
Submitted:
15 May 2024
Posted:
16 May 2024
You are already at the latest version
Abstract
The high flammability and lack of charrability of polystyrene (PS) pose significant limitations on its broader applications. Therefore, there is an urgent need for a straightforward and efficient method for the synthesis of flame-retardant PS, which still poses a considerable challenge. In this study, we present an efficient approach to enhance the flame-retardant properties of PS through direct phosphorylating aromatic C(sp2)-H bonds using commercially available and inexpensive manganese catalyst. A range of phosphonates served as reactive substrates to enable a tunable degree of polymer functionalization. The corresponding phosphorylated PS specimens were determined by means of 1H NMR, 31P NMR, FTIR spectroscopy and GPC. Microscale combustion calorimetry (MCC) tests indicate that this protocol indeed enhances the flame-retardant performance of PS. Moreover, other advantages associated with the incorporation of the phosphonate group have been observed, including improved thermal resistance and wettability. This cost-effective strategy of Mn-catalyzed C-H can also be utilized to directly obtain phosphonate modification of waste foamed PS and styrene acrylonitrile copolymer, providing a new method for the purpose of recycling and upgrading PS plastics.
Keywords:
Post-Polymerization Modification
; Mn-Catalyzed
; Phosphorylation
; Aromatic C(sp2)-H Bonds
; Polystyrene
1. Introduction
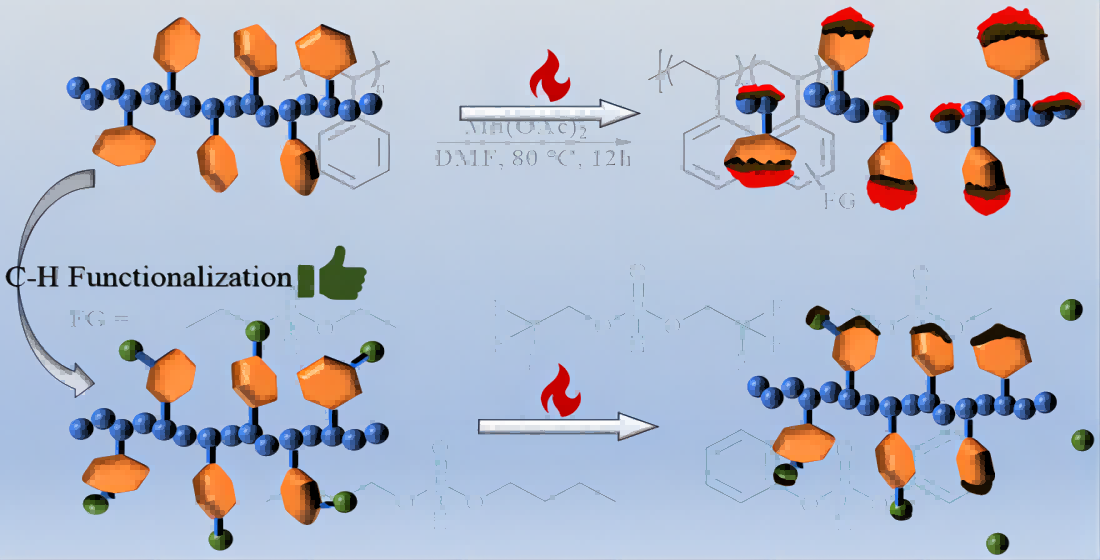
Polystyrene (PS) is ubiquitous and indispensable to our daily life, which is widely used in medical materials, consumer packaging, transportation, textiles, and energy due to its exceptional performances, including strong chemical resistant, mechanical strength, thermal stability and processing convenience. [1] Nevertheless, the high flammability and lack of charrability of PS greatly limited its broader applications. Consequently, the improvement of the flame-retardance capabilities of PS plays a crucial role in many desirable areas where fire safety standards are required. [2] Through targeted synthesis, the flame resistance of PS can traditionally be enhanced by introducing flame retardant additives such as halogen-containing compounds. [3] However, it has been established that halogen-containing compounds are highly recalcitrant, biologically accumulative, and hazardous to the ecosystem, animals as well as humans. [4] Over the past few years, phosphorus-containing compounds have been gaining attention as halogen-free flame retardants due to their versatility in chemistry, environmental friendly, multi-effect flame retardant mechanisms, and high flame resistance even at low concentrations. [5] Currently, two strategies are commonly employed to fabricate flame retarded PS composites, including physically mixing the polymer with flame retardant additives [6] and chemical incorporation of the phosphate ester group within the polymer via radical polymerisation [7] or metal-catalyzed co-polymerization. [8] Despite these significant achievements, several limitations remain. First, the former method requires high loading of flame retardant additives to obtain decent flame retardancy, which will adversely affect the physical and mechanical behavior of PS composite materials. Besides, several additives may loathsomely leach out of the composite material at an unacceptable rate. Second, the latter method needs re-engineering of polymerization processes and probably necessitates replacement of synthetic infrastructure on a large scale, which is both costly and labor consuming. In addition, co-polymerization strategies often suffer from in-homogeneous monomer reactivity, resulting in an-isotropic material attributes. Therefore, the design of a straightforward and efficient route for the synthetic preparation of flame-retardant PS is highly urgent, yet remains a significant challenge.
Post-polymerization modification (PPM) of mass-produced and cost-efficient PS provides a powerful and efficient platform to enable these universal polymers with new properties, higher value and broader applications. [9] However,the synthetic protocol involving the directed functionalization of PS remains quite limited owing to the low reactivity of aromatic C-H linkages. In the past decades, considerable efforts have been devoted to the functionalization of aromatic C(sp2)-H bonds in PS [10], including Friedel-Crafts alkylation [11]a or acylation [11]b, as well as halomethylation [11]c, perfluoroalkylation [11]d, alkyl-Li metalation [11],e potassium superbase metalation [11]f, sulfonation [11]g, and thianthrenation and thio-Suzuki-Miyaura coupling reaction [11]h. In spite of these elegant works, the majority of these reactions possess some disadvantages, including severe reaction environments and undesirable side reactions including chain breakage and cross-linking. Recently, transition metal-catalyzed activation of aromatic C(sp2)-H bonds of PS has been aroused much attention for the efficient installation of various functionalists in a step- and atom-economical manners. In 2007, Lee, Noh and Bae firstly developed Ir-catalyzed boronation of aromatic C(sp2)-H bonds of commercial polystyrene. [12] Fourteen years later, Azoulay et al. disclosed the first Au-catalyzed alkenylation of aromatic C(sp2)-H bonds of PS with methyl propiolate to synthesize the methyl acrylate functionalized PS, which could improve the physical properties such as Tg, elastic modulus, melt viscosity, and wettability. [13] More recently, Wang and co-workers demonstrated a highly efficient Au-catalyzed para-bromination of the aromatically C-H of PS and the resultant brominated PS could be converted to a variety of functional groups, such as NH2 and Bpin. [14] Despite these advances, the involvement of a precious metal iridium or gold was indispensable in these elegant transformations. Meanwhile, to our knowledge, the transition metal-catalyzed phosphorylation of aromatic C(sp2)-H bonds of PS has not been reported.
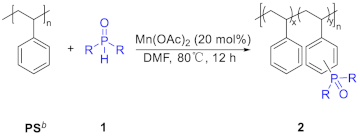
Considering the importance of the phosphorylated PS and previous work in Mn-catalyzed C-H activation [15], we assume that the commercially available and inexpensive manganese catalyst could be incorporate with phosphonates to achieve the phosphorylation of of PS. Herein, we explored a Mn-catalyzed phosphorylation of aromatically C(sp2)-H bonds of PS. The inexpensive and less toxic manganese complex was utilized as catalyst under mild conditions to deliver the the phosphorylated PS, which could enhance the flame-retardant properties of PS. A range of phosphonates served as reactive substrates to enable a customizable degree of polymer functionalization, which broadened the pathway for installation of phosphorus-containing compounds. The corresponding phosphorylated PS specimens were determined by 1H NMR, 31P NMR, FTIR spectrum and GPC. Microscale combustion calorimetry (MCC) tests suggest that this protocol indeed enhances the flame-retardant performance of PS. Additionally, we also observed extra benefits associated with the incorparation of the phosphonate group, including improved thermal stabilization and wettability. This method not only presents the first introduction of phosphorus into the PS backbone via C-H activation, but also provides a platform for the functionalization of the commercially available PS and industrial or post-consumer waste PS plastics [16].
2. Results and Discussion
We initially commenced our investigation of phosphorylation of PS C-H bonds by employing the commercial-grade PS (Figures S1–S3), whose number average molecular weight (Mn) is 151.0 kg·mol-1 and polydispersity (Ð) is 1.97. The diethyl phosphonate (DEP) was selected firstly as the model substrates (Table S1). Unfortunately, no desired product was observed when the reaction was performed by the employment of Mn(OAc)3·2H2O as catalyst in DMF at 80 °C under an air atmosphere according to the optimal condition of Zhang’s work [15] (Table S1, entry 1). After extensive optimization, including decreasing the ratio of [DEP]/[PS], increasing or decreasing both the amount of Mn(OAc)3·2H2O and temperature, and using halogenated solvents (DCE, DCM and CHCl3) or adding polar protic solvent (HOAc) in DMF, the best result (6 % phosphorylation by 1H NMR) was obtained when the reaction conducted in 0.2 equivalent Mn(OAc)3·2H2O (Table S1, entry 8). The desired phosphorylated product PS-DEP was isolated by precipitation from methanol and determined through 1H NMR, 31P NMR and FTIR analysis. Interestingly, replacement of Mn(OAc)3·2H2O with Mn(OAc)2 under above optimal condition gave the better result in 10 % phosphorylation by 1H NMR (Table 1, entry 1). Consequently, Mn(OAc)2 was chosen as the catalyst to conduct this reaction. The reaction was performed by the employment of Mn(OAc)2 as catalyst in N, N-dimethylformamide (DMF) at 80 ℃ under an air atmosphere (Table 1, entry 1). The desired phosphorylated product PS-DEP (Figures S13–S16) was isolated by precipitation from methanol and determined through 1H NMR, 31P NMR and FTIR analysis. To our delight, the reaction resulted in 10 % phosphorylation by 1H NMR (Figure S4). Further optimization revealed that increasing or decreasing the ratio of [DEP]/[PS] resulted in a reduced functionalization (entries 2-4). Surprisingly, as the amount of Mn(OAc)2 increases, the yield of the reaction shows a corresponding decrease (entries 5-9). The reaction could proceed smoothly when the reaction was carried out at 90 °C, whereas the catalytic reaction did not occur when the system was performed at 60 °C (entries 10-11). Other solvents that can dissolve the PS, including tetrahydrofuran (THF), toluene and halogenated solvent (DCE), turned out to be inferior to DMF (enties 12-14). Although the reaction does not occur with acetic acid (HOAc) alone as a solvent, the functionalization of the reaction can be obtained in 13% when using a mixture of HOAc and DMF (v/v = 1: 10) as solvent and 0.2 equivalent Mn(OAc)2 (Table 1, entries 15-16). However, a decrease in Mn of 92.9 kg/mol was detected via GPC, indicating that a main chain split. The prepared polymer samples were further characterized with NMR (1H NMR, 31P NMR, Figure 1 and Figure 2a) and the assistance of infrared (FT-IR, Figure 2b). The FT-IR spectrum did not show a P-H peak at 2425-2325 cm-1, while a P=O peak was observed at 1258 cm-1 independently, verifying that phosphorus was introduced into the polystyrene backbone. The introduction of phosphorus into the polystyrene backbone was further verified by 31P NMR. 1H NMR showed extensive overlapping peaks at 4.17-3.67 ppm for the chemical shifts of the hydrogen atoms of ethoxy (Figure 1). According to the previous literature [17]a, 31P NMR of C(sp3)-P bond exhibited at 27.65 ppm. However, peak of 20.31 ppm appear in the 31P NMR of this phosphorylated product, which is consistent with the peak of C(sp2)-P bond in the 31P NMR [17]b. These results indicated that the sites of the phosphorylation did not occur on the sp3 carbon of the polymer backbone. Meanwhile, we found that the reaction fluid in the reaction tube was clear and transparent, proving that the cross-linking did not occur in this phosphorylation of PS. Furthermore, radical trapping experiments were employed in the presence of 2.0 equivalent of 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) under the standard reaction conditions (Table 1, entries 17-18). It was found that this phosphorylation was completely inhibited, indicating that a radical process might be involved in this transformation.
Encouraged by the fact that PS was functionalized to 10 % under the originally established reaction conditions, we wanted to continue to explore whether it could be functionalized to a higher degree. Therefore, different phosphate esters were used to explore the possibility of PS functionalization (Table 2). The results showed that the degree of PS functionalization could be increased by adjusting the different phosphate esters. As shown in the Table 2, the reactions of bis(2, 2, 2,-trifluoromethyl) phosphite (BFP), dimethyl phosphite (DMP), dibutyl phosphite (DBP) and diphenyl phosphite (DPP) were carried out according to the originally established optimal conditions, the degree of functionalization was also calculated by 1H NMR and the structure was investigated through 31P NMR and FTIR. The results indicated an increase in the functionalization degree of DPP, its functionalization degree could reach 13.6 % (Table 2, entry 6) (Figure S26). At the same time, by GPC test, the Mn (176.2 kg/mol) of PS-DPP was slightly increased compared to the unmodified PS body, indicating that the chain degradation or cross-linking did not occur (Figure S12). [14]
The polymer samples were further studied by means of NMR (1H NMR, 31P NMR, Figure 4 and Figure 5a) and infrared spectroscopy (FT-IR, Figure 5b). The FT-IR analysis suggests the absence of P-H peak at 2425-2325 cm-1, while a distinct P=O peak at 1258 cm-1, confirming that functional phosphorus has been introduced into the polystyrene backbone. The 31P NMR further verifies the process. Meanwhile, 1H NMR showed the presence of the relevant characteristic peaks for all the other reactions, with the exception of PS-DPP, whose peaks basically overlapped on the starting benzene ring, all of which originated from the phenyl functionalisation of PS.
Furthermore, we also investigated the thermal resistance of the modified polymers and unmodified PS by thermogravimetric analysis (TGA) which is a general tool to study the decomposition of materials at different temperatures (Figure S29). [18] TGA was carried out under nitrogen atmosphere with a temperature ramp rate of 10 °C/min. The temperatures corresponding to at 5% mass loss (Td, 5%), 50% mass loss (Td, 50%), the fastest mass loss rate (Td, max), as well as the solid residual char yields at 600 °C can be used to estimate the thermal stability of polymers. The summarized data are shown in the Table 2.
For the PS sample, there was a single degradation pathway in a narrow temperature range between 363 °C and 446 °C due to the thermal depolymerization along the polymer backbone. The residue weight of the PS at 600 °C was at 2.48 %. For the phosphate-modified PS samples, the decomposition of phosphonate groups resulted in the thermal weight loss below 132 °C due to the lower bonding disassociation energy of the P-O-C and O=P-O bonds than that of the C-C bond. Therefore, the Td, 5% of PS-DEP, PS-DMP and PS-DBP were lower than that of PS . [19] The presence of the benzene ring structure of PS-DPP led to a delay in the thermal decomposition of the polymers, which made its Td, 5% higher than that of PS. [20] However, the decomposition of phosphonate groups also led to the creation of a carbonated layer that effectively isolated the polymer barrier from oxygen and prevented the evolution of inflammable gases. [5]a Hence, it was gratifying that the Td, 50%, Td, max and residual char yield of phosphate-functionalized PS samples were higher than that of PS. In particular, the residual char yield of PS-DPP was as high as 14.44 %.
Microscale combustion calorimetry (MCC) is a quantitative analytical experiment method for relatively rapid screening of polymer flammability. It requires only a small amount of sample to directly measure the heat of combustion for the gases in the heating sample process. [21] Relevant flammability parameters of polystyrene before and after modification were measured as shown in the Table 3, specifically HRC, pHRR, THR, TpHRR i.e. heat release capacity, peak heat release rate, total heat release and the temperature at pHRR, respectively.
The higher HRC, pHRR and THR of the studied samples indicate higher releases of flammable products. The HRC, pHRR of PS modified with phosphate ester were reduced by 40 % as compared to PS, indicating that the flame retardancy capacity of PS were enhanced after phosphate ester modification. The flame retardant properties of BFP was more effective in flame resistant compared to DPP and DMP due to the introduction of halogens in the phosphate ester.
In addition, the installation of the phosphate group altered the hydrophilicity of the PS (Figure S30). demonstrates that the functionalized polymers exhibit a lower water contact angle compared to the unmodified PS. This suggests an increase in surface wettability to water and greater hydrophilicity for the modified PS. The experiment found that the thermostability of two PS composites (PS: PS-DPP 95: 5 and 90: 10) (Table S2, Figure S31) remained unchangeable after recombination, indicating the formation of a strong covalent connection between PS and phosphate ester without any small molecule precipitation.
The method of Mn-catalyzed C-H activation can also be used to functionalized commercial PS products directly. Foamed polystyrene is a common type of polystyrene material. The waste foamed polystyrene could be directly connected to phosphate ester groups through the PS-DPP method mentioned above, as shown in Figure 6. The degree of functionalization could be up to 11.9 %, which shows that our reaction can be recycled on waste polystyrene. The industrial styrene-acrylonitrile copolymer (SAN) can also act as a compatible substrate, with a phosphorylated grafting rate of 6.25 % based on the above PS-DEP method. Meanwhile, 31P NMR further confirmed the introduction of phosphorus in waste foamed PS and SAN. The introduction of phosphorus to waste foamed and SAN was also verified by FT-IR spectroscopy (Figure 6c). The P-H peaks at 2425-2325 cm-1 were not observed, whereas SAN-DEP exhibited a P=O peak at 1258 cm-1; FPS-DPP displayed a P-O-C characteristic peak at 1110-1140 cm-1.
3. Materials and Methods
3.1. Materials
All reactions were performed under air atmosphere in a 25 mL sealed tube. The materials and solvents were purchased from common commercial sources and used without additional purification, if there is no special version.
3.2. General Procedure for the Phosphorylation
polystyrene (PS) (1.0 equive), phosphate (1.0 equive), Manganous acetate (0.2 equive) and solvent (1.0 mL) were added to a Schlenk reaction tube. The solution was reacted at 80 ℃ for 12 h. After cooling to room temperature, the solution was dissolved with dichloromethane and transferred to the pear-shaped liquid funnel. The organic phase purified by washing the water three to five times, dried with anhydrous sodium sulfate, concentrated and precipitated into methanol, filtered to obtain solid sediment and dried under vacuum. If necessary, the precipitation process was repeated one more time to ensure complete removal of any small molecules trapped in the polymer.
3.3. Characterizations
NMR spectra were recorded using a Bruker DRX 400MHz spectrometer. Chemical shifts δ (ppm) are referenced to tetramethylsilane (TMS) using the residual solvent as an internal standard (1H and 13C) or using the unified scale relative to the absolute frequency for 1H of 0.1% TMS in CDCl3 (31P) unless otherwise stated. For 1H NMR: CDCl3 7.27 ppm; For 13C NMR: CDCl3, 77.16 ppm. Infrared (IR) spectra were obtained using Thermo Scientific Nicolet iS20 spectrometer. IR data are reported in wavenumbers within the range of 4000–500 cm-1.Gel permeation chromatography (GPC) using THF as the solvent was used to characterize the molecular weight of polymers with a machine of Waters Model 1525 HPLC pump and a detector of Waters Model 2414 refractive index (RI) (Thermo Fisher).Thermogravimetric analysis, Decomposition onset temperatures (Td) and maximum decomposition temperatures (Tmax) of precipitated and dried polymer samples were measured by thermal gravimetric analysis (TGA) on a TA Instruments Q50 Thermogravimetric Analyzer. Polymer samples were heated from ambient temperatures to 600 ℃ at a heating rate of 10 ℃/min. Values of Td (temperature at 5% weight loss and temperature at 50% weight loss) were obtained from wt % vs. temperature (℃) plots.Static water contact angles were measured from 7 μL droplets of RO water using a Rame Hartgoniometer equipped with DropImageCA software. Reported values are the average of at least three droplets, placed in different regions of the surface.Microscale combustion calorimetry (MCC) tests were carried out in triplicate by C. Chong and S. Kumar (Interscience Fire Laboratory). The tests were carried out following the procedure specified in ASTM D 7309-19 standard test method for “Determining Flammability Characteristics of Plastics and Other Solid Materials Using Microscale Combustion Calorimetry”.
4. Conclusions
In summary, the post-polymerization modification of polystyrene has been successfully achieved through Mn-catalyzed phosphorylation of aromatic C(sp2)-H bond, resulting in a significant enhancement of the flame-retardant properties towards PS. This reaction is gentle, with virtually no change in the dispersity of the source polymer, and is compatible with a number of commercial available phosphonates. The corresponding phosphorylated PS materials were analyzed with 1H NMR, 31P NMR, IR spectroscopy and GPC. Compared to initial PS, the phosphonated PS exhibited higher Tmax and T50% and higher residue char yields, suggesting higher thermal stability. Two stages of thermal dissociation have been found in the TGA plots of phosphonated PS, while only a narrow decomposition process has been detected for PS. The MCC experimental results have confirmed that phosphonatd PS shows a better flame resistance effect, with a 40 % decrease in HRC, pHRR. In addition, we also found that the wettability of the polymer can be regulated after phosphorylation based Mn-catalyzed C-H activation. This economic catalytic method can be used directly to modify waste foamed PS and SAN undustrial plastics with phosphate esters, which provide a new way to recycle and upgrade PS plastics.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 1H NMR (400 MHz, CDCl3) spectrum of PS; Figure S2: 13C NMR (100 MHz, CDCl3) spectrum of PS; Figure S3: FT-IR spectra of PS; Figure S4: Calculation of the functionalization degree; Figure S5: Calculation of the functionalization degree; Figure S6: (a) Picture of Polystyrene (PS). (b) Picture of the reaction fluid in the reaction tube; Figure S7: (a) 13C NMR (125.7 MHz, CDCl3) spectrum of Diethyl phenylphosnate. (b) 13C NMR (100 MHz, CDCl3) spectrum of PS-DEP. (c) 31P NMR (CDCl3) spectrum of Diethyl phenylphosnate. (d) 31P NMR (162 MHz, CDCl3) spectrum of PS-DEP. (e) 31P NMR (CDCl3) spectrum of AC2NP2. (f) 31P NMR (162 MHz, CDCl3) spectrum of PS-DMP; Figure S8: GPC traces for PS, entries 1, 2, 3 and 5 of Table1; Figure S9: GPC traces for entries 11, 12 and 13 of Table1; Figure S10: GPC traces for entries 7 and 16 of Table1; Figure S11: GPC traces of PS, PS-DEP, PS-BFP, PS-DMP and PS-DBP of Table2; Figure S12: GPC trace of PS-DPP of Table2; Figure S13: 1H NMR (400 MHz, CDCl3) spectrum of PS-DEP; Figure S14: 13C NMR (100 MHz, CDCl3) spectrum of PS-DEP; Figure S15: 31P NMR (162 MHz, CDCl3) spectrum of PS-DEP; Figure S16: FT-IR spectra of PS-DEP; Figure S17: 1H NMR (400 MHz, CDCl3) spectrum of PS-BFP; Figure S18: 31P NMR (162 MHz, CDCl3) spectrum of PS-BFP; Figure S19: FT-IR spectra of PS-BFP; Figure S20: 1H NMR (400 MHz, CDCl3) spectrum of PS-DMP; Figure S21: 31P NMR (162 MHz, CDCl3) spectrum of PS-DMP; Figure S22: FT-IR spectra of PS-DMP; Figure S23: 1H NMR (400 MHz, CDCl3) spectrum of PS-DBP; Figure S24: 31P NMR (162 MHz, CDCl3) spectrum of PS-DBP; Figure S25: FT-IR spectra of PS-DBP; Figure S26: 1H NMR (400 MHz, CDCl3) spectrum of PS-DPP; Figure S27: 31P NMR (162 MHz, CDCl3) spectrum of PS-DPP; Figure S28: FT-IR spectra of PS-DPP; Figure S29: TGA(a) and DTG(b) curves of PS, PS-DEP, PS-DMP, PS-DBP and PS-DPP under nitrogen atmosphere; Figure S30: Selected images of water droplets on (a) PS. (b) PS-DEP (10.0 % functionality). (c) PS-BFP (8.5 % functionality). (d) PS-DMP (8.3 % functionality). (e) PS-DBP (11.0 % functionality) and (f) PS-DPP (13.6 % functionality); Figure S31: TGA (a) and DTG (b) curves of PS and blended PS with PS-DPP polymers under nitrogen atmosphere; Figure S32: 1H NMR (400 MHz, CDCl3) spectrum of waste expandable FPS; Figure S33: 13C NMR (100 MHz, CDCl3) spectrum of waste expandable FPS; Figure S34: FT-IR spectra of FPS; Figure S35: 1H NMR (400 MHz, CDCl3) spectrum of FPS-DPP; Figure S36: FT-IR spectra of FPS-DPP; Figure S37: 1H NMR (400 MHz, CDCl3) spectrum of SAN; Figure S38: 13C NMR (100 MHz, CDCl3) spectrum of SAN; Figure S39: FT-IR spectra of SAN; Figure S40: 1H NMR (400 MHz, CDCl3) spectrum of SAN-DEP; Figure S41: FT-IR spectra of SAN-DEP. Table S1: Screening the optimal reaction conditions with Diethyl phosphite (DEP) under Mn(OAc)3·2H2O catalysis; Table S2. TGA data of reference polymers;.
Author Contributions
Conceptualization, R.-X.L., C.L. and J.Z.; methodology, R.-X.L. and C.L.; formal analysis, R.-X.L.; investigation, R.-X.L., C.L., Y.-Q.Z, W.-H.S., Y.-P.Y. and L.S.; data curation, R.-X.L., Y.-B.Z. and J.Z.; writing – original draft, R.-X.L.;writing – review and editing, C.L. and J.Z.; visualization, R.-X.L. and Y.-B.Z.; supervision, C.L., Y.-Q.Z., W.-H.S. and L.S.; project administration, R.-X.L., C.L. and Y.-B.Z.; funding acquisition, C.L. and L.S. All authors have read and agreed to the published version of the manuscript.
Funding
The work was funded by the Natural Science Foundation of China (No. 51963010 and 52363018), the Natural Science Fundation of Jiangxi Province (No. 20202BABL213005 and 20212BAB203008).
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- (a) Maul, J.; Frushour, B. G.; Kontoff, J. R.; Eichenauer, H.; Ott, K-H.; Schade, C. Polystyrene and Styrene Copolymers. Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH 2007, 29, 475-522. (b) Geyer, R.; Jambeck, J. R.; Law, K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. (c) Tullo, A. Polystyrene marks a milestone; parting with old annual reports and product brochures. C&EN Global Enterprise. 2021, 99(23), 32. (d) Ghoshal, T.; Parmar, P. R.; Bhuyan, T.; Bandyopadhyay, D. Polystyrene Foams: Materials, Technology, and Applications. ACS Symp. Ser. 2023, 1439, 121-141.
- (a) Levchik, S. V.; Weil, E. D. New developments in flame retardancy of styrene thermoplastics and foams. Polym. Int. 2008, 57, 431-448. (b) Wang, Y.; Zhang, J. Thermal stabilities of drops of burning thermoplastics under the UL 94 vertical test conditions. J. Hazard. Mater. 2013, 246-247, 103-109. (c) Sun, P.; Huang, X.; Xu, C. Flashpoint and burning of thin molten plastic pool above hot boundary. Appl. Therm. Eng. 2022, 215, 118931.
- (a) Rani, M.; Shim, W. J.; Han, G. M.; Jang, M.; Song, Y. K.; Hong, S. H. Hexabromocyclododecane in polystyrene based consumer products: An evidence of unregulated use. Chemosphere 2014, 110, 111-119. (b) Beach, M. W.; Kearns, K. L.; Davis, J. W.; Stutzman, J. R.; Lee, D.; Lai, Y.; Monaenkova, D.; Kram, S.; Hu, J.; Lukas, C. Stability Assessment of a Polymeric Brominated Flame Retardant in Polystyrene Foams under Application-Relevant Conditions. Environ. Sci. Technol. 2021, 55, 3050-3058.
- (a) Ezechiáš, M.; Covino, S.; Cajthaml, T. Ecotoxicity and biodegradability of new brominated flame retardants: A review. Ecotoxicol. Environ. Saf. 2014, 110, 153-167. (b) Guo, L-C.; Lv, Z.; Zhu, T.; He, G.; Hu, J.; Xiao, J.; Liu, T.; Yu, S.; Zhang, J.; Zhang, H.; Ma, W. Associations between serum polychlorinated biphenyls, halogen flame retardants, and renal function indexes in residents of an e-waste recycling area. Sci. Total Environ. 2023, 858, 159746. (c) Wang, N.; Lai, C.; Xu, F.; Huang, D.; Zhang, M.; Zhou, X.; Xu, M.; Li, Y.; Liu, S.; Huang, X.; Nie, J.; Li, H. A review of polybrominated diphenyl ethers and novel brominated flame retardants in Chinese aquatic environment: Source, occurrence, distribution, and ecological risk assessment. Sci. Total Environ. 2023, 904, 166180.
- (a) Veen, I.; Boer, J. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere 2012, 88, 1119-1153. (b) Velencoso, M. M.; Battig, A.; Markwart, J. C.; Schartel, B.; Wurm, F. R. Molecular Firefighting-How Modern Phosphorus Chemistry Can Help Solve the Challenge of Flame Retardancy. Angew. Chem. Int. Ed. 2018, 57, 10450-10467. (c) Zhang, C.; Jiang,Y.; Li, S.; Huang, Z.; Zhan, X-Q.; Ma, N.; Tsai, F-C. Recent trends of phosphorus-containing flame retardants modified polypropylene composites processing. Heliyon 2022, 8, e11225.
- (a) Yan, Y-W.; Huang, J-Q.; Guan, Y-H.; Shang, K.; Jian, R-K.; Wang, Y-Z. Flame retardance and thermal degradation mechanism of polystyrene modified with aluminum hypophosphite. Polym. Degrad. Stab. 2014, 99, 35-42. (b) Qian, X.; Zheng, K.; Lu, L.; Wang, X.; Wang, H. A novel flame retardant containing calixarene and DOPO structures: Preparation and its application on the fire safety of polystyrene. Polym. Adv. Technol. 2018, 29, 2715-2723. (c) Wang, X.; Tu, H.; Xiao, H.; Lu, J.; Xu, J.; Gu, G. A novel halogen-free flame-retardant fabrication for the study of smoke suppression and flame retardancy of polystyrene. Polymer 2023, 283, 126240.
- (a) Dumitrascu, A.; Howell, B. A.; Flame-retarding vinyl polymers using phosphorus-functionalized styrene monomers. Polym. Degrad. Stab. 2011, 96, 342-349. (b) Tai,Q.; Song, L.; Hu, Y.; Yuen, R. K. K.; Feng, H.; Tao, Y. Novel styrene polymers functionalized with phosphorus–nitrogen containing molecules: Synthesis and properties. Mater. Chem. Phys. 2012, 134, 163-169. (c) Sun, J.; Wang, C.; Hong, Y-L.; Tan, Z-W.; Liu, C-M. Phosphine Oxide-Containing Multifunctional Polymer via RAFT Polymerization and Its High-Density Post-Polymerization Modification in Water. ACS Appl. Polym. Mater. 2021, 3, 3214-3226. (d) Baby, A.; Tretsiakova-McNally, S.; Joseph, P.; Arun, M.; Zhang, J.; Pospiech, D. The influence of phosphorus- and nitrogen- containing groups on the thermal stability and combustion characteristics of styrenic polymers. J. Therm. Anal. Calorim. 2023, 148, 229-241.
- Thomas, G. L.; Böhner C.; Ladlow, M.; Spring, D. R. Synthesis and utilization of functionalized polystyrene resins. Tetrahedron 2005, 61, 12153-12159. [CrossRef]
- (a) Gauthier, M. A.; Gibson, M. I.; Klok, H-A. Synthesis of Functional Polymers by Post-Polymerization Modification. Angew. Chem. Int. Ed. 2009, 48, 48-58. (b) Günay, K. A.; Theato, P.; Klok, H-A. Standing on the Shoulders of Hermann Staudinger: Post-polymerization Modification from Past to Present. J. Polym. Sci. A Polym. Chem. 2013, 51, 1-28. (c) Blasco, E.; Sims, M. B.; Goldmann, A. S.; Sumerlin, B. S.; Barner-Kowollik, C. 50th Anniversary Perspective: Polymer Functionalization. Macromolecules 2017, 50, 5215-5252. (d) Ohn, N.; Kim, J. G.; Mechanochemical Post-Polymerization Modification: Solvent-Free Solid-State Synthesis of Functional Polymers. ACS Macro Lett. 2018, 7, 561-565.
- (a) Williamson, J. B.; Lewis, S. E.; Johnson Ⅲ, R. R.; Manning, I. M.; Leibfarth, F. A. C-H Functionalization of Commodity Polymers. Angew. Chem. Int. Ed. 2019, 58, 8654-8668. (b) Zhao, Y.; Li, D.; Jiang X. Chemical Upcycling of Polyolefins through C-H Functionalization. Eur. J. Org. Chem. 2023, 26, e202300664 (1 of 15).
- (a) Isono, T.; Baba, E.; Tanaka, S.; Miyagi, K.; Dazai, T.; Li, F.; Yamamoto, T.; Tajima, K.; Satoh, T. Installation of the adamantyl group in polystyrene-block-poly(methyl methacrylate) via Friedel-Crafts alkylation to modulate the microphase-separated morphology and dimensions. Polym. Chem. 2023, 14, 2675-2684. Zhang, Z.; Zhang, Y.; Zeng, R.; Photoinduced iron-catalyzed C-H alkylation of polyolefins. Chem. Sci. 2023, 14, 9374-9379. (b)Tizpar, S.; Abbasian, M.; Taromi, F. A.; Entezami, A. A. Grafting of Poly(methyl methacrylate) or Polyacrylonitrile onto Polystyrene Using ATRP Technique. J. Appl. Polym. Sci. 2006, 100, 2619-2627. (c) Niu, M.; Li, T.; Xu, R.; Gu, X.; Yu, D.; Wu, Y. Synthesis of PS-g-POSS Hybrid Graft Copolymer by Click Coupling via “ Graft Onto” Strategy. J. Appl. Polym. Sci. 2013, 129, 1833-1844. (d) Lewis, S. E.; Jr, W. B. E.; Leibfarth, F. A. Upcycling aromatic polymers through C-H fluoroalkylation. Chem. Sci. 2019, 10, 6270-6277. (e) Chalk, A. J. Abnormal Reactions of Polystyrene Metalated with Butyllithium-N, N, N’, N’ -tetramethylethylenediamine. J. Polym. Sci. B: Polym. Lett. 1968, 6, 649-651. (f) Lochmann, L.; Fréchet, J. M. J. Controlled Functionalization of Polystyrene: Introduction of Reactive Groups by Multisite Metalation with Superbase and Reaction with Electrophiles. Macromolecules 1996, 29, 1767-1771. (g) Fahs, G. B.; Benson, S. D.; Moore, R. B. Blocky Sulfonation of Syndiotactic Polystyrene: A Facile Route toward Tailored Ionomer Architecture via Postpolymerization Functionalization in the Gel State. Macromolecules 2017, 50, 2387-2396. Si, J.; Hao, N.; Zhang, M.; Cheng, S.; Liu, A.; Li, L.; Ye, X. Universal Synthetic Strategy for the Construction of Topological Polystyrenesulfonates: The Importance of Linkage Stability during Sulfonation. ACS Macro Lett. 2019, 8, 730-736. (h) Xue, B.; Huang, P-P.; Zhu, M-Z.; Fu, S-Q.; Ge, J-H.; Li, X.; Liu, P-N. Highly Efficient and para-Selective C-H Functionalization of Polystyrene Providing a Versatile Platform for Diverse Applications. ACS Macro Lett. 2022, 11, 1252-1257.
- Shin, J.; Jensen, S. M.; Ju, J.; Lee, S.; Xue, Z.; Noh, S. K.; Bea, C. Controlled Functionalization of Crystalline Polystyrenes via Activation of Aromatic C-H Bonds. Macromolecules 2007, 40, 8600-8608.
- King, E. R.; Hunt, S. B.; Hamernik, L. J.; Gonce, L. E.; Wiggins, J. S.; Azoulay, J. D. Gold-Catalyzed Post-Polymerization Modification of Commodity Aromatic Polymers. JACS Au 2021, 1, 1342-1347.
- Dou, B.; Xu, Y.; Wang, J. Gold-Catalyzed Precise Bromination of Polystyrene. J. Am. Chem. Soc. 2023, 145, 10422-10430.
- Xu, W.; Zou, J-P.; Zhang, W. Manganese(III)-mediated direct phosphonylation of arenes. Tetrahedron Lett. 2010, 51, 2639-2643.
- (a) Moulay, S. Functionalized Polystyrene and Polystyrene-Containing Material Platforms for Various Applications. Polym. Plast. Technol. Eng. 2018, 57, 1045-1092. (b) McCoy, M.; Recycling polystyrene. C&EN 2018, 96, 13. (c) Billiet, S.; Trenor, S. R. 100th Anniversary of Macromolecular Science Viewpoint: Needs for Plastics Packaging Circularity. ACS Macro Lett. 2020, 9, 1376-1390. (d) Jehanno, C.; Alty, J. W.; Roosen, M.; Meester, S. D.; Dove, A. P.; Chen, E. Y.-X.; Leibfarth, F. A.; Sardon, H. Critical advances and future opportunities in upcycling commodity polymers. Nature 2022, 603, 803-814. (e) Goring, P. D.; Priestley, R. D. Polymer Recycling and Upcycling: Recent Developments toward a Circular Economy. JACS Au 2023, 3, 2609-2611.
- (a) G. Keglevich, E. Jablonkai and L. B. Bal’azs, A “green” variation of the Hirao reaction: the P–C coupling of diethyl phosphite, alkyl phenyl-Hphosphinates and secondary phosphine oxides with bromoarenes using a P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions RSC Adv. 2014, 4, 22808-22816. (b) Q. Tai, L. Songa, Y. Hua, R. K.K. Yuen, H. Feng and Y. Tao, Novel styrene polymers functionalized with phosphorus–nitrogen containing molecules: Synthesis and properties: Mater. Chem. Phys. 2012, 134, 163-169.
- Shapiro, A, J.; O’Dea, R. M.; Epps, T. H. III. Thermogravimetric Analysis as a High-Throughput Lignocellulosic Biomass Characterization Method. ACS Sustainable Chem. Eng. 2023, 11, 17216-17223.
- (a) Wang, X.; Hu, Y.; Song, L.; Xing, W.; Lu, H.; Lv, P.; Jie, G. Flame retardancy and thermal degradation mechanism of epoxy resin composites based on a DOPO substituted organophosphorus oligomer. Polymer 2010, 51, 2435-2445. (b) Blake, N.; Turner, Z. R.; Buffet, J-C.; O’Hare, D. Flame retardant phosphonate-functionalised polyethylenes. Polym. Chem. 2023, 14, 3175-3185.
- Yang, B.; Wang, L.; Guo, Y.; Zhang, Y.; Wang, N.; Cui, J.; Tian, L. Synthesis of a novel phosphate-containing highly transparent PMMA copolymer with enhanced thermal and flame retardant properties. Polym. Adv. Technol. 2020, 31, 472-481. [CrossRef]
- (a) Wilkie, C. A.; Chigwada, G.; Gilman, Sr. J. W.; Lyon, R. E. High-throughput techniques for the evaluation of fire retardancy. J. Mater. Chem. 2006, 16, 2023-2030. (b) Lyon, R. E.; Walters, R. N.; Stoliarov, S. I. Screening Flame Retardants for Plastics Using Microscale Combustion Calorimetry. Polym. Eng. Sci. 2007, 47, 1501-1510. (c) Price, D.; Cunliffe, L. K.; Bullett, K. J.; Hull, T. R.; Milnes, G. J.; Ebdon, J. R.; Hunt, B. J.; Joseph, P. Thermal behaviour of covalently bonded phosphate and phosphonate flame retardant polystyrene systems. Polym. Degrad. Stab. 2007, 92, 1101-1114.
Figure 1.
1H NMR spectra of entry 1 and PS.
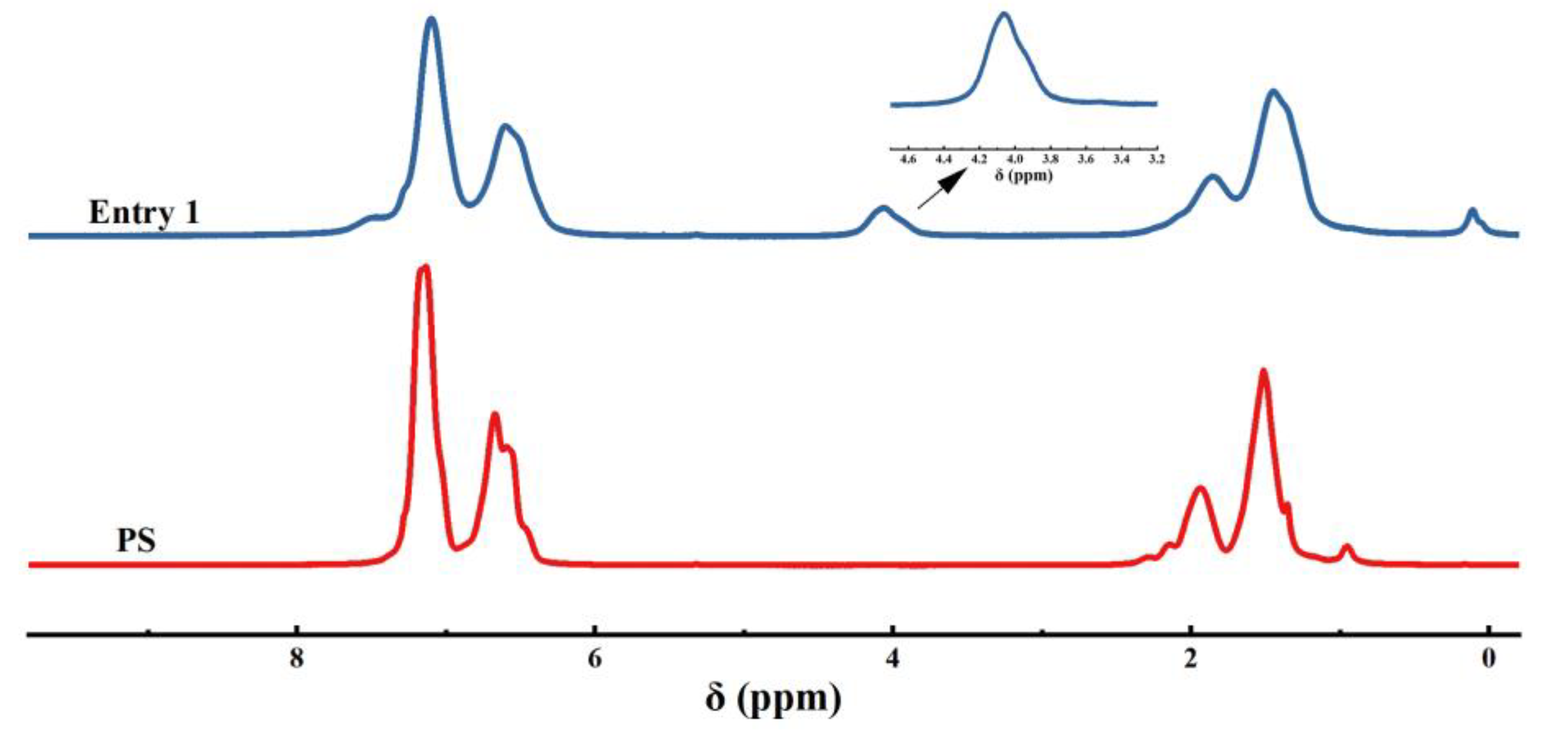
Figure 2.
(a) 31P NMR spectra of entry 1 and PS. (b) FT-IR spectra of entry 1 and PS.
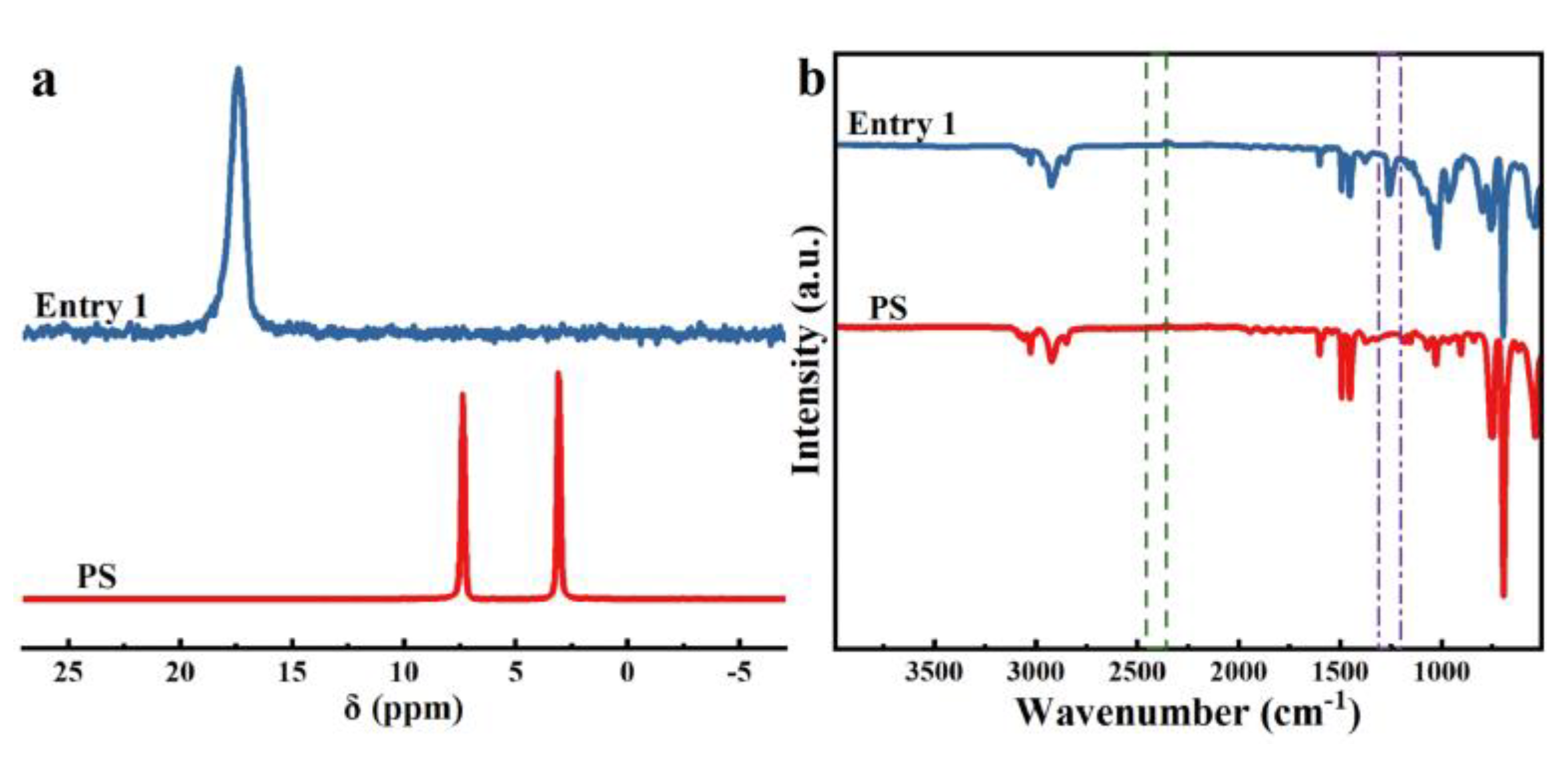
Figure 3.
The structures of phosphate esters.
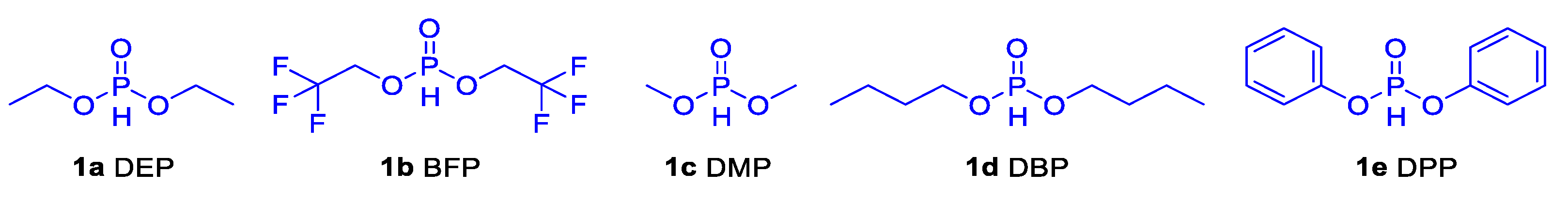
Figure 4.
1H NMR spectra of PS, PS-DEP, PS-BFP, PS-DMP, PS-DBP and PS-DPP.
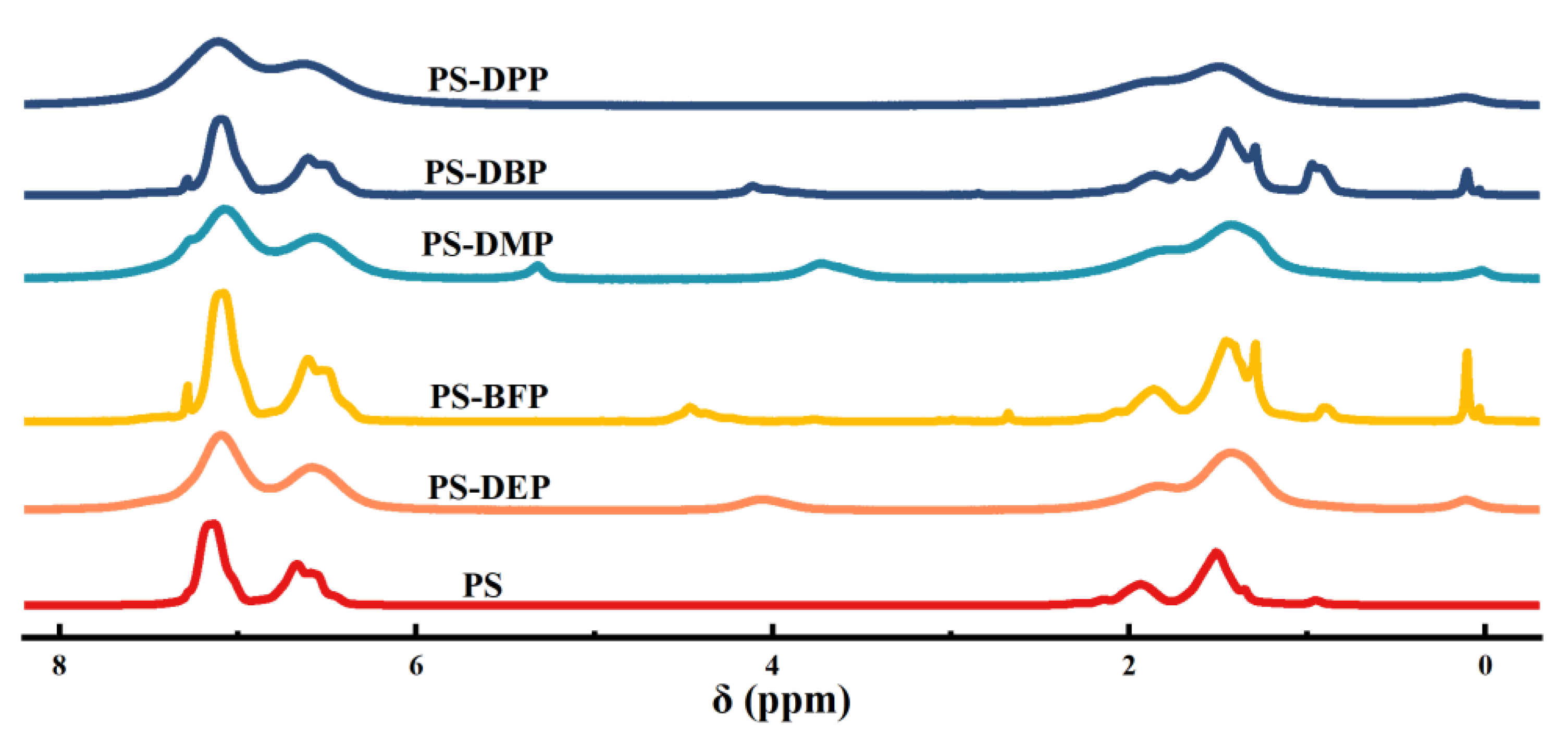
Figure 5.
(a) 31P NMR spectra of DEP, PS-DEP, BFP, PS-BFP, DMP, PS-DMP, DBP, PS-DBP, DPP and PS-DPP. (b) FT-IR spectra of PS, PS-DEP, PS-BFP, PS-DMP, PS-DBP and PS-DPP.
Figure 5.
(a) 31P NMR spectra of DEP, PS-DEP, BFP, PS-BFP, DMP, PS-DMP, DBP, PS-DBP, DPP and PS-DPP. (b) FT-IR spectra of PS, PS-DEP, PS-BFP, PS-DMP, PS-DBP and PS-DPP.
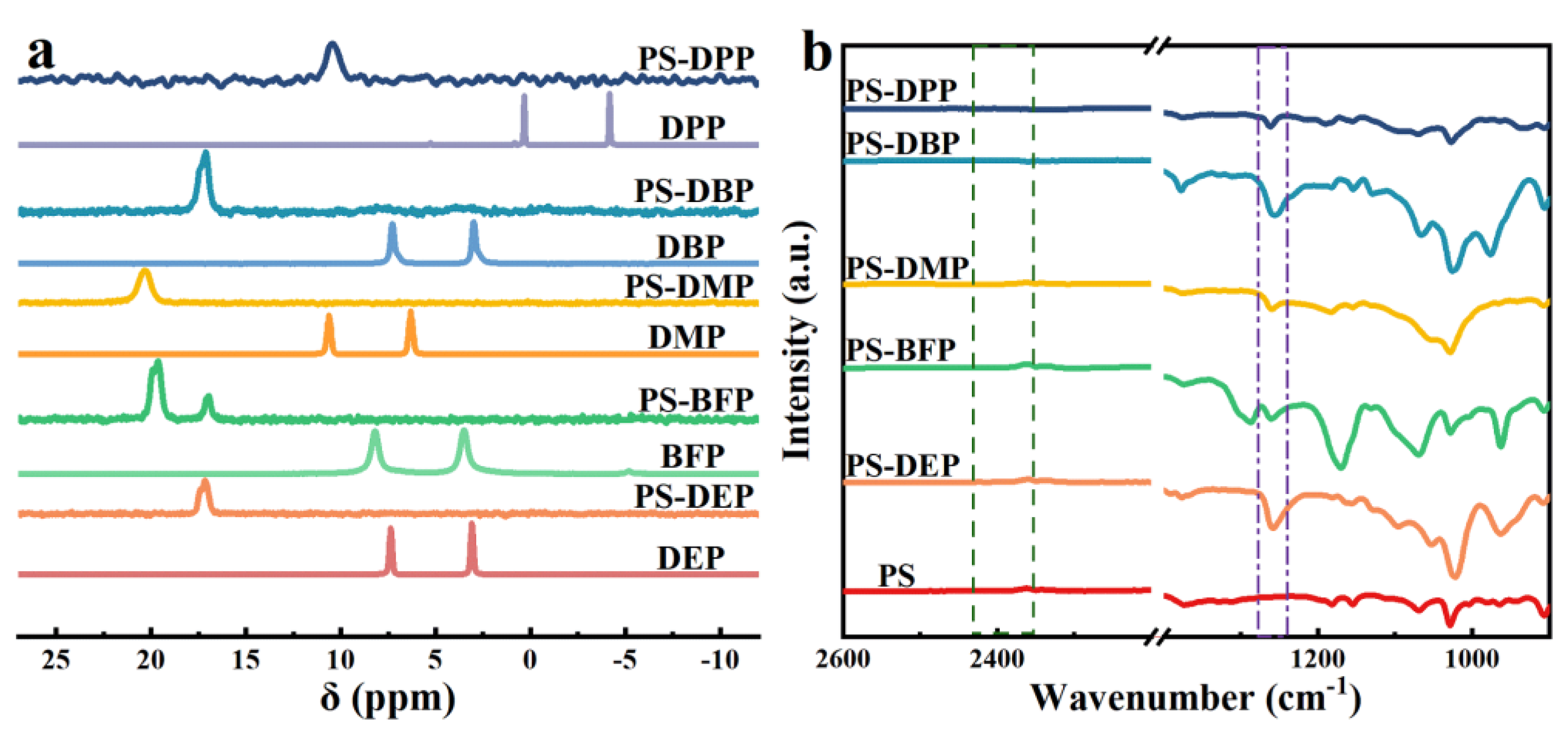
Figure 6.
(a) 1H NMR spectra of FPS, FPS-DPP, SAN and SAN-DEP. (b) 31P NMR spectra of DPP, FPS-DPP, DEP and SAN-DEP. (c) FT-IR spectra of FPS, FPS-DPP, SAN and SAN-DEP.
Figure 6.
(a) 1H NMR spectra of FPS, FPS-DPP, SAN and SAN-DEP. (b) 31P NMR spectra of DPP, FPS-DPP, DEP and SAN-DEP. (c) FT-IR spectra of FPS, FPS-DPP, SAN and SAN-DEP.
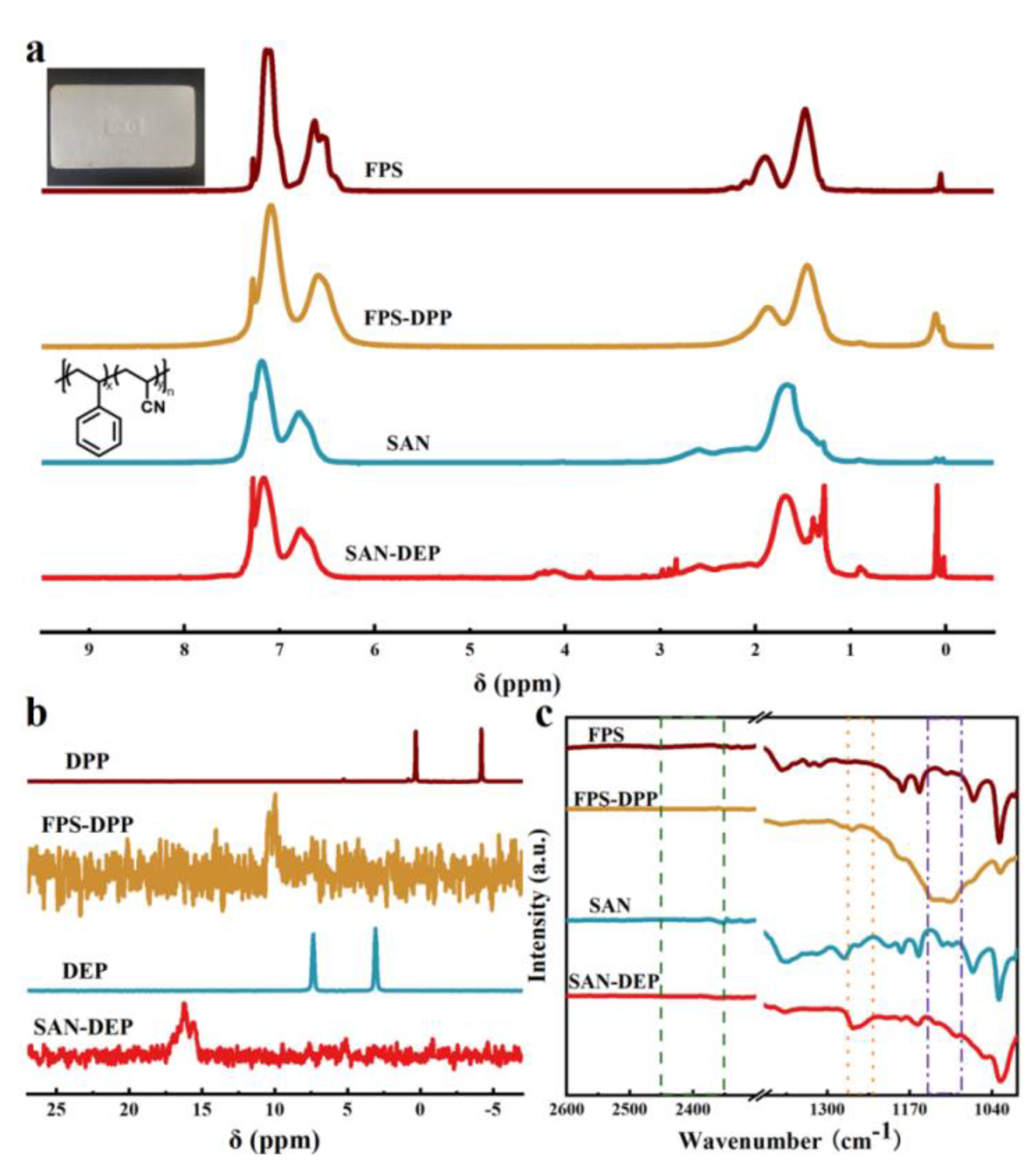

Table 1.
Screening the optimal reaction conditions with diethyl phosphite (DEP).
| PS-DEP c | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | [DEP]/[PS] | [Mn(OAc)2] |
T (℃) |
t (h) |
Mn (kg·mol-1) |
Ð | Functionalization d (%) |
| 1 | DMF | 1 | 0.2 | 80 | 12 | 135.0 | 1.88 | 10 |
| 2 | DMF | 0.75 | 0.2 | 80 | 12 | 156.2 | 1.91 | 5 |
| 3 | DMF | 2 | 0.2 | 80 | 12 | 147.7 | 1.99 | 6.75 |
| 4 | DMF | 3 | 0.2 | 80 | 12 | - | - | 0 |
| 5 | DMF | 1 | 0.4 | 80 | 12 | 142.8 | 1.86 | 7.75 |
| 6 | DMF | 1 | 1 | 80 | 12 | - | - | 1 |
| 7 | DMF | 2 | 1 | 80 | 12 | 146.0 | 1.66 | 6 |
| 8 | DMF | 2 | 2 | 80 | 12 | - | - | 0 |
| 9 | DMF | 2 | 3 | 80 | 12 | - | - | 0 |
| 10 | DMF | 1 | 0.2 | 60 | 12 | - | - | 0 |
| 11 | DMF | 1 | 0.2 | 90 | 12 | 152.0 | 1.88 | 4.25 |
| 12 | THF | 1 | 0.2 | 80 | 12 | 192.2 | 1.74 | 1 |
| 13 | toluene | 1 | 0.2 | 80 | 12 | 23.8 | 3.73 | 2.25 |
| 14 | DCE | 1 | 0.2 | 80 | 12 | - | - | 0 |
| 15 | HOAc | 1 | 0.2 | 80 | 12 | - | - | 0 |
| 16 | DMF: HOAc = 10: 1 | 1 | 0.2 | 80 | 12 | 92.9 | 2.09 | 13 |
| 17e | DMF | 1 | 0.2 | 80 | 12 | - | - | 0 |
| 18 f | DMF | 1 | 0.2 | 80 | 12 | - | - | 0 |
aConditions: PS (0.483 mmol), DEP, Solvent (1.0 mL). bPS [Mn = 151.0 kg·mol-1, Ð = 1.97]. cMn and Ð were determined by gel permeation chromatography (GPC) against a PS standard at 35 ℃ in THF. dThe functionalization of PS was determined by the 1H NMR analysis. eConditions: PS (0.483 mmol), DEP (0.483 mmol), DMF (1.0 mL). 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (TEMPO, 0.966 mmol). fConditions: PS (0.483 mmol), DEP (0.483 mmol), DMF (1.0 mL). butylated hydroxytoluenel (BHT, 0.966 mmol).
Table 2.
Screening of phosphate esters.
| 2 c | Nitrogen atmosphere e | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 1 | 2 |
Mn (kg·mol-1) |
Ð | Functionalization d (%) |
Td, 5% (℃) |
Td, 50% (℃) |
Td ,max (℃) |
Residue (%) |
| 1 | - | PS | - | - | - | 377.5 | 418.5 | 424 | 2.48 |
| 2 | DEP | PS-DEP | 134915 | 1.88 | 10 | 282.8 | 444.7 | 354, 463 | 10.36 |
| 3 | BFP | PS-BFP | 133.4 | 2.00 | 8.5 | n.d | n.d | n.d | n.d |
| 4 | DMP | PS-DMP | 140.8 | 1.97 | 8.3 | 131 | 424.9 | 426 | 12.15 |
| 5 | DBP | PS-DBP | 139.9 | 1.74 | 11 | 245.9 | 430.3 | 440 | 2.57 |
| 6 | DPP | PS-DPP | 176.2 | 1.74 | 13.6 | 395 | 424.5 | 424 | 14.44 |
aConditions: PS (1.0 equiv), 1 (1.0 equiv), DMF (1.0 mL). bPS [Mn = 151.0 kg·mol-1, Ð = 1.97]. cMn and Ð were determined by gel permeation chromatography (GPC) against a PS standard at 35 ℃ in THF. dThe functionalization of PS was determined by the 1H NMR analysis. eTGA tests were performed between 30-600 ℃ under nitrogen with a heating rate of 10 ℃·min-1.
Table 3.
MCC Data of PS and Phosphonate Functionalised PS.
| Sample | HRC (J/g·K) |
pHRR (W/g) |
THR (kJ/g) |
TpHRR (℃) |
|---|---|---|---|---|
| PS | 1043.4 | 1051.1 | 46.5 | 433.5 |
| PS-BFP | 536.7 | 539.2 | 35.9 | 458.1 |
| PS-DMP | 596.5 | 600.4 | 40.5 | 437.8 |
| PS-DPP | 642.3 | 647.1 | 40.6 | 434.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



