
Submitted:
17 May 2024
Posted:
20 May 2024
You are already at the latest version
Abstract
G-protein coupled estrogen receptor (GPER) was described to exerts several cardioprotective effects. However, the exact mechanism involved in cardiac protection remains unclear. The aim of this study is to investigate the role of GPER activation on excitation-contraction coupling (ECC), and the possibility that such effect participates in cardioprotection. Cardiac myocytes of male Wistar rats were isolated with digestive buffer and loaded with Fura-2-AM for measurement of intracellular calcium transients (CaT). Sarcomere shortening (SS) and L-type calcium current (ICaL) were also registered. Confocal technique was used to measure nitric oxide (NO) production in cells loaded with DAF-FM-diacetate. Cardiac myocytes exposed to E2 (10 nM), or G-1 (1 µM) for fifteen minutes decreased CaT, SS and ICaL. These effects were prevented using G-36 (antagonist of GPER, 1 µM), L-Name (NO synthase inhibitor, 100 nM), or wortmannin (phosphoinositide-3-kinase inhibitor, 100 nM). Moreover, G1 increased NO production, and this effect was abolished in the presence of wortmannin. We concluded that the selective activation of GPER with E2 or G1 in isolated cardiac myocytes of male rats induced a negative inotropic effect due to the reduction of ICaL and the decrease in CaT. Finally, the pathway that we proposed to be implicated in these effects is PI3K-NOS-NO.
Keywords:
Wistar rats
; cardiomyocytes
; G-protein coupled estrogen receptor (GPER)
; L-type calcium current
; Calcium transients
; cell shortening.
Introduction
The G-protein coupled estrogen receptor (GPER) was initially named GPR30 and was characterized as an orphan receptor [1,2]. In 2006, a selective agonist of GPR30 named G1 was synthesized by molecular screening and computational testing [3]. Finally, the receptor was renamed GPER by IUPHAR (International Union of Basic and Clinical Pharmacology) in 2007 [4,5]. As G1 is a non-steroidal compound with high selectivity to GPER and not to ERs [3], it represents a useful strategy to investigate the implications of GPER activation.
It is well known that E2 activates complex pathways involving both genomic and non-genomic pathways within different cell types. In the heart, while the genomic mechanisms are well characterized, being mediated by the classical estrogen receptor [6,7], GPER present non-genomics effects, either with E2 or G1 [8,9]. The selective activation of GPER triggers several intracellular pathways like ERK, PI3K, cAMP, PKA, eNOS, sGC, and AKT [8,9,10,11,12,13]. In 2015 we demonstrated that G1 binding to GPER can transactivate to the epidermal growth receptor (EGFR), leading to the activation of the PI3K-AKT pathway, which finally activates the electrogenic isoform of the sodium/bicarbonate cotransporter (eNBC) in isolated ventricular myocytes [14].
It was fully demonstrated that GPER activation affords cardioprotection. Chronic GPER activation decreases blood pressure [15], prevents diastolic dysfunction [16,17], and also the progression to heart failure in rats treated with isoproterenol [18]. Moreover, GPER activation prevents ischemic reperfusion injury with less myocardial inflammation [19], reducing infarct size with the activation of PI3K-AKT and MEK1/2-Erk1/2-GSK-3β pathways to inhibit mPTP opening [20] and by reducing mitochondrial dysfunction, mitophagy and regulation of the mPTP opening [21]. Accordingly, our laboratory recently demonstrated, in a rat ovariectomy model, that the chronic administration of G1 enhanced cardiac mitochondrial function, improved cardiac function, and reduced infarct size after ischemia [22].
Interestingly, another possibility to prevent cardiac dysfunction after ischemic/reperfusion damage is L-type calcium channel (LTCC) inhibition [23,24,25]. LTCC is located in cardiac T tubules [26,27,28], and calcium influx activates ryanodine receptors (RyR) of the sarcoplasmic reticulum (SR), triggering calcium-induced calcium-release (CICR) mechanism. This calcium transient is responsible for activating the contractile apparatus in the cardiomyocyte (excitation-contraction coupling, ECC). Modification in LTCC activity could induce changes in ECC, triggering pathologic events like cardiac arrhythmias, heart failure, and cardiac hypertrophy [29,30]. Several studies demonstrated that LTCC function decreases after treatment with high concentration of estrogen [31,32] and in OVX model [33]. Even with deletion of classics ERs, E2 decreases calcium current [34], indicating that another receptor acts in the presence of E2. The activation of GPER prevents the effect of isoproterenol on calcium current with E2 or G1, decreasing it [32,35]. However, the signaling behind the effect of GPER on LTCC remains unclear.
We have previously demonstrated that GPER is present in T tubules of cardiomyocytes [36], suggesting that their signaling can be relevant to ECC. Thus, in the present study, we tested the hypothesis that the activation of GPER and its downstream intracellular pathway PI3K and NOS, alters calcium handling by inhibiting LTCC and subsequently affecting ECC.
Results
E2 and G1 Reduce Cardiomyocyte Contractility through GPER Signaling
In order to investigate the effect of GPER on cardiac contractility, isolated ventricular myocytes were exposed to E2 (10 nmol/L) for 15 minutes. Figure 1A shows a representative continuous sarcomere shortening recording of one cardiomyocyte subjected to the acute application of E2 (10 nmol/L). Figure 1B depicts the average values of the negative inotropic effect of E2. Moreover, this effect was prevented when the cells were preincubated with the GPER blocker G36 (1 µmol/L, Figure 1C,D), suggesting that it is mediated by GPER activation and not by stimulation of the canonical E2 receptor (ER). Although we cannot confirm it, due to the rapid onset of this negative inotropic effect, it is also feasible to suggest that it is a non-genomic pathway. As shown in Figure 2A,B, the acute administration of the selective GPER agonist G1 (1 µmol/L) decreased sarcomere shortening in a similar magnitude to E2, reinforcing the idea that the activation of GPER is involved in the reduction of cardiac contractility. As expected, the effect of G1 was prevented by G36 (Figure 2C,D). To determine if the GPER-induced negative inotropic effect is a consequence of intracellular Ca2+ reduction, we recorded calcium transients (CaT) in the presence of E2 or G1. As shown in Figure 3, CaT decreased with E2 (panel A) or G1 (panel C), and these effects were prevented when the myocytes were preincubated with G36 (panels B and D). One possibility might be that a reduction in SR Ca2+ reuptake leads to a reduction in SR Ca2+ content. To investigate this, a caffeine-induced calcium transient was performed at the end of each experiment. However, G1 had no significant effect on caffeine-induced calcium transient amplitude (Figure 3E). Therefore, activation of GPER signaling reduces contractility by diminishing Ca2+ handling, without affecting SR Ca2+ content.
Activation of GPER Decreases L-Type Calcium Current (ICaL) in Cardiac Myocytes
SR Ca2+ release through ryanodine receptors (RyR) induced by the Ca2+ influx produced by the opening of the sarcolemmal L-type voltage calcium channel during the action potential plateau represents the basis of excitation-contraction coupling. Thus, we employed the patch-clamp technique in the whole-cell configuration to assess the participation of ICaL in the GPER-induced negative inotropic effect observed in the previous experiments. E2 or G1 reduced ICaL recorded at 0 mV by approximately 25 % (Figure 4A and Figure 5A, respectively). This effect was prevented when the cells were preincubated with G36 (Figure 4B and Figure 5B), indicating that E2 and G1 exert their effect through the activation of GPER. The reduction in ICaL induced by E2 or G1 was observed at different test voltages, as depicted in the IV plots of Figure 4C and Figure 5C. Overall, these data demonstrate that GPER activation reduces ICaL, affecting ECC and producing a negative inotropic effect in cardiac myocytes.
PI3K and NOS Are Involved in the GPER-Triggered Intracellular Pathway
It is known that GPER activation stimulates the PI3K and NOS pathways, modulating diverse cardiac functions [8,10,14]. Thus, we used wortmannin (100 nM) and L-Name (100 nM) to block PI3K and NOS, respectively. The inhibitors were used before and after exposing cardiomyocytes to G1 and SS, CaT and ICaL were measured. Figure 6 shows that the effect of G1 is abolished in the presence of PI3K or NOS blockade indicating that GPER intracellular pathway involves PI3K and NOS activation.
Finally, to understand if the effect of GPER initiates with the activation of NOS or with the stimulation of PI3K, we measured NO production with the fluorescent dye DAF-FM diacetate. The cells were perfused with G1 or wortmannin plus G1. As shown in Figure 7, G1 increased the production of NO. This effect is prevented when the cardiomyocytes were pretreated with wortmannin (Figure 4A,D), indicating that GPER acts by activating PI3K, which subsequently induces an increase in NO production by NOS stimulation.
Discussion
In the present paper, we demonstrated for the first time that cardiac GPER activation with E2 or G1 induced a rapid negative inotropic effect through downregulation of LTCC, which decreases ICaL and CaT, subsequently reducing cardiomyocyte contractility. In association with this mechanism, a previous report from our laboratory has demonstrated that GPER is located in T tubules of cardiomyocytes, suggesting that its signaling could be relevant for ECC [36]. Although it is not clear at this time whether this effect of GPER activation and decrease in Ca2+ is cardioprotective, it could be speculated that in certain conditions, for example reperfusion following an ischemia where there is a sudden intracellular Ca2+ overload, our proposed mechanism protects the cardiomyocyte. Accordingly, previous studies have demonstrated favorable results using different LTCC blockers [23,24,25], suggesting the important contribution of Ca2+ influx across these channels during reperfusion.
The idea of using GPER as a therapeutic target to prevent the development and progression of several cardiovascular diseases is still under investigation and needs further research. Different works have demonstrated that chronic activation of GPER produces cardiovascular protection like blood pressure reduction in an ovariectomy (OVX) model [15]; improvement of heart function after ischemia-reperfusion injury, independently of animal sex [10], and prevention of diastolic dysfunction in salt-induced hypertension [16]; among other publications. Consistently, we have recently reported that OVX rats exert a deteriorated mechanical response after ischemia and reperfusion injury and bigger infarct size, possibly due to an impaired mitochondrial function, which was successfully prevented with the chronic treatment with G1 [22]. Interestingly, acute application of E2 during reperfusion after ischemia has also been demonstrated as a cardioprotective alternative [21]. In this case, the authors proposed that GPER activation, increasing NO production and activating ERK 1/2 kinase can prevent mitochondrial transition pore aperture. This acute cardioprotective effect of GPER activation was also reported by Rocca et al., when identified PI3K-AKT-eNOS pathway to improve mitochondrial survival [37]. Nevertheless, the GPER-induced modulation of ECC reported herein might represent a novel non-genomic cardioprotective pathway that deserves future elucidation.
Several publications have demonstrated that the activation of GPER involves the increased activity of PI3K and NOS [8,10,13,14]. To determine whether this pathway is responsible for contractility and Ca2+ handling, we use inhibitors for each possible candidate. When we used L-Name to inhibit NOS GPER did not generate an effect on ECC, indicating that NOS represents one actor in GPER actions. However, since L-Name is a general blocker of NO production, we cannot elucidate which NOS is implicated in this effect. Several reports have described that the activity of Ca2+ channels could be modulated by NO [38,39]. Consistently with our results, α1C subunit have nitrosylation sites that negatively modulate channel activity [38]. In addition, previous reports have shown that specific inhibition of nNOS induced increment of CaL activity [40]. Moreover, transgenic mice overexpressing nNOS evidenced a negative inotropic effect associated with CaL block [41]. However, another study described that NO donors increase calcium current [42]. One possibility to explain this controversy is that the final effect of NO on the channel depends on the dose of this gas. Nevertheless, we can speculate that the pathway involved herein could be mediated by a possible nitrosylation of the channel, with the subsequent depression of its activity. However, we cannot discard the NO-induced activation of the guanylate cyclase-cGMP-PKG pathway, as this kinase was also shown that phosphorylate the channel and then inhibit it [42]. Thus, more experiments with blockers of this pathway are necessary to give new insights into this issue.
Recently, Francis et al. have described an anti-arrhythmic effect of GPER activation on OVX guinea pigs, without changes in ICaL [43]. The difference with the present work, in addition to the animal model, is the time that the cells were exposed to G1. In Francis’s paper, the cells were exposed for 2 hours to G1 1 μmol/L. It might be possible that our time exposition was sufficient to induce a rapid effect of GPER and after this time this effect disappeared due to modifications of GPER expression in the cell membrane by desensitization, like occurs in the regulation of other G-protein coupled receptors [44,45,46].
Finally, PI3K was another actor on the pathway studied. Using wortmannin during experiments of SS, CaT, and ICaL, we obtained that inhibition of PI3K avoided the effect of the selective activation of GPER. Since is known that PI3K phosphorylates AKT and this kinase stimulated NOS [14,47], we propose to evaluate if this sequence of events is triggered after the activation of GPER. Thus, we determined NO production using DAF-FM diacetate with or without wortmannin. G1 increased NO production and the inhibition of PI3K prevented this effect. These results indicate that the pathway triggered by GPER activation is PI3K/NOS/NO.
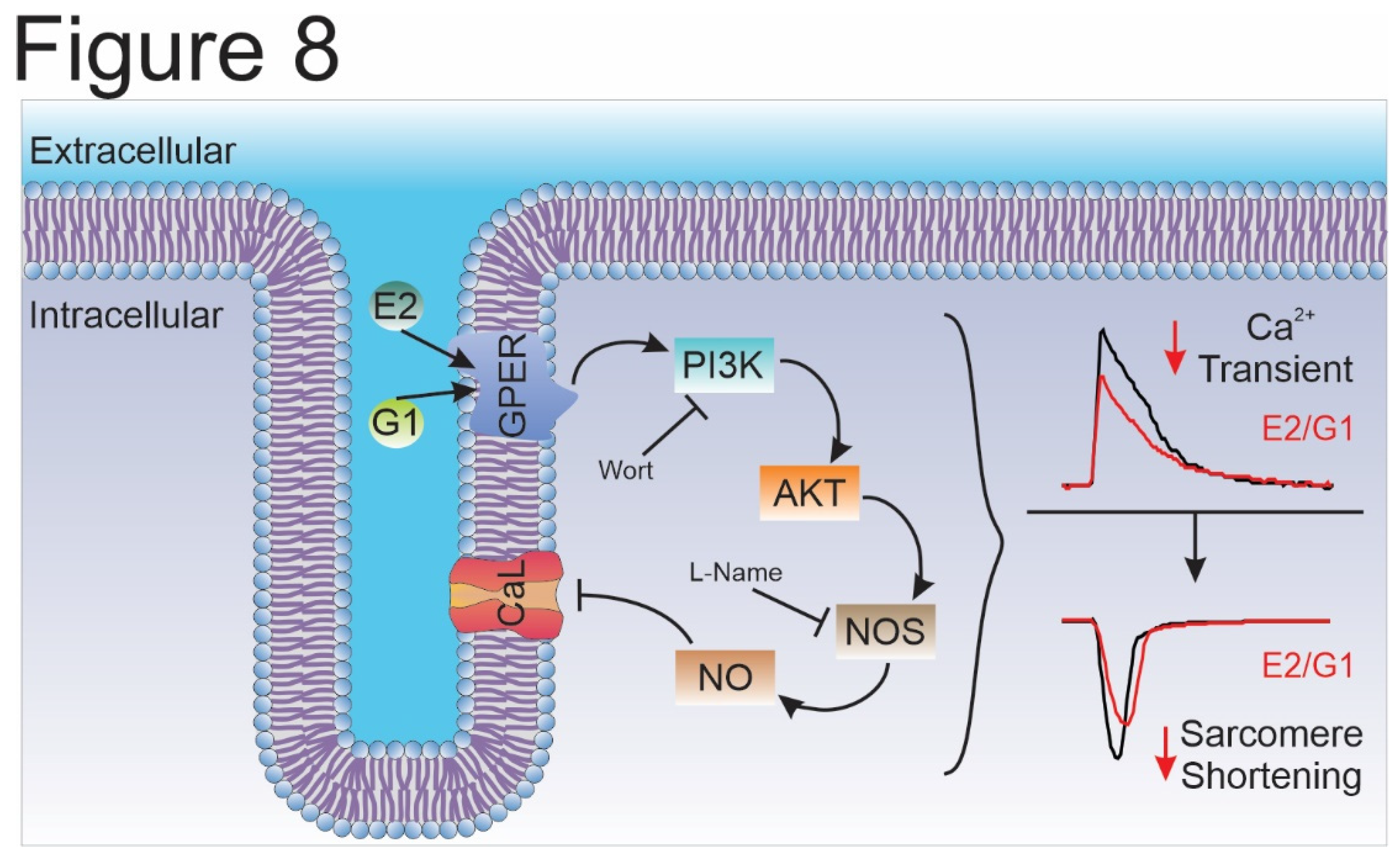
Overall, we conclude that the selective activation of GPER decreases CaT amplitude and consequently cardiomyocyte contractility by an ICaL reduction in Wistar male rats. In summary, we are reporting herein that the GPER-activated PI3K/NOS/NO pathway decreased CaL current and altered the contractile mechanism of the cardiomyocyte (Figure 8). Further experiments are needed to elucidate how NO specifically decreases ICaL.
Materials and Methods
Animals
All experiments were performed following the Guide for Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 2011) and approved by the Ethics committee of the Faculty of Medicine, La Plata, Buenos Aires, Argentina. Male Wistar rats of 12-week-old were used for all experiments.
Ventricular Myocytes Isolation
Ventricular myocytes were isolated according to the technique previously described [48] with some modifications. The heart was attached via the ascendent aorta to a cannula, excised, and mounted in a Langendorff apparatus. Then a retrograde perfused at 37ºC with perfusion buffer (P-B) of the following composition (in mmol/L): 146.2 NaCl, 4.7 KCl, 1 CaCl2, 10 HEPES, 0.35 NaH2PO4, 1 MgSO4, and 11 glucose (pH adjusted to 7.4 with NaOH). The solution was continuously bubbled with 100% O2. After a stabilization period of 4 minutes, the perfusion was switched to a nominally Ca2+-free P-B for 4-5 minutes. The hearts were then perfused with collagenase (140 units/ml) in P-B containing 50 µmol/L of CaCl2for 12-15 minutes or until it became flaccid. The heart was then removed from the perfusion apparatus by cutting at the atrioventricular junction. The desegregated myocytes were separated from the undigested tissue and the CaCl2 concentration of perfusion buffer was increased in 5 steps to 1 mmol/L at room temperature (22-25ºC).
Measure of Calcium Transient and Cell Shortening
Calcium transient recordings were performed as previously described [49]. Briefly, isolated myocytes were loaded with 3 μmol/L Fura-2 AM (Thermo Fisher Scientific) for 10 minutes. After a wash of the exceeding fluorescent dye, Fura-2 fluorescence kept in the myocytes was measured on an inverted microscope adapted for epifluorescence by IonOptix setup (IonOptix, Massachusetts, USA). The perfusion chamber with cells was continuously perfused at a flow rate of 1 ml/min and stimulated via 2-platinum electrodes on either side of the chamber at 0.5 Hz. The perfused solution is equal to P-B described previously (see above). The ratio of the Fura-2 fluorescence (at 510nm) obtained after exciting the dye at 340 and 380nm was taken as an index of intracellular Ca2+ (Ca2+i). Cell shortening was detected as sarcomere shortening (SS) using a video-based motion detection and stored by software for an off-line analysis using IonWizard (IonOptix). For sarcoplasmic-reticulum (SR) Ca2+ content measurement, a solution containing 15 mmol/L caffeine was rapidly applied to cells. The amplitude of the caffeine-induced Ca2+ transient was used to estimate SR Ca2+ content.
Patch-Clamp Recording
L-type calcium current (ICaL) was recorded with a whole-cell configuration of the Patch-Clamp technique in voltage-clamp mode, filtered at 2 kHz. AxoPatch 200B amplifier and analog-to-digital converter Digidata 1322A (Molecular Devices, California, USA) were used to acquire ICaL recorded with Clampex 9.2 Software (Molecular Devices). Borosilicate patch pipettes were pulled with P-97 puller (Sutter Instruments, California, USA) to a final resistance of 2.5-3.5 MΩ. The pipette was filled with (in mmol/L): 135 CsCl, 1 MgCl2, 4 Na2ATP, 5 EGTA, 10 HEPES (pH 7.2 with CsOH). Isolated rat ventricular myocytes were placed in a perfusion chamber and perfused at flow rate of 1 ml/min with bath solution (HEPES Buffer) contained (in mmol/L): 5 CsCl, 122 NaCl, 1 MgCl2, 1 CaCl2, 10 tetraethylammonium chloride, 5 4-aminopyridine, 5 glucose, 10 HEPES (pH 7.4 with NaOH). For ICaL recordings the following voltage protocol was applied: cardiomyocytes were depolarized from a holding potential of -80 to -40 mV for 200 msec to inactivate sodium current, and then to different test potentials ranging from -50 to +70 mV in 10 mV increments for 500 msec, delivered at 0.1 Hz. To obtain IV curves, the steady state current was subtracted from the peak inward current corresponding to each test pulse. The peak inward current was normalized to cell capacitance (pA/pF). The patch clamp data was processed and analyzed with ClampFit 10.3 (Molecular Device).
Nitric Oxide Production
Cardiac myocytes were loaded with 5 μmol/L DAF-FM diacetate (Thermo Fisher Scientific) for 30 minutes at room temperature and imaged by epifluorescence on a Zeis 410 inverted confocal microscope (LSM Tech, Pennsylvania, USA). Excitation at 488 nm was provided by an argon laser and emission was collected in a range of 500-550 nm. Photographs were taken every 30 seconds for 25 minutes. Image J software was used for the analysis. Results were expressed as a percentage of time zero and slope fitted with linear regression. Slopes were normalized using the difference between treated and control conditions.
Treatments
All experiments were performed at room temperature (20-25ºC) and with bath solution with or without: 0.01 % DMSO (Sigma, solvent for all reagents); 10 nM E2 (Invitrogen); 1 µM G1 (GPER agonist, Cayman Chemicals), 1 µM G36 (GPER antagonist, Cayman Chemicals), 100 nM Wortmannin (Invitrogen) or 100 nM L-NAME (Sigma) for 15 minutes after stabilization with the corresponding control solution for each set of experiments.
Statistics
GraphPad Prism 8 (GraphPad, California, USA) was used for all the statistics analysis. Data were expressed as means ± SEM. The Shapiro-Wilk test was used to test normality. Data were compared with paired Student t-test or paired t-test with Wilcoxon test if variances could not be assumed to be equal. When more than 2 groups were evaluated, 1-way ANOVA with the adequate post-test was performed to every particular situation. A value of P<0.05 was considered statistically significant.
Author Contributions
Conceptualization, E. A. A.; Methodology, L. A. D. Z., M. S. E., A. M. I., M. E. R. and L. E. P.; Validation, L. A. D. Z., M. S. E., V. C. DG and E. A. A.; Formal Analysis, L. A. D. Z. and M. S. E.; Investigation, L. A. D. Z., M. S. E. and E. A. A.; Writing—Original Draft Preparation, L. A. D. Z. and M. S. E.; Writing—Review and Editing, V. C. DG and E. A. A.; Supervision, V. C. DG and E. A. A.; Funding Acquisition, E. A. A. L. A. D. Z and M. S. E. authors contributed equally to this work in methodology (Patch-Clamp and IonOptix methodology) and formal analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Agencia Nacional de Promoción Científica y Tecnología de Argentina (ANCPyT), PICT 2020 #2427 and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), PIP 1781 to E. A. A.
Acknowledgments
We greatly appreciate the contribution of Dr. Luis Gonano for contribution of DAF-FM-diacetate.
Conflicts of Interest
No conflicts of interest are declared.
References
- Kvingedal, A.M. and E.B. Smeland, A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett, 1997. 407(1): p. 59-62.
- Carmeci, C., et al., Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics, 1997. 45(3): p. 607-17.
- Bologa, C.G., et al., Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol, 2006. 2(4): p. 207-12.
- Prossnitz, E.R. and J.B. Arterburn, International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev, 2015. 67(3): p. 505-40.
- Barton, M., et al., Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J Steroid Biochem Mol Biol, 2018. 176: p. 4-15.
- Frump, A.L., et al., 17beta-Estradiol and estrogen receptor alpha protect right ventricular function in pulmonary hypertension via BMPR2 and apelin. J Clin Invest, 2021. 131(6).
- Mihai, M.C., et al., Mechanism of 17beta-estradiol stimulated integration of human mesenchymal stem cells in heart tissue. J Mol Cell Cardiol, 2019. 133: p. 115-124.
- Filice, E., et al., A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J Physiol Pharmacol, 2009. 60(4): p. 3-10.
- Holm, A., et al., The G protein-coupled estrogen receptor 1 (GPER1/GPR30) agonist G-1 regulates vascular smooth muscle cell Ca(2)(+) handling. J Vasc Res, 2013. 50(5): p. 421-9.
- Deschamps, A.M. and E. Murphy, Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol, 2009. 297(5): p. H1806-13.
- Yu, X., et al., Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am J Physiol Endocrinol Metab, 2011. 301(5): p. E882-8.
- Yu, X., et al., G protein-coupled estrogen receptor 1 mediates relaxation of coronary arteries via cAMP/PKA-dependent activation of MLCP. Am J Physiol Endocrinol Metab, 2014. 307(4): p. E398-407.
- Lindsey, S.H., L. Liu, and M.C. Chappell, Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids, 2014. 81: p. 99-102.
- De Giusti, V.C., et al., Aldosterone stimulates the cardiac sodium/bicarbonate cotransporter via activation of the g protein-coupled receptor gpr30. J Mol Cell Cardiol, 2015. 89(Pt B): p. 260-7.
- Lindsey, S.H., et al., Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology, 2009. 150(8): p. 3753-8.
- Jessup, J.A., et al., Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One, 2010. 5(11): p. e15433.
- Wang, H., et al., Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res, 2012. 94(1): p. 96-104.
- Kang, S., et al., Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS One, 2012. 7(10): p. e48185.
- Weil, B.R., et al., Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery, 2010. 148(2): p. 436-43.
- Kabir, M.E., et al., G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion. PLoS One, 2015. 10(9): p. e0135988.
- Feng, Y., et al., Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol, 2017. 174(23): p. 4329-4344.
- Ibanez, A.M., et al., Chronic GPER activation prevents ischemia/reperfusion injury in ovariectomized rats. Biochim Biophys Acta Gen Subj, 2022. 1866(2): p. 130060.
- Moreyra, A.E., et al., Chronic administration of nicardipine attenuates myocardial stunning in isolated rabbit hearts. J Mol Cell Cardiol, 1994. 26(8): p. 979-84.
- de Cingolani, G.E., et al., Chronic nifedipine treatment diminishes cardiac inotropic response to nifedifine: functional upregulation of dihydropyridine receptors. J Cardiovasc Pharmacol, 1996. 27(2): p. 240-6.
- Pardo, A.C., et al., Cardioprotective effects of N-methylacetazolamide mediated by inhibition of L-type Ca(2+) channel current. Biochim Biophys Acta Gen Subj, 2022. 1866(5): p. 130098.
- Shaw, R.M. and H.M. Colecraft, L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res, 2013. 98(2): p. 177-86.
- Kamp, T.J. and J.W. Hell, Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res, 2000. 87(12): p. 1095-102.
- Carl, S.L., et al., Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J Cell Biol, 1995. 129(3): p. 673-82.
- Hool, L.C., Elucidating the role of the L-type calcium channel in excitability and energetics in the heart: The ISHR 2020 Research Achievement Award Lecture. J Mol Cell Cardiol, 2022. 172: p. 100-108.
- Mukherjee, R. and F.G. Spinale, L-type calcium channel abundance and function with cardiac hypertrophy and failure: a review. J Mol Cell Cardiol, 1998. 30(10): p. 1899-916.
- Grohe, C., et al., Modulation of hypertensive heart disease by estrogen. Steroids, 1996. 61(4): p. 201-4.
- Meyer, R., et al., Rapid modulation of L-type calcium current by acutely applied oestrogens in isolated cardiac myocytes from human, guinea-pig and rat. Exp Physiol, 1998. 83(3): p. 305-21.
- Yang, H.Y., et al., Effect of ovariectomy on intracellular Ca(2+) regulation in guinea pig cardiomyocytes. Am J Physiol Heart Circ Physiol, 2017. 313(5): p. H1031-H1043.
- Ullrich, N.D., et al., Genomic deletion of estrogen receptors ERalpha and ERbeta does not alter estrogen-mediated inhibition of Ca2+ influx and contraction in murine cardiomyocytes. Am J Physiol Heart Circ Physiol, 2008. 294(6): p. H2421-7.
- Whitcomb, V., et al., Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem Pharmacol, 2020. 171: p. 113727.
- Orlowski, A., et al., The cardiac electrogenic sodium/bicarbonate cotransporter (NBCe1) is activated by aldosterone through the G protein-coupled receptor 30 (GPR 30). Channels (Austin), 2016. 10(5): p. 428-434.
- Rocca, C., et al., Notch1 Mediates Preconditioning Protection Induced by GPER in Normotensive and Hypertensive Female Rat Hearts. Front Physiol, 2018. 9: p. 521.
- Carnes, C.A., et al., Atrial glutathione content, calcium current, and contractility. J Biol Chem, 2007. 282(38): p. 28063-73.
- Sun, J., et al., Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res, 2006. 98(3): p. 403-11.
- Wang, Y., et al., Modulation of L-type Ca(2)(+) channel activity by neuronal nitric oxide synthase and myofilament Ca(2)(+) sensitivity in cardiac myocytes from hypertensive rat. Cell Calcium, 2015. 58(3): p. 264-74.
- Burkard, N., et al., Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ Res, 2007. 100(3): p. e32-44.
- Gonzalez, D.R., et al., S-Nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol, 2009. 54(3): p. 188-95.
- Francis, A.J., et al., GPER limits adverse changes to Ca(2+) signalling and arrhythmogenic activity in ovariectomised guinea pig cardiomyocytes. Front Physiol, 2022. 13: p. 1023755.
- Marchese, A., et al., G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol, 2008. 48: p. 601-29.
- Scita, G. and P.P. Di Fiore, The endocytic matrix. Nature, 2010. 463(7280): p. 464-73.
- Jean-Alphonse, F. and A.C. Hanyaloglu, Regulation of GPCR signal networks via membrane trafficking. Mol Cell Endocrinol, 2011. 331(2): p. 205-14.
- Qin, W., L. Cao, and I.Y. Massey, Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol Cell Biochem, 2021. 476(11): p. 4045-4059.
- Aiello, E.A., et al., Evidence for an electrogenic Na+-HCO3- symport in rat cardiac myocytes. J Physiol, 1998. 512 ( Pt 1)(Pt 1): p. 137-48.
- Espejo, M.S., et al., The reduced myofilament responsiveness to calcium contributes to the negative force-frequency relationship in rat cardiomyocytes: role of reactive oxygen species and p-38 map kinase. Pflugers Arch, 2017. 469(12): p. 1663-1673.
Figure 1.
E2 reduces cardiomyocyte contractility through GPER signaling. A and C. Representative traces of cardiac cell contraction in presence of E2 after ethanol or G36 plus E2 after G36 condition, respectively. Below, average traces of cell shortening at the end of each condition. B and D. Average data of sarcomere shortening for each condition. p < 0.05 indicated significantly different. N/n indicates animals and cells number, respectively.
Figure 1.
E2 reduces cardiomyocyte contractility through GPER signaling. A and C. Representative traces of cardiac cell contraction in presence of E2 after ethanol or G36 plus E2 after G36 condition, respectively. Below, average traces of cell shortening at the end of each condition. B and D. Average data of sarcomere shortening for each condition. p < 0.05 indicated significantly different. N/n indicates animals and cells number, respectively.
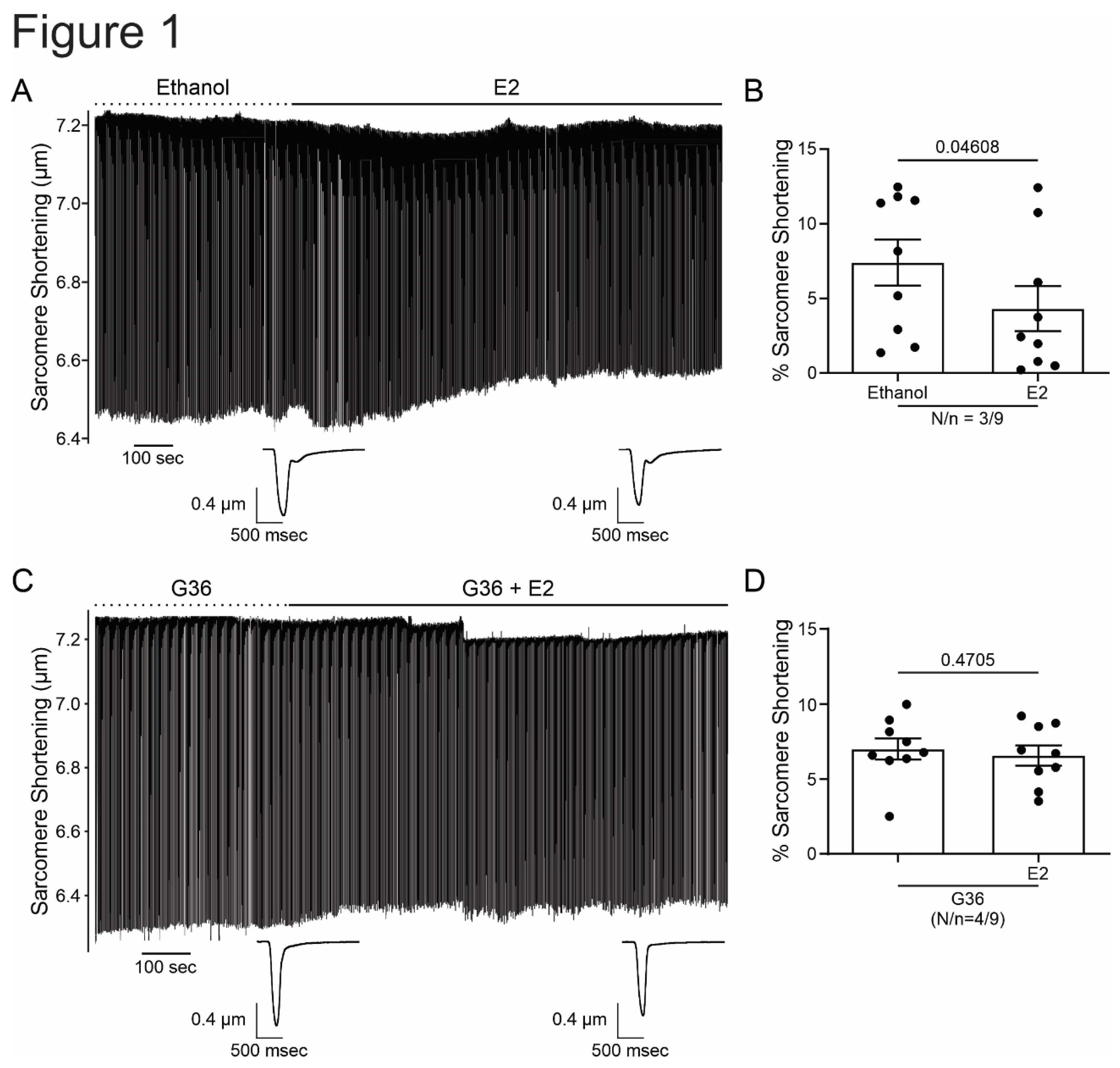
Figure 2.
G1 reduces cardiomyocyte contractility through GPER signaling. A and C. Representative traces of cardiac cell contraction in presence of G1 after DMSO or G36 plus G1 after G36 condition, respectively. Below, average traces of cell shortening at the end of each condition. B and D. Average data of sarcomere shortening for each condition. p < 0.05 indicated significantly different. N/n indicates animals and cells number, respectively.
Figure 2.
G1 reduces cardiomyocyte contractility through GPER signaling. A and C. Representative traces of cardiac cell contraction in presence of G1 after DMSO or G36 plus G1 after G36 condition, respectively. Below, average traces of cell shortening at the end of each condition. B and D. Average data of sarcomere shortening for each condition. p < 0.05 indicated significantly different. N/n indicates animals and cells number, respectively.
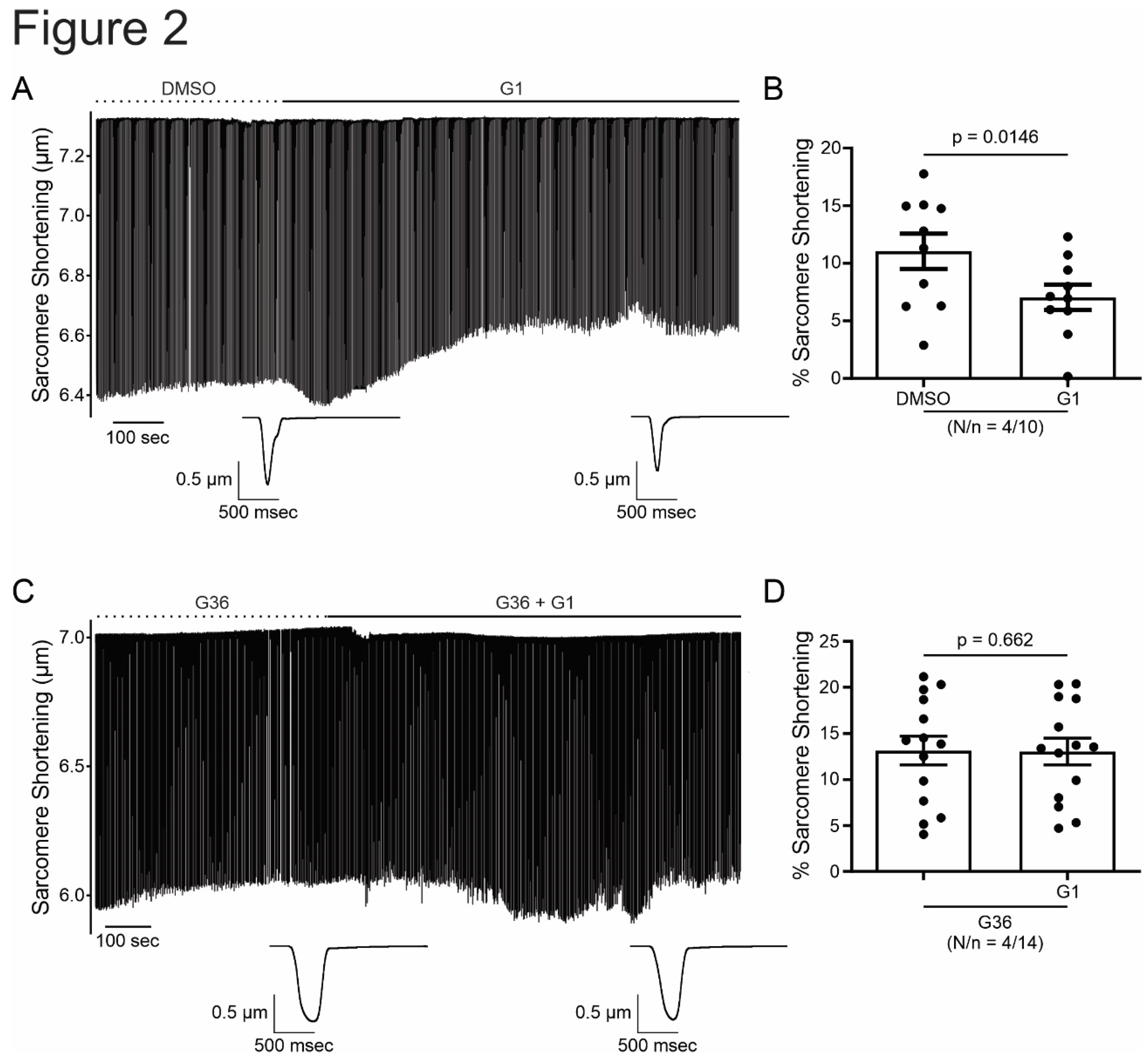
Figure 3.
Activation of GPER with E2 or G1 decrease calcium transient in isolated cardiomyocytes, without changing SR calcium content. A to D. Representative traces of calcium transient measured with Fura-2 in presence of E2, G36 plus E2, G1 or G36 plus G1, respectively, after each control condition (left). Right panels, representative average data of calcium transients for of all the treatment applied. E. Representative caffeine pulse induced-calcium transient (left) in presence of DMSO (up) or G1 (down) condition. Right. Representative average data of caffeine pulse induced-calcium transients. p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
Figure 3.
Activation of GPER with E2 or G1 decrease calcium transient in isolated cardiomyocytes, without changing SR calcium content. A to D. Representative traces of calcium transient measured with Fura-2 in presence of E2, G36 plus E2, G1 or G36 plus G1, respectively, after each control condition (left). Right panels, representative average data of calcium transients for of all the treatment applied. E. Representative caffeine pulse induced-calcium transient (left) in presence of DMSO (up) or G1 (down) condition. Right. Representative average data of caffeine pulse induced-calcium transients. p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
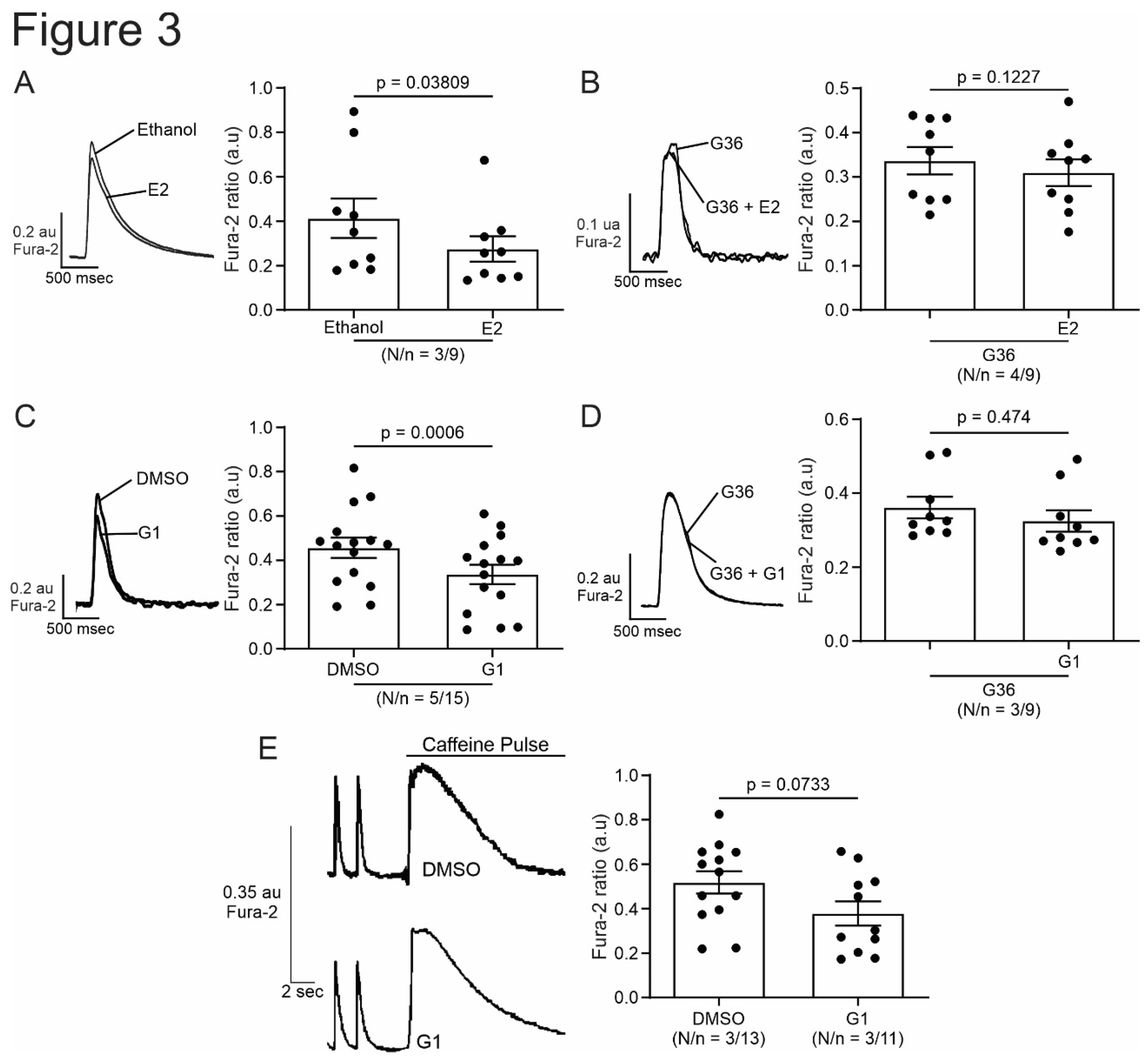
Figure 4.
Activation of GPER with E2 decrease L-type calcium current in cardiomyocytes. A and B. Top, Voltage protocol and average traces of calcium current at 0 mV in presence of E2 after ethanol or G36 plus E2 after G36 condition, respectively. Down, average data of calcium current at 0 mV and linked dot blot (inset) of each condition. C. IV plot of L-type calcium current with ethanol or E2 treatment, and voltage protocol (inset). p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
Figure 4.
Activation of GPER with E2 decrease L-type calcium current in cardiomyocytes. A and B. Top, Voltage protocol and average traces of calcium current at 0 mV in presence of E2 after ethanol or G36 plus E2 after G36 condition, respectively. Down, average data of calcium current at 0 mV and linked dot blot (inset) of each condition. C. IV plot of L-type calcium current with ethanol or E2 treatment, and voltage protocol (inset). p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
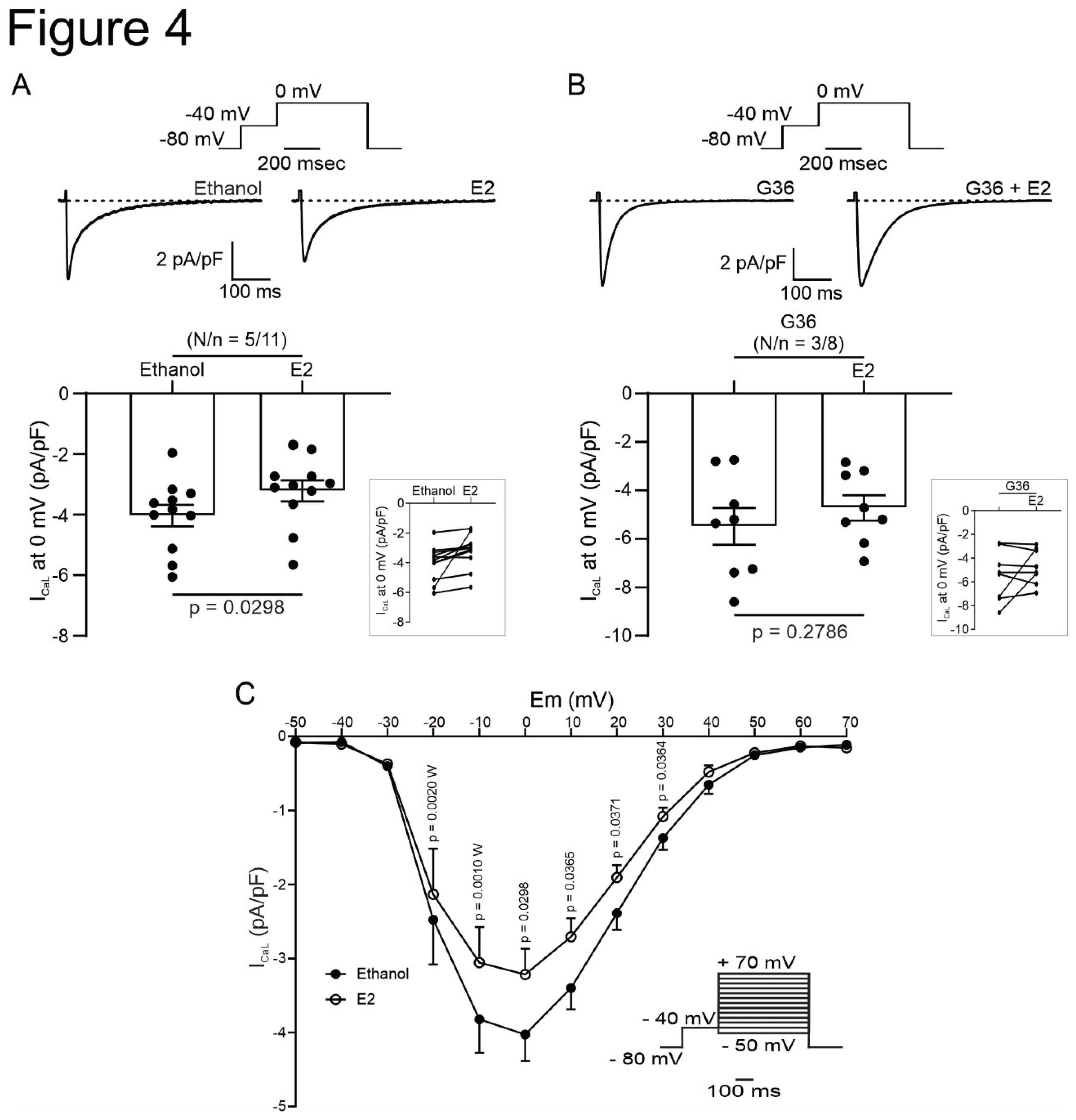
Figure 5.
Activation of GPER with G1 decrease L-type calcium current in cardiomyocytes. A and B. Top, Voltage protocol and average traces of calcium current at 0 mV in presence of G1 after DMSO or G36 plus G1 after G36 condition, respectively. Down, average data of calcium current at 0 mV and linked dot blot (inset) of each condition. C. IV plot of L-type calcium current with DMSO or G1 treatment, and voltage protocol (inset). p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
Figure 5.
Activation of GPER with G1 decrease L-type calcium current in cardiomyocytes. A and B. Top, Voltage protocol and average traces of calcium current at 0 mV in presence of G1 after DMSO or G36 plus G1 after G36 condition, respectively. Down, average data of calcium current at 0 mV and linked dot blot (inset) of each condition. C. IV plot of L-type calcium current with DMSO or G1 treatment, and voltage protocol (inset). p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.

Figure 6.
PI3K and NOS are involved in the GPER-triggered intracellular pathway. A and B. Average data of sarcomere shortening (left) and calcium transient (right) of cell treatment with wortmannin (PI3K blocker) or L-Name (NOS blocker) before exposing the cells to G1. C and D. Average traces (left) and average data (right) of calcium current of cell treated with wortmannin (PI3K blocker) or L-Name (NOS blocker) before exposing the cells to G1. p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
Figure 6.
PI3K and NOS are involved in the GPER-triggered intracellular pathway. A and B. Average data of sarcomere shortening (left) and calcium transient (right) of cell treatment with wortmannin (PI3K blocker) or L-Name (NOS blocker) before exposing the cells to G1. C and D. Average traces (left) and average data (right) of calcium current of cell treated with wortmannin (PI3K blocker) or L-Name (NOS blocker) before exposing the cells to G1. p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively.
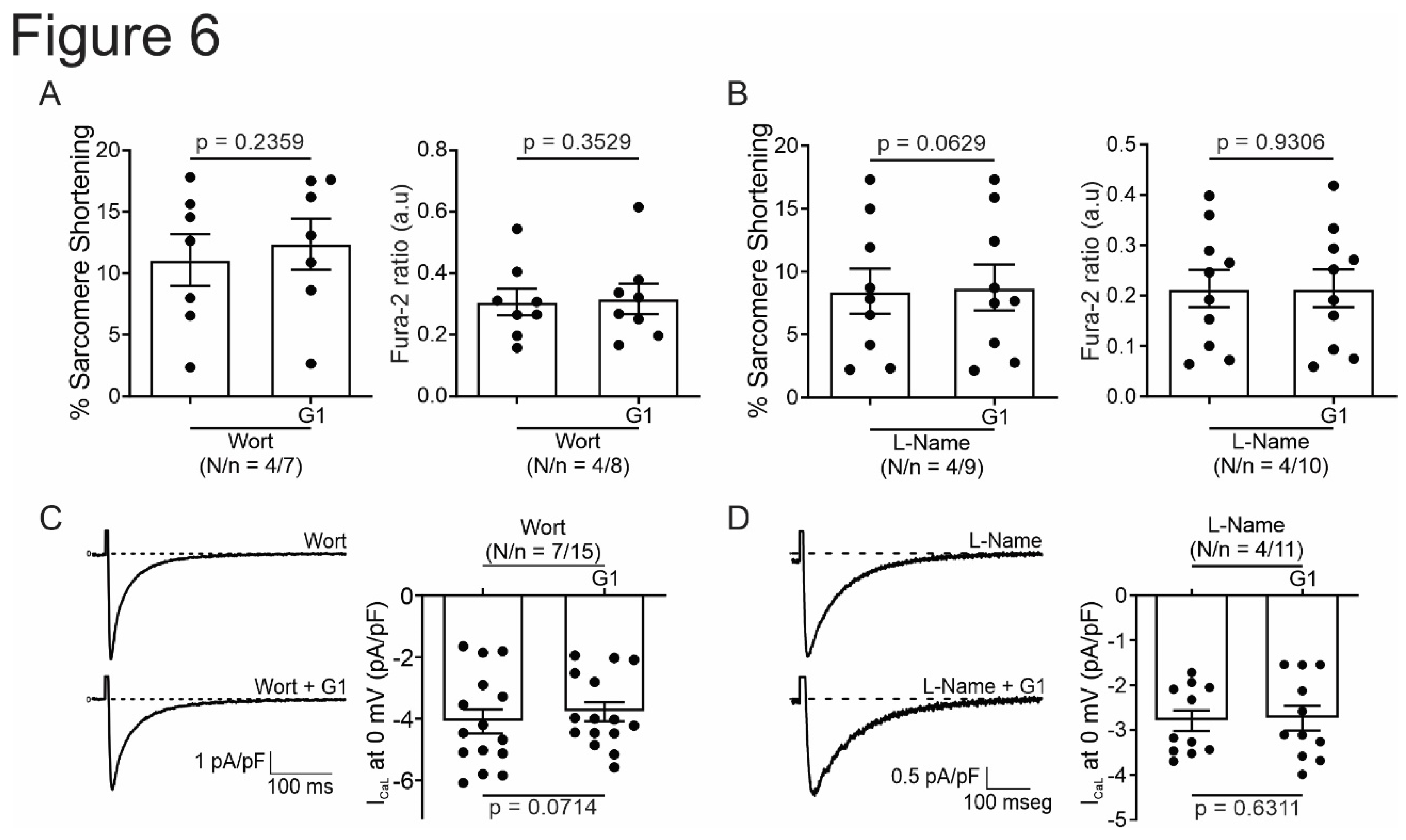
Figure 7.
GPER increases NO production activating PI3K before NOS activation. A. DAF-FM fluorescence respect to initial fluorescence of cells treated with G1 or wortmannin plus G1 after control condition (DMSO or wortmannin, respectively). B. Average slope of G1 or wortmannin plus G1. p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively. Since these values do not follow a normal distribution, Mann-Whitney test analysis was performed.
Figure 7.
GPER increases NO production activating PI3K before NOS activation. A. DAF-FM fluorescence respect to initial fluorescence of cells treated with G1 or wortmannin plus G1 after control condition (DMSO or wortmannin, respectively). B. Average slope of G1 or wortmannin plus G1. p < 0.05 indicates significantly different. N/n indicates animals and cells number, respectively. Since these values do not follow a normal distribution, Mann-Whitney test analysis was performed.
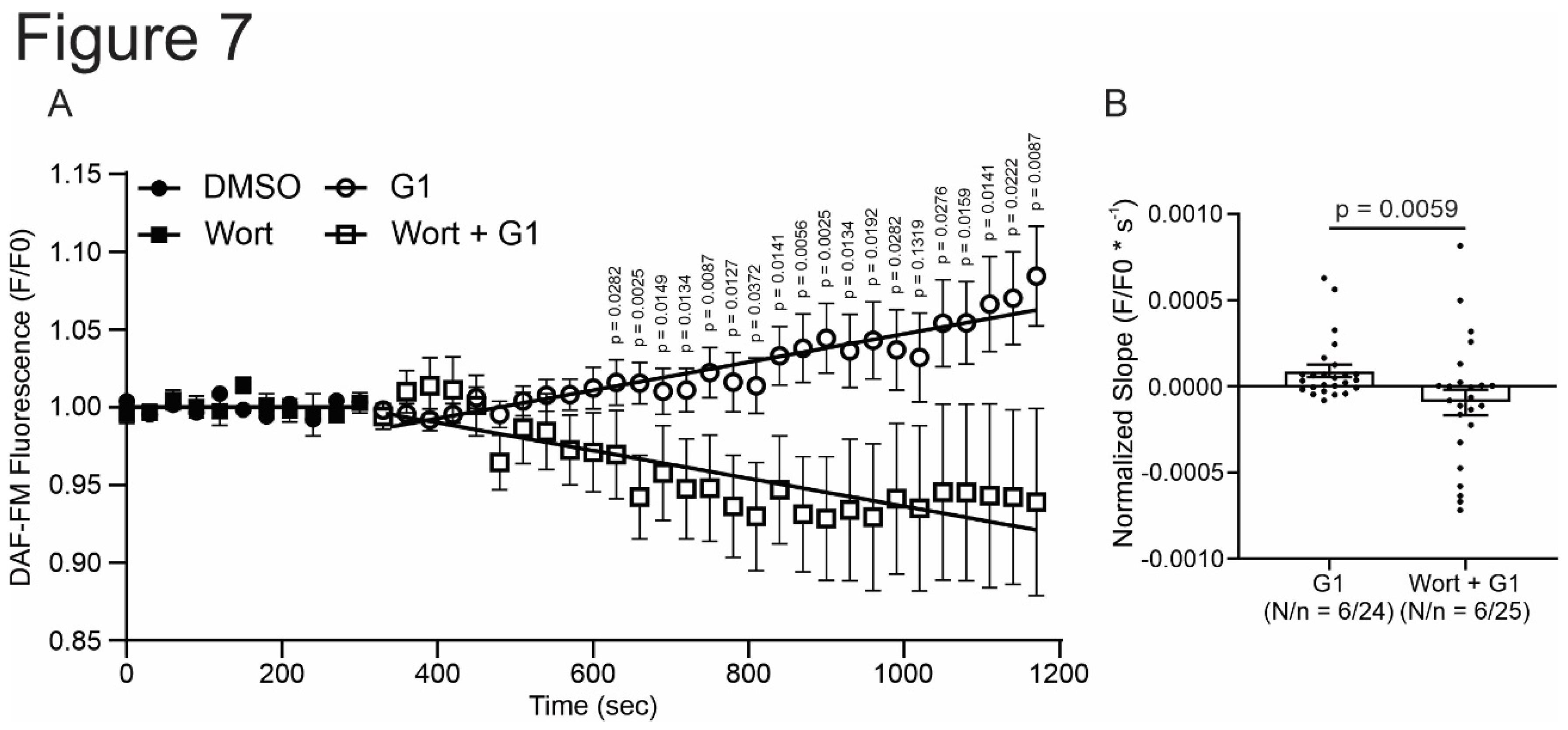
Figure 8.
Schematic summary of the molecular events proposed to underlie GPER activation. Briefly, GPER activation with E2 or G1 activates PI3K, which stimulates NOS, possibly, through AKT pathway, and then increases myocardial NO production. NO would be responsible for L-type calcium current inhibition that impact on calcium transient and, therefore, cell shortening. E2, estradiol; G1, agonist of GPER; GPER, G-protein coupled estrogen receptor; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; NOS, nitric oxide synthase; NO, nitric oxide; Wort, wortmannin, L-Name, NOS blocker; CaL, L-type calcium channel.
Figure 8.
Schematic summary of the molecular events proposed to underlie GPER activation. Briefly, GPER activation with E2 or G1 activates PI3K, which stimulates NOS, possibly, through AKT pathway, and then increases myocardial NO production. NO would be responsible for L-type calcium current inhibition that impact on calcium transient and, therefore, cell shortening. E2, estradiol; G1, agonist of GPER; GPER, G-protein coupled estrogen receptor; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; NOS, nitric oxide synthase; NO, nitric oxide; Wort, wortmannin, L-Name, NOS blocker; CaL, L-type calcium channel.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



