvy to be bound by Evvy’s privacy policy and access restrictions. Additional data can be made available through a Data Transfer Agreement that protects the privacy of participants’ data; interested researchers may make requests by contacting kthomasw@evvy.com. The information provided by interested researchers will be used to generate a Data Transfer Agreement (DTA). The DTA protects the privacy of the participants' data, and will need to be signed by both the institution and Evvy before data can be transferred.
Acknowledgments
The authors acknowledge the rest of the Evvy team for their support, and all the women who suffer from recurrent infections and have shared their stories with us.
Conflicts of Interest
This study was funded and conducted by Evvy. Thomas-White, Olmschenk, Gong, and Navarro are employed by Evvy. Phillips, Jarvis, Sanford, and White are employed by Microgen Diagnostics.
References
- A Muzny, C.; Balkus, J.; Mitchell, C.; Sobel, J.D.; Workowski, K.; Marrazzo, J.; Schwebke, J.R. Diagnosis and Management of Bacterial Vaginosis: Summary of Evidence Reviewed for the 2021 Centers for Disease Control and Prevention Sexually Transmitted Infections Treatment Guidelines. Clin. Infect. Dis. 2022, 74, S144–S151. [CrossRef]
- Peebles, K.; Velloza, J.; Balkus, J.E.; McClelland, R.S.; Barnabas, R.V. High Global Burden and Costs of Bacterial Vaginosis: A Systematic Review and Meta-Analysis. Sex. Transm. Dis. 2019, 46, 304–311. [CrossRef]
- Foxman, B., et al., Prevalence of recurrent vulvovaginal candidiasis in 5 European countries and the United States: results from an internet panel survey. J Low Genit Tract Dis, 2013. 17(3): p. 340-5.
- Flagg, E.W.; Meites, E.; Phillips, C.B.; Papp, J.; Torrone, E.A. Prevalence of Trichomonas vaginalis Among Civilian, Noninstitutionalized Male and Female Population Aged 14 to 59 Years: United States, 2013 to 2016. Sex. Transm. Dis. 2019, 46, e93–e96. [CrossRef]
- Amabebe, E.; Anumba, D.O.C. The Vaginal Microenvironment: The Physiologic Role of Lactobacilli. Front. Med. 2018, 5, 181. [CrossRef]
- Sobel, J.D.; Subramanian, C.; Foxman, B.; Fairfax, M.; Gygax, S.E. Mixed Vaginitis—More Than Coinfection and With Therapeutic Implications. Curr. Infect. Dis. Rep. 2013, 15, 104–108. [CrossRef]
- Nugent, R.P.; A Krohn, M.; Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [CrossRef]
- Amsel, R., et al., Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med, 1983. 74(1): p. 14-22.
- Schwiertz, A., et al., Throwing the dice for the diagnosis of vaginal complaints? Ann Clin Microbiol Antimicrob, 2006. 5: p. 4.
- Muzny, C.A.; Cerca, N.; Elnaggar, J.H.; Taylor, C.M.; Sobel, J.D.; Van Der Pol, B. State of the Art for Diagnosis of Bacterial Vaginosis. J. Clin. Microbiol. 2023, 61, e0083722. [CrossRef]
- Lebeer, S.; Ahannach, S.; Gehrmann, T.; Wittouck, S.; Eilers, T.; Oerlemans, E.; Condori, S.; Dillen, J.; Spacova, I.; Donck, L.V.; et al. A citizen-science-enabled catalogue of the vaginal microbiome and associated factors. Nat. Microbiol. 2023, 8, 2183–2195. [CrossRef]
- Ravel, J.; Moreno, I.; Simón, C. Bacterial vaginosis and its association with infertility, endometritis, and pelvic inflammatory disease. Am. J. Obstet. Gynecol. 2021, 224, 251–257. [CrossRef]
- Langille, M.G., et al., Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol, 2013. 31(9): p. 814-21.
- Trevethan, R. Sensitivity, Specificity, and Predictive Values: Foundations, Pliabilities, and Pitfalls in Research and Practice. Front. Public Heal. 2017, 5, 307–307. [CrossRef]
- Shi, Y.; Wang, G.; Lau, H.C.-H.; Yu, J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int. J. Mol. Sci. 2022, 23, 2181. [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [CrossRef]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4680–4687. [CrossRef]
- Romero, R.; Theis, K.R.; Gomez-Lopez, N.; Winters, A.D.; Panzer, J.J.; Lin, H.; Galaz, J.; Greenberg, J.M.; Shaffer, Z.; Kracht, D.J.; et al. The Vaginal Microbiota of Pregnant Women Varies with Gestational Age, Maternal Age, and Parity. Microbiol. Spectr. 2023, 11, e0342922. [CrossRef]
- Paavonen, J.A.; Brunham, R.C. Vaginitis in Nonpregnant Patients: ACOG Practice Bulletin Number 215. Obstet. Gynecol. 2020, 135, 1229–1230. [CrossRef]
- Healthcare, A. 2022 Survey of Physician Appointment Wait Times and Medicare and Medicaid Acceptance Rates. 2022; Available from: https://www.wsha.org/wp-content/uploads/mha2022waittimesurveyfinal.pdf.
- Peterson, C.-L.; Alexander, D.; Chen, J.C.-Y.; Adam, H.; Walker, M.; Ali, J.; Forbes, J.; Taboada, E.; Barker, D.O.R.; Graham, M.; et al. Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool. Microorganisms 2022, 10, 441. [CrossRef]
- Mu, S.; Hu, L.; Zhang, Y.; Liu, Y.; Cui, X.; Zou, X.; Wang, Y.; Lu, B.; Zhou, S.; Liang, X.; et al. Prospective Evaluation of a Rapid Clinical Metagenomics Test for Bacterial Pneumonia. Front. Cell. Infect. Microbiol. 2021, 11. [CrossRef]
- P, V.-B., S. CK, and S. JD, International Society for theStudy of Vulvovaginal Disease recommendations for the diagnosis andtreatment of vaginitis. Lisbon: Admedic, 2023.
- K, T.-W.; F, W.; P, N. Incidence and Symptom Profiling of Vaginitis Containing Aerobic and Anaerobic Pathogens. Am. J. Obstet. Gynecol. 2024, 230, S641–S642. [CrossRef]
- Skafte-Holm, A.; Humaidan, P.; Bernabeu, A.; Lledo, B.; Jensen, J.S.; Haahr, T. The Association between Vaginal Dysbiosis and Reproductive Outcomes in Sub-Fertile Women Undergoing IVF-Treatment: A Systematic PRISMA Review and Meta-Analysis. Pathogens 2021, 10, 295. [CrossRef]
- Zhou, Q.; Yu, Y.; Zhou, J.; Liu, J.; Gao, J. Relationship of Lactobacillus Vaginal Microbiota Changes and the Risk of Preterm Birth: A Systematic Review and Meta-Analysis. J. Women's Heal. 2024, 33, 228–238. [CrossRef]
- Kosti, I.; Lyalina, S.; Pollard, K.S.; Butte, A.J.; Sirota, M. Meta-Analysis of Vaginal Microbiome Data Provides New Insights Into Preterm Birth. Front. Microbiol. 2020, 11, 476. [CrossRef]
- Torcia, M.G. Interplay among Vaginal Microbiome, Immune Response and Sexually Transmitted Viral Infections. Int. J. Mol. Sci. 2019, 20, 266. [CrossRef]
- Łaniewski, P.; Herbst-Kralovetz, M.M. Connecting microbiome and menopause for healthy ageing. Nat. Microbiol. 2022, 7, 354–358. [CrossRef]
- Sharifian, K.; Shoja, Z.; Jalilvand, S. The interplay between human papillomavirus and vaginal microbiota in cervical cancer development. Virol. J. 2023, 20, 1–9. [CrossRef]
- Yang, X.; Da, M.; Zhang, W.; Qi, Q.; Zhang, C.; Han, S. Role of Lactobacillus in cervical cancer. Cancer Manag. Res. 2018, ume 10, 1219–1229. [CrossRef]
Figure 1.
The Evvy VMB test workflow.
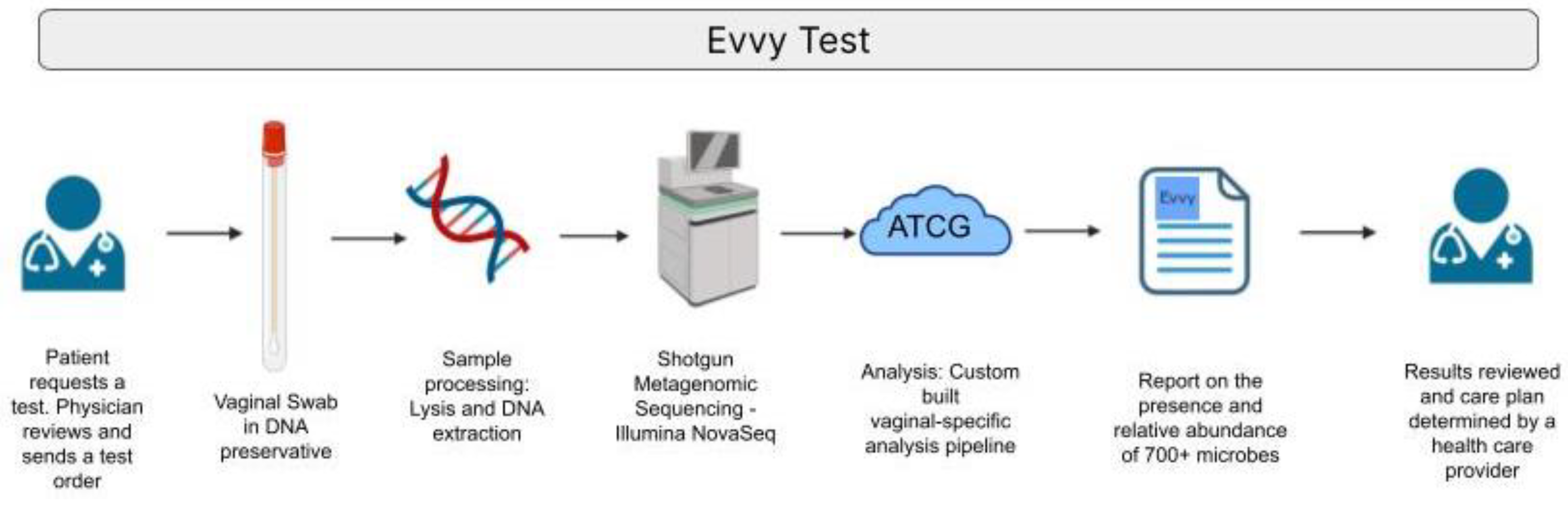
Figure 2.
Shotgun Metagenomics Resolution of Mock Community samples: (A) Of 1:10 dilution of mock community performed in the wet lab and (B) an in-silico subsampling of the initial mock community sample down to a 4 log decrease.
Figure 2.
Shotgun Metagenomics Resolution of Mock Community samples: (A) Of 1:10 dilution of mock community performed in the wet lab and (B) an in-silico subsampling of the initial mock community sample down to a 4 log decrease.
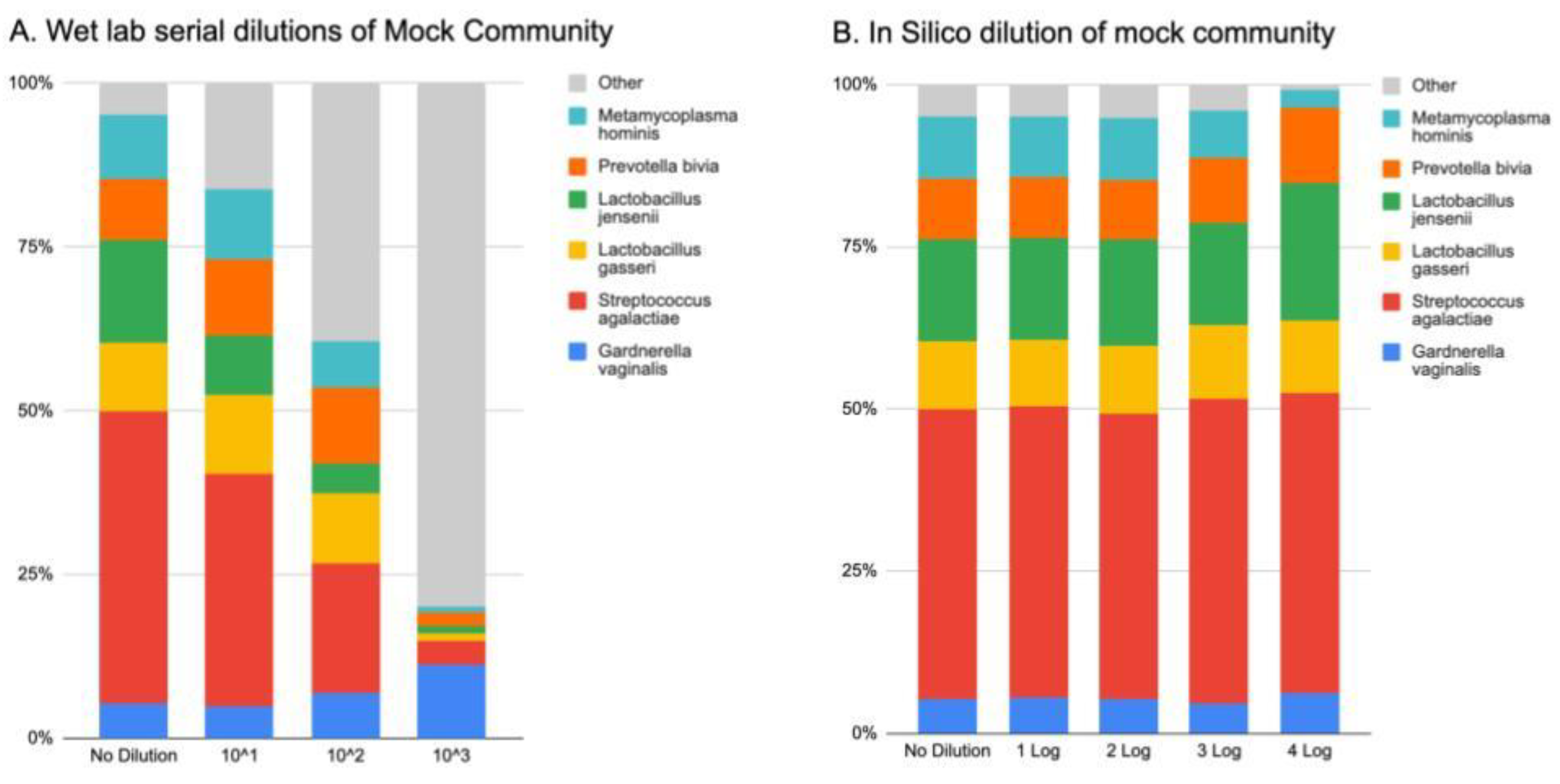
Figure 3.
Relative Abundance Graphs of Representative Vaginal Samples. These profiles represent 5 randomly selected samples containing the top 10 species detected by frequency and relative abundance listed in Table 2.
Figure 3.
Relative Abundance Graphs of Representative Vaginal Samples. These profiles represent 5 randomly selected samples containing the top 10 species detected by frequency and relative abundance listed in Table 2.
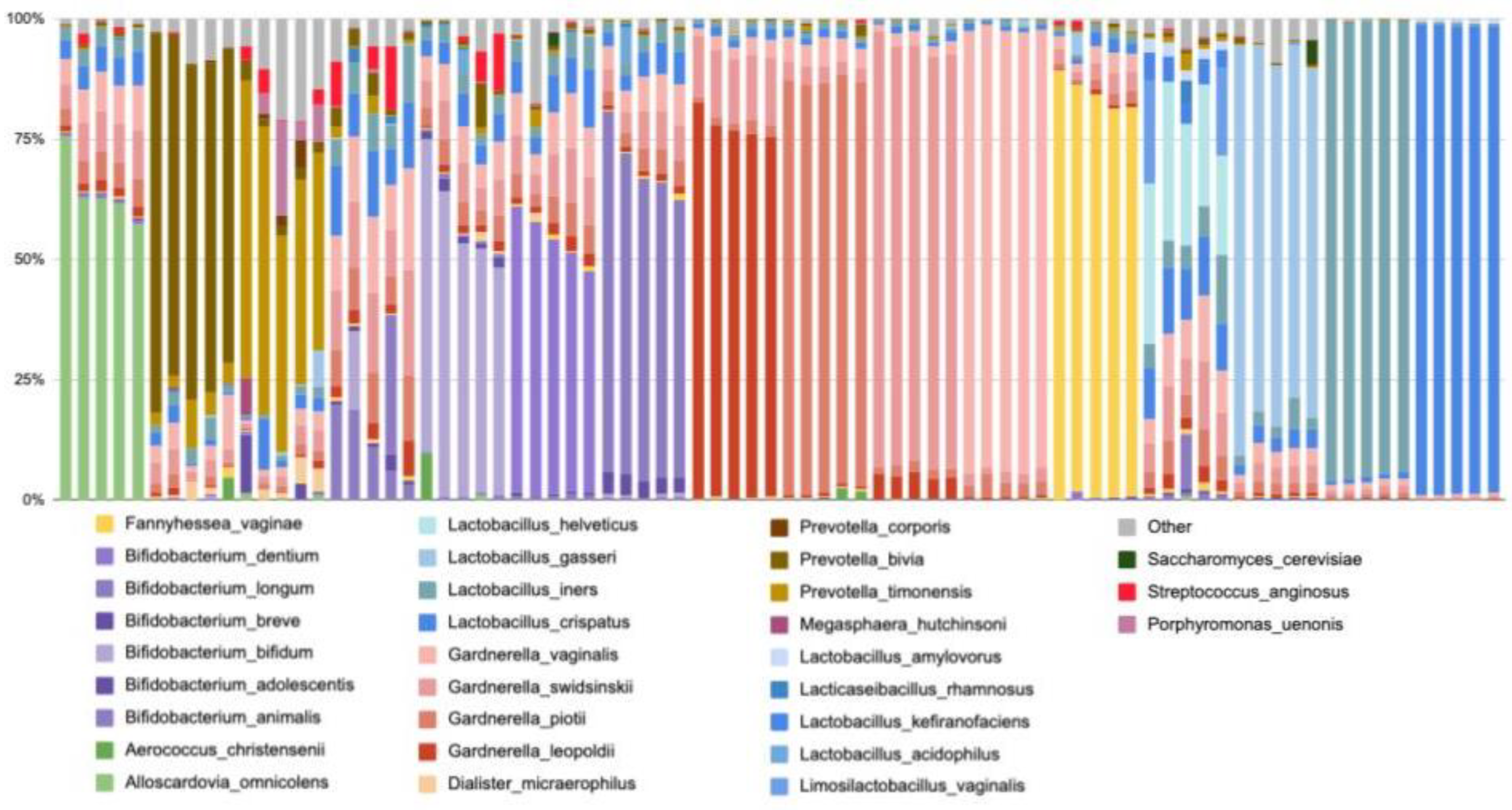
Table 1.
Sensitivity and specificity based on sequencing 168 ATCC isolates.
| Positive | Negative | Total | Predictive Value | ||
|---|---|---|---|---|---|
| NovaSeq | Positive | 95 | 11 | 106 | 89.6% (95/106) PPV |
| Negative | 7 | 99 | 106 | 93.4% (99/106) NPV | |
| Total | 102 | 110 | |||
| Sensitivity 93.1% (95/102) |
Specificity 90% (99/110) |
ATCC
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



