
Submitted:
15 May 2024
Posted:
15 May 2024
You are already at the latest version
Abstract
Molecular dynamics simulations were performed to examine the impact of temperature (ranging from 250K to 350K) on the structural behavior of cellulose chains when dissolved in an aqueous cuprammonium hydroxide Cuam solvent system. By monitoring the root mean square deviation (RMSD) and radius of gyration (Rg) values at each temperature, we found that the geometric deformation of the cellulose chains changed significantly at T=300K. In contrast, Rg and RMSD values remained stable at temperatures in the range of 250K, 270K and the range of 330K, 350K. Calculating the interaction energy between the solvent and cellulose chains revealed that temperature significantly influences the cellulose dissolution process in the Cuam solvent system and that T=300K is an efficient temperature for its dissolution. The number of intra- and interchain hydrogen bonds was also calculated as a function of temperature, and the analysis confirmed that there is no breakage of these bonds at temperatures below 300 K (i,e: 250K, 270K) and above 310 K (I,e, : 330K and 350K) either between two native chains or between a native chain and another derivative.
Keywords:
MD simulation
; Temperature Behavior of Cellulose
; Cellulose Dissolution
; Aqueous cuprammonium hydroxide solution
; Hydrogen bonding
; Cellulose
; The Interaction energy between Cellulose and solvent “Cuam”
1. Introduction
Cellulose stands as one of the most frequent biological materials on Earth, it is a material employed for thousands of years across diverse realms of human endeavors including the manufacture of paper, Film Production (Cellophane), Packaging, textile, and more [1,2,3]. Additionally, it serves as the primary raw material for the industrial production of liquid fuels using biomass conversion technologies and is a subject of considerable current interest [4]. Cellulose represents a syndiotactic biopolymer comprising anhydroglucose units (AGU) (Figure 1) linked by (1,4) β- glycosidic bonds [5]. It possesses fascinated physicochemical characteristics and appealing properties, including biocompatibility, biodegradability, and non-toxicity. [6,7]. This substance can form at least six distinct crystalline polymorphs and exists in less organized structures, commonly termed amorphous or paracrystalline cellulose [8]. It is categorized as a semi-crystalline polymer, incorporating disordered (amorphous) and well-ordered (crystalline) regions.
Inside the crystalline regions, cellulose chains form robust and complex intra- and inter-molecular hydrogen bonds acting as the cohesion among cellulose molecules, while in the amorphous domains, distribution occurs uniformly along fascinatingthe microfibers. [9,10]. The stability and compactness of the cellulose fiber arise from intra- and interchain hydrogen bonds, forming a network that significantly contributes to cellulose’s insolubility in both organic and non-organic solvents. This network also plays a crucial role in the resistance to natural microbial and enzymatic cellulose degradation [11].
Cellulose causes challenges in terms of dissolution, as it remains insoluble in both water and organic solvents, with limited solubility observed in selected classes of solvents. Available results from the literature show that there are some very important factors influencing the solubility of cellulose, such as the kinetic and thermodynamic control of the dissolution [12].
From a thermodynamic stand point, the dissolution process will occur spontaneously when the free energy change is negative [13]. That is, the process of a polymer dissolving in a solvent, is governed by the free energy of mixing where the entropy component can play a predominant role. This is particularly true in the case of semi-crystalline polymers such as cellulose where the translational and rotational entropy of polymer chains increase by dissolution of the polymer. Thus, cellulose in solution may have a much greater translational and rotational entropy than when it is enclosed in crystal form [4]. As a consequence, the dissolution of cellulose should be favored by increasing the temperature because of the increase of the entropic component of the free energy. However, several studies have shown that lowering the temperature favors the dissolution of cellulose, which suggests that the temperature vs volumetric fraction (ϕ2) profile of cellulose dissolution correspond to a lower critical solution temperature (LCST) [14]. The mechanism of this unusual behavior is still controversial, but there is strong evidence for the link between conformational changes and temperature. Indeed, segments with conformational flexibility around the C-C bond in O-CH2CH2-O can change their conformation depending on temperature. At higher temperatures, they adopt a less polar state around the C-C bond, while at lower temperatures, they move more polar states [15]. As a result, when the temperature decreases and polar states become prevalent and favorable interactions with the polar solvent are promoted by facilitating the dissolution of cellulose [12,16].
Another factor generally involved in the insolubility of cellulose is its amphiphilic property (Figure 2), which arises from the hydrophilic nature of the three hydroxyl groups situated at the equatorial positions of the ring and the hydrophobic nature of the C-H bonds positioned at the axial positions. Therefore, many studies have hypothesized that disturbing the equilibrium between the hydrophilic and hydrophobic nature of cellulose can significantly impact its dissolution [17,18]. Among the works approving such a hypothesis, Bergenstrahle et al [4] recently employed molecular dynamics simulations to calculate mean force potentials for the dissociation of cellulose oligomers in an aqueous solution.
The free energy calculation showed a significant hydrophobic pairing energy, at about 2.0 kcal/mol/residue, which favors the association of cellulose oligomer chains in a manner comparable to that found in the different crystal structures of cellulose.
It is noteworthy to mention that, according to Lindman et al [12] hydrophobic cellulose interactions are often disregarded in favor of the more robust hydrophilic hydrogen bonds, which are considered to be more crucial. From this point of view, the cellulose intra- and inter chain hydrogen bonds are considered to be a key factor for a better understanding of various physico-chemical properties associated with the solubility of cellulose and cellulose-based materials [20,21,22]. Therefore, tracking alterations in these hydrogen bonds may offer a viable approach for studying the cellulose dissolution process.
There are several classifications of cellulose solvents, according to their nature (aqueous or non-aqueous) and their effect on the cellulose structure (derivatizing or non-derivatizing) [12,23]. Derivatizing solvents are systems that modify the cellulose structure with the formation of an intermediate such as an ester, ether, or acetal to facilitate the decomposition of the cellulose and its dissolution. Among the derivatizing solvents, we can mention the DMF/N2O4 (N, N- dimethylformamide/dinitrogen tetraoxide) system, which leads to the formation of cellulose trinitrate [20,24], while the mixture of DMSO/paraformaldehyde leads to the formation of methyl cellulose soluble in non-aqueous solvents [20,25]. Non-derivatizing solvants are systems that dissolve cellulose without any chemical modification. As an exemple, The LiCl/DMAc (dimethylacetamide) [26,27], ZnCl2/H2O [28], LiSCN/H2O [26,29], LiBr/H2O, NaOH/H2O [30,31], this class of solvents dissolve the cellulose chains by breaking the intra- and interchain hydrogen bonds without any structural modification.
On the other hand, the literature review showed that the addition of urea as co-solvents or ZnO in the NaOH/H2O system reinforces the dissolution of cellulose by decreasing the hydrophobic interaction between the hydrocarbon part of the AGU [32]. Furthermore, solution of transition metal cations such as Ni (‘Nioxam’) [33], Zn (‘Zincoxen’) [34], Cd (‘Cadoxen’) [35], constitute an important class of solvent systems used in industrial processes for dissolving cellulose. The best known are cuprammonium hydroxide (Cuam) [36,37] and cupriethylene diamine hydroxide (Cuen) [38]. In this type of solvent system, complexes of cellulose AGU units and the transition metal cation is generally observed and the complexed moeity is considered as non-derivatizing intermediates. Thus, Cellulose with a degree of polymerization not exceeding 1000 AGU unit can be dissolved with this type of solvent.
Nowadays many studies have been focused on understanding the mechanism of cellulose’s solubility using molecular dynamics (MD) in various experimental conditions of solvent systems, temperature and pressure. Cai et al [39] investigated the interplay between an individual cellulose chain and a urea-water solvent mixture under different temperature conditions. The MD simulation result revealed a preference for the cellulose chains to engage in hydrogen bond formation with urea molecules more than with water molecules and maintain their stability even at low temperatures. However, when increasing the temperature in the range of 265 to 283 K the interaction between urea and cellulose decreases suggesting that urea molecules are forming an inclusive layer around the cellulose chain, thereby reinforcing the dissolution of cellulose in the urea-containing solvent mixture by minimizing self-interactions among cellulose chains. In the same theme, Bregado et al [40], examined the effect of temperature on dissollution of a 36-chain cellulose Iβ micro-fibril crystal model at 25 MPa in the range of temperature between 298 to 660 K. The results of this MD investigation showed that the cellulose chains dissolved completely at temperatures close to 600 K. The mechanism of the cellulose dissolution starts between 560 K and 580 K where the initial hydration layer separates from the outer chains situated on the hydrophilic planes through hydrogen bond interactions, which promotes the relaxation of the crystal lattice. Both effects potentially contribute to the advantageous dissolution of cellulose in compressed water. Ramakrishnan et al [41] performed MD simulations of cellulose in the mixture of an ionic liquid 1-ethyl-3-methylimidazolium acetate [C2C1Im][OAc] with water at two distinct temperatures, 300K and 433K. The results showed that the RMSD, and the number of cellulose intra and interchain bonds during the dissolution process are primarily influenced by the temperature. Thus, at T=300K the cellulose chains remain intact whereas at 433K a disruption of the chains takes place. The decrease in the number of H-bonds at higher temperature was due to the weakening of the interactions inherent in cellulose H-bonds and the interaction accumulated by IL with cellulose chains at higher temperatures.
The present work aims to report the results obtained in a molecular dynamics study of the effect of temperature on the dissolution of cellulose in Cuam solvent system. Our primary emphasis is on the Cuam solvent system and its mechanism of solubilizing cellulose typically characterized by the formation of cuprammonium complexes with AGU units of cellulose. These complexes lead to the deprotonation of the hydroxyl groups in the coordination bond connected to the C2 and C3 hydroxyl groups of each AGU. [42] (Figure 3) leading to a disruption in the interchain hydrogen bonds network of cellulose and therefore the segregation of the close packed chains of the cristalline structure.
2. Material and Methods
Throughout this study, molecular dynamics (MD) simulations at different temperatures were performed with the Schrödinger suite software (2018.4). The cellulose structure used in these simulations was extracted from an Iβ crystal structure constructed using a cellulose builder [44]. Parameterised by the OPLS-2005 force field [45] This model structure consists of 6 chains with 8 glucose units each. The Cuam solvent system contains water molecules modeled according to the TIP3P model. OH-, SO42-, and Na+ molecules were simulated using the Material package integrated in the Schrödinger 2018.4 software with the same force field. For all simulations, a cubic box of dimensions (8.12 nm × 8.12 nm × 8.12 nm) was used with periodic boundary conditions (PBC). This box contains 15901 atoms. Each simulation was assigned a different temperature, ranging from 250K to 350K. The pressure was controlled with a Berendsen barostat P=1bar, with a time of 10ns and a time interval of 2.0 fs, under a canonical ensemble (NVT) and the simulations were performed using the Desmond package.
2.1. Root Mean Square Deviation RMSD
The root mean square deviation RMSD indicates the mean deviation of a particle from a reference position. It is an important parameter that allows the control of structural changes during simulation processes and also to follow the diffusion of particles in a given solvent [46]. Several authors have studied the average RMSD of cellulose polymer in different solvent systems; as an example Manna and Ghosh [47] showed that the dissolution of cellulose in water-[C2mim][OAc] (1-ethyl-3-methylimidazolium (Emim+)) solvent is associated with high RMSD values.
In this study, the comparison between RMSD of cellulose chains containing 8 units of glucose dissolved in an aqueous Cuam solution at different temperatures ranging from 250k to 350K showed that the average values fluctuate around (1-3.5Ǻ) at T=250K, 270K, and 280K (Figure 4A). The time-dependent variation of the RMSD indicates stability across all systems, with minor fluctuations remaining below 3Å, suggesting the absence of significant dissolution at these temperatures. On the other hand, at higher temperatures, from 320K to 350K (Figure 4B), the cellulose also faces stability of RMSD values which fluctuated around (2-4Ǻ), indicating that no dissolution occurs.
However, between 300K and 310K (Figure 4C), the RMSD curve indicates an initial increase in RMSD vs of simulation’s time. For example, at a temperature of T=300K, the RMSD value increases to (22.5 Å) before stabilizing. Therefore, a temperature of 300K is considered an effective temperature for the dissolution of cellulose in the Cuam system.
According to Manna and Ghosh [47], the high average RMSD values obtained in our study indicate a modification in the conformation of the cellulose crystal structure within the Cuam solvent system leading to the dissolution of cellulose. This observation is confirmed by the visual representations of the final structures in the simulations at 300K, 330K, 350K, 280 and 250K (Figure 5), which show an aggregated form of the cellulose chains at 280K, 250K, 330K and 350K, while a significant separation of the chains is observed at T=300K.
2.2. Radius of Gyration Rg
In addition to the RMSD results, we also studied the radius of gyration (Rg) of cellulose in Cuam solvent system at different temperatures from 250K up to 350K, to observe the effect of temperature in the dissolution processus during simulation. The radius of gyration (Rg) is a measure defined as the root mean square distance of the atoms of a molecule from their common center of mass [48]. The radius of gyration (Rg) provides a general overview of the compactness of a structure by applying the following formula [49]:
The simulation of cellulose chains at low temperatures between 250 to 280K reveals that the Rg values (12-12.6 Ǻ) are very stable, with a fluctuation around 0.6 Ǻ (Figure 6A). Therefore, the stability of the values indicates that the cellulose is still compact, and no dissolution has occurred in the Cuam solvent system at low temperature. The Rg value increases to 30 Ǻ (Figure 6C) at a temperature between 300K and 310K. Due to the dissolution of cellulose chains which induces significant changes of the conformational arrangement of cellulose crystalline structure.
Above 310 K, the Rg values of the system decrease significantly (Figure 6B) and range in the same order than for temperatures below 300 K. It is noted that when the temperature is increased to values higher than T=310K, the cellulose becomes more compact due to cellulose insolubility.
2.3. Interaction Energy
The dissolution of cellulose is the process of breaking the chain bonds of cellulose into individual chains. Indeed, the interaction energy represents the bonding strength between the cellulose chains and the increase in this interaction energy, leads to a close packing of the cellulose chains [24]. Therefore, to get a better understanding of the effect of temperature in the dissolution of cellulose in the Cuam solvent system, we examined the interaction energy between the cellulose chains and solvent at different temperatures along the simulation process (Figure 7).
According to the presented curve (Figure 7A), it can be observed that the interaction energy between the Cuam solvent and cellulose chains remains stable at temperatures of 280K, 270K, and 250K. However, at temperatures ranging from 300K to 310K (Figure 7B), the interaction energy starts to decrease, from a value of -300 Kcal/mol to -350 Kcal/mol, indicating that cellulose chains have a preference for interaction with the solvent within this temperature range. In contrast, when the temperature increases from 320K to 350K (Figure 7C), the cellulose-solvent interaction energy remains stable compared to the T=300K and T=310K. This means that the binding force between cellulose chains is stronger, and therefore, cellulose is not dissolved in the solvent. The cellulose-solvent interaction energy at different temperatures is in agreement with the analysis of Rg cellulose-solvent and RMSD calculations, where cellulose interacts more with the solvent at T=300K than with the solvent at low and high temperatures.
2.4. Hydrogen Bonds
Intermolecular hydrogen bonds, along with numerous C-H...O inter-shells and van der Waals interactions, contribute to the stabilization of cellulose crystal Iβ [12,20]. Thus, during the dissolution process, the initial step involves breaking these hydrogen bonds to initiate the dissolution process. This investigation delves into the variation in the average number of hydrogen bonds focusing on two key intrachain hydrogen bonding patterns and two primary interchain patterns between two cellulose chains (native and derivatized one and between two natives’ ones).
In our study, for temperatures below 300 K (i.e. 250K and 270K) and above 310 K (i.e. 330K and 350K), we observed that the number of interchain hydrogen bonds remained constant (see Figure 8A–C), as the intrachain hydrogen bonds remain approximately stable arround the mean value of 8 H-bonds, whether between two native chains or between a native chain and a derivatized chain (see Figure 8D–F). This stability is attributed to the absence of breakage of H-Bond bonds at these temperatures, which is due to insolubility of cellulose in the Cuam solution.
On the contrary, at T = 300 K, a significant change in the intra-chain and inter-chain hydrogen bonding patterns between a native chain and a derivatized chain during simulation was observed. From (Figure 9A), which represents the variation in the number of interchain bonds throughout the simulation time, we can see a decrease in the number of H bonds over the simulation time. This decrease can be attributed to the breaking of interchain bonds. Analysis of the intrachain hydrogen bonds (Figure 9B) also shows a decrease in the number of H bonds. An increase in the disruption of interchain and intrachain hydrogen bonds leads to greater accessibility for hydroxyl groups to interact with the solvent, thus facilitating the dissolution process.
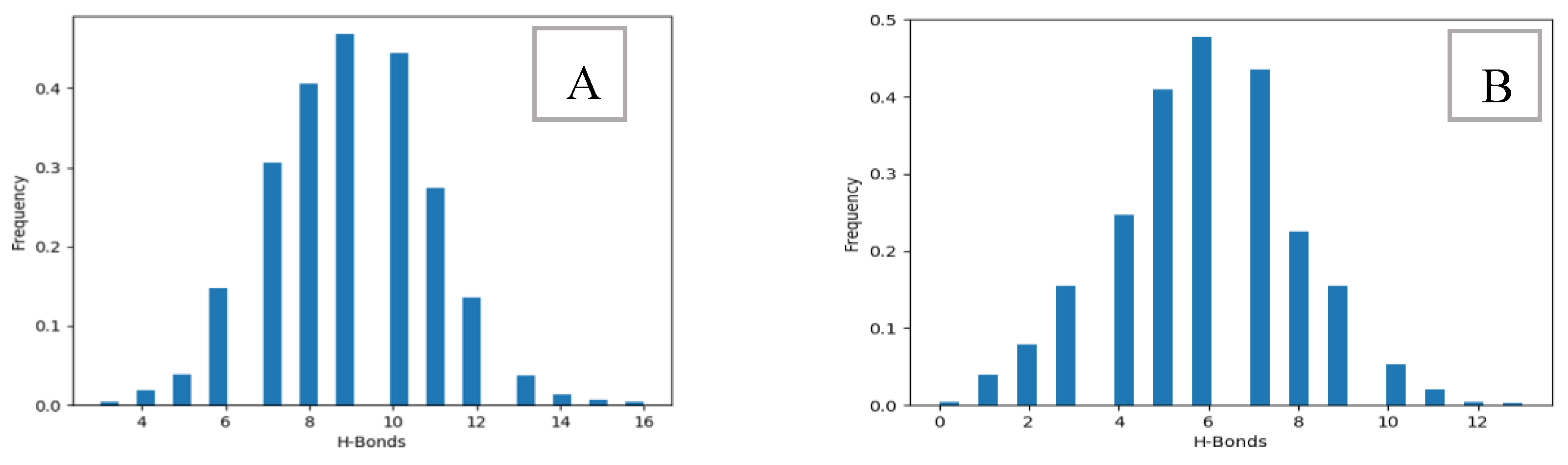
To better understand the changes in H bonds at T = 300K, we measured the number of interchain hydrogen bonds. By presenting the frequency of each occurrence in histograms (Figure 11A,B) it is possible to get an insight of the behavior of the cellulose crystals. For interchain bonds between two native chains, the histogram shows values ranging from 1 to 16, and between the native and the derivative, the values are lower, ranging from 1 to 12 in (Figure 11B).
Both frequency distributions exhibit a symmetrical shape, demonstrating a normal distribution with distinct peaks. In (Figure 11A), the peak is centered at 9 H-bonds, while in (Figure 11B), it is centered at 6 H-bonds. As per existing literature, the stable conformation of cellulose involves intermolecular hydrogen bonds (C-O---HO) in a parallel arrangement. The dissolution of cellulose results from the solvent’s ability to disrupt these intermolecular hydrogen bonds (C-O---HO) among cellulose molecules and eliminate hydrophobic interactions (C-H--- C). In our study at T=300K, the organization of native/derived cellulose chains underwent significant changes, making them less inclined to align in parallel due to the replacement of hydrogen with Cu(NH3)2. As explained in our prior article [36], the new Cuam complex can permeate native cellulose because of increased chain rotation. Consequently, new, albeit weaker, hydrogen bonds are formed, primarily (N-H----O-CH) and (CH2-CH-O NH), explaining the ease of breaking these bonds and subsequently dissolving cellulose. This work is the continuation of others ones in our group [50,51,52].
3. Conclusions
In this study, molecular dynamics simulation was conducted using cellulose (Iβ) in the Cuam solvent at different temperatures to gain a molecular-level understanding of the role of temperature in the efficient dissolution of cellulose. Cellulose dissolution occurred at temperatures of 300K and 310K, however, insolubility of cellulose was noted when the temperature was increased or decreased beyond the temperature range between 300K and 310K. Comparison of the RMSD and Rg values of the system at different temperatures suggests that deconstruction of cellulose chains occurs at 300K, while at temperatures between 250K and 280K, as well as between 320K and 350K, the values remain stable, indicating no conformational changes in these temperature ranges. The analysis of the interaction energy between the solvent and cellulose chains is also consistent with the RMSD and Rg results. The interaction energy between cellulose and the Cuam solvent decreases at T=300K-310K, indicating that cellulose chains prefer to interact with the solvent. Counting the number of intrachain and interchain hydrogen bonds during the simulation at T=300K-310K revealed a decrease in the number of interchain hydrogen bonds due to complexation of hydroxyl groups (linked to the C2 and C3 carbons of each anhydroglucose) by Cu(NH3)2, leading to chain deformation and disruption in the interchain hydrogen bond network of the cellulose crystal.
Author Contributions
LB: Conceptualization, Methodology, Software, Validation, Formal Analysis, Investigation, Resources, Data Curation, Writing - Original Draft, Writing - Review & Editing. RA, MEM: Investigation, Resources, Writing - Original Draft. MM, HB: Methodology, Software, Formal Analysis. BB, RT, LE: Project Administration, Validation, Writing - Review & Editing. AC: Project Administration, Supervision, Validation, Writing - Review & Editing. SBM and AYA: Funding and Editing.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through the large Group Research Project under grant number (RGP.2/344/45).
Conflicts of Interest
the authors declare no conflict of interest.
References
- Gericke, M., Fardim, P., & Heinze, T. (2012). Ionic liquids - Promising but challenging solvents for homogeneous derivatization of cellulose. In Molecules (Vol. 17, Issue 6, pp. 7458–7502). [CrossRef]
- Pinkert, A., Marsh, K. N., & Pang, S. (2010). Reflections on the solubility of cellulose. Industrial and Engineering Chemistry Research, 49(22), 11121–11130. [CrossRef]
- Venkatarajan, S., & Athijayamani, A. (2020). An overview on natural cellulose fiber reinforced polymer composites. Materials Today: Proceedings, 37(Part 2), 3620–3624. [CrossRef]
- Bergenstråhle, M., Wohlert, J., Himmel, M. E., & Brady, J. W. (2010). Simulation studies of the insolubility of cellulose. Carbohydrate Research, 345(14), 2060–2066. [CrossRef]
- Heinze, T. (2015). Cellulose: Structure and properties. Advances in Polymer Science, 271, 1–52. [CrossRef]
- Fu, L. H., Qi, C., Ma, M. G., & Wan, P. (2019). Multifunctional cellulose-based hydrogels for biomedical applications. In Journal of Materials Chemistry B (Vol. 7, Issue 10, pp. 1541–1562). Royal Society of Chemistry. [CrossRef]
- Seddiqi, H., Oliaei, E., Honarkar, H., Jin, J., Geonzon, L. C., Bacabac, R. G., & Klein-Nulend, J. (2021). Cellulose and its derivatives: towards biomedical applications. In Cellulose (Vol. 28, Issue 4, pp. 1893–1931). Springer Science and Business Media B.V. [CrossRef]
- Ruel, K., Nishiyama, Y., & Joseleau, J. P. (2012). Crystalline and amorphous cellulose in the secondary walls of Arabidopsis. Plant Science, 193–194, 48–61. [CrossRef]
- Liu, R., Yu, H., & Huang, Y. (n.d.). Structure and morphology of cellulose in wheat straw. [CrossRef]
- Poletto, M., Pistor, V., & J., A. (2013). Structural Characteristics and Thermal Properties of Native Cellulose. In Cellulose - Fundamental Aspects. InTech. [CrossRef]
- Bochek, A. M. (2003). Effect of Hydrogen Bonding on Cellulose Solubility in Aqueous and Nonaqueous Solvents.In Nauka/Interperiodica] Russian Journal of Applied Chemistry (Vol. 76, Issue 11). [CrossRef]
- Lindman, B., Karlström, G., & Stigsson, L. (2010). On the mechanism of dissolution of cellulose. Journal ofMolecular Liquids, 156(1), 76–81. [CrossRef]
- Medronho, B., & Lindman, B. (2014). Competing forces during cellulose dissolution: From solvents to mechanisms.In Current Opinion in Colloid and Interface Science (Vol. 19, Issue 1, pp. 32–40). Elsevier Ltd. [CrossRef]
- Väisänen, S., Ajdary, R., Altgen, M., Nieminen, K., Kesari, K. K., Ruokolainen, J., Rojas, O. J., & Vuorinen, T. (2021). Cellulose dissolution in aqueous NaOH–ZnO: cellulose reactivity and the role of ZnO. Cellulose, 28(3), 1267–1281. [CrossRef]
- Lindman, B., & Karlström, G. (2009). Nonionic polymers and surfactants: Temperature anomalies revisited. In Comptes Rendus Chimie (Vol. 12, Issues 1–2, pp. 121–128). [CrossRef]
- Medronho, B., Romano, A., Miguel, M. G., Stigsson, L., & Lindman, B. (2012). Rationalizing cellulose (in)solubility: Reviewing basic physicochemical aspects and role of hydrophobic interactions. Cellulose, 19(3), 581–587. [CrossRef]
- Biermann, O., Hädicke, E., Koltzenburg, S., Müller-Plathe, F., Müller-Plathe, F., Biermann, Dipl.-C. O., Hädicke, E., & Koltzenburg, S. (2001). Hydrophilicity and Lipophilicity of Cellulose Crystal Surfaces. In Angew.Chem. Int. Ed (Vol. 40, Issue 20). [CrossRef]
- Miyamoto, H., Umemura, M., Aoyagi, T., Yamane, C., Ueda, K., & Takahashi, K. (2009). Structural reorganization of molecular sheets derived from cellulose II by molecular dynamics simulations. Carbohydrate Research, 344(9), 1085–1094. [CrossRef]
- Yamane, C., Aoyagi, T., Ago, M., Sato, K., Okajima, K., & Takahashi, T. (2006). Two different surface properties of regenerated cellulose due to structural anisotropy. Polymer Journal, 38(8), 819–826. [CrossRef]
- Wohlert, M., Benselfelt, T., Wågberg, L., Furó, I., Berglund, L. A., & Wohlert, J. (2022). Cellulose and the role of hydrogen bonds: not in charge of everything. In Cellulose (Vol. 29, Issue 1). Springer Science and Business Media B.V. [CrossRef]
- Nishiyama, Y., Johnson, G. P., French, A. D., Forsyth, V. T., & Langan, P. (2008). Neutron crystallography, molecular dynamics, and quantum mechanics studies of the nature of hydrogen bonding in cellulose Iβ. Biomacromolecules, 9(11), 3133–3140. [CrossRef]
- Yano, S., & Hatakeyama, H. (1976). Effect of Hydrogen Bond Formation on Dynamic Mechanical Properties of Amorphous Cellulose (Vol. 20). [CrossRef]
- Thomas Heinze, A. K. (2005). Solvents Applied in the Field of Cellulose Chemistry - A Mini Review. Polímeros: Ciência e Tecnologia, . 15, 84–90. [CrossRef]
- Thomas Heinze, A. K. (2005). Solvents Applied in the Field of Cellulose Chemistry - A Mini Review. Polímeros: Ciência e Tecnologia, . 15, 84–90. [CrossRef]
- Ghasemi, M., Tsianou, M., & Alexandridis, P. (2017). Assessment of solvents for cellulose dissolution. Bioresource Technology, 228, 330–338. [CrossRef]
- Dawsey, T. R., & McCormick, C. L. (1990). The lithium chloride/dimethylacetamide solvent for cellulose: A literature review. Journal of Macromolecular Science, Part C, 30(3–4), 405–440. [CrossRef]
- Mohd, N., Draman, S. F. S., Salleh, M. S. N., & Yusof, N. B. (2017). Dissolution of cellulose in ionic liquid: A review. AIP Conference Proceedings, 1809. [CrossRef]
- Leipner, H., Fischer, S., Brendler, E., & Voigt, W. (2000). Structural changes of cellulose dissolved in molten salt hydrates. In Macromol. Chem. Phys (Vol. 201). [CrossRef]
- Liebert, T. (2010). Cellulose solvents-remarkable history, bright future. ACS Symposium Series, 1033, 3–54. [CrossRef]
- Kihlman, M., Medronho, B. F., Romano, A. L., Germgård, U., & Lindman, B. (2013). Cellulose dissolution in an alkali based solvent: Influence of additives and pretreatments. Journal of the Brazilian Chemical Society, 24(2), 295–303. [CrossRef]
- Yang, Y. J., Shin, J. M., Kang, T. H., Kimura, S., Wada, M., & Kim, U. J. (2014). Cellulose dissolution in aqueous lithium bromide solutions. Cellulose, 21(3), 1175–1181. [CrossRef]
- Zhang, S., Li, F. X., Yu, J. yong, & Hsieh, Y. Lo. (2010). Dissolution behaviour and solubility of cellulose in NaOH complex solution. Carbohydrate Polymers, 81(3), 668–674. [CrossRef]
- Wang, S., Lu, A., & Zhang, L. (2016). Recent advances in regenerated cellulose materials. In Progress in Polymer Science (Vol. 53, pp. 169–206). Elsevier Ltd. [CrossRef]
- Klufers, P., Mayer, P., & Schuhmacher, J. (1995). Coordination Equilibria in Transition Metal Based Cellulose Solvents” 1. Modifying the Polysaccharide Hydrogen Bond System: Classes of Cellulose Solvents. In Akzo AG, Obernburg and Wuppertal.-Part (Vol. 99). Huthig & Wepf Verlag.
- Achwal, W. B., & Gupta, A. B. (1968). Studies in a modified cadoxen solvent. In Die Angewandte Makromolekulare Chemie (Vol. 2). [CrossRef]
- Bourassi, L., Challioui, A., Merzouki, M., Abidi, R., Bouammali, B., Elfarh, L., & Amin Bouammali, M. (2022). A molecular dynamics (MD) simulation of the solubility behaviours of cellulose in aqueous cuprammonium hydroxide solution. Materials Today: Proceedings. [CrossRef]
- Sayyed, A. J., Deshmukh, N. A., & Pinjari, D. V. (2019). A critical review of manufacturing processes used in regenerated cellulosic fibres: viscose, cellulose acetate, cuprammonium, LiCl/DMAc, ionic liquids, and NMMO based lyocell. In Cellulose (Vol. 26, Issue 5, pp. 2913–2940). Springer Netherlands. [CrossRef]
- Eckelt, J., Knopf, A., Röder, T., Weber, H. K., Sixta, H., & Wolf, B. A. (2011). Viscosity-molecular weight relationship for cellulose solutions in either NMMO monohydrate or cuen. Journal of Applied Polymer Science, 119(2), 670–676. [CrossRef]
- Cai, L., Liu, Y., & Liang, H. (2012). Impact of hydrogen bonding on inclusion layer of urea to cellulose: Study of molecular dynamics simulation. Polymer, 53(5), 1124–1130. [CrossRef]
- Bregado, J. L., Tavares, F. W., Secchi, A. R., & Segtovich, I. S. V. (2021). Molecular dynamics of dissolution of a 36-chain cellulose Iβ microfibril at different temperatures above the critical pressure of water. Journal of Molecular Liquids, 336. [CrossRef]
- Parthasarathi, R., Balamurugan, K., Shi, J., Subramanian, V., Simmons, B. A., & Singh, S. (2015). Theoretical Insights into the Role of Water in the Dissolution of Cellulose Using IL/Water Mixed Solvent Systems. Journal of Physical Chemistry B, 119(45), 14339–14349. [CrossRef]
- Dias, Y. J., Kolbasov, A., Sinha-Ray, S., Pourdeyhimi, B., & Yarin, A. L. (2020). Theoretical and experimental study of dissolution mechanism of cellulose. Journal of Molecular Liquids, 312. [CrossRef]
- Parthasarathi, R., Balamurugan, K., Shi, J., Subramanian, V., Simmons, B. A., & Singh, S. (2015). Theoretical Insights into the Role of Water in the Dissolution of Cellulose Using IL/Water Mixed Solvent Systems. Journal of Physical Chemistry B, 119(45), 14339–14349. [CrossRef]
- Comput. Chem. 33 (2012) 1338–1346. [CrossRef]
- Kevin J. Bowers. (2006). Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. [CrossRef]
- Sánchez-Badillo, J. A., Gallo, M., Rutiaga-Quiñones, J. G., & López-Albarrán, P. (2021). Solvent behavior of an ionic liquid set around a cellulose Iβ crystallite model through molecular dynamics simulations. Cellulose, 28(11), 6767–6795. [CrossRef]
- Manna, B., & Ghosh, A. (2019). Dissolution of cellulose in ionic liquid and water mixtures as revealed by molecular dynamics simulations. Journal of Biomolecular Structure and Dynamics, 37(15), 3987–4005. [CrossRef]
- Li, W., Huang, S., Xu, D., Zhao, Y., Zhang, Y., & Zhang, L. (2017). Molecular dynamics simulations of the characteristics of sodium carboxymethyl cellulose with different degrees of substitution in a salt solution. Cellulose, 24(9), 3619–3633. [CrossRef]
- Roe, D. R., & Cheatham, T. E. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. [CrossRef]
- Meziane, H.; Laita, M.; Azzaoui, K.; Boulouiz, A.; Neffa, M.; Sabbahi, R.; Nandiyanto, ABD.; EL Idrissi, A.; Abidi, N.; Siaj, M.; Touzani, R. (2024) Nanofibers from cellulosic materials: A comprehensive review about preparation, characterization and applications. Moroccan J. Chemistry. 12(1) 305-343. [CrossRef]
- El Yousfi R., Brahmi M., Dalli M., Achalhi N.3, Azougagh O., Tahani A., Touzani R., El Idrissi A. (2023). Recent Advances in Nanoparticle Development for Drug Delivery: A Comprehensive Review of Polycaprolactone-Based Multi-Arm Architectures. Polymers. 15(8)1835. [CrossRef]
- Benahmed A., Azzaoui K., El Idrissi A., Belkheir H., Said Hassane SO., Touzani R., Rhazi L. (2022) Cellulose Acetate-g-Polycaprolactone Copolymerization Using Diisocyanate Intermediates and the Effect of Polymer Chain Length on Surface, Thermal, and Antibacterial Properties. Molecules. 27(4)1408. [CrossRef]
Figure 1.
Unite anhydroglucose (AGU).
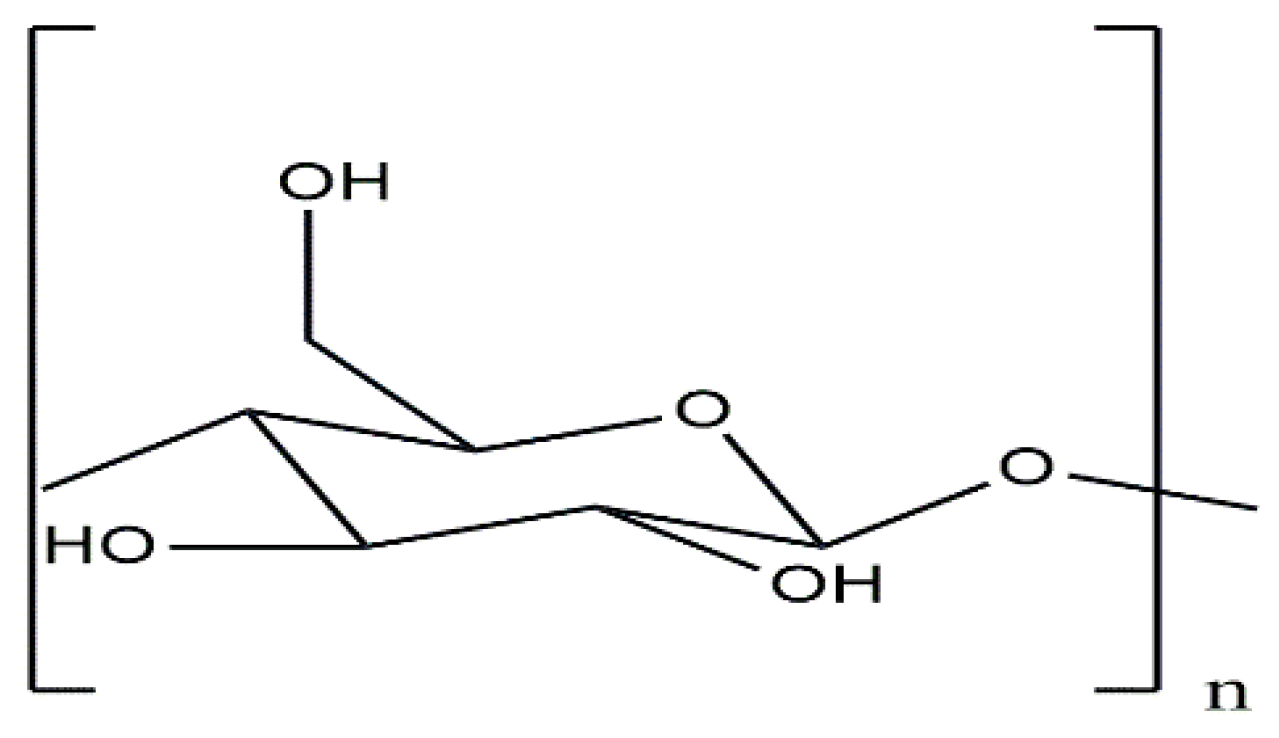
Figure 2.
(a) The hydrophobic regions of the cellulose molecule, (b) The hydrophilic regions of the cellulose molecule [19].
Figure 2.
(a) The hydrophobic regions of the cellulose molecule, (b) The hydrophilic regions of the cellulose molecule [19].
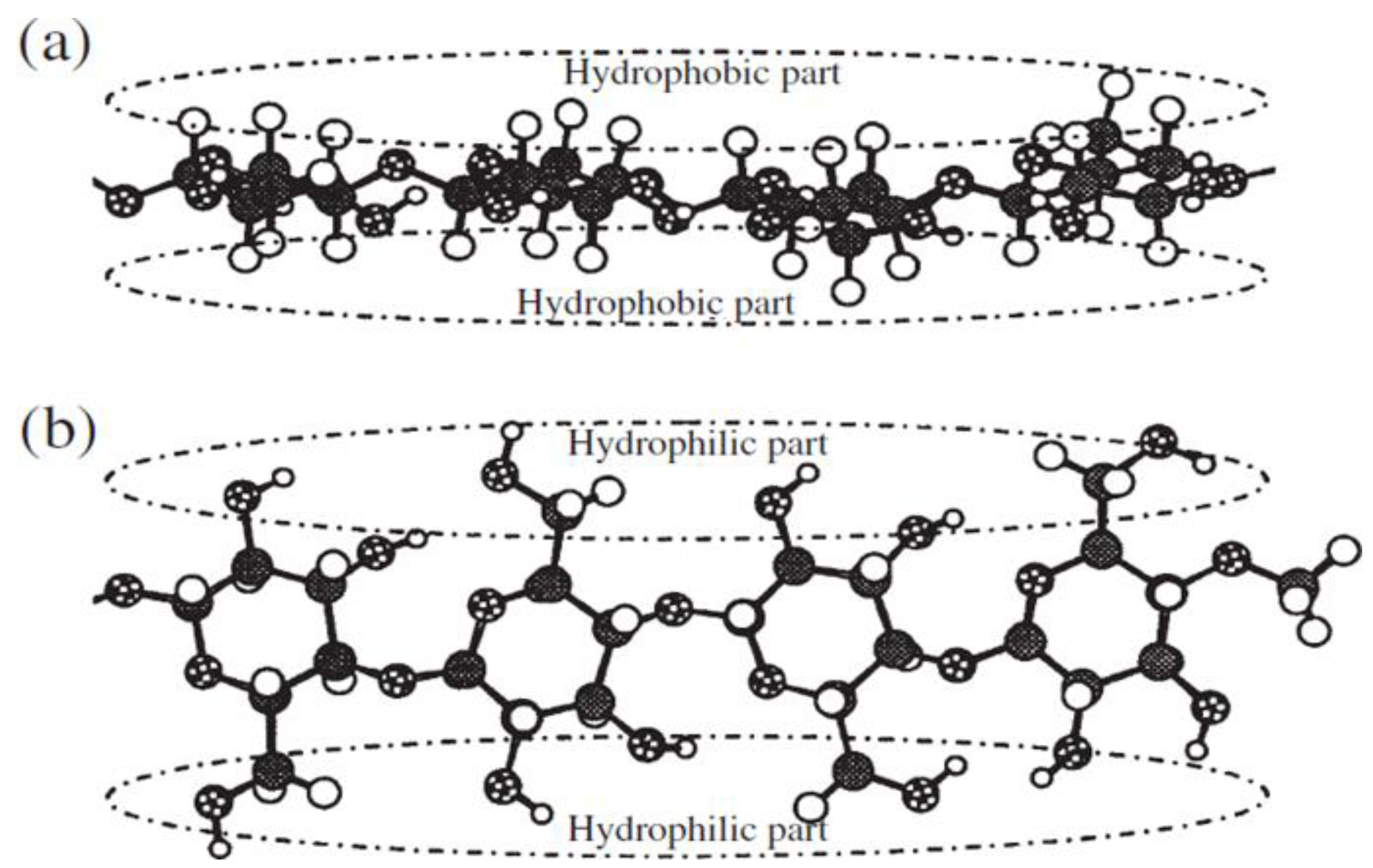
Figure 3.
Complex formation of cellulose in cuprammonium hydroxide (Cuam) [43].
Figure 3.
Complex formation of cellulose in cuprammonium hydroxide (Cuam) [43].
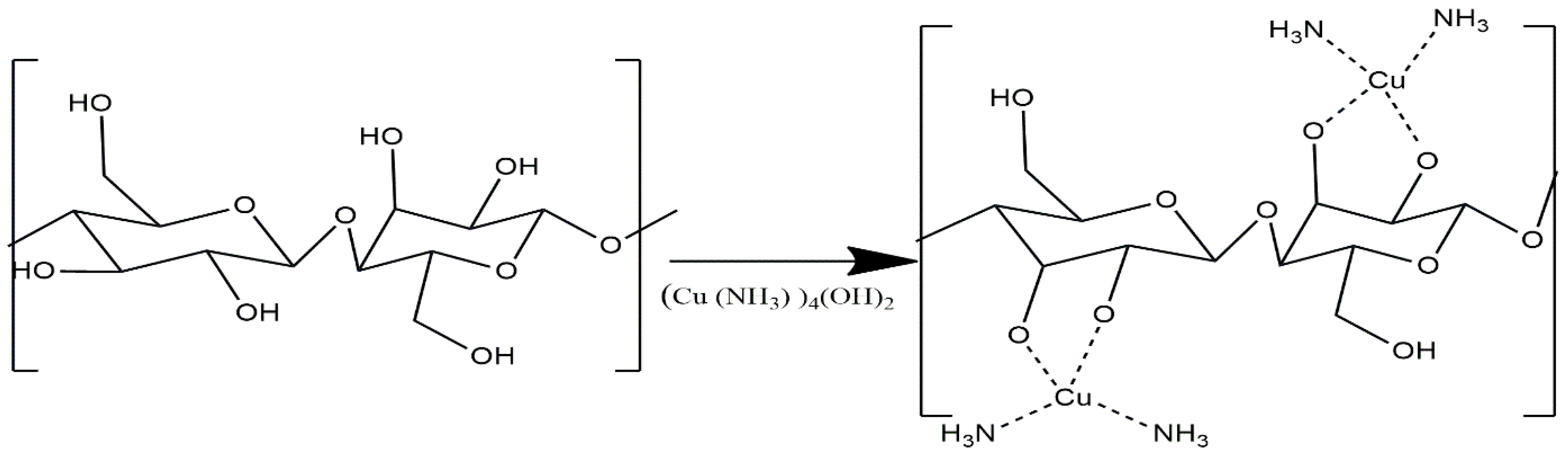
Figure 4.
(A): Root Mean Square Deviation RMSD for MD trajectories of cellulose at different temperatures 280K, 270K, 280K, (B): 330K,335K and 350k, (C): 300K and 310K.
Figure 4.
(A): Root Mean Square Deviation RMSD for MD trajectories of cellulose at different temperatures 280K, 270K, 280K, (B): 330K,335K and 350k, (C): 300K and 310K.

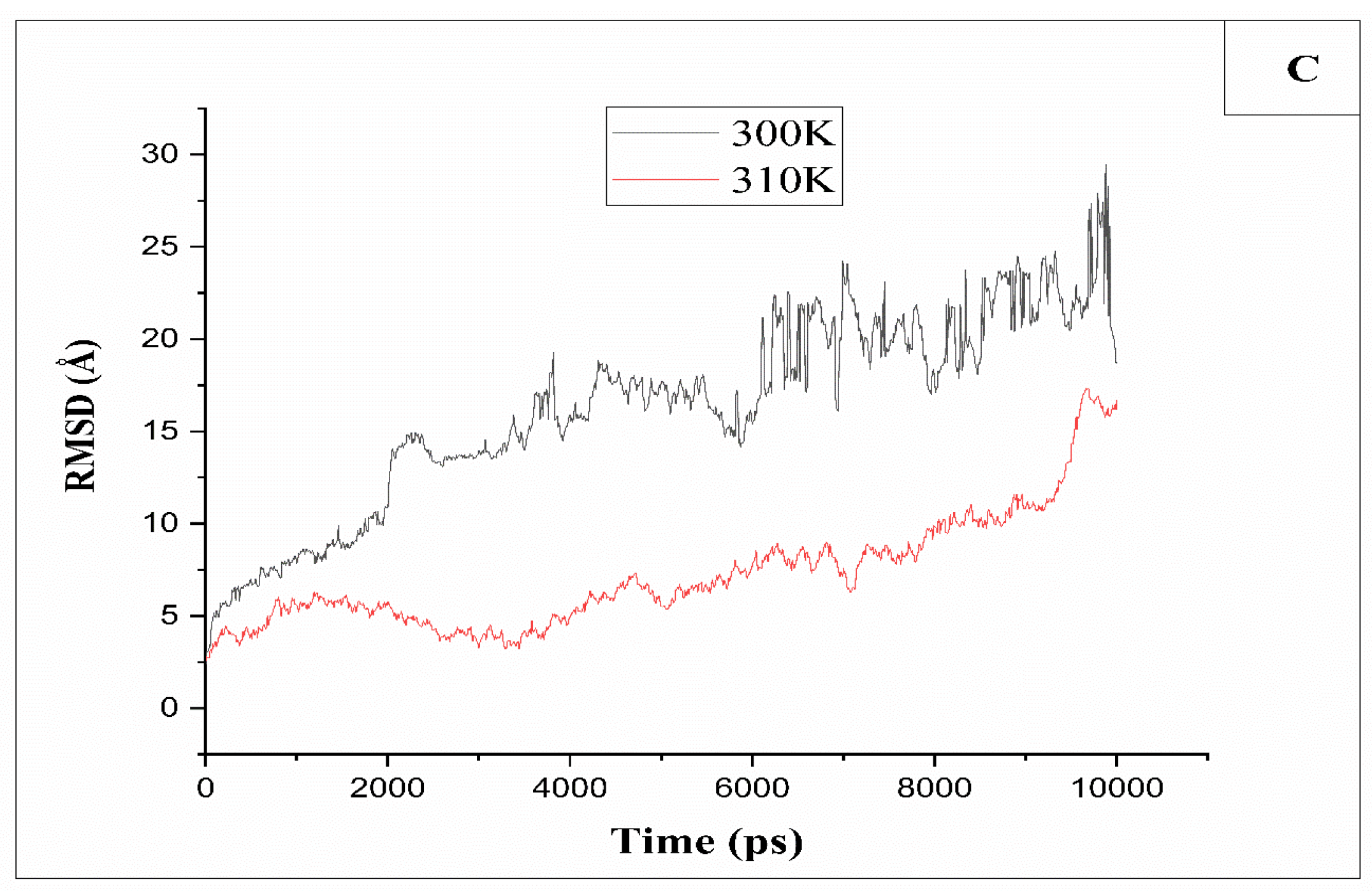
Figure 5.
Images capturing the cellulose structures at the end of the simulation under varying temperatures.
Figure 5.
Images capturing the cellulose structures at the end of the simulation under varying temperatures.
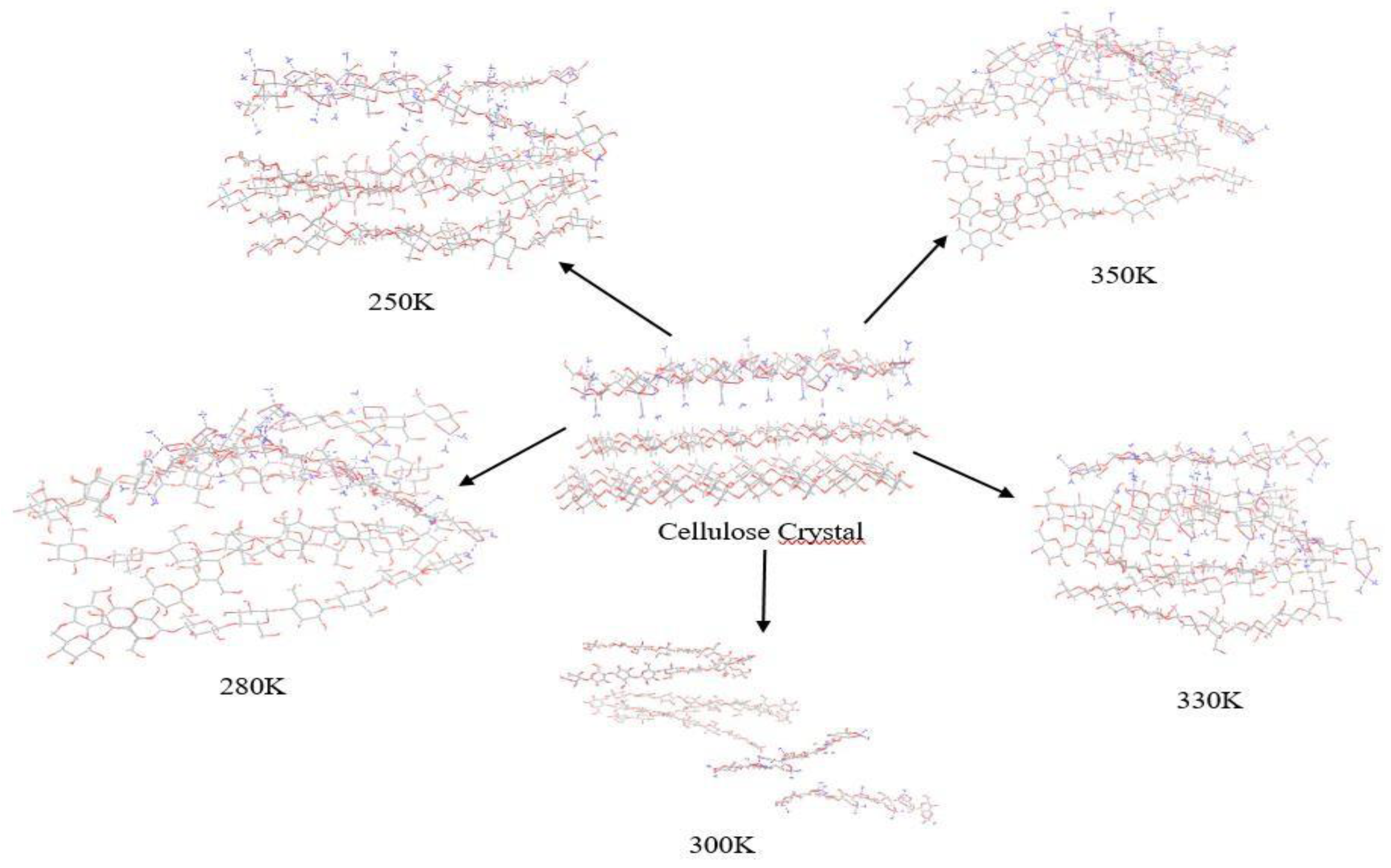
Figure 6.
(A): Radius of gyration (Rg) for MD trajectories of cellulose at different temperatures. 280K, 270K, and 250K, (B): T=335K and 350K, (C): T=300K and 310K.
Figure 6.
(A): Radius of gyration (Rg) for MD trajectories of cellulose at different temperatures. 280K, 270K, and 250K, (B): T=335K and 350K, (C): T=300K and 310K.
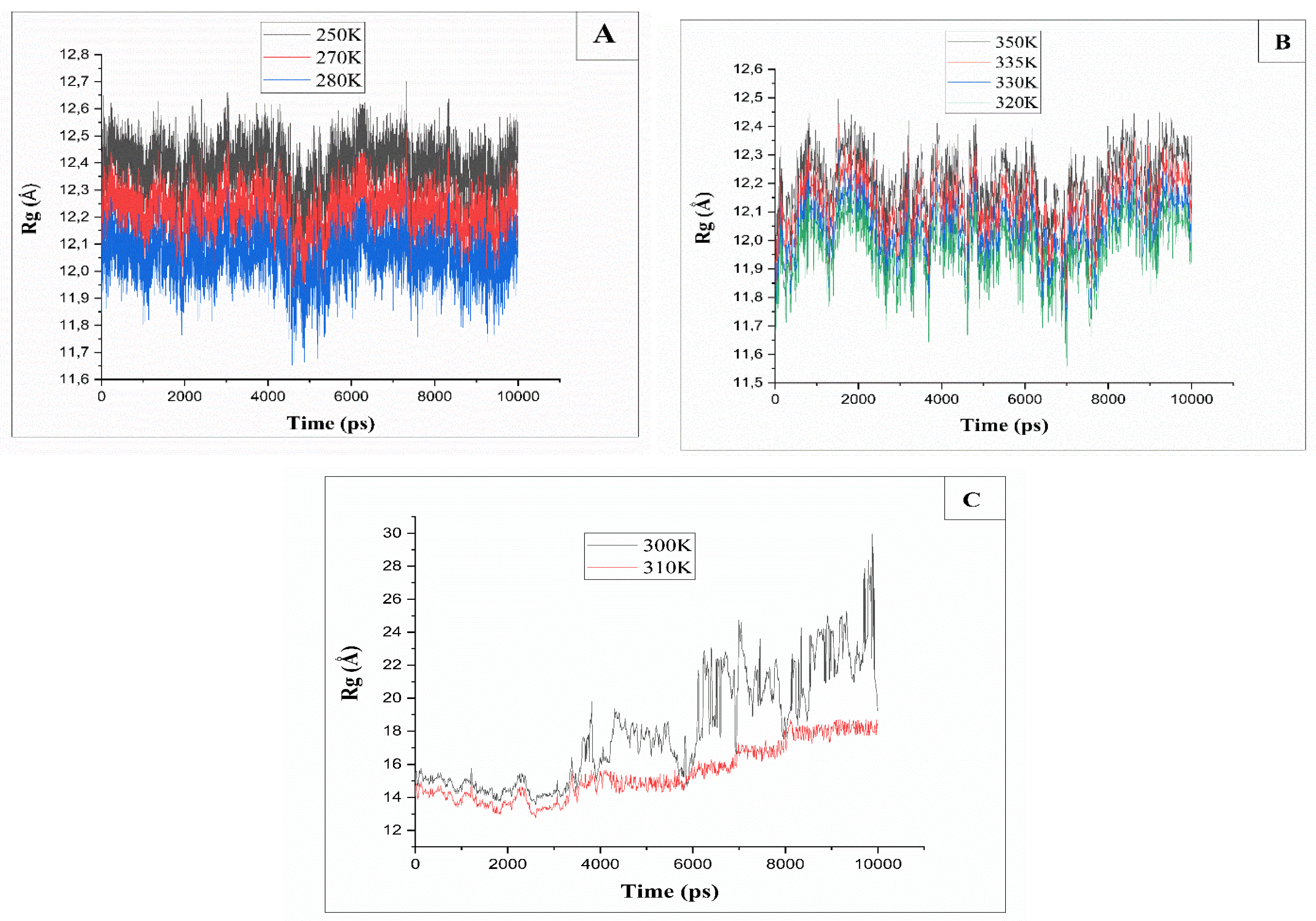
Figure 7.
(A): Interaction energy (I.E) of cellulose with different temperatures. 310k, 300K, 280K, 270K and 250K. (B): Interaction energy (I.E) of cellulose with different temperatures. 310K and 300K. (C): Interaction energy (I.E) of cellulose with different temperatures. 300K, 310K, 320K, 330K, 335K and 350K.
Figure 7.
(A): Interaction energy (I.E) of cellulose with different temperatures. 310k, 300K, 280K, 270K and 250K. (B): Interaction energy (I.E) of cellulose with different temperatures. 310K and 300K. (C): Interaction energy (I.E) of cellulose with different temperatures. 300K, 310K, 320K, 330K, 335K and 350K.
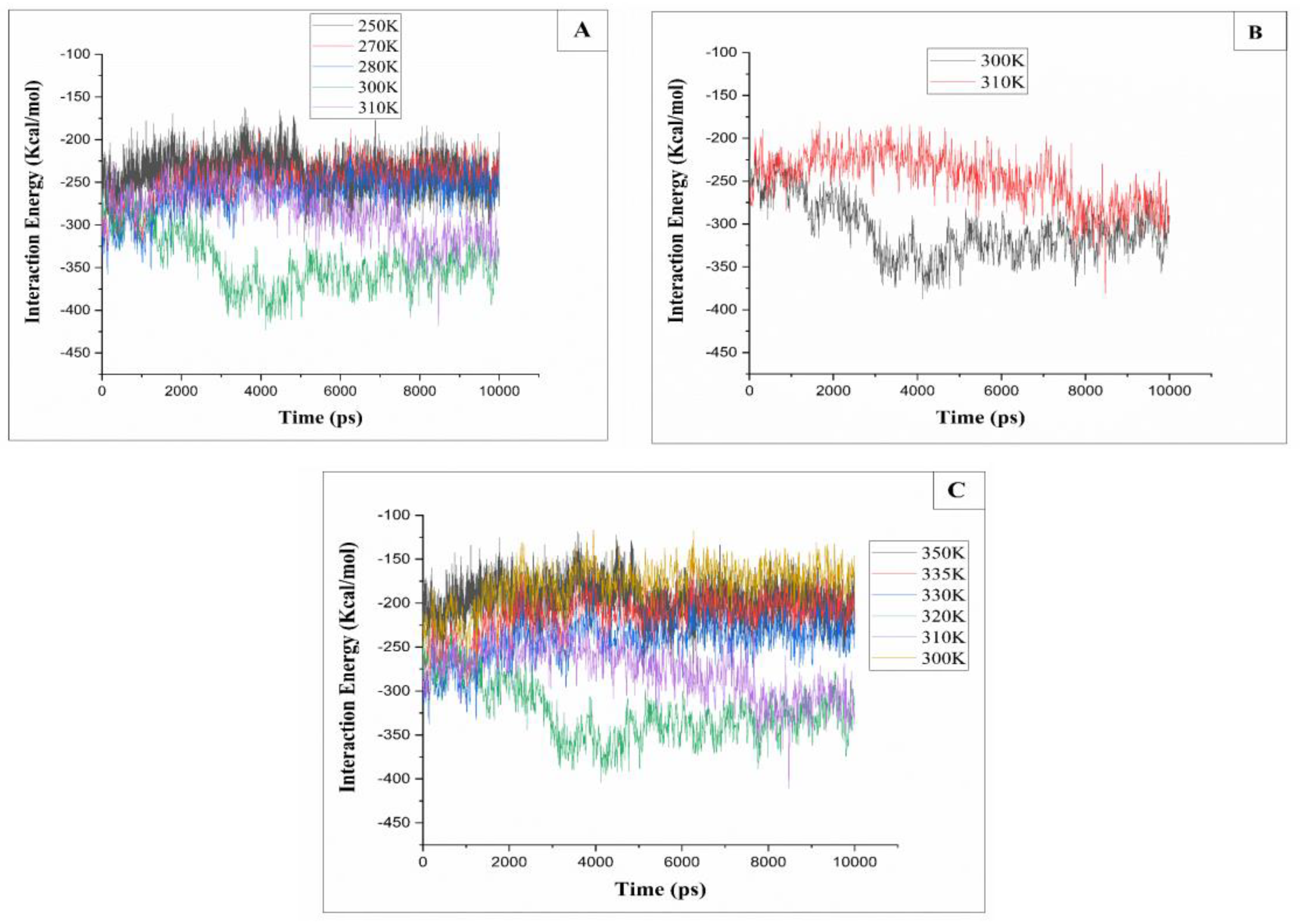
Figure 8.
The progression of intermolecular hydrogen bonds between native and derivatized cellulose chains at (A)T=250 K, (B): T=280K, (C): T=350K, (D): intramolecular hydrogen bonds between native and derivatized chains at T=250K, (E): T=280K, (F): T= 350K.
Figure 8.
The progression of intermolecular hydrogen bonds between native and derivatized cellulose chains at (A)T=250 K, (B): T=280K, (C): T=350K, (D): intramolecular hydrogen bonds between native and derivatized chains at T=250K, (E): T=280K, (F): T= 350K.
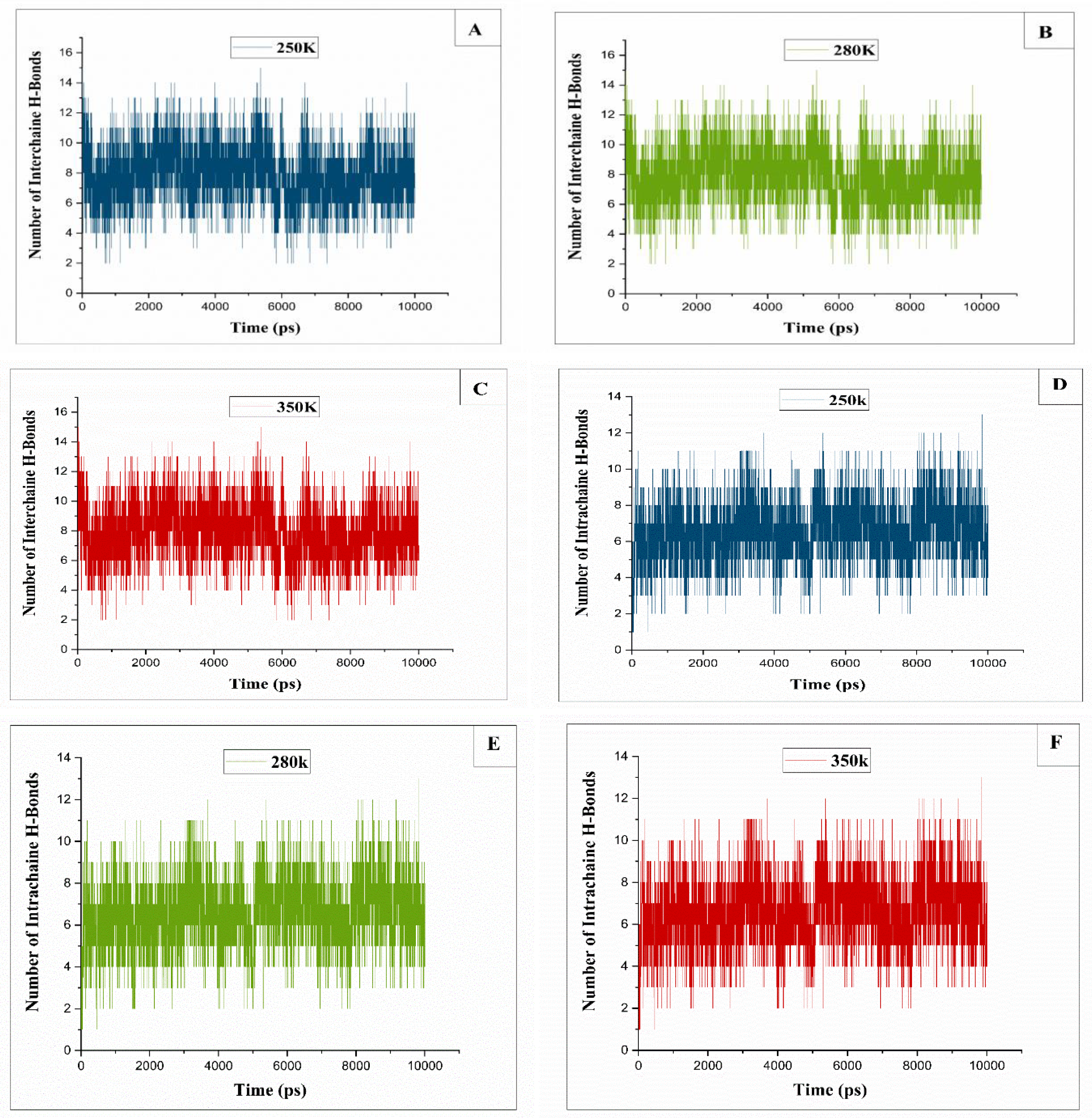
Figure 9.
(A): The evolution of intermolecular hydrogen bonds counted between native and derived cellulosic chains at T=300K, (B): The evolution of intramolecular hydrogen bonds counted between native and derived cellulosic chains at T=300K.
Figure 9.
(A): The evolution of intermolecular hydrogen bonds counted between native and derived cellulosic chains at T=300K, (B): The evolution of intramolecular hydrogen bonds counted between native and derived cellulosic chains at T=300K.
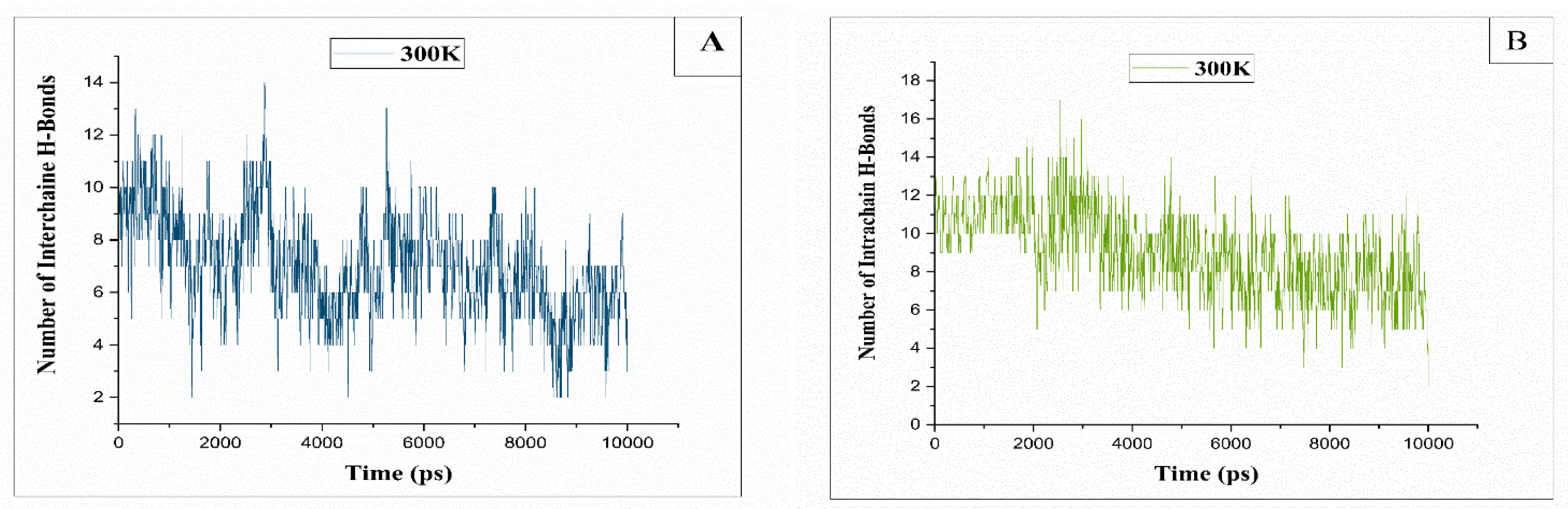
Figure 10.
(A): The evolution of intermolecular hydrogen bonds counted between 2 native cellulosic chains at T=300K, (B): The changes in intramolecular hydrogen bonds between two native cellulosic chains observed at a temperature of 300K.
Figure 10.
(A): The evolution of intermolecular hydrogen bonds counted between 2 native cellulosic chains at T=300K, (B): The changes in intramolecular hydrogen bonds between two native cellulosic chains observed at a temperature of 300K.
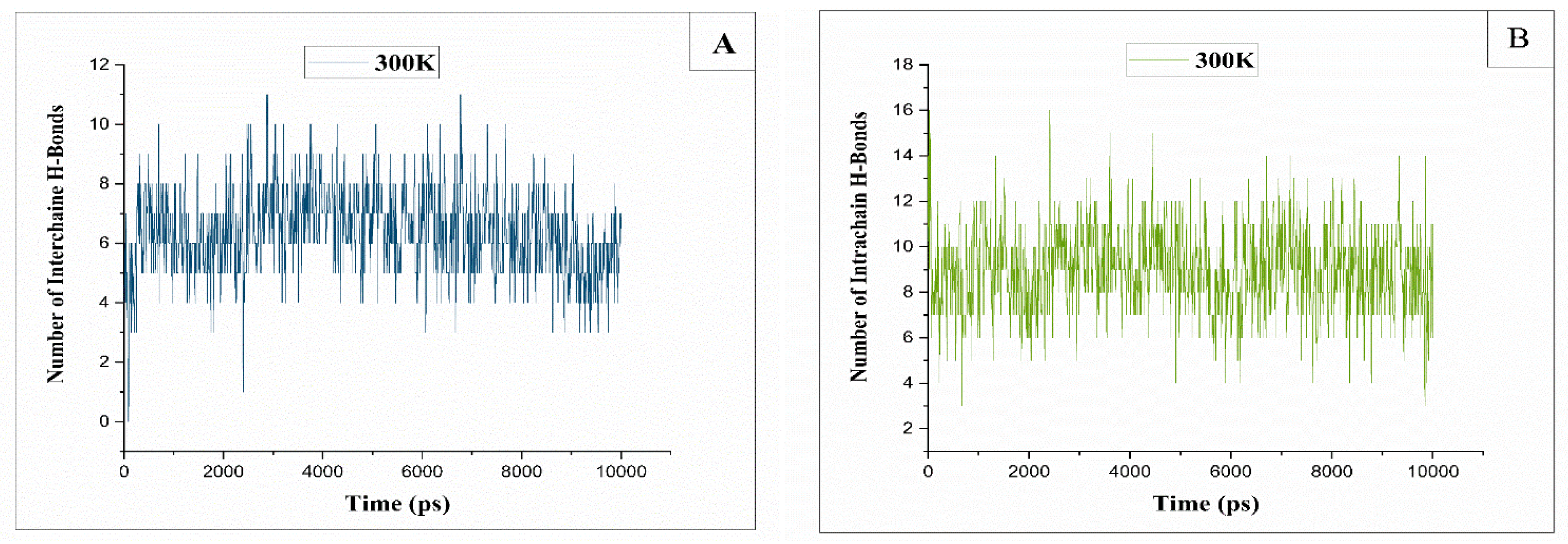
Figure 11.
The histogram illustrates the count of intermolecular hydrogen bonds. (A): Between two native chains at a temperature of 300K, (B): Between native chains and the derivatized one at T=300K.
Figure 11.
The histogram illustrates the count of intermolecular hydrogen bonds. (A): Between two native chains at a temperature of 300K, (B): Between native chains and the derivatized one at T=300K.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



